

Abstract
INTRODUCTION:
The aim of this study was to investigate the epigenetic regulation and underlying mechanism of NRIP3 in colorectal cancer (CRC).
METHODS:
Eight cell lines (SW480, SW620, DKO, LOVO, HT29, HCT116, DLD1, and RKO), 187 resected margin samples from colorectal cancer tissue, 146 cases with colorectal adenomatous polyps, and 308 colorectal cancer samples were used. Methylation-specific PCR, Western blotting, RNA interference assay, and a xenograft mouse model were used.
RESULTS:
NRIP3 exhibited methylation in 2.7% (5/187) of resected margin samples from colorectal cancer tissue, 32.2% (47/146) of colorectal adenomatous polyps, and 50.6% (156/308) of CRC samples, and the expression of NRIP3 was regulated by promoter region methylation. The methylation of NRIP3 was found to be significantly associated with late onset (at age 50 years or older), poor tumor differentiation, lymph node metastasis, and poor 5-year overall survival in CRC (all P < 0.05). In addition, NRIP3 methylation was an independent poor prognostic marker (P < 0.05). NRIP3 inhibited cell proliferation, colony formation, invasion, and migration, while induced G1/S arrest. NRIP3 suppressed CRC growth by inhibiting PI3K-AKT signaling both in vitro and in vivo. Methylation of NRIP3 sensitized CRC cells to combined PI3K and ATR/ATM inhibitors.
DISCUSSION:
NRIP3 was frequently methylated in both colorectal adenomatous polyps and CRC. The methylation of NRIP3 may potentially serve as an early detection, late-onset, and poor prognostic marker in CRC. NRIP3 is a potential tumor suppressor. NRIP3 methylation is a potential synthetic lethal marker for combined PI3K and ATR/ATM inhibitors.
KEYWORDS: NRIP3, DNA methylation, PI3K-AKT signaling, late-onset colorectal cancer, synthetic lethality
INTRODUCTION
Colorectal cancer (CRC) ranks as the third most common cancer and the second leading cause of cancer-related mortality globally (1,2). Notably, there is an increasing trend in early-onset CRC (patients younger than 50 years), which currently accounts for approximately 10% of all new diagnoses (3). Patients with early-onset CRC are more likely to present with hereditary syndromes compared with their late-onset counterparts (1,4). High-frequency microsatellite instability caused by germline mutations occurs more frequently in early-onset CRC, while sporadic high-frequency microsatellite instability tumors caused by epigenetic inactivation of MLH1 occur more frequently in late-onset CRC (1,5–7). Sporadic CRC happens more frequently than hereditary CRC. The association of epigenetic changes in other genes was not observed in early-onset/late-onset CRC.
Nuclear receptors (NRs) usually play important roles in regulating gene expression, cell growth, and development by functioning as transcription factors (8). NR-interacting protein 1 (NRIP1) has been documented to act as either an activator or inhibitor in various cancer types (8–10). NR-interacting protein 3 (NRIP3) is a novel transcriptional coregulator of NR, which is located on chromosome 11p15.4, a fragile site associated with cancer. A recent study conducted by Wolfe et al (11) reported that defects of this region were related to embryonal tumors. However, the precise regulatory mechanisms and expression patterns of NRIP3 in CRC remain elusive. Therefore, the objective of this investigation was to analyze the epigenetic changes and elucidate the underlying mechanisms of NRIP3 in human CRC, with the ultimate aim of uncovering innovative therapeutic approaches.
METHODS
Human tissue samples and cell lines
To assess the possibility of gene expression regulation, we used the TCGA and GTEx databases (http://gepia.cancer-pku.cn/index.html). To ensure the exclusion of tissue-specific methylation, 6 samples of colorectal mucosa were collected from noncancerous individuals. To further investigate the methylation status of NRIP3 in CRC, a total of 187 resected margin samples from CRC tissue, 146 cases of colorectal adenomatous polyps, and 308 CRC tissue samples were examined by methylation-specific PCR (MSP). All samples were obtained from the Chinese PLA General Hospital. Patient medical records were reviewed to collect following information: age, sex, polyp characteristics (number, size, pathology, and location), colorectal cancer characteristics (stage, size, location, differentiation, and lymph node metastasis), and treatment variables (chemotherapy and radiotherapy). All tumors were classified according to TNM staging system (eighth). All CRC cells were previously derived from primary colorectal cancer, including DKO, SW480, LOVO, HT29, HCT116, DLD1, RKO, and SW620. The institutional review board of the Chinese PLA General Hospital granted approval for the procedure.
Lentiviral expression vectors and stable expression cells for NRIP3
For functional study, human NRIP3 CDS (NM_020645.3) was constructed using the expression vector pCDH-CMV-MCS-puro and amplificated using the specified primers: 5′-CCGGAATTCATGTTTTACTCAGGGCTCCTCACTG-3′ (F) and 5′-GCGGGATCCTTATGCTTCTGAAGTGTTGTCTTCATTC-3′ (R). NRIP3-expressing lentiviral or empty vectors were packaged according to previously described methods (12). The lentivirus was introduced into DLD1, RKO, and HCT116 cells, by adding it to the growth medium. NRIP3 stably expressed cells were selected with 2.0 μg/mL (DLD1), 1.0 μg/mL (RKO), and 1.5 μg/mL (HCT116) of puromycin for 3 days.
SiRNA knockdown technique
Specific sequences for NRIP3 siRNA and scrambled control duplex are listed in Supplementary Table 2 (see Supplementary Digital Content 3, http://links.lww.com/CTG/B68). SiRNA oligonucleotide and SiRNA scrambled control duplex (Gene Pharma, Shanghai, China) were transfected into NRIP3 highly expressed cells. The efficiency of transfection was assessed, and siRNA#3 was found to be the most effective.
Xenograft mouse model
NRIP3 stably expressed and unexpressed DLD1 (4 × 106 cells) and RKO cells (6 × 106 cells) were suspended in 0.15 mL of phosphate-buffered saline and subcutaneously injected into the dorsal right side of 4-week-old BABL/c nude mice. The tumor volume was measured every 3 days for a total of 18 days, using the formula V = L × W2/2, where V represents volume (mm3), L represents the biggest diameter (mm), and W represents the smallest diameter (mm). All experimental procedures were conducted in accordance with the guidelines by the Animal Ethics Committee of the Chinese PLA General Hospital.
Evaluating the sensitivity of CRC cells to VE-822 and AZD0156
The IC50 value was evaluated using an MTT assay. CRC cells, both with and without NRIP3 expression, were seeded into 96-well plates at a density of 2000 cells/well. The cells were treated by 0.1 μM cisplatin (#S1166; Selleck, Houston, TX) combination with 0, 0.2, 0.4, 0.8, 1.6, 3.2, 6.4, 12.8, and 25.6 μM VE-822 (#S7102; Selleck) or 0, 1, 2, 4, 8, 16, 32, 64, and 128 μM AZD0156 (#S8375; Selleck) for 48 hours. For synthetic lethality analysis, NRIP3 unexpressed DLD1, RKO, and HCT116 cells were treated with 0.2 μM VE-822/combined with 50 nM NVP-BEZ235 (#S1009; Selleck) or treated with 0.5 μM AZD0156/combined with 50 nM NVP-BEZ235 under 0.1 μM cisplatin treatment for DNA damage. Each experiment was repeated 3 times.
Additional materials and methods
RNA isolation, RT-PCR, methylation detection, immunohistochemistry, MTT assay, colony formation, cell cycle, transwell assay, gene expression array analysis, and Western blot approaches were described in Supplementary Materials and Methods (see Supplementary Digital Content 1, http://links.lww.com/CTG/B66). Primer sequences are listed in Supplementary Table 1 (see Supplementary Digital Content 2, http://links.lww.com/CTG/B67).
Statistical analysis
SPSS software (version 22.0; IBM, Armonk, NY) was used in this study, and the χ2 test was used for independent dichotomous variables. The Student t test was applied for mean ± SD analysis. Kaplan-Meier plots and log-rank tests were performed to evaluate overall survival (OS). The association with 5-year OS was analyzed using univariate and multivariate Cox proportional hazards regression models. P < 0.05 is considered as significant difference.
RESULTS
The expression of NRIP3 is regulated by promoter region methylation
The expression of NRIP3 was reduced in CRC samples compared with that in normal colonic mucosa from noncancerous patients (P < 0.05, Figure 1a). Furthermore, we observed an inverse association between the mRNA levels of NRIP3 and the methylation status around the transcription start site in 307 cases of available CRC data (cg03963327, cg03323636, cg03666741, all P < 0.05, Figure 1b and c).
Figure 1.

The expression and methylation status of NRIP3 in CRC. (a) The levels of NRIP3 in CRC tumor tissue and normal tissue samples (TCGA + GTEx datasets). num, number; T, tumor; N, normal; COAD, colorectal adenocarcinoma cancer; READ, rectal adenocarcinoma cancer. (b) The association of NRIP3 expression and methylation status of CpG sites close to TSS site in the promoter region. TSS, transcription start site. (c) Scatter plots show representative CpG sites methylation status and NRIP3 expression association (cg03963327, cg03323636 and cg03666741). (d) Semiquantitative RT-PCR shows the expression of NRIP3 in CRC cells. 5-Aza, 5-aza-2′-deoxycytidine; GAPDH, internal control of RT-PCR; H2O, double distilled water; (−), absence of 5-Aza; (+), presence of 5-Aza. (e) MSP results of NRIP3 in CRC cells. U, unmethylated alleles; M, methylated alleles; IVD, in vitro methylated DNA, serves as methylation control; NL, normal lymphocytes DNA, serves as unmethylation control; H2O, double distilled water. (f) BSSQ results of NRIP3 in DLD1, RKO, HCT116, and DKO cells. The size of unmethylated MSP products is 183 bp. Bisulfite sequencing was performed in a 261-bp region around the NRIP3 transcription start site (from −354 bp to −94 bp). Filled circles, methylated CpG sites; open circles, unmethylated CpG sites; TSS, transcription start site.
Notably, NRIP3 exhibited high expression in DKO cells, no expression in LOVO, HT29, HCT116, DLD1, and RKO cells, and reduced expression in SW480 and SW620 cells (Figure 1d). NRIP3 was unmethylated in DKO cells, completely methylated in LOVO, HT29, HCT116, DLD1, and RKO cells, and partially methylated in SW480 and SW620 cells (Figure 1e). The regulation of NRIP3 expression by methylation was further validated by 5-aza-2′-deoxycytidine (5-Aza) treatment. The expression of NRIP3 was restored in LOVO, HT29, HCT116, DLD1, and RKO cells, increased in SW620 and SW480 cells, but remained unchanged in DKO cells (Figure 1d). These findings suggested that the expression of NRIP3 is regulated by promoter methylation. NRIP3 was densely methylated in DLD1, RKO, and HCT116 cells and unmethylated in DKO cells (Figure 1f). Consistent results were observed in bisulfite sequencing and MSP.
NRIP3 is frequently methylated in human primary CRC
As shown in Figure 2a, tissue-specific methylation was excluded by detection of 6 cases of normal colorectal mucosa. Among the margin samples obtained from resected CRC tissues, NRIP3 methylation was observed in 5 of 187 cases (2.7%). Notably, all the methylated cases were older than 69 years. Although no statistical significance was observed, the findings imply a potential relationship between NRIP3 methylation and old age. NRIP3 was methylated in 32.2% (47/146) of colonic adenomatous polyps and 50.6% (156/308) of CRC (P < 0.01, Figure 2a and b), suggesting that NRIP3 may be an early detection marker for CRC. The median age of adenomatous polyp patients was 60 years (28–88 years), including 93 cases of male and 53 cases of female individuals. The number of cases were 64 in proximal colon (28 in ascending colon and 36 transverse colon) and 82 in distal colon (17 in descending colon, 41 in sigmoid colon and 24 rectum). A significant association was observed between NRIP3 methylation and older age (age 50 years or older, P < 0.05, see Supplementary Table 3, Supplementary Digital Content 4, http://links.lww.com/CTG/B69), while no significant associations were found with gender, polyp size, or location (all P > 0.05, see Supplementary Table 3, Supplementary Digital Content 4, http://links.lww.com/CTG/B69).
Figure 2.

The methylation and expression status of NRIP3 in normal colorectal mucosa, colonic polyps, and primary CRC. (a) Representative MSP results of NRIP3 in normal colorectal mucosa, colonic adenomatous polyps, and primary CRC samples. N, normal colorectal mucosa; CRP, colonic adenomatous polyp samples; CRC, primary CRC samples. IVD, in vitro methylated DNA, serves as methylation control; NL, normal lymphocytes DNA, serves as unmethylation control; H2O, double distilled water. (b) NRIP3 methylation frequency in colonic adenomatous polyps and primary CRC samples. **P < 0.01. (c) The association of NRIP3 methylation and 5-year OS of CRC. (d) NRIP3 staining in CRC and adjacent tissue samples (top: 200×; bottom: 400×). (e) Box plots for NRIP3 expression, horizontal lines represent the median score; the bottom and top of the boxes represent the 25th and 75th percentiles, respectively; vertical bars represent expression levels. (f) Bar diagram: NRIP3 expression and case numbers; white: unmethylation; gray: methylation.
Among patients with CRC, the median age was 60 years (29–86 years), including 192 cases of male and 116 cases of female individuals. The number of cases were 80 in proximal colon (68 in ascending colon and 12 in transverse colon) and 228 in distal colon (19 in descending colon, 59 in sigmoid colon, and 150 in rectum). No chemotherapy or radiotherapy was performed before surgery for all patients. NRIP3 methylation was significantly associated with older age (age 50 years or older), tumor differentiation, and lymph node metastasis (all P < 0.05, Table 1). No significant associations were observed between NRIP3 methylation and gender, tumor size, TNM stage, and tumor location (all P > 0.05, Table 1). In 190 cases of CRC with available OS data, NRIP3 methylation was found significantly associated with poor 5-year OS (P < 0.05, Figure 2c) and was an independent prognostic factor for poor 5-year OS (P < 0.05, Table 2).
Table 1.
Clinical factors and NRIP3 methylation status in patients with colorectal cancer
| Clinical parameter | n = 308 | Methylation status | P value | |
| Unmethylated (n = 152, 49.4%) | Methylated (n = 156, 50.6%) | |||
| Gender | ||||
| Male | 192 | 101 | 91 | 0.1417 |
| Female | 116 | 51 | 65 | |
| Age | ||||
| <50 | 56 | 35 | 21 | 0.0296a |
| ≥50 | 252 | 117 | 135 | |
| Differentiation | ||||
| Well/moderately | 239 | 130 | 109 | 0.0010a |
| Poorly | 69 | 22 | 47 | |
| TNM stage | ||||
| I/II | 188 | 99 | 89 | 0.1460 |
| III/IV | 120 | 53 | 67 | |
| Lymph node metastasis | ||||
| Negative | 203 | 110 | 93 | 0.0182a |
| Positive | 105 | 42 | 63 | |
| Tumor size | ||||
| <5 cm | 163 | 81 | 82 | 0.8985 |
| ≥5 cm | 145 | 71 | 74 | |
| Tumor location | ||||
| Proximal | 80 | 35 | 45 | 0.2442 |
| Distal | 228 | 117 | 111 | |
P values are obtained from the χ2 test, significant difference (P < 0.05).
Table 2.
Univariate and multivariate analyses of NRIP3 methylation status with 5-year overall survival in patients with CRC
| Clinical parameter | Univariate analysis | Multivariate analysis | ||
| HR (95% CI) | P value | HR (95% CI) | P value | |
| Gender (male vs female) | 1.345 (0.692–2.615) | 0.382 | ||
| Age (≥50 vs <50 yr) | 1.418 (0.730–2.757) | 0.303 | ||
| Tumor size (≥4 vs <4 cm) | 1.211 (0.635–2.307) | 0.561 | ||
| Differentiation (low vs high or middle differentiation) | 2.420 (1.231–4.757) | 0.010a | 1.476 (0.730–2.984) | 0.278 |
| TNM stage (III/IV vs I/II) | 4.831 (2.384–9.791) | 0.000b | 11.185 (3.008–41.587) | 0.000b |
| Lymph node metastasis (positive vs negative) | 3.358 (1.727–6.530) | 0.000b | 0.341 (0.097–1.195) | 0.093 |
| NRIP3 (methylation vs unmethylation) | 2.396 (1.203–4.773) | 0.013a | 2.256 (1.069–4.761) | 0.033a |
| Tumor location (distal vs proximal) | 0.648 (0.330–1.272) | 0.208 | ||
CI, confidence interval; HR, hazard ratio.
P < 0.05.
P < 0.001.
To assess NRIP3 levels, an IHC assay was used in 72 cases of available CRC and matched margin samples. The expression of NRIP3 was higher in margin tissue samples compared with CRC tissues (P < 0.001, Figure 2d and e), with staining observed in both the nucleus and cytoplasm. The reduction in NRIP3 levels was associated with the methylation of its promoter region (P < 0.001, Figure 2f), indicating the epigenetic regulation of NRIP3 expression in CRC.
NRIP3 suppresses CRC cell proliferation
To assess the impact of NRIP3 on cell proliferation, MTT assay and colony formation were used. The OD values before and after reexpression of NRIP3 in DLD1, RKO, and HCT116 cells were 1.500 ± 0.135 vs 1.175 ± 0.094, 1.086 ± 0.086 vs 0.702 ± 0.075, and 1.406 ± 0.038 vs 1.042 ± 0.032, respectively (Figure 3a). The restoration of NRIP3 expression led to a significant decrease in the OD value, suggesting that NRIP3 suppresses CRC cell proliferation (all P < 0.05). The siRNA technique was used to validate the effect of NRIP3 on cell proliferation. The OD value in NRIP3 expression DKO cells and siRNA knocking down cells was 0.987 ± 0.092 vs 1.513 ± 0.111 (P < 0.01, Figure 3a), further demonstrating the inhibitory effect of NRIP3 on CRC cell growth. The clone number before and after reexpression of NRIP3 in DLD1, RKO, and HCT116 cells was 210.0 ± 7.34 vs 146.7 ± 7.41, 241.0 ± 4.55 vs 153.7 ± 6.13, and 150.7 ± 5.25 vs 100.0 ± 5.72, respectively. The clone number exhibited a significant reduction after reexpression of NRIP3 in CRC cells (all P < 0.01, Figure 3b). To further validate the effect of NRIP3 on clonogenicity, the siRNA knockdown technique was used. In DKO cells, the number of clones was 82.0 ± 5.10 vs 128.7 ± 2.62 in the control and siRNA knockdown groups. The clone number was increased significantly by knockdown of NRIP3 in DKO cell (P < 0.01, Figure 3b). These results demonstrated that NRIP3 inhibits CRC cell proliferation.
Figure 3.

The effect of NRIP3 on cell proliferation and cell cycles. (a) Growth curves represent the effects of NRIP3 in CRC cells. *P < 0.05, **P < 0.01, and ***P < 0.001. (b) Colony formation results. The average number of tumor clones is represented by the bar diagram. **P < 0.01, ***P < 0.001. (c) The effect of NRIP3 on cell phase. The bar diagram represents the percentage. *P < 0.05, **P < 0.01, and ***P < 0.001. (d) Western blots show the effects of NRIP3 knockdown. Scramble: siRNA negative control; siRNA#1, siRNA#2, and siRNA#3: 3 different siRNA for NRIP3. (e) Western blot shows the levels of NRIP3, cyclin E1, cyclin A2, and cyclinD1. Actin: internal control. Scramble: siRNA negative control; siRNA#3: siRNA targeting NRIP3.
NRIP3 induces G1/S phase arrest
The effects of NRIP3 on the cell cycle were assessed using flow cytometry. In NRIP3 unexpressed and reexpressed DLD1 cells, the percentages of cell phase were 61.08 ± 0.60% vs 64.67 ± 0.84% in G0/G1 phase (P < 0.01), 28.23 ± 0.05% vs 25.88 ± 0.34% in S phase (P < 0.01), and 10.68 ± 0.57% vs 9.45 ± 0.52% in G2/M phase. For NRIP3 unexpressed and reexpressed RKO cells, the percentages of cell phase were 44.15 ± 0.04% vs 46.97 ± 0.98% in G0/G1 phase (P < 0.01), 35.65 ± 0.42% vs 32.64 ± 0.98% in S phase (P < 0.05), and 20.20 ± 0.44% vs 20.39 ± 1.05% in G2/M phase. Similarly, for NRIP3 unexpressed and reexpressed HCT116 cells, the percentages of cell phase were 42.75 ± 0.48% vs 48.98 ± 1.79% in G0/G1 phase (P < 0.01), 39.22 ± 1.14% vs 32.18 ± 0.99% in S phase (P < 0.01), and 18.03 ± 1.30% vs 18.84 ± 0.82% in G2/M phase. These findings suggest that NRIP3 induces G1/S phase arrest (Figure 3c). The cell phase distribution was 64.96 ± 0.48% vs 55.31 ± 1.17% in G0/G1 phase (P < 0.01), 22.49 ± 0.29% vs 33.78 ± 0.55% in S phase (P < 0.01), and 12.55 ± 0.44% vs 10.92 ± 0.65% in G2/M phase before and after knockdown of NRIP3 in DKO cells (Figure 3c), further validating the abovementioned results. In addition, the reexpression of NRIP3 in DLD1, RKO, and HCT116 cells led to a decrease in the levels of cyclin D1, cyclin A2, and cyclin E1. Conversely, NRIP3 knockdown in DKO cells resulted in an increase in the levels of cyclin D1, cyclin A2, and cyclin E1 (Figure 3d and e). These observations further support that NRIP3 induces G1/S arrest.
NRIP3 suppresses cell migration and invasion
The role of NRIP3 on cell migration and invasion was examined by transwell assay. In NRIP3 unexpressed and reexpressed DLD1, RKO, and HCT116 cells, the number of migratory cells was 224.00 ± 13.74 vs 178.08 ± 7.97, 176.92 ± 6.69 vs 102.5 ± 9.71, and 232.63 ± 11.55 vs 181.40 ± 18.65, respectively. Notably, the reexpression of NRIP3 in CRC cells resulted in a significant reduction in the number of migratory cells (all P < 0.01, Figure 4a). Furthermore, the knockdown of NRIP3 in DKO cells led to an increase in the number of migratory cells from 151.29 ± 14.78 to 173.90 ± 35.39. The knockdown of NRIP3 in DKO cells resulted in a significant increase in the number of migratory cells (P < 0.001, Figure 4a). The abovementioned results demonstrate that NRIP3 suppresses CRC cells migration.
Figure 4.

The effect of NRIP3 on cell migration and invasion, PI3K-AKT signaling in CRC cells. (a) Migration and invasion results. Bar diagram represent the average number of migration and invasion cells. ***P < 0.001. (b) Western blot shows the levels of NRIP3, MMP2, MMP7, and MMP9. (c) The KEGG enrichment analysis in unexpressed vs reexpressed RKO cells. (d) Western blot shows the levels of NRIP3, PI3K, AKT, p-AKT, mTOR, and p-mTOR in CRC cells. (e) MTT assay shows cell growth curves. Empty vector: NRIP3 unexpressed cells; Empty vector +: NRIP3 unexpressed cells plus NVP-BEZ235 treatment (50 nM); NRIP3: NRIP3 reexpressed cells; NRIP3+: NRIP3 expression plus NVP-BEZ235 treatment. Scramble: siRNA negative control; Scramble+: Scramble siRNA plus NVP-BEZ235 treatment; siRNA#3: the most effective NRIP3 targeting siRNA; siRNA#3+: siNRIP3 plus NVP-BEZ235 treatment. ***P < 0.001. (f) Western blot shows the levels of NRIP3, PI3K, AKT, p-AKT, mTOR, and p-mTOR, before and after NVP-BEZ235 treatment, with or without NRIP3 expression.
The number of invasive cells was 176.50 ± 9.40 vs 129.58 ± 4.84, 177.08 ± 10.63 vs 115.17 ± 9.61, and 193.63 ± 6.89 vs 137.33 ± 27.33 in NRIP3 silencing and forcing–expressed DLD1, RKO, and HCT116 cells, respectively, demonstrating that NRIP3 inhibits CRC cell invasion (all P < 0.01, Figure 4a). The number of invasive cells was 80.14 ± 7.92 vs 103.40 ± 12.82 in NRIP3 highly expressed and siRNA knocking down DKO cells. The knockdown of NRIP3 resulted in a significant increase in the number of invasive cells (P < 0.01, Figure 4a). The abovementioned data demonstrate that NRIP3 suppresses cell invasion. Figure 4b illustrates that the levels of MMP2, MMP7, and MMP9 decreased after reexpression of NRIP3 in DLD1, RKO, and HCT116 cells and increased after NRIP3 knockdown in DKO cells, further validating the results at the molecular level.
NRIP3 inhibits PI3K-AKT signaling
To understand the mechanism of NRIP3 in the development of CRC, a microarray analysis was conducted to compare the gene expression profiles of RKO cells with and without NRIP3 expression. The results were analyzed using KEGG pathway enrichment, revealing significant alterations in the PI3K-AKT signaling (Figure 4c). NRIP3 was reported to be involved in PI3K-AKT signaling in gastric mucosa in obese patients and patients with obesity-related diabetes (13). Increased expression of NRIP3 was reported in PIK3CA gene mutated breast cancer (14). Thus, we focused on the role of NRIP3 in PI3K-AKT signaling. PI3K, p-AKT, and p-mTOR levels were reduced after reexpression of NRIP3 in DLD1, RKO, and HCT116 cells, while no changes were observed in the total levels of AKT and mTOR. In DKO cells, the knockdown of NRIP3 resulted in an increase in PI3K, p-AKT, and p-mTOR levels, while no obvious changes of total AKT and mTOR were noticed (Figure 4d). This observation suggests that NRIP3 plays an inhibitory role in the PI3K-AKT signaling pathway.
To further validate the involvement of NRIP3 in the PI3K-AKT-mTOR signaling pathway, NVP-BEZ235, a PI3K-mTOR inhibitor, was used. The OD values before and after NVP-BEZ235 treatment were 1.306 ± 0.088 vs 0.857 ± 0.055, 1.154 ± 0.013 vs 0.642 ± 0.018, and 0.888 ± 0.034 vs 0.740 ± 0.032 in NRIP3 unexpressed DLD1, RKO, and HCT116 cells, respectively. These results demonstrate a significant decrease in OD values on inhibition of the PI3K-mTOR signaling pathway (all P < 0.01, Figure 4e). While after reexpression of NRIP3 in DLD1, RKO, and HCT116 cells, the OD values were 0.915 ± 0.095 vs 0.908 ± 0.147, 0.631 ± 0.030 vs 0.581 ± 0.067, and 0.578 ± 0.017 vs 0.559 ± 0.005 under the conditions of NVP-BEZ235 untreatment and treatment, respectively. No significant differences were observed between groups treated with the PI3K-mTOR inhibitor and the untreated groups (all P > 0.05). Before and after NVP-BEZ235 treatment, the OD values were 0.640 ± 0.024 vs 0.617 ± 0.013 in the siRNA scramble control DKO group (P > 0.05). In siNRIP3 knockdown DKO cells, the OD values were 0.755 ± 0.024 vs 0.501 ± 0.027 before and after NVP-BEZ235 treatment, respectively, indicating a significant decrease in OD value by NVP-BEZ235 treatment (P < 0.01, Figure 4e). These results imply that the defect of NRIP3 activated PI3K-AKT pathway.
NVP-BEZ235 resulted in decreased levels of PI3K, p-AKT, and p-mTOR in NRIP3 unexpressed DLD1, RKO, and HCT116 cells, but no significant changes were observed in NRIP3 forced expression cells (Figure 4f). In DKO cells, NVP-BEZ235 led to decreased levels of PI3K, p-AKT, and p-mTOR by knocking down NRIP3, while no obvious difference was observed in scramble DKO cells (Figure 4f). These data further demonstrate that PI3K-AKT signaling is inhibited by NRIP3 in CRC.
CRC cell xenografts were suppressed by NRIP3
In DLD1 and RKO cell xenografts with or without NRIP3 expression, the tumor volume was 688.07 ± 125.32 mm3 vs 444.08 ± 52.76 mm3 (P < 0.01) and 511.53 ± 70.90 mm3 vs 239.33 ± 55.05 mm3 (P < 0.01), respectively. Tumor volume was reduced in NRIP3 reexpressed xenografts compared with NRIP3 unexpressed xenografts (Figure 5a and b). The xenograft weight was 0.500 ± 0.080 g vs 0.254 ± 0.048 g, and 0.121 ± 0.014 g vs 0.037 ± 0.016 g in NRIP3 silenced and reexpressed DLD1 and RKO cells, respectively. Restoration of NRIP3 expression significantly reduced the tumor weight (all P < 0.01, Figure 5c). The abundance of PI3K, p-AKT, and p-mTOR was assessed using IHC in NRIP3 unexpressed and expressed xenografts. The expression of NRIP3 resulted in a reduction of PI3K, p-AKT, and p-mTOR levels, indicating the inhibitory role of NRIP3 in PI3K-AKT signaling in vivo (Figure 5d).
Figure 5.

The effect of NRIP3 on PI3K/AKT signaling in CRC cell xenografts. (a) Representative results of NRIP3 unexpressed and reexpressed cell xenografts. (b) Growth curves of NRIP3 unexpressed and reexpressed cell xenografts. **P < 0.01, ***P < 0.001. (c) Tumor weight of NRIP3 unexpressed and reexpressed cell xenografts. Bars represent the tumor weight. ***P < 0.001. (d) Representative immunohistochemistry results for xenografts.
NRIP3 methylation is a sensitive marker of combined PI3K-Akt-mTOR inhibitor with ATR/ATM inhibitors
PI3K has been reported to be involved in DNA damage repair, and combination of its inhibitor with a CHK1 inhibitor enforced antitumor efficiency (15,16). Our study demonstrated that epigenetic silencing of NRIP3 activates the PI3K pathway in CRC. Thus, we assessed the sensitivity of NRIP3 expressed and silenced CRC cells to VE-822 (an ATR inhibitor) and AZD0156 (an ATM inhibitor). Under the low levels of cisplatin treatment, the IC50 of VE-822 was 2.540 ± 0.411 μM vs 0.839 ± 0.179 μM, 3.779 ± 0.473 μM vs 0.479 ± 0.088 μM, and 10.272 ± 0.607 μM vs 0.659 ± 0.083 μM in NRIP3 unexpressed and force-expressed DLD1, RKO, and HCT116 cells, respectively. Reduced IC50 value was observed by expression of NRIP3 (all P < 0.01, Figure 6a). Similarly, under low-level cisplatin induction, the IC50 values of AZD0156 were 9.980 ± 1.137 μM vs 5.211 ± 0.817 μM, 13.270 ± 2.010 μM vs 7.973 ± 0.672 μM, and 18.479 ± 0.305 μM vs 10.640 ± 1.470 μM in DLD1, RKO, and HCT116 cells without expression and forced expression of NRIP3, respectively. The IC50 value of AZD0156 was significantly reduced by NRIP3 (all P < 0.05, Figure 6a). These results demonstrated that NRIP3 enhances the sensitivity of CRC cells to ATR and ATM inhibitors.
Figure 6.

Loss of NRIP3 expression sensitized cells to NVP-BEZ235/VE-822 and NVP-BEZ235/AZD0156. (a) The IC50 curve of VE-822 and AZD0156 under cisplatin treatment in CRC cells. (b) The levels of ATR, p-ATR, ATM, p-ATM, CHK1, p-CHK1, CHK2, and p-CHK2 in NRIP3 unexpressed and reexpressed CRC cells with or without cisplatin treatment (0.1 μM). (c) MTT assay shows the sensitivity of CRC cells to VE-822, AZD0156, or NVP-BEZ235. VE-822: 0.2 μM; AZD0156: 0.5 μM; NVP-BEZ235: 50 nM. VE-822 + NVP-BEZ235: VE-822 plus NVP-BEZ235 treatment; AZD0156 + NVP-BEZ235: AZD0156 plus NVP-BEZ235 treatment. *P < 0.05, ***P < 0.001.
To further validate the role of NRIP3 in ATR/CHK1 or ATM/CHK2 signaling, the levels of ATR, p-ATR, CHK1, p-CHK1, ATM, p-ATM, CHK2, and p-CHK2 were assessed through Western blotting. Under cisplatin treatment, restoration of NRIP3 expression in CRC cells resulted in elevated levels of p-ATR, p-CHK1, p-ATM, and p-CHK2 (Figure 6b), indicating the activation of ATR/CHK1 and ATM/CHK2 signaling by NRIP3.
Then, the MTT assay was used to analyze the synthetic role of NVP-BEZ235 with VE-822/AZD0156 in NRIP3 silenced DLD1, RKO, and HCT116 cells. The OD values in NRIP3 silenced DLD1, RKO, and HCT116 cells treated with VE-822 or combined NVP-BEZ235 with VE-822 were 0.9341 ± 0.027 vs 0.643 ± 0.017, 0.989 ± 0.098 vs 0.836 ± 0.042, and 0.996 ± 0.097 vs 0.797 ± 0.027, respectively. A reduction in OD value was observed when NVP-BEZ235 and VE-822 were combined (all P < 0.05, Figure 6c). Similarly, in NRIP3 unexpressed DLD1, RKO, and HCT116 cells treated with NVP-BEZ235 or combined NVP-BEZ235 with AZD0156, the OD values were 0.427 ± 0.007 vs 0.245 ± 0.017, 0.491 ± 0.023 vs 0.404 ± 0.015, and 0.531 ± 0.009 vs 0.452 ± 0.009, respectively. A decrease in OD value was observed with the combination of NVP-BEZ235 and AZD0156 compared with that with AZD0156 alone (all P < 0.01, Figure 6c). These results indicate that NRIP3 methylation is a potential synthetic lethal marker for combined PI3K with ATR/ATM inhibitors.
DISCUSSION
Synthetic lethality provides an approach to selectively target cancer cells harboring undruggable mutations (17). The successful application of poly (ADP-ribose) polymerase (PARP) inhibitors in BRCA1/2 mutated cells is a paradigmatic model (18). BRCAness has broadened its usage to abnormal changes in other DNA damage repair (DDR) genes in cancer (19,20). In addition, therapeutic targeting of cell fate–associated signaling pathways represents another avenue for cancer treatment (21,22). Epigenetic-based synthetic lethality is a novel strategy to target partners of epigenetic changed DDR or cell fate genes in cancer (23,24). By discovering new DDR/cell fate–related genes may develop more cancer therapy strategies. Epigenetic dysregulation of these components will offer more opportunities for synthetic lethal therapy.
NRs are intracellular transcription factors that regulate the activity of complex gene networks. This superfamily is typically subdivided into 3 families, namely the steroid receptor, retinoid, and orphan receptor families (25). Within the NRIP subfamily, there exist 3 members. NRIP1 was originally identified in breast cancer as a modulator of ERα activity. Thereafter, it was found to interact with and inhibit other transcription factors, including NRs and E2F (26). In CRC, NRIP1 inhibits WNT signaling, and reduced expression of NRIP1 is associated with poor OS (9). NRIP1 mainly acts as a transcriptional repressor through its 4 inhibitory domains (27). NRIP2 has been reported to stabilize and activate β-catenin in injured podocytes, and it is upregulated in CRC initiating cells to modulate WNT signaling (28,29). NRIP3 is highly expressed and confers resistance to chemoradiotherapy in human esophageal cancer (30). Epigenetic heterogeneity and cancer type–specific DNA methylation have been previously reported (22,31,32).
By analyzing TCGA and GTEx databases, the levels of NRIP3 expression were found to be reduced in CRC, and the reduced expression was associated with the methylation status of a few CpG sites in the promoter region, reminding the epigenetic regulation of NRIP3 expression. Then, we analyzed the expression and epigenetic regulation of NRIP3 in CRC cell lines. NRIP3 expression was lost or reduced in all detected CRC cells, and its expression was regulated by promoter region methylation. The tissue type–specific methylation was excluded by detecting 6 cases of normal colonic mucosa from noncancerous patients. The epigenetic regulation of NRIP3 was validated by analyzing immunohistochemical staining and methylation status in primary CRC. NRIP3 was methylated only in 5 cases among 187 margin samples from resected CRC, and the methylated patients were all aged 69 years or older. NRIP3 was methylated in 32.2% of colonic adenomatous polyps and 50.6% of CRC. Notably, a significant association was found between methylation and older age (50 years or older) in both tissue types (all P < 0.05), implying that NRIP3 methylation is a potential late-onset marker for CRC. NRIP3 methylation was associated with tumor differentiation and lymph node metastasis in CRC and is an independent prognostic factor for poor 5-year OS. The abovementioned data implicate the malignant character of NRIP3 methylation.
A genomic model of CRC is well established (33). Patients with early-onset colorectal cancer are more likely to have a hereditary syndrome while later-onset patients are mostly sporadic cases, with no identifiable cause (1). Accumulation of promoter region methylation has been observed during esophageal carcinogenesis (31,34). We found that the NRIP3 methylation ratio was higher in invasive CRC than in precancerous adenomatous polyps. The interplay between genetics and epigenetics is a common event in cancer, and the environment is regarded as a major contributor to epigenetic changes (22,35,36). Thus, it is plausible to hypothesize that aberrant epigenetic changes may contribute to the development of late-onset tumors. NRIP3 methylation may potentially serve as an early detection, later-onset, and independent poor prognostic factor. However, it is necessary to increase the case numbers and validate the results by separate groups to strengthen our assumptions. Functional study found that NRIP3 suppresses CRC cell growth both in vitro and in vivo, suggesting it is a potential tumor suppressor. Through comparing the expression of NRIP3 in CRC cells that were unexpressed and overexpressed, significant changes in gene expression were observed in the PI3K-AKT signaling. Given that activation of the PI3K-AKT signaling was reported to be related to poor prognosis (37,38), and our results indicate that NRIP3 methylation is an independent poor prognostic factor for CRC. Then, we focused on PI3K-AKT signaling pathway. The results demonstrated that NRIP3 inhibits the activity of PI3K-AKT signaling pathway in both CRC cells and xenografts.
Previous studies have linked NRIP3 to chemoresistance in esophageal cancer, and PI3K-AKT signaling is known to be involved in DDR (15,16,30). Although regulation network of PI3K-AKT signaling is highly complex (39,40), our study primarily focused on ATR/CHK1 and ATM/CHK2 signaling pathways using a low-dose cisplatin treatment CRC cell model. The results indicate that NRIP3 activates ATR/CHK1 and ATM/CHK2 signaling pathways. PI3K-AKT signaling was reported to inhibit ATR/CHK1 and ATM/CHK2 signaling pathways (39,41,42). It is supposed that epigenetic silencing NRIP3 may activate PI3K-AKT signaling to inhibit ATR/CHK1 and ATM/CHK2 signaling. To answer this question, further study was performed to test the efficiency of ATR and ATM inhibitors in both NRIP3 expressed and unexpressed CRC cells. An increased sensitivity to ATR and ATM inhibitors was observed in cells expressing NRIP3. In addition to inhibition of ATR and ATM signaling pathways, PI3K-AKT signaling plays more complex roles in DDR (39,43), we assess PI3K inhibitor in NRIP3 silenced CRC cells. The sensitivity of NRIP3 silenced cell to ATR and ATM inhibitors was enhanced by addition of a PI3K inhibitor. These findings indicate that synthetic lethal efficiency may be caused by PI3K and ATR/ATM inhibitors in NRIP3 methylated cells (Figure 7).
Figure 7.
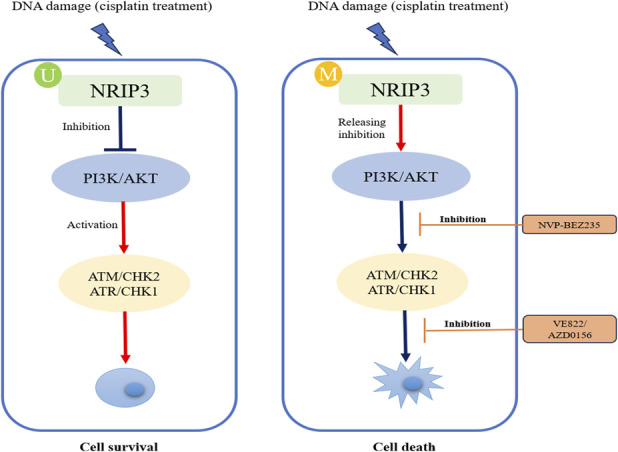
A working model for synthetic lethality of PI3K and ATR/ATM inhibitors in NRIP3 epigenetic silenced CRC cells. U, unmethylation; M, methylation; DDR, DNA damage repair.
This study offers an insight into understanding of epigenetic-based targeting therapy in CRC. By using the rationale of BRCAness, our previous studies have explored epigenetic-based synthetic lethality in esophageal and lung cancers (20,21,23,24). Epigenetic-based synthetic lethality is becoming a novel strategy for cancer therapy and will broaden the scope of precision medicine. However, it is important to acknowledge that this study has certain limitations. PI3K-AKT and DDR signaling pathways exhibit considerable complexity, and the DNA damage model used in this study is limited to low-dose cisplatin treatment. Thus, further comprehensive investigations are necessary to acquire practical approaches for cancer therapy.
In conclusion, NRIP3 methylation is a potential late-onset, poor prognosis, and early detection marker in CRC. NRIP3 suppresses CRC growth by inhibiting PI3K-AKT signaling pathway both in vitro and in vivo. Epigenetic silencing of NRIP3 sensitizes CRC cells to PI3K and ATR/ATM inhibitors.
CONFLICTS OF INTEREST
Guarantor of the article: Mingzhou Guo, MD, PhD.
Specific author contributions: M.Z. and X.L. performed experiments. A.G., Q.W., Y.Y., and F.S. helped analyze the data. M.Z. and M.G. wrote the manuscript. M.G. designed the study and edited the manuscript. J.G.H. and K.H. interpreted the data. All the authors have read and approved the final version of the manuscript.
Financial support: This work was supported by grants from National Key Research and Development Program of China (2018YFA0208902, 2020YFC2002705); National Science Foundation of China (82272632, 81672138, U1604281); Beijing Science Foundation of China (7171008); Youth Innovation Science Foundation of Chinese PLA general hospital (22QNCZ027).
Potential competing interests: None to report.
Institutional review board statement: All procedures were performed in accordance with the ethics committee of the authors' institution.
Study Highlights.
WHAT IS KNOWN
✓ The role of NRIP3 in colorectal cancer remains unclear.
✓ Synthetic lethality is a novel model for cancer therapy.
WHAT IS NEW HERE
✓ NRIP3 methylation is significantly associated with late onset (at age 50 years or older) and is an independent poor prognostic marker.
✓ NRIP3 is a potential tumor suppressor.
✓ NRIP3 methylation is a synthetic lethal marker for combined PI3K and ATR/ATM inhibitors.
Supplementary Material
ACKNOWLEDGMENT
DKO cells with DNMT1 and DNMT3b double knockouts from HCT116 cells were a generous gift from Stephen B. Baylin.
Footnotes
SUPPLEMENTARY MATERIAL accompanies this paper at http://links.lww.com/CTG/B66, http://links.lww.com/CTG/B67, http://links.lww.com/CTG/B68, http://links.lww.com/CTG/B69
Meiying Zhang and Xiaoyun Li contributed equally to this work.
Contributor Information
Meiying Zhang, Email: zhangmeiying.1988@163.com.
Xiaoyun Li, Email: fqmyy@163.com.
James G. Herman, Email: Hermanj3@upmc.edu.
Aiai Gao, Email: aiaigao@outlook.com.
Qian Wang, Email: wangqian333666@163.com.
Yuanxin Yao, Email: yaoyx301@163.com.
Fangfang Shen, Email: ffshen2010@163.com.
Kunlun He, Email: kunlunhe@301hospital.com.cn.
REFERENCES
- 1.Sinicrope FA. Increasing incidence of early-onset colorectal cancer. N Engl J Med 2022;386(16):1547–58. [DOI] [PubMed] [Google Scholar]
- 2.Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2018;68(6):394–424. [DOI] [PubMed] [Google Scholar]
- 3.Siegel RL, Miller KD, Goding Sauer A, et al. Colorectal cancer statistics, 2020. CA Cancer J Clin 2020;70(3):145–64. [DOI] [PubMed] [Google Scholar]
- 4.Cercek A, Chatila WK, Yaeger R, et al. A comprehensive comparison of early-onset and average-onset colorectal cancers. J Natl Cancer Inst 2021;113(12):1683–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pearlman R, Frankel WL, Swanson B, et al. Prevalence and spectrum of germline cancer susceptibility gene mutations among patients with early-onset colorectal cancer. JAMA Oncol 2017;3(4):464–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stoffel EM, Koeppe E, Everett J, et al. Germline genetic features of young individuals with colorectal cancer. Gastroenterology 2018;154(4):897–905.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sinicrope FA. Lynch syndrome-associated colorectal cancer. N Engl J Med 2018;379(8):764–73. [DOI] [PubMed] [Google Scholar]
- 8.Triki M, Lapierre M, Cavailles V, et al. Expression and role of nuclear receptor coregulators in colorectal cancer. World J Gastroenterol 2017;23(25):4480–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lapierre M, Bonnet S, Bascoul-Mollevi C, et al. RIP140 increases APC expression and controls intestinal homeostasis and tumorigenesis. J Clin Invest 2014;124(5):1899–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lapierre M, Castet-Nicolas A, Gitenay D, et al. Expression and role of RIP140/NRIP1 in chronic lymphocytic leukemia. J Hematol Oncol 2015;8:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wolfe DM, Webster Carrion A, Masukhani MM, et al. Concurrent hepatoblastoma and Wilms tumor leading to diagnosis of Beckwith-Wiedemann syndrome. J Pediatr Hematol Oncol 2023;45(4):e525–e529. [DOI] [PubMed] [Google Scholar]
- 12.He T, Zhang M, Zheng R, et al. Methylation of SLFN11 is a marker of poor prognosis and cisplatin resistance in colorectal cancer. Epigenomics 2017;9(6):849–62. [DOI] [PubMed] [Google Scholar]
- 13.Wen X, Qian C, Zhang Y, et al. Key pathway and gene alterations in the gastric mucosa associated with obesity and obesity-related diabetes. J Cell Biochem 2019;120(4):6763–71. [DOI] [PubMed] [Google Scholar]
- 14.Cizkova M, Cizeron-Clairac G, Vacher S, et al. Gene expression profiling reveals new aspects of PIK3CA mutation in ERalpha-positive breast cancer: Major implication of the Wnt signaling pathway. PLoS One 2010;5(12):e15647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang TT, Brill E, Nair JR, et al. Targeting the PI3K/mTOR pathway augments CHK1 inhibitor-induced replication stress and antitumor activity in high-grade serous ovarian cancer. Cancer Res 2020;80(23):5380–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Juvekar A, Hu H, Yadegarynia S, et al. Phosphoinositide 3-kinase inhibitors induce DNA damage through nucleoside depletion. Proc Natl Acad Sci USA 2016;113(30):E4338–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hahn WC, Bader JS, Braun TP, et al. An expanded universe of cancer targets. Cell 2021;184(5):1142–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005;434(7035):917–21. [DOI] [PubMed] [Google Scholar]
- 19.Lord CJ, Ashworth A. BRCAness revisited. Nat Rev Cancer 2016;16(2):110–20. [DOI] [PubMed] [Google Scholar]
- 20.Hu Y, Guo M. Synthetic lethality strategies: Beyond BRCA1/2 mutations in pancreatic cancer. Cancer Sci 2020;111(9):3111–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gao A, Guo M. Epigenetic based synthetic lethal strategies in human cancers. Biomarker Res 2020;8:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guo M, Peng Y, Gao A, et al. Epigenetic heterogeneity in cancer. Biomarker Res 2019;7:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Du W, Gao A, Herman JG, et al. Methylation of NRN1 is a novel synthetic lethal marker of PI3K-Akt-mTOR and ATR inhibitors in esophageal cancer. Cancer Sci 2021;112(7):2870–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li H, Yang W, Zhang M, et al. Methylation of TMEM176A, a key ERK signaling regulator, is a novel synthetic lethality marker of ATM inhibitors in human lung cancer. Epigenomics 2021;13(17):1403–19. [DOI] [PubMed] [Google Scholar]
- 25.Bain DL, Heneghan AF, Connaghan-Jones KD, et al. Nuclear receptor structure: Implications for function. Annu Rev Physiol 2007;69:201–20. [DOI] [PubMed] [Google Scholar]
- 26.Docquier A, Harmand PO, Fritsch S, et al. The transcriptional coregulator RIP140 represses E2F1 activity and discriminates breast cancer subtypes. Clin Cancer Res 2010;16(11):2959–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Christian M, Tullet JM, Parker MG. Characterization of four autonomous repression domains in the corepressor receptor interacting protein 140. J Biol Chem 2004;279(15):15645–51. [DOI] [PubMed] [Google Scholar]
- 28.Hou Q, Le W, Kan S, et al. Nuclear receptor interacting protein-2 mediates the stabilization and activation of β-catenin during podocyte injury. Front Cell Dev Biol 2021;9:781792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wen Z, Pan T, Yang S, et al. Up-regulated NRIP2 in colorectal cancer initiating cells modulates the Wnt pathway by targeting RORβ. Mol Cancer 2017;16(1):20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Suo D, Wang L, Zeng T, et al. NRIP3 upregulation confers resistance to chemoradiotherapy in ESCC via RTF2 removal by accelerating ubiquitination and degradation of RTF2. Oncogenesis 2020;9(8):75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guo M, Ren J, Brock MV, et al. Promoter methylation of HIN-1 in the progression to esophageal squamous cancer. Epigenetics 2008;3(6):336–41. [DOI] [PubMed] [Google Scholar]
- 32.Zhu C, Zhang M, Wang Q, et al. Intratumor epigenetic heterogeneity: A panel gene methylation study in thyroid cancer. Front Genet 2021;12:714071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vogelstein B, Papadopoulos N, Velculescu VE, et al. Cancer genome landscapes. Science (New York, NY) 2013;339(6127):1546–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guo M, Ren J, House MG, et al. Accumulation of promoter methylation suggests epigenetic progression in squamous cell carcinoma of the esophagus. Clin Cancer Res 2006;12(15):4515–22. [DOI] [PubMed] [Google Scholar]
- 35.Yan W, Herman JG, Guo M. Epigenome-based personalized medicine in human cancer. Epigenomics 2016;8(1):119–33. [DOI] [PubMed] [Google Scholar]
- 36.You JS, Jones PA. Cancer genetics and epigenetics: Two sides of the same coin? Cancer Cell 2012;22(1):9–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ye Y, Huang Z, Zhang M, et al. Synergistic therapeutic potential of alpelisib in cancers (excluding breast cancer): Preclinical and clinical evidences. Biomed Pharmacother 2023;159:114183. [DOI] [PubMed] [Google Scholar]
- 38.Malinowsky K, Nitsche U, Janssen KP, et al. Activation of the PI3K/AKT pathway correlates with prognosis in stage II colon cancer. Br J Cancer 2014;110(8):2081–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xu N, Lao Y, Zhang Y, et al. Akt: A double-edged sword in cell proliferation and genome stability. J Oncol 2012;2012:951724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Plo I, Laulier C, Gauthier L, et al. AKT1 inhibits homologous recombination by inducing cytoplasmic retention of BRCA1 and RAD51. Cancer Res 2008;68(22):9404–12. [DOI] [PubMed] [Google Scholar]
- 41.Xu N, Hegarat N, Black EJ, et al. Akt/PKB suppresses DNA damage processing and checkpoint activation in late G2. J Cell Biol 2010;190(3):297–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yakovlev VA, Sullivan SA, Fields EC, et al. PARP inhibitors in the treatment of ARID1A mutant ovarian clear cell cancer: PI3K/Akt1-dependent mechanism of synthetic lethality. Front Oncol 2023;13:1124147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Park JH, Jung KH, Kim SJ, et al. Radiosensitization of the PI3K inhibitor HS-173 through reduction of DNA damage repair in pancreatic cancer. Oncotarget 2017;8(68):112893–906. [DOI] [PMC free article] [PubMed] [Google Scholar]