

Abstract

The HIV-1 assembly process begins with a newly synthesized Gag polyprotein being targeted to the inner leaflet of the plasma membrane of the infected cells to form immature viral particles. Gag–membrane interactions are mediated through the myristoylated (Myr) N-terminal matrix (MA) domain of Gag, which eventually multimerize on the membrane to form trimers and higher order oligomers. The study of the structure and dynamics of peripheral membrane proteins like MA has been challenging for both experimental and computational studies due to the complex transient dynamics of protein–membrane interactions. Although the roles of anionic phospholipids (PIP2, PS) and the Myr group in the membrane targeting and stable membrane binding of MA are now well-established, the cooperative interactions between the MA monomers and MA-membrane remain elusive in the context of viral assembly and release. Our present study focuses on the membrane binding dynamics of a higher order oligomeric structure of MA protein (a dimer of trimers), which has not been explored before. Employing time-lagged independent component analysis (tICA) to our microsecond-long trajectories, we investigate conformational changes of the matrix protein induced by membrane binding. Interestingly, the Myr switch of an MA monomer correlates with the conformational switch of adjacent monomers in the same trimer. Together, our findings suggest complex protein dynamics during the formation of the immature HIV-1 lattice; while MA trimerization facilitates Myr insertion, MA trimer–trimer interactions in the immature lattice can hinder the same.
Introduction
The human immunodeficiency virus type 1 (HIV-1) assembly process is mediated primarily by the retroviral group-specific antigen (Gag) polyprotein.1,2 Membrane binding of Gag to the inner leaflet of the plasma membrane (PM) of the host cell is a crucial step in the assembly process. The Gag polyprotein consists of the matrix (MA), capsid (CA), spacer-peptide 1 (SP1), nucleocapsid (NC), spacer-peptide 2 (SP2), and p6 domains. While the CA and NC domains contribute to Gag multimerization and RNA encapsidation during the viral assembly,3−5 the p6 domain plays an important role in viral budding. Membrane targeting to the PM and recruitment of the envelope (Env) protein is mediated by the MA domain, which is post-translationally acylated with a myristate (Myr) group in its N-terminus.6−10 Structural studies have shown that MA proteins primarily oligomerize into trimeric structures, which then undergo higher order oligomerization to form a lattice of hexamer-of-trimers.11 MA binding to the PM is facilitated by several key factors, including electrostatic interactions of its highly basic region (HBR) with negatively charged lipid headgroups, hydrophobic interactions of the Myr group, membrane internal structure, etc.11−20 Moreover, myristoylated MA ((+)Myr-MA) prefers binding specifically to membrane microdomains enriched in PI(4,5)P2 and cholesterol.21,22 The relative contribution of MA–lipid interactions is nontrivial to determine; however, it likely depends on the multimerization state of Gag and the specific protein–protein and protein–membrane interfaces.
MA protein is a peripheral membrane protein (PMP) whose biological activity is predominantly determined by its ability to switch between a soluble and membrane-bound state. Unlike integral membrane proteins, specific protein–lipid binding site structures of PMPs are hard to determine both for experiment and simulation studies due to the unique nature of PMP–membrane interactions, which are often transient.12,18,23−29 A few long-time scale all-atom molecular dynamics (AAMD) simulations aimed to explore this nonequilibrium process of PMP binding with the membrane.30−35 Although the strategy of AAMD simulations to study the membrane binding of PMP seems simple, it is computationally expensive even for a small monomeric PMP as enough sampling is needed to assess protein–membrane interfaces. Moreover, the HIV-1 MA assembly process involves MA binding and aggregation that modify the local composition and lipid dynamics of the membrane.
The PM of eukaryotic cells has asymmetric lipid distribution and diverse lipid unsaturation. The inner PM leaflet is enriched in unsaturated phospholipids, like phosphatidylethanolamine (PE), phosphatidylserine (PS), phosphatidylcholine (PC), a small amount of phosphatidylinositol (PI) lipids, and cholesterol, whereas the outer PM leaflet mainly contains phosphatidylcholine (PC), saturated sphingomyelin lipids (SM), and cholesterol. Cholesterol helps counteract unfavorable packing of saturated and unsaturated lipids in membranes in general and maintains structural integrity and membrane fluidity. Experimental studies on HIV-1 assembly dynamics have suggested that Gag multimerization on the PM induces clustering of specific lipid species like PIP2 and cholesterol in the inner leaflet, which in turn remodels the outer leaflet through trans-bilayer coupling.15,36−39 The relationship between MA multimerization and membrane binding is still unclear along with its impact on the lateral organization of lipids in each PM leaflet.40,41
A cryoelectron tomography (cryo-ET) experimental study by Briggs and co-workers has resolved the MA trimer–trimer interacting interface in immature virus particles.12 Based on the cryo-ET density map, they suggested that no Myr group remains in the sequestered position in the immature MA lattice (Figure 1). MA trimer–trimer interactions in the fitted atomic model (PDB ID: 7OVQ) are mediated by N-terminal residues, namely α-helix 1(H1) and 310 helix (shown in Figure 1E). Another experimental study by Saad and co-workers studied such MA–MA interactions in crystal structures using X-ray crystallography and NMR spectroscopy.20 In a previous AAMD study, we characterized the membrane binding mechanism of MA monomers and preformed trimers, along with details of Myr insertion and the initial membrane response to protein binding.31 It was observed that the trimer conformation enhances lipid rearrangement and accelerates Myr insertion. An enrichment of PIP2 at the MA-binding site has been suggested several times before.11,42,43 Although it is well accepted now that MA trimerization and PIP2 recruitment enhance Myr exposure,10,18 the effect of MA trimer–trimer interactions on Myr insertion is yet to be explored. The effect of protein–lipid interactions on protein conformational changes of MA trimers during membrane binding is the focus of the present study.
Figure 1.

Side view of the membrane-bound MA monomers in two different states of Myr switch. Key regions are labeled: H1, HBR, H2, 310 helix, H3, H4, and H5 (in the order of appearance in the protein sequence). (A) Myr group is sequestered into the hydrophobic pocket and (B) Myr group is inserted into the membrane. (C, D) Top view of the final structures of the MA dimer of trimers at 5 μs of simulations. (E) Interactions at the trimer–trimer interface of the immature-like MA dimer of trimers.
We performed and report here microsecond-long AAMD simulations of two MA trimers initially placed above an asymmetric membrane model. Our realistic membrane model aims to accelerate sampling of protein–lipid interactions, mimicking the membrane environment at MA assembly sites. Our previous AAMD study with MA monomers showed the impact of the membrane model on capturing relevant dynamics for this system.31 Our asymmetric model is based on lipidomic analysis of HIV-1 particles produced in HeLa cells reported by Lorizate et al.36 Using different initial configurations of two formed MA trimers, we observe a correlation between MA–MA interactions, membrane binding dynamics, and Myr insertion.
Simulation Models and Methods
Membrane Model and Protein–Membrane System Setup
The crystallographic structure of trimeric (−)Myr-MA was retrieved from PDBID:1HIW in the Protein Data Bank (https://www.rcsb.org).7 The monomeric (+)Myr-MA structure (PDB ID: 1UPH)10 with all MA residues starting from residue 1 (MYR+GLY) to residue 131(TYR) was then superimposed to the trimer 1HIW to obtain the final trimeric (+)Myr-MA structure. Then, the immature-like MA dimer model was built by fitting the atomic model of the MA trimers to the cryoET density map (EMDB: 13087).12 For the random dimer of trimer simulations, the PDB structure of trimeric MA (PDB ID:1HIW) was lipidated using the CHARMM_GUI Web server. Details of protein–protein interactions (PPI) in these systems are provided in Figures 1 and 3.
Figure 3.

Identification of TTIs in two MA complexes: minimum distance of the adjacent trimer from the residues of trimer 1 and trimer 2 in (A) Immature-like dimer complex and (B) random dimer complex. The last 200 ns of 5 μs simulations is considered here to compute the average minimum distance. Binding domains are shown in blue boxes. Time evolution of TTI during the assembly formation in the random dimer model is monitored using the parameters: (C) minimum distance between two trimers and (D) number of Cα contacts (rcut = 1 nm).
The membrane model designed for the study was based on lipidomics analysis of HIV-1 particles reported by Lorizate et al.36Table 1 contains the molecular fraction of each lipid component per leaflet. This model mimics the asymmetric nature of the PM. The asymmetric bilayers were initially built as two symmetric bilayers, each with the corresponding lipid composition for the desired asymmetric leaflets. These symmetric models were equilibrated for 100 ns before merging one leaflet per model into an asymmetric bilayer. Given the difference in the lipid content, each leaflet had a different surface area. The smaller leaflet patch, modeling the outer leaflet of the PM, was replicated to match the surface area of the leaflet modeling the inner leaflet. Lipid molecules outside of the area covering the larger leaflet patch were deleted to render the asymmetric model with leaflets of the same dimensions. The asymmetric bilayer was further equilibrated for 250 ns before merging the equilibrated protein coordinates 1.0 nm above the bilayer using VMD software.44 Fully hydrated bilayers and an MA trimer in water were built and equilibrated separately using CHARMM-GUI Membrane Builder and Quick-Solvator, respectively.45−49 For immature-like dimer simulations, initially, two MA trimers are arranged where N-terminal residues, H1 and 310 helices, of an MA monomer of trimer1 were in close proximity to the same protein domains of another MA monomer of trimer2 (as reported by the recent cryoET experiment for the MA lattice in immature virions, EMDB: 13087).12 All systems were rendered neutral using a 0.15 M KCl salt solution. The total size of the immature-like dimer systems was ∼590,000 atoms, with a box size of 16.3 nm × 16.5 nm × 22.3 nm. The random dimer system size was ∼490,000 atoms, and the box size was 18.6 nm. Two replicas were run for 5 μs for each system on the Anton2 machine, for a total of 20 μs.
Table 1. Lipid Content for the Membrane Model Used in This Study.
| lipid species | DOPC | LSM | Chol | PIP2 | DOPS | POPE | |
|---|---|---|---|---|---|---|---|
| lipid fraction per leaflet | inner leaflet | 0.4 | 0.0 | 0.15 | 0.15 | 0.15 | 0.15 |
| outer leaflet | 0.3 | 0.35 | 0.35 | 0.0 | 0.0 | 0.0 |
AAMD Simulation Settings and Trajectory Analysis
All simulations used the CHARMM36m force field;50 equilibration runs were performed in GROMACS 2019,51 and microsecond production runs were carried out on the ANTON2 machine.52 Energy minimization was performed using the steepest descent integrator until the force was reduced to 1000 kJ mol–1 nm–1. The initial relaxation of each system was carried out using the default steps obtained from CHARMM-GUI, the platform used to build the initial simulation coordinates. The default relaxation scripts run first in the constant NVT ensemble and then in the constant NPT ensemble for a total of 2 ns. During this phase, the Cα backbone of the protein is harmonically restrained with a force constant of 1000 kJ mol–1 nm–2. Afterward, constant NPT dynamics were used for the equilibration and production runs without any positional restraints on the system. The temperature was kept constant at 310.15 K using the Nose–Hoover thermostat with 1.0 ps coupling constant.53,54 The pressure was set at 1 bar and controlled using the Parinello–Rahman barostat semi-isotropically due to the presence of the membrane; the compressibility factor was set at 4.5 × 10–5 bar–1 with a coupling time constant of 5.0 ps.55,56 Throughout the trajectories in GROMACS, van der Waals interactions were computed using a force-switching function between 1.0 and 1.2 nm, long-range electrostatics were evaluated using particle mesh Ewald,57 and hydrogen bonds were constrained using the LINCS algorithm.58
All final systems were run for at least 200 ns in GROMACS prior to transferring them to the Anton2 machine. Simulation parameters for the Anton2 runs were set by internal ark guesser files; as such, the cutoff values to compute interactions between neighboring atoms are selected automatically during system preparation. Long-range electrostatics in this machine were computed using the Gaussian Split Ewald algorithm,59 and hydrogen bonds were constrained using the SHAKE algorithm.60 Finally, the Nose–Hoover thermostat and MTK barostat controls are used to run NPT dynamics using the Multigrator integrator algorithm on Anton2.61
Analysis of the trajectories was performed using GROMACS, MDAnalysis,62,63 MDTraj,64 and VMD Tcl scripts.44 Time-lagged independent component analysis (tICA)65−68 was run as a dimensionality reduction technique to quantify the protein conformational change during membrane binding; this analysis was done using MDTraj and MSMBuilder.69
Results
Membrane Binding and Myr Insertion of Matrix (MA) Trimer Assembly
We have carried out AAMD simulations with a pair of myristoylated-MA (Myr-MA) trimers in proximity to an asymmetric bilayer. To mimic the plasma membrane environment during HIV-1 assembly, we have carefully chosen the lipid composition of our membrane model. The inner leaflet is enriched with PS, PIP2, and PE lipids, while the outer leaflet contains SM and cholesterol (for details, see the Simulation Models and Methods section). Depending on the initial arrangement of two MA trimers, we have simulated multiple replicas of two different model systems, each for 5 μs, to investigate Myr insertion of the MA trimers and the role of MA trimer–trimer interactions in that. Two systems are shown in Figure 1: (a) Immature-like MA dimer-of-trimers: The MA trimeric interface is stabilized by the interactions of N terminal residues, α-helix 1 (H1) and 310 helix, as seen in the immature virus particles12 (Figure 1E). Initially, the MA dimer of trimers is kept separated from the asymmetric membrane by at least 1 nm. (b) Random MA dimer of trimers: At t = 0, two MA trimers are kept separated from each other at a random arrangement near the asymmetric membrane. Throughout the article, we often refer to these models as “immature-like dimer” and “random dimer”.
We have analyzed detailed trimer–trimer interactions in these systems, which will be discussed later. The structures of membrane-bound MA monomers before and after the Myr switch are shown in Figure 1A,B highlighting key regions (5 α-helices, HBR domain, and 310 helix). Domains are rearranged among themselves and α-helix structures are perturbed depending on the Myr group position, whether it is sequestered into the hydrophobic pocket of MA or inserted into the membrane. Furthermore, with our unbiased AAMD simulations, we could study spontaneous MA conformational change induced by membrane binding.
A previous AAMD study by Monje-Galvan et al. reported membrane insertion events of the Myr moiety of HIV-1 matrix protein in detail.31 According to that study, for a single MA monomer, Myr insertion takes place only from the ‘open bound conformation’ (helix1 and HBR domain of the MA interacts with the membrane) when the domain between the Lid (310 helix) and H1 opens up. The first carbon of the Myr tail, C2, binds the membrane first and the rest of the tail explores the membrane surface for accessible large-enough lipid packing defect suitable for insertion. It was concluded that Myr insertion never occurs in a diving fashion, where the last carbon of the Myr tail, C14, inserts first.31 Although this mechanism of Myr insertion appears to be valid in our current simulations, the conformational exploration step is expected to be influenced by MA–MA interactions present in the system.
Our simulated data in this work suggest that MA trimer–trimer interactions (as observed in immature virions) can inhibit Myr insertion. Figure 2 shows membrane binding events in the two different systems of interest. Within 5 μs, only 2 Myr groups insert into the membrane in the immature-like MA dimer-of-trimer model. None of the Myr groups at the trimer–trimer interface exhibit Myr insertion although they are in exposed states. On the other hand, in the random dimer model (Figure 2), 4 Myr groups are at a stable inserted state at the end of the 5 μs trajectory. In both simulations, at least one Myr group remained in the sequestered state. Figure 2C,D presents the time evolution of the z-component of the distances of Myr group atoms from the membrane center (the membrane normal is oriented along the z-axis). The two carbon atoms of the Myr tail being considered here are the first carbon, C2, which is attached to the N-terminal GLY residue of MA, and the last carbon atom, C14, which remains very close to the bilayer center upon Myr insertion. The switch of the C14 membrane center distance from ∼40 to ∼5–10 Å in Figure 2C,D indicates the time instant of Myr insertion. In the immature-like MA dimer-of-trimer model, Myr insertion events of monomer 5 (p5) and monomer 6 (p6) occur at ∼3400 ns and ∼4700 ns, respectively. In the random MA dimer-of-trimer model, three Myr insertion events of p1, p2, and p4 occur within 500 ns of the trajectory and another (p5) occurs at ∼4000 ns. For the Myr residues in the sequestered state (p1 in the immature-like dimer and p3 in the random dimer model), the z-distances of C2 and C4 from the membrane center remain at ∼40 Å or greater. The Myr insertion pattern in multiple independent trajectories of these systems suggests that the Myr switch is more feasible in the random dimer model for two probable reasons: (1) MA trimers are able to move independently on the membrane surface and (2) NTD conformations of MA proteins are not restrained due to MA trimer–trimer interactions. Thus, our findings reveal a competition between MA trimer–trimer interactions and the Myr switch of MA proteins.
Figure 2.

(A, B) Snapshots showing the progression of events for the immature-like and random dimer systems. (C, D) Corresponding time evolution of the distance between the first carbon (C2) and the last carbon (C14) of the Myr group and the membrane center. Each protein unit is denoted as p1-p6, and each trimer is denoted as T1 and T2; p1, p2, and p3 constitute trimer 1, and p4, p5, and p6 belong to trimer 2.
MA trimer–trimer interface interactions in the two systems are quite different compared to each other at the final step of the 5 μs trajectory. In the immature-like dimer, the MA trimer–trimer interface is stabilized by the interactions of N-terminal residues and the N terminus of helix 1 with themselves and the 310 helices. The simulation started with this interface structure (where there were no MA-membrane contacts in the initial structure) and this MA–MA interface remained stable during membrane binding of the complex throughout the simulation. Therefore, this MA trimer–trimer interaction is observed to be not solely mediated by the membrane but stable in the solution phase also. However, in this scenario, PPI through N-terminal residues in the immature-like dimer hinder the conformational flexibility of the N-terminal domain (NTD) required for Myr insertion, as mentioned before.
MA Trimer–Trimer Interactions (TTIs) in Two Assembly Structures
As mentioned earlier, we have carried out simulations with and without a stable trimer–trimer interface at the initial protein structures. In the random dimer model, starting from two separated membrane-bound MA trimers, trimer–trimer assembly formation occurs spontaneously in our simulations, driven by the membrane and water-mediated interactions. However, in the Gag assembly process at the producer cell plasma membrane, CASP1 assembly, IP6, and RNA binding promote and accelerate the MA assembly process. On the other hand, immature-like dimers maintained the interface during the membrane targeting and binding process in our simulated trajectories. This observation suggests that the dimerization of MA trimers in an immature-like assembly is not purely lipid-mediated, which agrees with recent NMR studies by Saad and co-workers.20
Here, we have characterized TTIs in two MA complexes. We have computed the minimum distance of residues of trimer 1 and trimer 2 from the other trimer and plotted the distances against the residue numbers of the former (Figure 3A,B). If the magnitude of the minimum distance for a residue is lower than 3 Å, it signifies that the residue participates in the trimer–trimer interactions. Residue IDs 1, 3, 5, and 7 in trimer 1 of the immature-like dimer model correspond to a minimum distance of less than 2.5 Å. This indicates that these particular residues of trimer 1 are in close contact with trimer 2 residues. Figure 3A,B clearly distinguishes between TTIs for two different dimers of trimers, as sampled in the last 200 ns of the 5 μs trajectories. While in the immature-like dimer model, (among others), N-terminal residues are involved in TTIs (Figure 3A), in the random dimer model (among others), HBR domain residues are involved in TTIs (Figure 3B). Also, unlike the immature-like dimer, the trimer–trimer interface in the random dimer system is not symmetric; two TTI profiles for trimer 1 and trimer 2 in Figure 3B are different. In the random dimer system, the progression of the MA trimer association process is investigated using the minimum distance between two trimers (Figure 3C) and the number of Cα contacts between trimer 1 and trimer 2 (for a cutoff radius of 1 nm) (Figure 3D). While after ∼300 ns, the assembled state is stable for a random dimer complex, the trimer–trimer assembly samples over the different PPI networks and the number of contacts between two trimers keeps increasing throughout the trajectory.
Membrane-Induced Conformational Switch of MA Monomers
Protein–protein interactions (PPI) within or on cellular membranes are often a key event in cellular signaling.70,71 Prediction of PPI remains a challenging task for computer simulations as it is influenced by protein environments, such as macromolecular crowding, lipid molecules, and even the solvent environment,72−75 specific ion, and buffer.76,77 Therefore, it is expected that understanding PPI in a peripheral membrane protein complex is prohibitively difficult from both experimental and computational perspectives. We first aimed to understand the conformational transition in individual protein units in the complex, and then, we explored how membrane binding-induced structural change of protein impacts PPI in the MA multimeric complex or vice versa.
The membrane binding process results in protein conformational heterogeneity in different monomers of the MA complex depending on the Myr group position, either in the fully inserted configuration or sequestered in the hydrophobic pocket of the protein or at the membrane surface. The neutron reflectometry study confirms that the MA domain adopts different configurations on the membrane surface to aid the Myr group in choosing the proper orientation for the insertion.29 Six protein units with different conformations at the end of our 5 μs trajectory of the immature-like dimer are shown in Figure 4. During membrane binding, prior to Myr insertion, helix1(H1) and HBR interact with the anionic membrane surface. However, in the initial binding phase of the MA complex, depending on the PIP2/PS lipid accessibility, protein–membrane interactions exhibit significant diversity. In p4-p6, HBR interacts closely with the membrane, whereas for p1–3, the MA–membrane interface looks different. The H2 helix plays an important role in membrane binding for these. Moreover, the conformations of H1 and H2 helices are perturbed in some of these protein units.
Figure 4.

(A) Structural heterogeneity of the six MA monomers (p1–6) at the end of the simulation trajectory of the immature-like dimer (5200 ns). Distances between Helix1 and 310 helices (dH1–310) are highlighted by green dashed lines and the values (in nm) are shown in green boxes. (B) Time series of the dH1–310 distance for individual MA monomers in the immature-like dimer.
Fluctuation in the distance between the H1 helix and 310 helix (dH1–310) plays an important role during Myr exposure.31 In the final structure of our simulation, dH1–310 for six monomers is observed to range from 1.97 to 2.54 nm (values of dH1–310(in nm) are shown in Figure 4A in green boxes). For p6, in its stable Myr-inserted state, dH1–310 is minimum, whereas for p2, at an intermediate state of Myr insertion, this distance is maximum. The time evolution of the distances, dH1–310, for individual monomers is plotted in Figure 4B, which shows a significantly different behavior of p2 and p3 throughout the trajectory. dH1–310 for p5 fluctuates around 2 nm during the entire trajectory and undergoes a transition to >2.3 nm at the end of the trajectory. This kind of structural heterogeneity (with respect to the distance between the H1 helix and the 310 helix) is also observed in other trajectories of both model systems. Furthermore, the conformational transition in one protein unit is correlated with the dynamics of adjacent monomers. This aspect will be investigated later in more detail.
In our effort to understand the membrane binding mechanism of the MA–protein complex, we further looked into the structural transition of the protein units along the trajectory. Root mean-squared deviation (RMSD) and distance RMSD (DRMSD) analyses yield a direct measure of conformational fluctuation. Both are calculated with respect to the initial MA structure (at t = 0). Fluctuating domains like helix5 (H5) and N-terminal Myr are not considered in these calculations. DRMSD is calculated for the pair of atoms within 0.8 nm. In the immature-like dimer, protein unit 2 (p2) of trimer 1 and p4 of trimer 2 are at the trimer–trimer interface and the initial membrane anchoring of the protein complex is mediated by p2 around 30 ns (Figure 5). RMSD analysis shows that p2 and the adjacent p3 monomer (RMSD > 0.4 nm) undergo a structural transition at the beginning of the binding process, whereas most other monomers exhibit a lower RMSD value throughout the simulation. As mentioned before, the p2 monomer first encounters the membrane and makes contact with the anionic lipid molecules (PIP2, shown in green color) through its HBR domain. Although RMSD analysis could not differentiate the conformational change of the p2 monomer from the adjacent p3 monomer, pairwise DRMSD could capture the structural transition of the p2 monomer at the moment of its membrane anchoring (Figure 5B). The distance between the H1 helix and the 310 helix increases for p2 at that point as it attains the “open conformation” (Figure 5D, Figure 4B), described in ref (31)(31).
Figure 5.

Conformational sampling of MA monomers in the immature-like MA dimer of trimers. (A, B) RMSD and distance RMSD (DRMSD) suggest that upon initial binding (∼30 ns) of p2, which initiates the binding process, the p2 monomer undergoes a structural transition that does not occur for the remaining monomers. (C) Side view of the system at the instant of initial membrane anchoring of p2. PIP2 lipids are shown in green color. (D) Structural transition of p2 (t ∼ 30 ns, in blue) with respect to its initial conformation (t = 0 ns; in faded green). The Myr group of this unit at t ∼ 30 ns is shown in red. The distance between H1 and 310 helices increases at ∼30 ns, and the Myr group comes out of the sequestered state.
As captured by the pairwise RMSD, the p5 monomer undergoes maximum conformational fluctuation throughout the trajectory and after Myr insertion of the adjacent p6 monomer (around 4600 ns, Figure 2C), its RMSD value reaches the maximum value (0.45 nm). This correlation between Myr insertion of one monomer and conformational change of the adjacent monomer requires more attention and will be analyzed, in detail, in the next section, employing the time–structure-based independent component analysis (tICA) method.65−68
Cooperativity of MA Monomers during Myristoyl Switch
Encouraged by the RMSD profile of p5 (Figure 5A) that captures the structural transition
of the p5 monomer at the time of Myr insertion of the adjacent p6
monomer, we aimed to explore this further by performing a dimensional
reduction technique using tICA. This method can quantify large conformational
changes in complex biomolecules by identifying the slowest collective
degrees of freedom. For a d-dimensional trajectory 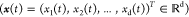 with Cartesian coordinates x1,x2,···,xd, tICA determines independent collective degrees
of freedom,
with Cartesian coordinates x1,x2,···,xd, tICA determines independent collective degrees
of freedom, 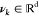 , by maximizing the autocorrelation function
for a given lag time. For a chosen lag time, τ, the time-lagged
covariance matrix, is computed with tICA variables (x(t)), and the vectors (νk) are determined
by solving the generalized eigenvalue problem.78,79νk denotes the tICA eigenvectors and yk(t)(=νk·x(t)) represents the projections of the trajectory onto
the tICA eigenvectors or tICA components (tICs).
, by maximizing the autocorrelation function
for a given lag time. For a chosen lag time, τ, the time-lagged
covariance matrix, is computed with tICA variables (x(t)), and the vectors (νk) are determined
by solving the generalized eigenvalue problem.78,79νk denotes the tICA eigenvectors and yk(t)(=νk·x(t)) represents the projections of the trajectory onto
the tICA eigenvectors or tICA components (tICs).
We have considered the variable, pairwise distances between MA residues to compute the covariance matrix. A lag time of 10 ns is chosen for this purpose. Cα atoms of MA residues in helix 1 to helix 4 were considered in the tIC computation. This tICA variable can capture MA conformational transition. tICA coordinates do not include any lipid molecules or the protein–lipid distances. We compute tIC vectors of individual MA monomers in the protein complexes. The resulting tICA heatmap for p5 as 2D projections along the first and second tIC vectors (tIC1, tIC2) is shown in Figure 6B. The slowest relaxation modes (first two, tIC1 and tIC2) of p5 could capture the conformational switch of the MA monomer in the immature-like dimer undergoing a transition from the unbound to the membrane-bound state. In the encounter complex, membrane binding is accomplished mainly by the HBR domain, and the Myr group is still not inserted into the membrane, which occurs at the bound state. The second component (tIC2) of p5 and p6 is related to the conformational transition during the initial membrane encounter. Although neither of the first two slowest components of p5 and p6 exhibits a transition at the instant of the Myr insertion event leading to the membrane-bound state of p5 protein (∼3400 ns), tIC1 of p5 shows a signature of conformational transition at the time of Myr insertion of the adjacent monomer p6 around 4700 ns (Myr dynamics data are shown in Figure 6C). This reveals the role of trimeric MA–MA interactions in the membrane binding process of MA protein.
Figure 6.

Conformational switch of an MA monomer (p5) at the instant of Myr switch of an adjacent monomer (p6) is depicted with the help of the two slowest time–structure-based independent components (tICs). (A) MA trimer2 with p5 and p6 at the end of the simulation, (B)2D tICA heatmap of MA monomer 5 (p5) distinguishes all the three states during membrane binding, and (C) time evolution of the vertical component for the distance of the last carbon of Myr for both p5 and p6 monomers from the bilayer center (denoted by MYR5 and MYR6, respectively). (D) Projection of the trajectory with respect to the two slowest tIC vectors. A conformational switch of p5 is captured by tIC1 at ∼4700 ns during Myr insertion of p6. The vertical dashed line indicates the time of Myr insertion of p5.
We have added another evidence of such a correlation for a trajectory of the random dimer model (Figures S1 and S2). In that simulation, 5 Myr groups undergo spontaneous Myr insertion within 5 μs. We have performed tICA on that trajectory and a conformational switch of both p5 and p6 is observed at ∼600 ns during the Myr insertion of p5 (Figure S2). As discussed earlier, the Myr insertion event has been identified by plotting the distance of the last carbon atom (C14) of the Myr group from the bilayer center (Figure S2A) and the conformational transitions of p5 and p6 have been captured by tIC1 of p6 (Figure S2B) and tIC2 of p5 (Figure S2C).
Discussion and Conclusions
The present study interrogates the role of protein–protein interactions (PPIs) in the Myr dynamics of the membrane-bound higher order oligomeric structure of MA protein (a dimer of trimers). The contribution of PPIs versus protein–lipid interactions triggering Myr exposure is yet to be understood. We have investigated the Myr insertion of the MA complex for two different systems with or without a stable immature virion-like trimer–trimer interface at the initial structure. Our findings reveal that the dynamics of Myr groups are impacted by the trimer–trimer interface structure. When N-terminal residues are involved in TTIs in the immature-like MA dimer of trimers, Myr groups at the trimeric interface although exposed do not show Myr insertion within simulation time (5 μs). The loss of flexibility in the NTD due to the trimer–trimer interactions (like immature virion) hampers the Myr insertion process. In the random dimer model, a greater number of Myr groups insert into the membrane within the same simulation time (Figure 2). TTIs in these two systems are reported in Figure 3. For the random dimer model, another trajectory shown in Figure S1 sampled 5 Myr insertion events within 5 μs. On the other hand, no trajectories for immature-like dimer models exhibited more than 2 Myr insertions (which are not at the trimer–trimer interface).
We analyzed the structural heterogeneity of MA monomers in the membrane-bound immature-like dimer and the resulting MA–lipid interactions. Unlike other monomers, the monomer that anchors the membrane first exhibits a conformational switch during membrane targeting, which was detected by DRMSD analysis (Figure 5). At the final step of the simulations, depending on the Myr group position, i.e., either inserted or sequestered or at the membrane surface, we observed a diverse nature of MA configurations and MA–lipid interactions (Figure 4). Furthermore, RMSD analysis shows interesting structural dynamics of the p5 monomer, during the Myr switch of p6. To explore this further, we have analyzed the tICA projections. The dimensionally reduced data of MA conformations revealed the cooperativity of events (Figure 6). Myr insertion of one monomer is observed to induce a conformational change of an adjacent MA monomer. This correlated conformational change through the trimeric interface occurs in other trajectories as well. In this scenario, two monomers from the same trimer affect the behavior of one another. This result adds to the previous experimental prediction of Myr exposure being facilitated by the MA trimerization10 and suggests the role of MA trimerization in promoting the Myr insertion event, i.e., the transition from the Myr exposed state to the membrane-inserted state. However, we do not see this conformational correlation across trimers in the 5 μs of simulation per replica. Please note that tICA on the individual trajectories of MA monomers captured the correlated conformational transition at the instant of Myr insertion. Due to the structural heterogeneity and divergent membrane environment of the peripheral membrane proteins during the membrane binding process, the same tICA vectors are not produced for the conformational transition of different MA monomers. The data of structural heterogeneity, correlated MA conformational transitions during Myr insertion, captured by tICA, have been reproduced by using other trajectories.
Overall, our simulated data support a mechanism of the Gag assembly process where membrane-bound MA forms trimers, the Myr insertion happens, and, finally, trimeric MA with membrane-inserted Myr groups assembles further to form a dimer of trimers and hexamer of trimers. On the other hand, our data cannot rule out the possibility of the formation of dimeric contacts in the higher order oligomers followed by a slow kinetics of Myr insertion. However, as explored in another set of simulations (see details of the simulation method in the Supporting Information), MA monomers at the dimeric interface can sample a domain-swapped Myr group configuration when Myr groups are not inserted into the membrane before the formation of dimeric contacts (Figure 7). In this configuration, the Myr group of an MA subunit of trimer 1 is sequestered into the hydrophobic pocket of the adjacent MA monomer across the dimeric interface. The pocket contains hydrophobic residues of the N-terminal domain (VAL6), helix2 (ILE33, VAL34), 310 helix (LEU50), and helix4 (ILE81, LEU84). This Myr-swapped configuration is in agreement with the recent experimental data by Samal et al.20 The X-ray structure of immature-like MA crystals revealed Myr swapping through the dimeric interface. In that configuration, Myr groups are sequestered in the hydrophobic cavity containing similar residues of helix2, helix4, and 310 helix as shown in Figure 7C. As observed in our simulation, the lifetime of the Myr-swapped state is greater than 3 μs. Together, our simulations suggest that (1) the cooperativity of Myr insertion is associated with the trimerization of MA and (2) the preformed dimeric interface of MA prior to Myr insertion can induce a Myr-swapped state. Our simulated data do not provide any information about the switch between the Myr-swapped state and Myr-inserted state or the reversibility of Myr insertion.
Figure 7.

(A) Side view of a membrane-bound MA dimer of trimers at the end of 4 μs simulation (see details in the Supporting Information). Solvent molecules have been excluded for clarity. 2 N-terminal Myr groups of trimer 1 (shown in green) are inserted into the cytoplasmic leaflet of the membrane and 1 Myr is sequestered in the hydrophobic cavity of the MA subunit across the twofold axis. (B, C) Domain-swapped Myr group of the MA subunit of trimer 1 in the hydrophobic cavity of the MA subunit of trimer 2 (shown in pink).
Finally, we investigate the membrane response to the initial binding of the MA complex. The time evolution of local lipid count data for the inner-leaflet lipid molecules are shown in Figure S3. For these calculations, we have considered a distance cutoff of 10 Å between the phosphorus (P) atoms of phospholipid headgroups of inner-leaflet PIP2, PS, PC, and PE lipids and the Cα atoms of the MA monomers.80−85 After the initial encounter of all the monomers in the protein complex with the membrane (t ∼ 200 ns, as marked in the dotted gray line), there is a visible increase in the MA-bound PS and PE lipids and the number of MA-bound PC lipid is decreased with time (from ∼40 at 1–1.5 μs to ∼30 at 4–4.5 μs). It has been well accepted that the specific interaction of the Gag matrix domain and PI(4,5)P2 lipid plays a crucial role in the membrane binding process of matrix protein. Our simulation data suggest that MA binds to the PIP2-rich domain of the inner leaflet as the specific interactions trigger the membrane targeting process. Later, nonspecific interactions with charged lipid PS help stabilize the protein–membrane binding as seen from the increase in PS lipid count data with time. A detailed investigation of membrane lateral organization around MA multimers is beyond the scope of this work and has been reported in our other study.86
As mentioned earlier, several factors contribute to the membrane binding of HIV-1 MA protein to the inner leaflet of the PM: (i) nonspecific electrostatic interactions between the HBR of MA and anionic membrane lipids (PS, PI), (ii) specific interactions between MA protein domains and phosphatidylinositol-4,5-bisphosphate [PI(4,5)P2], and (iii) hydrophobic interactions between the lipidated N-terminus of MA (Myr group) and the bilayer. Although several experimental studies claimed that the myristoylation of Gag is essential for the Gag assembly process, the exact role of the Myr group is still debated. Recently, AAMD simulations by Monje-Galvan and Voth proposed that the Myr group is important for lipid sorting and membrane reorganization dynamics at the protein assembly site and yet not for PM targeting or MA monomer binding events themselves.31 On the other hand, NMR studies proposed that MA trimerization aids in the transition from the Myr-sequestered state to the Myr-exposed state in the solution.10 Later, it was shown that PI(4,5)P2 binding to MA induces a conformational change to facilitate Myr exposure.18 Our present study suggests a correlation between Myr insertion into the membrane and the conformational switch of an adjacent monomer in the MA trimer.
There is still much to be learned about MA–MA and MA-membrane interactions, lipid redistribution, membrane remodeling, and MA lattice formation during viral assembly. In our simulations, we did not obtain membrane binding of the MA multimeric complex with all the Myr residues inserted into the membrane spontaneously within the simulation time scale; in fact, there was at least one Myr group remaining in a sequestered position. Further studies are therefore required to explore the dynamics of Myr insertion in MA multimeric complexes and the factors that facilitate or hinder binding cooperativity of additional protein units as the viral assembly sites evolve.
Acknowledgments
Research reported in this publication was supported by the Behavior of HIV in Viral Environments (B-HIVE) Center of the National Institutes of Health under award number U54AI170855. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Computational resources were partially provided by the Pittsburgh Super Computing Center through the Anton 2 machine (Grant R01GM116961 from the National Institutes of Health) and the specific allocation PSCA17046P. The Anton 2 machine at PSC was generously made available by D.E. Shaw Research. Part of this work was also completed with resources from the University of Chicago Research Computing Center, the Extreme Science and Engineering Discovery Environment (XSEDE), and the NIH-funded Beagle-3 computer (NIH award 1S10OD028655).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jpcb.3c06222.
Time series of Myr insertion in another trajectory of the random MA dimer-of-trimer model, tICA results for the random dimer model, time series of local lipid count for inner-leaflet lipids to study membrane response to the initial binding of the MA dimer-of-trimer complex, and simulation details of the immature-like MA dimer of trimers (using different simulation setups) sampling myristoyl-swapped configuration (PDF)
Author Present Address
† Department of Chemical and Biological Engineering, University at Buffalo, Buffalo, New York 14260, United States
The authors declare no competing financial interest.
Supplementary Material
References
- Freed E. O. HIV-1 assembly, release and maturation. Nat. Rev. Microbiol. 2015, 13 (8), 484–496. 10.1038/nrmicro3490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chukkapalli V.; Ono A. Molecular determinants that regulate plasma membrane association of HIV-1 Gag. J. Mol. Biol. 2011, 410 (4), 512–524. 10.1016/j.jmb.2011.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schur F. K. M.; Obr M.; Hagen W. J.; Wan W.; Jakobi A. J.; Kirkpatrick J. M.; Sachse C.; Kräusslich H.-G.; Briggs J. A. An atomic model of HIV-1 capsid-SP1 reveals structures regulating assembly and maturation. Science 2016, 353 (6298), 506–508. 10.1126/science.aaf9620. [DOI] [PubMed] [Google Scholar]
- Pak A. J.; Gupta M.; Yeager M.; Voth G. A. Inositol Hexakisphosphate (IP6) Accelerates Immature HIV-1 Gag Protein Assembly toward Kinetically Trapped Morphologies. J. Am. Chem. Soc. 2022, 144 (23), 10417–10428. 10.1021/jacs.2c02568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pak A. J.; Grime J. M.; Sengupta P.; Chen A. K.; Durumeric A. E.; Srivastava A.; Yeager M.; Briggs J. A.; Lippincott-Schwartz J.; Voth G. A. Immature HIV-1 lattice assembly dynamics are regulated by scaffolding from nucleic acid and the plasma membrane. Proc. Natl. Acad. Sci. U.S.A. 2017, 114 (47), E10056–E10065. 10.1073/pnas.1706600114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalonde M. S.; Sundquist W. I. How HIV finds the door. Proc. Natl. Acad. Sci. U.S.A. 2012, 109 (46), 18631–18632. 10.1073/pnas.1215940109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill C. P.; Worthylake D.; Bancroft D. P.; Christensen A. M.; Sundquist W. I. Crystal structures of the trimeric human immunodeficiency virus type 1 matrix protein: implications for membrane association and assembly. Proc. Natl. Acad. Sci. U.S.A. 1996, 93 (7), 3099–3104. 10.1073/pnas.93.7.3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang C.; Ndassa Y.; Summers M. F. Structure of the N-terminal 283-residue fragment of the immature HIV-1 Gag polyprotein. Nat. Struct. Biol. 2002, 9 (7), 537–543. 10.1038/nsb806. [DOI] [PubMed] [Google Scholar]
- Dick R. A.; Vogt V. M. Membrane interaction of retroviral Gag proteins. Front. Microbiol. 2014, 5, 187 10.3389/fmicb.2014.00187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang C.; Loeliger E.; Luncsford P.; Kinde I.; Beckett D.; Summers M. F. Entropic switch regulates myristate exposure in the HIV-1 matrix protein. Proc. Natl. Acad. Sci. U.S.A. 2004, 101 (2), 517–522. 10.1073/pnas.0305665101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfadhli A.; Barklis R. L.; Barklis E. HIV-1 matrix organizes as a hexamer of trimers on membranes containing phosphatidylinositol-(4, 5)-bisphosphate. Virology 2009, 387 (2), 466–472. 10.1016/j.virol.2009.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu K.; Ke Z.; Zila V.; Anders-Össwein M.; Glass B.; Mücksch F.; Müller R.; Schultz C.; Müller B.; Kräusslich H.-G.; Briggs J. A. G. Maturation of the matrix and viral membrane of HIV-1. Science 2021, 373 (6555), 700–704. 10.1126/science.abe6821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waheed A. A.; Freed E. O. The role of lipids in retrovirus replication. Viruses 2010, 2 (5), 1146–1180. 10.3390/v2051146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono A.; Ablan S. D.; Lockett S. J.; Nagashima K.; Freed E. O. Phosphatidylinositol (4, 5) bisphosphate regulates HIV-1 Gag targeting to the plasma membrane. Proc. Natl. Acad. Sci. U.S.A. 2004, 101 (41), 14889–14894. 10.1073/pnas.0405596101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brügger B.; Glass B.; Haberkant P.; Leibrecht I.; Wieland F. T.; Kräusslich H.-G. The HIV lipidome: a raft with an unusual composition. Proc. Natl. Acad. Sci. U.S.A. 2006, 103 (8), 2641–2646. 10.1073/pnas.0511136103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercredi P. Y.; Bucca N.; Loeliger B.; Gaines C. R.; Mehta M.; Bhargava P.; Tedbury P. R.; Charlier L.; Floquet N.; Muriaux D.; et al. Structural and molecular determinants of membrane binding by the HIV-1 matrix protein. J. Mol. Biol. 2016, 428 (8), 1637–1655. 10.1016/j.jmb.2016.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tedbury P. R.; Novikova M.; Ablan S. D.; Freed E. O. Biochemical evidence of a role for matrix trimerization in HIV-1 envelope glycoprotein incorporation. Proc. Natl. Acad. Sci. U.S.A. 2016, 113 (2), E182–E190. 10.1073/pnas.1516618113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saad J. S.; Miller J.; Tai J.; Kim A.; Ghanam R. H.; Summers M. F. Structural basis for targeting HIV-1 Gag proteins to the plasma membrane for virus assembly. Proc. Natl. Acad. Sci. U.S.A. 2006, 103 (30), 11364–11369. 10.1073/pnas.0602818103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yandrapalli N.; Muriaux D.; Favard C. Lipid domains in HIV-1 assembly. Front. Microbiol. 2014, 5, 220 10.3389/fmicb.2014.00220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samal A. B.; Green T. J.; Saad J. S. Atomic view of the HIV-1 matrix lattice; implications on virus assembly and envelope incorporation. Proc. Natl. Acad. Sci. U. S. A. 2022, 119 (23), e2200794119 10.1073/pnas.2200794119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favard C.; Chojnacki J.; Merida P.; Yandrapalli N.; Mak J.; Eggeling C.; Muriaux D. HIV-1 Gag specifically restricts PI (4, 5) P2 and cholesterol mobility in living cells creating a nanodomain platform for virus assembly. Sci. Adv. 2019, 5 (10), eaaw8651 10.1126/sciadv.aaw8651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen Y.; Feigenson G. W.; Vogt V. M.; Dick R. A. Mechanisms of PI (4, 5) P2 Enrichment in HIV-1 Viral Membranes. J. Mol. Biol. 2020, 432 (19), 5343–5364. 10.1016/j.jmb.2020.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai C.-L.; Landgraf K. E.; Voth G. A.; Falke J. J. Membrane docking geometry and target lipid stoichiometry of membrane-bound PKCα C2 domain: a combined molecular dynamics and experimental study. J. Mol. Biol. 2010, 402 (2), 301–310. 10.1016/j.jmb.2010.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai C.-L.; Srivastava A.; Pilling C.; Chase A. R.; Falke J. J.; Voth G. A. Molecular mechanism of membrane binding of the GRP1 PH domain. J. Mol. Biol. 2013, 425 (17), 3073–3090. 10.1016/j.jmb.2013.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahelin R. V.Monitoring peripheral protein oligomerization on biological membranes. In Methods in Cell Biology; Elsevier, 2013; Vol. 117, pp 359–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen A. H.; John L. H.; Sansom M. S.; Corey R. A. Specific interactions of peripheral membrane proteins with lipids: what can molecular simulations show us?. Biosci. Rep. 2022, 42 (4), BSR20211406 10.1042/BSR20211406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monje-Galvan V.; Klauda J. B. Peripheral membrane proteins: Tying the knot between experiment and computation. Biochim. Biophys. Acta, Biomembr. 2016, 1858 (7), 1584–1593. 10.1016/j.bbamem.2016.02.018. [DOI] [PubMed] [Google Scholar]
- Pu M.; Orr A.; Redfield A. G.; Roberts M. F. Defining specific lipid binding sites for a peripheral membrane protein in situ using subtesla field-cycling NMR. J. Biol. Chem. 2010, 285 (35), 26916–26922. 10.1074/jbc.M110.123083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eells R.; Barros M.; Scott K. M.; Karageorgos I.; Heinrich F.; Lösche M. Structural characterization of membrane-bound human immunodeficiency virus-1 Gag matrix with neutron reflectometry. Biointerphases 2017, 12 (2), 02D408 10.1116/1.4983155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogaski B.; Klauda J. B. Membrane-binding mechanism of a peripheral membrane protein through microsecond molecular dynamics simulations. J. Mol. Biol. 2012, 423 (5), 847–861. 10.1016/j.jmb.2012.08.015. [DOI] [PubMed] [Google Scholar]
- Monje-Galvan V.; Voth G. A. Binding mechanism of the matrix domain of HIV-1 gag on lipid membranes. eLife 2020, 9, e58621 10.7554/eLife.58621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q.; Pechersky Y.; Sagawa S.; Pan A. C.; Shaw D. E. Structural mechanism for Bruton’s tyrosine kinase activation at the cell membrane. Proc. Natl. Acad. Sci. U.S.A. 2019, 116 (19), 9390–9399. 10.1073/pnas.1819301116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryckbosch S. M.; Wender P. A.; Pande V. S. Molecular dynamics simulations reveal ligand-controlled positioning of a peripheral protein complex in membranes. Nat. Commun. 2017, 8 (1), 6 10.1038/s41467-016-0015-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui H.; Ayton G. S.; Voth G. A. Membrane binding by the endophilin N-BAR domain. Biophys. J. 2009, 97 (10), 2746–2753. 10.1016/j.bpj.2009.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blood P. D.; Voth G. A. Direct observation of Bin/amphiphysin/Rvs (BAR) domain-induced membrane curvature by means of molecular dynamics simulations. Proc. Natl. Acad. Sci. U.S.A. 2006, 103 (41), 15068–15072. 10.1073/pnas.0603917103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorizate M.; Sachsenheimer T.; Glass B.; Habermann A.; Gerl M. J.; Kräusslich H. G.; Brügger B. Comparative lipidomics analysis of HIV-1 particles and their producer cell membrane in different cell lines. Cell. Microbiol. 2013, 15 (2), 292–304. 10.1111/cmi.12101. [DOI] [PubMed] [Google Scholar]
- Mücksch F.; Citir M.; Lüchtenborg C.; Glass B.; Traynor-Kaplan A.; Schultz C.; Brügger B.; Kräusslich H.-G. Quantification of phosphoinositides reveals strong enrichment of PIP2 in HIV-1 compared to producer cell membranes. Sci. Rep. 2019, 9 (1), 17661 10.1038/s41598-019-53939-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta P.; Seo A. Y.; Pasolli H. A.; Song Y. E.; Johnson M. C.; Lippincott-Schwartz J. A lipid-based partitioning mechanism for selective incorporation of proteins into membranes of HIV particles. Nat. Cell Biol. 2019, 21 (4), 452–461. 10.1038/s41556-019-0300-y. [DOI] [PubMed] [Google Scholar]
- Sengupta P.; Lippincott-Schwartz J. Revisiting membrane microdomains and phase separation: a viral perspective. Viruses 2020, 12 (7), 745. 10.3390/v12070745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumner C.; Ono A. Relationship between HIV-1 Gag multimerization and membrane binding. Viruses 2022, 14 (3), 622. 10.3390/v14030622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee P.; Voth G. A. Conformational transitions of the HIV-1 Gag polyprotein upon multimerization and gRNA binding. Biophys. J. 2023, 123, 42–56. 10.1016/j.bpj.2023.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barros M.; Heinrich F.; Datta S. A.; Rein A.; Karageorgos I.; Nanda H.; Lösche M. Membrane binding of HIV-1 matrix protein: dependence on bilayer composition and protein lipidation. J. Virol. 2016, 90 (9), 4544–4555. 10.1128/JVI.02820-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlier L.; Louet M.; Chaloin L.; Fuchs P.; Martinez J.; Muriaux D.; Favard C.; Floquet N. Coarse-grained simulations of the HIV-1 matrix protein anchoring: revisiting its assembly on membrane domains. Biophys. J. 2014, 106 (3), 577–585. 10.1016/j.bpj.2013.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. VMD: visual molecular dynamics. J. Mol. Graphics 1996, 14 (1), 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Jo S.; Kim T.; Im W. Automated builder and database of protein/membrane complexes for molecular dynamics simulations. PLoS One 2007, 2 (9), e880 10.1371/journal.pone.0000880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S.; Kim T.; Iyer V. G.; Im W. CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29 (11), 1859–1865. 10.1002/jcc.20945. [DOI] [PubMed] [Google Scholar]
- Brooks B. R.; Brooks C. L. III; Mackerell A. D. Jr; Nilsson L.; Petrella R. J.; Roux B.; Won Y.; Archontis G.; Bartels C.; Boresch S.; et al. CHARMM: the biomolecular simulation program. J. Comput. Chem. 2009, 30 (10), 1545–1614. 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu E. L.; Cheng X.; Jo S.; Rui H.; Song K. C.; Dávila-Contreras E. M.; Qi Y.; Lee J.; Monje-Galvan V.; Venable R. M.; et al. CHARMM-GUI Membrane Builder toward Realistic Biological Membrane Simulations; Wiley Online Library, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.; Cheng X.; Swails J. M.; Yeom M. S.; Eastman P. K.; Lemkul J. A.; Wei S.; Buckner J.; Jeong J. C.; Qi Y.; et al. CHARMM-GUI input generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM simulations using the CHARMM36 additive force field. J. Chem. Theory Comput. 2016, 12 (1), 405–413. 10.1021/acs.jctc.5b00935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J.; Rauscher S.; Nawrocki G.; Ran T.; Feig M.; De Groot B. L.; Grubmüller H.; MacKerell A. D. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14 (1), 71–73. 10.1038/nmeth.4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abraham M. J.; Murtola T.; Schulz R.; Páll S.; Smith J. C.; Hess B.; Lindahl E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
- Shaw D. E.; Grossman J.; Bank J.; Batson B.; Butts J.; Chao J.; Deneroff M.; Dror R.; Even A.; Fenton C.; et al. Anton 2: raising the bar for performance and programmability in a special-purpose molecular dynamics supercomputer. Int. Conf. High-Perform. 2014, 41–53. [Google Scholar]
- Hoover W. G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31 (3), 1695. 10.1103/PhysRevA.31.1695. [DOI] [PubMed] [Google Scholar]
- Nosé S. A molecular dynamics method for simulations in the canonical ensemble. Mol. Phys. 1984, 52 (2), 255–268. 10.1080/00268978400101201. [DOI] [Google Scholar]
- Nosé S.; Klein M. Constant pressure molecular dynamics for molecular systems. Mol. Phys. 1983, 50 (5), 1055–1076. 10.1080/00268978300102851. [DOI] [Google Scholar]
- Parrinello M.; Rahman A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52 (12), 7182–7190. 10.1063/1.328693. [DOI] [Google Scholar]
- Darden T.; York D.; Pedersen L. Particle mesh Ewald: An N· log (N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98 (12), 10089–10092. 10.1063/1.464397. [DOI] [Google Scholar]
- Hess B.; Bekker H.; Berendsen H. J.; Fraaije J. G. LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18 (12), 1463–1472. . [DOI] [Google Scholar]
- Shan Y.; Klepeis J. L.; Eastwood M. P.; Dror R. O.; Shaw D. E. Gaussian split Ewald: A fast Ewald mesh method for molecular simulation. J. Chem. Phys. 2005, 122 (5), 054101 10.1063/1.1839571. [DOI] [PubMed] [Google Scholar]
- Ryckaert J.-P.; Ciccotti G.; Berendsen H. J. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23 (3), 327–341. 10.1016/0021-9991(77)90098-5. [DOI] [Google Scholar]
- Lippert R. A.; Predescu C.; Ierardi D. J.; Mackenzie K. M.; Eastwood M. P.; Dror R. O.; Shaw D. E. Accurate and efficient integration for molecular dynamics simulations at constant temperature and pressure. J. Chem. Phys. 2013, 139 (16), 164106 10.1063/1.4825247. [DOI] [PubMed] [Google Scholar]
- Gowers R. J.; Linke M.; Barnoud J.; Reddy T. J.; Melo M. N.; Seyler S. L.; Domanski J.; Dotson D. L.; Buchoux S.; Kenney I. M.. et al. MDAnalysis: a Python Package for the Rapid Analysis of Molecular Dynamics Simulations. In Proceedings of the 15th Python in Science Conference; SciPy: Austin, TX, 2016; Vol. 98, p 105. [Google Scholar]
- Michaud-Agrawal N.; Denning E. J.; Woolf T. B.; Beckstein O. MDAnalysis: a toolkit for the analysis of molecular dynamics simulations. J. Comput. Chem. 2011, 32 (10), 2319–2327. 10.1002/jcc.21787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGibbon R. T.; Beauchamp K. A.; Harrigan M. P.; Klein C.; Swails J. M.; Hernández C. X.; Schwantes C. R.; Wang L.-P.; Lane T. J.; Pande V. S. MDTraj: a modern open library for the analysis of molecular dynamics trajectories. Biophys. J. 2015, 109 (8), 1528–1532. 10.1016/j.bpj.2015.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molgedey L.; Schuster H. G. Separation of a mixture of independent signals using time delayed correlations. Phys. Rev. Lett. 1994, 72 (23), 3634. 10.1103/PhysRevLett.72.3634. [DOI] [PubMed] [Google Scholar]
- Naritomi Y.; Fuchigami S. Slow dynamics in protein fluctuations revealed by time-structure based independent component analysis: the case of domain motions. J. Chem. Phys. 2011, 134 (6), 02B617 10.1063/1.3554380. [DOI] [PubMed] [Google Scholar]
- Schwantes C. R.; Pande V. S. Improvements in Markov state model construction reveal many non-native interactions in the folding of NTL9. J. Chem. Theory Comput. 2013, 9 (4), 2000–2009. 10.1021/ct300878a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Hernández G.; Paul F.; Giorgino T.; De Fabritiis G.; Noé F. Identification of slow molecular order parameters for Markov model construction. J. Chem. Phys. 2013, 139 (1), 015102 10.1063/1.4811489. [DOI] [PubMed] [Google Scholar]
- Harrigan M. P.; Sultan M. M.; Hernández C. X.; Husic B. E.; Eastman P.; Schwantes C. R.; Beauchamp K. A.; McGibbon R. T.; Pande V. S. MSMBuilder: statistical models for biomolecular dynamics. Biophys. J. 2017, 112 (1), 10–15. 10.1016/j.bpj.2016.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore D. T.; Berger B. W.; DeGrado W. F. Protein-protein interactions in the membrane: sequence, structural, and biological motifs. Structure 2008, 16 (7), 991–1001. 10.1016/j.str.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews E. E.; Zoonens M.; Engelman D. M. Dynamic helix interactions in transmembrane signaling. Cell 2006, 127 (3), 447–450. 10.1016/j.cell.2006.10.016. [DOI] [PubMed] [Google Scholar]
- Mondal B.; Reddy G. Cosolvent effects on the growth of protein aggregates formed by a single domain globular protein and an intrinsically disordered protein. J. Phys. Chem. B 2019, 123 (9), 1950–1960. 10.1021/acs.jpcb.8b11128. [DOI] [PubMed] [Google Scholar]
- Banerjee P.; Mondal S.; Bagchi B. Effect of ethanol on insulin dimer dissociation. J. Chem. Phys. 2019, 150 (8), 084902 10.1063/1.5079501. [DOI] [PubMed] [Google Scholar]
- Banerjee P.; Bagchi B. Dynamical control by water at a molecular level in protein dimer association and dissociation. Proc. Natl. Acad. Sci. U.S.A. 2020, 117 (5), 2302–2308. 10.1073/pnas.1908379117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee P.; Mondal S.; Bagchi B. Insulin dimer dissociation in aqueous solution: A computational study of free energy landscape and evolving microscopic structure along the reaction pathway. J. Chem. Phys. 2018, 149 (11), 114902 10.1063/1.5042290. [DOI] [PubMed] [Google Scholar]
- Brudar S.; Hribar-Lee B. Effect of buffer on protein stability in aqueous solutions: A simple protein aggregation model. J. Phys. Chem. B 2021, 125 (10), 2504–2512. 10.1021/acs.jpcb.0c10339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts D.; Keeling R.; Tracka M.; Van Der Walle C.; Uddin S.; Warwicker J.; Curtis R. Specific ion and buffer effects on protein–protein interactions of a monoclonal antibody. Mol. Pharmaceutics 2015, 12 (1), 179–193. 10.1021/mp500533c. [DOI] [PubMed] [Google Scholar]
- Schultze S.; Grubmüller H. Time-lagged independent component analysis of random walks and protein dynamics. J. Chem. Theory Comput. 2021, 17 (9), 5766–5776. 10.1021/acs.jctc.1c00273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwantes C. R.; Pande V. S. Modeling molecular kinetics with tICA and the kernel trick. J. Chem. Theory Comput. 2015, 11 (2), 600–608. 10.1021/ct5007357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karmakar S.; Klauda J. B. Modeling the membrane binding mechanism of a lipid transport protein Osh4 to single membranes. Biophys. J. 2022, 121 (8), 1560–1575. 10.1016/j.bpj.2022.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W.; Corey R. A.; Ansell T. B.; Cassidy C. K.; Horrell M. R.; Duncan A. L.; Stansfeld P. J.; Sansom M. S. PyLipID: A Python package for analysis of protein–lipid interactions from molecular dynamics simulations. J. Chem. Theory Comput. 2022, 18 (2), 1188–1201. 10.1021/acs.jctc.1c00708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corradi V.; Sejdiu B. I.; Mesa-Galloso H.; Abdizadeh H.; Noskov S. Y.; Marrink S. J.; Tieleman D. P. Emerging diversity in lipid–protein interactions. Chem. Rev. 2019, 119 (9), 5775–5848. 10.1021/acs.chemrev.8b00451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez R. X.; Campbell O.; Pradhan A. J.; Atilla-Gokcumen G. E.; Monje-Galvan V. Modeling the molecular fingerprint of protein-lipid interactions of MLKL on complex bilayers. Front. Chem. 2023, 10, 1088058 10.3389/fchem.2022.1088058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakaash D.; Fagnen C.; Cook G. P.; Acuto O.; Kalli A. C. Molecular dynamics simulations reveal membrane lipid interactions of the full-length lymphocyte specific kinase (Lck). Sci. Rep. 2022, 12 (1), 21121 10.1038/s41598-022-25603-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corey R. A.; Stansfeld P. J.; Sansom M. S. The energetics of protein–lipid interactions as viewed by molecular simulations. Biochem. Soc. Trans. 2020, 48 (1), 25–37. 10.1042/BST20190149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee P.; Qu K.; Briggs J. A.; Voth G. A. Molecular dynamics simulations of HIV-1 matrix-membrane interactions at different stages of viral maturation. Biophys. J. 2024, 123 (3), 389–406. 10.1016/j.bpj.2024.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.