

Significance
Development in a metazoan requires that the division and differentiation of diverse cells be coordinated with nutrient availability. We show that one mechanism by which this occurs in Caenorhabditis elegans is through signaling by the neuronal IL-17 cytokine, ILC-17.1, and its control over p53/CEP-1. In the presence of food, ILC-17.1 release suppresses p53/CEP-1 and allows reproductive growth; decreased ILC-17.1 signaling activates p53/CEP-1-dependent transcription and metabolic programs, leading to the reversible arrest of larvae as quiescent dauers. Our studies suggest that an ancestral function of IL-17 is linking nutrient availability to energy metabolism and growth. They reveal a DNA damage–independent function of p53/CEP-1 in invertebrate development. Finally, our studies support the existence of a previously undescribed dauer pathway in C. elegans.
Keywords: p53/cep-1, C. elegans, dauer, immunometabolomic, IL-17
Abstract
During metazoan development, how cell division and metabolic programs are coordinated with nutrient availability remains unclear. Here, we show that nutrient availability signaled by the neuronal cytokine, ILC-17.1, switches Caenorhabditis elegans development between reproductive growth and dormancy by controlling the activity of the tumor suppressor p53 ortholog, CEP-1. Specifically, upon food availability, ILC-17.1 signaling by amphid neurons promotes glucose utilization and suppresses CEP-1/p53 to allow growth. In the absence of ILC-17.1, CEP-1/p53 is activated, up-regulates cell-cycle inhibitors, decreases phosphofructokinase and cytochrome C expression, and causes larvae to arrest as stress-resistant, quiescent dauers. We propose a model whereby ILC-17.1 signaling links nutrient availability and energy metabolism to cell cycle progression through CEP-1/p53. These studies describe ancestral functions of IL-17 s and the p53 family of proteins and are relevant to our understanding of neuroimmune mechanisms in cancer. They also reveal a DNA damage–independent function of CEP-1/p53 in invertebrate development and support the existence of a previously undescribed C. elegans dauer pathway.
During metazoan development, nutrient availability is coordinated with the division, growth, and metabolic activity of individual cells through cell–cell communication. This is also the case in the invertebrate Caenorhabditis elegans, a free-living bacterivore, which displays a dramatic developmental plasticity to ensure that its growth and reproduction match available resources (1–10). When C. elegans larvae hatch under optimal conditions (at 20 °C, low population densities, on abundant food), they develop continuously into reproducing adults. However, if they hatch under suboptimal conditions, such as in the paucity of food, at high population densities, or high ambient temperatures, larvae implement an alternative developmental program and arrest as quiescent, stress-resistant larvae called “dauer” larvae. Dauer larvae display metabolic and organismal phenotypes specialized for dispersal and survival and can remain arrested in this state for months to resume development into reproductive adults when favorable conditions return (1–10). Previous studies have identified molecular pathways that mediate the dauer decision, showing that growth-promoting molecules like insulins, transforming β growth factor (TGFβ/DAF-7), and lipid-based dafachronic acid hormones are released by sensory neurons and other cells to license continued development; adverse environments inhibit these growth-promoting signals and trigger dauer arrest (1–11). A number of quantitative trait loci (QTL) also modulate dauer (12). Yet, how the dauer entry decision results in a coordinated change in cell fates across different tissues and is linked with the systemic shutdown of anabolic pathways remains poorly understood.
An important group of proteins that mediate cell–cell communication and metabolism in metazoa are secreted proteins called cytokines (13, 14). The IL-17 cytokines constitute a family of proinflammatory cytokines, highly conserved across animal phyla. In mammals, these cytokines are released by specialized immune cells to activate immune surveillance, enhance barrier function, promote wound healing, and play crucial immunometabolic roles in maintaining energy homeostasis (15). In humans, IL-17s also promote cancers and autoimmune diseases such as psoriasis (16, 17). Here, we show that the C. elegans IL-17 ortholog, ILC-17.1, signals food availability and coordinates cell division with metabolism by controlling the activity of the C. elegans tumor suppressor p53 ortholog, CEP-1. Specifically, neuronal ILC-17.1 suppresses CEP-1/p53 activity in the presence of food to license growth. Upon the loss of ILC-17.1 signaling, CEP-1/p53 is activated, and remarkably, this switches whole organism development from continuous growth to dormancy. The p53-like tumor suppressor genes are found in all multicellular animals where they prevent the transmission of damaged DNA by activating a multifaceted program that controls cell cycle checkpoints, mediates reversible growth arrest or apoptosis, and controls metabolic flux (18–22). Our studies show that these functions of CEP-1/p53 also act, in the absence of DNA damage, to control developmental quiescence of C. elegans, suggesting that the developmental function of the p53 gene family could have shaped their evolution (23–25).
Results
ILC-17.1 Is Released by C. elegans Amphid Neurons in Response to Food and Prevents Dauer Arrest.
C. elegans express three IL-17 orthologs (26, 27). Of these, ILC-17.1 is a neuromodulator that is expressed by a subset of specialized sensory neurons with epithelial properties called amphid neurons, that are exposed to the environment (26–28). We found that a deletion in ilc-17.1, syb5296, that removes almost all the coding sequence (2173 bp out of 2980 bp; SI Appendix, Fig. S1A) and abolishes mRNA expression (SI Appendix, Fig. S1B) caused larvae to constitutively enter the dauer state (Fig. 1 A and B and SI Appendix, Fig. S1 C and D). Dauer larvae can be identified by their growth arrest and distinct morphology characterized by specialized cuticular structures called alae, a constricted pharynx, arrested germline, decreased pharyngeal pumping rates, and detergent (1% SDS) resistance (3–9). ilc-17.1(syb5296)X dauer larvae displayed all these features (SI Appendix, Fig. S1 E–J). Even at 20 °C, ilc-17.1-deleted larvae entered the dauer state transiently: Approximately 30% (31.2 ± 5%) were SDS resistant 48 h posthatching on OP50 bacteria, and most larvae exited dauer and became reproductive adults by 72 h (SI Appendix, Fig. S1 C and D). Under the same conditions, none of the wild-type larvae were detergent resistant, nor did they arrest as dauers. However, as with other mutations that promote dauer entry (3, 4, 29), the dauer arrest of ilc-17.1 deletion mutants was dramatically accentuated at the slightly higher ambient temperature of 25 °C, which still supported the growth of all wild-type larvae into reproductive adults, but caused practically 100% of larvae lacking ilc-17.1 to enter and persist in the arrested dauer state for days (Fig. 1 A–D).
Fig. 1.

ILC-17.1 signaling of food availability by larval amphid neurons modulates dauer arrest. (A) Schematic of experiment. For each experiment, independent samples of 10 d-one adults of each strain laid eggs for 2 to 4 h at 20 °C on OP50 lawns. Adults were removed, embryos transferred to 25 °C, larval phenotype scored after approximately 48 h, and categorized as dauers or stage 4 larvae (L4) and/or young adults. SDS resistance was used to confirm dauers. Extent of crowding on plates was comparable [Representative average numbers of embryos/plates are 64, 68, 56, 46, and 65 for the wild type (N2), ilc-17.1(syb5296) X, ilc-17.1(syb5297) X, ilc-17.1(syb5296) rescued with ilc-17.1p::ilc-17.1, and ilc-17.1(syb5296) rescued with unc-54p::ilc-17.1, respectively]. ilc-17.1 rescued (ilc-17.1p) are ilc-17.1 deletion mutants expressing extrachromosomal ilc-17.1 cDNA driven by ilc-17.1 promoter. ilc-17.1 rescued (unc-54p) are ilc-17.1 deletion mutants expressing extrachromosomal ilc-17.1 cDNA driven by unc-54 promoter. Pearson's Chi-squared test with Yates' continuity correction was used to compare samples in the manuscript, unless otherwise stated. Chi-squared, df, and P values for the whole experiment and P values for pair-wise comparisons are reported. ***P < 0.001, **P < 0.01, *P < 0.05, and ns, not significant. (B) Percentage larvae that arrest as dauers, (n = 3 to 12 experiments; Chi-squared = 2359.6, df = 4, P-value < 2.2e-16). (C and D). Representative micrographs 48 h posthatching at 25 °C: (C) wild type (N2) are L4s; (D) ilc-17.1(syb5296) are dauer. (Scale bar, 1 mm.) (E) Percentage dauers among wild-type (N2) and ilc-17.1 mutants on control (L4440) and ilcr-2 RNAi, (n = 3 to 10 experiments. Chi-squared = 1815.1, df = 7, P-value < 2.2e-16). (F) Schematic of the AID experiment to degrade ILCR-2. (G) Percentage dauers following tissue-specific degradation of ILCR-2. X-axis: tissue-specific TIR1 expression (n = 4 to 6 experiments; Chi-squared = 335.56, df =9, P-value < 2.2e-16). (H) Schematic of the experiment to localize ilc-17.1 mRNA and ILC-17.1 protein in the presence and absence of food (OP50). Embryos hatched at 20 °C and imaged after 24 to 36 h (L1 stage larvae): (I and K), Representative micrographs of z-section of pharyngeal region of larvae showing mCherry as proxy for ilc-17.1 mRNA expression in amphid neurons. (J and L) z-section projection showing immunolocalization of ILC-17.1::HA protein. (I and J) Larvae in the absence of food and (K and L) presence of food. (M and N) Controls (N2) that do not express HA or mCherry to indicate the specificity of the fluorescence signal (n > 3 experiments, 4 to 5 larvae each). (Scale bar, 5 µm.) (O) KEGG enrichments associated with differentially down-regulated genes, and (P) differentially up-regulated genes in ilc-17.1 mutant larvae. p.adjust < 0.05. RNA-seq on 32 to 36 h larvae. (Q) Heatmap depicting expression levels (log2 normalized counts) of the major dauer-specific collagens in L2 wild-type (N2) larvae en route to adult development, ilc-17.1 deletion mutants en route to dauer entry and ilc-17.1 deletion mutants rescued from dauer with unc-54p::ilc-17.1. Bars show mean ± S.E.M. Individual data points in the bar graphs in B and E and G represent the % dauers/experiment, and the bar graph depicts the mean of these percentages. ***P < 0.001, **P < 0.01, *P < 0.05, and ns, not significant.
The ilc-17.1 gene has not been identified in previous genetic screens for dauer pathway components (3, 5, 30–32). Therefore, although no off-target effects using CRISPR have been observed in C. elegans even after whole genome sequencing (33–35), we conducted additional experiments to confirm the role of ilc-17.1 in dauer entry by i) examining the developmental phenotype of another independently generated CRISPR deletion in ilc-17.1, syb5297 (loss of 2188 bp of 2980 bp; SI Appendix, Fig. S1A), ii) backcrossing ilc-17.1 (syb5296) mutants, and iii) down-regulating ilc-17.1 and its receptor (26, 27) in a wild-type (N2) background using RNA interference (RNAi) and auxin-induced degradation (AID). Like ilc-17.1 (syb5296), ilc-17.1 (syb5297) created using a different guide RNA also displayed a completely penetrant dauer arrest at 25 °C (Fig. 1 A and B; Materials and Methods). Not surprisingly, CRISPR alone did not trigger dauer arrest, and a similarly large CRISPR-induced deletion in a related gene F25D1.3, syb7367 (loss of 1475 bp of 1642 bp) did not trigger dauer arrest (SI Appendix, Fig. S1K). ilc-17.1 (syb5296) larvae continued to arrest as dauers after they were backcrossed twice into a wild-type background (SI Appendix, Fig. S1K). In addition, RNAi-mediated downregulation of ilc-17.1 in wild-type (N2) animals, but not control RNAi treatment, caused a small but significant number of larvae to arrest as SDS-resistant dauers (SI Appendix, Fig. S1L). ILC-17.1 signals through cytokine receptors which in C. elegans consists of ILCR-1/ILCR-2 (26, 27). The ILCR-2 subunit is widely expressed and readily visible in the pharynx, hypodermis, intestine, neurons, and germline, as determined using a bicistronic SL2 cassette to tag the ilcr-2 receptor at its endogenous locus with GFP (SI Appendix, Fig. S2 A–G). Downregulation of ilcr-2 through RNAi in wild-type larvae also caused over one-third, 39.6% ± 4%, to arrest as dauers and did not alter the dauer entry of ilc-17.1-deleted larvae (Fig. 1E). RNAi is generally insufficient to induce dauer arrest even with known dauer pathway genes (3). Therefore, the RNAi-induced dauer entry of larvae exposed to ilc-17.1 or ilcr-2 RNAi was not only statistically significant but also strongly supportive of the role of the ilc-17.1 pathway in dauer.
ilcr-2 RNAi down-regulated ilcr-2 expression across all somatic and germline tissues, albeit to variable extents (SI Appendix, Fig. S2 H–M). Therefore, as additional confirmation, and to identify the tissues where ILCR-2 signaling was required to promote continuous development, we used the AID system to degrade the endogenous ILCR-2 receptor in individual tissues (Fig. 1F). We targeted the nervous system (using the rgef-1p promoter), pharynx (myo-2p), hypodermis (col-10p), muscle (unc-54p), all somatic cells (eft-3p) and the germline (mex-5p) individually, using animals that expressed Transport Inhibitor Response 1 (TIR1) in these tissues and endogenous ilcr-2 fused to an AID degron. Degradation of ILCR-2 in the pharynx or in the nervous system alone caused some larvae to arrest as dauers (Fig. 1G). Degradation of ILCR-2 in the muscle, hypodermis or germline alone had no effect (Fig. 1G). Blue Fluorescent Protein (BFP), which served as a proxy for AID, was reduced to undetectable levels upon auxin exposure in a similar percentage of larvae in all cases (SI Appendix, Fig. S2N), and thus did not explain the differences in dauer entry seen upon ILCR-2 degradation in the different tissues. Control larvae of the same genotype, not exposed to auxin, did not arrest as dauers. Thus, the loss of ILCR-2 receptors in the nervous system or pharynx was sufficient to induce the dauer arrest in a subset of animals.
Surprisingly, degrading ILCR-2 using TIR1 expressed under the pan-somatic eft-3 promoter, which is expressed in somatic cells such as neurons, pharyngeal cells, intestine, hypodermis, and muscle, but not in the germline, was less effective in inducing dauer arrest than ilcr-2 RNAi, or degradation in the pharynx or nervous system alone: Only 1.9 ± 0.8 % arrested as dauers compared to 39.6% ± 4% with ilcr-2 RNAi and 16.3 ± 1.1 % and 11.3 ± 1.5 % upon neuronal or pharyngeal AID (Fig. 1G). A logical hypothesis which requires future testing is that downregulation of ILCR-2 elicits complex tissue cross talk, whereby germline-specific ILCR-2 degradation alone, while insufficient to trigger dauer arrest (e.g., mex-5p::TIR1), might synergize with knock-down in somatic tissue to promote dauer arrest mirroring the effects observed with ilcr-2 RNAi. Likewise, decreased ILC-17.1 signaling in some tissues upon pan-somatic ILCR-2 degradation using the eft-3 promoter, might antagonize the dauer-inducing effects of degradation in the pharynx or nervous system. Although more in-depth studies are required to understand the tissue requirements, taken together, these observations support the role of ILC-17.1 signaling through the ubiquitously expressed ILCR-2 receptors in the dauer pathway. They also suggest that during normal development ILC-17.1 signaling may elicit complex intertissue cross talk to promote the growth of larvae into reproducing adults or trigger their dauer arrest.
ilc-17.1 mRNA expression is restricted to a small subset of neurons. Only a pair of amphid sensory neurons (ASE) and two pairs of interneurons (AUA and RMG) expressed transgenic ilc-17.1 reporter constructs in a previous study (26) and a few dye-filling amphid neurons expressed mCherry as a bicistronic SL2 cassette along with ilc-17.1 mRNA from the endogenous ilc-17.1 locus in our studies (Fig. 1H and SI Appendix, Fig. S3 A–G). The dauer phenotype of ilc-17.1 deletion mutants could be rescued by re-expressing ilc-17.1 under the control of its own promoter (Fig. 1B and SI Appendix, Fig. S3H; see SI Appendix, Fig. S3I for expression levels of rescue construct in comparison to the endogenous gene). The extent of rescue was significant, although modest on OP50 (Fig. 1B), but was higher on HT115 bacteria where ILC-17.1 re-expression rescued approximately 63.6% of the ilc-17.1-deleted larvae from dauer arrest (SI Appendix, Fig. S3H ). Re-expressing ilc-17.1 ectopically in ilc-17.1-deleted larvae, remote from its normal site of expression, using the muscle-specific unc-54 promoter (SI Appendix, Fig. S3 J–T ), also rescued dauer arrest of ilc-17.1-deleted larvae (Fig. 1B and SI Appendix, Fig. S3H), and RNAi mediated downregulation of ilcr-2 reduced the percentage of ilc-17.1 larvae that were rescued by ectopically re-expressing ILC-17.1 (SI Appendix, Fig. S3H ). The diet-dependence of rescue was not observed with ectopically expressed ilc-17.1 (Fig. 1B and SI Appendix, Fig. S3H ). The effects of AID-induced knockdown of ILCR-2 in different tissues were also similar when larvae were fed HT115 bacteria instead of OP50 (SI Appendix, Fig. S3U and Fig. 1G). This suggested that the role of ILC-17.1 signaling in dauer might be related to food, a prominent modulator of the dauer decision (3–9), and that the ILC-17.1 ligand could be responsive to the food signals.
To directly examine whether ILC-17.1 responded to food, we immunolocalized ILC-17.1 protein tagged at its endogenous locus using a HA-tag, in larvae that were hatched in the absence or presence of food (OP50; Fig. 1 H–L). While the site of ilc-17.1 expression was not apparently altered by the presence of food, as detected by mCherry expression that remained restricted to amphid neurons (Fig. 1 I and K), ILC-17.1 protein was present outside the amphid neurons, concentrated in the pharynx when larvae were exposed to food (Fig. 1L), but not when larvae hatched in the absence of food (Fig. 1J, quantification in SI Appendix, Fig. S3V; negative controls Fig. 1 M and N). This suggested that ILC-17.1 protein was released from its site of expression in amphid neurons, to be either selectively translated or retained by the pharynx in response to food signals [we could not reliably detect ILC-17.1 protein in distal tissues such as the intestine or coelomocytes]. The secretion of ILC-17.1 protein was also supported by immunolocalization studies on the muscle-expressed ILC-17.1 protein which, although expressed under a well-characterized muscle-specific promoter (i.e., unc-54), could be constitutively detected outside muscle cells, throughout the animal (SI Appendix, Fig. S3 M–T). This release of the ectopically expressed ILC-17.1 was not obviously affected by the absence of food (SI Appendix, Fig. S3 Q–T). These studies together showed that ILC-17.1 was released from its locus of expression in response to food, consistent with its being secreted, and that the ILC-17.1 signal, transduced through the ILCR-2 receptors in the neurons and/or pharyngeal muscle, acted cell nonautonomously to control the growth of C. elegans.
Sequencing total RNA (RNA-seq) extracted from bleach synchronized larvae grown for 32 to 36 h at 25 °C when ilc-17.1 mutant larvae were en route to dauer arrest, and wild-type larvae were late L2s en route to developing into reproducing adults, showed that ilc-17.1 mutant larvae expressed the molecular signatures of dauer larvae (6, 36–38) that were distinct from wild-type larvae and from ilc-17.1 deletion mutants that re-expressed ILC-17.1 (SI Appendix, Fig. S4A and Datasets S1–S3). This signature consisted of the downregulation of anabolic processes such as DNA replication and ribosome biogenesis (Fig. 1O), upregulation of catabolic processes and stress responses such as autophagy, fatty acid metabolism, glutathione metabolism, and xenobiotic defense pathways (Fig. 1P) and the expression of dauer-specific collagen genes that differ from the collagens expressed during continuous development (39) (Fig. 1Q and Dataset S4). We evaluated whether the gene expression profile of the ilc-17.1 deleted dauers was similar to previously described C. elegans dauers that accumulate in the population upon starvation or crowding by comparing our RNA-seq data with published datasets (38). Consistent with a role for ILC-17.1 in signaling nutrient availability, the expression profile of ilc-17.1-deleted larvae was more closely correlated to dauer larvae generated by starvation (38) (SI Appendix, Fig. S4B), compared to ascaroside-induced dauers (37) (SI Appendix, Fig. S4C and Datasets S5 and S6). These differences were surprising and suggest that the trigger and route of dauer entry could determine their gene expression profiles (40, 41).
ILC-17.1 Loss Activates CEP-1/p53 to Trigger Dauer Entry through the Activation of DAF-16/FOXO, DAF-3/SMAD-DAF-5/Ski Complex, and Steroid Hormone Pathways.
To understand how the loss of ILC-17.1 was triggering dauer entry, we first considered the possibility that because of its role as a neuromodulator (26), the lack of ilc-17.1 prevented larvae from finding or ingesting food. However, this did not seem to be the case (SI Appendix, Fig. S5; see SI Appendix, Extended Text 1 for details). Therefore, to answer how the loss of ILC-17.1 triggered dauer entry, we adopted a genetic approach (2–11, 29). In C. elegans, insulins released in the presence of food act through the sole insulin-like receptor, DAF-2, antagonize the activation of the Forkhead transcription factor DAF-16, and promote continuous growth: Loss of DAF-2 signaling leads to DAF-16 activation and dauer entry (2–11, 29). Likewise, TGFβ/DAF-7 ligands antagonize the DAF-3/SMAD-DAF-5/Ski transcription factor complex to permit continuous growth: Inhibition of DAF-7 activates DAF-3/SMAD-DAF-5/Ski and leads to dauer arrest (2–11, 29). Both insulins and TGF-β promote the synthesis of sterol-based dafachronic acid (DA) hormones that act through the nuclear hormone receptor, DAF-12, to support adult development (2–11, 29). Loss of DA pathway components also triggers dauer arrest, and providing exogenous DA to, or decreasing DAF-12 activity in, animals deficient in insulin or TGFβ signaling, can bypass their dauer arrest (3, 42). Finally, a mutation in a transmembrane guanylyl cyclase daf-11 that causes chemosensory defects also triggers dauer arrest (3, 43). ilc-17.1 deletion mutants lacked obvious chemosensory defects (SI Appendix, Fig. S5 and SI Appendix, Extended Text 1) and continued to arrest as dauers on 5 mM 8-Bromo-cGMP, which supresses daf-11-associated dauer formation (3, 43) (SI Appendix, Fig. S6A). Therefore, to identify signal transduction pathways through which the loss of ILC-17.1 signaling controlled the dauer decision, we examined its genetic interactions with daf-16, daf-5, and DA signaling.
A daf-16 loss of function mutation, mu86 (44), completely suppressed dauer formation in animals lacking ILC-17.1, indicating that activation of DAF-16 was one mechanism responsible for their dauer arrest (Fig. 2A). The activation of DAF-16 in animals lacking ILC-17.1 could be quantified by an increase in the proportion of larvae with DAF-16::GFP (45) positive intestinal nuclei even at 20 °C (Fig. 2 B–D), and the upregulation of DAF-16 target genes (46, 47) both at 20 °C (SI Appendix, Fig. S6B) and 25 °C (Fig. 2E). DAF-16 activation could be suppressed by re-expressing ILC-17.1 in ilc-17.1 deletion mutants under its endogenous promoter, or ectopically, under the muscle-specific promoter (Fig. 2E). Surprisingly, a loss of function mutation in daf-5, e1386 (48) also completely suppressed the dauer arrest of ilc-17.1 mutant larvae (Fig. 2F), and exposing ilc-17.1 deficient larvae to 50 nM exogenous Δ7-dafachronic acid (42), partially but significantly suppressed their dauer arrest (Fig. 2G). The activation of DAF-16/FOXO and DAF-3/SMAD-DAF-5/Ski by the loss of ILC-17.1 was not simply because ilc-17.1-deleted larvae were deficient in the expression of insulin signaling and TGFβ/ DAF-7 signaling pathway components, as determined by expression levels of the signaling pathway components and transcriptional reporters (SI Appendix, Fig S7; Datasets S7 and S8, see SI Appendix, Extended Text 2 for details). The dauer arrest of daf-2 (e1370) (49), could not be rescued by ILC-17.1 overexpression, suggesting that ILC-17.1 did not act genetically downstream of daf-2 (SI Appendix, Fig. S8A). Thus, it appeared that ILC-17.1 acted upstream of both DAF-16/FOXO and DAF-3/SMAD-DAF-5/Ski (and likely, DAF-9/DAF-12), but not downstream of DAF-2, to modulate dauer entry.
Fig. 2.

Reduced ILC-17.1 signaling acts through DAF-16/FOXO, DAF-3/SMAD-DAF-5/Ski complex, steroid hormone signaling, and CEP-1/p53 to control dauer arrest. (A) Percent dauers. X-axis: genotype (n = 4 to 8 experiments, Chi-squared = 497.65, df = 1, P-value < 2.2e-16). (B) Percent 36 h larvae, grown at 20 °C showing nuclear DAF-16::GFP. n = 3 experiments with 10 to 15 larvae each. Each data point represents the average no. of larvae with nuclear DAF-16::GFP/experiment (unpaired t test). Note: Experiments were conducted at 20 °C, since DAF-16::GFP localized constitutively in intestinal nuclei even in wild-type larvae at 25 °C. (C and D) Representative micrographs of DAF-16::GFP (boxed) in wild-type and ilc-17.1 deletion mutant larvae. (Scale bar, 5 µm.) (E) Average sod-3, mtl-1, and lys-7 mRNA levels in larvae 32 to 36 h posthatching at 25 °C. mRNA levels are relative to pmp-3 and normalized to wild-type (N2) values. n = 4 to 10 experiments (unpaired t test). (F) Percent dauers. X-axis: genotype (n = 3 to 5 experiments, Chi-squared = 787.3, df = 3, P-value < 2.2e-16). (G) Percent dauers in ilc-17.1 mutants on control (H2O) and exogenous 50 nM Δ7-dafachronic acid (DA) (n = 3 to 5 experiments, Chi-squared = 784.32, df = 3, P-value < 2.2e-16). (H) Percent dauers, X-axis: genotype (n = 3 to 5 experiments, Chi-squared = 1029.2, df = 3, P-value < 2.2e-16). (I and J) Representative micrographs of I, ilc-17.1-deleted larvae arrested as dauers, and J ilc-17.1;cep-1 that grew to L4s, 48 h posthatching at 25 °C. (Scale bar, 1 mm). Bars show the mean ± S.E.M. Individual data points in bar graphs in A, F–H, represent the average % dauers/experiment. ***P < 0.001, **P < 0.01, *P < 0.05, and ns, not significant
This was curious since DAF-16/FOXO and DAF-3/SMAD-DAF-5/Ski are thought to act independently, and in parallel to modulate the C. elegans dauer decision (3, 5). Therefore, to identify the mechanism(s) that could activate both pathways in ilc-17.1-deleted larvae, we conducted RNAi knockdown of candidate genes known to interact with these dauer effectors (50–57), and asked which, if any, also rescued the dauer entry of ilc-17.1 larvae (SI Appendix, Fig. S8B). We considered only the positive hits, due to the variable penetrance of RNAi. Among the positive candidates, we found most interesting, cep-1, the C. elegans ortholog of p53 (58, 59), since p53 family of proteins restrain cell cycle progression and alter metabolism, both cardinal features of dauer entry. Indeed, a deletion in cep-1, gk138, rescued the dauer arrest of ilc-17.1 deletion mutants and nearly all (98%) of cep-1;ilc-17.1 double mutant larvae grew into reproductive adults (Fig. 2 H–J), suggesting that CEP-1/p53 was also responsible for the dauer phenotype of ilc-17.1 mutants, and was activated.
In mammalian cells, p53 activation occurs through an increase in p53 protein levels due to its escape from constitutive degradation by the E3 ubiquitin ligase MDM2 (18, 20, 60). Although the C. elegans genome lacks MDM2 orthologs, CEP-1/p53 activation through translational and posttranslational modifications has been reported (61, 62). We therefore used Western analysis of FLAG-tagged endogenous C. elegans CEP-1/p53 to test whether CEP-1/p53 was activated in ilc-17.1 mutant larvae, as implied by the rescue. This was the case: In ilc-17.1-deleted larvae, CEP-1::FLAG levels increased by twofold, levels similar to that seen upon gamma irradiation, which also activates CEP-1/p53, and was used as a positive control (Fig. 3A) (63, 64). Accordingly, overexpressing CEP-1 alone, even to a modest 1.5-fold level over the wild type (see SI Appendix, Fig. S9A for cep-1 o/e mRNA levels), was sufficient to arrest growth and promote an almost completely penetrant dauer phenotype (Fig. 3 B–D). CEP-1 overexpression was achieved by crossing out the cep-1 lg12501 deletion mutation from a widely used transgenic strain expressing functional, fluorescently tagged, CEP-1, CEP-1::GFP, under its own promoter (61, 65, 66). In the cep-1(lg12501) strain, CEP-1::GFP is the only source of CEP-1/p53, rescues cep-1 (lg12501) apoptosis defects and is expressed at half the levels of the wild type (SI Appendix, Fig. S9A; cep-1 mRNA levels). CEP-1::GFP did not induce dauer arrest in the cep-1, lg12501 deletion background (Fig. 3B), indicating that cep-1 expression levels, and not the GFP tag, was responsible for the dauer arrest of CEP-1::GFP overexpressing larvae. The overexpression of CEP-1/p53 phenocopied the ilc-17.1 deletion, not only causing all larvae to arrest as dauers at 25 °C but also prompting transient dauer entry at 20 °C, as assessed by SDS-resistance (SI Appendix, Fig. S9B). RNA-seq analysis showed that genes differentially expressed in CEP-1/p53 overexpressing larvae en route to their dauer arrest significantly overlapped with those expressed in larvae lacking ILC-17.1 (Fig. 3E, Padj. < 0.05; Datasets S9 and S10). CEP-1/p53 overexpressing larvae also up-regulated dauer-specific collagens (SI Appendix, Fig. S9C and Dataset S4), down-regulated anabolic pathways (SI Appendix, Fig. S9D), and up-regulated catabolic pathways (SI Appendix, Fig. S9E). In addition, between 15 h to 32 h posthatching at 25 °C, CEP-1/p53 overexpressing larvae and ilc-17.1 deletion mutants showed a remarkably constant gene expression profile, and only 340 and 34 genes respectively changed expression (P < 0.05), compared to the >10,000 genes that were either up-regulated or down-regulated in wild-type larvae and cep-1;ilc-17.1 double mutant larvae that were en route to becoming reproductive adults (PCA; SI Appendix, Fig. S9F and Dataset S11). The upregulation of known CEP-1/p53 targets (63, 64, 67, 68), the BH3-only proteins egl-1 and ced-13 that occurred in the ilc-17.1 deletion mutants, in a cep-1-dependent manner, at both 20 °C and 25 °C, further confirmed CEP-1/p53 activation in the absence of ILC-17.1 (Fig. 3F and SI Appendix, Fig. S10A). The upregulation of CEP-1/p53 targets in ilc-17.1 deletion mutants could be suppressed by re-expressing ILC-17.1 under its endogenous-, or muscle-specific promoters (Fig. 3F and SI Appendix, Fig. S10A). In addition, Chromatin Immunoprecipitation followed by quantitative PCR (ChIP-qPCR) using FLAG tagged cep-1 animals showed that the loss of ilc-17.1 increased CEP-1/p53 binding at promoter regions of egl-1 and ced-13, and the increased occupancy was abrogated upon re-expressing ILC-17.1 (Fig. 3 G and H).
Fig. 3.

CEP-1/p53 is activated by reduced ILC-17.1 signaling. (A) Top: Representative western blot showing CEP-1::FLAG and tubulin. Bands on western correspond to numbers in data bars. cep-1 RNAi used for specificity. Samples constitute 32 to 36 h larvae grown at 25 °C, except for positive control, irradiated sample (#4; adults). Bottom: Average CEP-1 levels quantified relative to tubulin and normalized to control (Ctrl; CEP-1::FLAG in wild-type background). n = 4 experiments (unpaired t test). (B) Percent dauers. X-axis: genotype (n = 3 to 4 experiments, Chi-squared = 569.35, df = 2, P-value < 2.2e-16). mRNA expression levels in cep-1 OE and cep-1 (lg12501); CEP-1::GFP were ~1.5X and ~0.5X wild-type levels, respectively, see SI Appendix, Fig. S9A. (C and D) Representative micrographs 48 h posthatching at 25 °C, of C, CEP-1/p53 overexpressing larvae arrested as dauers and D, cep-1 (lg12501); CEP-1::GFP L4s. (Scale bar, 1 mm.) (E) Venn diagram depicting overlap between differentially expressed genes (P < 0.05) in ilc-17.1 deletion mutants and CEP-1/p53 overexpressing larvae, 32 to 36 h posthatching at 25 °C. P < 0.0; hypergeometric test. (F) Average egl-1 and ced-13 mRNA levels in 32 to 36 h larvae grown at 25 °C. n = 6 to 7 experiments. mRNA levels relative to pmp-3 and normalized to wild-type (N2) values (unpaired t test). (G and H) CEP-1 occupancy (percent input; CEP-1::FLAG was immunoprecipitated with anti-FLAG antibody) at the promoter-proximal regions of G, egl-1 and H, ced-13 in 32 to 36 h larvae grown at 25 °C (Top, schematic). X-axis: genotype. n = 4 experiments (unpaired t test). (A, G, and H) Endogenous cep-1 was FLAG tagged at its C terminus using CRISPR/Cas9, crossed into backgrounds noted, and probed with anti-FLAG antibody. Bars show the mean ± S.E.M. Individual points in bar graphs in B represent the % dauers/experiment. ***P < 0.001, **P < 0.01, *P < 0.05, and ns, not significant.
Like in the ILC-17.1 deficient animals, the dauer arrest of larvae overexpressing CEP-1 was dependent on DAF-16, DAF-5 and the steroid hormone pathway: down-regulating daf-16 expression by RNAi (Fig. 4A), crossing CEP-1/p53 overexpressing larvae into daf-5, e1386, or treatment with 50 nM exogenous Δ7-dafachronic acid, all suppressed their dauer arrest (Fig. 4B). In accordance, DAF-16 target genes were also up-regulated in larvae overexpressing CEP-1/p53 at both 25 °C (Fig. 4C) and 20 °C (SI Appendix, Fig. S10B). Differentially expressed genes in CEP-1/53 overexpressing larvae overlapped with published gene expression profiles of daf-2 (e1370) and active DAF-16 (SI Appendix, Fig. S10 C and D and Datasets S12 and S13). In addition, and most convincing, the upregulation of daf-16 target genes in ilc-71.1 deletion mutant larvae was cep-1 dependent at both 25 °C and 20 °C (Fig. 4C and SI Appendix, Fig. S10B). CEP-1/p53 activation-induced dauer entry appeared to be somewhat specific to reduced ILC-17.1 signaling, as the dauer arrest of daf-2(e1370) larvae that occurred due to reduced insulin signaling did not depend on cep-1 (SI Appendix, Fig. S10E). Moreover, in agreement with previous reports (69, 70), cep-1 was not required for dauer induction at high temperatures as cep-1 (gk138) larvae could enter dauer at 27 °C just like wild-type animals (SI Appendix, Fig. S10F). Taken together, these results showed that ILC-17.1 normally acted to repress CEP-1/p53 transcriptional activity. Upon loss of ILC-17.1 signaling, CEP-1/p53 was activated, in turn activated DAF-16/ FOXO (and perhaps DAF-3/SMAD-DAF-5/Ski and DAF-12) to trigger the dauer arrest of ilc-17.1 deletion mutants. However, the ILC-17.1/CEP-1 dauer axis appeared independent of the insulin-like receptor, daf-2, per se.
Fig. 4.
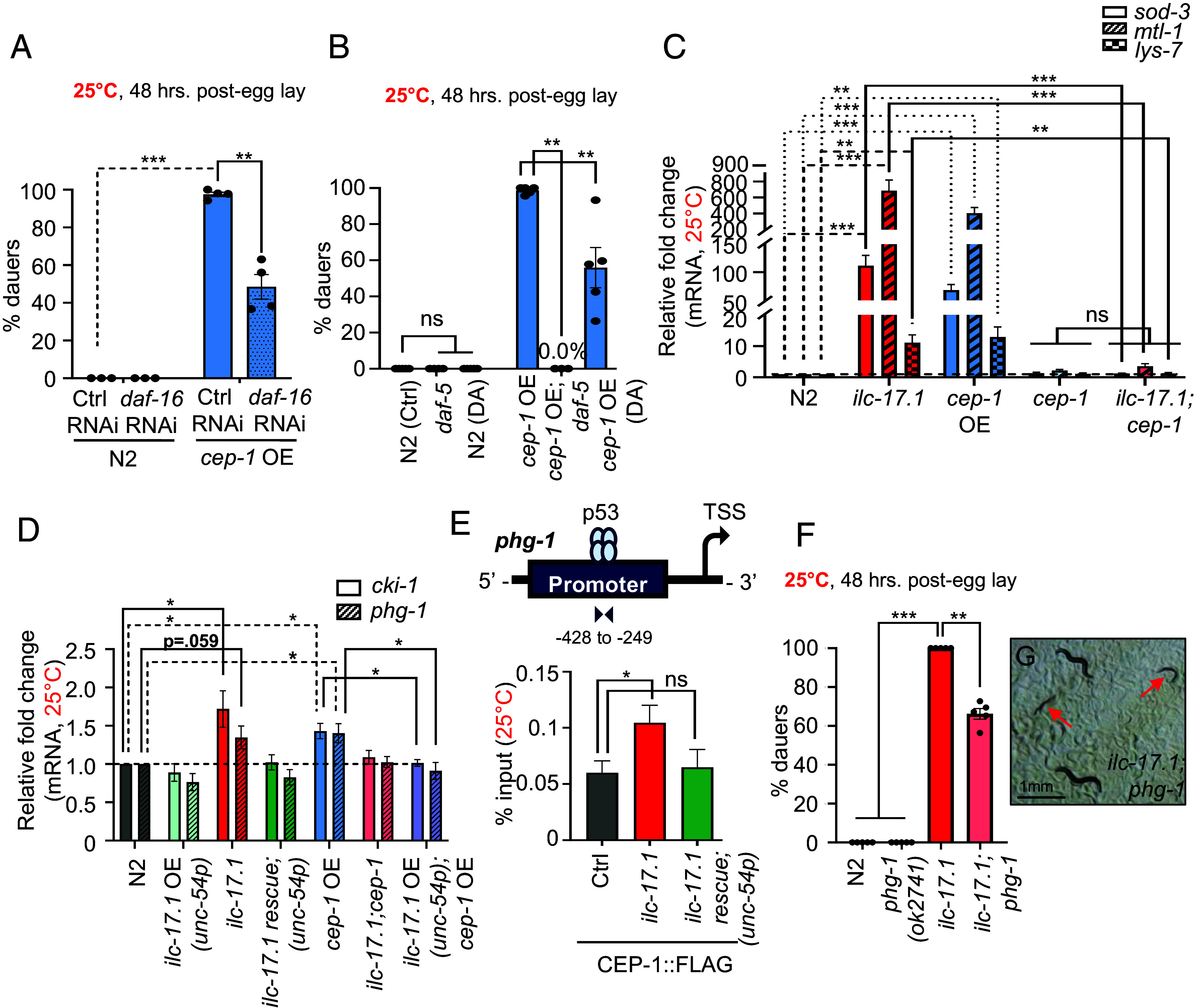
CEP-1/p53 acts through DAF-16/FOXO, DAF-3/SMAD-DAF-5/Ski complex, and cell cycle inhibitors to control dauer entry. (A) Percent dauers. X-axis: genotype and RNAi treatment: Ctrl, L4440 or daf-16 (n = 3 to 4 experiments, Chi-squared = 494.22, df = 3, P-value < 2.2e-16). (B) Percent dauers. X-axis: genotype and treatment: water (control) and 50 nM Δ7-dafachronic acid (DA) (n = 3 to 4 experiments, Chi-squared = 982.66, df = 5, P-value < 2.2e-16). (C) Average sod-3, mtl-1, and lys-7 mRNA levels in larvae 32 to 36 h posthatching at 25 °C. n = 4 to 10 experiments (unpaired t test). (D) Average cki-1 and phg-1 mRNA levels in 32 to 36 h larvae grown at 25 °C. (C and D) mRNA levels were determined relative to pmp-3 and normalized to wild-type (N2) values. n = 4 to 6 experiments. *P < 0.05 (unpaired t test). (E) CEP-1 occupancy (percent input) in 32 to 36 h larvae grown at 25 °C, at the promoter proximal region of phg-1 (Top, schematic). X-axis: genotype. n = 4 experiments (unpaired t test). (F) Percent dauers. X-axis: genotype (n = 6 experiments, Chi-squared = 826.95, df = 3, P-value < 2.2e-16). (G) Representative micrograph 48 h posthatching at 25 °C of ilc-17.1;phg-1 double mutants, showing dauers (arrows) and L4s. (Scale bar, 1 mm.) (A, B, and F), Bar graphs: Bars show the mean ± S.E.M, and individual points represent the % dauers/experiment. ***P < 0.001, **P < 0.01, *P < 0.05, and ns, not significant.
CEP-1/p53 Directly or Indirectly Controls Cell Cycle Progression.
A conserved function of the p53 family of proteins is to restrain cell cycle progression. In C. elegans, most cell divisions are completed during embryogenesis, but a subset of somatic and germline cells divide and differentiate postembryonically, and these cells arrest during dauer entry (71–86). We therefore tested whether CEP-1/p53 activation upon loss of ILC-17.1 was participating in the arrest of cell cycle progression during postembryonic development. Indeed, ilc-17.1-deleted larvae up-regulated mRNA levels of cell cycle inhibitors cki-1, one of the two p21 homologs, and phg-1, the C. elegans homologue of Growth arrest-specific 1 (Gas1) (Fig. 4D). These are known to regulated by CEP-1/p53 (87). phg-1, but not cki-1, was a direct target of CEP-1/p53 and ilc-17.1 deletion mutants displayed increased CEP-1/p53 occupancy at the phg-1 promoter (Fig. 4E and SI Appendix, Fig. S11A). Again, as with the other CEP-1/p53 targets, the increased expression of cki-1 and phg-1, and the increased occupancy at the phg-1 promoter was restored to wild-type levels upon re-expressing ILC-17.1 (Fig. 4 D and E). Surprisingly, downregulation of phg-1 and cki-1 by RNAi modestly, but significantly, rescued dauer arrest in ilc-17.1 deletion mutants and CEP-1/p53 overexpressing larvae and allowed a small number of larvae to develop into adults (SI Appendix, Fig. S11B). Crossing ilc-17.1 deletion mutants into phg-1 deletion mutants, ok2741 also resulted in a rescue and ~40% escaped dauer arrest and grew into reproductive adults (Fig. 4 F and G). In addition, a proportion of ilc-17.1 deletion mutant and CEP-1/p53 overexpressing larvae that appeared morphologically dauer, displayed modest but significant non-dauer physiological traits on phg-1 and cki-1 RNAi such as increased rates of sporadic pumping (SI Appendix, Fig. S11C; wild-type L2 were used as controls), suggesting there may also occur a wider, albeit incomplete, dauer rescue in some tissues. The dauer arrest of daf-2 mutant larvae which did not depend on cep-1, was also not rescued upon down-regulating phg-1 or cki-1, nor were the pumping rates of daf-2 dauers altered (SI Appendix, Fig. S11 B and C). Although we cannot distinguish at this time, whether the downregulation of the cell cycle inhibitors allowed larvae to exit dauer, or suppressed dauer entry and complete arrest, these data showed that loss of ILC-17.1 signaling or overexpressing CEP-1/p53 up-regulated CEP-1/p53-dependent cell cycle inhibitors, and this increase appeared to be one mechanism responsible, at least in part, for the dauer arrest.
ILC-17.1 Links Glucose Metabolism to CEP-1/p53 Activity.
Mammalian p53 plays a central roles in energy metabolism and, in turn, can be activated by energy deprivation or limited glucose, through signaling by AMP-activated protein kinase (AMPK) or mammalian target of rapamycin (mTOR) pathways (88, 89). Therefore, the responsiveness of ILC-17.1 to food signals suggested the intriguing possibility that the loss of ILC-17.1 signaling activated CEP-1/p53 because of energy deprivation, and this could link nutrient availability to metabolism and cell cycle progression. AMPK did not appear to be activated in ilc-17.1 deleted animals (SI Appendix, Fig. S12A), nor was mTOR activity obviously reduced (SI Appendix, Fig. S12B). However, ILC-17.1 signaling did appear to link glucose utilization to developmental progression, since down-regulating fgt-1, the main Glucose transporter (GLUT) responsible for glucose absorption in C. elegans (90), decreased the extent of dauer-rescue conferred by re-expressing ILC-17.1 in ilc-17.1 mutant larvae (Fig. 5A), and increased the numbers of ilc-17.1 deletion mutant larvae that arrested as dauers at 20 °C (Fig. 5B). Conversely, extra glucose (91) suppressed the dauer arrest of approximately 30% ilc-17.1 deficient larvae (Fig. 5C and SI Appendix, Fig. S12C), suggesting that the normal amounts of glucose available in the diets of C. elegans larvae was insufficient for ilc-17.1 larvae to develop into adults. Consistently, decreasing glucose import through fgt-1 RNAi once again, lowered the percentage of larvae that were rescued by the extra glucose (SI Appendix, Fig. S12D), and no rescue was observed on the nonhydrolyzable glucose analog 2-Deoxy-d-glucose (2-DOG; SI Appendix, Fig. S12E) which cannot undergo glycolysis and does not enter the metabolic pathway. The same concentrations of glucose could not rescue daf-2 larvae that arrested as dauers (Fig. 5C). We ruled out nonspecific effects of glucose rescue by testing the role of increased ROS or other supplements (SI Appendix, Fig. S12 E and F; see SI Appendix, Extended Text 3 for details). Down-regulating fgt-1 in wild-type larvae, while not sufficient to trigger dauer arrest, modestly increased the expression of CEP-1/p53 target egl-1 (but not ced-13; SI Appendix, Fig. S12G). Thus, these data supported the possibility that the activation of CEP-1/p53 upon the loss of ILC-17.1 could, at least in part, occur due an impairment in the ilc-17.1-deleted larvae’s ability to utilize glucose.
Fig. 5.

Impaired glucose utilization in ILC-17.1 deficient larvae activates CEP-1/p53 and alters the expression of OXPHOS genes. (A) Top: Schematic of experimental design. Right: Percent dauers. X-axis: genotype. Larve treated with control (L4440) and fgt-1 RNAi (n = 5 experiments, Chi-squared = 1817.1, df = 5, P-value < 2.2e-16). (B) Bottom: Schematic of experimental design. Right: Percent SDS (1%)-resistant dauers at 48 and 72 h posthatching and grown at optimal conditions of 20 °C. X-axis: genotype. Larve treated with control (L4440) and fgt-1 RNAi (n = 3 experiments, Chi-squared = 206.82, df = 7, P-value < 2.2e-16). (C) Top: Schematic of experimental design. Bottom: Percent dauers. X-axis: genotype and treatment: control (H2O) and 50 mM glucose (n = 3 to 6 experiments, Chi-squared = 2882.3, df = 5, P-value < 2.2e-16). (D) Average phg-1, sod-3, and mtl-1 mRNA levels in 32 to 36 h larvae that arrest as dauers (on H2O, control) and larvae that bypass dauer (50 mM glucose) at 25 °C. mRNA levels are relative to pmp-3 and normalized to wild-type (N2) values. n = 3 experiments (unpaired t test). (E) Average cyc-2.2 mRNA levels in 32 to 36 h larvae. mRNA levels are relative to pmp-3 and normalized to the wild-type (N2) values. n = 4 to 6 experiments (unpaired t test). Bars show the mean ± S.E.M. Individual points in the bar graphs in A–C represent the % dauers/experiment. ***P < 0.001, **P < 0.01, *P < 0.05, and ns, nonsignificant.
Glucose supplementation also rescued dauer arrest of CEP-1/p53 overexpressing larvae (Fig. 5C and SI Appendix, Fig. S10C), arguing that decreased glucose intake in the absence of ILC-17.1 was not a consequence of CEP-1/p53 activation, but instead acted logically upstream of CEP-1/p53. In agreement, extra glucose suppressed the increased expression of phg-1 the direct target of CEP-1/p53, and decreased DAF-16/FOXO activation (Fig. 5D). Surprisingly, not only did CEP-1/p53 appear to be activated by the inability of ilc-17.1 to utilize glucose normally but CEP-1/p53 activity in turn decreased the expression levels of key enzymes involved in glucose metabolism (92): phosphofructokinase-1.2 (pfk-1.2), the rate-limiting enzyme in glycolysis, and cytochrome c (cyc-2.2), the complex IV subunit of the mitochondrial electron transport chain responsible. This was visible in the RNA-seq data (SI Appendix, Fig. S13 A–F and Dataset S14) and confirmed by qRT-PCR (SI Appendix, Fig. S13G and Fig. 5E). Moreover, although the downregulation of pfk-1.2 or cyc-2.2 alone was not sufficient to induce dauer in wild-type animals, RNAi knockdown of cyc-2.2 in ilc-17.1-deleted larvae increased the percentage of dauers under optimal conditions of 20 °C, suggesting that CEP-1/p53-dependent decrease of cyc-2.2 could feedback onto ilc-17.1 loss to increase dauer propensity (SI Appendix, Fig. S13H). These data, together, suggested that the loss of ILC-17.1 impaired glucose utilization and this activated CEP-1/p53, which, then further limited glucose utilization by down-regulating genes important for glycolysis and OXPHOS. Thus, ILC-17 secretion from amphid neurons that occurred in response to food availability appeared to link cell fate decisions with energy metabolism through CEP-1/p53.
ILC-17.1 Suppresses CEP-1/p53 in C. elegans and Human Epithelial cells.
Our data pointed toward a model whereby ILC-17.1 signaling suppressed CEP-1/p53. Therefore, we tested this directly. Indeed, overexpressing ILC-17.1 in animals also overexpressing CEP-1/p53 inhibited their dauer arrest (Fig. 6A). In addition, since the most-accepted and canonical function of C.elegans CEP-1/p53 is in triggering apoptosis in the adult germline in response to DNA-damage, we also examined whether ILC-17.1 repressed CEP-1/p53’s function in apoptosis. As has been previously shown (67), the loss of CEP-1/p53 in cep-1 (gk138) adults has negligible effects on physiological apoptosis, but suppresses the increased apoptosis that occurs in response to gamma irradiation (Fig. 6B). CEP-1/p53 overexpression, surprisingly, caused a significant increase in physiological apoptosis, and a marked increase in irradiation-induced apoptosis, both of which could be suppressed by also overexpressing ILC-17.1 (Fig. 6B). Overexpressing ilc-17.1 in a wild-type background also decreased physiological and irradiation-induced apoptosis, but this effect was variable and did not reach significance (average nos. of apoptotic cells in nonirradiated N2 v ILC-17.1 overexpressing animals were 3.56 ± 1.3 and 2.53 ± 1.1 cells, and upon irradiation, increased to 7.1 ± 1.8 in N2, and 5.72 ± 2.2 in IL-17 overexpressing animals). For reasons that remain to be understood, ilc-17.1 deletion did not alter apoptosis in the adult germline, perhaps reflecting a different function postdevelopment. IL-17 could also modestly repress p53, autonomously, in mammalian cells (SI Appendix, Fig. S14 A and B; see SI Appendix, Extended Text 4 for details). Together, these data suggest that the inhibition of CEP-1/p53’s activity could be a conserved function of IL-17 s.
Fig. 6.

ILC-17.1 suppresses CEP-1/p53. (A) Percent dauers. X-axis: genotype (n = 4 experiments, Chi-squared = 1245.4, df = 2, P-value < 2.2e-16). (B) Top: Schematic of germline-specific apoptosis (apoptotic cells in red) scored with Acridine Orange. Bottom: Average numbers of apoptotic cells in day-2 adult animals under control, nonirradiated conditions, and upon irradiation with 75 Gy (day-1 adults were irradiated). n = 3 experiments and 12 gonad arms/experiment (ANOVA with Tukey's correction, df = 13). Note: P values between N2 vs. ilc-17.1 OE (unc-54p), nonirradiated and irradiated, are P = 0.0192 and P = 0.0623, if compared by themselves (unpaired t test), but do not rise to significance when corrected for the multiple comparisons. Data in all graphs show mean ± S.E.M. ***P < 0.001 and **P < 0.01.
Discussion
Here, we show roles for the interleukin 17 (IL-17) family of cytokines and p53 in the development of an invertebrate species, C. elegans. Our data support a model whereby ILC-17.1 signaling of food availability by amphid neurons ensures the continuous growth of larvae into reproductive adults under favorable conditions through suppression of CEP-1/p53. IL-17s play broad-ranging immunometabolic roles in mammals and are one of the most conserved proinflammatory cytokines among animal phyla. Thus, their ancestral role in signaling nutrient availability as in C. elegans may be broadly conserved. Our data also reveal that reduced ILC-17.1 signaling, expected to occur when food is scarce, activates CEP-1/p53, to trigger dauer arrest through the well-characterized dauer activating pathways, DAF-16/FOXO, DAF-3/SMAD-DAF-5/Ski and also, perhaps steroid hormone signaling. In its role in promoting dauer arrest in C. elegans, CEP-1/p53 inhibits cell cycle progression and modulates metabolic flux by directly or indirectly controlling the levels of phosphofructokinase (pfk-1) and cytochrome C (cyc-2.2). Notably, these are also mechanisms by which p53 restrains tumorigenesis. In mammals too, uncontrolled p53 activity is associated with a developmental syndrome through its effects on cell cycle progression, apoptosis, and the migration of neural crest cells (25). We therefore cautiously propose that the ancestral roles of the p53 gene family in modulating development could have the driving force in their evolution.
While there remain several puzzling aspects associated with this study, our data indicate that there may exist a previously undiscovered C. elegans dauer pathway. The dauers formed by ilc-17.1 deletion, or the modest 1.5X increase in CEP-1/p53 expression, display all the cardinal morphological and physiological features of dauers, including SDS-resistance, decreased pumping rates, presence of alae, changes in collagens, upregulation of catabolic and downregulation of anabolic genes, and the ability to remain arrested for days to weeks as dauers. Nevertheless, they also differ in some respects from the well-studied dauers formed through decreased insulin signaling (daf-2), and other pathways, in that, unlike previously studied dauers, a modest, but significant percentage of ilc-17.1 dauers larvae bypass or are rescued from dauer upon down-regulating cell cycle inhibitors such as phg-1, or when fed extra glucose. This suggests that there may exist different “types” of dauer larvae. The discovery of new C. elegans dauer pathway genes is surprising given the extensive genetic screens that have been conducted (3, 5, 30–32). However, genes that require a complete deletion to confer a dauer phenotype, or genes that control development through complex and antagonistic tissue-specific effects, as possible for ilcr-2, could have been missed. In this regard, two other deletions of the ilc-17.1 exist in the community (tm5218 and tm5124). These do not arrest as dauers, but are also not null alleles and express exons 1 and 2 of ilc-17.1 transcript (SI Appendix, Fig. S14C). The tm5218 deletion, however, removes the predicted beta strands required for the canonical cysteine knot fold of IL-17s and might have arguably been expected to result in the loss of ILC-17.1 signaling capability, mimic ilc-17.1 deletion mutants, and arrest as dauers. Although this is not the case, and all tm5218 homozygotes escape dauer arrest, a variable, but significant percentage of trans-heterozygotes for ilc-17.1(tm5218) and ilc-17.1(syb5296) larvae arrest as dauers, as confirmed by PCR genotyping, indicating noncomplementation (SI Appendix, Fig. S14D; results from one experiment; dauer arrest varied between 0 and 75% of F1 trans-heterozygotes; n > 10 experiments). This observation adds confidence to the role of ilc-17.1 in dauer, but also reinforces our lack of understanding of the nature and stoichiometry of the IL-17 homo/heterodimers and IL-17-ILCR-2 signaling assemblies in C. elegans.
We hypothesize that ILC-17.1 is secreted upon sensory neuronal activity to control CEP-1/p53 activity, but this remains to be shown, and the neuronal circuits identified (93). Nutrient scarcity has shaped much of evolution, and the ability of cells and organisms to sense nutrient availability and prepare in anticipation to pause cell cycle progression and growth and maintain a quiescent state until resources are optimal, has unquestionable selective advantages. For C. elegans, like for most organisms, the availability of food is not guaranteed. Neuronal ILC-17.1 signaling appears to be one mechanism which allows the animal to match the cell fate programs and metabolic requirements of the developing larvae with resource availability. Neurons also control dauer entry through TGFβ and insulin signaling. In addition, other stress-responsive transcriptional programs that can be activated autonomously by cells, are also controlled through neuronal signaling in C. elegans, allowing the nervous system to coordinate tissue-specific transcriptional and epigenetic responses with organismal physiology and behavior (94–97). The extent to which IL-17 s function similarly in mammals remains to be understood (98, 99).
Materials and Methods
Descriptions of strains and methods are included in SI Appendix, Extended Methods. Strains will be available upon request after publication. The data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GSE218596, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE218596, and GSE229132, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE229132 (100, 101).
Supplementary Material
Appendix 01 (PDF)
Dataset S01 (XLSX)
Dataset S02 (XLSX)
Dataset S03 (XLSX)
Dataset S04 (XLSX)
Dataset S05 (XLSX)
Dataset S06 (XLSX)
Dataset S07 (XLSX)
Dataset S08 (XLSX)
Dataset S09 (XLSX)
Dataset S10 (XLSX)
Dataset S11 (XLSX)
Dataset S12 (XLSX)
Dataset S13 (XLSX)
Dataset S14 (XLSX)
Acknowledgments
We thank the V.P. laboratory, Drs. Sarit Smolikove, Josep Comeron, Anna Malkova and Michael Petrascheck for comments, Dr. Peter Ratcliffe, Oxford, for his generous gift of CeHIF-1antibody, and Dr. Mario de Bono, Institute of Science and Technology Austria for C. elegans strains. Nematode strains were provided by the Caenorhabditis Genetics Center (funded by the NIH Infrastructure Programs P40 OD010440). This work was supported by NIH R01 AG060616 (V.P.).
Author contributions
V.P. designed and funded research; P.D.I. collaborated in research design; A.G., S.M., S.D., J.C.-C., A.D., K.M.-W., P.D.I., and V.P. performed research; V.P. contributed new reagents/analytic tools; A.G., S.M., S.D., J.C.-C., A.D., K.M.-W., P.D.I., and V.P. analyzed data; and A.G., S.M., J.C.-C., A.D., P.D.I., and V.P. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission. M.P. is a guest editor invited by the Editorial Board.
Data, Materials, and Software Availability
RNA-seq data have been deposited in NCBI’s Gene Expression Omnibus (GSE218596 and GSE229132) (100, 101). All other data are included in the manuscript and/or supporting information.
Supporting Information
References
- 1.Golden J. W., Riddle D. L., The Caenorhabditis elegans dauer larva: Developmental effects of pheromone, food, and temperature. Dev. Biol. 102, 368–378 (1984). [DOI] [PubMed] [Google Scholar]
- 2.Swanson M. M., Riddle D. L., Critical periods in the development of the Caenorhabditis elegans dauer larva. Dev. Biol. 84, 27–40 (1981). [DOI] [PubMed] [Google Scholar]
- 3.Hu P. J., Dauer. WormBook 1–19 (2007). http://www.wormbook.org. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Karp X., Working with dauer larvae. WormBook 2018, 1–19 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fielenbach N., Antebi A., C. elegans dauer formation and the molecular basis of plasticity. Genes Dev. 22, 2149–2165 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burnell A. M., Houthoofd K., O’Hanlon K., Vanfleteren J. R., Alternate metabolism during the dauer stage of the nematode Caenorhabditis elegans. Exp. Gerontol. 40, 850–856 (2005). [DOI] [PubMed] [Google Scholar]
- 7.Rashid S., Wong C., Roy R., Developmental plasticity and the response to nutrient stress in Caenorhabditis elegans. Dev. Biol. 475, 265–276 (2021). [DOI] [PubMed] [Google Scholar]
- 8.Cohen S. M., Wrobel C. J. J., Prakash S. J., Schroeder F. C., Sternberg P. W., Formation and function of dauer ascarosides in the nematodes Caenorhabditis briggsae and Caenorhabditis elegans. G3 (Bethesda) 12, jkac014 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Braendle C., Milloz J., Felix M. A., Mechanisms and evolution of environmental responses in Caenorhabditis elegans. Curr. Top. Dev. Biol. 80, 171–207 (2008). [DOI] [PubMed] [Google Scholar]
- 10.Diaz S. A., Viney M., The evolution of plasticity of dauer larva developmental arrest in the nematode Caenorhabditis elegans. Ecol. Evol. 5, 1343–1353 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ewald C. Y., Landis J. N., Porter Abate J., Murphy C. T., Blackwell T. K., Dauer-independent insulin/IGF-1-signalling implicates collagen remodelling in longevity. Nature 519, 97–101 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harvey S. C., Shorto A., Viney M. E., Quantitative genetic analysis of life-history traits of Caenorhabditis elegans in stressful environments. BMC Evol. Biol. 8, 15 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhu J., Thompson C. B., Metabolic regulation of cell growth and proliferation. Nat. Rev. Mol. Cell Biol. 20, 436–450 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Altan-Bonnet G., Mukherjee R., Cytokine-mediated communication: A quantitative appraisal of immune complexity. Nat. Rev. Immunol. 19, 205–217 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Douglas A., Stevens B., Lynch L., Interleukin-17 as a key player in neuroimmunometabolism. Nat. Metab. 5, 1088–1100 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mills K. H. G., IL-17 and IL-17-producing cells in protection versus pathology. Nat. Rev. Immunol. 23, 38–54 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li X., Bechara R., Zhao J., McGeachy M. J., Gaffen S. L., IL-17 receptor-based signaling and implications for disease. Nat. Immunol. 20, 1594–1602 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hall P. A., Lane D. P., Tumor suppressors: A developing role for p53? Curr. Biol. 7, R144–147 (1997). [DOI] [PubMed] [Google Scholar]
- 19.Jain A. K., Barton M. C., p53: Emerging roles in stem cells, development and beyond. Development 145, dev158360 (2018). [DOI] [PubMed] [Google Scholar]
- 20.Levine A. J., p53: 800 million years of evolution and 40 years of discovery. Nat. Rev. Cancer 20, 471–480 (2020). [DOI] [PubMed] [Google Scholar]
- 21.Rutkowski R., Hofmann K., Gartner A., Phylogeny and function of the invertebrate p53 superfamily. Cold Spring Harb. Perspect. Biol. 2, a001131 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pfister N. T., Prives C., Transcriptional regulation by wild-type and cancer-related mutant forms of p53. Cold Spring Harb. Perspect. Med. 7, a026054 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lu W. J., Amatruda J. F., Abrams J. M., p53 ancestry: Gazing through an evolutionary lens. Nat. Rev. Cancer 9, 758–762 (2009). [DOI] [PubMed] [Google Scholar]
- 24.Bowen M. E., Mulligan A. S., Sorayya A., Attardi L. D., Puma- and Caspase9-mediated apoptosis is dispensable for p53-driven neural crest-based developmental defects. Cell Death Differ. 28, 2083–2094 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Van Nostrand J. L., Attardi L. D., Guilty as CHARGED: p53’s expanding role in disease. Cell Cycle 13, 3798–3807 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen C., et al. , IL-17 is a neuromodulator of Caenorhabditis elegans sensory responses. Nature 542, 43–48 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Flynn S. M., et al. , MALT-1 mediates IL-17 neural signaling to regulate C. elegans behavior, immunity and longevity. Nat. Commun. 11, 2099 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lillis M., Zaccardi N. J., Heiman M. G., Axon-dendrite and apical-basolateral sorting in a single neuron. Genetics 221, iyac036 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Golden J. W., Riddle D. L., A pheromone-induced developmental switch in Caenorhabditis elegans: Temperature-sensitive mutants reveal a wild-type temperature-dependent process. Proc. Natl. Acad. Sci. U.S.A. 81, 819–823 (1984). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thomas J. H., Birnby D. A., Vowels J. J., Evidence for parallel processing of sensory information controlling dauer formation in Caenorhabditis elegans. Genetics 134, 1105–1117 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vowels J. J., Thomas J. H., Genetic analysis of chemosensory control of dauer formation in Caenorhabditis elegans. Genetics 130, 105–123 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ailion M., Thomas J. H., Isolation and characterization of high-temperature-induced Dauer formation mutants in Caenorhabditis elegans. Genetics 165, 127–144 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li W., Ou G., The application of somatic CRISPR-Cas9 to conditional genome editing in Caenorhabditis elegans. Genesis 54, 170–181 (2016). [DOI] [PubMed] [Google Scholar]
- 34.Paix A., et al. , Scalable and versatile genome editing using linear DNAs with microhomology to Cas9 Sites in Caenorhabditis elegans. Genetics 198, 1347–1356 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xu S., The application of CRISPR-Cas9 genome editing in Caenorhabditis elegans. J. Genet. Genomics 42, 413–421 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Holt S. J., Staying alive in adversity: Transcriptome dynamics in the stress-resistant dauer larva. Funct. Integr. Genomics 6, 285–299 (2006). [DOI] [PubMed] [Google Scholar]
- 37.Cohen S. M., Sun J. J., Schroeder F. C., Sternberg P. W., Transcriptional response to a dauer-inducing ascaroside cocktail in late L1 in C. elegans. MicroPubl. Biol., 10.17912/micropub.biology.000397 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang J., Kim S. K., Global analysis of dauer gene expression in Caenorhabditis elegans. Development 130, 1621–1634 (2003). [DOI] [PubMed] [Google Scholar]
- 39.Wirick M. J., et al. , daf-16/FOXO blocks adult cell fate in Caenorhabditis elegans dauer larvae via lin-41/TRIM71. PLoS Genet. 17, e1009881 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kyani-Rogers T., et al. , Developmental history modulates adult olfactory behavioral preferences via regulation of chemoreceptor expression in Caenorhabditis elegans. Genetics 222, iyac143 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ow M. C., Borziak K., Nichitean A. M., Dorus S., Hall S. E., Early experiences mediate distinct adult gene expression and reproductive programs in Caenorhabditis elegans. PLoS Genet. 14, e1007219 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Motola D. L., et al. , Identification of ligands for DAF-12 that govern dauer formation and reproduction in C. elegans. Cell 124, 1209–1223 (2006). [DOI] [PubMed] [Google Scholar]
- 43.Vowels J. J., Thomas J. H., Multiple chemosensory defects in daf-11 and daf-21 mutants of Caenorhabditis elegans. Genetics 138, 303–316 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lin K., Dorman J. B., Rodan A., Kenyon C., daf-16: An HNF-3/forkhead family member that can function to double the life-span of Caenorhabditis elegans. Science 278, 1319–1322 (1997). [DOI] [PubMed] [Google Scholar]
- 45.Libina N., Berman J. R., Kenyon C., Tissue-specific activities of C. elegans DAF-16 in the regulation of lifespan. Cell 115, 489–502 (2003). [DOI] [PubMed] [Google Scholar]
- 46.Lee S. S., Kennedy S., Tolonen A. C., Ruvkun G., DAF-16 target genes that control C. elegans life-span and metabolism. Science 300, 644–647 (2003). [DOI] [PubMed] [Google Scholar]
- 47.Kenyon C., Murphy C. T., Enrichment of regulatory motifs upstream of predicted DAF-16 targets. Nat. Genet. 38, 397–398 (2006). [DOI] [PubMed] [Google Scholar]
- 48.da Graca L. S., et al. , DAF-5 is a Ski oncoprotein homolog that functions in a neuronal TGF beta pathway to regulate C. elegans dauer development. Development 131, 435–446 (2004). [DOI] [PubMed] [Google Scholar]
- 49.Gems D., et al. , Two pleiotropic classes of daf-2 mutation affect larval arrest, adult behavior, reproduction and longevity in Caenorhabditis elegans. Genetics 150, 129–155 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zecic A., Braeckman B. P., DAF-16/FoxO in Caenorhabditis elegans and its role in metabolic remodeling. Cells 9, 109 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Oh S. W., et al. , JNK regulates lifespan in Caenorhabditis elegans by modulating nuclear translocation of forkhead transcription factor/DAF-16. Proc. Natl. Acad. Sci. U.S.A. 102, 4494–4499 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Greer E. L., et al. , An AMPK-FOXO pathway mediates longevity induced by a novel method of dietary restriction in C. elegans. Curr. Biol. 17, 1646–1656 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Leiser S. F., Begun A., Kaeberlein M., HIF-1 modulates longevity and healthspan in a temperature-dependent manner. Aging Cell 10, 318–326 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Perrin A. J., et al. , Noncanonical control of C. elegans germline apoptosis by the insulin/IGF-1 and Ras/MAPK signaling pathways. Cell Death Differ. 20, 97–107 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gurkar A. U., et al. , Dysregulation of DAF-16/FOXO3A-mediated stress responses accelerates oxidative DNA damage induced aging. Redox Biol. 18, 191–199 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Baruah A., et al. , CEP-1, the Caenorhabditis elegans p53 homolog, mediates opposing longevity outcomes in mitochondrial electron transport chain mutants. PLoS Genet. 10, e1004097 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ewald C. Y., Castillo-Quan J. I., Blackwell T. K., Untangling longevity, dauer, and healthspan in Caenorhabditis elegans insulin/IGF-1-signalling. Gerontology 64, 96–104 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Derry W. B., Putzke A. P., Rothman J. H., Caenorhabditis elegans p53: Role in apoptosis, meiosis, and stress resistance. Science 294, 591–595 (2001). [DOI] [PubMed] [Google Scholar]
- 59.Schumacher B., Hofmann K., Boulton S., Gartner A., The C. elegans homolog of the p53 tumor suppressor is required for DNA damage-induced apoptosis. Curr. Biol. 11, 1722–1727 (2001). [DOI] [PubMed] [Google Scholar]
- 60.Prives C., Signaling to p53: Breaking the MDM2-p53 circuit. Cell 95, 5–8 (1998). [DOI] [PubMed] [Google Scholar]
- 61.Schumacher B., et al. , Translational repression of C. elegans p53 by GLD-1 regulates DNA damage-induced apoptosis. Cell 120, 357–368 (2005). [DOI] [PubMed] [Google Scholar]
- 62.Sendoel A., Kohler I., Fellmann C., Lowe S. W., Hengartner M. O., HIF-1 antagonizes p53-mediated apoptosis through a secreted neuronal tyrosinase. Nature 465, 577–583 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gartner A., Boag P. R., Blackwell T. K., Germline survival and apoptosis. WormBook 1–20 (2008). http://www.wormbook.org. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schumacher B., et al. , C. elegans ced-13 can promote apoptosis and is induced in response to DNA damage. Cell Death Differ. 12, 153–161 (2005). [DOI] [PubMed] [Google Scholar]
- 65.Levi-Ferber M., et al. , It’s all in your mind: Determining germ cell fate by neuronal IRE-1 in C. elegans. PLoS Genet. 10, e1004747 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hicks T., et al. , R-loop-induced irreparable DNA damage evades checkpoint detection in the C. elegans germline. Nucleic Acids Res. 50, 8041–8059 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Greiss S., Schumacher B., Grandien K., Rothblatt J., Gartner A., Transcriptional profiling in C. elegans suggests DNA damage dependent apoptosis as an ancient function of the p53 family. BMC Genomics 9, 334 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schertel C., Conradt B., C. elegans orthologs of components of the RB tumor suppressor complex have distinct pro-apoptotic functions. Development 134, 3691–3701 (2007). [DOI] [PubMed] [Google Scholar]
- 69.Arum O., Johnson T. E., Reduced expression of the Caenorhabditis elegans p53 ortholog cep-1 results in increased longevity. J. Gerontol. A, Biol. Sci. Med. Sci. 62, 951–959 (2007). [DOI] [PubMed] [Google Scholar]
- 70.Lee C. C., et al. , Mutation of a Nopp140 gene dao-5 alters rDNA transcription and increases germ cell apoptosis in C. elegans. Cell Death Dis. 5, e1158 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sulston J. E., Horvitz H. R., Post-embryonic cell lineages of the nematode, Caenorhabditis elegans. Dev. Biol. 56, 110–156 (1977). [DOI] [PubMed] [Google Scholar]
- 72.Sternberg P. W., Vulval development. WormBook 1–28 (2005). http://www.wormbook.org. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Horvitz H. R., Sternberg P. W., Nematode postembryonic cell lineages. J. Nematol. 14, 240–248 (1982). [PMC free article] [PubMed] [Google Scholar]
- 74.Chisholm A. D., Hardin J., Epidermal morphogenesis. WormBook 1–22 (2005). http://www.wormbook.org. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Herman M. A., Hermaphrodite cell-fate specification. WormBook 1–16 (2006). http://www.wormbook.org. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Costa M., Weir M., Coulson A., Sulston J., Kenyon C., Posterior pattern formation in C. elegans involves position-specific expression of a gene containing a homeobox. Cell 55, 747–756 (1988). [DOI] [PubMed] [Google Scholar]
- 77.Euling S., Ambros V., Reversal of cell fate determination in Caenorhabditis elegans vulval development. Development 122, 2507–2515 (1996). [DOI] [PubMed] [Google Scholar]
- 78.Braendle C., Felix M. A., Plasticity and errors of a robust developmental system in different environments. Dev. Cell 15, 714–724 (2008). [DOI] [PubMed] [Google Scholar]
- 79.Karp X., Greenwald I., Control of cell-fate plasticity and maintenance of multipotency by DAF-16/FoxO in quiescent Caenorhabditis elegans. Proc. Natl. Acad. Sci. U.S.A. 110, 2181–2186 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tenen C. C., Greenwald I., Cell non-autonomous function of daf-18/PTEN in the somatic gonad coordinates somatic gonad and germline development in C. elegans dauer larvae. Curr. Biol. 29, 1064–1072.e1068 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hong Y., Roy R., Ambros V., Developmental regulation of a cyclin-dependent kinase inhibitor controls postembryonic cell cycle progression in Caenorhabditis elegans. Development 125, 3585–3597 (1998). [DOI] [PubMed] [Google Scholar]
- 82.Karp X., Ambros V., Dauer larva quiescence alters the circuitry of microRNA pathways regulating cell fate progression in C. elegans. Development 139, 2177–2186 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.O’Keeffe C., Greenwald I., EGFR signal transduction is downregulated in C. elegans vulval precursor cells during dauer diapause. Development 149, dev201094 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Colella E., Li S., Roy R., Developmental and cell cycle quiescence is mediated by the nuclear hormone receptor coregulator DIN-1S in the Caenorhabditis elegans dauer larva. Genetics 203, 1763–1776 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Narbonne P., Roy R., Inhibition of germline proliferation during C. elegans dauer development requires PTEN, LKB1 and AMPK signalling. Development 133, 611–619 (2006). [DOI] [PubMed] [Google Scholar]
- 86.Wong C., Kadekar P., Jurczak E., Roy R., Germline stem cell integrity and quiescence are controlled by an AMPK-dependent neuronal trafficking pathway. PLoS Genet. 19, e1010716 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Derry W. B., et al. , Regulation of developmental rate and germ cell proliferation in Caenorhabditis elegans by the p53 gene network. Cell Death Differ. 14, 662–670 (2007). [DOI] [PubMed] [Google Scholar]
- 88.Vousden K. H., Ryan K. M., p53 and metabolism. Nat. Rev. Cancer 9, 691–700 (2009). [DOI] [PubMed] [Google Scholar]
- 89.Jones R. G., et al. , AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol. Cell 18, 283–293 (2005). [DOI] [PubMed] [Google Scholar]
- 90.Feng Y., Williams B. G., Koumanov F., Wolstenholme A. J., Holman G. D., FGT-1 is the major glucose transporter in C. elegans and is central to aging pathways. Biochem. J. 456, 219–229 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Schulz T. J., et al. , Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab. 6, 280–293 (2007). [DOI] [PubMed] [Google Scholar]
- 92.Walker M. D., et al. , WormPaths: Caenorhabditis elegans metabolic pathway annotation and visualization. Genetics 219, iyab089 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bargmann C. I., Horvitz H. R., Control of larval development by chemosensory neurons in Caenorhabditis elegans. Science 251, 1243–1246 (1991). [DOI] [PubMed] [Google Scholar]
- 94.Das S., Min S., Prahlad V., Gene bookmarking by the heat shock transcription factor programs the insulin-like signaling pathway. Mol. Cell 81, 4843–4860.e4848 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Das S., et al. , Serotonin signaling by maternal neurons upon stress ensures progeny survival. eLife 9, e55246 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tatum M. C., et al. , Neuronal serotonin release triggers the heat shock response in C. elegans in the absence of temperature increase. Curr. Biol. 25, 163–174 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ooi F. K., Prahlad V., Olfactory experience primes the heat shock transcription factor HSF-1 to enhance the expression of molecular chaperones in C. elegans. Sci. Signal 10, eaan4893 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Venkatesh H., Monje M., Neuronal activity in ontogeny and oncology. Trends Cancer 3, 89–112 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Curry R. N., Glasgow S. M., The role of neurodevelopmental pathways in brain tumors. Front. Cell Dev. Biol. 9, 659055 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Cruz J., Godthi A., Das S., Prahlad V., Neuronal IL-17 controls C. elegans developmental diapause through p53/CEP-1. Gene Expression Omnibus (GEO). https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE218596. Deposited 22 November 2022.
- 101.Cruz J., Godthi A., Prahlad V., Il-17 p53 Developmental Series. Gene Expression Omnibus (GEO). https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE229132. Deposited 6 April 2023.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Dataset S01 (XLSX)
Dataset S02 (XLSX)
Dataset S03 (XLSX)
Dataset S04 (XLSX)
Dataset S05 (XLSX)
Dataset S06 (XLSX)
Dataset S07 (XLSX)
Dataset S08 (XLSX)
Dataset S09 (XLSX)
Dataset S10 (XLSX)
Dataset S11 (XLSX)
Dataset S12 (XLSX)
Dataset S13 (XLSX)
Dataset S14 (XLSX)
Data Availability Statement
RNA-seq data have been deposited in NCBI’s Gene Expression Omnibus (GSE218596 and GSE229132) (100, 101). All other data are included in the manuscript and/or supporting information.