

Abstract
Objective
Alzheimer's disease neuropathologic change and alpha‐synucleinopathy commonly co‐exist and contribute to the clinical heterogeneity of dementia. Here, we examined tau epitopes marking various stages of tangle maturation to test the hypotheses that tau maturation is more strongly associated with beta‐amyloid compared to alpha‐synuclein, and within the context of mixed pathology, mature tau is linked to Alzheimer's disease clinical phenotype and negatively associated with Lewy body dementia.
Methods
We used digital histology to measure percent area‐occupied by pathology in cortical regions among individuals with pure Alzheimer's disease neuropathologic change, pure alpha‐synucleinopathy, and a co‐pathology group with both Alzheimer's and alpha‐synuclein pathologic diagnoses. Multiple tau monoclonal antibodies were used to detect early (AT8, MC1) and mature (TauC3) epitopes of tangle progression. We used linear/logistic regression to compare groups and test the association between pathologies and clinical features.
Results
There were lower levels of tau pathology (β = 1.86–2.96, p < 0.001) across all tau antibodies in the co‐pathology group compared to the pure Alzheimer's pathology group. Among individuals with alpha‐synucleinopathy, higher alpha‐synuclein was associated with greater early tau (AT8 β = 1.37, p < 0.001; MC1 β = 1.2, p < 0.001) but not mature tau (TauC3 p = 0.18), whereas mature tau was associated with beta‐amyloid (β = 0.21, p = 0.01). Finally, lower tau, particularly TauC3 pathology, was associated with lower frequency of both core clinical features and categorical clinical diagnosis of dementia with Lewy bodies.
Interpretation
Mature tau may be more closely related to beta‐amyloidosis than alpha‐synucleinopathy, and pathophysiological processes of tangle maturation may influence the clinical features of dementia in mixed Lewy‐Alzheimer's pathology.
Introduction
The neuropathologic diagnosis of Lewy body disease (LBD) is characterized by the accumulation of alpha‐synuclein (a‐syn) aggregates (Lewy bodies and neurites), 1 and Alzheimer's disease neuropathologic change (ADNC) is characterized by aggregates of beta‐amyloid plaques and neurofibrillary tau tangles. 2 Most ADNC patients (>50%) have co‐existing a‐syn pathology along with plaques and tangles. 3 Moreover, ~ >50% of individuals with LBD have a secondary neuropathological diagnosis of intermediate‐ or high‐level ADNC 4 (Fig. 1A). Tau pathology correlates strongly with several clinical features in LBD 5 , 6 , 7 ; conversely, co‐occurring a‐syn pathology and ADNC contributes to the heterogeneity of Alzheimer's disease (AD) dementia. 8 , 9 , 10
Figure 1.
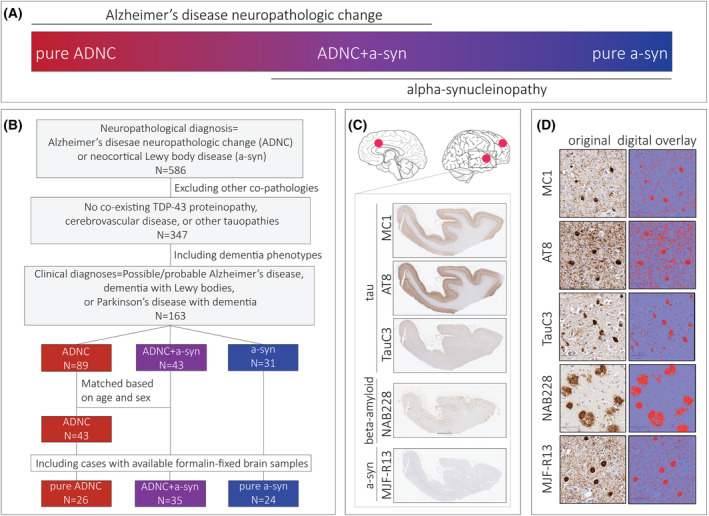
Study design. (A) The diagram illustrates the overlap between Alzheimer's disease neuropathologic change and alpha‐synucleinopathy groups in the study, (B) Flow diagram of inclusion and exclusion criteria for selection of participants, (C) Serial sections sampled from superior/middle temporal gyrus, middle frontal gyrus, and anterior cingulate gyrus were immunohistochemically stained for tau, beta‐amyloid, and a‐syn. (D) Digital overlay of percent area‐occupied (red) is shown for each stain.
Previous autopsy work finds correlation among a‐syn, tau, and beta‐amyloid pathologies in LBD 11 ; however, overall, there is lower tau burden in both in vivo PET studies 12 and postmortem studies 13 of LBD compared to AD. These autopsy studies mainly focused on phosphorylated tau reactivity which largely labels all forms of ADNC tau pathology; however, three stages of neurofibrillary tangle (NFT) maturation (i.e., pretangles, mature tangles, and ghost tangles) have been characterized that correspond to post‐translational modifications of tau (PTMs), recognized by specific monoclonal antibodies. 14 , 15 Thus, there is a clinicopathological spectrum of ADNC and a‐syn pathologies; however, the discrete pathological contributions of NFT maturation to the clinical features in the spectrum of AD and LBD are not clearly understood.
Previous in vivo and in vitro studies indicate mechanistic interactions between a‐syn, beta‐amyloid, and tau pathologies. 16 , 17 , 18 , 19 Indeed, a recent study by our group 17 identified a unidirectional interaction where a‐syn can influence aggregation and spread of tau pathology in a murine model, while tau had no significant effect on seeding and accumulation of a‐syn. Interestingly, cerebral beta‐amyloidosis can accelerate both tau and a‐syn pathology in vivo, suggesting complex synergistic interactions between beta‐amyloid, tau, and a‐syn at a molecular level 18 ; however, examining these interactions within the human brain presents challenges. The study of tau PTMs can give us insight into the temporal progression of tau pathology and cellular vulnerabilities to tau‐mediated neurodegeneration to further study molecular interactions of a‐syn and tau pathology in the human brain 20 and their contributions to clinical features of dementia.
In this study, we address these gaps by testing the hypothesis that advanced stages of tau maturation are more closely associated with beta‐amyloid than a‐syn and relate most closely to AD dementia than clinical features of dementia with Lewy bodies (DLB). We used a panel of antibodies preferentially reactive to different maturation stages of tau pathology with validated digital histology methods 21 in a unique autopsy cohort consisting of neuropathologically defined groups of pure ADNC, pure a‐syn, and a co‐pathology group of ADNC+a‐syn. We first tested the associations between pathologies across neuropathologically defined groups and then tested the association of tau conformational epitopes with clinical features of dementia. Our findings provide new insights into the role of tau maturation in the clinical expression of pathology that can inform future work to define biological subgroups of individuals with mixed ADNC and a‐syn pathology.
Methods
Participants
We focused our study to individuals with dementia and cortical pathology by selecting those with neuropathological diagnoses of ADNC and/or diffuse/neocortical stages of LBD from the Penn Integrated Neurodegenerative Disease Database (INDD). Individuals with any evidence of TDP‐43 co‐pathology, cerebrovascular disease (i.e., any evidence of gross brain infarcts or two or more microinfarcts 2 ), or other tauopathies on autopsy were excluded to avoid the confounding effect of co‐existing pathologies. Since the study is focused on dementia, only individuals with dementia phenotypes at death (i.e., possible/probable AD, 22 Parkinson's disease with dementia (PDD) 23 and DLB 24 ) were included. We found 163 individuals that met our pathological and clinical criteria in INDD. Next, we categorized participants into three groups based on neuropathological diagnoses and ADNC level 25 : those with no evidence of a‐syn pathology were categorized as pure ADNC, individuals with no to low level ADNC were categorized as pure a‐syn, and participants with intermediate‐high level ADNC were classified into the co‐pathology group (ADNC+a‐syn). We then selected a subgroup of ADNC individuals without any evidence of alpha‐synucleinopathy as a reference group (N = 43) that was matched to the ADNC+a‐syn group based on age at death and sex. Finally, we excluded individuals without tissue available from regions of interest (i.e., anterior cingulate cortex, superior/middle temporal cortex, and middle frontal cortex) at the Center for Neurodegenerative Disease Research (CNDR) brain bank to arrive at our final cohort (N = 85) (Fig. 1B). A summary of demographic, pathological, and clinical features of the final cohort is shown in Table 1.
Table 1.
Demographic and pathological characteristics.
| Pure ADNC (N = 26) | ADNC+a‐syn (N = 35) a | Pure a‐syn (N = 24) | |
|---|---|---|---|
| Sex [Male] | 22 | 28 | 22 |
| Age at onset b | 62.3 [11.01] | 65.26 [9.64] | 60.56 [6.97] |
| Age at death | 70.38 [10.85] | 75.2 [9.09] | 73.12 [6.28] |
| Disease duration b | 8.08 [4.14] | 10.6 [5.92] | 13.04 [5.81] |
| Dementia duration | 8.08 [4.14] | 7.54 [4.84] | 6.34 [3.62] |
| Motor dementia interval | −6.6 [5.12] c | 2.92 [6.42] | 5.78 [7.44] |
| Clinical diagnosis | |||
| Alzheimer's disease | 26 | 12 | |
| Parkinson's disease with dementia | 11 | 17 | |
| Dementia with Lewy bodies | 12 | 7 | |
| MMSE score d | 7.25 (N = 19) | 17.57 (N = 20) | 18 (N = 9) |
| Braak stage | |||
| B1 | 20 | ||
| B2 | 17 | 4 | |
| B3 | 26 | 18 | |
| Thal phase | |||
| A0 | 7 | ||
| A1 | 8 | ||
| A2 | 1 | 9 | 5 |
| A3 | 25 | 26 | 4 |
| CERAD score | |||
| C0 | 15 | ||
| C1 | 2 | 2 | |
| C2 | 6 | 3 | |
| C3 | 26 | 27 | 4 |
| Cerebral amyloid angiopathy | 21 | 30 | 7 |
| ARTAG e | 9 | 11 | 8 |
| APOE e4 allele carriers | 18 (69.2%) | 22 (62.8%) | 9 (37.5%) |
| GBA variant | 1 (2.8%) | 9 (37.5%) | |
Note: Mean [Standard deviation] reported for age of onset, age at death, dementia duration, and motor dementia interval.
There was one individual with Presenilin‐1 and one individual with LRRK2 mutation in the mixed ADNC+a‐syn group.
Age of onset was unavailable for one individual in the a‐syn and one individual in the ADNC+a‐syn group.
Five participants in the pure ADNC group developed motor features.
Average of MMSE score closest to autopsy reported. (N = 5 individuals have converted MoCA score; N = 37 missing data).
Nine participants have not been evaluated for age‐related tau astrogliopathy (ARTAG).
Neuropathologic assessment
Fresh brain tissue from autopsy was fixed in neutral buffered formalin overnight as previously reported. 21 Tissue was processed and embedded in paraffin for preparation of 6 μm sections for diagnostic evaluation at the CNDR 26 in accordance with published criteria. 1 , 2 All procedures were compliant with the University of Pennsylvania institutional review board.
Digital neuropathologic assessment
Adjacent tissue sections were immunohistochemically stained with MC1, AT8, TauC3, NAB228, and MJF‐R13 in the Penn Digital Neuropathology Lab according to the protocol described previously 27 (Fig. 1C, Table 2). We focused on MC1 and TauC3 as important markers of early and mature NFTs, respectively, 14 , 28 and included AT8 as a measure of pathology‐specific NFTs across a range of early‐to‐intermediate maturities. 29 MC1 detects a noncontinuous epitope of tau ranging from aa 7–9 and 312–322, 30 and reactivity is largely observed in diffuse threads and pretangles. TauC3 marks D421‐truncated tau that selectively reacts with mature intra‐ and extra‐cellular tangles and threads that co‐localize with amyloid‐binding dyes in AD. 15
Table 2.
List of antibodies.
| Antibody | Epitope | Species | Dilution | Antigen retrieval method | Source |
|---|---|---|---|---|---|
| AT8 (Phosphorylated tau) | Tau, Phospho S202, Phospho T205 | Mouse, monoclonal | 1:1 K | – | Thermo‐Fisher (#MN1020) |
| MC1 (Early tau conformation) | Tau, aa 7–9, and 312–322 | Mouse, monoclonal | 1:500 | – | Dr. Peter Davies |
| TauC3 (Mature Tau Conformation) | Tau, aa 412–421 | Mouse, monoclonal | 1:20 K | 88% Formic acid | Dr. Nicholas Kannan |
| MJF‐R13 (a‐syn) | α‐synuclein, Phospho S129 | Rabbit, monoclonal | 1:25 K | Proteinase K | Abcam (ab209421) |
| NAB228 (AB oligomers) | Beta‐amyloid, aa 1–11 | Mouse, monoclonal | 1:40 K | 88% Formic acid | CNDR |
Note: Epitopes, hosts, dilutions, and the method of antigen retrieval indicated for each antibody.
Stained histology slides were digitally scanned (Aperio AT2, Leica Biosystem, Wetzlar, Germany) at 20x magnification. Digital histology analysis was performed using QuPath software (version 0.2.3).
We applied a semi‐automated image analysis approach using a modified belt‐transect method to segment gray matter and measure percent area‐occupied (%AO) by pathology in histologic sections as described previously. 21 Figure 1D illustrates an example of digital overlay of positive pixel detection for each antibody.
Clinical assessment
We performed a structured review of clinical charts blinded to neuropathologic diagnosis to collect detailed clinical data relevant to this study. The clinical features collected consisted of core features associated with DLB 24 including presence or absence of cognitive fluctuations (i.e., mention of specific features indicative of cognitive fluctuations, 31 excessive daytime drowsiness, episodes of disorganized speech, or staring into space), recurrent visual hallucinations (i.e., spontaneous, well‐formed visual hallucinations in more than one clinical visit or confirmed recurrent by informant), rapid eye movement (REM) sleep behavior disorder (i.e., confirmed by polysomnogram or report of features strongly predictive of REM sleep behavior disorder (RBD) 32 including a clear history of dream enactment, repeated episodes of sleep‐related vocalization and/or complex motor behaviors), and parkinsonism (one or more of the cardinal features including resting tremor, bradykinesia, and rigidity) (Table S1).
If a clinical feature was not described in clinical notes, it was considered absent for the purpose of analysis. Nine participants in the ADNC+a‐syn group had missing clinical data, and two individuals with end‐stage clinical diagnosis of probable AD were found to have nonamnestic presentations of logopenic variant primary progressive aphasia or behavioral variant of frontotemporal dementia at onset and were excluded from the analyses that tested the association between pathology and clinical features. We used the available clinical data to confirm the clinical diagnosis of participants and test associations between pathology and relevant clinical features.
Additionally, Mini‐Mental State Examination (MMSE) scores within the 5 years from autopsy were collected from the INDD. For those who had available Montreal cognitive assessment scores near autopsy (N = 5), scores were converted to MMSE using a validated conversion method. 33
Statistical analysis
We used Kruskal–Wallis and pairwise Mann–Whitney‐Wilcoxon tests to compare demographic features across groups.
We averaged %AO measurements for each pathology from the three regions sampled to obtain a cortical average %AO value for analysis. Missing data from damaged/exhausted tissue were excluded from the cortical average for a given stain (missing data = 10% AT8, 11% MC1, 12% TauC3, 12% beta‐amyloid, 12% a‐syn sections). %AO values were natural log transformed to obtain a normal distribution. In order to log transform data points with %AO = 0 in our dataset, we added a constant value (%AO + 10−5) to all data prior to applying the log transform.
AD and LBD are clinicopathologic terms, and for clarity, we differentiate neuropathologic entities from clinical phenotypes for analyses. We use the term ADNC for neuropathologic diagnosis of beta‐amyloid and tau pathology, 2 a‐syn for neuropathologic diagnosis of alpha‐synucleinopathy, and ADNC+a‐syn for ADNC with concomitant alpha‐synucleinopathy. Since our groups are solely defined by neuropathologic diagnoses, the ADNC+a‐syn group includes individuals with clinical diagnoses of AD, PDD, and DLB. We use the terms AD 22 and LBD 34 (i.e., PDD 23 and DLB 24 ) to denote the clinical diagnoses associated with these pathologies.
Our main findings are presented in four sections:
In the first section of the results, we compare pathology %AO between pure and mixed pathology groups. Linear models with averaged cortical %AO as the dependent variable were used to examine group differences while covarying for age at death and sex. A sub‐analysis was performed for each model covarying for tau Braak stage (B0‐3) as a continuous variable.
The second section examines the relationship between pathologies among individuals within the ADNC and alpha‐synucleinopathy spectrum separately. Linear models covarying for age and sex tested the correlation between pathologies.
The third section compared %AO by pathology between clinical groups using similar linear models as in the first section with clinical groups as the independent variable. We also tested the association between pathology and presence/absence of specific clinical features in individuals with mixed ADNC+a‐syn pathology using logistic regression models with %AO as the independent variable and presence/absence of a clinical feature of DLB as the dependent variable.
Finally, we used data‐driven methods of clustering analyses with tau pathology %AO data in our total cohort to define clinically and biologically meaningful subgroups of individuals across AD and LBD to further inform our clinicopathologic findings. We used unsupervised hierarchical clustering to categorize individuals based on the averaged cortical %AO with MC1, AT8, and TauC3. One case with missing MC1 %AO was excluded from the analysis. D‐index and Hubert statistics indicated k = 3 as the optimal number of clusters. Additionally, we used the silhouette index to validate the overall clustering quality (Fig. S1).
All statistical analyses were performed using R statistical software (R version 4.2.2) and a statistical threshold of α < 0.05. All missing data are reported in tables and figures.
Results
Demographics and group characteristics
Characteristics of our groups are summarized in Table 1. Individuals with pure a‐syn were significantly younger at symptom onset compared to participants with ADNC+a‐syn (p = 0.017). There was no significant difference at age of death (p = 0.12) or dementia‐death interval (p = 0.29); however, individuals in the pure a‐syn group had a longer overall disease duration compared to the pure ADNC (p = 0.0007) and ADNC+a‐syn groups (p = 0.02). Participants in the ADNC+a‐syn group had a shorter motor dementia interval than the pure a‐syn group, but the difference was not significant (p = 0.1).
The latest MMSE score for the ADNC group was significantly lower compared to the ADNC+a‐syn group (p = 0.00004) and pure a‐syn group (p = 0.0004). Ten individuals carried a GBA mutation: 9 in the pure a‐syn group and one with ADNC co‐pathology.
Comparing pathology burdens between pure and mixed pathology groups
First, we compared the average cortical pathology burdens between ADNC+a‐syn and pure ADNC (Fig. 2). There was greater tau pathology in the pure ADNC group compared to the ADNC+a‐syn for all tau epitopes including MC1 (β = 2.82, SE = 0.58, p < 0.001), AT8 (β = 2.96, SE = 0.51, p < 0.001), and TauC3 (β = 1.86, SE = 0.37, p < 0.001). After controlling for tau Braak staging, this finding remained significant for all tau pathologies detected by MC1 (p = 0.02), AT8 (p = 0.002), and TauC3 (p = 0.003). However, ADNC+a‐syn and pure ADNC had similar levels of beta‐amyloid (p = 0.09) (Fig. 3A).
Figure 2.
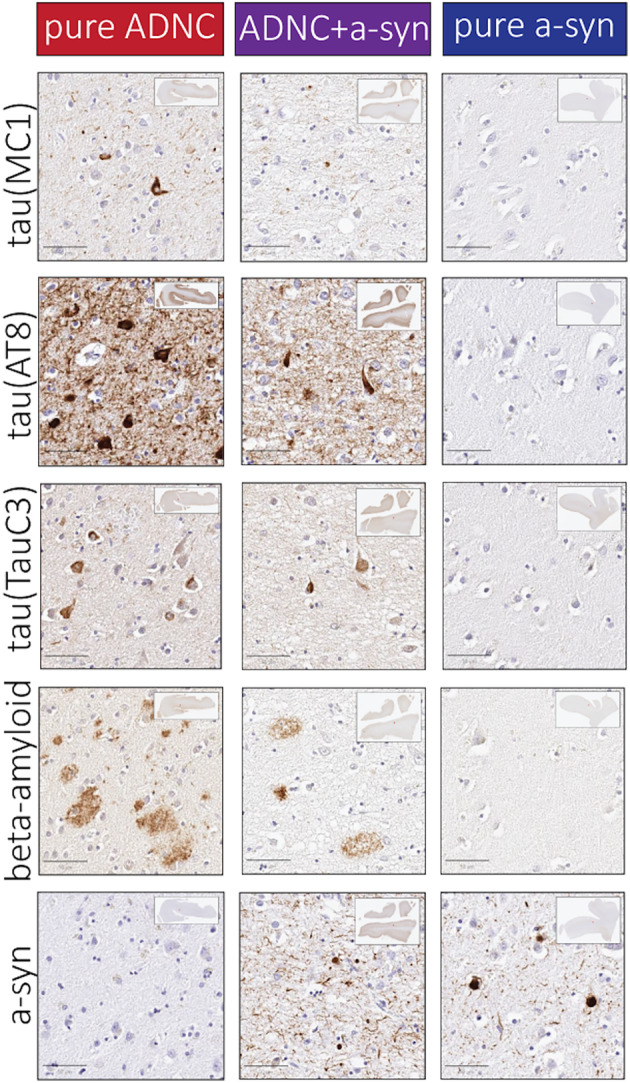
Representative photomicrographs. Photomicrographs from layer V in superior/middle temporal cortex are shown in individuals with either pure ADNC, ADNC+a‐syn, or pure a‐syn groups, respectively. Scale bar: 50 μm.
Figure 3.
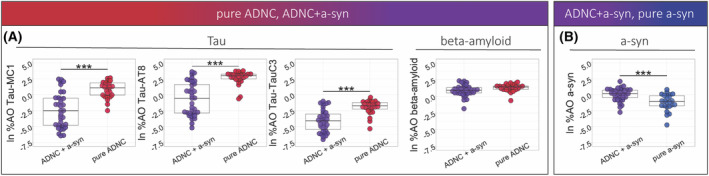
Tau, beta‐amyloid, and a‐syn pathology in pure and mixed pathology groups. (A) Percent area‐occupied (%AO) was compared between the ADNC+a‐syn and the pure ADNC group for beta‐amyloid and tau pathology marked with MC1, AT8, and TauC3, (B) Percent area‐occupied (%AO) was compared between the ADNC+a‐syn and the pure a‐syn group for a‐syn pathology, Color indicates pathologic groups, (***p‐value < 0.001, **p‐value < 0.01, *p‐value < 0.05).
Next, we compared pathology burdens for tau, beta‐amyloid, and a‐syn between groups of pure a‐syn and ADNC+a‐syn (Fig. 2). As expected, the pure a‐syn group showed lower MC1 (β = −3.29, SE = 0.58, p < 0.001), AT8 (β = −3.49, SE = 0.55, p < 0.001), TauC3 (β = −1.08, SE = 0.42, p = 0.014), and beta‐amyloid (β = −3.29, SE = 0.56, p < 0.001) %AO in comparison with the ADNC+a‐syn group. Furthermore, average cortical burden of a‐syn was lower in the pure a‐syn group in comparison with the ADNC+a‐syn (β = −1.28, SE = 0.3, p < 0.001) (Fig. 3B).
-
II
Association between tau, beta‐amyloid, and a‐syn pathologies
We tested the association between tau, beta‐amyloid, and a‐syn pathologies among individuals within the ADNC and alpha‐synucleinopathy spectrums (Fig. 4).
Figure 4.
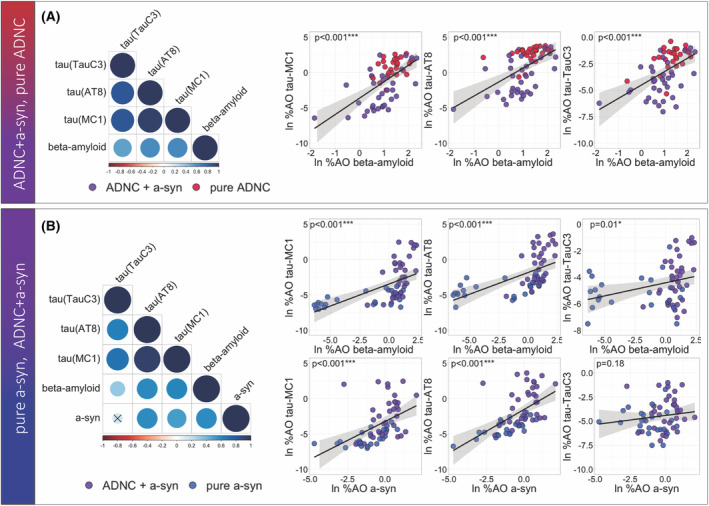
Tau, beta‐amyloid, and a‐syn pathology in ADNC spectrum and alpha‐synucleinopathies spectrum. (A) Correlation matrix and scatterplots show the correlation between tau (MC1, AT8, and TauC3) and beta‐amyloid pathologies in the ADNC spectrum (B) Correlation matrix and scatterplots show the correlation between tau (MC1, AT8, and TauC3), beta‐amyloid, and a‐syn pathologies in the alpha‐synucleinopathies spectrum, Color values for individual data points indicate pathologic group; correlation line is based on simple linear correlation; and p‐values were extracted from the multivariate models (***p‐value < 0.001, **p‐value < 0.01, *p‐value < 0.05).
Among individuals within the ADNC spectrum (i.e., pure ADNC and ADNC+a‐syn), there were positive correlations between beta‐amyloid and all tau epitopes of MC1 (β = 2.14, SE = 0.4, p < 0.001), AT8 (β = 1.65, SE = 0.38, p < 0.001), and TauC3 (β = 1.08, SE = 0.27, p < 0.001). Moreover, we found all tau pathologies correlated with each other (p < 0.001) (Fig. 4A).
Among individuals within the alpha‐synucleinopathy spectrum (i.e., pure a‐syn and ADNC+a‐syn), there was a positive association between beta‐amyloid pathology and all tau epitopes recognized with MC1 (β = 0.64, SE = 0.11, p < 0.001), AT8 (β = 0.63, SE = 0.11, p < 0.001), and TauC3 (β = 0.21, SE = 0.08, p = 0.013). Additionally, there were strong associations between a‐syn %AO and MC1 (β = 1.2, SE = 0.23, p < 0.001) and AT8 %AO (β = 1.37, SE = 0.21, p < 0.001), while there was no significant correlation between a‐syn %AO and TauC3 (p = 0.18). (Fig. 4B).
-
III
Pathological correlates of clinical symptoms associated with mixed ADNC+a‐syn
We focused on the mixed ADNC+a‐syn pathology group, where clinicopathologic diagnosis is most challenging, to examine the associations between pathology and clinical features. The comparisons of pathology burden between the subgroups with clinical syndromes of AD and LBD within the mixed ADNC+a‐syn pathology group suggested overall less tau pathology in the clinical LBD group compared to the clinical AD across all tau markers of MC1 (β = −2.06, SE = 0.87, p = 0.024), AT8 (β = −2.12, SE = 0.8, p = 0.013), and TauC3 (β = −1.23, SE = 0.58, p = 0.042). After controlling for AD tau Braak staging, there was no significant difference for tau %AO between the clinical groups of AD and LBD (p > 0.21). Additionally, the clinical LBD group showed lower burden of beta‐amyloid compared to the clinical AD group (β = −0.83, SE = 0.25, p = 0.003), while the two groups had similar levels of a‐syn pathology (p = 0.98) (Fig. 5A).
Figure 5.
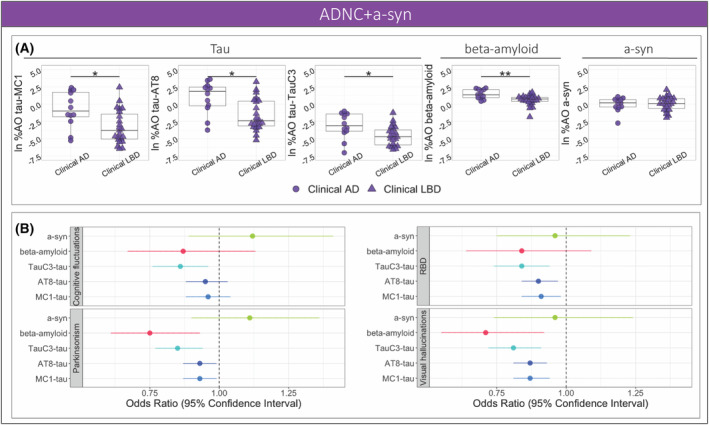
Clinical correlates of pathologies the ADNC+a‐syn group. (A) Percent area‐occupied (%AO) was compared between the clinical groups of AD and LBD with mixed ADNC+a‐syn pathology for MC1‐, AT8‐, and TauC3‐immunoreactive tau, beta‐amyloid, and a‐syn. (B) The forest plot illustrates the odds ratio and 95% confidence intervals for the association between pathologies and core clinical features of dementia with Lewy bodies based on logistic regression models; shape indicates clinical syndrome (***p‐value < 0.001, **p‐value < 0.01, *p‐value < 0.05).
To examine the effect of tau maturation on specific clinical outcomes, we performed logistic regression models to test the predictive value of pathology burden on the presence or absence of core features of DLB among individuals with ADNC+a‐syn (Table S2). Notably, TauC3 was the tau epitope with the most consistent and lowest odds ratio associated with all clinical DLB features studied. Beta‐amyloid %AO also had negative associations with clinical parkinsonism and visual hallucinations, while a‐syn %AO did not associate with clinical DLB features (Fig. 5B).
-
IV
Tau pathology in the full spectrum of ADNC, ADNC+a‐syn, and a‐syn
Finally, we studied clinical and genetic associations of tau pathology in the total cohort by performing a clustering analysis based on tau immunoreactivity recognized by MC1, AT8, and TauC3.
Using hierarchical clustering based on MC1, AT8, TauC3 %AO data, we identified three groups of individuals with varying amounts of tau pathology (Fig. 6A). The three clusters were characterized by overall low tau %AO in cluster 1 (N = 40), high MC1 %AO, high AT8 %AO, and low TauC3 %AO in cluster 2 (N = 13), and high MC1, AT8, and TauC3 %AO in cluster 3 (N = 31). Interestingly, the frequency of cluster groups reflected neuropathological categories, where the pure a‐syn group consisted exclusively with cluster 1 with low tau %AO for all markers, while the pure ADNC group was largely enriched (92%) with cluster 3 with high pathology for all three tau markers (Fig. 6B). Moreover, individuals with GBA mutation conferring risk of a‐syn were largely categorized in cluster 1 with low tau (N = 9/10).
Figure 6.
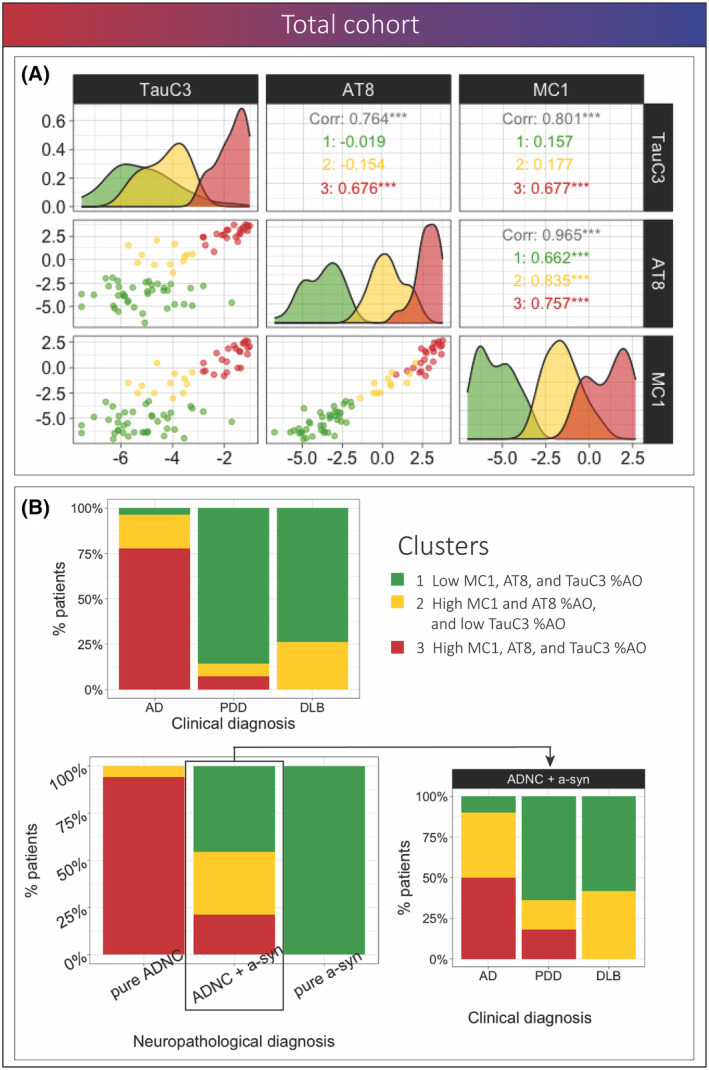
Clustering analysis using pathology data in the full cohort. (A) Scatterplot matrix shows the association between pathologies and distribution of clusters for each of the variables, the colors represent the clusters, and the values show the correlation between MC1‐, AT8‐, and TauC3‐tau within the total cohort and each cluster (B) Bar plots represent the percentage of individuals from each cluster within pathological groups and percentage of individuals from each cluster within each clinical subgroup among individuals in the total cohort and within the ADNC+a‐syn group; color indicates clusters.
The mixed ADNC+a‐syn group was composed of ~77% of individuals in clusters 1 or 2 with low mature tau (TauC3), and the remaining ~23% of the mixed pathology group was classified into cluster 3 with high tau %AO for all three markers.
Since the pure ADNC and pure a‐syn groups consisted of homogenous clinical syndromes of clinical AD and LBD, respectively, we examined the clinical syndrome associations within the mixed ADNC+a‐syn group which had heterogenous clinical presentations across AD, PDD, and DLB (Fig. 6B); those with DLB phenotype were solely distributed in cluster 1 and 2, demonstrating a conspicuous absence of mature TauC3 immunoreactive pathology in clinical DLB. Moreover, greater than 80% of the PDD group with mixed pathology were categorized in cluster 1 or 2 with low TauC3 as well. In contrast, over half of the participants with clinical AD in the mixed pathology group were in the cluster 3 with high TauC3 (Fig. 6B).
Discussion
Here, we comprehensively examined the inter‐relationships and clinical influence of a‐syn, beta‐amyloid, and three tau PTMs marking various maturation stages of tau pathology in a unique cohort of autopsy‐confirmed individuals within the clinicopathological spectrum of AD and LBD. We report several important findings relevant to the biological contributions to clinical expressions in individuals with mixed ADNC and a‐syn pathology. First, we find a positive correlation between early tau epitopes and a‐syn among individuals with alpha‐synucleinopathies. In contrast, TauC3, which is selective for mature tangles, was exclusively associated with beta‐amyloid and not a‐syn. Furthermore, using both clinical subgroup comparisons and data‐driven methods, we found TauC3 had a strong, consistent negative association with all core clinical features of DLB in individuals with mixed ADNC+a‐syn pathology. Moreover, participants with clinical DLB and those with GBA mutations associated with alpha‐synucleinopathy risk, had minimal mature tau pathology in the cortex measured by TauC3, compared to clinical AD with mixed pathology. These data suggest that tau tangle maturation in the cortex is influential to the clinical expression of dementia in individuals with mixed ADNC+a‐syn and may mark a clinically meaningful biological subgroup of individuals with mixed pathology driven by ADNC‐related pathophysiology.
In our pathological group comparisons between pure ADNC and mixed ADNC+a‐syn, we found higher tau pathology for all three markers in pure ADNC (Fig. 3A). This is consistent with previous studies, which found the overall burden of total tau pathology is significantly lower in individuals with mixed ADNC+a‐syn compared to pure Alzheimer's disease. 13 , 35 One interpretation could be that with mixed pathology, there are added functional impairments 8 causing earlier death prior to the full maturation of tau observed in individuals with pure ADNC. Indeed, pure ADNC participants had lower MMSE scores near death (Table 1) suggesting the cortical neurodegenerative process is more advanced compared to individuals with ADNC+a‐syn.
Furthermore, consistent with previous reports, 5 , 36 , 37 we found higher a‐syn pathology in mixed ADNC+a‐syn compared to pure a‐syn, suggesting a‐syn pathology may be potentiated by ADNC among individuals with alpha‐synucleinopathy. The combined observations of greater tau in pure ADNC vs mixed ADNC+a‐syn (Fig. 3A) and greater a‐syn in mixed ADNC+a‐syn vs pure a‐syn groups (Fig. 3B) may appear paradoxical. However, this phenomenon could be explained by considering the possibility of a differential temporal sequence of synergistic interactions between these pathologies. Indeed, previous research in model systems find directional relationships between tau, beta‐amyloid, and a‐syn, where a‐syn may induce tau formation but tau has limited capacity to induce a‐syn aggregation, 17 which may inform our observations. There are model data suggestive of molecular interactions between a‐syn and tau 18 that are reflective of postmortem correlations of both pathologies in LBD brains. 17 , 38 , 39 Moreover, several in vivo and in vitro studies support a synergistic interaction between α‐syn and beta‐amyloid. 19 , 40 Some data suggest beta‐amyloid42 induces phosphorylation of a‐syn and beta‐amyloid can promote a‐syn pathology by modulating protein clearance. 19 Additionally, postmortem human studies find a‐syn co‐pathology is common in both sporadic and familial ADNC, 3 , 41 and beta‐amyloid pathology is associated with higher a‐syn aggregations in LBD. 11 , 36 The combination of these findings with our observations of different patterns of associations between tau and a‐syn across the ADNC spectrum and alpha‐synucleinopathies spectrum could potentially further support the notion of distinct temporal sequences for synergistic interactions between these pathologies. Future work employing in vivo seeding aggregation assays for a‐syn, 42 in conjunction with AD biomarkers and postmortem examination, could potentially offer a more comprehensive understanding of the chronological sequence of these pathologies in individuals within the AD‐LBD spectrum.
Here, among individuals with alpha‐synucleinopathy, we found positive correlations between all pathologies measured, with the exception of the lack of association between mature TauC3 and a‐syn %AO (Fig. 4B). The finding of positive correlation between a‐syn and phosphorylated tau is supportive of previous research that have revealed close association between tau and a‐syn. 17 , 38 , 39 Additionally, we add new data including the observation of this association of a‐syn with early conformational MC1 epitope. Interestingly, while we found no significant association between TauC3 %AO and a‐syn, TauC3 pathology correlated with beta‐amyloid, suggesting tau maturation may be more closely linked to AD pathophysiology than alpha‐synucleinopathy. One previous study found colocalization of D421‐truncated tau within Lewy bodies in LBD. 43 Thus, we cannot entirely exclude a synergistic interaction of D421 truncation of tau induced by alpha‐synucleinopathy at a molecular level; however overall, in our cortical sampling of cellular pathology across the spectrum of ADNC and alpha‐synucleinopathies, we did not detect an association between the burden of TauC3 and a‐syn pathology. This observation is further supported by our clinicopathological correlations and clustering approaches below, which highlights strong clinical and genetic associations of mature TauC3 pathology.
Examination of clinical correlates in the mixed ADNC+a‐syn pathology group with heterogenous clinical phenotypes found lower tau and beta‐amyloid pathology in clinical LBD compared to clinical AD, while the two groups had strikingly similar levels of a‐syn pathology (Fig. 5A). This finding suggests that tau and beta‐amyloid pathology might be the primary factors driving the clinical phenotype of AD among individuals with alpha‐synucleinopathy, regardless of the levels of a‐syn. Nevertheless, it is important to acknowledge that the variances in tau levels between the clinical AD and LBD groups may also be attributable to lower tau Braak stages. Indeed, it is difficult to differentiate the effects of topographical spread and tau maturity since both are highly correlated. 44 However, beyond categorical designation of AD vs LBD clinical syndromes, we also found differential associations of pathology with discrete clinical features of LBD. Overall, lower levels of tau, particularly low TauC3 %AO, were associated with a lower likelihood of clinical core features of DLB (Fig. 5B). Using the entire cohort, we also found additional data to support a strong clinical influence of mature TauC3 reactive NFTs using a data‐driven approach. Our cluster analysis based on tau antibody reactivity data found that the high tau cluster (i.e., high %AO for MC1, AT8, and TauC3) was enriched for clinical AD and lacked clinical DLB. Moreover, GBA mutation carriers were almost exclusively in the low TauC3 clusters. Taken together, these data suggest that mature NFT pathology may be a beta‐amyloid‐driven process that strongly influences the phenotypic presentation of dementia with mixed ADNC+a‐syn pathology.
Our findings are consistent with pathological diagnostic criteria 25 , 45 suggesting higher ADNC pathology lowers diagnostic accuracy of DLB, 46 and a recent in vivo biomarker work finding a lower frequency of DLB clinical features associated with tau positivity. 47 , 48 Importantly, we find new data suggesting that mature cortical tau marked by TauC3 is a relatively specific marker of clinical AD in individuals with mixed tau and a‐syn pathology. Current biomarkers cannot distinguish between morphological forms of tau pathology in living individuals and future work is needed to confirm these findings over the course of disease when in vivo biomarkers for mature vs early tau are developed.
We used a unique strategy to select our participants for study across the spectrum of AD and LBD (Fig. 1B) and analyzed over 1100 postmortem sections to measure mixed pathologies and tau conformations comprehensively and objectively with validated digital histopathologic methods, which facilitated our novel computational analyses. However, our study has several limitations. Due to our focus on relatively “pure” contributions of tau, beta‐amyloid, and a‐syn, we excluded individuals with other age‐related co‐pathologies such as TDP‐43 pathology which are important contributors to age‐related dementia. 49 We focused on cortical pathology and dementia syndromes, but future larger‐scale studies with longitudinal neuropsychological testing can more comprehensively model these interactions by also including subcortical and limbic regions, as well as individuals with brainstem and amygdala/limbic‐only alpha‐synucleinopathy. Moreover, considering the missing clinical data and the biases resulting from retrospective chart data rather than prospective method of clinical data collection, it is important to acknowledge the potential impact of these limitations on the interpretation of the results. Finally, since a‐syn pathology may not persist after neurons are degenerated compared to “ghost pathology” in tau NFTs, we cannot rule out an influence of neuron loss on our a‐syn %AO measurements and future studies will more comprehensively model a‐syn pathology with digital measures of neuronal density as these tools are developed.
Despite these limitations, we find novel evidence from human brain histopathology data to suggest there is a biological subgroup of mixed ADNC+a‐syn pathology marked by high levels of cortical mature NFTs and a relative absence of DLB clinical features and genetic risk. These data can guide future efforts to further refine the biological contributions of mixed pathologies to aging and the clinical expression of mixed pathologies across AD and LBD.
Author Contributions
The authors confirm contribution to the paper as follows: Study conception and design: SA, MG, DJI; Acquisition and analysis of data: SA, ML, MC, KAQC, EBL, DJI; Drafting manuscript: SA, KAQC, DTO, MC, JG, JSP, CTM, KCL, AD, MAS, TFT, DW, DAW, VL, ASC, EBL, DJI. All authors reviewed the results and approved the final version of the manuscript.
Conflict of Interest Statement
The authors have no conflict of interest to declare.
Supporting information
Data S1.
Acknowledgements
We would like to thank Theresa Schuck and John Robinson for their assistance in autopsy coordination. We are grateful to Dr. Nicholas Kanaan for providing TauC3 antibody. We particularly appreciate individuals and families whose participation in research and brain donation made this work possible. We would like to acknowledge the late Dr. John Trojanowski and Dr. Murray Grossman who have made enormous personal and societal impacts on improving the diagnosis and treatment of neurodegenerative disease. Grants: U19AG062418‐01A1, R01‐NS‐109260, P01‐AG‐066597, P30‐AG‐072979, Penn IOA, and The Barnstone Foundation.
Funding Statement
This work was funded by Barnstone Foundation; Institute on Aging, University of Pennsylvania ; National Institute of Health grants P01‐AG‐066597, P30‐AG‐072979, R01‐NS‐109260, and U19AG062418‐01A1.
Data Availability Statement
The corresponding author can provide the data supporting the findings of this study upon request.
References
- 1. Attems J, Toledo JB, Walker L, et al. Neuropathological consensus criteria for the evaluation of Lewy pathology in post‐mortem brains: a multi‐centre study. Acta Neuropathol (Berl). 2021;141:159‐172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hyman BT, Phelps CH, Beach TG, et al. National Institute on aging‐Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease. Alzheimers Dement J Alzheimers Assoc. 2012;8:1‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hamilton RL. Lewy bodies in Alzheimer's disease: a neuropathological review of 145 cases using alpha‐synuclein immunohistochemistry. Brain Pathol Zurich Switz. 2000;10:378‐384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Irwin DJ, Lee VM‐Y, Trojanowski JQ. Parkinson's disease dementia: convergence of α‐synuclein, tau and amyloid‐β pathologies. Nat Rev Neurosci. 2013;14:626‐636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Irwin DJ, Grossman M, Weintraub D, et al. Neuropathological and genetic correlates of survival and dementia onset in synucleinopathies: a retrospective analysis. Lancet Neurol. 2017;16:55‐65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Coughlin D, Xie SX, Liang M, et al. Cognitive and pathological influences of tau pathology in Lewy body disorders. Ann Neurol. 2019;85:259‐271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Coughlin DG, Hurtig HI, Irwin DJ. Pathological influences on clinical heterogeneity in Lewy body diseases. Mov Disord. 2020;35:5‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chung EJ, Babulal GM, Monsell SE, Cairns NJ, Roe CM, Morris JC. Clinical features of Alzheimer disease with and without Lewy bodies. JAMA Neurol. 2015;72:789‐796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Connor DJ, Salmon DP, Sandy TJ, Galasko D, Hansen LA, Thal LJ. Cognitive profiles of autopsy‐confirmed Lewy body variant vs pure Alzheimer disease. Arch Neurol. 1998;55:994‐1000. [DOI] [PubMed] [Google Scholar]
- 10. Olichney JM, Galasko D, Salmon DP, et al. Cognitive decline is faster in Lewy body variant than in Alzheimer's disease. Neurology. 1998;51:351‐357. [DOI] [PubMed] [Google Scholar]
- 11. Miller RL, Dhavale DD, O'Shea JY, et al. Quantifying regional α ‐synuclein, amyloid β, and tau accumulation in Lewy body dementia. Ann Clin Transl Neurol. 2022;9:106‐121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kantarci K, Lowe VJ, Boeve BF, et al. AV‐1451 tau and β‐amyloid positron emission tomography imaging in dementia with Lewy bodies. Ann Neurol. 2017;81:58‐67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hansen LA, Samuel W. Criteria for Alzheimer's disease and the nosology of dementia with lewy bodies. Neurology. 1997;48:126‐132. [DOI] [PubMed] [Google Scholar]
- 14. Moloney CM, Lowe VJ, Murray ME. Visualization of neurofibrillary tangle maturity in Alzheimer's disease: a clinicopathologic perspective for biomarker research. Alzheimers Dement J Alzheimers Assoc. 2021;17:1554‐1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Binder LI, Guillozet‐Bongaarts AL, Garcia‐Sierra F, Berry RW. Tau, tangles, and Alzheimer's disease. Biochim Biophys Acta. 2005;1739:216‐223. [DOI] [PubMed] [Google Scholar]
- 16. Giasson BI, Forman MS, Higuchi M, et al. Initiation and synergistic fibrillization of tau and alpha‐synuclein. Science. 2003;300:636‐640. [DOI] [PubMed] [Google Scholar]
- 17. Bassil F, Meymand ES, Brown HJ, et al. α‐Synuclein modulates tau spreading in mouse brains. J Exp Med. 2021;218:e20192193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Clinton LK, Blurton‐Jones M, Myczek K, Trojanowski JQ, LaFerla FM. Synergistic interactions between Abeta, tau, and alpha‐synuclein: acceleration of neuropathology and cognitive decline. J Neurosci. 2010;30:7281‐7289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Marsh SE, Blurton‐Jones M. Examining the mechanisms that link β‐amyloid and α‐synuclein pathologies. Alzheimers Res Ther. 2012;4:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Arezoumandan S, Xie SX, Cousins KAQ, et al. Regional distribution and maturation of tau pathology among phenotypic variants of Alzheimer's disease. Acta Neuropathol (Berl). 2022;144:1103‐1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Irwin DJ, Byrne MD, McMillan CT, et al. Semi‐automated digital image analysis of Pick's disease and TDP‐43 Proteinopathy. J Histochem Cytochem. 2016;64:54‐66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement J Alzheimers Assoc. 2011;7:263‐269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Emre M, Aarsland D, Brown R, et al. Clinical diagnostic criteria for dementia associated with Parkinson's disease. Mov Disord. 2007;22:1689‐1707; quiz 1837. [DOI] [PubMed] [Google Scholar]
- 24. McKeith IG, Boeve BF, Dickson DW, et al. Diagnosis and management of dementia with Lewy bodies: fourth consensus report of the DLB consortium. Neurology. 2017;89:88‐100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Montine TJ, Phelps CH, Beach TG, et al. National Institute on Aging‐Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathol (Berl). 2012;123:1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Toledo JB, Van Deerlin VM, Lee EB, et al. A platform for discovery: the University of Pennsylvania Integrated Neurodegenerative Disease Biobank. Alzheimers Dement J Alzheimers Assoc. 2014;10:477‐484.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Coughlin DG, Coslett HB, Peterson C, et al. Lateralized ante mortem and post mortem pathology in a case of Lewy body disease with corticobasal syndrome. Alzheimers Dement N Y N. 2022;8:e12294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Guillozet‐Bongaarts AL, Garcia‐Sierra F, Reynolds MR, et al. Tau truncation during neurofibrillary tangle evolution in Alzheimer's disease. Neurobiol Aging. 2005;26:1015‐1022. [DOI] [PubMed] [Google Scholar]
- 29. Mercken M, Vandermeeren M, Lübke U, et al. Monoclonal antibodies with selective specificity for Alzheimer tau are directed against phosphatase‐sensitive epitopes. Acta Neuropathol (Berl). 1992;84:265‐272. [DOI] [PubMed] [Google Scholar]
- 30. Jicha GA, Bowser R, Kazam IG, Davies P. Alz‐50 and MC‐1, a new monoclonal antibody raised to paired helical filaments, recognize conformational epitopes on recombinant tau. J Neurosci Res. 1997;48:128‐132. [DOI] [PubMed] [Google Scholar]
- 31. Ferman TJ, Smith GE, Boeve BF, et al. DLB fluctuations: specific features that reliably differentiate DLB from AD and normal aging. Neurology. 2004;62:181‐187. [DOI] [PubMed] [Google Scholar]
- 32. Boeve BF, Molano JR, Ferman TJ, et al. Validation of the Mayo sleep questionnaire to screen for REM sleep behavior disorder in an aging and dementia cohort. Sleep Med. 2011;12:445‐453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. van Steenoven I, Aarsland D, Hurtig H, et al. Conversion between mini‐mental state examination, Montreal cognitive assessment, and dementia rating scale‐2 scores in Parkinson's disease. Mov Disord. 2014;29:1809‐1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lippa CF, Duda JE, Grossman M, et al. DLB and PDD boundary issues: diagnosis, treatment, molecular pathology, and biomarkers. Neurology. 2007;68:812‐819. [DOI] [PubMed] [Google Scholar]
- 35. Gearing M, Lynn M, Mirra SS. Neurofibrillary pathology in Alzheimer disease with lewy bodies: two subgroups. Arch Neurol. 1999;56:203‐208. [DOI] [PubMed] [Google Scholar]
- 36. Halliday GM, Holton JL, Revesz T, Dickson DW. Neuropathology underlying clinical variability in patients with synucleinopathies. Acta Neuropathol (Berl). 2011;122:187‐204. [DOI] [PubMed] [Google Scholar]
- 37. Jellinger KA, Attems J. Prevalence and impact of vascular and Alzheimer pathologies in Lewy body disease. Acta Neuropathol (Berl). 2008;115:427‐436. [DOI] [PubMed] [Google Scholar]
- 38. Lashley T, Holton JL, Gray E, et al. Cortical alpha‐synuclein load is associated with amyloid‐beta plaque burden in a subset of Parkinson's disease patients. Acta Neuropathol (Berl). 2008;115:417‐425. [DOI] [PubMed] [Google Scholar]
- 39. Lee VM‐Y, Giasson BI, Trojanowski JQ. More than just two peas in a pod: common amyloidogenic properties of tau and alpha‐synuclein in neurodegenerative diseases. Trends Neurosci. 2004;27:129‐134. [DOI] [PubMed] [Google Scholar]
- 40. Bassil F, Brown HJ, Pattabhiraman S, et al. Amyloid‐Beta (Aβ) plaques promote seeding and spreading of alpha‐synuclein and tau in a mouse model of Lewy body disorders with Aβ pathology. Neuron. 2020;105:260‐275.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Leverenz JB, Fishel MA, Peskind ER, et al. Lewy body pathology in familial Alzheimer disease: evidence for disease‐ and mutation‐specific pathologic phenotype. Arch Neurol. 2006;63:370‐376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Arnold MR, Coughlin DG, Brumbach BH, et al. α‐Synuclein seed amplification in CSF and brain from patients with different brain distributions of pathological α‐synuclein in the context of Co‐pathology and non‐LBD diagnoses. Ann Neurol. 2022;92:650‐662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Newman J, Rissman RA, Sarsoza F, et al. Caspase‐cleaved tau accumulation in neurodegenerative diseases associated with tau and alpha‐synuclein pathology. Acta Neuropathol (Berl). 2005;110:135‐144. [DOI] [PubMed] [Google Scholar]
- 44. Basurto‐Islas G, Luna‐Muñoz J, Guillozet‐Bongaarts AL, Binder LI, Mena R, García‐Sierra F. Accumulation of aspartic acid421‐ and glutamic acid391‐cleaved tau in neurofibrillary tangles correlates with progression in Alzheimer disease. J Neuropathol Exp Neurol. 2008;67:470‐483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. McKeith IG. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the consortium on DLB international workshop. J Alzheimers Dis. 2006;9:417‐423. [DOI] [PubMed] [Google Scholar]
- 46. Chin KS, Yassi N, Churilov L, Masters CL, Watson R. Prevalence and clinical associations of tau in Lewy body dementias: a systematic review and meta‐analysis. Parkinsonism Relat Disord. 2020;80:184‐193. [DOI] [PubMed] [Google Scholar]
- 47. Ferreira D, Przybelski SA, Lesnick TG, et al. β‐Amyloid and tau biomarkers and clinical phenotype in dementia with lewy bodies. Neurology. 2020;95:e3257‐e3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hansen L, Salmon D, Galasko D, et al. The Lewy body variant of Alzheimer's disease: a clinical and pathologic entity. Neurology. 1990;40:1‐8. [DOI] [PubMed] [Google Scholar]
- 49. Schneider JA, Arvanitakis Z, Bang W, Bennett DA. Mixed brain pathologies account for most dementia cases in community‐dwelling older persons. Neurology. 2007;69:2197‐2204. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1.
Data Availability Statement
The corresponding author can provide the data supporting the findings of this study upon request.