

Significance
The distinct oxidative reactivities reported here for the TMC-anti and TMC-syn isomers are surprising and require the examination of what factors can give rise to such differences for this class of iron complexes. In particular, our observation that the oxygen atom transfer reactivity of a nonheme FeIV=O unit can be so significantly enhanced by a simple flip in its orientation relative to its macrocyclic tetraamine host is truly remarkable and suggests that there is much more that can be learned for catalyst design by paying attention to the effect of ligand topology on the iron active site.
Keywords: nonheme iron(IV)-oxo complexes, topological isomers, olefin epoxidation, tetramethylcyclam
Abstract
TMC-anti and TMC-syn, the two topological isomers of [FeIV(O)(TMC)(CH3CN)]2+ (TMC = 1,4,8,11-tetramethyl-1,4,8,11-tetraazacyclotetradecane, or Me4cyclam), differ in the orientations of their FeIV=O units relative to the four methyl groups of the TMC ligand framework. The FeIV=O unit of TMC-anti points away from the four methyl groups, while that of TMC-syn is surrounded by the methyl groups, resulting in differences in their oxidative reactivities. TMC-syn reacts with HAT (hydrogen atom transfer) substrates at 1.3- to 3-fold faster rates than TMC-anti, but the reactivity difference increases dramatically in oxygen-atom transfer reactions. R2S substrates are oxidized into R2S=O products at rates 2-to-3 orders of magnitude faster by TMC-syn than TMC-anti. Even more remarkably, TMC-syn epoxidizes all the olefin substrates in this study, while TMC-anti reacts only with cis-cyclooctene but at a 100-fold slower rate. Comprehensive quantum chemical calculations have uncovered the key factors governing such reactivity differences found between these two topological isomers.
Oxoiron(IV) species are key intermediates in biological and abiological oxidations like C–H hydroxylation and halogenation, as well as olefin epoxidation (1–4). A significant effort by various groups has led to the characterization of synthetic nonheme oxoiron(IV) complexes that serve as spectroscopic and functional mimics of nonheme iron oxygenases to enhance our understanding of these natural processes (4). Thus far, over a hundred such synthetic nonheme oxoiron(IV) complexes have been documented since the report of the crystal structure of anti-[FeIV(O)(TMC)(CH3CN)]2+ (TMC = 1,4,8,11-tetramethyl-1,4,8,11-tetraazacyclotetradecane, or Me4cyclam), the first example for an oxoiron(IV) complex in a nonheme ligand environment (5). Much effort has since been invested in increasing our understanding of HAT (hydrogen-atom transfer) reactions that result in the hydroxylation of C–H bonds (6). However, much less attention has thus far been paid to OAT (oxygen-atom transfer) reactions, although some examples of olefin epoxidation have been reported for a handful of nonheme iron enzymes (7–9). Similarly, only a few examples of synthetic nonheme oxoiron(IV)-mediated epoxidation reactions have thus far been reported (10–14). In this article, we have explored the HAT and OAT reactivity of the anti- and syn-isomers of [FeIV(O)(TMC)(CH3CN)]2+ (Fig. 1) and find significantly faster OAT rates for R2S oxidation and olefin epoxidation by the syn complex relative to its anti-counterpart. Ligand topology would thus appear to be a critical factor in enhancing such oxidative reactivity, and our insights are reported in this paper.
Fig. 1.

Top: Generation of anti and syn-isomers of [FeIV(O)(TMC)(CH3CN)]2+ complexes in CH3CN by respective treatment of the FeII precursor with oxo-transfer agents PhIO and the sterically bulkier s-ArIO at 25 °C. Bottom: van der Waals radii space-filling models for the anti (Left) and syn (Right) isomers showing greater substrate accessibility to the oxo atom in the syn isomer.
Results and Discussion
Oxoiron(IV) complexes supported by the TMC macrocycle can be generated as two discrete forms in CH3CN, namely anti-, and syn-isomers, as determined by the orientation of the FeIV=O moiety relative to the four methyl groups on the macrocycle. TMC-anti or anti-[FeIV(O)(TMC)(CH3CN)]2+, reported in 2003, represents the first nonheme oxoiron(IV) complex to have been crystallographically characterized (5). It is formed by the reaction of [FeII(TMC)(OTf)](OTf) with PhIO in CH3CN. Its Fe=O unit points in the direction opposite to that of the four methyl groups of the TMC ligand, which surround the sixth ligand, namely CH3CN. Twelve years later the corresponding TMC-syn isomer was obtained by using the sterically bulkier 2-tBuSO2-C6H4IO (s-ArIO) oxidant, resulting in the FeIV=O unit pointing in the opposite direction and surrounded by the four methyl groups (15).While it is not yet fully understood why different topological isomers TMC-anti and TMC-syn are generated with PhIO and s-ArIO, respectively, it is clear that they are formed using these two distinct oxidants. When the FeII(TMC)(OTf)2 precursor is reacted with PhIO, the only product isolated is the TMC-anti isomer, which is shown by XRD to adopt the trans-I (R,S,R,S) configuration with all four methyl groups arrayed on one side of the macrocycle and the oxo atom bound to the anti-face of the TMC ligand (5). On the other hand, when FeII(TMC)(OTf)2 is reacted with s-ArIO, an attack occurs at the less hindered syn-face to give TMC-syn (15). These observations show that the two isomers are formed by clearly distinct pathways with very little cross-contamination of the other isomer if at all. [Note: A complex we originally reported to be the TMC-syn isomer in 2008 (16) was later deduced to be the fluoride derivative of TMC-anti (15)].
A triflate anion has been found to be bound trans to the oxo atom in the crystal structure of TMC-syn (SI Appendix, Fig. S2). However, it is displaced by CH3CN upon dissolution into the solvent, as confirmed by its 19F-NMR spectrum showing a free triflate peak at ~79 ppm (SI Appendix, Fig. S3). Despite their structural differences, the two isomers exhibit quite similar UV-Vis and Raman features (Table 1) but give rise to distinct 1H-NMR spectra with paramagnetically shifted ligand resonances that are opposite in sign for the corresponding ligand protons in the two isomers (SI Appendix, Fig. S2), reflecting the opposite orientations of the paramagnetic Fe=O unit relative to the TMC macrocycle (15).
Table 1.
Experimental (and DFT-derived) properties of TMC-anti and TMC-syn complexes
| Properties |
TMC-anti S = 1 |
TMC-anti S = 2 (DFT) |
TMC-syn S = 1 |
TMC-syn S = 2 (DFT) |
|---|---|---|---|---|
| r(Fe–O) (Å) | 1.646 (1.636) | 1.633 | 1.625 (1.624) | 1.623 |
| r(Fe–Nave) (Å) | 2.091 (2.131) | 2.220 | 2.068 (2.123) | 2.217 |
| r(Fe–L) (Å) | 2.058 (2.026) | 2.012 | 2.146 (2.093) | 2.076 |
| r(Fe-oop)* (Å) | −0.033 (−0.027) | −0.026 | +0.063 (+0.106) | +0.155 |
| λmax (nm) | 825 (706) | 608 | 815 (691) | 604 |
| ν (Fe=O) (cm−1) | 839 (864) | 877 | 856 (892) | 903 |
| δ (mm s−1) | 0.17 (0.152) | 0.143 | 0.16 (0.149) | 0.149 |
| ΔEQ (mm s−1) | 1.24 (0.962) | −0.948 | 1.55 (1.274) | −0.599 |
*Fe-oop = out-of-plane distance between the Fe and the plane defined by the four TMC-N atoms (see SI Appendix, Computational Details, for information on how these values were obtained). Note that all the italicized texts in this table represent results from DFT calculations.
H-Atom Transfer Reactivity Differences between TMC-anti and TMC-syn Isomers.
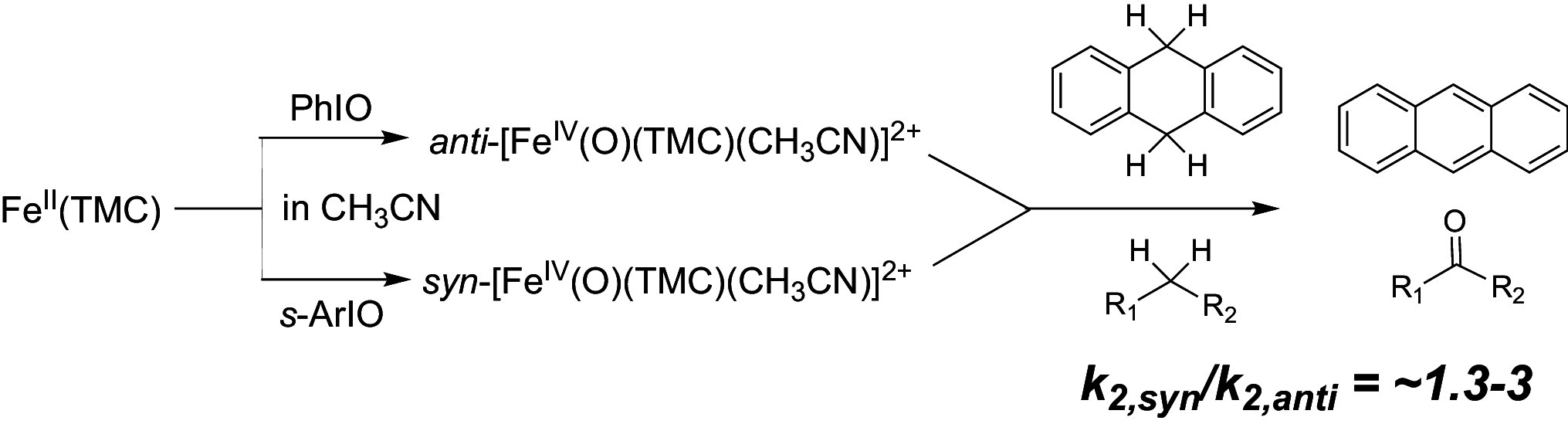
Perhaps not too surprisingly, the structural differences between the TMC-anti and TMC-syn isomers give rise to distinct HAT (H-atom transfer) and OAT (O-atom transfer) reactivity. As C–H bond cleavage is the reaction most commonly catalyzed by nonheme iron enzymes, earlier studies on synthetic nonheme FeIV=O complexes have focused mostly on HAT and the related concerted proton-coupled electron transfer (cPCET) reactions (17). Distinguishing between these two mechanisms can be done by inspection of the change in orbitals along the reaction path (18) or by computing the charge displacement function at the transition state (19). Here we used the latter approach to show (SI Appendix, Fig. S1) that the reactions studied here involve an HAT mechanism, and not cPCET. These studies compared TMC-anti with related complexes to assess relative rates of hydrocarbon oxidation that reveal a systematic decline in oxidation rates with an increase in C–H bond dissociation energy (3, 20). Parallel studies show that TMC-syn also exhibits a similar pattern of decreasing rates with substrates having stronger C–H bonds but at rates 1.3- to 3-fold higher than TMC-anti (Table 2 and SI Appendix, Fig. S5). Prior to doing these experiments, we had expected the methyl groups surrounding the Fe=O unit in TMC-syn to impose a greater steric barrier around the Fe=O unit and thus hinder the approach of the target substrate C–H bond to decrease its HAT reactivity. However, TMC-syn actually has an Fe−O bond 0.02 Å shorter than that of TMC-anti based on X-ray crystallography as well as a Raman Fe=O stretch that is 17 cm−1 higher in frequency as observed in CH3CN solution (15) (Table 1). Both effects are corroborated by our DFT calculations at the B97-D3/TZ2P (21, 22) [COSMO (23–25), ZORA (26)] level, which show a 0.012 Å shorter Fe=O bond with a 28 cm−1 higher Fe=O frequency (Table 1).
Table 2.
Comparison of TMC-anti/TMC-syn HAT and OAT rates [k2 (M−1s−1) at 25 °C]
| Substate | TMC-anti | TMC-syn | syn/anti | Products (yield) |
|---|---|---|---|---|
| HAT | ||||
| Xanthene | 0.67 | 0.92 | 1.4 | Xanthone (1.0) |
| 9,10-DHA | 0.23 | 0.33 | 1.5 | Anthracene (0.56) |
| 1,4-CHD | 0.22 | 0.29 | 1.3 | Benzene (0.52) |
| Fluorene | 0.01 | 0.02 | 2 | Fluorenone (0.60) |
| Cyclohexene | 0.002 | 0.006 | 3 | Cyclohexenol (0.70) |
| OAT (R2S) | ||||
| c-C4H8S | 0.14 | 19 | 135 | 1-C4H8S(O) (0.33) |
| Me2S | 0.097 | 13 | 135 | Me2S(O) (0.45) |
| PhSMe | 0.0096 | 8.2 | 855 | PhS(O)Me (0.50) |
| Ph2S | N. R. | 0.96 | extreme | Ph2S(O) (0.63) |
Reaction conditions: 1.0 mM [FeII(TMC)(OTf)](OTf) in CH3CN, 1.5 mM PhIO/s-ArIO, and >100 mM substrate reacted under Ar at 25 °C. N. R. denotes no reaction under these conditions.
The higher rates for TMC-syn can be rationalized in part by the greater access of the substrate to the oxo atom on its Fe=O moiety relative to that of the corresponding TMC-anti due to its larger out-of-plane distance (Fe-oop) relative to the plane spanned by the equatorial nitrogen atoms of the TMC macrocycle (Table 1). For TMC-anti, the iron center is found to be below the N4 plane (−0.03 Å, as shown by X-ray crystallography), moving closer to the axial acetonitrile ligand. In contrast for TMC-syn, the iron is found to be above the N4 plane (+0.06 Å, observed by X-ray crystallography), moving away from the acetonitrile, thereby increasing the exposure of the oxo atom (Fig. 1, Bottom) and enhancing its oxidative reactivity. In fact, the van der Waals radii space-filling model in Fig. 1 (SI Appendix, Fig. S14) shows the oxo atom of TMC-syn to be actually more accessible to the substrate, as the CH3 groups are in fact more spread out in space. In contrast, the oxo atom of TMC-anti is surrounded by well-packed H-atoms that make it less accessible (Fig. 1, Bottom, and SI Appendix, Fig. S14). We have estimated the exposed surface area with the Connolly surface area approach (27) and indeed observed a slightly larger area of the oxo atom that is exposed in TMC-syn (33.44 Å2) than in TMC-anti (31.33 Å2).
We have investigated the HAT reactions with 9,10-dihydroanthracene (DHA) and cyclohexene (c-C6H10) using DFT calculations at the B97-D3/TZ2P level to shed light on the higher reactivity observed experimentally for TMC-syn relative to TMC-anti. Note that we have designated the corresponding barriers with respect to the S = 1 reactant complex, which is the spin ground state of the reactants. Subsequently, we observe the usual involvement of the σ-reaction channel (28–30) and two-state reactivity (31–33) (Scheme 1) whereby the reaction starts on the S = 1 surface and then switches over to the S = 2 surface further along on the reaction coordinate, as the latter has a lower barrier (SI Appendix, Fig. S17).
Scheme 1.

Switching between two spin states in the two-state reactivity model (LS is low spin, and HS is high spin), with the switching region shown in red; the energy of the minimum-energy crossing point (MECP) is indicated by the horizontal line.
Apart from the structural changes surrounding the Fe(IV)=O reactant leading to its larger exposure during the reaction pathway (vide supra), we find yet another aspect of the TMC-syn complex that contributes to its increased reactivity. At the transition state of the HAT reactions of TMC-syn, the Fe=O unit moves further out of the plane with respective Fe-oop distances of +0.22 Å and +0.25 Å (Fig. 2 and SI Appendix, Table S1). Interestingly, these differences already exist at the reactants stage in the S = 2 state, where the metal center is found to be 0.16 Å above the TMC-syn plane. So, this is clearly an effect of switching to the higher spin state at the transition state. At the same time, the axial acetonitrile moves away ca. 0.15 Å from the plane spanned by the TMC-syn ligand (Fig. 2), in the direction opposite from the Fe=O unit, resulting in the generation of a 5-coordinate Fe=O center.
Fig. 2.

Out-of-plane distances of the Fe=O and the CH3CN units relative to the N4 plane (B97-D3/TZ2P) for S = 1 and S = 2 states of TMC-anti and TMC-syn, for Fe=O reactants, and transition states of HAT (DHA, c-C6H10) and OAT (PhSMe, styrene) substrates. Shown with dotted lines are the distances between iron and the axial CH3CN. Also indicated (in red, next to the Fe=O unit) are the Fe-oop distances.
In great contrast, the metal center in TMC-anti remains in the same position below the TMC-anti plane (ca. −0.03 Å) for both S = 1 and S = 2 species. Furthermore, the Fe=O unit for the HAT transition states with TMC-anti are found to move only slightly, by ca. +0.04 Å as compared to the syn counterpart (where it moved by 0.11 to 0.14 Å; see Fig. 2 and SI Appendix, Table S1). Similarly, the movement of acetonitrile at the TS (ca. 0.06 to 0.07 Å) in TMC-anti is smaller than observed in TMC-syn (0.07 to 0.10 Å, see SI Appendix, Table S1). Consequently, the slightly altered structures lead us to a relatively small reactivity difference (1.3- to 3-fold) between TMC-syn and TMC-anti for HAT reactions. To our great surprise, the difference in OAT rates observed for TMC-anti and TMC-syn are significantly larger, emphasizing distinctions in the mechanisms of these two transformations.
A compelling rationale for the out-of-plane movement of the axial CH3CN ligand and the consequent weakening of the axial ligand field for TMC-syn can be found by inspection of the frontier molecular orbitals (Fig. 3, Top). As previously noted (30, 34, 35), the dxy(β) acceptor orbital of the high-spin S = 2 complex is found to be at a lower energy than in its S = 1 analog, bringing it closer to the highest-occupied MO of the substrate. As a result, electron transfer from the substrate to the metal is facilitated, leading to a lower barrier. In the case of the TMC-syn reactant, this acceptor orbital is found to be 0.03 eV lower in energy than for TMC-anti, consistent with its higher HAT reactivity. Dissociation of the bound CH3CN would lower the energy of this orbital even further, but this would be accompanied by the loss of favorable metal–ligand bonding interactions. Hence, there is a subtle balance between lowering the acceptor orbital for enhanced electron transfer and the energetic penalty associated with it. Our results show that for HAT, the metal–ligand interactions are strong enough that only a mild dissociation of CH3CN takes place.
Fig. 3.

Top: Energies of the frontier MOs for the S = 2 configuration of TMC-syn. Indicated in red is the decrease in the energy of the σ* and dxy orbitals upon lengthening of the Fe–NCCH3 bond to 2.5 Å. Corresponding figures for S = 1 and S = 2 of TMC-anti can be found in SI Appendix, Fig. S16. Bottom: Transition state structures calculated for TMC-anti (Left) and TMC-syn (Right) in their reactions with PhSMe (H-atoms omitted for clarity) where the CH3CN ligand becomes detached from the Fe center upon reaction of TMC-syn with substrate, but NOT in the case of TMC-anti.
OAT Reactivity Differences between TMC-anti and TMC-syn Isomers.
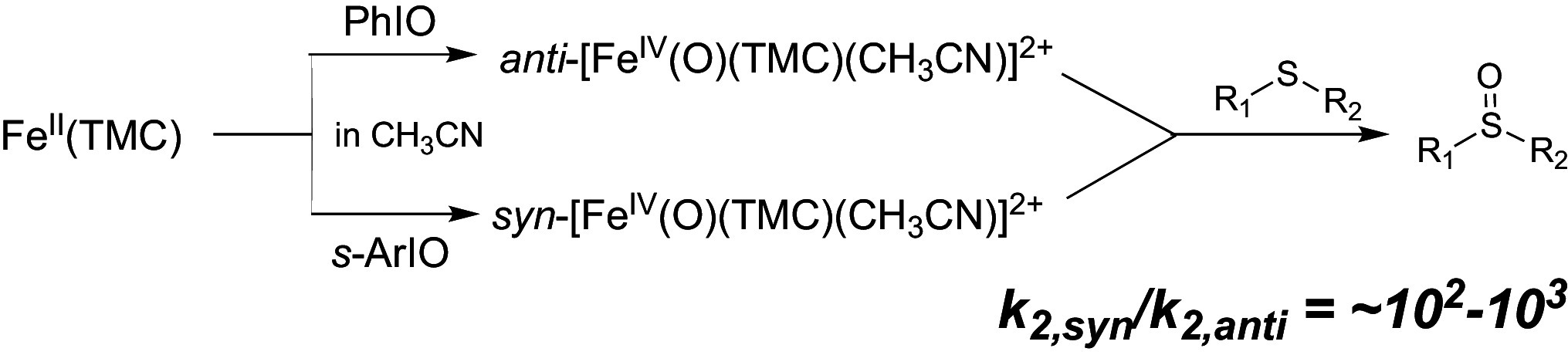
OAT to sulfides.
More dramatic reactivity differences are observed between the anti and syn isomers in OAT transformations (Table 2). At 298 K, TMC-syn oxidizes dialkyl sulfides at rates ~130-fold faster than TMC-anti, a difference which grows to ~850-fold for alkyl aryl sulfides (Table 2 and SI Appendix, Fig. S6). This trend suggests that electronic factors play a much bigger role than steric considerations in controlling OAT reactivity (vide supra).
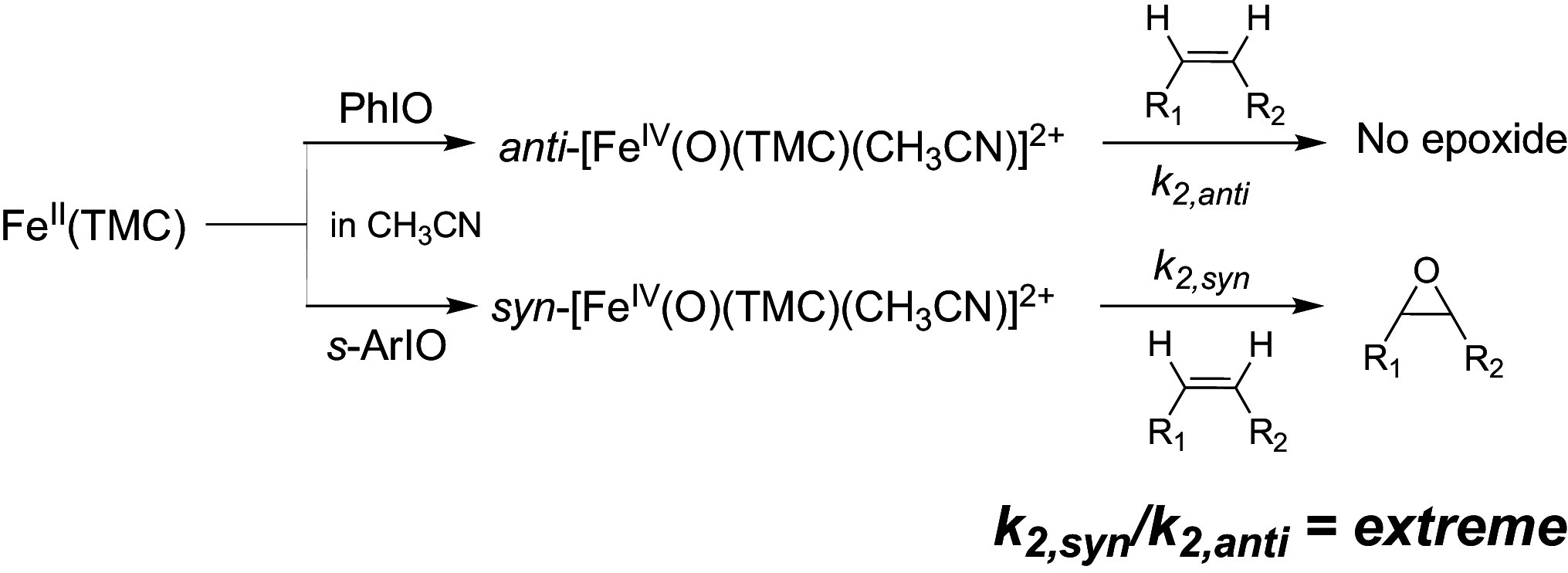
Olefin epoxidation.
Extension of our reactivity explorations to olefin epoxidation reveals even further surprises. Quite intriguingly, we have observed the epoxidation of a series of olefins, but only by the TMC-syn isomer (Table 3). Indeed TMC-anti is essentially unreactive with the olefins we have tested, except for cis-cyclooctene, the most electron-rich olefin in our list, which is epoxidized by TMC-anti but at a rate 100-fold slower than the corresponding TMC-syn isomer. This comparison emphasizes the effectiveness of TMC-syn as an olefin epoxidation agent. As shown in Fig. 4, Inset, epoxidation rates span a range of two orders of magnitude, from 1.5 × 10−3 M−1s−1 for 1-octene to 0.13 M−1s−1 for trans-stilbene, and in between is styrene with an intermediate rate of 1.6 × 10−2 M−1s−1 that is further modulated by introducing substituents on the phenyl ring (Table 3 and SI Appendix, Fig. S7). Clearly, conjugation to the double bond lowers the barrier for epoxidation to enhance the reaction rate. Intriguingly, a linear correlation is obtained between the second-order rate constants (log k2) vs. their 1-e oxidation potentials (Eox) with a slope of −1.1 (Fig. 4, Inset), suggesting that epoxidation likely occurs in two steps: initial inner-sphere electron transfer from olefin to the Fe=O unit, followed by a radical rebound step. This hypothesis is supported by the partial cis-to-trans isomerization observed for the two open-chain cis-disubstituted olefins cis-2-heptene (cis:trans = 2.8:1) and cis-stilbene (cis:trans = 1:2), with the fraction of trans epoxide formed enhanced by the introduction of aromatic substituents that increase the lifetime of the 1-e-oxidized substrate. The correlation of the epoxidation rates to oxidation potentials of the olefin substrates is reminiscent of the observation of such a complex in olefin epoxidation by oxo-iron porphyrins (36).
Table 3.
Olefin epoxidation rates and yields for nonheme FeIV=O complexes, at 25 °C unless otherwise noted
| Fe=O Complex | k2 (M−1s−1) | Epoxide yield |
|---|---|---|
| TMC-syn (this work) | ||
| 1-octene | 0.0015 | 0.32 |
| trans-4-octene | 0.005 | 0.36 |
| trans-2-heptene | 0.009 | 0.29 |
| cis-2-heptene | 0.013 | 0.38 (cis:trans 2.8:1) |
| cis-cyclooctene | 0.014 | 0.70 |
| styrene | 0.016 | 0.38 |
| 4-Me-styrene | 0.03 | 0.45 |
| 4-MeO-styrene | 0.08 | 0.50 |
| 4-Cl-styrene | 0.007 | 0.21 |
| cis-stilbene | 0.011 | 0.41 (cis:trans 1:2) |
| trans-stilbene | 0.13 | 0.45 |
| [FeIV(O)(PyMAC)]2+ * | ||
| Cyclooctene | 0.45 | 0.81 |
| [FeIV(O)(N3S2)]2+ † | ||
| Styrene | 0.026 | 0.60 |
| [FeIV(O)(N2Py2Q)]2+‡ | ||
| 4-X-styrenes | 0.186 to 2.53 | |
| cis-stilbene | 0.139 | |
| trans-stilbene | 1.12 | |
| [FeIV(O)(cyclam)]2+ § | at −20 °C | |
| 1-octene | 0.013 | 0.33 |
| cyclohexene | 0.015 | 0.60 |
| cis-stilbene | 0.023 | 0.30 |
| [FeIV(O)(Me3NTB)]2+ ¶ | at −40 °C | |
| 4-X-styrenes | 5.8 to 29 | 0.89 (for styrene) |
| [FeIV(O)(TQA)]2+ # | at −40 °C | |
| Cyclooctene | 3.3 | 0.55 |
| 1-octene | 5.3 | 0.35 |
| 4-X-styrenes | 1.4 to 12 | 0.92 (for styrene) |
Ligand abbreviations: PyMAC = 2,7,12-trimethyl-3,7,11,17-tetraaza-bicyclo[11.3.1]-heptadeca-1(17),13,15-triene, N3S2 = 2,6-bis(2-methylthiophenylimino-methyl)pyridine, N2Py2Q = bis(pyridyl)methyl bis(quinolylmethyl)amine, Me3NTB = tris((1-methyl-1H-benzo[d]imidazol-2yl)methyl)-amine, TQA = tris(quinolin-2-yl-methyl)amine.
*From ref. 12.
†From ref. 11.
‡From ref. 14.
§From ref. 37.
¶From ref. 13.
Fig. 4.

PhSMe reaction rates with TMC-syn and TMC-anti observed at 25 °C. (Inset) Plot of log k2 obtained at 25 °C vs. Eox of olefins with 1-e oxidation potentials from ref. 39.
Indeed, in our DFT study, we do find that the reaction proceeds through a two-step mechanism, following the σ-reaction channel (28–30) in which the oxo moiety binds to one of the carbons of the double bond in the first (inner-sphere electron transfer) step and then subsequently in the second step makes the bond to the second carbon to form the epoxide product. This result is in line with previous computational studies (40, 41) on OAT mechanisms that showed such a two-step mechanism for olefin epoxidation.
Interestingly, cis-stilbene is an order of magnitude less reactive than its trans counterpart, despite having an Eox value just higher by 0.2 V. Its significantly lower reactivity may result from the cis disposition of the two phenyl substituents on the double bond that may hinder the approach of the Fe=O moiety. Our DFT studies suggest this to be the case. The Ph–C=C–Ph dihedral angle for trans-stilbene (180° in substrate) changes relatively little at the TS (167.9° with TMC-syn, 155.5° with TMC-anti). Instead, for cis-stilbene (8° in substrate), it changes to 33.4° (TMC-syn) and 39.0° (TMC-anti) at the TS. Furthermore, our DFT calculations indicate that the first barrier in the mechanism is indeed lower for trans-stilbene with TMC-syn (ΔG 9.43 trans-stilbene vs. 10.52 kcal·mol−1 cis-stilbene) (SI Appendix, Figs. S28 and S29). Moreover, cis-stilbene affords a mixture of cis- and trans-epoxide products with a ratio of 1:2 in favor of the trans-epoxide, suggesting that the proposed rebound step occurs more slowly in the case of this substrate. The only other cis-di-substituted-olefin in this study, namely cis-2-heptene, also yields a mixture of epoxide products, but with the cis-epoxide favored 2.8:1 over its trans counterpart. The difference in the two epoxidation results is likely correlated with the lifetime of the putative 1-e-oxidized olefin derivative, which should be longer for cis-stilbene due to the presence of phenyl groups that can conjugate with the 1e-oxidized olefin to extend the lifetime of the 1-e-oxidized radical cation intermediate. Indeed, according to our DFT calculations, the 1-e-oxidized intermediate is 10.8 kcal·mol−1 more stable than the corresponding S = 2 TS with cis-stilbene (with TMC-syn); in contrast, for trans-stilbene, the intermediate is located only 5.7 kcal·mol−1 below the S = 2 TS. At this intermediate, the Ph–C=C–Ph dihedral angle for trans-stilbene has lowered to 144.5° (i.e., a change of 36° compared to reactants), while for cis-stilbene it has increased to 53.4° (a change of 45° compared to reactants). In the latter case, a small rotational barrier (ca. 3.8 kcal·mol−1 at electronic energy) might then easily move it over to the trans-stilbene orientation, leading to the trans-epoxide product. The second step in the process to epoxide has smaller barriers than the first step (7.6 kcal·mol−1 for cis-stilbene, 5.9 kcal·mol−1 for trans-stilbene), in which the bond between oxygen and the distal carbon is already partially formed (O–C trans-stilbene: 1.785 Å for TMC-syn, 1.895 Å for TMC-anti) (SI Appendix, Fig. S22).
Hammett parameters.
The OAT reactivity of the TMC-syn isomer has been further studied with a series of para-substituted thioanisoles and styrenes, showing that electron-donating substituents display enhanced second-order rate constants (k2) compared to their electron-withdrawing counterparts (Tables3 and 4). The (k2X/k2H) value was used for linear free energy correlation analysis (SI Appendix, Fig. S8) where log(k2X/k2H) values vs. σρ+ values of para-substituted thioanisoles and styrenes were plotted and Hammett ρ value of −1.3 for para-substituted thioanisoles and −2.0 for para-substituted styrenes were obtained. The relatively higher value of ρ indicated the buildup of a positive charge on the olefinic carbon in the transition state. Although we have not explicitly studied the para-substituted styrenes computationally, we did explore the buildup of charge on the olefinic carbons of styrene in going from the reaction complex to the transition state. These data showed that for the proximal carbon (making a bond to oxo in the first step of the reaction) the MDC-d (42) charge increases along the pathway, from 0.07 e at the RC (S = 1 and S = 2) to 0.23 e at the TS (S = 2). It is accompanied by creation of radical character on the substrate (SI Appendix, Fig. S20), which is located primarily on the distal olefin carbon atom of styrene (ca. 0.27 e), cis/trans-stilbene (ca. 0.27/0.31 e) and cis-cyclooctene (ca. 0.27 e). Moreover, for styrene and stilbene, a significant radical character is observed at the para-position of the phenyl group (ca. 0.09/0.10 e) that is bound to this distal carbon (SI Appendix, Fig. S20).
Table 4.
Oxidative reactivity of oxoiron(IV) complex pairs with different topologies
| Isomer pairs | Reactivity differences between isomers | Ref. |
|---|---|---|
| [FeIV(O)(BQCN)(CH3CN)]2+ cis-α > cis-β (ΔE = 0.11 V) | HAT (cumene): 20x OAT (p-CN-C6H4SMe): 100x | (43) |
| [FeIV(O)(BP)]2+ BP1 > BP2 (ΔE = 0.25 V) | HAT: 10 to 20x OAT: sulfoxidation 100x | (44) |
|
FeIV(O)(Me2EBC)(CH3CN)]2+ > TMC-anti (ΔE = 0.06 V) |
HAT: ~30 to 100x OAT: sulfoxidation ~400x |
(20) |
|
[FeIV(O)(TMC)(CH3CN)]2+ TMC-syn > TMC-anti (Ered = 0.36 & 0.39 V vs. SCE @ 25 °C) |
HAT: 1.5-3x OAT: sulfoxidation 100-850x cyclooctene epoxidation 100x Epoxidation only by TMC-syn: 1-octene, 4-R-styrene (R = H, Me, MeO, Cl) trans-stilbene (no cis epoxide product) cis-stilbene (1:2 cis:trans epoxide) |
This work |
ChemDraw pictures for these complexes can be found in Fig. 5.
Fig. 5.

Oxoiron(IV) complexes with different ligand topologies showing HAT and OAT reactivity differences.
Comparing with other topological isomeric pairs.
Previously published work provides hints that ligand topology can play a role in modulating reactivity, as observed for two pairs of isomers among the nonheme oxoiron(IV) complexes (Table 4). In the first two pairs of isomers compared by Nam and Comba, the cis-α isomers are found [FeIV(O)(BQCN)(CH3CN)]2+ (BQCN = N,N′-dimethyl-N,N′-bis(8-quinolyl)-cyclohexanediamine) (with the quinoline rings-oriented trans to each other) to be more reactive than the cis-β counterparts (with the quinoline rings oriented cis to each other) by 10- to 20-fold with respect to HAT and 100-fold with respect to OAT (43). Similarly, Comba and coworkers have compared the reactivities of a second pair of complexes consisting of [FeIV(O)(BP1)]2+ and [FeIV(O)(BP2)]2+, (BP = 3,7-diazabicyclo[3.3.3]nonane), which also differ in the relative orientations of the two pyridine rings; the BP2 isomer with the two pyridine rings perpendicular to the Fe=O bond is more reactive by 20- to 40-fold (HAT) and 100-fold (OAT) than the BP1 isomer (44).
The reactivity differences within each pair of complexes can be qualitatively rationalized by differences in the FeIV/III reduction potentials between the two members of each pair, 0.11 V for the BQCN pair and 0.25 V for the BP pair. In both cases, the isomer with the more positive reduction potential is the more reactive complex. On the other hand, two structurally related complexes [FeIV(O)(Me2EBC)(CH3CN)]2+ (Me2EBC = 4,11-dimethyl-1,4,8,11-tetraazabicyclo-[6.6.2]hexadecane) and anti-[FeIV(O)(TMC)(CH3CN)]2+ exhibit large differences in reactivity (~50-fold for HAT and ~400-fold for OAT) despite having a small difference in FeIV/III potentials (0.06 V). We also carried out cyclic voltammetry experiments to obtain FeIV/III reduction potentials for TMC-syn and TMC-anti in CH3CN at 25 °C. Unfortunately, the resulted cyclic voltammograms are irreversible having one prominent cathodic peak with a small difference in Ep,c values (33 mV) between them (SI Appendix, Fig. S9). When we performed spectral redox titrations using ferrocene the same difference was obtained (SI Appendix, Fig. S10). The Ered value of TMC-syn was determined to be Ered = 0.36 V vs. SCE at 25 °C. A value of 0.39 V vs. SCE was reported for TMC-anti at 25 °C (45). So, the difference in reduction potential is only 30 mV, a factor of 4-8-fold smaller than found the BQCN and BP pairs of complexes. So, it is difficult to apply the reduction potential argument to rationalize the observed reactivity difference.
What Factors, Then, Are responsible for Extreme OAT Differences in Reactivity between TMC-syn and TMC-anti?
Our DFT calculations at B97-D3/TZ2P indicate that the higher reactivity of TMC-syn could partially be explained by the higher accessibility of its Fe=O unit. However, further examination of the transition states for the OAT reactions shows that more factors are involved in the higher reactivity of TMC-syn. In the case of OAT to thioanisole with TMC-syn, the Fe=O unit moves further out of the plane defined by the equatorial nitrogen atoms of TMC (Fig. 2). More importantly, the axial CH3CN dissociates from the Fe=O unit of TMC-syn in the transition state during OAT reactions, while it stays bound within TMC-anti (Fig. 2). We have explored this effect in more detail and performed constrained transition-state searches and geometry optimizations, in which we kept the Fe-N(CH3CN) distance fixed at 2.20 Å. These constrained TS searches increase the barrier for OAT with TMC-syn by ca. 6.3 kcal·mol−1, which recovers part of the difference in barrier between TMC-syn and TMC-anti (11.0 kcal·mol−1), but not completely. Apart from thioanisole oxidation, we also computed the reaction pathways with diphenyl sulfide and dimethyl sulfide. Our DFT results (SI Appendix, Table S2) corroborate the experimental values from Table 2 that show a decrease in reaction rates in the order Me2S > PhSMe > Ph2S, reflecting an increase of the reaction barrier along this series. For PhSMe, the transition barrier (ΔG‡) for TMC-anti is 15.7 kcal·mol−1 at the S = 2 spin state and is even higher for the S = 1 spin state at 27.1 kcal·mol−1 (in both cases with respect to the reactant complex in the S = 1 state). On the other hand, the transition barriers for TMC-syn are 4.6 kcal·mol−1 and 15.6 kcal·mol−1 for the S = 2 and S = 1 spin states, respectively. However, for both TMC-syn and TMC-anti, a spin-state switching needs to go from the S = 1 state at the reactant complex to the S = 2 state at the TS. Therefore, we also located the minimum energy crossing points (MECP) (46–48), which are those points in space where the S = 1 and S = 2 states have the same electronic energy. In the case of TMC-syn, this MECP is found at +8.2 kcal·mol−1 above the S = 1 RC (and 1.7 kcal·mol−1 below the corresponding S = 2 TS). Hence, a major part of the barrier involves the spin barrier for switching over to S = 2, and subsequently, only a small chemical barrier remains. Note that the chemical barrier for TMC-anti is considerably higher (vide supra), and the MECP to go from S = 1 to S = 2 is located well below it (+7.2 kcal·mol−1 above the S = 1 RC, and 13.7 kcal·mol−1 below the corresponding S = 2 TS) (SI Appendix, Fig. S19).
In the case of OAT for olefin substrates, the transition barrier is smallest (ca. 9.4 kcal·mol−1) for the reaction of TMC-syn (S = 2) with trans-stilbene, corresponding to the highest epoxidation rate observed. On the other hand, TMC-anti has a significantly higher transition barrier of 17.4 kcal·mol−1. The experimental results show that TMC-anti is only reactive to cis-cyclooctene but at a much slower rate, which is corroborated by our DFT calculations that show the smallest barrier for this olefin (14.4 kcal·mol−1) (SI Appendix, Fig. S21C). Nevertheless, it is still 2 to 5 kcal·mol−1 larger than the corresponding barriers with TMC-syn. Our DFT study shows that the epoxidation of styrene is a two-step process involving two transition states. In the first step, the oxo attacks the proximal carbon, leading to a 1e-oxidized intermediate, which in the second step of the mechanism closes the epoxide ring (SI Appendix, Fig. S21D). The rate-determining step in all cases is the making of the C–O bond in the first step (9.4 kcal·mol−1 for trans-stilbene), with the second step typically involving a barrier that is half as large (5.7 kcal·mol−1 for trans-stilbene). An important aspect in this enhanced reactivity picture of TMC-syn is the axial acetonitrile, which detaches from iron during the reaction (Fig. 2).
A Rationale For The Relationship Between The Axial Acetonitrile And Oxidative Reactivity.
The detachment of the acetonitrile takes place during OAT in the case of TMC-syn, but not with TMC-anti. We have estimated the effect of acetonitrile dissociation by performing constrained optimizations of TMC-anti and TMC-syn, where we increase the Fe–N(CH3CN) distance from 2.0 to 2.5 Å in steps of 0.1 Å. Interestingly, the dxy(β) orbital decreases in energy by only a small amount (from −5.52 eV to −5.66 eV for TMC-syn) as the axial acetonitrile distance increases from ca. 2.0 to 2.5 Å. More affected however is the σ*(α) orbital, which drops by ca. 0.5 eV (from −4.9 eV to −5.4 eV). The combination of these two effects now allows us to propose a rationale for the effect of the orientation of the Fe=O unit within the TMC ligand (anti or syn) on reactivity. HAT reactions are one-electron processes, which involve the lower-lying dxy(β) orbital, so acetonitrile dissociation has only a limited effect on lowering of the orbital energy (and with it, increased overlap with the substrate occupied orbital for more efficient electron transfer). Therefore, there is a limited amount of dissociation observed and relatively small effects on reaction rates (Fig. 6).
Fig. 6.

Rationalizing the different outcomes found for the axial CH3CN of TMC-syn following the 1-e HAT reaction with DHA (Left) and the 2-e OAT reaction with trans-stilbene (Right), where CH3 CN dissociation occurs only in the latter case. Shown in gray are the electron transfers from the substrate MOs to the Fe-complex MOs. A red arrow indicates the lowering of the σ* orbital, which facilitates the enhanced OAT reactivity for TMC-syn.
On the other hand, OAT reactions are two-electron processes that require involvement of the σ*(α) orbital, which is highly sensitive to the positioning of the CH3CN axial ligand, due to the occupation of the anti-bonding dz2 orbital on the metal center. The further the acetonitrile moves away from the Fe=O center, the lower the orbital energy of σ*(α), leading to the corresponding favorable effect on the reactivity in the S = 2 state. Indeed, we do see enhanced reactivity for OAT with TMC-syn that is accompanied by the gradual dissociation of the acetonitrile ligand. Although the lowering of the σ*(α) orbital is also seen for TMC-anti, the energetic cost for CH3CN dissociation is substantial, much more than for TMC-syn. This partial dissociation of the acetonitrile to a Fe–N(NCCH3) distance of 2.5 Å costs ca. 8.5 kcal·mol−1 for TMC-anti, but only ca. 4 kcal·mol−1 for TMC-syn. This energetic penalty might be related to the weak interactions between the bound CH3CN and the N-bound methyl groups in the TMC-anti ligand (in syn fashion with respect to CH3CN). In the case of TMC-syn, these methyl groups are located on the opposite side of the ligand and hence have no such stabilizing interaction with CH3CN. Therefore, the favorable HAT/OAT reactions with TMC-syn, and not TMC-anti, can be traced back to a combination of weak (dispersion) interactions that lower the energy of the σ* orbital as well as the dissociation of the axial CH3CN ligand.
These results reinforce the balance between stability and reactivity: The thermodynamically more stable Fe(IV)-oxo complex (TMC-anti) shows more favorable weak interactions with the axial ligand (CH3CN), which disfavor HAT and OAT reactions compared to the kinetic Fe(IV)-oxo (TMC-syn) isomer. In the latter case, the axial ligand detaches from the metal center during the course of the OAT reaction, considerably enhancing the OAT reactivity of TMC-syn. This comparison of the two isomers suggests that the design of ligands for biomimetic oxidation chemistry may require a rethink when OAT reactions are targeted. Preferentially, the axial coordination site should be labile, allowing the metal-based oxidant to switch between pseudo-octahedral and square pyramidal symmetry for enhanced reactivity.
Conclusions
Herein, we have described two topological isomers of [FeIV(O)(TMC)(CH3CN)]2+, TMC-anti and TMC-syn, that differ in the orientation of the FeIV=O moiety relative to the four methyl groups of the TMC ligand, resulting in differences in their oxidative reactivities. TMC-syn reacts with HAT substrates at 1.5- to 3-fold faster rates than TMC-anti. More strikingly, there is a much bigger difference in the OAT rates. R2S substrates are oxidized into R2S=O products by TMC-syn at 102- to 103-fold faster rates than TMC-anti. Even more remarkably, TMC-syn epoxidizes all the olefin substrates examined in this study, while TMC-anti reacts only with cis-cyclooctene, the most electron-rich of the olefins studied here, but at a 100-fold slower rate than found for TMC-syn. Our DFT calculations show that the significant reactivity difference between the two Fe(O)TMC isomers derives from the 0.02 Å shorter Fe=O bond found for TMC-syn, the greater exposure of the TMC-syn Fe=O unit due to its larger out-of-plane distance from the TMC-N4 plane and the consequent longer distance of the trans axial CH3CN ligand that facilitates subsequent dissociation of CH3CN in the transition state to generate a much more reactive five-coordinate Fe(O)TMC oxidant in the course of substrate oxidation.
Materials and Methods
A detailed Experimental and Theoretical Methods can be found in SI Appendix. These sections include detailed experimental procedures for the synthesis of our complexes, kinetic measurements, and product analysis, together with computational details, spin-resolved charge-displacement function, and definition of the out-of-plane distance. Additional figures and tables containing supporting data to the main text are also provided, including characterization of our complexes by UV–Vis and NMR spectroscopies, determination of kinetic measurements, cyclic voltammograms, GC–MS spectra, DFT-derived spin densities, and potential energy surfaces.
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
This work was supported by the NIH (grant R35 GM-131721 to L.Q.), AEI/MCIN (grant PID2020-114548GB-I00 to M.S.), GenCat (PhD fellowship 2022 FI_B 00118 and grant 2021SGR00487 to F.A.), and a developer’s license to M.S. by SCM. We thankfully acknowledge the computer resources at Universidad de Málaga and the technical support provided by Picasso (QH-2023-1-0006). We thank Dr. Johannes Klein (University of Groningen) and Dr. Lorenzo D’Amore (Janssen Pharmaceuticals) for insightful discussions.
Author contributions
B.C., M.S., and L.Q. designed research; B.C., F.A., and Y.S. performed research; B.C., F.A., Y.S., M.S., and L.Q. analyzed data; and B.C., F.A., M.S., and L.Q. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
Reviewers: A.S.B., University of California Irvine; D.P.G., Johns Hopkins University; and J.T.G., Princeton University.
Contributor Information
Marcel Swart, Email: marcel.swart@gmail.com.
Lawrence Que, Jr., Email: larryque@umn.edu.
Data, Materials, and Software Availability
Computational data have been deposited in IOCHEM-BD (10.19061/iochem-bd-4-65) (49). All other data are included in the manuscript and/or SI Appendix.
Supporting Information
References
- 1.Costas M., Mehn M. P., Jensen M. P., Que L., Dioxygen activation at mononuclear nonheme iron active sites: Enzymes, models, and intermediates. Chem. Rev. 104, 939–986 (2004). [DOI] [PubMed] [Google Scholar]
- 2.Jasniewski A. J., Que L., Dioxygen activation by nonheme diiron enzymes: Diverse dioxygen adducts, high-valent intermediates, and related model complexes. Chem. Rev. 118, 2554–2592 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guo M., Corona T., Ray K., Nam W., Heme and nonheme high-valent iron and manganese oxo cores in biological and abiological oxidation reactions. ACS Cent. Sci. 5, 13–28 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abelson C. S., Aboelenen A. M., Rasheed W., Que L., “Synthetic nonheme high-valent iron-oxo complexes structures and oxidative function” in Comprehensive Coordination Chemistry III (Elsevier, 2021), pp. 412–454. [Google Scholar]
- 5.Rohde J.-U., et al. , Crystallographic and spectroscopic characterization of a nonheme Fe(IV)=O complex. Science 299, 1037–1039 (2003). [DOI] [PubMed] [Google Scholar]
- 6.Nam W., Lee Y.-M., Fukuzumi S., Hydrogen atom transfer reactions of mononuclear nonheme metal-oxygen intermediates. Acc. Chem. Res. 51, 2014–2022 (2018). [DOI] [PubMed] [Google Scholar]
- 7.Li J., et al. , Epoxidation catalyzed by the nonheme iron(II)- and 2-oxoglutarate-dependent oxygenase, AsqJ: Mechanistic elucidation of oxygen atom transfer by a ferryl intermediate. J. Am. Chem. Soc. 142, 6268–6284 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thibodeaux C. J., Chang W., Liu H., Enzymatic chemistry of cyclopropane, epoxide, and aziridine biosynthesis. Chem. Rev. 112, 1681–1709 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hollenhorst M. A., et al. , The nonribosomal peptide synthetase enzyme DdaD tethers Nβ-fumaramoyl-l-2,3-diaminopropionate for Fe(II)/α-ketoglutarate-dependent epoxidation by DdaC during dapdiamide antibiotic biosynthesis. J. Am. Chem. Soc. 132, 15773–15781 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Balland V., et al. , Spectroscopic characterization of an FeIV intermediate generated by reaction of XO − (X = Cl, Br) with an Fe II complex bearing a pentadentate non-porphyrinic ligand−hydroxylation and epoxidation activity. Eur. J. Inorg. Chem. 2004, 301–308 (2004). [Google Scholar]
- 11.Annaraj J., et al. , An iron(II) complex with a N3S2 thioether ligand in the generation of an iron(IV)-oxo complex and its reactivity in olefin epoxidation. Inorg. Chim. Acta. 362, 1031–1034 (2009). [Google Scholar]
- 12.Ye W., Ho D. M., Friedle S., Palluccio T. D., Rybak-Akimova E. V., Role of Fe(IV)-oxo intermediates in stoichiometric and catalytic oxidations mediated by iron pyridine-azamacrocycles. Inorg. Chem. 51, 5006–5021 (2012). [DOI] [PubMed] [Google Scholar]
- 13.Bae S. H., et al. , Mononuclear nonheme high-spin (S = 2) versus intermediate-spin (S = 1) iron(IV)–oxo complexes in oxidation reactions. Angew Chem. Int. Ed. 55, 8027–8031 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Biswas J. P., et al. , Effect of the ligand backbone on the reactivity and mechanistic paradigm of non-heme iron(IV)-oxo during olefin epoxidation. Angew. Chem. Int. Ed. 60, 14030–14039 (2021). [DOI] [PubMed] [Google Scholar]
- 15.Prakash J., Rohde G. T., Meier K. K., Münck E., Que L., Upside down! Crystallographic and spectroscopic characterization of an [Fe IV (O syn) (TMC)] 2+ complex. Inorg. Chem. 54, 11055–11057 (2015). [DOI] [PubMed] [Google Scholar]
- 16.Ray K., et al. , An inverted and more oxidizing isomer of [FeIV (O)(tmc)(NCCH 3)] 2+. Angew. Chem. Int. Ed. 47, 8068–8071 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tyburski R., Liu T., Glover S. D., Hammarström L., Proton-coupled electron transfer guidelines, fair and square. J. Am. Chem. Soc. 143, 560–576 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Klein J. E. M. N., Knizia G., cPCET versus HAT: A direct theoretical method for distinguishing X-H bond-activation mechanisms. Angew. Chem. Int. Ed. 57, 11913–11917 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.D’Amore L., Belpassi L., Klein J. E. M. N., Swart M., Spin-resolved charge displacement analysis as an intuitive tool for the evaluation of cPCET and HAT scenarios. Chem. Commun. 56, 12146–12149 (2020). [DOI] [PubMed] [Google Scholar]
- 20.England J., et al. , Oxoiron(IV) complex of the ethylene-bridged dialkylcyclam ligand Me 2 EBC. Inorg. Chem. 54, 7828–7839 (2015). [DOI] [PubMed] [Google Scholar]
- 21.Grimme S., Antony J., Ehrlich S., Krieg H., A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010). [DOI] [PubMed] [Google Scholar]
- 22.Grimme S., Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 27, 1787–1799 (2006). [DOI] [PubMed] [Google Scholar]
- 23.Swart M., Rösler E., Bickelhaupt F. M., Proton affinities in water of maingroup-element hydrides—Effects of hydration and methyl substitution. Eur. J. Inorg. Chem. 2007, 3646–3654 (2007). [Google Scholar]
- 24.Klamt A., Schüürmann G., COSMO: A new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J. Chem. Soc., Perkin Trans. 2, 799–805 (1993). [Google Scholar]
- 25.Pye C. C., Ziegler T., An implementation of the conductor-like screening model of solvation within the Amsterdam density functional package. Theor. Chem. Acc. 101, 396–408 (1999). [Google Scholar]
- 26.Lenthe E. V., Baerends E. J., Snijders J. G., Relativistic regular two-component Hamiltonians. J. Chem. Phys. 99, 4597–4610 (1993). [Google Scholar]
- 27.Connolly M. L., Solvent-accessible surfaces of proteins and nucleic acids. Science 221, 709–713 (1983). [DOI] [PubMed] [Google Scholar]
- 28.Neidig M. L., et al. , Spectroscopic and electronic structure studies of aromatic electrophilic attack and hydrogen-atom abstraction by non-heme iron enzymes. Proc. Natl. Acad. Sci. U.S.A. 103, 12966–12973 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Geng C., Ye S., Neese F., Analysis of reaction channels for alkane hydroxylation by nonheme iron(IV)–oxo complexes. Angew. Chem. Int. Ed. 49, 5717–5720 (2010). [DOI] [PubMed] [Google Scholar]
- 30.Kazaryan A., Baerends E. J., Ligand field effects and the high spin-high reactivity correlation in the H abstraction by non-heme iron(IV)–oxo complexes: A DFT frontier orbital perspective. ACS Catal. 5, 1475–1488 (2015). [Google Scholar]
- 31.Schröder D., Shaik S., Schwarz H., Two-state reactivity as a new concept in organometallic chemistry. Acc. Chem. Res. 33, 139–145 (2000). [DOI] [PubMed] [Google Scholar]
- 32.Usharani D., Wang B., Sharon D. A., Shaik S., “Principles and prospects of spin‐states reactivity in chemistry and bioinorganic chemistry” in Spin States in Biochemistry and Inorganic Chemistry, Swart M., Costas M., Eds. (Wiley, 1st ed., 2015), pp. 131–156. [Google Scholar]
- 33.Shaik S., De Visser S. P., Ogliaro F., Schwarz H., Schröder D., Two-state reactivity mechanisms of hydroxylation and epoxidation by cytochrome P-450 revealed by theory. Curr. Opin. Chem. Biol. 6, 556–567 (2002). [DOI] [PubMed] [Google Scholar]
- 34.Bernasconi L., Louwerse M. J., Baerends E. J., The role of equatorial and axial ligands in promoting the activity of non-heme oxidoiron(IV) catalysts in alkane hydroxylation. Eur. J. Inorg. Chem. 2007, 3023–3033 (2007). [Google Scholar]
- 35.Janardanan D., Usharani D., Shaik S., The Origins of dramatic axial ligand effects: Closed-shell MnV O complexes use exchange-enhanced open-shell states to mediate efficient H abstraction reactions. Angew. Chem. Int. Ed. 51, 4421–4425 (2012). [DOI] [PubMed] [Google Scholar]
- 36.Groves J. T., Watanabe Y., The mechanism of olefin epoxidation by oxo-iron porphyrins. Direct observation of an intermediate. J. Am. Chem. Soc. 108, 507−508 (1986). [DOI] [PubMed] [Google Scholar]
- 37.Engelmann X., et al. , Trapping of a highly reactive oxoiron(IV) complex in the catalytic epoxidation of olefins by hydrogen peroxide. Angew. Chem. Int. Ed. 58, 4012–4016 (2019). [DOI] [PubMed] [Google Scholar]
- 38.Biswas A. N., et al. , Modeling TauD-J: A high-spin nonheme oxoiron(IV) complex with high reactivity toward C-H bonds. J. Am. Chem. Soc. 137, 2428–2431 (2015). [DOI] [PubMed] [Google Scholar]
- 39.Wang B., Lee Y., Seo M. S., Nam W., Mononuclear nonheme iron(III)-iodosylarene and high-valent iron-oxo complexes in olefin epoxidation reactions. Angew. Chem. Int. Ed. 54, 11740–11744 (2015). [DOI] [PubMed] [Google Scholar]
- 40.Dhuri S. N., et al. , Interplay of experiment and theory in elucidating mechanisms of oxidation reactions by a nonheme Ru IV O Complex. J. Am. Chem. Soc. 137, 8623–8632 (2015). [DOI] [PubMed] [Google Scholar]
- 41.Terencio T., et al. , Chemoselectivity in the oxidation of cycloalkenes with a non-heme iron(IV)-oxo-chloride complex: Epoxidation vs. hydroxylation selectivity. J. Am. Soc. Mass Spectrom. 30, 1923–1933 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Swart M., Van Duijnen PTh., Snijders J. G., A charge analysis derived from an atomic multipole expansion. J. Comput. Chem. 22, 79–88 (2001). [Google Scholar]
- 43.Hong S., et al. , Ligand topology effect on the reactivity of a mononuclear nonheme iron(IV)-oxo complex in oxygenation reactions. J. Am. Chem. Soc. 133, 11876–11879 (2011). [DOI] [PubMed] [Google Scholar]
- 44.Wang D., et al. , Nonheme oxoiron(IV) complexes of pentadentate N5 ligands: Spectroscopy, electrochemistry, and oxidative reactivity. Chem. Sci. 4, 282–291 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee Y.-M., Kotani H., Suenobu T., Nam W., Fukuzumi S., Fundamental electron-transfer properties of non-heme oxoiron(IV) complexes. J. Am. Chem. Soc. 130, 434–435 (2008). [DOI] [PubMed] [Google Scholar]
- 46.Bearpark M. J., Robb M. A., Bernhard Schlegel H., A direct method for the location of the lowest energy point on a potential surface crossing. Chem. Phys. Lett. 223, 269–274 (1994). [Google Scholar]
- 47.Takayanagi T., Nakatomi T., Automated reaction path searches for spin-forbidden reactions. J. Comput. Chem. 39, 1319–1326 (2018). [DOI] [PubMed] [Google Scholar]
- 48.Harvey J. N., Aschi M., Schwarz H., Koch W., The singlet and triplet states of phenyl cation. A hybrid approach for locating minimum energy crossing points between non-interacting potential energy surfaces. Theor. Chem. Acc. 99, 95–99 (1998). [Google Scholar]
- 49.Ahsan F., Swart M., OAT_TP_KP collection. IOChem-BD. 10.19061/iochem-bd-4-65. Deposited 20 October 2023. [DOI]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
Computational data have been deposited in IOCHEM-BD (10.19061/iochem-bd-4-65) (49). All other data are included in the manuscript and/or SI Appendix.