

Abstract
AP endonuclease-1/Redox factor-1 (APE1/Ref-1 or Ref-1) is a multifunctional protein that is overexpressed in most aggressive cancers and impacts various cancer cell signaling pathways. Ref-1′s redox activity plays a significant role in activating transcription factors (TFs) such as NFκB, HIF1α, STAT3 and AP-1, which are crucial contributors to the development of tumors and metastatic growth. Therefore, development of potent, selective inhibitors to target Ref-1 redox function is an appealing approach for therapeutic intervention. A first-generation compound, APX3330 successfully completed phase I clinical trial in adults with progressing solid tumors with favorable response rate, pharmacokinetics (PK), and minimal toxicity. These positive results prompted us to develop more potent analogs of APX3330 to effectively target Ref-1 in solid tumors. In this study, we present structure-activity relationship (SAR) identification and validation of lead compounds that exhibit a greater potency and a similar or better safety profile to APX3330. In order to triage and characterize the most potent and on-target second-generation Ref-1 redox inhibitors, we assayed for PK, mouse and human S9 fraction metabolic stability, in silico ADMET properties, ligand-based WaterLOGSY NMR measurements, pharmacodynamic markers, cell viability in multiple cancer cell types, and two distinct 3-dimensional (3D) cell killing assays (Tumor-Microenvironment on a Chip and 3D spheroid). To characterize the effects of Ref-1 inhibition in vivo, global proteomics was used following treatment with the top four analogs. This study identified and characterized more potent inhibitors of Ref-1 redox function (that outperformed APX3330 by 5–10-fold) with PK studies demonstrating efficacious doses for translation to clinic.
Keywords: Ref-1 redox function, Novel targeted inhibitors, Potency, Nuclear Factor kappa B (NFκB), Hypoxia Inducible Factor 1 (HIF1α), Cancers, PDAC, MPNST
1. Introduction
Cancer is the second most common cause of death in the United States with a total of 1,958,310 new cancer cases and 609,820 deaths expected in 2023. Although the cancer death rate is reported to have declined in the past 28 years due to early diagnosis, chemotherapy before or after surgery, and combination treatments, there are several cancers in which progress has been rather stagnant including soft tissue sarcomas (STSs) and pancreatic cancer [1–5]. In the past decade, approximately 40 new targeted therapies have been approved by the FDA for 12 different cancers, but only a small percentage of patients are eligible for such targeted therapies [6]. This is due to several factors such as slow drug development process, cancer genome heterogeneity, drug resistance, druggability gap, and cost of treatment [7,8].
Cancer progression and metastasis is regulated by altered transcriptional networks through gain- or loss- of function in critical signaling pathways, genomic deletions and mutations, or epigenetic modifications [9,10]. Dysregulation of gene expression by activation of transcription factors (TFs) frequently occurs in cancer and plays a role in the production of many onco-proteins [11,12]. Aggressive cancers like pancreatic ductal adenocarcinoma (PDAC) [13,14], breast cancer (BC) [15,16], and malignant peripheral nerve sheath tumor (MPNST) [17] are characterized by a hypoxic, nutrient poor and dense inflammatory stroma [18]. NFκB (nuclear factor κB), HIF-1α (hypoxia inducible factor-1α), AP-1 (activator protein-1), TGFβ / SMADs (transforming growth factor β / Suppressor of Mothers against Decapentaplegic) are some of the TFs that contribute to these characteristics as well as to the crosstalk between tumor and its microenvironment (TME) [19,20]. Due to their role in maintaining the transformed cancer phenotype and sustained crosstalk within the TME, these TFs and the proteins that regulate them offer potential nodes for therapeutic intervention. Even though direct blockade of TF activity is a viable therapeutic approach, intensive crosstalk and redundancy among TFs poses a challenge. Therefore, targeting upstream regulators of the TFs and thereby effecting downstream transcriptional networks may have application in the clinical treatment of aggressive cancers [10,11,21].
AP endonuclease-1 / Redox factor-1 (APE1/Ref-1 or Ref-1) is one such regulatory protein that is upstream of numerous TFs such as NFκB, HIF1α, STAT3, AP-1 (Fos/Jun) and stimulates their DNA binding activity [22,23]. Ref-1, a multifunctional protein with redox signaling and DNA repair as its major functions, and additional activities including RNA processing, DNA maintenance, RNA quality control, miRNA metabolism, regulation of stress-response gene expression and mitochondrial DNA (mtDNA) repair is dysregulated in cancer cells [23]. Its endonuclease function is the rate-limiting step in the Base Excision Repair (BER) pathway and is considered indispensable for cell survival [24]. As a redox factor, Ref-1 reduces the cysteine (Cys) residues within the TFs in a thiol-mediated reaction. Ref-1 is an unusual redox factor in that, it must adopt a partially unfolded state in order to expose Cys65, which is critical for its redox activity and does not have a consensus sequence as most other redox proteins do [25–28].
Ref-1 plays a role in many aspects of cancer signaling including transcription, response to oxidative stress, and metabolism and is overexpressed in multiple aggressive cancers like PDAC, MPNST, breast, prostate, and others. Ref-1 is expressed in both stromal as well as tumor cells with its expression being higher in tumor cells [29–31]. The DNA repair function of Ref-1 is independent of the redox function and therefore, development of small molecules that target only its redox function alone is feasible for cancer therapy. We have been pursuing Ref-1 as a target in pancreatic cancer and MPNST, based on the role of hypoxia and HIF signaling in both diseases [32,33]. For example, parent compound APX3330 is a Ref-1 redox inhibitor that successfully completed phase I clinical trial with a 32% response rate, no significant toxicities, a favorable PK, predicted target engagement, and an identified phase II dose. Patient biopsies showed a decrease in genes regulated by Ref-1-activated TFs like NFκB, HIF1α, STAT3 and AP-1 [34–36]. While the phase I data is encouraging, potency of APX3330 in preclinical models is 50–75 μM in cell-based proliferation assays. Therefore, identification of second-generation compounds with increased efficacy and potency that also demonstrate a similar safety profile as APX3330 is of great interest. Through a structure-activity relationship (SAR) study, chemical modifications of second-generation naphthoquinones have led to the identification of 13 second-generation Ref-1 inhibitors out of more than 350 molecules.
Identified lead compounds were further evaluated for chemical and metabolic stability, improved drug-like properties, pharmacokinetics (PK), mouse and human S9 fraction metabolic stability, in silico ADMET (absorption, distribution, metabolism, and excretion–toxicity) properties, and WaterLOGSY NMR for ligand interaction. Through these studies, four compounds were prioritized for target engagement and efficacy. Using both pancreatic cancer and MPNST cells, transcription factor driven luciferase activity and mitochondrial plate-based assays were used to analyze Ref-1 redox specific inhibition [32]. Further, cytotoxicity in monolayer and 3D co-culture assays and in vivo tumor proteomics were employed to identify most effective second-generation Ref-1 redox inhibitors.
Collectively, our data indicates that the performance characteristics of these Ref-1 inhibitors meet and exceed the critical success factors for advancement to preclinical toxicology studies. Hence, these novel oral Ref-1 inhibitors offer promising strategies for the treatment of not only cancer, but also other diseases where Ref-1 has been implicated as a therapeutic target, e.g., ocular and inflammatory bowel disease (IBD) [37].
2. Methods
2.1. Synthesis of second-generation Ref-1 inhibitors
2.1.1. Scheme 1. Synthetic scheme for APX2009, APX2014, and APX2053
Scheme 1.
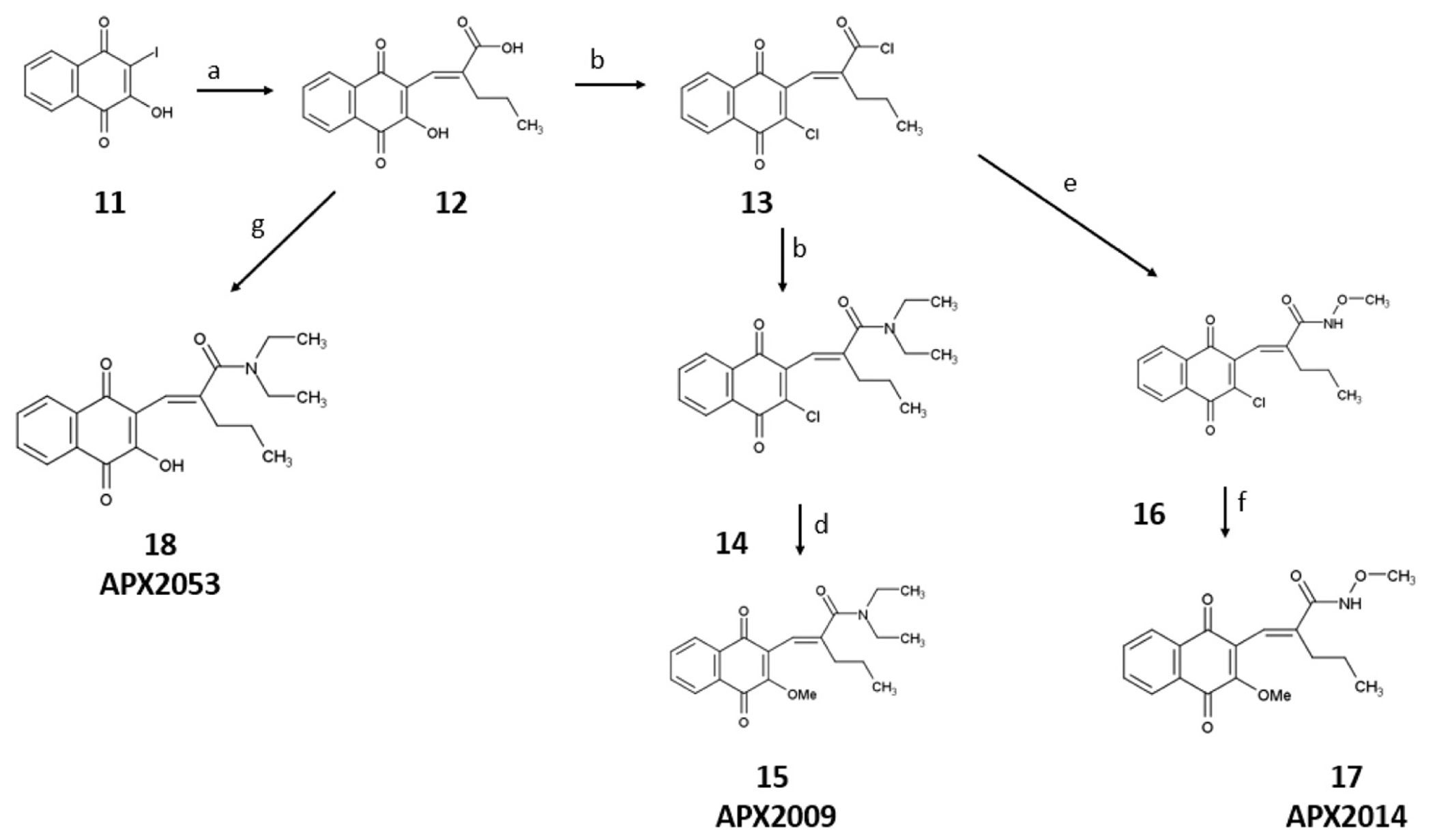
Reagents and conditions for the synthesis of APX2009, APX2014, and APX2053: a) 2-propylacrylic acid, potassium carbonate, palladium(II)acetate, argon, 100 °C,1 h, 72%; b) dimethylformamide, oxalyl chloride, room temperature, 18 h, 100% crude; c) dichloromethane, diethylamine hydrochloride, diisopropylethylamine, room temperature, 15 min, 62%; d) methanol, sodium methoxide, argon, 30 min, 96%; e) dichloromethane, methoxyamine HCl, diisopropylethylamine, argon, room temperature, 30 min, 71%; f) methanol, sodium methoxide, argon, 30 min, 83%; g) dichloromethane, argon, diethylethylamine, 4-dimethylaminopyridine, 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide, reflux, 4 h, 8%.
2.1.2. Scheme 2. Synthesis of APX2051 and APX2044
Scheme 2.
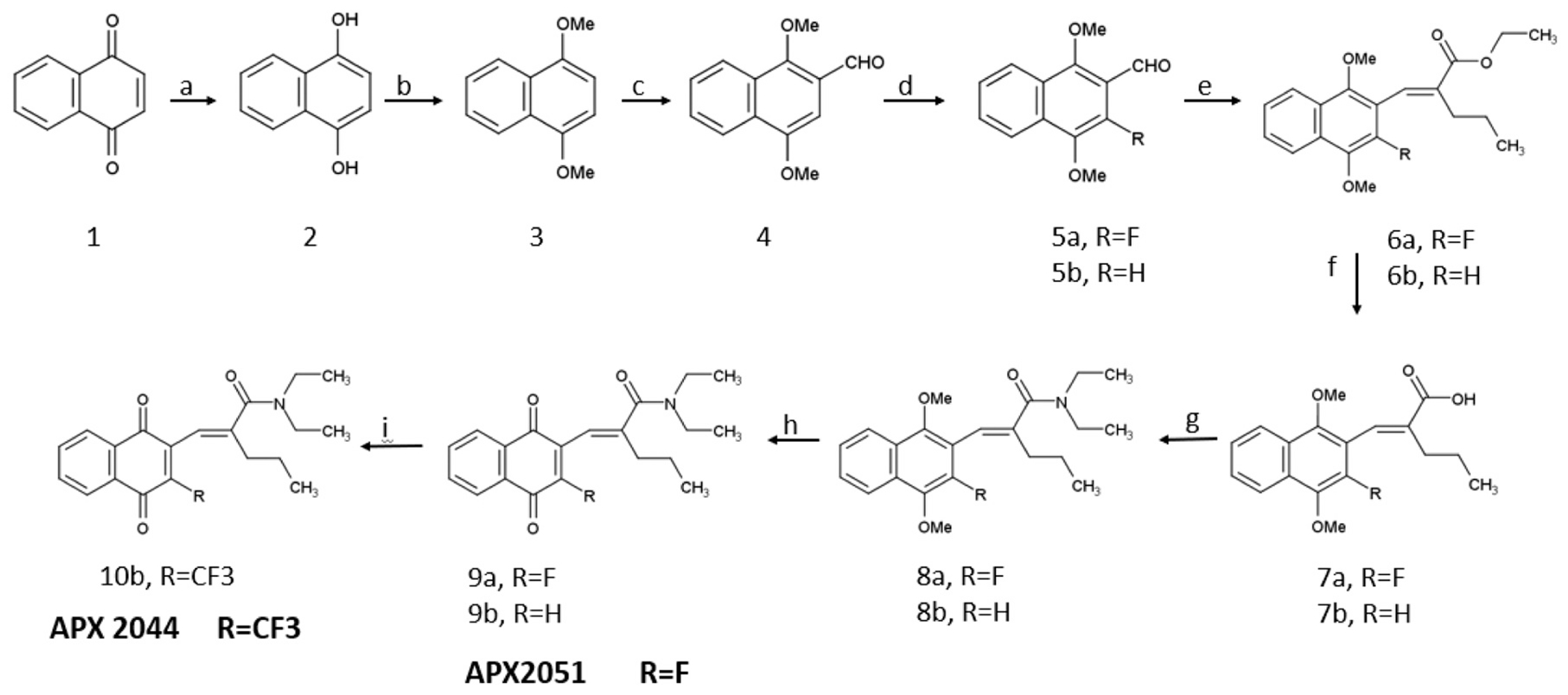
Reagents and conditions for the synthesis of APX2044 and APX2051: a) diethyl ether, sodium dithionite, room temperature, 6 h, 100% crude; b) dimethylformamide, potassium carbonate, room temperature, 30 min, methyl iodide; room temperature, 20 h, 53%; c) chloromethyl methyl ether, dichloromethane, tin(IV) tetrachloride; 0 °C, 1 h, room temperature, 16 h, 46%; d) acetonitrile, Selectfluor, 90 °C, 8 h, 23%; e) sodium hydride, ethyl-2-(diethoxy-phosphoryl)-pentanoate 19, 60 °C, 8 h, 41% (6a) and 53% (6b); f) sodium hydroxide, water, ethanol, 100 °C, 6 h, 98% (7a) and 76% (7b); g) dichloromethane, HATU (Hexa-fluorophosphate Azabenzotriazole Tetramethyl Uronium), diisopropylethylamine, diethylamine, room temperature, 3 h, 51% (8a) and 72% (8b); h) nitric acid, silver oxide, acetic acid, ethyl acetate, room temperature, 1 h, 48% (9a) and 70% (9b); i) copper iodide, TEMPO (2,2,6,6-tetramethylpiperdin-1-yl)oxidanyl), 1-(Trifluoromethyl)– 1,2-benziodoxol-3(1 H)-one (Togni Reagent II), argon, t-butanol, dichloromethane, 4%.
2.1.3. Scheme 3. Synthetic scheme for intermediates
Scheme 3.
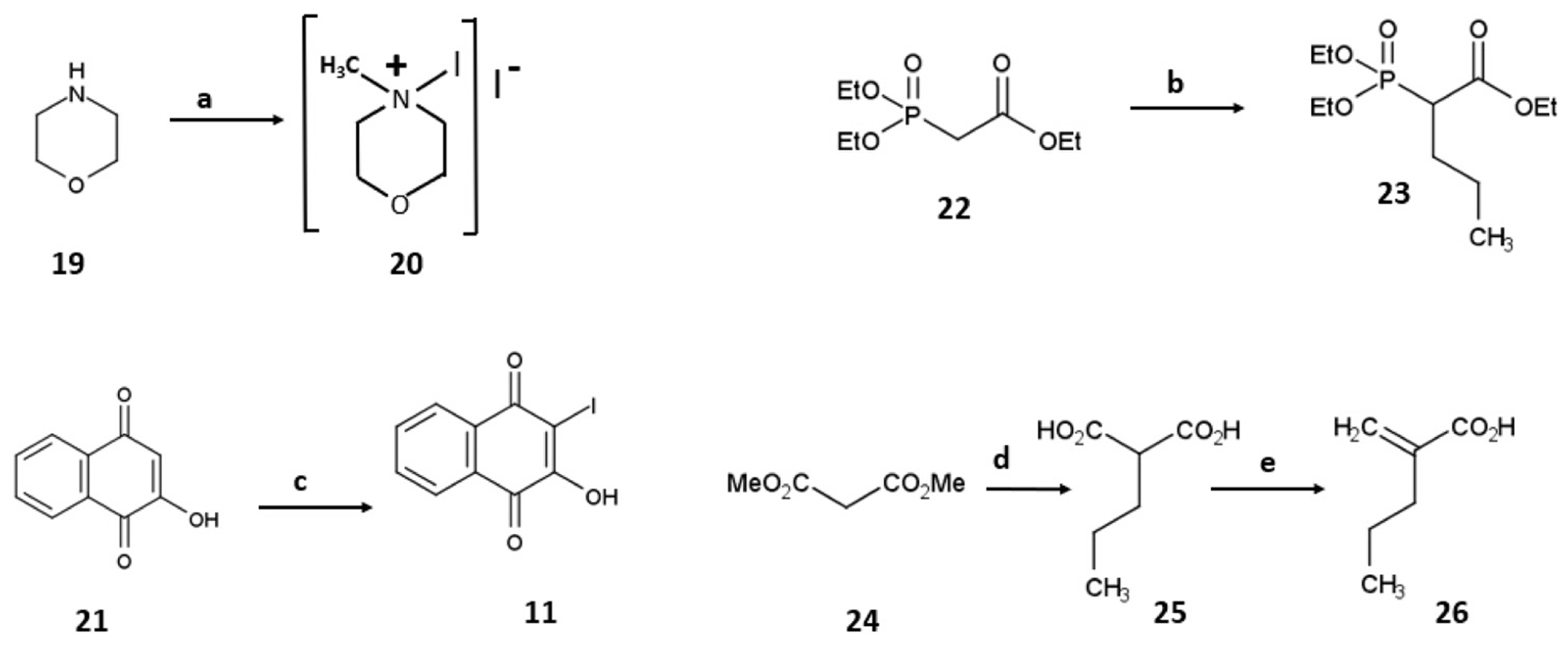
Reagents and conditions for the synthesis of APX2009, APX2024, and APX2053: a) morpholine, methanol, argon, iodine, room temperature, 2 h, 72%; b) dimethyl sulfoxide, potassium t-butoxide, 0 °C, 30 min, n-propyl iodide, room temperature 60 °C, 2 h, 84% (crude); c) water, argon, potassium carbonate, 4-iodomorpholin-4-ium iodide complex 20, room temperature, 1 h, then phosphoric acids, room temperature, 24 h, 82%; d) sodium hydride, tetrahydrofuran, dimethyl malonate, room temperature, 30 min, then 2 M sodium hydroxide, reflux, 4 h, 33%; e) ethyl acetate, diethylamine, formaldehyde, room temperature, 4 h, 74%.
2.2. PK studies
2.2.1. Standard curve and quality control samples
Solutions of APX2009, APX2014, APX2044, APX2051, and APX2053 were prepared separately in polypropylene tubes by adding DMSO: Methanol (50:50; v/v) to create a 1 mg/mL solution. A solution of temazepam was prepared in a polypropylene tube by adding methanol to create a 1 mg/mL solution. All solutions were stored at − 20 °C. The standard curve and QC samples were prepared daily from methanolic dilutions of the 1 mg/mL stock solutions. These dilutions were mixed with non-supplemented plasma (total volume of 200 mL) to prepare the standard curve and QC samples. The standard curve for all APX compounds was: 0, 1, 3, 10, 30, 100, 300, 1000, 3000, and 10000 ng/mL in plasma. The QC samples had the following concentrations: 3 ng/mL (low QC), 100 ng/mL (medium QC), and 8000 ng/mL (high QC).
2.2.2. Standard curve for thermodynamic solubility
Solutions of APX2009, APX2014, APX2044, APX2051, and APX2053 were prepared separately in polypropylene tubes by adding DMSO: Methanol (50:50; v/v) to create a 1 mg/mL solution. A solution of temazepam was prepared in a polypropylene tube by adding methanol to create a 1 mg/mL solution. All solutions were stored at − 20 °C. The standard curve was prepared daily from methanolic dilutions of the 1 mg/mL stock solutions. These dilutions were mixed with acetonitrile (total volume of 200 mL) to prepare the standard curve. The standard curve for all compounds was: 0, 100, 300, 1000, 3000, 10000, 30000, 100000, 300000, and 1000000 ng/mL in acetonitrile.
2.2.3. Conditions for HPLC-MS/MS
Chromatographic analysis was performed using a Sciex Exion UHPLC and Sciex 6500 + QTRAP MS/MS. Reverse phase chromatography was achieved at ambient temperature using an Inertsil ODS-2 150 × 4.6 mm 5 μm column. The mobile phase consisted of 0.2% formic acid and acetonitrile and was delivered via gradient at a flow rate of 600 μL/min for 13 min. The column effluent was monitored using a Sciex 6500 + QTRAP triple-quadrupole mass spectrometer (Framingham, MA) equipped with an electrospray probe in positive ionization mode. It was controlled by Analyst software version 1.7.2. in conjunction with Windows 10 Enterprise LTSC. A flow injection analysis was performed, after the standard infusions, on each analyte to maximize sensitivity. The analytes were optimized at a source temperature of 450 °C, under unit resolution for quadrupole 1 and 3, and were given a dwell time of 100 msec and a settling time of 0 msec. The ion spray voltage was 4500 V and the interface heater was on. Optimal gas pressures for all three analytes were: collision gas medium, curtain gas 20, ion source gas (1) 50, ion source gas (2) 40. Multiple reaction monitoring was used to measure Q1/Q3 transitions. Mass spectrometry settings for the Q1/Q3 transitions and voltages are listed in (Table 1).
Table 1.
Mass spectrometry settings for the Q1/Q3 transitions and voltages.
| ID | Q1 Mass | Q3 Mass | DP (volts) | EP (volts) | CE (volts) | CXP (volts) |
|---|---|---|---|---|---|---|
| APX2009 | 356.318 | 283.1 | 70 | 10 | 25 | 25 |
| APX2009 HQ | 358.318 | 285.1 | 70 | 10 | 25 | 25 |
| APX2014 | 330.239 | 283.0 | 60 | 12 | 20 | 30 |
| APX2014HQ | 332.239 | 285.0 | 60 | 8 | 20 | 20 |
| APX2044 | 394.029 | 321.0 | 30 | 4 | 25 | 18 |
| APX2044HQ | 396.029 | 323.0 | 30 | 4 | 30 | 18 |
| APX2051 | 344.044 | 243.1 | 20 | 4 | 30 | 18 |
| APX2051HQ | 346.044 | 245.1 | 20 | 4 | 30 | 18 |
| APX2053 | 342.136 | 269.0 | 101 | 10 | 21 | 14 |
| APX2053HQ | 344.136 | 271.0 | 101 | 10 | 21 | 14 |
| Temazepam | 301.008 | 255.1 | 80 | 10 | 15 | 5 |
2.2.4. Sample preparation for PK analysis
Frozen plasma samples (stored in a −80 °C freezer) were thawed to ambient temperature. A 20 μL aliquot of the sample was placed in a 12 × 75 mm polypropylene tube. A volume of 20 μL of 0.1 ng/μL of internal standard, temazepam, was added to the tube along with 2 mL of ethyl acetate. Only APX2053 samples had 50 μL of citric acid buffer pH 3.0 added. The tube was vortex mixed for 20 s. After centrifugation at 3000 rpm at ambient temperature for three minutes, the upper organic phase was transferred to a clean 12 × 75 polypropylene tube and evaporated to dryness. A 200 μL volume of acetonitrile was added to each tube and the tube was mixed for 20 s on a vortex mixer. A 5 μL aliquot of each sample was injected into the HPLC.
2.3. Metabolic stability, plasma stability and thermodynamic stability studies
2.3.1. Sample preparation for phase 1 metabolic stability
Human liver S9, mouse liver S9 fragments, human liver microsomes, and mouse liver microsomes were thawed on ice. Fresh solutions of 5 mM MgCl2 in phosphate buffer pH 7.4 and 4 mM NADPH in phosphate buffer pH 7.4 with 5 mM MgCl2 were prepared. A 1 μM aliquot of compound was added to a 1.1 mL microtube in a 96-well plate and were evaporated to dryness under nitrogen digital dry bath. A 10 μL aliquot of human liver S9, mouse liver S9 fragments, human liver microsomes, or mouse liver microsomes (20 mg/mL) was added to the tubes along with 140 μL of 5 mM MgCl2 in phosphate buffer pH 7.4. Samples pipetted gently to mix. Reaction was initiated by the addition of 50 μL of 4 mM NADPH in phosphate buffer pH 7.4 with 5 mM MgCl2. Samples were shaken at 400 rpm and incubated for 0, 5, 10, 20, 40, and 60 min. After 0, 5, 10, 20, 40, and 60 min, 700 μL of ice-cold acetonitrile was added to stop the reaction. A volume of 20 μL of 0.1 ng/μL of internal standard, Temazepam, was added to the tube. Tubes were shaken for 30 s at 2400 rpm. After centrifugation at 4000 rpm at ambient temperature for three minutes, 450 μL of supernatant were transferred to a 96-well plate. Samples were dried under nitrogen digital dry bath until dryness. A 100 μL volume of acetonitrile was added to each well and the plate was shaken at 2400 rpm for one minute. A 5 μL aliquot of each sample was injected into the HPLC.
2.3.2. Sample preparation for phase 2 metabolic stability
Human liver S9 and mouse liver S9 fragments were thawed on ice. UGT reaction mix solution (A&B) was prepared. A 1 μM aliquot of compound was added to a 1.1 mL microtube in a 96-well plate and were evaporated to dryness under nitrogen digital dry bath. A 10 μL aliquot of human liver S9 or mouse liver S9 fragments was added to the tubes along with 190 μL of UGT mix. Samples pipetted gently to mix. Samples were shaken at 400 rpm and incubated for 0, 5, 10, 20, 40, and 60 min. After 0, 5, 10, 20, 40, and 60 min, 500 μL of ice -cold acetonitrile was added to stop the reaction. A volume of 20 μL of 0.1 ng/μL of internal standard, temazepam, was added to the tube. Tubes were shaken for 30 s at 2400 rpm. After centrifugation at 3000 rpm at ambient temperature for five minutes, 450 μL of supernatant were transferred to a 96-well plate. Samples were dried under nitrogen digital dry bath for two hours. A 100 μL volume of acetonitrile was added to each well and the plate was shaken at 2400 rpm for one minute. A 5 μL aliquot of each sample was injected into the HPLC.
2.3.3. Sample preparation for plasma stability
Mouse and human plasma were pre-warmed in 37 °C water bath. A 1 μM aliquot of compound was added to a 1.1 mL microtube 96-well plate and evaporated to dryness under nitrogen digital dry bath. The 96-well plate was placed on a 37 °C heat block and 200 μL of prewarmed plasma was added to each tube. After 0, 5, 10, 20, 40, and 60 min, 500 μL of cold acetonitrile was added to stop the reaction. A volume of 20 μL of 0.1 ng/μL of internal standard, temazepam, was added to the tube. Tubes were shaken for 30 s at 2400 rpm. After centrifugation at 3000 rpm at ambient temperature for five minutes, 450 μL of supernatant were transferred to a 96-well plate. Samples were dried under nitrogen digital dry bath for two hours. A 100 μL volume of acetonitrile was added to each well and the plate was shaken at 2400 rpm for one minute. A 5 μL aliquot of each sample was injected into the HPLC.
2.3.4. Sample preparation for thermodynamic solubility
FaSSIF, FeSSIF, and FaSSGF solutions were prepared as directed by Biorelevant and heated to 37 °C in water bath. FaSSIF, FeSSIF, or FaSSGF solution was added to APX compounds to create a 1 mg/mL sample. All samples were shaken at 40 rpm in a 37 °C water bath for 24 h. At 24 h, samples were spun at 20 g for 3 min. Samples were filtered through a Whatman miniprep filter. A 10 μL aliquot of the sample was placed in a 12 × 75 polypropylene tube. A volume of 10 μL of 100 ng/μL of internal standard, temazepam, was added to the tube along with 980 μL of acetonitrile. The tube was vortex mixed for 20 s. Samples further diluted by adding 20 μL to 180 μL of acetonitrile and vortexed to mix. A 5 μL aliquot of each sample was injected into the HPLC.
2.4. WaterLOGSY NMR studies
All NMR spectroscopy was performed at Indiana University School of Medicine on a Bruker Avance-III-600 NMR spectrometer operating at 600.2 MHz (1 H), 61.081 MHz (15 N), and 150.9 MHz (13 C) and equipped with a TCI cryoprobe. Experiments were conducted at 303 K in 5 mm diameter NMR tubes with a sample volume of 500 μL. Compounds (APX2009, APX2014, APX2044, APX2051, APX2053, and RN7–58) were prepared as 50 mM stock solutions in CD3CN, except for APX2051, for which a 25 mM stock was prepared due to limited solubility. Solutions were buffered in 20 mM sodium phosphate, 0.1 M NaCl pH 6.5 with 90% H2O and 10% D2O. WaterLOGSY experiments were done with 20 μM Ref-1 (39–318/C138A) and 200 μM compound for each experiment. The mixing time was set to 1.5 s, and data were collected with 128 scans, a sweep width of 10 ppm, an acquisition time of 1.58 s, and a relaxation delay 5 s. Prior to Fourier transformation, the data were multiplied with an exponential function with a line broadening of 1 Hz.
2.5. Cell culture
Pancreatic cancer patient-derived tumor cell line, Pa03C, and cancer-associated fibroblast cell lines, CAF19 (from Dr. Anirban Maitra at The Johns Hopkins University), and CAF2 were isolated using the outgrowth method and were then transduced with TdTomato for tumor cells or EGFP for CAFs as previously described [38,39]. Pa08x cell line was developed in house (Indiana University Simon Comprehensive Cancer Center) as a xenoline from PDAC patient-derived xenograft as described in Supplement. MPNST cell line NF90–8 was received from Dr. Verena Staedtke (Johns Hopkins University). R-HT163 and R-HT172 cell lines were generated in the lab (Indiana University Simon Comprehensive Cancer Center) as xenolines from MPNST patient-derived xenografts as described in Supplement. Cells were maintained at 37 °C in 5% CO2 and grown in DMEM (Invitrogen; Carlsbad, CA) with 10% FBS (Hyclone; Logan, UT), or under hypoxic conditions of 1% O2 / 5% CO2 using a Ruskinn Invivo2 200 hypoxia workstation. Cell line identity was confirmed by DNA fingerprint analysis (IDEXX BioResearch, Columbia, MO) for species and baseline short-tandem repeat analysis testing. Cell lines were 100% human, and a nine-marker short tandem repeat analysis exists on file. They were also confirmed to be mycoplasma free and used at a passage below 10 for the low passage tumor cells and below 20 for the immortalized CAFs.
2.6. Luciferase reporter assay
The pGreenFire 2.0 NFκB or HIF1α reporter viruses (TR412VA-P & TR426VA-P) and negative control Lentivirus (TR411VA-P) were purchased from System Biosciences and transduced into Pa03C and NF90–8 cells as per manufacturer’s protocol. Following selection in puromycin, cells were sorted for high GFP expression and confirmed for luciferase activity following induction with TNF-α or hypoxia, respectively. The GFP-luciferase expressing cells were seeded and 24 h later treated with increasing doses of Ref-1 inhibitors for 24 h and assayed for cell death or luciferase activity, using alamarBlue™ (Invitrogen, Waltham, Mass) or a Promega One-Glo luciferase assay system (Promega # E4550). Basal activity was reported for NFκB and for HIF-1α, normoxic versus hypoxic (1% O2 / 5% CO2) conditions were assayed. Reporter activity following treatment was normalized to cells with no treatment (media only) for NFκB and to cells under normoxia for HIF1α.
2.7. Reverse transcription and real time PCR
Total RNA was extracted using Qiagen RNeasy Mini kit (Qiagen, Valencia, CA) and reverse-transcribed into cDNA using random hexamers and MultiScribe reverse transcriptase (Applied Biosystems, Foster City, CA) according to manufacturer’s protocol. Survivin gene expression was assayed for Ref-1 inhibitor treated Pa03C and NF90–8 cells using SYBR Green Real Time PCR master mix (Applied Biosystems, Foster City, CA) in a CFX96 Real Time detection system (BioRad, Hercules, CA). The reaction conditions used for qPCR were as published in Gampala et al. [32]. Quantitative analysis was performed using comparative Ct method using actin as the reference gene. The primer sequences (5′ to 3′) used are: Survivin Fw: CCACTGAGAACGAGCCAGACTT and Rv: GTATTACAGGCGTAAGCCACCG; Actin Fw: CACCATTGGCAATGAGCGGTTC and Rv: AGGTCTTTGCGGATGTCCACGT.
2.8. Mitochondrial plate-based assay
S-1 Mitoplates (Biolog, Hayward, CA) were used to investigate mitochondrial function as previously described [32]. Assays were performed as per manufacturer’s protocol. Briefly, plates were activated by adding the Assay Mix to the wells to dissolve the substrates, for at least 60 min at 37 °C. Following drug treatment (24 h), cells were collected, counted, resuspended in provided buffer and plated at 5 × 104 cells/well. Cells were added to the plate, which was immediately read at 590 nm kinetically at 5 min intervals for 4 h at 37 °C. Data was analyzed using Graphpad Prism 8, statistical significance was determined using the 2-way ANOVA and p-values < 0.05 were considered statistically significant.
2.9. NADP/NADPH assay
The NADP/NADPH Assay Kit (ab65349) from Abcam was utilized to measure the NADPH to NADP ratio in Pa03C cells as a readout of general redox state. The assay followed the manufacturer’s instructions. Briefly, cytoplasmic NADPH/NADP+ was extracted from 4 × 106 cells using 400 μL of the provided extraction buffer after treatment, processed and analyzed for NADPH or total NADP as described in our previous publication [32].
2.10. Kinetic fluorescence based APE1 endonuclease assay
The Ref-1 redox inhibitors were tested by an established APE1 endonuclease activity assay to determine if the Ref-1 redox inhibitors would affect the efficiency of APE1 to repair DNA. Briefly, APE1 endonuclease activity was measured by a fluorescent signal following cleavage 5′ of the apurinic/apyrimidinic (AP) site mimic tetrahydrofuran (THF) that subsequently released a measurable fluorescein-labeled product from the following annealed oligo substrate (Eurogentec, San Diego, CA):
5′ -(6-FAM)-GAATCC-(THF)-CCATACGTATTATATCCAATTCC- 3’ and.
5′ -GGAATTGGATATAATACGTATGGTGGATTC-(DABCYL)– 3′.
Prior to cleavage, the fluorescent signal was quenched due to its proximity to dabcyl in the complementary strand. APE1 enzyme (Georgiadis, IUSM) was optimized at concentrations of 0.07–0.1 nM before each test to produce a linear signal (495/530 nM) at 37 °C over a 5 min kinetic read on a Synergy H1 fluorescent plate reader (BioTek, Winooski, VT). The second-generation Ref-1 inhibitors were assayed at 20 μM concentration representing a dose that resulted in cell killing based on the viability assays of the new compounds. The rate of reaction for APE1 alone represented the maximum repair capacity, while an APE1 endonuclease repair inhibitor, APE1 Inhibitor III (Ari3; Millipore Sigma, Burlington, MA) represented specific repair inhibition, also at 20 μM. All compounds were tested in triplicate per test and replicated (N = 2). The averaged Vmax was normalized to APE1-alone Vmax.
2.11. Cell cytotoxicity and Ref-1 inhibitor screening
Patient-derived MPNST and pancreatic cancer cells as well as CAFs maintained in 10% FBS DMEM growth medium (Gibco™, Thermo-Fisher, Waltham, Mass) were plated at 2000 cells per well in poly-D-lysine treated 96-well plates (TPP®, Trasadingen, CH) and allowed to attach overnight. Cells were then treated with Ref-1 inhibitors, in triplicate, made up in DMEM medium containing 5% FBS at increasing concentrations serially diluted in a 5-point dose scheme. Cells were treated for 48–72 h in 5% CO2 at 37 °C, and a fluorescent metabolic viability indicator, alamarBlue™ (Invitrogen, Waltham, Mass) was added to each well at 10% final. After a 5 h incubation, plates were read on a Synergy H1 fluorescent plate reader (BioTek, Winooski, VT). For each drug dose, the relative fluorescence units (RFU) were averaged, and the background subtracted for potential autofluorescence. RFU was normalized to the media control wells for a final percent viability. The Ref-1 inhibitors were tested at least three times (N = 3), and IC50 statistical analysis was determined by utilizing the Cho method (Research Productivity Solutions®, H. Shannon) to ascertain an absolute IC50 for comparison.
2.12. 3D Co-culture assays
Ultra-low attachment 96-well plates (Corning Inc., Life Sciences) were used to generate 3-dimensional tumor spheroids in the presence of CAFs as reported in the results and as described previously [32]. Following plating, cells were treated on Days 4, 8, and 12 with media containing 5% serum, 3% growth factor reduced Matrigel, and inhibitors as indicated in the figure legends. On Days 4, 8, 12, and 14, spheroids were analyzed for fluorescence intensity using Thermo ArrayScan high-content imaging system. Images of 3D structures were captured by ArrayScan using a 2.5x objective for TdTomato and EGFP; then 2D projections were processed to quantify differences in total intensity of both CAFs and tumor.
2.13. Tumor-microenvironment-on-chip (T-MOC) assay
The tumor-microenvironment-on-chip (T-MOC) is a 3D in vitro microfluidic platform composed of three channels – an interstitial and two side channels. The interstitial channel was filled with cell-collagen mixture fulfilling the 3D culture condition while the side channels were filled with the culture medium. Details of fabrication and preparation were described previously [40]. Briefly, pancreatic cancer cells (Pa03C) and cancer-associated fibroblasts (CAF19) were mixed at a 1:1 cell ratio into the cell-collagen mixture. The initial cell concentration was 1.5 × 106 cells/mL for each cell type. After loading, the devices were incubated at 37 °C for 1 h for collagen gelation. Then, the culture medium was introduced through the side channels. The loading process was controlled by pressurizing the channels.
In order to assess drug efficacy, the PDAC T-MOC was cultured for 48 h before treatment. On day 0, drugs were perfused through the side channels. The drug solutions were prepared as 0 μM (control), 3, 10 and 30 μM of inhibitors in the culture medium. The channels were reperfused with the drug-containing medium every day for 6 days. The drug efficacy was evaluated on day 6 in two ways, cell growth and cell survival as detailed in the supplementary methods.
2.14. In vivo xenograft study
All animal studies were conducted under the guidelines of the National Institutes of Health and were approved by the Institutional Animal Care and Use Committee of Indiana University School of Medicine (approval Number: 21165). 4–6 week old male NSG (NOD.Cg-Prkdc scid Il2rg tm1Wjl/SzJ (NOD/SCID7γ(−/−))) mice were obtained from the In Vivo Therapeutics Core within the Preclinical Modeling and Therapeutics Core of the Indiana University Simon Comprehensive Cancer Center. Animals were maintained under pathogen-free conditions and a 12 h light-dark cycle. NSG mice were subcutaneously implanted with 2.5 × 106 Pa03C cells in the hind flank using a 200 μL volume of 50:50 solution of Matrigel:DMEM medium. When tumor volumes reached ~250 mm3, the mice were randomized into 5 groups of 5 mice before commencing treatment. The treatment regimen consisted of intra-tumoral administration of either vehicle (DMSO) or APX3330 (50 μM) or APX2009 (20 μM) or APX2014 (10 μM) or APX2051 (20 μM) for three times (8 h apart). 4 h after the third treatment, mice were sacrificed and tumor volumes and weights were noted. Flash frozen tumor samples were processed for proteomics.
2.15. Quantitative global proteomic comparison of protein levels
Sample preparation, mass spectrometry analysis, bioinformatics, and data evaluation for quantitative proteomics and phospho-proteomics experiments were performed in collaboration with the Indiana University Proteomics Center for Proteome Analysis at the Indiana University School of Medicine similar to previously published protocols [41].
Differential expression test via generalized linear model: Noting the proteomics data collected from three batches, a generalized linear model-based test was applied to identify the over or under expressed proteins and handle potential batch effects.
Specifically, the following linear model was utilized:
where y, , , , and ε represent gene expression, parameter of the treatment effect, Ref-1 inhibitor treatment (1) vs control (0), fixed effect led by batches and errors, respectively. Proteins show consistent differential expression between the four Ref-1 inhibitor treated cells vs DMSO controls and were tested by F test of the significance of parameter α. The glm(family=“binomial”) in R was implemented for the test.
Pathway enrichment analysis was conducted by using hypergeometric test against the MsigDB v6 canonical pathway set [42]. P < 0.05 was used as a significant cutoff. GSEA plot was utilized for visualization of significantly over- and under-expressed pathways.
2.16. Statistics
All experiments were conducted independently at least three times. To determine significance, either 2-way ANOVA (Luciferase reporter assay, mitochondrial plate-based assay, cytotoxicity assay and 3D spheroid assay) or one-way ANOVA (NADP assay), using Graph Pad Prism Version 9. For T-MOC assays, the statistical analysis of drug efficacy differences for each drug control was performed using the Tukey post hoc multiple comparison test provided in the ANOVA. Comparisons were made to appropriate controls as detailed in the figure legends and a difference was considered statistically significant when the p-value was less than 0.05.
3. Results
3.1. New generation Ref-1 inhibitors as anticancer agents: key data on lead compounds
Structure-activity relationship (SAR) efforts to identify second-generation compounds with improved potency in cancer models and drug-like properties, focused on the modifications to second-generation naphthoquinones. Based on initial SAR studies, we selected 13 s-generation compounds from > 350 that were further characterized for positive properties including efficacy in Ref-1 redox inhibition, in silico ADMET properties (Supplemental Table 1) and cellular studies (see Methods for all Synthesis details and synthesis schemas 1, 2, and 3 for five lead molecules). However, further screening of these compounds included using mouse and human S9 fraction metabolic stability, plasma half-life, thermodynamic solubility, metabolic stability studies and PK identified five to be the lead molecules – APX2009, APX2014, APX2044, APX2051, and APX2053. This triage was also supported by increased efficacy for cell killing using Pa03C PDAC cell line (Supplementary Fig. S4B).
Hypothesis-driven incorporation of the aromatic moiety on the left-hand side followed by replacement of the carboxylic acid resulted in the lead chemical scaffold represented by APX2009 and APX2014 (Fig. 1). The lead series showed acceptable molecular weights and partition coefficient logP. In-silico modeling (ADMET Predictor v.11.0) suggested acceptable solubility and high permeability with no red flag related to rat primary hepatocyte (RPH), CYP inhibition, or mutagenicity (Supplemental Table 1). To assess the correlation between these predictions and the correlating standard in vitro and in vivo systems, we evaluated several compounds with the goal of identifying potential drug-like property gaps. The aqueous solubility as well as the solubility in simulated fluids were acceptable for the lead compounds and consistent with in silico predictions. In addition, moderate to high CaCO2 bi-directional permeability was observed across the series (Table 2).
Fig. 1.
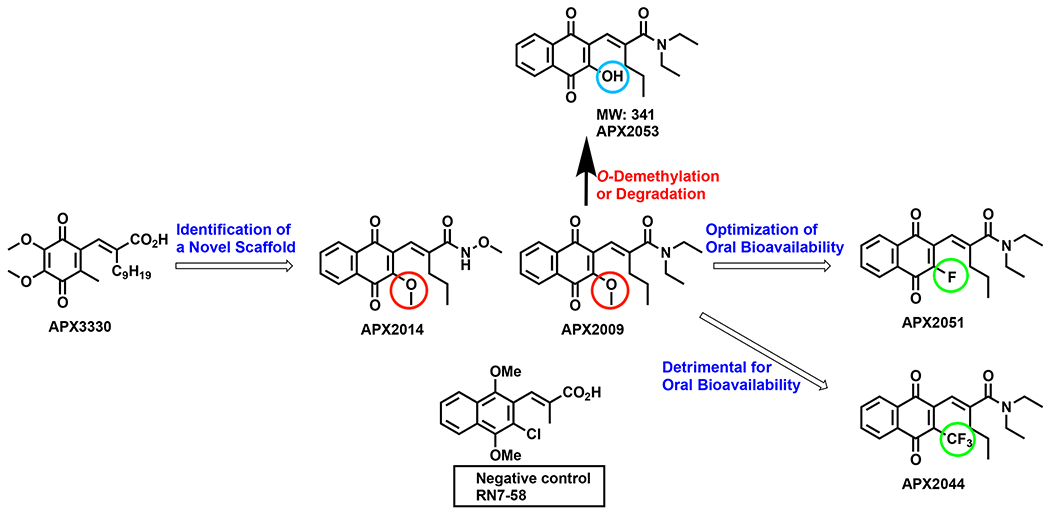
SAR Evolution towards optimization of pharmacokinetic properties and selectivity for Ref-1: Schematic representing lead chemical scaffolds with chemical moieties highlighted for modulation of pharmacokinetics and Ref-1 redox inhibition. Red circles highlight the methoxy groups in APX2009 and APX2014. Green circles indicate the trifluoromethoxy and fluoro groups in APX2044 and APX2051, respectively. Blue circle denotes the hydroxyl group of APX2053.
Table 2.
Profiling for Key Representative Compounds and Comparison to the Reference Standards APX3330.
| Compound | APX3330 | APX2009 | APX2014 | APX2044 | APX2051 | APX2053 | |
|---|---|---|---|---|---|---|---|
| Calculated Properties | MW | 378.5 | 355.4 | 329.4 | 393.4 | 343.4 | 341.4 |
| S + LogP | 4.4 | 2.8 | 2.2 | 3.5 | 3.0 | 2.9 | |
| S + Peff | 1.0 | 4.7 | 3.1 | 4.3 | 5.1 | 5.4 | |
| S + MDCK - LE | low | high | high | High | high | high | |
| RPH Cytolethality | Nonlethal | Nonlethal | Nonlethal | Nonlethal | Nonlethal | Nonlethal | |
| CYP3A4 Inhibition | < 50% | < 50% | < 50% | < 50% | < 50% | No (81%) | |
| Mutag | Non-mutag | Non-mutag | Non-mutag | Non-mutag | Non-mutag | Non-mutag | |
| Pgp Substrate | Yes-substrate | Non-substrate | Yes-substrate | Yes-substrate | Non-substrate | Yes-substrate | |
| In Vitro Activity | Red. EMSA (IC50 mM) | 16 | < 1 | < 1 | ND | ND | ND |
| DNA Repair | Neg. | Neg. | Neg. | Neg. | Slight. Inhib. | Slight. Inhib. | |
| NFκB (IC50 mM) | 39.3 | 5.1 | 5.9 | 5.6 | 11.9 | ND | |
| 3D tumor(Pa03C) EC50 (mM) | > 20 | 4.6 | 2.7 | 6.4 | 5.7 | 16.1 | |
| 3D CAF19 EC50 (mM) | > 20 | 7.8 | 10 | 10.6 | 8.6 | 14.3 | |
| Metabolic Stability CLINT (mL/min/kg body weight) | Human Liver | 31.8 | 4.3 | 40.6 | 36.3 | 2.9 | 2.3 |
| Microsome Mouse Liver | 67.4 | 19.3 | 23.4 | 141.8 | 24.5 | 19.0 | |
| Microsome Human Liver S9 Phase | 4.4 | 2.9 | 1.0 | 5.9 | 5.3 | 1.9 | |
| Mouse Liver S9 Phase I | 14.2 | 7.9 | 20.2 | 37.6 | 25.9 | 34.6 | |
| Human Liver S9 Phase II | 5.6 | ND | 4.7 | 23.2 | 3.7 | ND | |
| Mouse Liver S9 Phase II | 21.6 | ND | 18.0 | 56.0 | 8.2 | ND | |
| Plasma Stability | Human Plasma T1/2 (min) | 509 | 163 | 284 | 42 | 9900 | 25 |
| Mouse Plasma T1/2 (min) | 94 | 34 | 17 | 2 | > 9900 | 132 | |
| Thermodynamic Solubility | Phosphate buffer (pH=6.8) | 0.496 | 0.094 | 0.407 | 0.009 | 0.013 | |
| FasSIF (mg/mL) | 0.671 | 0.198 | 0.385 | 0.004 | 0.085 | 1.01 | |
| FedSIF (mg/mL) | 0.448 | 0.558 | 0.603 | 0.081 | 0.050 | 1.20 | |
| FasSGF (mg/mL) | 0.001 | 0.095 | 0.510 | 0.002 | 0.090 | 0.76 | |
| CaCO2 bidirectional permeability | A to B | 5.2 | 33.7 | 41.7 | ND | ND | 39.7 |
| Efflux | 0.65 | 0.75 | 0.88 | ND | ND | 1 | |
| In Vivo PK in mouse | Dose (mg/kg) Oral/IV | 25/5 | 25/5 | 25/5 | 25/5 | 25/5 | 25/1 |
| Vehicle Oral/IV | MC/ Cr: | MC/ Cr: | MC/ Cr: | MC/ Kolliphor | MC/ Kolliphor | PKT/ Kolliphor | |
| Oral T1/2 (h) | EtOH 3.8 | EtOH 1.8 | EtOH 22 | HS15 ND | HS15 3.7 | HS15 2.1 | |
| IV Clearance (mL/h) | 0.5 | 41 | 22 | > 100 | 2.5 (mL/min/kg) | 1.3 | |
| Oral AUC0-∞ (ng*mL-1*hr) | 77946 | 113 | 6100 | < 10 | 22765 | 25379 | |
| Oral Cmax (ng/mL) | 8815 | 27 | 476 | < 10 | 7450 | 5705 |
However, the metabolic and/or chemical stability were identified as potential risks for the compound series. Therefore, efforts were focused on understanding the competition between phase 1 and phase 2 metabolism and its impact on the in vivo clearance and ultimately the oral bioavailability. Stability studies were conducted in both plasma and in the presence of the enabling vehicle to help distinguish between degradation and metabolism.
Metabolic stability was examined in human and mouse liver microsomes as well as in human and mouse liver S9 showing moderate intrinsic clearance CLINT (Table 2). This was followed by metabolite identification studies (MetID) that were performed following the incubation of compounds in microsomes from mouse, rat, dog, or human. Analysis of metabolite from compound APX2009 showed predominantly the products of O-demethylation (APX2053, MW: 341) and its glucuronide, the loss of ethyl group from the carboxamide function as well as other glucuronides (Fig. 1).
Chemical stability studies on APX2009 in the presence of an enabling vehicle such as PKT (Propylene glycol: Kolliphor HS15: Tween 80) showed acceptable stability at room temperature after four days. However, stressing the mixture at 60 °C for 4 h led to the formation of significant amount of the hydroquinone (HQ) of APX2053 (MW: 243) that may have derived from the loss of methyl in APX2009, followed by conversion to the HQ derivative. The question was whether this degradation would happen in plasma affecting the total clearance and bioavailability.
Compounds were then administered to mice by oral routes (PO), intravenous (IV), and intraperitoneal (IP) either as suspensions or solutions in the adequate vehicles (Table 2). PK parameters such as the area under the curve (AUC0-last) and Cmax were at and below the in vitro target affinity respectively for compound APX2014 and lower for APX2009. In addition, the data showed high total clearance (CL = 22 and 41 mL/h respectively) that would potentially lead to a high projected human dose. Plasma stability studies on APX2009 and APX2014 show a very short half-life (34 and 17 min, respectively, Table 1) suggesting a similar mechanism of degradation, leading potentially to formation of the HQ of APX2053.
Since neither permeability nor solubility were identified as key limiting steps for these compounds, hypotheses were focused on metabolism and stability. This risk was mitigated through an issue-driven SAR, focused on identifying and fixing the main site of metabolism and degradation. For example, replacing the methoxy group in APX2009 with a fluorine blocked the site of metabolism resulting in the identification of compound APX2051 with significantly higher T1/2 after incubation in human plasma (T1/2 = 9900 min) and in mouse (T1/2 >9900 min). Such significant improvement in plasma stability led to low in vivo clearance, approximately 200-fold increase in oral AUC @ 25 mg/kg (APX2009: 113 ng*mL-1 *hr compared to APX2051: 22765 ng*mL-1 *hr), and sustained exposure with good oral bioavailability. The fluoro atom in this region of the molecule of APX2051 provides unique advantages in terms of metabolic stability and PK properties. Other substituents such as CF3 in APX2044 were detrimental for oral bioavailability. Therefore, the fluoro substitution seems to be adequate optimization in this domain.
Collectively, the performance characteristics of these compounds meet and exceed the critical success factors for advancement to pre-clinical toxicology studies and further development as novel oral Ref-1 inhibitors that might offer promising strategies for the treatment of cancer as well as other disease indications.
3.2. New lead compounds directly interact with Ref-1 as shown by 1D WaterLOGSY NMR
To determine whether second generation compounds interact directly with Ref-1, we pursued a sensitive ligand-based 1D NMR approach, waterLOGSY, which effectively probes the solvent accessibility (water) of small molecule protons in the presence and absence of a protein. Following assignment of the 1D NMR spectrum of the small molecule, protons that experience differences in water accessibility in the presence of Ref-1 were identified in the waterLOGSY difference spectrum (Supplementary Fig. S1) [43]. We have extensively characterized interactions of the parent compound, APX3330, with Ref-1 through a coupled NEM-fingerprinting, mass spectrometry assay [25–27,44]. All of the compounds, APX3330 [25], 2009, 2014, 2044, 2051, 2053, and a control compound RN7–58, had peaks in waterLOGSY difference spectra that were consistent with interactions with Ref-1 (Supplementary Fig. S1) [25–27,44]. Overall, the results suggest that protons within non-polar parts of these compounds interact with Ref-1 (Fig. 2). Specifically, in all of the naphthoquinone containing compounds and RN7–58, protons 1–4 exhibit changes in water accessibility in the presence of Ref-1 (Fig. 2). Compounds APX2009, 2044, 2051, and 2053 have similar structures, differing in a single moiety on the quinone ring of the compound, with -OCH3, -CF3, -F, and -OH, respectively, and in all of these compounds, protons associated with methyl groups 8, 11, and 12 interact with Ref-1. Although the right-side of these compounds are similar, the olefinic proton 5 interacts with Ref-1 only in APX2009. In APX2014, the right portion of the molecule includes a more polar group (NHOCH3); in this compound, the olefinic proton 5 and the propyl group methylene and methyl protons 6, 7, and 8 interact with Ref-1. While the negative control compound RN7–58 also shows protons that interact with Ref-1, they are found within more polar moieties, specifically the methoxy protons of the compound, suggesting that this compound may interact at a different site on Ref-1 than the other compounds.
Fig. 2.
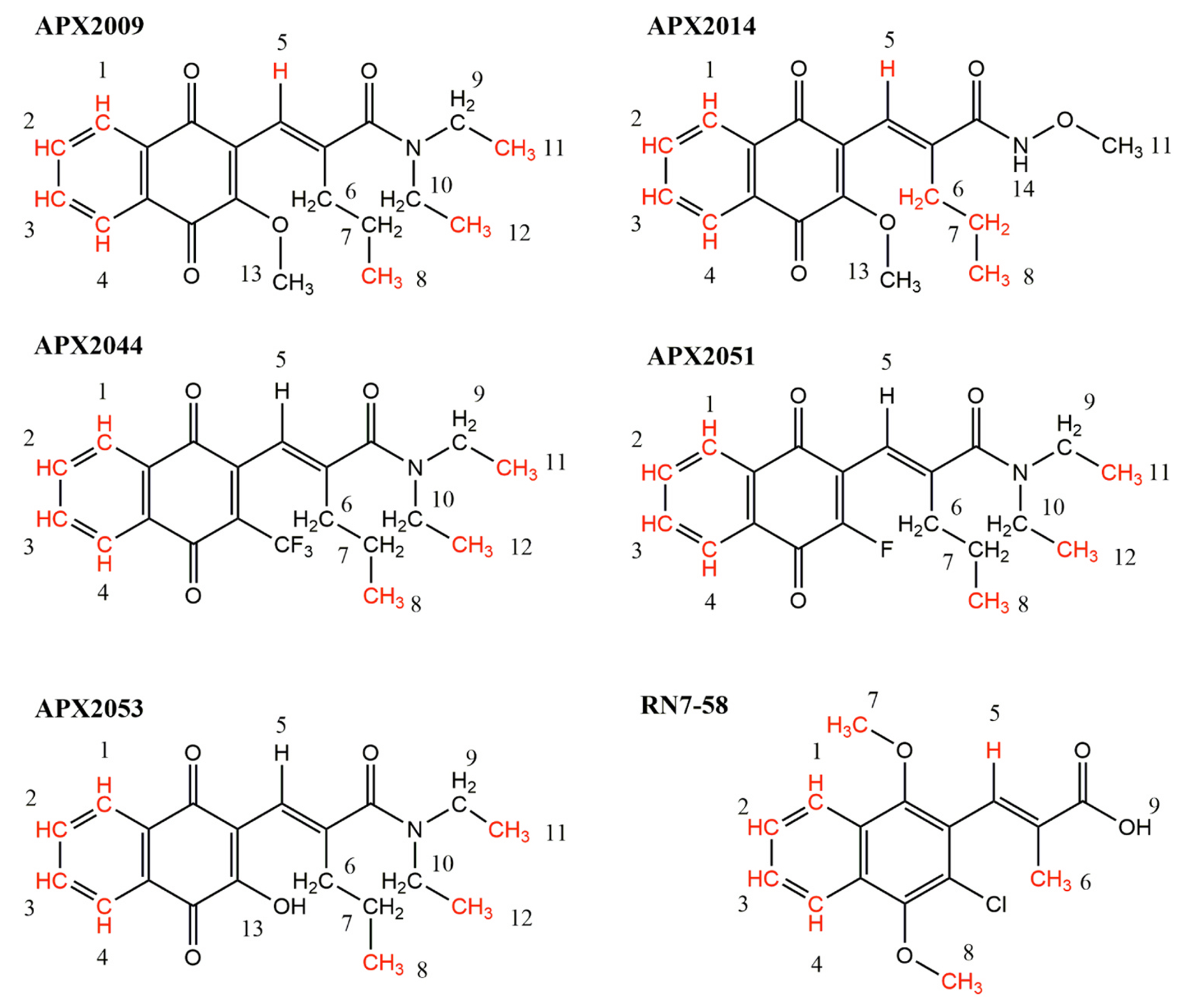
WaterLOGSY NMR to investigate direct binding of APX compounds to Ref-1. Protons (along with C atoms) in APX2009, 2014, 2044, 2051, 2053, and control compound RN7–58 that have altered solvent accessibility in the presence of Ref-1 were identified in 1D WaterLOGSY NMR experiments and are shown on the chemical structures highlighted in red. Numbers refer to the assignments of proton chemical shifts observed in the 1D 1 H and WaterLOGSY spectra for each compound (SI Fig. S1).
3.3. Ref-1 redox inhibition by selected compounds does not affect the AP endonuclease activity of Ref-1
We initially determined whether selected Ref-1 inhibitors exert any effect on the repair activity of Ref-1. We treated cell lysate from cancer cells with varying doses of the lead compounds and analyzed for Ref-1′s repair activity using a kinetic fluorescence-based endonuclease assay. This assay demonstrated that the compounds had no significant impact on Ref-1 endonuclease activity at 20 μM (Supplementary Fig. S2). APE1 repair inhibitor III (ARi3) was used as a positive control for inhibition of the endonuclease activity of Ref-1 and decreased Ref-1 endonuclease activity by 63% [45].
3.4. New Ref-1 redox inhibitors dose-dependently suppressed Ref-1 regulated transcription factor activity
New Ref-1 inhibitors were analyzed for their ability to block the activity of three Ref-1-regulated TFs: NFκB, HIF1α, and STAT3. For confirmation of Ref-1 redox activity inhibition, pancreatic cancer cells that stably express NFκB− or HIF1α− driven luciferase were utilized. To confirm the results in additional cancer cells, MPNST line NF90–8 was also stably transduced with both NFκB− or HIF1α− driven luciferase and treated with Ref-1 inhibitors. These assays were conducted at a time point in which a significant and dose-dependent decrease in luciferase activity could be observed prior to an impact on cell viability. At 24 h, a reduction in Ref-1-regulated transcription factor activity could be reliably assessed in live cancer cells. Following treatment with APX2009, 2014, 2044, and 2051, Pa03C (pancreatic cancer cells) and NF90–8 cells (MPNST cells) demonstrated a dose dependent reduction in both NFκB and HIF1α activity at 2 – 11-fold lower doses compared to parent compound APX3330. Data in Fig. 3A demonstrates a significant reduction in basal NFκB activity with APX2009, 2014, 2044, and 2051 significantly more potent than APX3330 and APX2044 being the most potent in two cancer cell lines. Interestingly, NFκB activity was inhibited at lower doses of second-generation compounds in the NF90–8 cells compared to the Pa03C cells. Negative control, RN7–58, and new analog, APX2053 do not decrease the NFκB activity or affect cell viability at any of the doses tested (Fig. 3A). Significant decreases in NFκB activity following treatment with these compounds implies that Ref-1 redox activity is blocked following treatment and that the effect is selective for Ref-1 as two analogs, RN7–58 and APX2053 did not have similar effects.
Fig. 3.
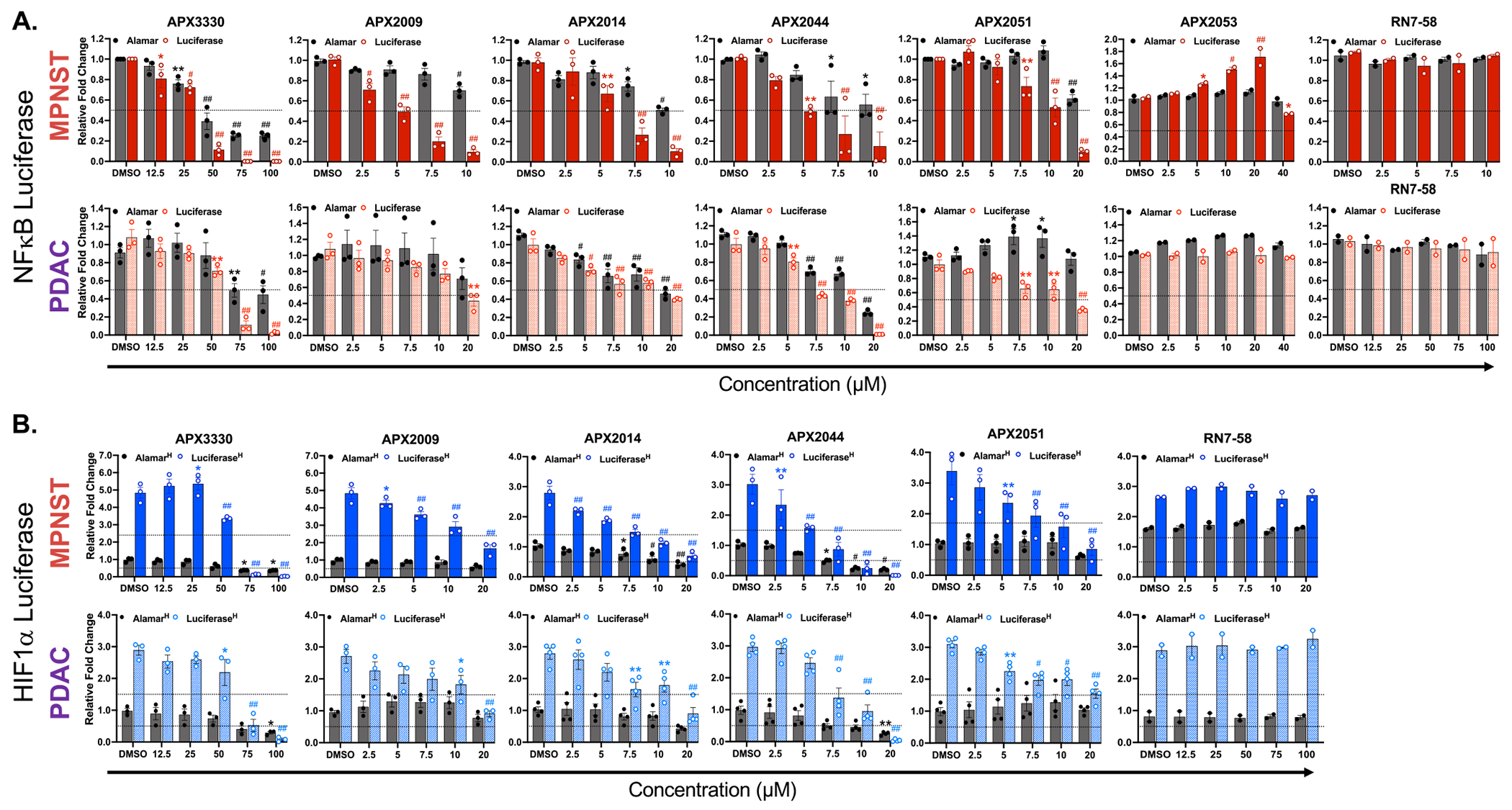
Target Engagement. Screening of Ref-1 inhibitors for inhibition of transcription factor driven luciferase activity. All grey bars indicate cell viability using alamar blue assay. A. Red solid and hashed bars indicate luciferase activity driven by NFκB for NF90–8 (MPNST) and Pa03C (PDAC) cells, respectively. B. Blue solid and hashed bars indicate luciferase activity driven by HIF1α for NF90–8 (MPNST) and Pa03C (PDAC) cells, respectively under 1% hypoxic conditions. All treatments were done in 5% FBS growth media for 24 h with indicated concentrations of the compounds (μM). Data represents Mean±SEM for biological repeats of n > 2 where p > 0.05, 0.01, 0.001 and 0.0001 are represented as * , * *, # and ## respectively.
The activity of a second TF regulated by Ref-1, HIF1α, was also assessed following treatment with these new analogs for additional confirmation of inhibition of Ref-1 redox activity. In stably expressing lines for HIF1α -driven luciferase activity, conditions of 1% hypoxia induced HIF1α activity by 3 - 5-fold depending on the cell line. Even with significant induction of HIF1α activity, Ref-1 redox analogs exert a significant and dose-dependent reduction in luciferase activity with 50% inhibition in the order of APX3330 > APX2009 > APX2051 > APX2014 > APX2044 for both MPNST and PDAC cells under 1% hypoxia (Fig. 3B, and normoxia data in Supplementary Fig. S3). Similar to NFκB reporter data, HIF1α activity and cell viability were not affected by RN7–58.
Table 3 includes the EC50 values for inhibition of NFκB and HIF1α activity and also shows that the slopes of the dose response curves comparing luciferase vs viability were significantly different. The steeper curve with the TF activity indicates that the Ref-1 redox inhibitors are blocking activity of the TFs in live cells. As expected, at higher concentrations of Ref-1 inhibitors, the impairment of TF activity does begin to impact cell viability. RN7–58 which is the inactive analog for Ref-1 inhibition was identified to be inactive against TF activity even at 100 μM concentration. Further, APX2053 which was the O-demethylated metabolite of APX2009 when tested up to 40 μM was ineffective at blocking Ref-1 activated TF activity (for NFκB).
Table 3.
Potency of Ref-1 inhibitors for inhibition of transcription factor driven luciferase activity and comparison to the Parent Compound APX3330.
| NF90-8 | |||||
|---|---|---|---|---|---|
| Ref-1 Redox Analog | NFκB Luciferase 24 hr EC50±SE (μM) | Significance of Slopes (95% CI) | Ref-1 Redox Analog | HIF1α Luciferase 24 hr EC50±SE (μM) | Significance of Slopes (95% CI |
|
| |||||
| APX3330 | 33.7 ± 2.81 | P = 0.0186 | APX3330 | 55.1 ± 0.44 | P < 0.0001 |
| APX2009 | 5.1 ± 0.3 | P < 0.0001 | APX2009 | 14.3 ± 0.25 | P < 0.0001 |
| APX2014 | 6.1 ± 0.5 | P < 0.0001 | APX2014 | 8.1 ± 0.53 | P < 0.0001 |
| APX2044 | 5.2 ± 0.2 | P = 0.0004 | APX2044 | 5.7 ± 0.6 | P = 0.0002 |
| APX2051 | 11.9 ± 0.81 | P = 0.0072 | APX2051 | 9.7 ± 1.21 | P < 0.0001 |
| RN7-58 | NA | P = 0.6612 | RN7-58 | NA | P = 0.6349 |
| Pa03C | |||||
| Ref 1 Redox Analog | NFκB Luciferase 24 hr EC50 ± SE (μM) | Significance of Slopes (95% CI) | Ref 1 Redox Analog | HIF1α Luciferase 24 hr EC50 ± SE (μM) | Significance of Slopes (95% CI) |
| APX3330 | 54.5 ± 1.97 | P = 0.0211 | APX3330 | 60.2 ± 4.13 | P < 0.0001 |
| APX2009 | 18.8 ± 1.35 | P = 0488 | APX2009 | 14.5 ± 1.06 | P < 0.0001 |
| APX2014 | 13.4 ± 1.26 | P = 0.0213 | APX2014 | 9.7 ± 1.24 | P < 0.0001 |
| APX2044 | 8.2 ± 0.43 | P = 0.1872 | APX2044 | 8.3 ± 1.23 | P = 0.0002 |
| APX2051 | 14.9 ± 0.77 | P = 0.0037 | APX2051 | 12.6 ± 2.11 | P < 0.0001 |
| RN7-58 | NA | P = 0.9872 | RN7-58 | NA | P = 0.9610 |
As another confirmation for Ref-1 redox specific activity of these compounds, we assayed STAT3 inhibition which we have previously demonstrated is also under Ref-1 redox signaling control [46]. Thus, expression of STAT3 target gene survivin was assayed using RT-qPCR. STAT3 activation induces survivin gene expression as reported in several cancers which leads to apoptotic cell death [47–49]. Therefore, survivin levels were assessed in two human cancer cell lines, PDAC (Pa03C) and MPNST (NF90–8) after treatment with the lead Ref-1 redox inhibitors. As expected, survivin gene expression is significantly downregulated with the new inhibitors at 3.75 - 5-fold lower concentration than APX3330 in both cell lines, implying that STAT3 activity is decreased following Ref-1 redox inhibition (Supplementary Figure 3C).
3.5. Ref-1 inhibition decreases tricarboxylic acid (TCA) cycle substrate utilization by tumor cells
We have previously published dramatic effects of Ref-1 redox activity on metabolic pathways through both gene expression changes and in mitochondrial function. Pa03C cells had dramatically reduced levels of four TCA cycle substrates (α-keto-glutaric acid, succinic acid, fumaric acid, and L-malic acid) following Ref-1 inhibition and this contributed to inhibitor-induced cell killing [32]. As validation of on-target effects with the new more potent Ref-1 redox inhibitors, mitochondrial function was evaluated in NF90–8 cells treated with APX3330, APX2009, APX2014, APX2044, and APX2051. Significantly reduced rates of reaction for the TCA cycle substrates were observed (30–80% for α-keto-glutaric acid, 40–90% succinic acid, 5–80% fumaric acid, and 20–80% L-malic acid) (Fig. 4A). APX2051 at 10 μM demonstrated the most potent inhibition of mitochondrial function while minimal effects were observed for APX3330 at doses of 50 μM. Further, the inhibitory impact on mitochondrial function was dose-dependent for these molecules as shown in Supplementary Fig. S4A. These results confirm that Ref-1 inhibition leads to mitochondrial dysfunction as was previously shown with single cell RNAseq following Ref-1 knockdown and the new analogs demonstrate a similar phenotype [32].
Fig. 4.
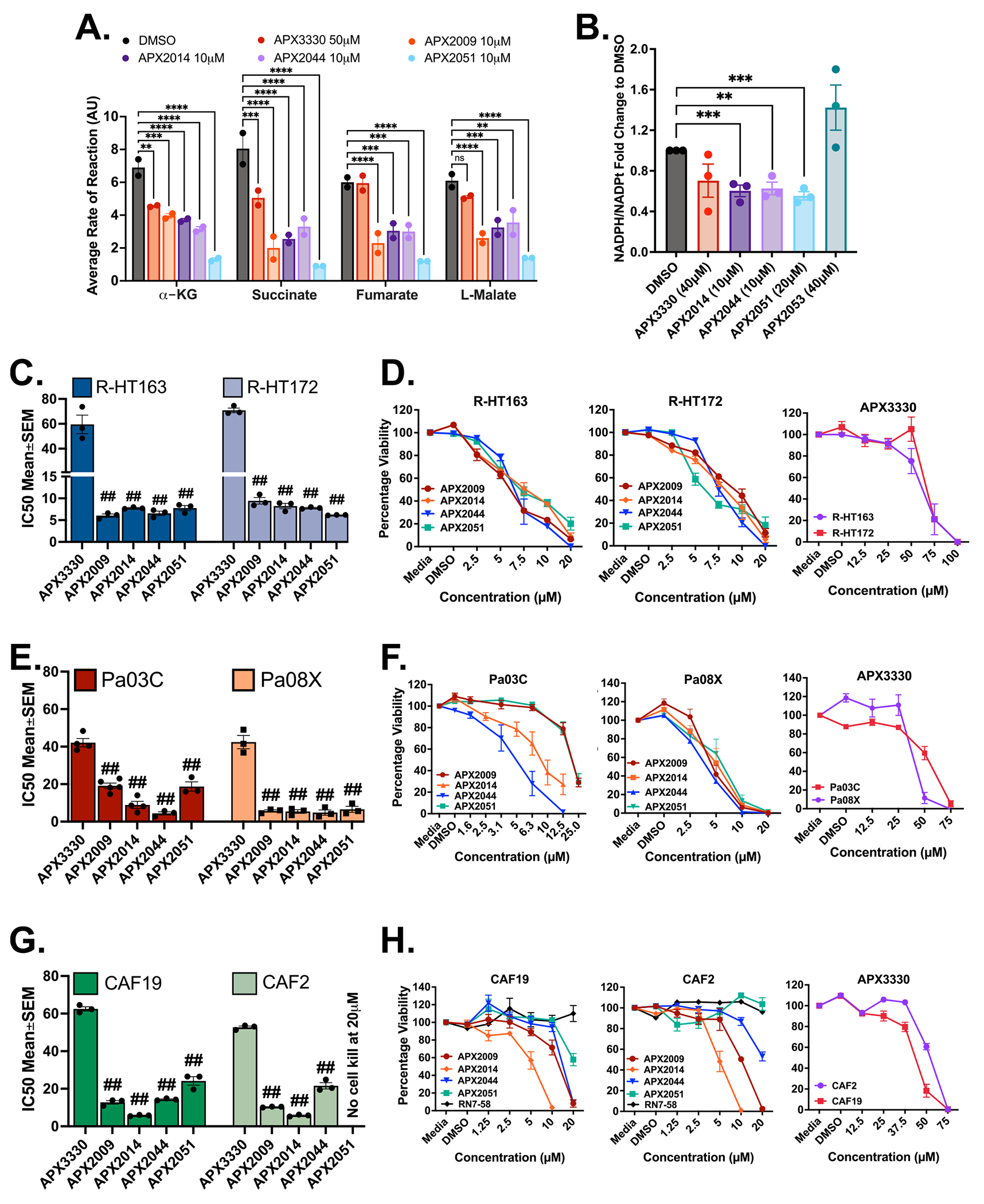
Phenotype Assays. Screening of Ref-1 inhibitors for increased potency. A. Graph showing decreased TCA cycle substrate utilization by NF90–8 cells in the presence of Ref-1 redox inhibitors (10 μM used for new compounds and 50 μM for APX3330). Cells were treated for 24 h with indicated concentrations of the compounds and data represented as Mean±SEM for biological repeats of n = 2. p < 0.05, 0.01, 0.001 and 0.0001 is represented as *, **, *** and **** respectively. B. Pa03C cells were treated for 6 h with indicated concentrations of the lead molecules and fold change of the ratio of NADPH/NADPt is represented as Mean±SEM (n = 3, p < 0.01, and 0.001 represented as ** and ***, respectively). C. Bar graphs showing differences in IC50 (μM) values for the new generation of Ref-1 redox inhibitors and their dose dependent cell kill represented as line graphs (0–20 μM for APX2009, 2014, 2044 and 2051; 0–100 μM for APX3330) (D) on two models of patient-derived MPNST cell lines, R-HT163 and R-HT172. Cells were treated for 72 h and data was represented as Mean IC50 ± SE for n = 3, p < 0.0001 represented as ## compared to APX3330. E. Bar graphs showing differences in IC50 (μM) values for the new generation of Ref-1 redox inhibitors and their dose dependent cell kill represented as line graphs (0–25 μM for APX2009, 2014, 2044 and 2051; 0–75 μM for APX3330) (F) on two models of patient-derived pancreatic cancer cells, Pa03C and Pa08X. Cells were treated for 48 and 72 h respectively. Data representative of Mean IC50 ± SE for n ≥ 3, p < 0.0001 represented as ## compared to APX3330. G. Graphs showing differences in IC50 values for the new generation of Ref-1 redox inhibitors and their dose-dependent cell kill represented as line graphs (0–20 μM for APX2009, 2014, 2044, 2051 and RN7–58; 0–75 μM for APX3330) (H) against two cell line models of patient-derived cancer-associated fibroblasts, CAF19 and CAF2 cells. Data representative of Mean IC50 ± SE for n = 3. p < 0.0001 is represented as ## relative to APX3330.
3.6. NADP/NADPH assay measuring the redox state of the cell following Ref-1 inhibition with the new inhibitors
To assess whether the reduced form of intracellular NADPH is decreased after Ref-1 redox inhibition, the ratio of NADPH to NADP+ was quantitated in Pa03C cells. As expected, upon blocking the redox activity of Ref-1 with APX2014, 2044, and 2051, we see a significant decrease in NADPH levels indicating there is a more oxidized environment (Fig. 4B). APX2053 which did not show any impact upon NFκB signaling (Fig. 3) also does not show reduced NADPH levels in this assay. After treatment with 40 μM APX3330, NADPH levels were not significantly decreased. We previously demonstrated that APX2009 has similar potency in this assay to the additional analogs [32].
3.7. Evaluation for potency of identified compounds using 2D and 3D cellular models revealed preferential inhibition of tumor cells over CAFs
After establishing Ref-1 protein interaction with the new redox inhibitors, blockade of the activity of two Ref-1 regulated TFs, and the expected decrease in mitochondrial function and NADPH levels, the top lead analogs were evaluated for growth inhibition in two low passage patient-derived cell lines for MPNST (R-HT163 and R-HT172) and pancreatic cancer (Pa03C and Pa08x) as well as cancer-associated fibroblast cells (CAF2 and CAF19). Monolayer data identified most efficacious compounds to be APX2009, APX2014, APX2044, and APX2051. These molecules showed variable potency on tumor and CAF cells for 50% growth reduction: 5–20 μM for tumor and 5–25 μM for CAFs. Further, the tumor growth inhibition was similar or greater than CAF growth inhibition as shown in Fig. 4C–H and Supplementary Fig. S4B, C, D. The order of 50% growth inhibition was noted to be:
R-HT163: APX3330 < 2014 = 2051 < 2044 < 2009,
R-HT172: APX3330 < 2009 < 2014 < 2044 < 2051,
Pa03C: APX3330 < 2009 < 2014 < 2051 < 2044,
Pa08x: APX3330 < 2014 < 2009 < 2051 < 2044,
CAF19: APX3330 < 2051 < 2044 < 2009 < 2014, and
CAF2: APX3330 < 2051 < 2044 < 2009 < 2014.
It is interesting to note that in monolayer APX2044 and APX2051 were most potent in the tumor lines but had little effect on the CAFs’ proliferation compared to the tumor cells. APX2009 and APX2014 seemed to affect cell growth more potently in CAFs compared to tumors.
Using two physiologically relevant 3D culture models, the lead Ref-1 inhibitors were evaluated for their effects on tumor and CAF cells in co-culture. First, confirmation of increased efficacy at lower doses compared to APX3330 was performed in 3D co-culture spheroids over a time period of 14 days with increasing doses and multiple treatments to better recapitulate the in vivo setting. This physiologically relevant 3D co-culture assay again confirms that APX2009, 2014, 2044 and 2051 look to be promising candidates for tumor cell growth inhibition through Ref-1 redox signaling blockade (Fig. 5A, B, C and Supplementary Fig. S5A, B). In this co-culture model, spheroid size was dramatically reduced following treatment with Ref-1 inhibitors. There were significant differences between the tumor cell IC50s and the CAF IC50s, but both cell populations were decreased following treatment (Fig. 5C). As a marker of myofibroblastic CAFs or myCAFs, CAF2 or CAF19 cells in monolayer or in 3D co-culture were stained with αSMA. As expected, there is variable expression of αSMA in the CAF cultures indicating various phenotypic differences in CAFs exist in these conditions (Supplementary Figure 5E, F).
Fig. 5.
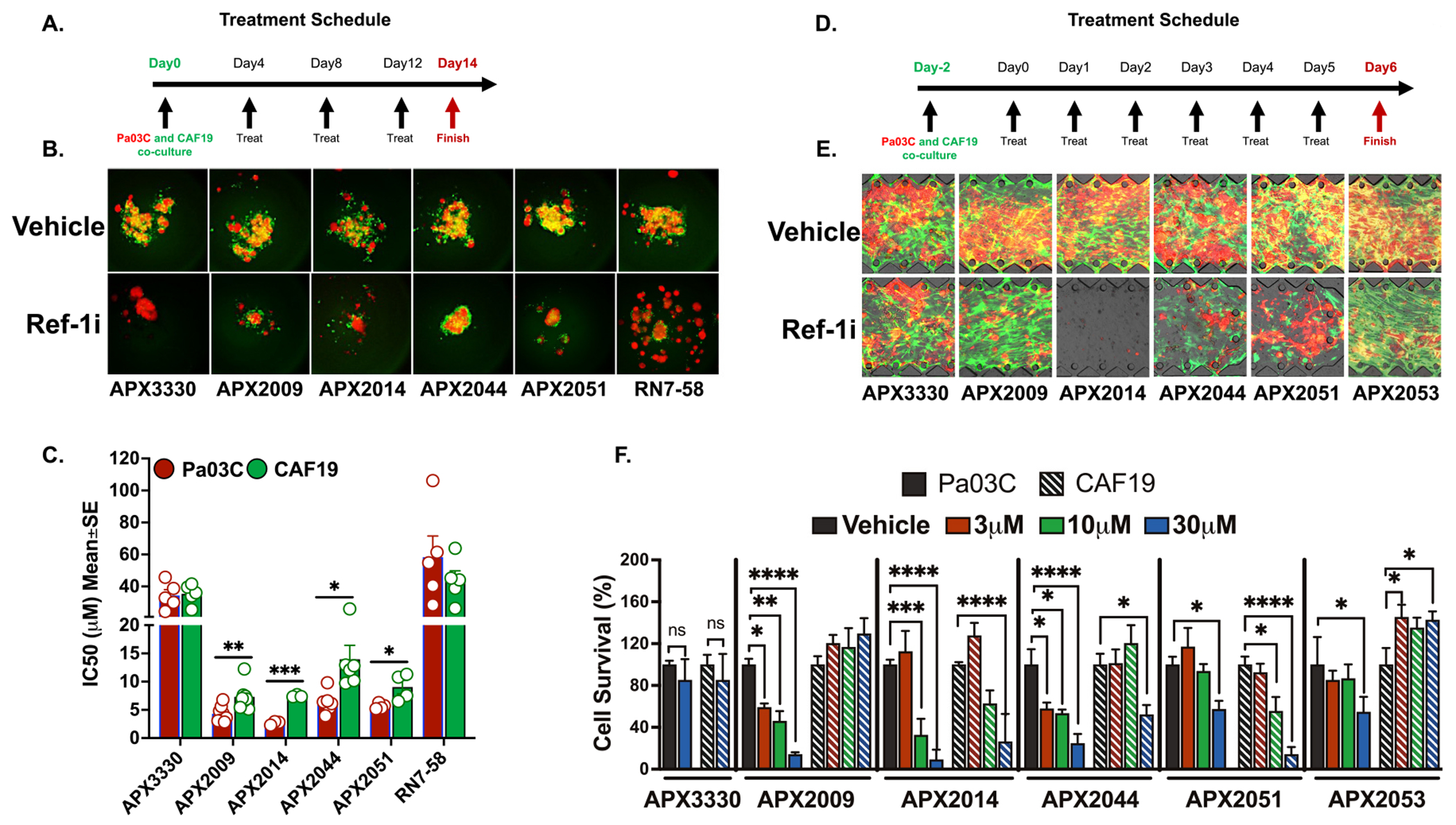
New generation Ref-1 redox inhibitors effectively reduce tumor growth in co-culture with CAFs. A. Schematic showing the treatment timeline for 3D coculture treatment with Ref-1 redox inhibitors. B. Representative pictures of low passage patient-derived cell line, Pa03C plated as 3D co-cultures with CAF19 cells at a ratio of 1:4. These co-cultures were treated with increasing concentrations of Ref-1 redox inhibitors on Days 4, 8 and 12 following intensity measurements on Days 4, 8, 12, and 14. Tumor cells were represented by red and the CAFs by green fluorescence (50 μM for APX3330 and RN7–58; 10 μM for APX2009, APX2014, APX2044, and APX2051). C. Graph representing differences in IC50 (μM) values for the new generation of Ref-1 redox inhibitors (0–20 μM for APX2009, 2014, 2044 and 2051; 0–75 μM for APX3330 and RN7–58) against Pa03C cells vs CAF19 cells. Data representative of Mean IC50 ± SE for n > 3. p < 0.05, 0.01, and 0.001 is represented as * , * *, and * ** respectively for comparison of CAF inhibition to tumor kill. D. Treatment timeline for PDAC T-MOC co-culture model. E. Representative fluorescent microscopic observation of Pa03C (red) and CAF19 (green) in PDAC T-MOC on Day 6 treated with vehicle control or 30 μM of Ref-1 redox inhibitors. F. Quantitation for survival of tumor and CAF cells in the T-MOC system treated with increasing concentrations of Ref-1 inhibitors (n ≥ 3, Mean±SEM. p < 0.05, 0.01, 0.001 and 0.0001 is represented as *, **, *** and **** respectively). APX3330 was used as the parent compound for comparison and RN7–58 as a Ref-1 redox inactive inhibitor compound in all these experiments.
A second 3D model that can recapitulate interstitial transport of the compounds within the tumors and aptly named tumor-microenvironment on chip model (T-MOC) was used for final in vitro validation. Both tumor cells and CAFs were seeded into interstitial channels with ECM to mimic in vivo conditions. The five most promising compounds inhibited tumor growth 11-fold in low passage PDAC patient-derived Pa03C cells compared to CAFs (Fig. 5D, E, F and Supplementary Fig. S5C, D). Interestingly, we again observed a differential response in CAFs vs tumor cells in these sophisticated 3D co-culture models, especially with APX2009 and 2044. In general, Ref-1 inhibitors show greater inhibition of tumor cell growth compared to CAFs. This is promising as it has been shown that inhibition of certain populations of CAF cells can lead to deleterious side effects, disease progression, and drug resistance [50].
3.8. Proteomic analysis identified key downstream effectors of Ref-1 inhibition
NSG mice were implanted with Pa03C cells and tumors were allowed to grow until tumor volumes reached ~250 mm3. Following treatment with Ref-1 inhibitors (APX3330, 2009, 2014, and 2051) as well as controls, tumors were harvested, and proteomics data collected. Differential expression analysis was first conducted to identify the proteins that are consistently over- or under-expressed after Ref-1 inhibitor treatment vs control by using a generalized linear model (see details in Methods). Among the 5457 human proteins identified from the proteomics experiment, we identified 115 proteins that increased in expression and 453 proteins that decreased in expression in the PDAC tumors following Ref-1 inhibition upon comparison to control treated tumors (Fig. 6A, Supplemental Table 4). Further, pathway enrichment analysis identified the proteins that increased after treatment significantly enrich in the matrisome, steroid hormone biosynthesis, extracellular matrix, and apoptosis-related pathways (Fig. 6B, D, E) while the proteins that were significantly decreased enrich in pathways involved with cell cycle, DNA repair, mitochondrial proteins, TGFβ signaling, electron transport chain, oxidative phosphorylation, and IL-2 and IL-6 signaling pathways (Fig. 6C and 6F–I). It is noteworthy that the pathways that show significant enrichment of over/under expressed proteins identified from this proteomics data strongly overlap with our published work derived from scRNA-seq data collected from Ref-1 knockdown vs controls of samples from the same cell line [32,51].
Fig. 6.
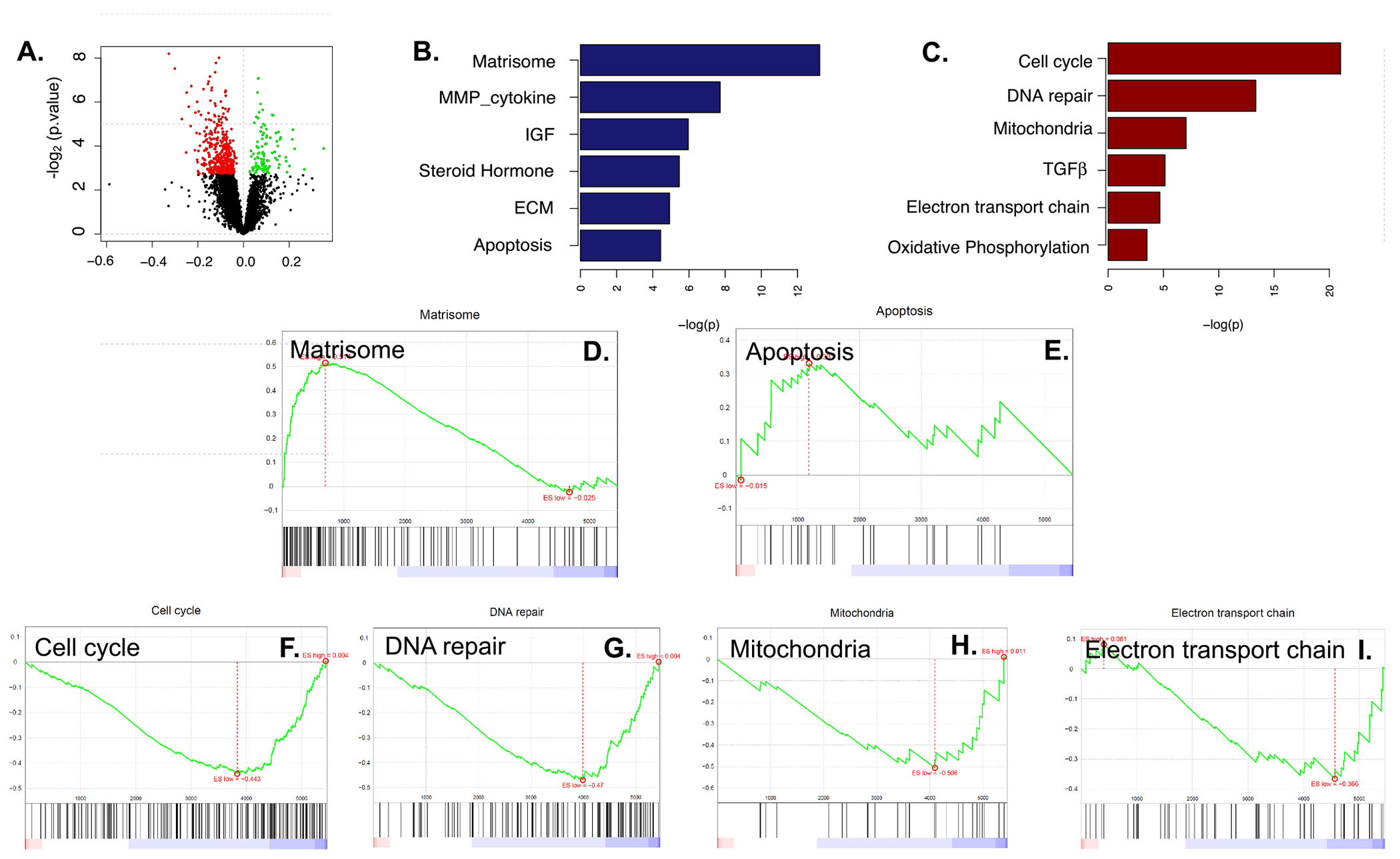
In Vivo Tumor Proteomics. A. Volcano plot of the differentially expressed proteins. Y-axis represents -log2 (p value) and X-axis represents the regression parameter of effect of Ref-1 inhibitor derived from glm model (see details in Methods); B. Selected pathways that are significantly enriched by proteins that were higher in the Ref-1 treated tumors; C. Selected pathways that are significantly enriched by proteins that were lower in the Ref-1 treated tumors; D-I. GSEA plots of selected pathways: upregulated (D,E) and downregulated (F-I).
4. Discussion and conclusions
In this study, an in-depth characterization of rationally designed Ref-1 redox signaling inhibitors was performed with SAR guidance to evaluate PK properties, safety, stability, target inhibition, and efficacy in monolayer and 3D against aggressive tumors like PDAC and MPNST. While Ref-1 is primarily known for its DNA repair function, its redox function leads to transcriptional activation of multiple TFs which plays a role in numerous diseases [52]. Ref-1 acts as a transcriptional regulator by reducing critical cysteines of several cancer-related TFs including NFκB, HIF1α, and STAT3. Oxidized TFs do not bind to DNA as effectively as reduced TFs, and therefore Ref-1 redox activity contributes to full activation of these TFs [53]. We along with others have shown that Ref-1 is overexpressed in many pathological conditions including cancer [22]. Its overexpression in cancers such as lung [54,55], breast [56], prostate [57], colorectal [32,58], prostate [57], colorectal [56], sarcoma [33] and pancreatic [32,58], suggests that Ref-1 contributes to tumor progression, chemoresistance, and poor prognosis.
Both functions of Ref-1 have been shown to contribute to above aggressive phenotypes [59]. Although inhibition of its DNA repair function is likely to dramatically impact cell survival, concern over the therapeutic window and the toxicity to normal cells has led us to focus on redox inhibition [60,61]. Hence targeting the redox function of Ref-1 using small molecule inhibitors that are selective and potent has implications against cancer, age-related macular degeneration (AMD), diabetic retinopathy (DR), and other HIF-driven diseases [37]. We developed a Ref-1 redox inhibitor, APX3330, that has completed Phase I clinical trial with good response, safety profile and target engagement [62]. It also completed Phase II trial in DR/DME with positive outcomes and a strong safety profile [63]. Even though APX3330 is proven to be an effective compound in cancer, its potency is lower in in vitro cellular assays and may be problematic for difficult to penetrate solid tumors such as PDAC. Hence, we identified second generation naphthoquinone derivatives and evaluated their potency for Ref-1 redox function and impact on cell survival of solid tumors and soft tissue tumors [62].
From a list of 350 compounds, we narrowed down the compounds to 13 and then identified five lead inhibitors (APX2009, APX2014, APX2044, APX2051, and APX2053). The compounds presented here differ from and show a number of advantages over parent compound, APX3330. Based on our SAR studies, the newer compounds have substituted dimethoxy substituents with an aromatic moiety on the left-hand side followed by replacement of the carboxylic acid with the more neutral amide moieties which has led to a new chemical scaffold for further alterations. In addition, we blocked the potent metabolic 3-methyl position of APX3330. Lastly, the very lipophilic nonyl side chain of APX3330 was modified to a small, less lipophilic propyl group. These new compounds are much more efficacious as demonstrated with improved drug-like properties, acceptable, partition coefficient log P, and polar surface area (PSA). In-silico modeling suggested acceptable solubility, permeability, and metabolic stability across species with no red flags related to rat primary hepatocyte (RPH) cytotoxicity, CYP inhibition, human either a go-no-go related Gene (hERG) inhibition, phospholipidosis, mutagenicity and P-glycoprotein substrate. The aqueous solubility as well as the solubility in simulated fluids were reasonable for the lead compounds and consistent with in-silico predictions. Moderate to high CaCO2 bi-directional permeability was observed across the series (Table 2).
Based on different functional side chains like methoxy (R-OCH3 in APX2009 with R-OCH3 and an amide in APX2014), trifluoromethyl (R-CF3 in APX2044), fluoro (R-F in APX2051) and hydroxyl (R-OH in APX2053) groups present (Fig. 1), there were differences in the activity of the inhibitors. Of note, O-demethylation of APX2009 resulting in compound APX2053 demonstrated a lack of inhibition of Ref-1 redox activity as demonstrated by NFκB-driven luciferase reporter assay (Fig. 3A), no significant change in the NADPH/NADP+ levels (Fig. 4B) and a decrease in cell killing effect in the 3D spheroid assay (APX2009: 4.3 μM and APX2053: 16 μM) (Supplementary Figure 5 A, C and D). Substitution of a fluorine (APX2051) or -CF3 (APX2044) at this same position gave increased inhibition of TF activity and also had potent cell killing effects, especially in the tumor cells. All five compounds had peaks in waterLOGSY difference spectra that were consistent with interactions with Ref-1. APX2053 and RN7–58 did not exhibit effective inhibition of Ref-1 activity but did bind to Ref-1 as assessed by waterLOGSY. The waterLOGSY results suggest that RN7–58 may interact at a different site on Ref-1 than the other compounds with the interactions at the methoxy protons of the compound. The fact that APX2053 interacts with Ref-1 but lacks activity suggests that interaction with Ref-1 is necessary but insufficient to guarantee redox inhibition. Further mechanistic studies will be required to determine why APX2053 with the hydroxyl group on the ring instead of -OCH3, F, or CF3 impacts activity.
Ref-1 interacts directly with TFs including NFκB and HIF1α through a thiol reaction between the cysteine groups (R-SH) of Ref-1 and the TFs, thereby activating the TF to bind to the promoter region of genes involved in cellular signaling pathways. Therefore, as a primary screen for target inhibition, we evaluated the inhibitors identified through SAR and PK studies against PDAC and MPNST cell lines stably expressing either NFκB- or HIF1α - luciferase reporter construct. These reporter assays confirmed dose-dependent inhibition of TF activity by APX2009, 2014, 2044, and 2051, but not 2053. Also, it was interesting to note that MPNST cells were more sensitive to Ref-1 inhibition than the PDAC cells within compared doses. This could be due to the influence of the TME in which the tumors evolved in in vivo and this would differ between cancers. For example, PDAC is known for its inflammatory, fibrotic, and dense TME [64]. Further, it is evident from the literature that HIF1α drives tumor progression in both MPNST and PDAC, but PDAC is considered more severely hypoxic [18]. Hence, we hypothesize that, the lead compounds exhibit differential activity against different disease types based on the level of transcription factor stimuli as demonstrated in Fig. 4A,B. APX2044 showed the highest potency against both NFκB and HIF1α TF activity at doses that were non-lethal to cells indicating high specificity for Ref-1 inhibition (Fig. 3 and Table 3). These exciting results demonstrate a 6-fold increase in potency of these compounds compared to APX3330 for reduction in Ref-1-regulated TF activity.
Ref-1 modulates cellular redox state in many ways: by regulating cellular signaling through binding with TFs, interactions with NRF2, and via inhibition of intracellular ROS production [65]. Multiple TFs regulated by Ref-1 in turn control several metabolic signals. To confirm redox-regulated metabolic downregulation with the new inhibitors, cancer cells were treated with the top inhibitors and the impact on metabolism was assessed using a plate-based mitochondrial functional assay and NADPH assay. The mitochondrial functional assay demonstrated that Ref-1 redox inhibition leads to significant downregulation of TCA cycle substrates, α-KG, succinate, malate, and fumarate through enhancement of oxidized cellular state (decrease in NADPH available for energy production) as well as changes in expression of proteins involved in the electron transport chain. In the current study, we observed a similar, but significantly enhanced inhibition of these TCA cycle substrates and NADPH/NADP+ ratio in the presence of the new analogs compared to APX3330 (used a >4-fold higher concentration) (Fig. 4A,B). As a result of inhibition of Ref-1 and the effects on mitochondrial metabolites and NADPH levels, a significant inhibition of tumor cell viability has been observed in 2D cytotoxicity assay. APX2051, APX2014, and APX2044 dramatically reduced the growth of MPNST patient tumor lines (R-HT163 and R-HT172) by 8–10-fold, and PDAC patient tumor lines (both Pa03C and Pa08X) by 2-, 4- and 10-fold respectively compared to APX3330 (Fig. 4C, D, E, F). Interestingly, APX2051 and APX2044 were less potent in CAFs than tumors (Fig. 4C–H). CAF proliferation was affected more at higher doses of these compounds whereas tumors demonstrated a more dose-dependent response to Ref-1 inhibition (Fig. 4D, F and H). CAFs and tumor cells interact and communicate via paracrine and autocrine secretion involving nutrient exchange which can impact survival under various stressors including hypoxia and nutrient scarcity. Hence targeting both populations of cells may be an effective strategy to target tumor growth and metastasis. However, ongoing research seeks to dissect and understand which CAFs are tumor-promoting and which CAFs are tumor-restraining and the role that Ref-1 may play in promoting either an iCAF (inflammatory-CAFs) or myCAF (myofibroblastic) phenotype. Perhaps iCAFs would be more sensitive to Ref-1 inhibition due to the expression of IL-6 and its role in stimulating STAT3 pathway which is under Ref-1 redox control. We would predict that Ref-1 inhibition would block iCAFs as Ref-1 has been shown to play a role in the regulation of inflammatory pathways through its regulation of NFκB and STAT3 [66] and hence could be part of a regimen to target the CAFs that enhance tumor growth [67]. This study identified promising Ref-1 redox inhibitors that are potentially more cytotoxic to tumor cells than CAFs warranting more studies to understand which subtype of CAFs rely on Ref-1 signaling.
Until recently, the majority of cell-based drug screening was performed using monolayer cell culture models. However, this screening platform does not clearly represent the complexity of tumor tissue and its microenvironment which likely contributes to the failure of translating drugs from pre-clinical to clinical setting. In these studies, three-dimensional (3D) cell culture technologies that more closely resemble in vivo cell environments were used for better precision of Ref-1 inhibitor activity [68,69]. To distinguish effects on tumor cells versus CAFs, cells were labeled with different fluorescent markers. A traditional spheroid model as well as tumor microenvironment on chip (T-MOC) model were further used to analyze the potency and cytotoxicity of the lead inhibitors. 3D co-culture spheroid model utilized here provides more physiologically similar environment with regions of hypoxia and deposition of extracellular matrix (ECM) components by the CAFs [31]. Identified lead compounds showed enhanced tumor killing compared to CAFs (>2-fold potent) even under increased hypoxic conditions observed within the spheroids [70]. There is a significant difference between the IC50s in the tumors versus CAFs with the spheroid model. However treatment with Ref-1 compounds do still induce a decrease in CAF proliferation over time (Fig. 5C and Supplementary Figures 5A, B). Future studies will interrogate if there is a difference in the gene signature or phenotype of the CAFs that remain following Ref-1 inhibition.
As a second 3D co-culture model for screening Ref-1 inhibitors, the microfluidic T-MOC platform was used to assess differences in potency of Ref-1 inhibitors against tumors and CAFs. The T-MOC system was chosen to effectively mimic the interstitial drug transport in PDAC in the presence of CAFs, while also providing 3D culture conditions with a high content of type I collagen and fluid flow [71]. Tissue fluid pressure governs the uptake of nutrients and fluids by the cells within an organism. This pressure increases substantially under neoplastic conditions compared to healthy tissues and thus an increase in interstitial pressure within these tumors leads to ineffective systemic therapy [72]. T-MOC system was chosen to effectively mimic the interstitial drug transport in PDAC in presence of CAFs, while also providing 3D culture conditions with a high content of type I collagen and fluid flow [71]. Strikingly, in the T-MOC model, we again observed a selectivity for tumor cell growth inhibition compared to CAFs similar to the 3D spheroid model (Fig. 5F and Supplementary Figures 5C, D).
As further confirmation of on-target effects of the new Ref-1 analogs, proteomic analysis of Ref-1 treated tumors identified several significantly downregulated pathways that have been previously implicated in Ref-1 signaling [32]. A significant reduction of proteins pertaining to cell cycle, DNA repair, electron transport chain, and oxidative phosphorylation was observed as reported in ours as well as others’ publications [32]. Interestingly, TGFβ signaling was also downregulated and this pathway plays an important role in PDAC signaling, therefore we will follow up on this finding in future work. Additionally, there was upregulation of some proteins in the tumors after Ref-1 inhibition. More proteins were downregulated as we are modulating a transcriptional regulator which should result in less TF binding and less transcription of downstream targets. Proteins associated with apoptosis were increased which is a favorable response as we try to enhance the killing of tumors. Induction of matrisomal proteins was observed in tumors treated with Ref-1 redox inhibitors (Fig. 6). Identifying proteins that increase following Ref-1 treatment may also provide us with mechanisms of therapeutic resistance which will be studied more in depth in future work.
Apart from its involvement in cancer, Ref-1 has also been implicated in the protection of Dorsal root ganglion (DRG) neurons after cisplatin oncotherapy and also enteric neurons in Inflammatory bowel disease model via increased repair of oxidative DNA damage [73,74]. Furthermore, Ref-1 is implicated in regulating inflammatory responses in diseases such as rheumatoid arthritis, inflammatory bowel disease, and sepsis, influencing the activity of NFκB and maintaining the balance between pro-inflammatory and anti-inflammatory signaling pathways [75,76]. We identified lead molecules that can specifically target Ref-1′s redox function, show increased efficacy in a series of in vitro assays, and display characteristics that exceed the critical success factors for advancement to preclinical toxicology studies. Further, development of these molecules as novel oral Ref-1 inhibitors offers promising strategies for the treatment of cancer as well as other disease indications. While we attempted to rank the lead compounds that were characterized, there are still challenges and additional studies to be done as we progress toward IND and the clinic. Such challenges include: further understanding of penetration of drug within the tumor, how inhibition of transcription through various TFs is blocking tumor growth and its impact on the TME, understanding how the various compounds bind to Ref-1 and thereby inhibit its redox activity. Future studies will also address any potential off-target effects. Our group recently published that pancreatic tumor cells that were modified by CRISPR-editing to express the Ref-1 redox deficient mutant displayed smaller primary tumors in the pancreas as well as a reduction in metastatic burden. This work further demonstrates that Ref-1 redox activity is a viable target in cancer and the importance of advancing these molecules [77]. In vivo efficacy studies are underway to enable us to move the top compounds to IND enabling studies for clinical trials. In conclusion, the top five Ref-1 redox inhibitors were specific to Ref-1 redox inhibition, potent against both solid tumors and soft tissue tumors, and had favorable drug characteristics, PK and metabolic profiles.
Supplementary Material
Acknowledgements
Pharmacokinetic data for characterization of the new Ref-1 inhibitors was provided by the Clinical Pharmacology Analytical Core at Indiana University School of Medicine; a core facility supported by the IU Simon Comprehensive Cancer Center (IUSCCC) Support Grant P30 CA082709. The mass spectrometry work performed in this work was done by the Indiana University School of Medicine Proteomics Core. Acquisition of the IUSM Proteomics core instrumentation used for this project was provided by the Indiana University Precision Health Initiative. The proteomics work was supported, in part, with support from the Indiana Clinical and Translational Sciences Institute funded, in part by Award Number UL1TR002529 from the National Institutes of Health, National Center for Advancing Translational Sciences, Clinical and Translational Sciences Award and the Cancer Center Support Grant for the IU Simon Comprehensive Cancer Center (Award Number P30CA082709) from the National Cancer Institute. Mice were purchased through the In Vivo Therapeutic Core within IUSCCC. MPNST cell lines, R-HT163 and R-HT172 were generated from the patient-derived xenograft tissue (Riley Children’s Hospital) kindly provided by Dr. Karen Pollok, Dr. Reza M Saadatzadeh and In Vivo Therapeutics core within IUSCCC. The graphical abstract was created using BioRender (BioRender.com).
Funding
This work was supported by National Institutes of Health grants from the National Cancer Institute. This work was supported by grants from the National Institute of Health, National Cancer Institute (R01CA167291 (M.L.F. and M.R.K.), R01CA254110 (M.L.F. and M.R. K.), R01CA264471 (M.L.F.), R01CA231267 (M.R.K.) and R01CA205166 (M.R.K.)) and the National Heart, Lung, and Blood Institute (U01HL143403 (M.L.F. and B.H.) and R01HL140961 (M.R.K.)) and U01 Pancreatic Ductal Adenocarcinoma (PDAC) Stromal Reprogramming Consortium (PSRC) (U01CA274304) (M.L.F. and B.H.). M.L.F. was additionally supported by the Riley Children’s Foundation and the IU Simon Comprehensive Cancer Center, P30CA082709. M.R.K. was also supported by National Eye Institute grant (R01EY031939).
Footnotes
Ethics approval and consent to participate
All animal studies were conducted under the guidelines of the National Institutes of Health and were approved by the Institutional Animal Care and Use Committee of Indiana University School of Medicine.
CRediT authorship contribution statement
Millie M. Georgiadis: Writing – review & editing, Supervision, Conceptualization. Christine Bach: Writing – original draft, Resources, Methodology, Investigation, Formal analysis. Whitney Smith-Kinnamen: Supervision, Methodology, Investigation, Formal analysis. Silpa Gampala: Writing – review & editing, Writing – original draft, Validation, Methodology, Investigation, Formal analysis, Conceptualization. Mark R. Kelley: Writing – review & editing, Writing – original draft, Supervision, Resources, Funding acquisition, Conceptualization. Melissa L. Fishel: Writing – review & editing, Writing – original draft, Visualization, Validation, Supervision, Resources, Methodology, Investigation, Funding acquisition, Formal analysis, Data curation, Conceptualization. Emma H. Doud: Writing – original draft, Validation, Software, Methodology, Investigation, Formal analysis. Ratan Rai: Writing – original draft, Methodology, Investigation, Formal analysis. Hye-ran Moon: Writing – review & editing, Visualization, Methodology, Investigation, Formal analysis. Amber L. Mosley: Supervision. Randall Wireman: Writing – review & editing, Methodology, Investigation, Formal analysis. Sara K. Quinney: Supervision. Jacqueline Peil: Validation. D. Wade Clapp: Supervision. Sonia Kiran: Validation. Chafiq Hamdouchi: Writing – original draft, Validation, Supervision, Methodology, Investigation, Conceptualization. Dana K. Mitchell : Methodology. James Wikel: Supervision, Conceptualization. Kylee Brewster: Investigation. Chi Zhang: Writing – original draft, Validation, Methodology, Investigation, Formal analysis. Henry Mang: Investigation. Bumsoo Han: Writing – review & editing, Supervision, Funding acquisition, Conceptualization. Andi Masters: Writing – original draft, Methodology, Investigation, Formal analysis.
Declaration of Competing Interest
None.
Mark R. Kelley has licensed APX3330 through Indiana University Research and Technology Corporation to Apexian Pharmaceuticals LLC. Second generation compounds also licensed to Apexian Pharmaceuticals. Apexian has licensed some compounds to Ocuphire Parma. Neither Apexian Pharmaceuticals nor Ocuphire Pharma had control nor oversight of the studies, interpretation, or presentation of the data in this manuscript. Authors do not have any conflict of interests.
Appendix A. Supporting information
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.phrs.2024.107092.
Data Availability
No data was used for the research described in the article.
References
- [1].Siegel RL, Miller KD, Fuchs HE, Jemal A, Cancer statistics, 2022, CA Cancer J. Clin 72 (2022) 7–33. [DOI] [PubMed] [Google Scholar]
- [2].Cronin KA, et al. , Annual report to the nation on the status of cancer, part 1: National cancer statistics, Cancer (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Henley SJ, et al. , Annual report to the nation on the status of cancer, part I: National cancer statistics, Cancer 126 (2020) 2225–2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Chhikara BS, Parang K, Global cancer statistics 2022: the trends projection analysis, Chem. Biol. Lett 10 (2022) 451. [Google Scholar]
- [5].Sun J, Russell CC, Scarlett CJ, McCluskey A, Small molecule inhibitors in pancreatic cancer, RSC Med. Chem 11 (2020) 164–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Smith CEP, Prasad V, Targeted cancer therapies, Am. Fam. Physician 103 (2021) 155–163. [PubMed] [Google Scholar]
- [7].Padma VV, An overview of targeted cancer therapy, Biomedicine 5 (2015) 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Zhang G, Zhang J, Gao Y, Li Y, Li Y, Strategies for targeting undruggable targets, Expert Opin. Drug Discov 17 (2022) 55–69. [DOI] [PubMed] [Google Scholar]
- [9].Yedida GR, Nagini S, Mishra R, The importance of oncogenic transcription factors for oral cancer pathogenesis and treatment, Oral Surg. Oral Med Oral Pathol. Oral Radio 116 (2013) 179–188. [DOI] [PubMed] [Google Scholar]
- [10].Ell B, Kang Y, Transcriptional control of cancer metastasis, Trends Cell Biol. 23 (2013) 603–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Baumgart S, Ellenrieder V, Fernandez-Zapico ME, Oncogenic transcription factors: cornerstones of inflammation-linked pancreatic carcinogenesis, Gut 62 (2013) 310–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Zboril E, Yoo H, Chen L, Liu Z, Dynamic interactions of transcription factors and enhancer reprogramming in cancer progression, Front. Oncol 11 (2021) 753051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Gupta VK, et al. , Hypoxia-driven oncometabolite L-2HG maintains stemness-differentiation balance and facilitates immune evasion in pancreatic cancer, Cancer Res. 81 (2021) 4001–4013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Shah VM, Sheppard BC, Sears RC, Alani AW, Hypoxia: friend or foe for drug delivery in pancreatic cancer, Cancer Lett. 492 (2020) 63–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Lama-Sherpa TD, Kammerud SC, Shevde LA, Immunology (2020), 115–115. [Google Scholar]
- [16].Tang K, et al. , Hypoxia-reprogrammed tricarboxylic acid cycle promotes the growth of human breast tumorigenic cells, Oncogene 38 (2019) 6970–6984. [DOI] [PubMed] [Google Scholar]
- [17].Fukushima S, et al. , Hypoxia-inducible factor 1 alpha is a poor prognostic factor and potential therapeutic target in malignant peripheral nerve sheath tumor, Plos One 12 (2017) eOl78064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Emami Nejad A, et al. , The role of hypoxia in the tumor microenvironment and development of cancer stem cell: a novel approach to developing treatment, Cancer Cell Int. 21 (2021) 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Naqvi AAT, Hasan GM, Hassan MI, Investigating the role of transcription factors of pancreas development in pancreatic cancer, Pancreatology 18 (2018) 184–190. [DOI] [PubMed] [Google Scholar]
- [20].van Roey R, Brabletz T, Stemmier MP, Armstark I, Deregulation of transcription factor networks driving cell plasticity and metastasis in pancreatic cancer, Front. Cell Dev. Biol 9 (2021) 753456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Islam Z, et al. , Transcription factors: the fulcrum between cell development and carcinogenesis, Front. Oncol 11 (2021) 681377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Caston RA, et al. , The multifunctional APE1 DNA repair-redox signaling protein as a drug target in human disease, Drug Discov. Today (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Mijit M, et al. , Identification of novel pathways regulated by APE1/Ref-1 in human retinal endothelial cells, Int. J. Mol. Sci 24 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Tell G, Fantini D, Quadrifoglio F, Understanding different functions of mammalian AP endonuclease (APE1) as a promising tool for cancer treatment, Cell. Mol. Life Sci (2010) 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Rai R. et al. , Chemically Induced Partial Unfolding of the Multifunctional Apurinic/apyrimidinic Endonuclease 1. bioRxiv (2023). [Google Scholar]
- [26].Luo M, et al. , Characterization of the redox activity and disulfide bond formation in apurinic/apyrimidinic endonuclease, Biochemistry 51 (2012) 695–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Zhang J, et al. , Inhibition of apurinic/apyrimidinic endonuclease I’s redox activity revisited, Biochemistry 52 (2013) 2955–2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Luo M, He H, Kelley MR, Georgiadis M, Redox regulation of DNA repair: implications for human health and cancer therapeutic development, Antioxid. Redox Signal 12 (2010) 1247–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kelley MR, Georgiadis MM, Fishel ML, APE1/Ref-1 role in redox signaling: translational applications of targeting the redox function of the DNA repair/redox protein APE1/Ref-1, Curr. Mol. Pharmacol 5 (2012) 36–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Kelley MR, Logsdon D, Fishel ML, Targeting DNA repair pathways for cancer treatment: what’s new? Future Oncol. 10 (2014) 1215–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Logsdon DP, et al. , Blocking HIF signaling via novel inhibitors of CA9 and APE1/Ref-1 dramatically affects pancreatic cancer cell survival, Sci. Rep 8 (2018) 13759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Gampala S, et al. , Ref-1 redox activity alters cancer cell metabolism in pancreatic cancer: exploiting this novel finding as a potential target, J. Exp. Clin. Cancer Res 40 (2021) 251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Gampala S, et al. , Exploring transcriptional regulators Ref-1 and STAT3 as therapeutic targets in malignant peripheral nerve sheath tumours, Br. J. Cancer 124 (2021) 1566–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Chu L, et al. , CTC enumeration and characterization as a pharmacodynamic marker in the phase I clinical study of APX3330, an APE1/Ref-1 inhibitor, in patients with advanced solid tumors, e14531-e14531, J. Clin. Oncol 37 (2019). e14531–e14531. [Google Scholar]
- [35].Fishel ML, Cheng H, Shahda S, & Kelley MR, in AACR-NCI-EORTC International Conference: Molecular Targets and Cancer Therapeutics (Mol Cancer Ther December 2015 14; B167, Boston, MA; 2015). [Google Scholar]
- [36].Shahda S, et al. , A phase I study of the APE1 protein inhibitor APX3330 in patients with advanced solid tumors, J. Clin. Oncol 37 (2019), 3097–3097. [Google Scholar]
- [37].Mijit M CR, Gampala S, Fishel ML, Fehrenbacher J, Kelley MR, APE1/Ref-1 – one target with multiple indications: emerging aspects and new directions, J. Cell. Signal 2 (2021) 151–161. [PMC free article] [PubMed] [Google Scholar]
- [38].Jones S, et al. , Core signaling pathways in human pancreatic cancers revealed by global genomic analyses, Science 321 (2008) 1801–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Richards KE, et al. , Cancer-associated fibroblast exosomes regulate survival and proliferation of pancreatic cancer cells, Oncogene 36 (2017) 1770–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Ozcelikkale A, et al. , Differential response to doxorubicin in breast cancer subtypes simulated by a microfluidic tumor model, J. Control. Release 266 (2017) 129–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Kumar B, et al. , Tumor collection/processing under physioxia uncovers highly relevant signaling networks and drug sensitivity, Sci. Adv 8 (2022) eabh3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Subramanian A, et al. , Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles, Proc. Natl. Acad. Sci 102 (2005) 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Raingeval C, et al. , 1D NMR WaterLOGSY as an efficient method for fragment-based lead discovery, J. Enzym. Inhib. Med. Chem 34 (2019) 1218–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Su D, et al. , Interactions of Apurinic/Apyrimidinic endonuclease with a redox inhibitor: evidence for an alternate conformation of the enzyme, Biochemistry (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Rai G, et al. , Synthesis, biological evaluation, and structure-activity relationships of a novel class of apurinic/apyrimidinic endonuclease 1 inhibitors, J. Med Chem 55 (2012) 3101–3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Cardoso AA, et al. , APE1/Ref-1 regulates STAT3 transcriptional activity and APE1/Ref-1-STAT3 dual-targeting effectively inhibits pancreatic cancer cell survival, Plos One 7 (2012) e47462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Gritsko T, et al. , Persistent activation of stat3 signaling induces survivin gene expression and confers resistance to apoptosis in human breast cancer cells, Clin. Cancer Res 12 (2006) 11–19. [DOI] [PubMed] [Google Scholar]
- [48].Al Zaid Siddiquee K, Turkson J, STAT3 as a target for inducing apoptosis in solid and hematological tumors, Cell Res. 18 (2008) 254–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Aoki Y, Feldman GM, Tosato G, Inhibition of STAT3 signaling induces apoptosis and decreases survivin expression in primary effusion lymphoma, Blood 101 (2003) 1535–1542. [DOI] [PubMed] [Google Scholar]
- [50].Ni Y, et al. , The role of tumor-Stroma interactions in drug resistance within tumor microenvironment, Front. Cell Dev. Biol 9 (2021) 637675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Wan C, et al. , LTMG: a novel statistical modeling of transcriptional expression states in single-cell RNA-Seq data, Nucleic Acids Res. 47 (2019) e111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Heisel C, et al. , APE1/Ref-1 as a novel target for retinal diseases, J. Cell Signal 2 (2021) 133–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Ayyildiz D, et al. , Architecture of the human APE1 interactome defines novel cancers signatures, Sci. Rep 10 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Yang X, et al. , The regulatory role of APE1 in epithelial-to-mesenchymal transition and in determining EGFR-TKI responsiveness in non-small-cell lung cancer, Cancer Med. (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Li Z, et al. , Apurinic endonuclease 1 promotes the cisplatin resistance of lung cancer cells by inducing Parkin-mediated mitophagy, Oncol. Rep 42 (2019) 2245–2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Codrich M, et al. , Inhibition of APE1-endonuclease activity affects cell metabolism in colon cancer cells via a p53-dependent pathway, DNA Repair 82 (2019) 102675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Mcllwain David W., Alexander Boos MLF, Kelley Mark R., Jerde Travis J., APE1/Ref-1 redox-specific inhibition decreases survivin protein levels and induces cell cycle arrest in prostate cancer cells, Oncotarget 9 (2018) 10962–10977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Choi Y-D, et al. , APE1 promotes pancreatic cancer proliferation through GFRα1/Src/ERK Axis-cascade signaling in response to GDNF, Int. J. Mol. Sci 21 (2020) 3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Kothandapani A, Sawant A, Dangeti VS, Sobol RW, Patrick SM, Epistatic role of base excision repair and mismatch repair pathways in mediating cisplatin cytotoxicity, Nucleic Acids Res. 41 (2013) 7332–7343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Meira LB, et al. , Heterozygosity for the mouse Apex gene results in phenotypes associated with oxidative stress, Cancer Res. 61 (2001) 5552–5557. [PubMed] [Google Scholar]
- [61].Xue Z, Demple B, Knockout and inhibition of APE1: roles of APE1 in base excision DNA repair and modulation of gene expression, Antioxidants 11 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Demin AA, et al. , XRCC1 prevents toxic PARP1 trapping during DNA base excision repair, Mol. Cell 81 (2021) 3018–3030, e3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Gonzalez VH, et al. , 25-LB: oral APX3330 reduces the DRSS worsening after 24-weeks of daily treatment—efficacy and safety results of the ZETA-1 Phase 2 trial in diabetic retinopathy, Diabetes 72 (2023). [Google Scholar]
- [64].Taniguchi K, Karin M, NF-kappaB, inflammation, immunity and cancer: coming of age, Nat. Rev. Immunol 18 (2018) 309–324. [DOI] [PubMed] [Google Scholar]
- [65].Abdel Hadi N, Reyes-Castellanos G, Carrier A, Targeting redox metabolism in pancreatic cancer, Int. J. Mol. Sci 22 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Oliveira TT, et al. , APE1/Ref-1 role in inflammation and immune response, Front. Immunol 13 (2022) 793096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Simon T, Salhia B, Cancer-associated fibroblast subpopulations with diverse and dynamic roles in the tumor microenvironment, Mol. Cancer Res 20 (2022) 183–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Langhans SA, Three-dimensional in vitro cell culture models in drug discovery and drug repositioning, Front. Pharm 9 (2018) 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Ajjarapu SM, Tiwari A, Kumar S, Applications and utility of three-dimensional in vitro cell culture for therapeutics, Future Pharmacol. 3 (2023) 213–228. [Google Scholar]
- [70].Logsdon DP, et al. , Regulation of HIF1alpha under hypoxia by APE1/Ref-1 impacts CA9 expression: dual targeting in patient-derived 3D pancreatic cancer models, Mol. Cancer Ther 15 (2016) 2722–2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Moon HR, et al. , An engineered pancreatic cancer model with intra-tumoral heterogeneity of driver mutations, Lab a chip 20 (2020) 3720–3732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].DuFort CC, et al. , Interstitial pressure in pancreatic ductal adenocarcinoma is dominated by a Gel-Fluid phase, Biophys. J 110 (2016) 2106–2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Jiang Y, Guo C, Vasko MR, Kelley MR, Implications of Apurinic/Apyrimidinic endonuclease in reactive oxygen signaling response after Cisplatin treatment of dorsal root ganglion neurons, Cancer Res. 68 (2008) 6425–6434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Sahakian L, et al. , APE1/Ref-1 as a therapeutic target for inflammatory Bowel disease, Biomolecules 13 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Kelley MR, Georgiadis MM, Fishel ML, APE1/Ref-1 role in redox signaling: translational applications of targeting the redox function of the DNA repair/redox protein APE1/Ref-1, Curr. Mol. Pharmacol 5 (2012) 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Tell G, Damante G, Caldwell D, Kelley MR, The intracellular localization of APE1/Ref-1: more than a passive phenomenon? Antioxid. Redox Signal 7 (2005) 367–384. [DOI] [PubMed] [Google Scholar]
- [77].Mijit M, et al. , In vitro and In vivo evidence demonstrating chronic absence of Ref-1 Cysteine 65 impacts Ref-1 folding configuration, redox signaling, proliferation and metastasis in pancreatic cancer, Redox Biol. 69 (2023) 102977. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
No data was used for the research described in the article.