

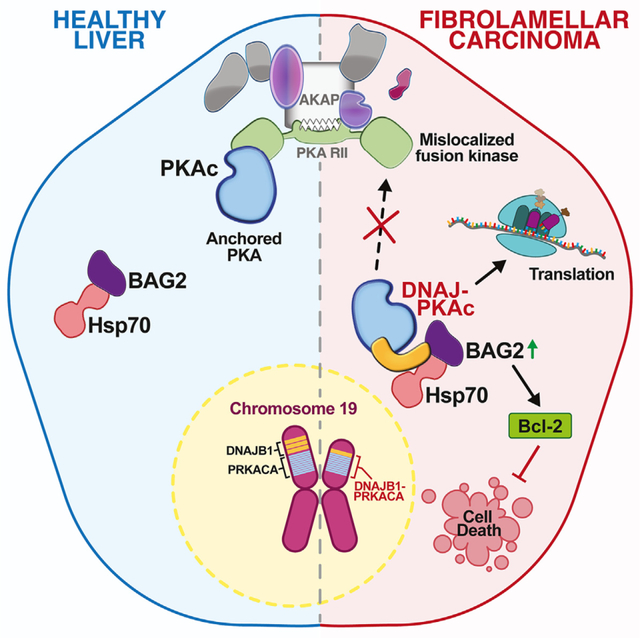
SUMMARY
The DNAJ-PKAc fusion kinase is a defining feature of fibrolamellar carcinoma (FLC). FLC tumors are notoriously resistant to standard chemotherapies, with aberrant kinase activity assumed to be a contributing factor. By combining proximity proteomics, biochemical analyses, and live-cell photoactivation microscopy, we demonstrate that DNAJ-PKAc is not constrained by A-kinase anchoring proteins. Consequently, the fusion kinase phosphorylates a unique array of substrates, including proteins involved in translation and the anti-apoptotic factor Bcl-2-associated athanogene 2 (BAG2), a co-chaperone recruited to the fusion kinase through association with Hsp70. Tissue samples from patients with FLC exhibit increased levels of BAG2 in primary and metastatic tumors. Furthermore, drug studies implicate the DNAJ-PKAc/Hsp70/BAG2 axis in potentiating chemotherapeutic resistance. We find that the Bcl-2 inhibitor navitoclax enhances sensitivity to etoposide-induced apoptosis in cells expressing DNAJ-PKAc. Thus, our work indicates BAG2 as a marker for advanced FLC and a chemotherapeutic resistance factor in DNAJ-PKAc signaling scaffolds.
In brief
Lauer et al. define molecular mechanisms of chemotherapeutic resistance that impede treatment of the adolescent liver cancer fibrolamellar carcinoma (FLC). Their studies implicate the athanogene BAG2 as a marker for advanced FLC and a chemotherapeutic resistance factor in DNAJ-PKAc fusion kinase signaling scaffolds.
Graphical Abstract
INTRODUCTION
Rare tumors represent approximately 20% of total cancer incidence.1 Gene fusions that involve protein kinases represent a broad class of oncogenes that drive a diverse set of human hematologic and solid tumors.2 Expression of a DNAJ-PKAc fusion kinase is an emblematic feature of a rare form of liver cancer known as fibrolamellar carcinoma (FLC).3 Approximately 500 new cases of FLC are diagnosed in the United States each year, typically in young adults between the ages of 15 and 35 with no history of risk factors for liver disease.4 Five-year survival rates for these patients range between 40% and 60%, with markedly worse outcomes for patients diagnosed with metastatic disease.5 FLC tumors are refractory to standard chemotherapies and radiation treatments.6,7 Chemotherapy often fails because tumor cells develop resistance to apoptosis, which leads to increased cancer invasion and progression to metastasis.8 Thus, understanding the molecular mechanisms of chemotherapy resistance in FLC represents an important line of investigation that may have bearing on other cancers.
The DNAJ-PKAc fusion kinase is detected in more than 90% of patients with FLC,3,9 with a few cases also bearing a PKA-RIα deletion.10 Exome sequencing reveals a 400-kb deletion in chromosome 19 that results in translation of this fusion kinase, in which the chaperone-binding domain of Hsp40 is fused in frame to exons 2–10 of the catalytic subunit of protein kinase A (PKAc).6,9,11 Structural studies show that DNAJ-PKAc adopts the conformation of an active kinase with an operative J domain.12,13 While overwhelming evidence links this chimeric kinase to FLC, the molecular mechanisms that underlie DNAJ-PKAc action are unclear.3,7,14 Identifying the pathological gains of function imparted by aberrant kinase activity or a new spectrum of protein-protein interactions that proceed through the DNAJ domain is important for understanding the signaling defects that occur in FLC. More recently DNAJ-PKAc chimeras have been described in cholangiocarcinoma, hepatocellular carcinoma (HCC), and oncocytic biliary tumors.15–17 Discovering druggable effectors and binding partners that function downstream of DNAJ-PKAc is a new strategy in the development of effective therapeutics for FLC and other rare cancers.
Compartmentalization through association with A-kinase anchoring proteins (AKAPs) is a mechanism that imparts specificity to PKA signaling.18,19 AKAPs also integrate subcellular signals from other kinases and effector enzymes at sites proximal to their substrates.20–23 Signaling through AKAPs limits the range and duration of information relay within cells.24–26 In this report, we show that DNAJ-PKAc preferentially interacts with a particular spectrum of binding partners. Provocative new findings here suggest that DNAJ-PKAc is largely excluded from AKAP signaling islands, thereby disrupting subcellular distribution of the enzyme. We find that substrate recognition by the chimeric kinase is unaffected. However, displacement from AKAP signaling islands affords the fusion enzyme access to aberrant substrates in cellular compartments not usually occupied by wild-type PKAc. One of these proteins is Bcl-2-associated athanogene 2 (BAG2), a co-chaperone that is recruited to the fusion kinase through association with Hsp70. BAG2 has been linked to poor prognosis and resistance to chemotherapeutic agents in certain cancers.27–29 Pharmacological studies with clinically relevant drugs implicate a pro-survival function of BAG2 at DNAJ-PKAc scaffolds and confirm a synergistic effect of etoposide and navitoclax on enhancing drug-induced cell death through Bcl-2 inhibition in cellular models of FLC.
RESULTS
Enzyme-catalyzed proximity labeling identifies molecular associations with DNAJ-PKAc
Little is known regarding the molecular mechanism of DNAJ-PKAc in FLC pathogenesis. We first used enzyme-catalyzed proximity labeling to identify the interacting partners and protein components involved in aberrant DNAJ-PKAc complexes (Figure 1A). AML12 hepatocytes expressing PKAc variants tagged with the biotin ligase miniTurbo were generated using lentiviral transduction.19,30,31 Single-cell clones were selected, grown in individual wells of 48-well plates, and then screened for expression of the PKAc-miniTurbo variants using immunofluorescence staining (Figure 1B). Doxycycline-inducible expression of the miniTurbo-fused bait proteins was adjusted to physiologically relevant levels of PKAc variants within the cells (Figure 1C, bottom). Neutravidin-horseradish peroxidase (HRP) detection of biotinylated proteins demonstrates strong labeling for both cell lines after live incubation with biotin for 2 h (Figure 1C, top). Two clones each of DNAJ-PKAc- and wild-type (WT) PKAc-miniTurbo-expressing cell lines were selected to proceed with proximity labeling and affinity purification-mass spectrometry. Quantitative analysis of the resulting liquid chromatography-tandem mass spectrometry (LC-MS/MS) data identified a total of 1,174 proteins with 261 significant hits (t test, p < 0.05). Proteins with a log2 fold change greater than 1.5 versus WT PKAc were labeled as either less proximal (Figure 1D, red dots) or more proximal (Figure 1D, blue dots) to the DNAJ-PKAc fusion. STRING analysis was used to create a network for proteins exhibiting greater association with DNAJ-PKAc. These include the co-chaperone proteins stress-induced phosphoprotein 1 (STIP1) and BAG2 (Figure 1E).32 Reactome pathway analysis of the DNAJ-PKAc proximitome using Enrichr revealed enrichment of pathways associated with mRNA processing and metabolism as well as pathways associated with collagen biosynthesis and cytoskeletal remodeling that are involved in fibrosis and metastasis, respectively (Figure 1F).33–38
Figure 1. Enzyme-catalyzed proximity labeling identifies molecular associations with DNAJ-PKAc.
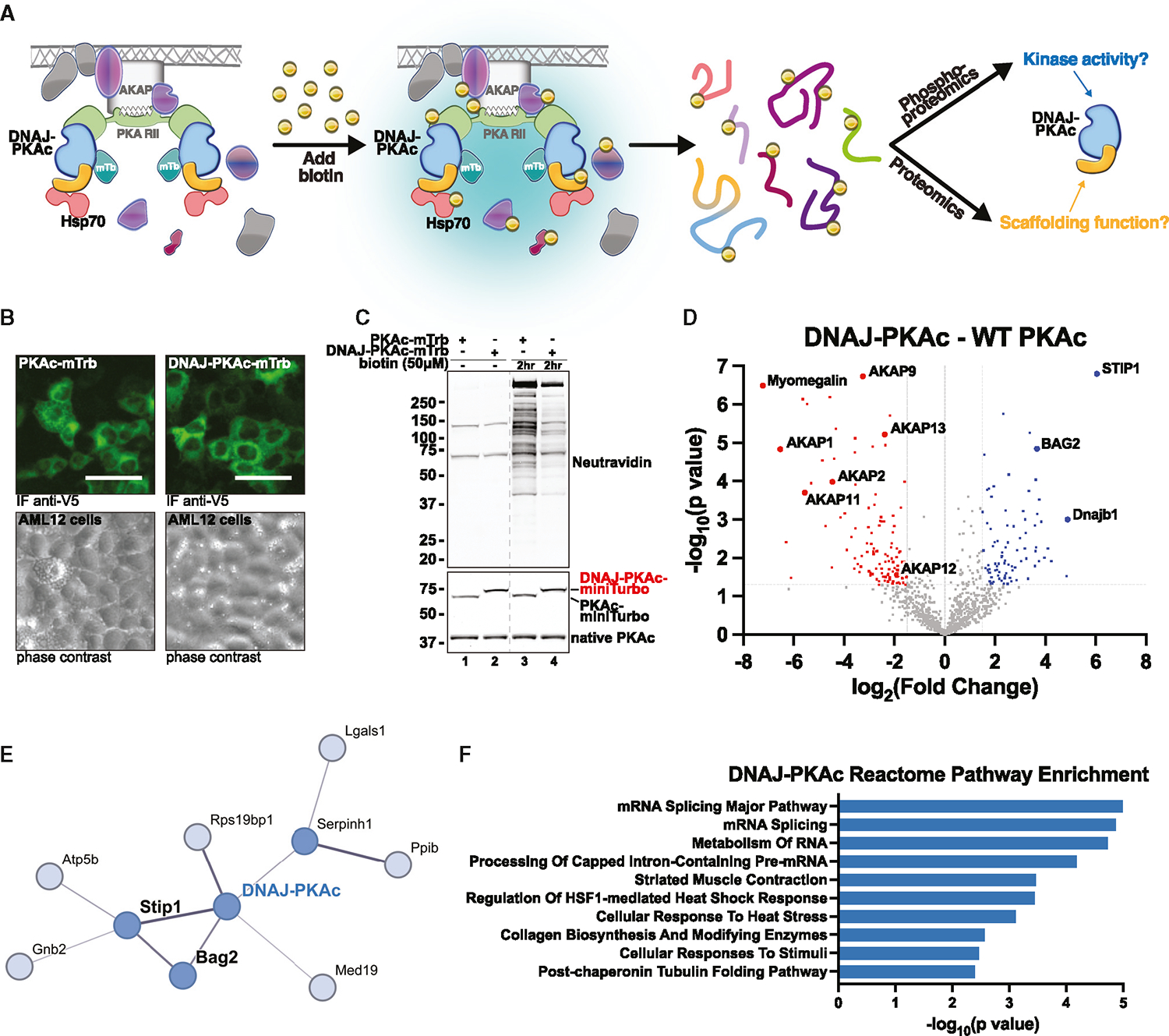
(A) Schematic of miniTurbo (mTrb)-driven proximity labeling workflow and study design. Addition of biotin allows labeling of proteins within a 5- to 10-nm radius of bait proteins. Following isolation, biotinylated or phosphorylated peptides were subjected to MS analysis.
(B) Immunofluorescence imaging of AML12 hepatocytes demonstrating inducible expression of PKAc-mTrb (green, top left) or DNAJ-PKAc-mTrb (green, top right) with corresponding phase contrast (bottom). Scale bars, 50 μm
(C) Immunoblot of cell lysates from stable AML12 lines treated with either DMSO or biotin (50 mM). Neutravidin-HRP (top) shows labeling of biotinylated proteins. PKAc (bottom) shows expression of mTrb-tagged PKAc variants (top band) over native PKAc (bottom band). The dashed line removes lanes from a separate experiment.
(D) Volcano plot of MS results, showing proteins with increased (blue) and decreased (red) association with DNAJ-PKAc compared with WT PKAc. Proteins with p > 0.05 and log2 fold change less than 1.5 are shown in gray. Six biological replicates.
(E) STRING network depiction of selected proteins with greater enrichment in DNAJ-PKAc versus WT PKAc.
(F) Bar chart of the top 10 enriched terms from the Reactome 2022 gene set library for proteins more associated with DNAJ-PKAc. Results are displayed based on −log10(p value).
Mislocalization of DNAJ-PKAc from AKAP signaling islands
Our proximity proteomics screen revealed that AKAPs were generally less associated with the DNAJ-PKAc fusion compared with WT PKAc. STRING analysis of proteins identified as having decreased association with DNAJ-PKAc revealed a network of AKAPs (Figure 2A). Quantitative analysis demonstrated reduced association of type II regulatory subunits alpha and beta (RIIα and RIIβ) with DNAJ-PKAc (Figure 2B). There was no significant difference in association with the type I regulatory subunit alpha between WT PKAc and DNAJ-PKAc. Decreased peptide counts for AKAPs suggest displacement of DNAJ-PKAc from AKAP signaling islands (Figure 2C). To confirm this, photoactivation live-cell microscopy assays were performed to assess in situ localization of PKAc variants (Figure 2D). AML12 hepatocytes were transiently transfected with variants of PKAc fused to photo-activatable (PA) mCherry (magenta), RIIα-iRFP (data not shown), and AKAP79-YFP (cyan) to recruit PKAc holoenzymes to the plasma membrane. Following photoactivation of a 2-μm locus on the cell membrane, we observed that WT PKAc remains predominantly colocalized with membrane-associated AKAP79 (Figure 2D, top row; Figure 2E, gray trace; Figure 2F, gray bar; Video S1). Conversely, DNAJ-PKAc rapidly diffuses into the cytosol, away from the membrane (Figure 2D, second row from top; Figure 2E, blue trace; Figure 2F, blue bar; Video S2). Quantification of cells across three independent experiments demonstrates that DNAJ-PKAc exhibits increased mobility compared with WT PKAc (Figures 2E and 2F). Interestingly, the RIIα-iRFP signal remains localized at the membrane, suggesting that DNAJ-PKAc mislocalization occurs due to interference with regulatory and catalytic subunit binding (Figure S1). To determine the mechanism of fusion kinase mislocalization, we repeated this assay with two additional controls, DNAJH33Q-PKAc, a point mutation to disrupt Hsp70 binding, and PKAcΔ14, which mimics the N-terminal truncation of PKAc in the fusion protein.3,39 The PKAcΔ14 truncation remains localized at the plasma membrane via AKAP79 anchoring (Figure 2D, bottom row; Figure 2E, green trace; Figure 2F, green bar; Video S3). This suggests that the N-terminal portion of the kinase is not necessary for AKAP association. Interestingly, DNAJH33Q-PKAc exhibits an intermediate phenotype, not demonstrating full retention at the plasma membrane but not diffusing completely into the cytosol (Figure 2D, second row from bottom; Figure 2E, pink trace; Figure 2F pink bar; Video S4). Together, these experiments indicate that DNAJ-PKAc is not confined within AKAP signaling islands due to disrupted association of the fusion protein with PKA regulatory subunits and that this effect is partially alleviated when Hsp70 association with the fusion kinase is disrupted.
Figure 2. Mislocalization of DNAJ-PKAc from AKAP signaling islands.
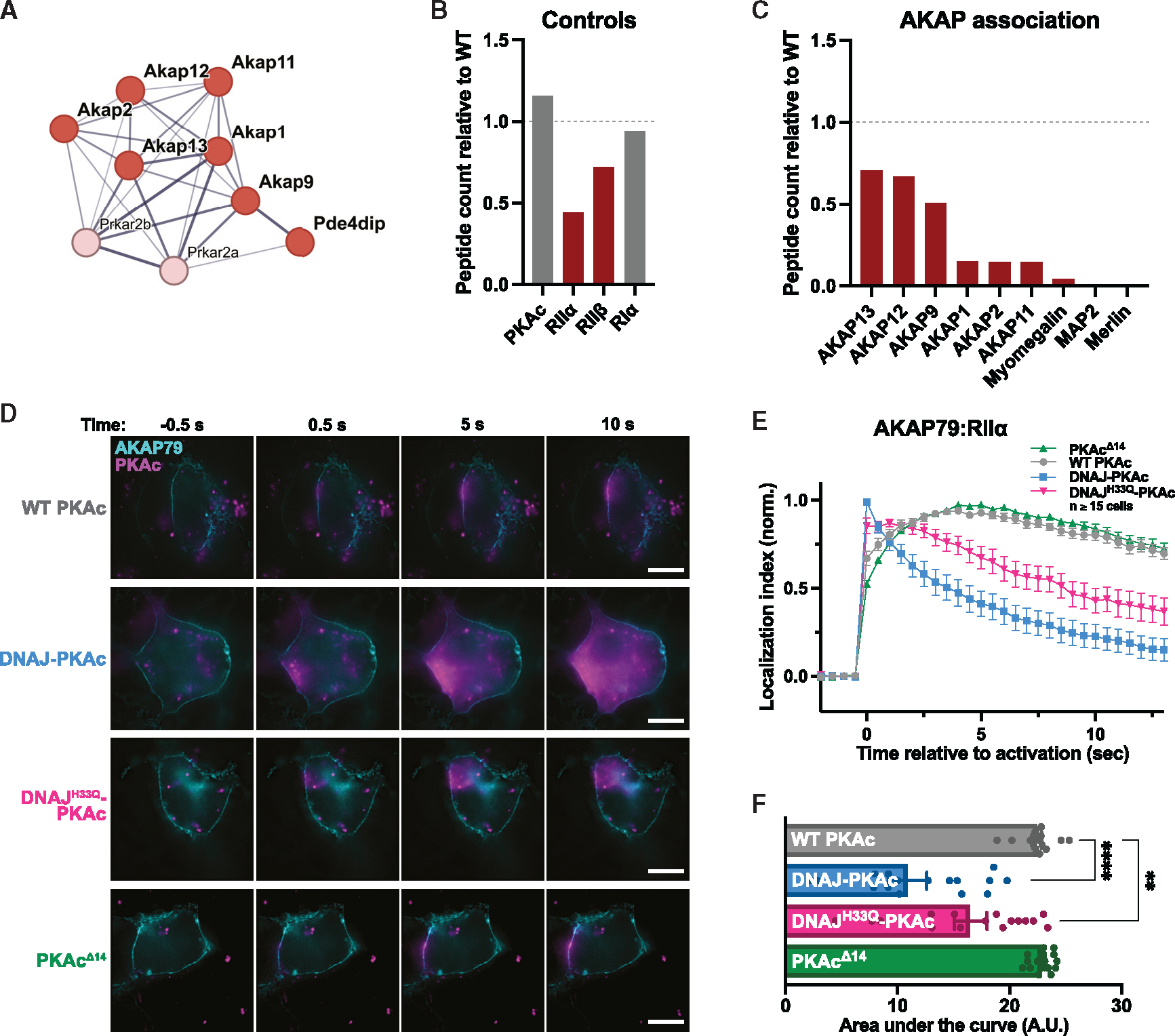
(A) STRING network depiction of selected proteins with lesser enrichment in DNAJ-PKAc versus WT PKAc.
(B and C) Quantification of DNAJ-PKAc association with components of the PKAc holoenzyme (B) and AKAPs (C) based on peptide count relative to WT PKAc. </p/>(D) Photoactivation time courses of AML12 hepatocytes expressing AKAP79-GFP, RIIα-iRFP, and either WT PKAc, DNAJ-PKAc, DNAJH33Q-PKAc, or PKAcΔ14 tagged with PA mCherry. Scale bars, 10 μm
(E) Quantification of (D). Data represent 3 experimental replicates.
(F) Integration of (E). Analyzed by one-way ANOVA and multiple comparisons corrected with Dunnett’s method; mean ± SEM, ****p ≤ 0.0001, **p ≤ 0.01.
Proximity phosphoproteomics uncovers distinct DNAJ-PKAc phosphorylation patterns
We next performed proximity phosphoproteomics enrichment to identify differences in substrate phosphorylation that occur due to mislocalization of DNAJ-PKAc. Following biotin incubation and harvest, biotinylated proteins were isolated from AML12 cells stably expressing either WT PKAc-, DNAJ-PKAc-, or DNAJ-PKAcK72H-miniTurbo. Each digested peptide sample was selectively enriched further for phosphorylated peptides using iron immobilized metal affinity chromatography (IMAC; Figure 3A). Labeling efficiency and expression levels of each PKAc variant were verified prior to extraction and MS analysis (Figures 3B, S2A, and S2B). Phospho-enriched MS results comparing the fusion with WT PKAc identified 385 phosphopeptides that significantly differed in abundance, of which 186 were less associated (Figure 3C, red dots) and 209 were more associated (Figure 3C, blue dots) with DNAJ-PKAc. Comparison of substrate motifs through alignment and logo analysis of basophilic phosphopeptides revealed only minor differences in kinase consensus sequences between WT PKAc and DNAJ-PKAc (Figures 3D and 3E).
Figure 3. Proximity phosphoproteomics uncovers distinct DNAJ-PKAc phosphorylation patterns.
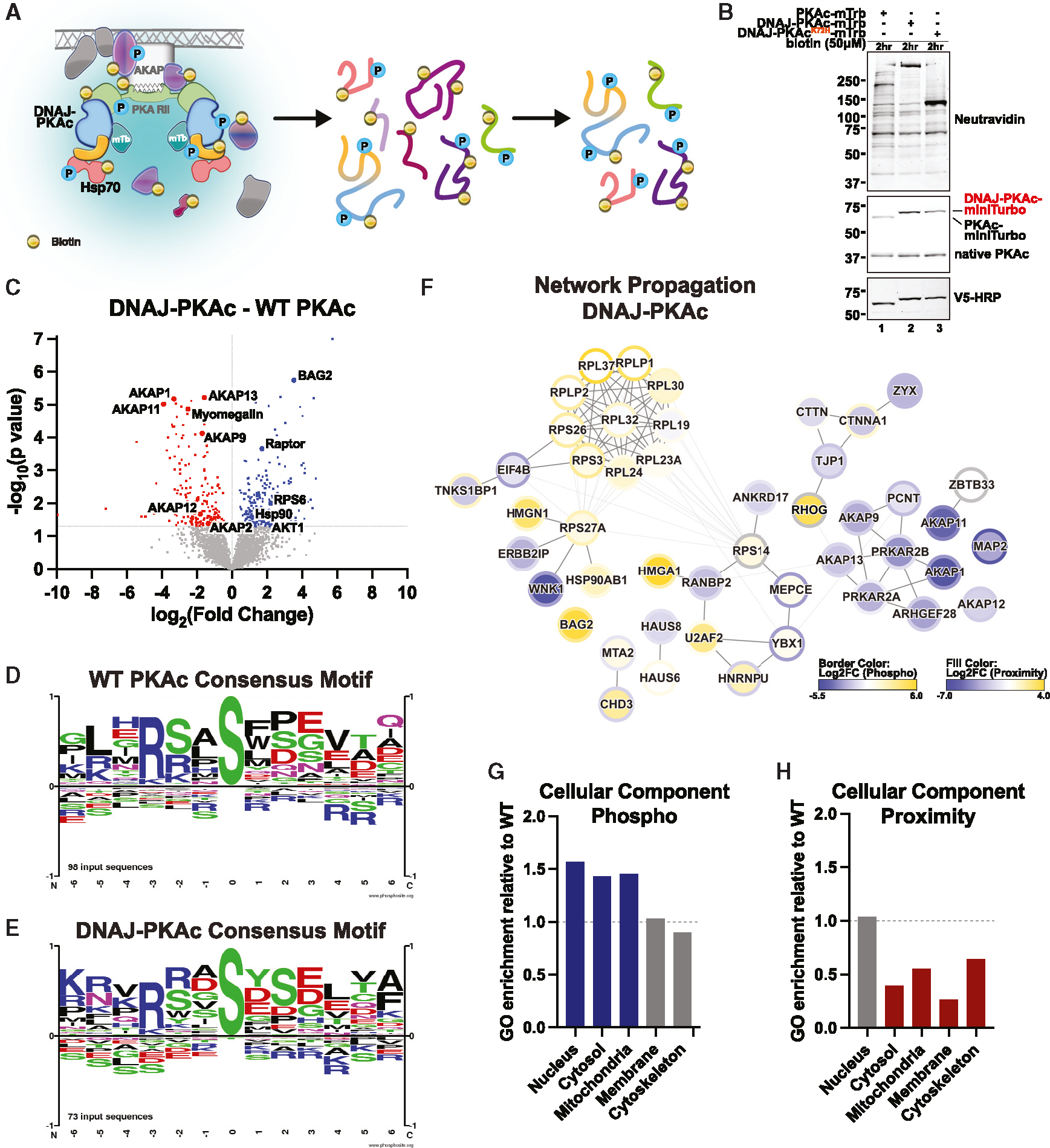
(A) Schematic of phosphoenrichment following mTrb-driven biotin labeling.
(B) Immunoblot of cell lysates from stable AML12 lines following biotin (50 μM) incubation. Neutravidin-HRP (top) shows labeling of biotinylated proteins. PKAc (center) shows expression of mTrb-tagged PKAc variants (top band) over native PKAc (bottom band). V5-HRP (bottom) shows specific expression of PKAc-mTrb variants.
(C) Volcano plot of MS results showing phosphopeptides with increased (blue) and decreased (red) association with DNAJ-PKAc compared with WT PKAc. Proteins with p > 0.05 are shown in gray. Four biological replicates.
(D and E) Logo analysis depicting basophilic substrate consensus motifs for WT PKAc (D) and DNAJ-PKAc (E).
(F) Selected results from ReactomeFI network propagation. Proximity proteomics is represented by node fill color. Proximity phosphoproteomics is represented by node border color. Degree of association is represented on a yellow (more associated) to blue (less associated) spectrum based on log2(fold change). False discovery rate (FDR) < 0.1.
(G and H) Gene Ontology (GO) enrichment scores for DNAJ-PKAc cell components relative to WT PKAc.
This analysis highlighted a close relationship between signaling changes resulting from AKAP mislocalization (Figures 2A and S2C) and chaperone/co-chaperone association similar to those observed in the proximity proteomics screen (Figure 1D). We have previously demonstrated an association between oncogenic PKA signaling and mRNA translation.40 Consistent with these findings, examination of the STRING network for phosphopeptides with increased DNAJ-PKAc association revealed Gene Ontology (GO) enrichment of biological processes often increased in cancer (i.e., mRNA processing, ribosome biogenesis, and translation) as well as members of the TORC1 signaling complex (Rps6, Akt, and Raptor; Figure S2D).41–44 Network propagation was performed on both the proximity proteomics (node fill color) and proximity phosphoproteomics (node border color) datasets to assess the links between alterations in DNAJ-PKAc localization and substrate phosphorylation.45 Integrated analysis of the two propagated proximity proteomics networks uncovered a cluster of significantly phosphorylated ribosomal proteins involved in translation as well as a cluster of AKAPs that were both less associated and less phosphorylated in the presence of the oncogenic fusion kinase (Figure 3F). These findings highlight how altered mRNA translation may be a consequence of DNAJ-PKAc mislocalization.
We next performed quantitative comparison of WT PKAc and DNAJ-PKAc GO enrichment scores for cellular component association. This revealed increased phosphorylation of proteins localized to the nucleus, cytosol, and mitochondria in cells expressing DNAJ-PKAc (Figure 3G). Importantly, these phos phorylation events occurred despite the chimeric fusion having near-equivalent association with proteins in the nucleus and over 40% less association with proteins in the cytosol and mitochondria (Figure 3H). Thus, while substrate selectivity of the fusion kinase is not substantially altered, mislocalization from AKAP signaling complexes allows DNAJ-PKAc aberrant access to substrates in different cellular compartments than those occupied by WT PKAc.
Substrates of DNAJ-PKAc are regulators of ribosome biogenesis and translation
To investigate changes in phosphorylation patterns due to DNAJ-PKAc, we compared the DNAJ-PKAc and kinase-dead DNAJ-PKAcK72H proximity phosphoproteomics datasets. Analysis identified 536 significant phosphopeptides, 239 of which had increased association and 297 of which had decreased association with the DNAJ-PKAc active fusion compared with the kinase-dead fusion (Figure 4A). Experiments with the DNAJ-PKAcK72H kinase-dead mutant were performed to eliminate phosphopeptides that were not direct substrates of the fusion protein. The total pool of phosphopeptides significantly more associated with DNAJ-PKAc (Figure 3C, blue dots) includes both substrates of the fusion and associated phosphoproteins that are phosphorylated by other kinases. Therefore, integrating the proximity phosphoproteomics datasets from Figures 3C and 4A allowed us to distinguish between interacting partners not phosphorylated by DNAJ-PKAc and likely substrates of DNAJ-PKAc. This subtractive method of analysis extracted a list of 67 phosphorylation sites across 53 putative substrates of the FLC fusion kinase (Figure 4B) that were significantly more associated with DNAJ-PKAc compared with both WT PKAc (Figure 3C, blue dots) and DNAJ-PKAcK72H (Figure 4A, blue dots). STRING network and GO term analysis of this phosphoprotein subset revealed increased association of DNAJ-PKAc with proteins involved in ribosome biogenesis and translation (Figure 4C). Peptides from regulators of these processes were at least 2-fold more phosphorylated in the presence of the DNAJ-PKAc fusion versus WT PKAc (Figures 4D and 4E). A puromycin-based incorporation assay was performed to assess global translation activity in AML12 cells expressing DNAJ-PKAc (Figures 4F and S3). Compared with WT AML12 cells, AML12DNAJ-PKAc cells showed significantly increased levels of protein synthesis (Figure 4G). This finding was further validated using a bicistronic translation reporter, where GFP is linked to mCherry by the hepatitis C virus (HCV) internal ribosomal entry sequence (IRES). The HCV IRES has been shown previously to enable translation even with full inhibition of eIF4A, allowing the use of mCherry levels as a control for total ribosomal capacity compared with GFP, which demonstrates cap-dependent mRNA translation.46 The results of this latter assay again demonstrated increased overall translational activity in cells expressing the DNAJ-PKAc fusion kinase (Figure 4H). This further supported an increase in cap-dependent translation, likely impacting genes such as MYC.40,47 Finally, to determine the impact of the kinase activity of the fusion on protein synthesis, we treated cells with BLU2864 to selectively inhibit PKA activity 1 h prior to addition of puromycin. These experiments revealed a dose-dependent decrease in translation corresponding to degree of PKA inhibition by BLU2864, with greater inhibition occurring at the higher dose (Figures 4I and 4J). Hence, the presence and activity of DNAJ-PKAc impacts processes involved in the translational capacity of the cell.
Figure 4. Substrates of DNAJ-PKAc are regulators of ribosome biogenesis and translation.
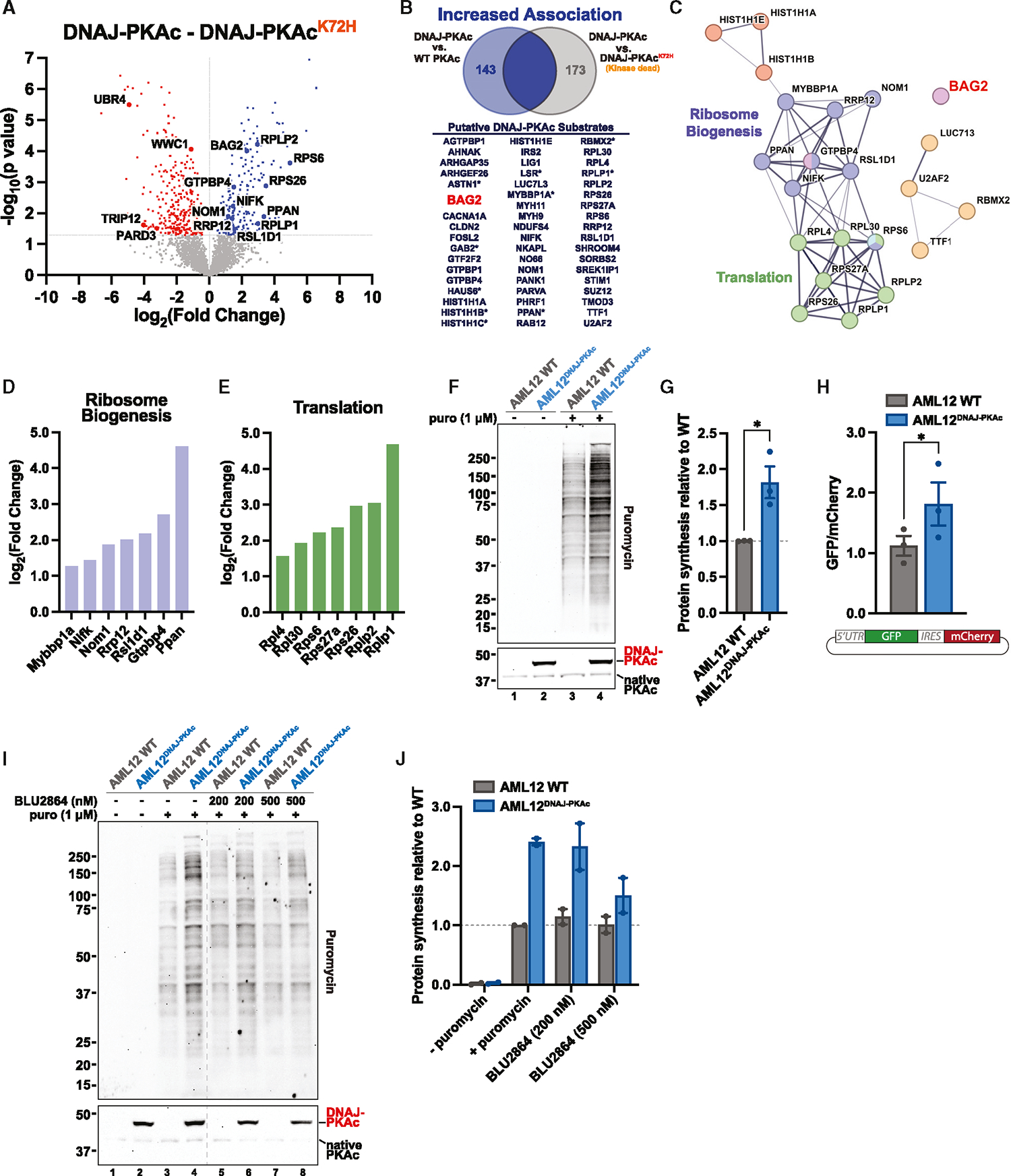
(A) Volcano plot of MS results showing phosphopeptides with increased (blue) and decreased (red) association with DNAJ-PKAc compared with DNAJ-PKAcK72H. Proteins with p >0.05 are shown in gray. Four biological replicates.
(B) Venn diagram with resulting list of putative DNAJ-PKAc substrates identified by overlapping phosphosites that have increased association with DNAJ-PKAc versus WT PKAc and DNAJ-PKAcK72H. An asterisk indicates two or more phosphosites identified on a corresponding protein.
(C) STRING network depicting selected functional clusters of putative DNAJ-PKAc substrates.
(D and E) Bar graphs showing log2(fold change) over WT PKAc for DNAJ-PKAc phosphoproteins associated with ribosome biogenesis (D) and translation (E).
(F) Immunoblot of cell lysates from WT AML12 and AML12DNAJ-PKAc treated with either vehicle or puromycin (1 μM). Puromycin conditions (top) show newly synthesized, puromycin-labeled proteins. PKAc (bottom) shows expression of DNAJ-PKAc (top band) over native PKAc (bottom band). See also Figure S3.
(G) Quantification of (F), measuring protein synthesis in AML12DNAJ-PKAc cells versus WT AML12 cells. Data represent 3 biological replicates. Mean ± SEM. *p ≤ 0.05.
(H) Measurement of mRNA translation in WT AML12 cells (gray) and AML12DNAJ-PKAc cells (blue) stably transduced with a bicistronic reporter. Spontaneously cycling cells were analyzed by fluorescence-activated cell sorting (FACS), and a normalized ratio of GFP/mCherry signal was determined. Data represent 3 biological replicates. Student’s t test. Mean ± SEM. *p ≤ 0.05.
(I) Immunoblot of cell lysates from WT AML12 and AML12DNAJ-PKAc cells treated with either vehicle, puromycin (1 μM), or puromycin (1 μM) plus BLU2864 (200 nM or 500 nM). Puromycin conditions (top) show newly synthesized, puromycin-labeled proteins. PKAc (bottom) shows expression of DNAJ-PKAc (top band) over native PKAc (bottom band). The dashed line removes lanes from a separate experiment.
(J) Quantification of (I), measuring protein synthesis in AML12DNAJ-PKAc cells versus WT AML12 cells. Data represent 2 biological replicates. Mean ± SEM.
BAG2 is recruited to DNAJ-PKAc and overexpressed in FLC tumors
BAG-family proteins are involved in a variety of important cellular functions, including cell survival and stress response.27 BAG2 is a known regulator of Hsp70-mediated protein refolding and CHIP-mediated ubiquitination that has been implicated in several different cancers.29,48–51 Our proximity labeling data uncovered BAG2 as an interaction partner and putative substrate of DNAJ-PKAc (Figure 5A). Assessment of the BAG2 phosphopeptides identified through MS revealed a basophilic kinase recognition motif surrounding the phosphorylated serine 20 (Figure 5B). The canonical PKA motif R-X-X-S/T* at this site is shared with MAPKAPK2, a p38 mitogen-activated protein kinase (MAPK) substrate known to phosphorylate BAG2 at Ser20.52–54 Phospho-BAG2 was also detected upon MS analysis of FLC tumors (Figures S4A and S4B).
Figure 5. BAG2 is recruited to DNAJ-PKAc and overexpressed in FLC tumors.
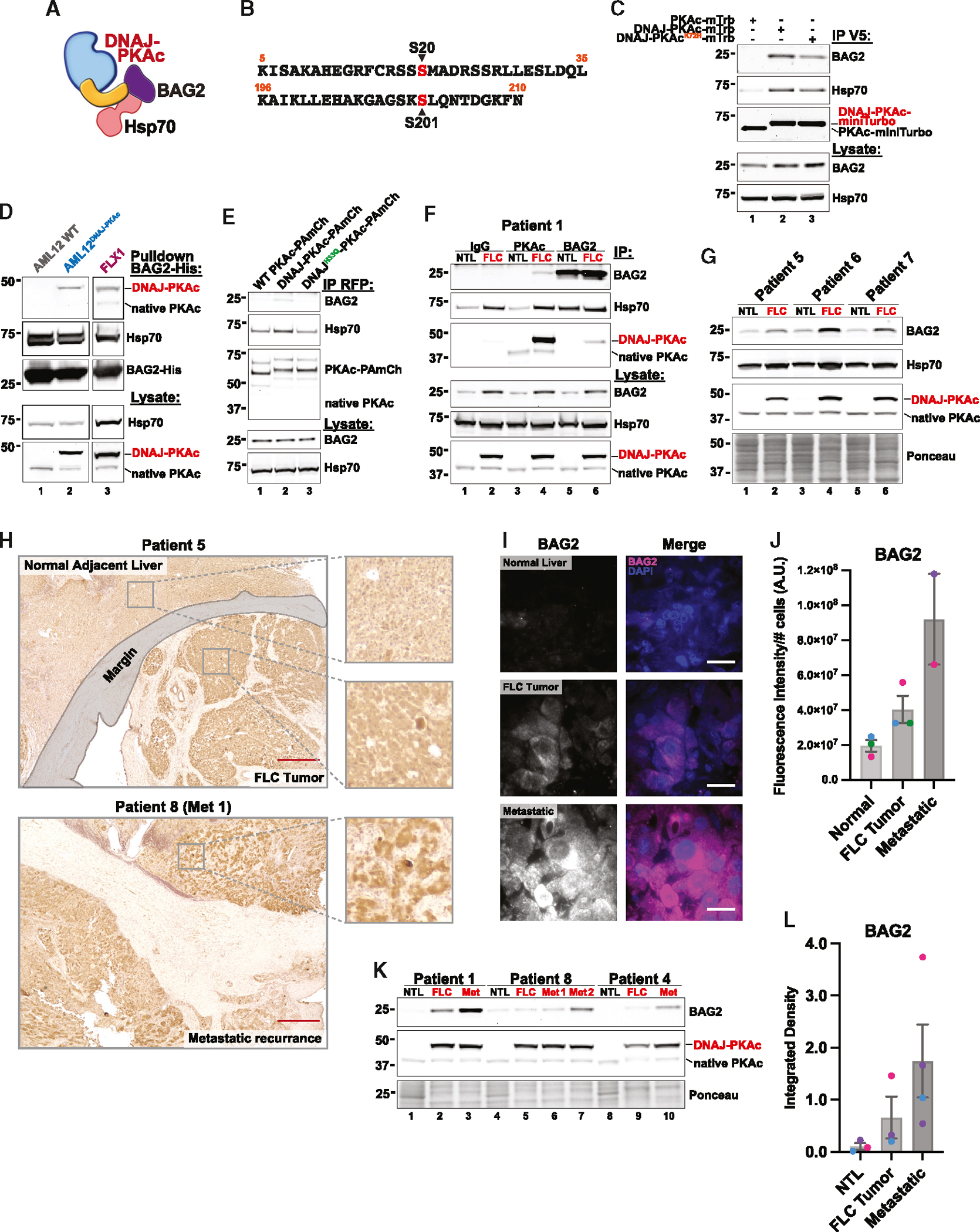
(A) Model of BAG2 interaction with Hsp70 and DNAJ-PKAc.
(B) Peptide sequences of BAG2 phosphosites identified in the MS screen. The phosphorylated residue is labeled in red.
(C) Immunoprecipitation of V5-tagged PKAc variants from stable AML12 cell lines; represents 3 replicate experiments.
(D) Pull-down of PKAc variants using BAG-His as bait in lysates from AML12 WT (lane 1), AML12DNAJ-PKAc (lane 2), and FLX1 (lane 3) cells; represents 3 replicate experiments for AML12 cells and 2 replicate experiments for FLX1 cells.
(E) Immunoprecipitation of mCherry-tagged PKAc variants from HEK293T cells; represents 3 replicate experiments.
(F) Immunoprecipitation with antibodies to immunoglobulin G (IgG) control (lanes 1 and 2), PKAc (lanes 3 and 4), and BAG2 (lanes 5 and 6) from paired non-tumor liver (NTL) and FLC tumor tissue lysates from a single patient.
(G) Immunoblot of paired NTL and FLC tumor tissue lysates from 3 patients. See also Figure S4C for additional data.
(H) Chromogenic immunohistochemistry of BAG2 in resected liver tissue from patients with FLC. Images are representative of 8 normal liver/FLC tumor pairs and 2 Met tumors. Scale bars, 500 μm
(I) Immunofluorescence staining of BAG2 in resected liver tissue from patients with FLC. Images are representative of 3 normal liver/FLC tumor pairs and 2 Met tumors. Scale bars, 20 μm
(J) Quantification of (H), with BAG2 fluorescence intensity normalized to the number of cells in the field; 10 images each per patient sample. Individual patients are designated by color. Mean ± SEM.
(K) Immunoblot of paired NTL, FLC tumor, and metastatic (Met) tumor tissue lysates from 3 patients.
(L) Quantification of (J). Individual patients are designated by color. Mean ± SEM.
We next validated BAG2 recruitment to DNAJ-PKAc biochemically. Immunoprecipitation of PKAc variants from AML12 cells revealed that both active DNAJ-PKAc and the kinase-dead mutant co-precipitate BAG2 (Figure 5C, lanes 2 and 3) whereas WT PKAc does not (Figure 5C, lane 1). Additional validation of this protein-protein interaction was conducted in cell lysates incubated with purified BAG2-His (Figure 5D). Pull-down experiments were performed using both AML12DNAJ-PKAc cells (Figure 5D, lane 2) and the FLX1 cell line established from a patient-derived xenograft (PDX) model (Figure 5D, lane 3).40 Control experiments conducted in WT AML12 lysates confirmed that BAG2-His does not interact with native PKAc (Figure 5D, lane 1). Importantly, BAG2-His pulled down DNAJ-PKAc and Hsp70 in both cellular contexts. Because it is known that BAG2 can associate with substrates both directly and indirectly via Hsp70, we wanted to determine whether association of BAG2 was primarily Hsp70 mediated.48 HEK293T cells were transiently transfected with either WT PKAc-, DNAJ-PKAc-, or DNAJH33Q-PKAc-PAmCherry. Subsequent immunoprecipitation of each PKAc variant demonstrated that the DNAJH33Q-PKAc mutant disrupted recruitment of not only Hsp70 but also BAG2 to the FLC fusion kinase (Figure 5E, lane 3). Importantly, immunoprecipitation of PKAc in 7 paired (normal liver and FLC primary tumor) and 4 metastatic patient tissue samples demonstrated that DNAJ-PKAc pulls down BAG2 in both primary (FLC) and metastatic (Met) tumors whereas WT PKAc in adjacent, non-tumor liver (NTL) does not (Figure 5F, lanes 3 and 4; Figures S4C and S4D). This result was further validated in a reciprocal immunoprecipitation of BAG2, which pulled down DNAJ-PKAc in FLC but not WT PKAc in FLC or NTL tissue (Figure 5F, lanes 5 and 6).
BAG2 overexpression has been linked to poor clinical outcome and prognosis in a variety of cancers.28,29,55,56 Interestingly, BAG2 mRNA is enriched in FLC tumors.57 Therefore, we compared BAG2 levels in normal liver and primary tumor tissue within each patient. Immunoblot analysis of lysates from 7 different patients demonstrated overall increased expression of BAG2 in FLC versus NTL tissue (Figure 5G; Figure S4C, lysate panels). To assess BAG2 expression and global distribution in clinical samples, we performed immunohistochemistry on patient tissue sections. Chromogenic and immunofluorescence detection of BAG2 in stained tissue revealed a qualitative increase in BAG2 levels from normal adjacent liver to FLC tumor and primary tumor to metastasis (Figures 5H and 5I). Quantification of immunofluorescence images from 3 NTL/FLC pairs and 2 Met tissue sections confirmed this result (Figure 5J). Furthermore, evaluation of BAG2 expression via immunoblot in paired samples from 3 patients with advanced disease showed a progressive increase in BAG2 levels from adjacent NTL to FLC primary tumor to Met recurrence within each patient (Figures 5K and 5L). Finally, to determine whether the presence of DNAJ-PKAc had a stabilizing effect on BAG2 and its partner, the anti-apoptotic protein Bcl-2, we treated cells with the protein synthesis inhibitor cycloheximide and collected lysates at 2-h time points from 0 to 8 h. These data showed that both BAG2 and Bcl-2 are stabilized in AML12 cells expressing DNAJ-PKAc compared with WT AML12 cells (Figures S4E–S4G). In sum, these findings suggest that BAG2 overexpression in FLC is correlated with disease progression and that BAG2 and Bcl-2 proteins are stabilized in the presence of the fusion kinase DNAJ-PKAc.
BAG2 promotes tumor cell survival and resistance to drug-induced cell death
BAG-family proteins regulate cell growth, stress response, and cell death through interaction with several signaling partners, including Bcl-2, an inhibitor of cell death.58–60 However, previous studies have suggested that the anti-apoptotic effects of BAG2 may be tumor type specific.56 Therefore, we wanted to determine whether BAG2 contributes to tumorigenesis and whether its association with Bcl-2 protects against cell death in the context of FLC (Figure 6A). CRISPR-Cas9 gene editing was employed to generate a knockout (KO) of BAG2 in AML12 cells expressing the DNAJ-PKAc fusion (Figure 6B). It has been demonstrated previously that AML12DNAJ-PKAc cells exhibit accelerated proliferation compared with WT AML12 cells.14 Therefore, we assessed whether knocking out BAG2 in AML12DNAJ-PKAc cells affects this fundamental property of tumorigenesis. Bromodeoxyuridine (BrdU) incorporation, evaluated using a colorimetric ELISA, showed reduced DNA synthesis in AML12DNAJ-PKAc cells lacking BAG2 compared with mock control AML12DNAJ-PKAc cells (Figure 6C). We next performed a puromycin-based incorporation assay to monitor global translation and found that AML12DNAJ-PKAc BAG2 KO cells demonstrated a trend of decreased protein synthesis compared with AML12DNAJ-PKAc and AML12DNAJ-PKAc mock control cell lines (Figures S5A and S5B). To assess the anti-apoptotic role of BAG2 in FLC, we induced cell death in WT AML12, AML12DNAJ-PKAc, and AML12DNAJ-PKAc BAG2 KO cells with the potent chemotherapeutic agent etoposide. Examination of cell density and morphology revealed that cells expressing DNAJ-PKAc were more resistant to etoposide-induced cell death, as evidenced by minimal effects of treatment on cell growth, size, and shape compared with WT AML12 cells (Figure 6D, top). Conversely, BAG2 KO AML12DNAJ-PKAc cells exhibited features such as swelling and blebbing, suggesting restored susceptibility to drug-induced cell death when subjected to etoposide (Figure 6D, bottom). A hallmark of cell death processes is PARP-1 activation and cleavage.61 Immunoblot analysis and quantification of cleaved PARP-1 following etoposide treatment corroborated our findings, demonstrating higher levels of the 89-kDa apoptotic fragment in WT AML12 and AML12DNAJ-PKAc BAG2 KO cells than in normal AML12DNAJ-PKAc cells (Figures 6E and 6F).
Figure 6. BAG2 promotes tumor cell survival and resistance to drug-induced cell death.
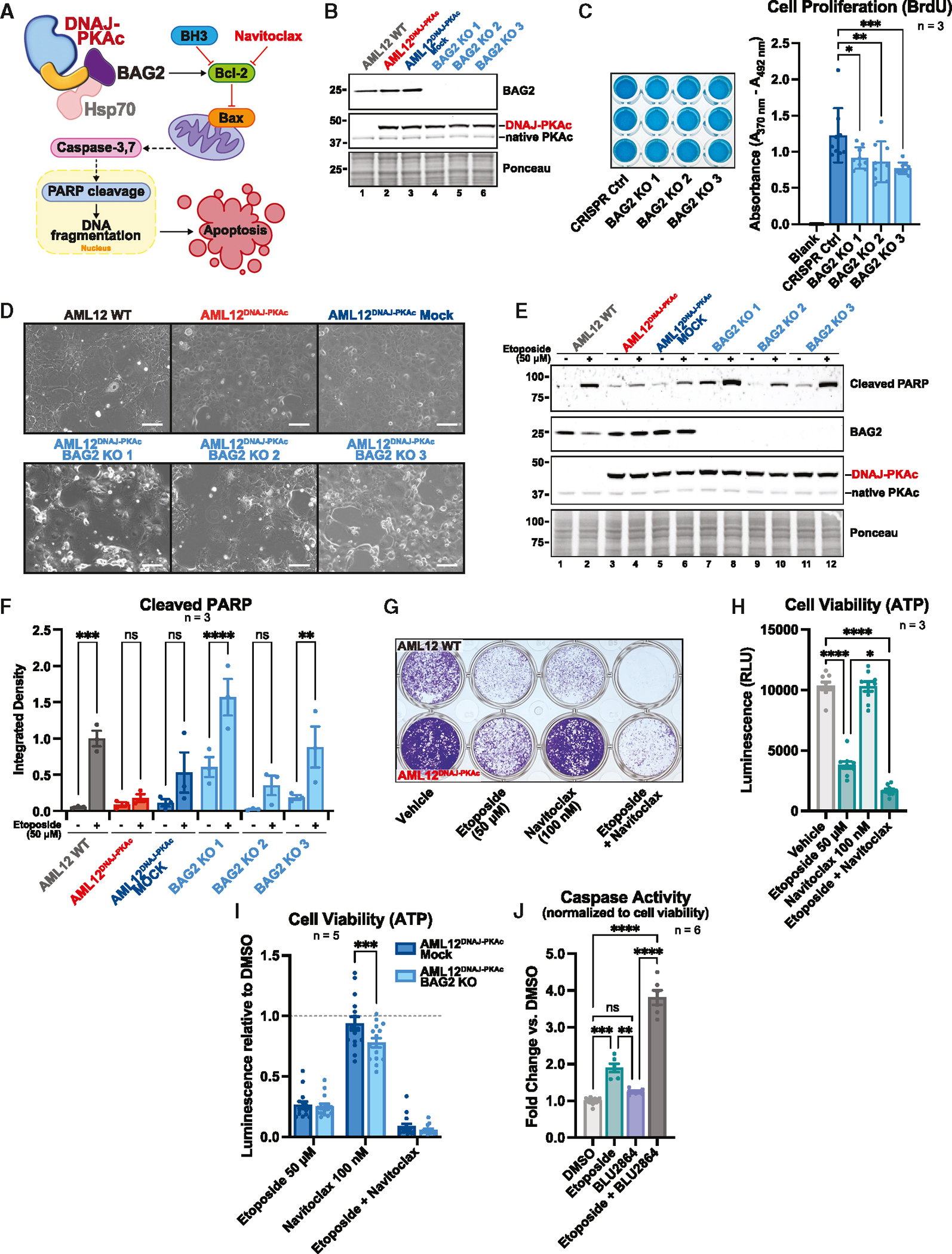
(A) Schematic showing BAG2 regulation of apoptosis in FLC.
(B) Immunoblot of three CRISPR-Cas9-generated BAG2 KOs in AML12DNAJ-PKAc clonal cell lines.
(C) ELISA BrdU incorporation assay measuring cell proliferation in three AML12DNAJ-PKAc BAG2 KO clones versus the AML12DNAJ-PKAc mock (CRISPR Ctrl) control line. Data represent three biological replicates. Mean ± SEM. ***p ≤ 0.001, **p ≤ 0.01, *p ≤ 0.05.
(D) Phase contrast images showing etoposide-induced cell death in WT AML12, AML12DNAJ-PKAc, AML12DNAJ-PKAc CRISPR mock control, and three clonal AML12DNAJ-PKAc BAG2 KO cell lines. Images are representative of 3 experimental replicates. Scale bars 100 μm
(E) Immunoblot of etoposide-induced PARP cleavage in WT AML12, AML12DNAJ-PKAc, AML12DNAJ-PKAc CRISPR mock control, and three clonal AML12DNAJ-PKAc BAG2 KO cell lines.
(F) Quantitation of (E). Data represents 3 biological replicates. Mean ± SEM. ****p ≤ 0.0001, ***p ≤ 0.001, **p ≤ 0.01.
(G) Crystal violet stain cell survival assay showing WT AML12 cells (top wells) and AML12DNAJ-PKAc cells (bottom wells) treated with vehicle, etoposide (50 μM), navitoclax (100 nM), or etoposide (50 μM) plus navitoclax (100 nM). The image is representative of 3 experimental replicates.
(H) Quantification of the CellTiter-Glo luminescence assay, showing cell viability of AML12DNAJ-PKAc cells following treatment with vehicle, etoposide (50 μM), navitoclax (100 nM), or etoposide (50 μM) plus navitoclax (100 nM). Data represent 3 experimental replicates. Mean ± SEM. ****p ≤ 0.0001, **p ≤ 0.01.
(I) Quantification of the CellTiter-Glo luminescence assay, showing cell viability of the AML12DNAJ-PKAc BAG2 KO clonal cell line versus the AML12DNAJ-PKAc mock control cell line following treatment with vehicle, etoposide (50 μM), navitoclax (100 nM), or etoposide (50 μM) plus navitoclax (100 nM). Data represent 5 experimental replicates. Mean ± SEM. ***p ≤ 0.001.
(J) Quantification of the Caspase-Glo 3/7 luminescence assay, showing apoptotic activity of FLX1 cells following treatment with vehicle, etoposide (50 μM), BLU2864 (5 μM), or etoposide (50 μM) and BLU2864 (5 μM). The CellTiter-Glo viability assay was run in parallel to determine cell density, and the Caspase-Glo result was normalized based on CellTiter-Glo results within each condition. Data represent 6 experimental replicates. Mean ± SEM. ****p ≤ 0.0001, ***p ≤ 0.001, **p ≤ 0.01.
Recently, BH3 mimetics have emerged as potent targeted therapeutics in the context of cancer.62,63 These molecules, such as ABT-199/venetoclax and its derivative, ABT-263/navitoclax, inhibit Bcl-2-family proteins to induce apoptosis and are used clinically.64 Therefore, we tested the effect of Bcl-2 inhibition on cell survival and susceptibility to induced cell death. Total cell density was monitored by performing crystal violet staining after treatment with etoposide alone, navitoclax alone, or a combination of the two compounds (Figure 6F). WT AML12 cells were susceptible to both etoposide and navitoclax and highly susceptible to the etoposide/navitoclax combination (Figure 6G, top wells), whereas cells expressing DNAJ-PKAc were moderately affected by etoposide treatment, completely resistant to navitoclax, and highly susceptible to the etoposide/navitoclax combination (Figure 6G, bottom wells). We further measured the impact of this drug combination using CellTiter-Glo to measure ATP levels, an indicator of cellular metabolism. Co-incubation with etoposide and navitoclax significantly and synergistically decreased AML12DNAJ-PKAc cell viability more effectively than either compound alone (0.450 ± .042 versus etoposide, 0.163 ± .012 versus vehicle; SEM, n = 3) (Figure 6H). Furthermore, cell viability assays evaluating the effects of etoposide and navitoclax on AML12DNAJ-PKAc BAG2 KO cells demonstrated a statistically significant enhancement of drug sensitivity to navitoclax in cells lacking BAG2 compared with AML12DNAJ-PKAc mock control cells (Figure 6I). Next, we determined whether the kinase activity of DNAJ-PKAc also contributes to drug resistance. FLX1 cells were treated with either etoposide, the PKAc inhibitor BLU2864, or a combination of the two compounds. Drug effects were evaluated by measuring cell viability and caspase activity, an indicator of apoptosis. While etoposide and BLU2864 individually reduced FLX1 cell viability in a CellTiter-Glo assay, combining the two agents had a limited additive effect (Figure S5C). In contrast, a Caspase-Glo 3/7 assay revealed minimal induction of apoptosis with either etoposide or BLU2864 alone. However, both compounds synergized to induce apoptosis (Figure 6J). Together, these findings imply a pro-survival function of BAG2 in FLC. Small-molecule studies further confirm a synergistic effect of drug-induced cell death and Bcl-2 inhibition in this disease and connect the anti-apoptotic function of BAG2 to the kinase activity of DNA-PKAc.
DISCUSSION
Cancer is a genetic disease, with most solid tumors marked by mutations in 2–6 driver genes.65 In contrast, the adolescent liver cancer FLC is driven by a single genetic lesion on chromosome 19 that generates the chimeric enzyme DNAJ-PKAc.3,66,67 Expression of a fusion kinase that encodes a chaperone binding domain joined in frame with the catalytic core of PKAc is a dominant oncogenic event in FLC.3 However, the molecular mechanisms by which this fusion kinase precipitates this aggressive and intractable liver cancer remain poorly understood. This is in part due to a scarcity of clinical samples available to investigate this rare tumor and limited access to animal models that faithfully recapitulate the molecular signature of FLC.67–70 Here, we used live-cell enzyme-catalyzed biotinylation to identify DNAJ-PKAc-associated proteins and dysregulated biological processes in a hepatocyte model of FLC. While aberrant kinase activity is assumed to be an initiating factor in this disease, the recruitment of DNAJ-PKAc-specific binding partners, such as the chaperone Hsp70, implies that the scaffolding function of DNAJ-PKAc is another disease-driving determinant.14,71 Abnormal enhancer activity, transcriptome remodeling, and altered translation have also been identified as factors contributing to FLC oncogenesis.40,72 Thus, a critical aspect of this work is the equivalent attention given to the catalytic actions, scaffolding functions, and downstream signaling impacts of this fusion kinase.
AKAPs are crucial for spatial regulation of protein kinase A and are responsible for directing the actions of this highly utilized multipurpose enzyme.18,25,73–76 Surprisingly, our proximity MS experiments identified a loss of DNAJ-PKAc association with AKAPs. This suggests that FLC tumor cells harbor an unrestrained fusion kinase that increases off-target phosphorylation of cellular proteins. Interestingly, our live-cell photoactivation studies demonstrated that recruitment of the chaperone Hsp70 is a factor in the displacement of DNAJ-PKAc. Hsp70 often works as part of a chaperone/co-chaperone complex and therefore could mediate the association of other proteins with the catalytic subunit of PKA via the added J domain.77 Indeed, the immunoprecipitation experiments in Figure 5E demonstrate that the Hsp70-binding mutant of DNAJ-PKAc fails to bind the co-chaperone BAG2. Together, these data suggest that disruption of AKAP interaction could be a consequence of recruiting binding partners that impede the geometric organization required for PKAc anchoring. Our previous biochemical studies suggested that the binding of DNAJ-PKAc to regulatory subunits was minimally affected.71 Thus, mislocalization of DNAJ-PKAc may be context dependent and subject to the availability of binding partners, such as Hsp70, that block association with regulatory subunits or AKAPs.
Displacement of DNAJ-PKAc from AKAP complexes is reminiscent of Cushing’s adenomas, where distinct mutations in the catalytic core of PKAc disrupt AKAP-dependent compartmentalization.78 In adrenal Cushing’s syndrome, mislocalization arises from perturbations of the protein-protein interface by which PKAc associates with regulatory subunits. In contrast, the acquired scaffolding function of DNAJ-PKAc confers enhanced cytoplasmic mobility in FLC. Furthermore, precision medicine approaches have shown that adrenal Cushing’s stems from a range of mutations in different regions of the catalytic subunit, whereas FLC normally arises from a single genetic lesion.71,79 Mutations in the catalytic core of Cushing’s kinases have been suggested to cause substrate rewiring that alters how the kinase recognizes basophilic substrate motifs.79–81 In contrast, our analyses of proximal phosphorylated peptides in Figures 3D and 3E indicate that native PKAc and the fusion kinase utilize virtually identical recognition motifs. Thus, we argue that displacement from AKAPs results in altered compartmentalization of DNAJ-PKAc, providing this promiscuous kinase access to additional compatible substrates. For instance, increased phosphopeptides from the TORC1 complex components AKT1 and Raptor echo previous reports of mTOR involvement in FLC, where hyperphosphorylation of S6K was observed.82 Moreover, three of the largest biological process clusters consisted of mRNA processing, ribosome biogenesis, and protein translation machinery (Figure S2D). This finding is consistent with the involvement of DNAJ-PKAc signaling in translation initiation, an oncogenic process effectively targeted using a clinically relevant eIF4A inhibitor in a patient-derived cellular model of FLC.40
A troubling feature of FLC is its resistance to systemic chemotherapies.83 Our study presents a likely mechanism for this challenging complication through the recruitment of BAG2. This cochaperone belongs to a family of proteins whose key functions center around the regulation of cell survival, apoptosis, and stress response.84–86 BAG2 is known to interact with Hsp70 and thus may be recruited into the DNAJ-PKAc signaling scaffold via protein-protein interactions with the chaperone and the fusion kinase.48,87 Future structural studies will be necessary to define the precise topology of this multiprotein configuration. Several studies have investigated the oncogenic action of BAG2 and proposed context-specific roles for this co-chaperone. A study of thyroid cancer found that BAG2 adopts a pro-apoptotic function following proteasome inhibition.88 Conversely, recent studies have identified a more prevalent role of BAG2 in promoting cell survival and tumorigenesis. For example, high levels of BAG2 in gastric cancer are linked to poor outcomes.89 Furthermore, BAG2 upregulation in both glioma and breast cancer confers chemoresistance and protection against apoptosis.28,51,55,90 Finally, research in HCC reports decreased overall survival in patients with elevated BAG2.56,91 Remarkably, each study describes a different strategy by which BAG2 contributes to disease progression. However, a common thread, regardless of mechanism, is that increased BAG2 expression is correlated with poor disease prognosis. In tissue from patients with FLC, we find that BAG2 protein levels correlate with severity of disease, increasing first in primary tumors and peaking in Met disease. In this way, BAG2 may be considered a marker for disease progression in FLC and may have utility as a prognostic and/or predictive biomarker.
As outlined above, the molecular mechanism of BAG2 action in tumorigenesis is multifaceted. Emerging data have linked BAG2 to elements of MAPK signaling in the context of cancer.89,91 BAG2 is directly phosphorylated at serine 20 by MAPKAPK2, a major downstream mediator of p38-dependent processes.54 Alterations in the p38/MAPK pathway have been shown to contribute to tumor growth and Met progression in certain cancers.92 In addition, association with Bcl-2, an inhibitor of the pro-apoptotic protein Bax, suggests an anti-apoptotic role of BAG-family proteins.93,94 Indeed, recent studies have established BAG2 as a promoter of cell survival in the regulation of apoptosis.89,95,96 Our results demonstrate that BAG2 is not only more associated with the fusion kinase but also more phosphorylated (at serine 20) in the presence of DNAJ-PKAc (Figures 3D and 5B). We further propose that upregulation of BAG2 may contribute to chemotherapy resistance through attenuation of apoptosis. Enhanced sensitivity to etoposide-induced cell death in FLC model cells lacking BAG2 supports this mechanism (Figures 6D and 6E). This also led to our hypothesis that intervention at the level of Bcl-2 with the BH3 mimetic navitoclax may circumvent BAG2-mediated suppression of and resistance to apoptosis. Both compounds have been clinically evaluated. Etoposide is a US Food and Drug Administration (FDA)-approved topoisomerase II inhibitor used alone or in combination for the treatment of various cancers, including small cell lung cancer, testicular cancer, and lymphoma.97 Navitoclax, the predecessor of the FDA-approved Bcl-2 inhibitor venetoclax, is currently in phase III clinical trials following promising phase II results as a combination therapy in myelofibrosis.98,99 The success of the combination drug experiments in our model cell line offers promise for therapeutically targeting apoptosis resistance in FLC tumors. Future investigation of the effects of etoposide/navitoclax combinations in FLC tumor PDX mice and hepatocyte organoid models expressing an allele of the fusion kinase will undoubtedly reveal additional details regarding the oncogenic action of DNAJ-PKAc.100
In conclusion, we discovered that the pathogenic PKA fusion kinase driving FLC is displaced from AKAP signaling islands, leading to abnormal phosphorylation of substrates residing in distal subcellular compartments. Additionally, we established that DNAJ-PKAc drives enhanced cell survival through interaction with BAG2 and that this oncogenic mechanism can be sensitized to pro-apoptotic drugs via BAG2 deletion or inhibition of Bcl-2. Future studies will undoubtably focus on discerning the therapeutic value of targeting the DNAJ-PKAc/BAG2/Bcl-2 axis in FLC.
Limitations of the study
There are limitations to the current study and clear directions for future investigation. Our data show that the scaffolding function of DNAJ-PKAc contributes to cancer progression, but an important unanswered question is to what extent the kinase activity of the fusion enzyme augments oncogenesis. Further studies with the highly selective PKAc inhibitor compound BLU2864 and the protein kinase inhibitor (PKI) protein are on the horizon. Although we demonstrate that BAG2 interacts with the fusion kinase, this athanogene is also a co-chaperone for heat shock protein 70 (Hsp70). In fact, the BAG domain, common to all BAG family members, promotes nucleotide release via interaction with the nucleotide binding domain of Hsp70. The three-dimensional organization of the DNAJ-PKAc/Hsp70/BAG2 axis is unresolved and is a topic for future structural analyses. A deeper molecular understanding of FLC has been hampered due to the scarcity of suitable animal models and lack of phospho-antibodies against BAG2. Existing mouse models of FLC do not accurately recapitulate disease onset. However, more reliable PDX models are just now becoming available. Future studies will also be able to take advantage of elegant CRISPR-engineered human hepatocyte organoids that express an engineered allele of DNAJ-PKAc. The relationship of BAG2 with Bcl-2 is an interesting discovery but a topic that requires more detailed study. Obvious questions that remain include how BAG2 impacts Bcl-2 action and how changes in the phosphorylation status of the anthanogene affect modulation of apoptosis.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, John D. Scott (scottjdw@uw.edu).
Materials availability
Plasmids and cell lines generated for this study are available upon request.
Data and code availability
The proximity biotinylation mass spectrometry proteomics and phosphoproteomics data have been deposited to MassIVE with the identifier MSV000092440 (ftp://massive.ucsd.edu/MSV000092440/) and are available as of the date of publication. Accession numbers are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| V5-Tag | Thermo Fisher |
R96025 RRID:AB_2556564 |
| Alexa Fluor™ 488, donkey anti-mouse | Thermo Fisher | A21202 RRID:AB_141607 |
| Pierce™ High Sensitivity NeutrAvidin™-HRP | Thermo Fisher | 31030 |
| PKAcα, mouse | BD Biosciences | 610981 |
| Alexa Fluor™ 647, donkey anti-rabbit | Thermo Fisher |
A31573 RRID:AB_2536183 |
| BAG2, polyclonal rabbit | Invitrogen | PA5-78853 |
| Hsp70, polyclonal rabbit | Proteintech | 10995-1-AP |
| V5-HRP | Invitrogen | 46–0708 |
| Puromycin, clone 12D10 | Millipore | MABE343 |
| RFP | Rockland | 200-101-379 |
| Bcl-2, monoclonal rabbit | Invitrogen | MA5-41210 |
| Cleaved PARP (Asp214) (D6X6X) | Cell Signaling Technology | 94885 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| Stable Competent Cells | NEB | C3040I |
| GC10 Competent Cells | Genesee | 42–659 |
| One Shot™ Stbl3™ Competent Cells | Invitrogen | C737303 |
|
| ||
| Biological samples | ||
|
| ||
| Human liver tissue (FLC and non-tumor liver) | Obtained via R.S.Y., UW Department of Surgery | N/A |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Blasticidin S HCl | Invitrogen | 46–1120 |
| Doxycycline hyclate | Sigma | D9891 |
| Biotin | Sigma | B4501 |
| Polybrene | Santa Cruz | sc-134220 |
| DMEM/F-12 Medium, HEPES, no phenol red | Life Technologies | 11039047 |
| DPBS (PBS) | Thermo Fisher | 14190144 |
| 0.25% Trypsin-EDTA, phenol red | Thermo Fisher | 25200056 |
| Paraformaldehyde, 16% Solution, EM Grade | Electron Microscopy Sciences | 15710 |
| Albumin, Bovine (BSA) | VWR | 0332 |
| Normal Donkey Serum | Jackson ImmunoResearch | 017-000-121 |
| ProLong® Diamond Antifade Mountant | Thermo Fisher | P36962 |
| Triton™ X-100 | Sigma | T9284 |
| Tween® 20 | Sigma | P2287 |
| DMSO | Sigma | D2438 |
| NanoLink® magnetic streptavidin beads | Tri-link Biotechnologies, Vector Labs | M-1002-020 |
| Urea | Fisher Scientific | BP169-10 |
| Tris HCl powder | Fisher Scientific | H5123 |
| TCEP-HCl | Goldbio | TCEP25 |
| 2-Chloroacetamide (CAM) | Sigma | C0267 |
| Triethylammonium bicarbonate buffer, 1 M, pH 8.5 | Sigma | T7408 |
| Endoproteinase LysC | NEB | P8109S |
| Pierce™ Trypsin protease, MS-grade | Thermo Fisher | 90057 |
| Sodium fluoride | Sigma | S7920 |
| Tergitol (NP-40) | Sigma | NP-40 |
| Sodium deoxycholate | Sigma | D6750 |
| Sodium dodecyl sulfate | Fisher | BP166-500 |
| AEBSF | Sigma | A8456 |
| Benzamidine hydrochloride hydrate | Sigma | B6506 |
| Leupeptin | Sigma | L2884 |
| Pepstatin A | MP Biomedicals | 195368 |
| β-Glycerophosphate | Sigma | G6251 |
| 2-mercaptoethanol | Millipore | M3148 |
| Cellstar Easystrainers 40 mm | VWR | 89508–342 |
| DAPI solution (1 mg/mL) | Life Technologies | 62248 |
| TBST (10x) | Cell Signaling Technologies | 9997S |
| Sodium hydroxide | Fisher | S318-500 |
| Hydrochloric acid | Fisher | A144S-500 |
| 0.2 μm sterile filter | Fisher | 09-741-04 |
| GemCell Fetal Bovine Serum | Gemini | 100–500 |
| DMEM | Life Technologies | 11965–118 |
| Insulin, Transferrin, and Selenium (ITS) | Lonza | 17-838Z |
| Mirus Trans-IT LT | Mirus Bio | MIR 2305 |
| Lipofectamine™ 3000 Transfection Reagent | Invitrogen | L3000008 |
| Sodium chloride | Fisher | S671-3 |
| EDTA | Invitrogen | 15576–028 |
| Acetic acid, glacial | Fisher Scientific | 0714 |
| Formic acid | Sigma | F4636 |
| High-Select Fe-NTA Phosphopeptide Enrichment Kit | Thermo Scientific | A32992 |
| Puromycin dihydrochloride from Streptomyces alboniger | Sigma | P7255-25MG |
| Ponceau S solution | Sigma | P7170 |
| Protein G Agarose, Fast Flow | Millipore | 16–266 |
| cOmplete His-Tag Purification Resin | Roche | 08778850001 |
| Re-Blot Plus Strong Solution (10x) | Fisher | 2504 |
| Cycloheximide | Sigma | C1988-1G |
| Etoposide (VP-16) | Santa Cruz | sc-3512B |
| Navitoclax | Selleckchem | S1001 |
| BLU2864 | Med Chem Express | HY-150076 |
| Crystal Violet | Sigma | C3886-26 |
| RPMI | Thermo Fisher | 11875093 |
| Recombinant Human HGF (HEK293 derived) | PeproTech | 100-39H |
| Recombinant Human BAG2 His Protein | Novus Biologicals | NBP1-51025 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Pierce™ BCA Protein Assay Kit | Thermo Fisher | PI23227 |
| Cell Proliferation ELISA, Brdu (colorimetric) | Roche | 11647229001 |
| CellTiter-Glo® Luminescent Cell Viability Assay | Promega | G7572 |
| Caspase-Glo® 3/7 Assay System | Promega | G8091 |
|
| ||
| Deposited data | ||
|
| ||
| Proximity biotinylation mass spectrometry proteomics/phosphoproteomics | This paper | MassIVE: MSV000092440 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| HEK293T cell line | GE Lifesciences | HCL4517 |
| AML12 cell line | ATCC; obtained via K.J.R. by way of Nelson Fausto lab (original depositor; Wu et al.)101 | CRL-2254 RRID:CVCL_0140 |
| AML12DNAJ–PKAc cell line | Generated by R.E.T. (Turnham et al.)14 | N/A |
| FLX1 cell line | Obtained via J.D.G. by way of Bardeesy lab (original source) | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| Cloning Primer SL_AgeI_DP_fwd (TATTATACC GGTCACCATGGGTAAAGACTACTACCAGAC) | This paper | N/A |
| Cloning Primer SL_AgeI_DP_rev (TATTATACC GGTAAACTCAGAAAACTCCTTGCCACACTTC) | This paper | N/A |
| Cloning Primer ds_DP_k72h_fwd (CACATCCT CGACAAACAGAAGGTGG) | This paper | N/A |
| Cloning Primer ds_DP_k72h_rev (CATGGCATAGTGGTTCCCGG) | This paper | N/A |
| Cloning Primer SL_AgeI_mTurboGSG_fwd (TATTATACCGGTGGTTCAGGCGGCAAG CCCATCCCCAAC) |
This paper | N/A |
| Cloning Primer SL_MluI_mTurbo_rev (TATTATACGCGTTCATTCGGCAGACCGC) | This paper | N/A |
| Sequencing Primer mTrb_nTerm_R (CGACCACAGGCAGGACTG) | This paper | N/A |
| Cloning Primer SL_NheI_DP_fwd (ATTATTGCTAGCCACCATGGGTAA AGACTACTACCAGAC) |
This paper | N/A |
| Cloning Primer SL_ggAgeI_DP_rev (TATTATACCGGTCCAAACTCAGAAAA CTCCTTGCCACACTTC) |
This paper | N/A |
| Cloning Primer SL_DP_H33Q_fwd (CAACCGGACAAGAACAAGGAGC) | This paper | N/A |
| Cloning Primer SL_DP_H33Q_rev (GTAGCGCAGCGCCTG) | This paper | N/A |
| Cloning Primer SL_d14PKAc_fwd (GTGAAAGAATTCTTAGCCAAAGCC) | This paper | N/A |
| Cloning Primer SL_d14PKAc_rev (CATGGTGCCAACIIIIIIGTAC) | This paper | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| pCW57-MCS1-P2A-MCS2 (Blast) | (Barger etal.)102 | Adam Karpf; Addgene Plasmid #80921 |
| pMD2.G | Trono Lab (unpublished) | Didier Trono; Addgene Plasmid #12259 |
| psPAX2 | Trono Lab (unpublished) | Didier Trono; Addgene Plasmid #12260 |
| BAG-2 CRISPR Plasmids (m) | Santa Cruz | sc-431882 |
| pEYFP-N1_AKAP79 | Generated by M.H.O. (Omar et al.)78 | N/A |
| piRFP-N1_RIIa | Generated by M.H.O. (Omar et al.)78 | N/A |
| pPAmCherry1-N1 | (Subach et al.)103 | Vladislav Verkhusha; Addgene Plasmid #31928 |
| pPAmCherry-N1 _PKAcWT | This paper | N/A |
| pPAmCherry-N1_DNAJPKAc | This paper | N/A |
| pPAmCherry-N1_ DNAJH33QPKAc | This paper | N/A |
| pPAmCherry-N1 _PKAcΔ14 | This paper | N/A |
| pCW57-PKAcWT-V5-miniTurbo | This paper | N/A |
| pCW57-DNAJPKAc-V5-miniTurbo | This paper | N/A |
| pCW57-DNAJPKAcK72H-V5-miniTurbo | This paper | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| Perseus | https://maxquant.net/perseus/ | N/A |
| ImageJ (FIJI) | https://ImageJ.net/downloads | N/A |
| GraphPad Prism 9 | GraphPad Prism (https://graphpad.com) | N/A |
| MaxQuant | https://www.maxquant.org/ | N/A |
| STRING Database | https://string-db.org/ | N/A |
| The Gene Ontology Resource powered by PANTHER | https://geneontology.org/ | N/A |
| Enrichr | https://maayanlab.doud/Enrichr/ | N/A |
| PhosphoSitePlus® | https://www.phosphosite.org/homeAction.action | N/A |
| Cytoscape | https://cytoscape.org/ | N/A |
|
| ||
| Other | ||
|
| ||
| DynaMag™-2 Magnet | Thermo Fisher | 12321D |
| Thermo Scientific™ Low Protein Binding Collection Tubes, 1.5 mL | Thermo Fisher | 90410 |
| Eppendorf ThermoMixer® F1.5 | Eppendorf | 5384000020 |
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Human liver tissue
Human FLC tumor and paired normal liver were consented for tissue donation under IRB-approved protocols (#31281 and #51710) and obtained in collaboration with the Yeung lab in the UW Department of Surgery. De-identified patient tissue samples were either formalin-fixed, paraffin-embedded and mounted on glass slides for imaging, or fresh-frozen and stored at −80°C until homogenization for either immunoprecipitation in lysis buffer containing 2 mM EDTA, 20 mM NaF, 130 mM NaCl, 50 mM Tris pH 7.5 (at 4°C), and 1% Triton X-100, with protease inhibitors (10 μM leupeptin/pepstatin, 1 mM benzamidine, and 1 mM AEBSF) and phosphatase inhibitor (10 mM sodium β-glycerophosphate), or Western blot in RIPA lysis buffer containing (1% NP-40 Tergitol, 0.5% deoxycholate, 0.1% SDS, 130 mM NaCl, 20 mM NaF, 2 mM EDTA, and 20 mM Tris pH 7.5 (at 4°C) along with 1 mM AEBSF, 10 μM leupeptin/pepstatin, and 1 mM benzamidine).
Cell lines and cell culture
HEK293T cells for lentiviral production were obtained from GE Lifesciences and maintained in DMEM containing 10% Gemini FBS. Wildtype AML12 hepatocytes were obtained from the Riehle lab via ATCC and were developed by the Nelson Fausto lab.101 AML12DNAJ–PKAc cells were generated previously by Rigney Turnham.14 All AML12 cell lines were maintained in DMEM/F12 with 10% Gemini FBS, 1 mL 500X ITS supplement (Lonza 17–838Z; 5 μg/mL insulin, 5 μg/mL transferrin, 5 ng/mL selenium), 50 μg/mL gentamycin, and 0.1 μM dexamethasone. FLX1 cells were derived in the Bardeesy lab and were maintained in RPMI with 50 ng/mL hepatocyte growth factor, 10% FBS, 100 U/mL penicillin, and 100 U/mL streptomycin. All cell lines used in this study were grown at 37°C with 5% CO2.
Microbe strains
Amplification of non-viral mammalian expression plasmids was performed in GC10 competent cells (Genesee) and grown at 37°C. Amplification of viral vectors was performed in either Stbl3 (Invitrogen) or Stable (NEB) competent cells and grown at 30°C.
METHOD DETAILS
Antibodies
The following antibodies were used in our studies: V5-tag Thermo Fisher R96025 (IF, IP); PKAc BD 610981 (IF, WB, IP); NeutrAvidin-HRP Pierce 31030 (WB); Puromycin Millipore (MABE343); PKAc CST 5842 (WB); BAG2 Invitrogen PA5–78853 (IF, WB, IP, IHC); Hsp70 Proteintech 10995–1-AP (WB); RFP Rockland 200–101-379 (IP); RFP GenScript A00682 (WB); Bcl-2 Invitrogen MA5–41210 (WB); Cleaved PARP CST 94885 (WB).
Plasmid generation
Specific plasmids are listed in the key resources table. Standard cloning was performed using PCR (35 μL ddH2O, 10 μL 5x HF Phusion buffer (NEB), 1 μL of 10 mM mixed dNTPs, 2.5 μL combined primers at 10 mM each, 1 μL template DNA at 10 ng/mL, and 0.5 μL Hot-Start Phusion polymerase (NEB)) in a Bio-Rad thermocycler. Thermocycling protocols varied depending on primer conditions and length of target region (30 s/kb). For mutagenesis protocols, DpnI restriction enzyme and polynucleotide kinase were used (NEB). Some constructs were made using the Gateway cloning system (Thermo Fisher). Ligation was performed with T4 DNA ligase (NEB) for 10–20 min at RT or at 4°C overnight using manufacturer’s recommendations. Transformation into competent DNA (see Microbe strains above) was performed on ice for 15–30 min before heat shock for 30 s at 42°C.
Generation of BAG2 KO AML12DNAJ-PKAc cells using CRISPR/Cas9 gene editing
A pool of vectors encoding 3 different Bag2-specific gRNAs (sequences available from manufacturer upon request), Cas9 enzyme, and GFP were purchased from Santa Cruz Biotechnology, Inc (Dallas, TX). AML12 hepatocytes containing the DNAJ-PKAc mutation were transfected with the pooled plasmids using Lipofectamine 3000 Reagent. 24 h after transfection, single cells were sorted according to GFP fluorescence using FACS into 96-well plates. Clones were screened by immunoblot for loss of BAG2 expression.
Immunofluorescent staining
Cells with inducible expression of miniTurbo-fused PKAc subunit variants were plated in 48 well tissue culture dishes. 16–24 h later, doxycycline was added to induce overexpression of bait proteins. 48 h post-induction, cells were fixed with 4% paraformaldehyde in PBS for 15 min at 25°C and washed 3x with PBS. Cells were then blocked for 1 h at 25°C in 3% BSA and 0.3% Triton X-100 in PBS and primary antibodies diluted in blocking solution were applied to the cells at 4°C for 12–16 h. Following 3x washes with PBS, cells were incubated at 25°C with fluorescent secondary antibodies (used at 1:1000) and DAPI (~1:10,000). Cells were washed three more times with PBS and imaged on a Keyence BZ-X710 microscope.
Immunoblotting
Cell lysates were made using RIPA lysis buffer (1% NP-40 Tergitol, 0.5% deoxycholate, 0.1% SDS, 130 mM NaCl, 20 mM NaF, 2 mM EDTA, and 20 mM Tris pH 7.5 (at 4°C) along with 1 mM AEBSF, 10 mM leupeptin/pepstatin, and 1 mM benzamidine). Human liver protein extracts were made by homogenizing fresh frozen tissue sections in RIPA buffer. For experiments to detect S/T phosphoproteins, 10 mM β-glycerophosphate was added. Samples were incubated 5 min on ice and spun at 15,000 × g for 10 min at 4°C. Protein concentration was measured by BCA (Thermo Scientific). Gels were loaded with 15–20 mg protein after heating for 10 min at 80°C with PAGE sample buffer containing 3% (final) b-mercaptoethanol. Proteins were transferred to nitrocellulose, incubated with ponceau S to measure total protein loading, blocked in 5% milk TBST for at least 30 min at RT, and probed with antibodies in 5% BSA TBST or 5% milk TBST overnight at 4°C. Membranes were washed 3 times in TBST and then incubated with secondary antibodies conjugated to HRP diluted in 5% milk TBST for 1–2 h at RT. Following secondary antibody incubation, membranes were washed again 3 times in TBST and signals were visualized with SuperSignal West Pico Chemiluminescent Substrate (Thermo Fisher) on an Invitrogen iBright FL1000 Imaging System. Quantification was performed with ImageJ analysis software (FIJI) by measuring signal minus background for each band and dividing by the appropriate control signal, as indicated in each figure.
Proximity biotinylation and sample prep for MS
Stable AML12 cell lines were made using lentivirus encoding a tetracycline-responsive promoter and variants of PKAc tagged with V5 and miniTurbo biotin ligase at the C terminus. Doxycycline (0.5–1 μg/mL) was used to induce optimized overexpression of the bait constructs, as determined by PKAc immunoblotting. miniTurbo-tagged variant expression was induced for 48 h prior to application of 50 mM biotin in DMSO. Cells were incubated with biotin for 2 h at 37°C, washed 2 times for 1 min using 10 mL PBS to deplete excess biotin, and then lysed using RIPA buffer (as described above). Protein concentrations were measured by BCA and samples were diluted to 1 mL of 0.5 mg/mL in RIPA buffer in low protein binding collection tubes (Thermo Fisher) containing 25 μL of NanoLink magnetic streptavidin beads. Tubes were rotated 1 h at RT and placed on a magnet. Supernatant was saved for diagnostics and samples were washed in RIPA 2 times, 2 M urea in 20 mM Tris 2 times, and 25 mM Tris 2 times. For normal mass spectrometry analysis, samples were resuspended in 8 M urea in 100 mM Tris pH 8.5 with 5 mM tris(2-carboxyethyl)phosphine hydrochloride (TCEP) and 10 mM chloroacetamide (CAM) and then incubated at 37°C for 1 h. For phosphopeptide mass spectrometry analysis, samples were resuspended in 20% trifluoroethanol 25 mM Tris pH 7.8 with 5 mM TCEP and 10 mM CAM and incubated at 95°C for 5 min104,105 For digestion, samples were diluted 2-fold with 100 mM TEAB and 1 μg LysC was added before incubation for 2 h shaking at 37°C. Samples were again diluted with 100 mM TEAB and 1 μg Trypsin was added before incubation overnight shaking at 37°C. In the morning, normal mass spectrometry samples were acidified to 1% formic acid and loaded on C18 StageTips. Samples for phosphoproteomics were subjected to IMAC phosphopeptide enrichment using a Thermo Scientific High-Select Fe-NTA Phosphopeptide Enrichment Kit prior to StageTip loading.105–107
LC-MS analysis
Peptides were eluted from StageTips using elution buffer (40% acetonitrile, 1% FA) and then loaded on a self-pulled 360 mm OD × 100 mm ID 20 cm column with a 7 μm tip packed with 3 μm Reprosil C18 resin (Dr. Maisch, Germany). For pull-down experiment, peptides were analyzed by nanoLC-MS in a 90 min gradient from 15% to 38% solvent B (for phosphopeptides 6%–35% solvent B) at 300 nL/min using a Thermo EASY nLC 1200 system (solvent A: 0.1% acetic acid; solvent B: 0.1% acetic acid, 80% acetonitrile). Mass spectra were collected from an Orbitrap Fusion Lumos Tribrid Mass Spectrometer using the following settings. For MS1, Orbitrap FTMS (R = 60,000 at 200 m/z; m/z 350–1600; 7e5 target; max 20 ms ion injection time); For MS2, Top Speed data-dependent acquisition with 3 s cycle time was used, HCD MS2 spectra were collected using the Orbitrap mass analyzer(R = 30,000 at 200 m/z; 31% CE; 5e4 target; max 100 ms injection time) an intensity filter was set at 2.5e4 and dynamic exclusion for 45 s.
Mass spectrometry data analysis
Mass spectra were searched against the UniProt human reference proteome downloaded on July 06th, 2016 using MaxQuant v1.6.2.6. Detailed MaxQuant settings: for phosphopeptide analysis, samples were set to fraction 1 and 5 for WT and mutant, respectively, to allow within-group “match between run”; for pull-down, “Label-free quantification” was turned on, but not “match between run”, no fractionation was set; Trypsin/P was selected in digestion setting. Other settings were kept as default. Protein network prediction and gene ontology analysis were performed using STRING database version 11.5 and gene ontology enrichment analysis was performed using The Gene Ontology Resource powered by PANTHER. Reactome pathway analysis was performed using Enrichr. For substrate motif predictions, PhosphoSitePlus sequence logo analysis was performed on significantly enriched phosphosites for each PKAc variant. Network propagation was performed as described previously.40 Briefly, p values from all identified proteins in the proximity dataset and the most significantly altered phosphopeptide from each protein in the proximity phosphorylation dataset were mapped onto the Reactome Functional Interaction (FI) network.108 The −log10 transformed p values were then propagated and empiric network p values determined after 20,000 permutations. Results were visualized with Cytoscape, showing the propagated p value for effect size and using the −log2(fold change) from the initial proteomics results to determine the direction of the effect.
Photoactivation assay
Wildtype AML12 hepatocytes were grown in glass bottom 35 mm dishes and transfected using Lipofectamine 3000 48 h before imaging. Mammalian expression plasmids with CMV promoters and encoding AKAP79-YFP, RIIα-iRFP, and either WT PKAc, DNAJ-PKAc, DNAJH33Q-PKAc, or PKAcΔ14 tagged with photoactivatable mCherry were used. Imaging was performed using a GE OMX SR system. Exposure and laser intensity were optimized for each experimental replicate and held constant among experimental conditions. Photoactivation laser duration was kept under 50 ms to activate a discrete area with minimal spread in the first image collected after activation. Images were collected at 2 Hz in 3 channels. A baseline of 4 images was taken prior to activation of the PKAc fluorophore. Cells were selected for imaging only when RIIα-iRFP signal was colocalized with AKAP79 signal. Secondary screening for this was performed posthoc. Timecourses were measured using ImageJ analysis software (FIJI). A localization index (intensity of the activated region divided by intensity of cytosolic region 6–8 mm distal) was used to interrogate change in fluorescent signal localization over time (mobility). For representative images and videos, deconvolution and alignment of green and far-red channels were performed using OMX software.
Puromycin translation assay
Cells were seeded at 400,000 cells/well in a 6-well plate. 48 h after plating, media was replaced with fresh media containing puromycin (1 μM) and cells were returned to 37°C incubator. After 30 min, cells were washed twice with DPBS for 1 min and cells were lysed in RIPA buffer (as described above) for immunoblot analysis. For cells treated with BLU2864, drug was added at either 200 nM or 500 nM final concentration 1 h prior to puromycin treatment.
Reporter assay
WT AML12 and AML12DNAJ–PKAc cells were transduced with a lentiviral bicistronic reporter (Kind gift of Professor Kevan Shokat, UCSF) and stably selected with puromycin. Cells were plated in triplicate at <50% confluence to ensure active proliferation and harvested via trypsinization 16–24 h later. They were then acquired on a FACSCanto2 (Becton Dickinson).
Immunoprecipitation
Cell lysates were made using lysis buffer containing 1% Triton X-100, 130 mM NaCl, 20 mM NaF, 2 mM EDTA, and 50 mM Tris pH 7.5 (at 4°C) along with 1 mM AEBSF, 10 μM leupeptin/pepstatin, and 1 mM benzamidine. Lysates were incubated 5 min on ice and spun at 15,000 × g for 10 min at 4°C. Protein concentration was measured by BCA (Thermo Scientific) and adjusted to 0.5 mg/mL (or 1 mg/mL for human tissue using lysis buffer. Samples (500 mL) were precleared by rotating with 20 μL protein G agarose beads for 30 min at 4°C. Supernatants were then incubated with 1–2 μg of the appropriate antibody overnight. In the morning, 30 μL of protein G agarose beads were added and samples were returned to 4°C rotation for 1 h. Beads were washed with lysis buffer 3 times and centrifuged at 5000 × g following each wash, then aspirated with a 27G needle before resuspending in 1x PAGE sample buffer (3% β-mercaptoethanol, final) and heating at 80°C for 10 min. Figures are representative for at least 3 experimental replicates.
Recombinant BAG2-His pulldown
Cells lysates were made using lysis buffer containing 0.5% Triton X-100, Tris pH 7.4 (at RT), 10 mM imidazole, and 2 mM EDTA, along with 1 mM AEBSF, 10 μM leupeptin/pepstatin, and 1 mM benzamidine. Lysates were incubated with recombinant BAG2-His overnight at 4°C. In the morning, cOmplete His-Tag Purification Resin pre-charged with Ni2+ (Roche) was added and the samples were returned to 4°C rotation for 1 h. Beads were washed with lysis buffer 3 times and centrifuged at 5000 × g following each wash, then aspirated with a 27G needle before resuspending in 1x PAGE sample buffer (3% b-mercaptoethanol, final) and heating at 80°C for 10 min. Figures are representative for at least 3 experimental replicates.
Immunohistochemistry
Formalin-fixed, paraffin-embedded normal, FLC, or metastatic liver tissue sections were deparaffinized by placing slides in 100% xylenes once for 10 min and once for 5 min. Samples were then rehydrated by placing slides in 100% ethanol twice for 10 min each, followed by 95% ethanol for 10 min, 80% ethanol for 10 min, and deionized water two times for 5 min each. Antigen retrieval was performed by placing slides in a chamber with pre-boiled 10 mM sodium citrate buffer (pH 6.0). The chamber was then placed inside of a vegetable steamer for 1 h. Slides were placed under cold running water for 10 min before permeabilization in 0.4% Triton X-100/PBS for 7 min. Blocking was carried out in 5% BSA and 10% donkey serum in PBST (containing 0.05% Tween) for 2 h at RT. For fluorescent IHC, slides were incubated with primary antibodies in 5% BSA in PBST overnight at 4°C. Cells were washed 3x in PBST for 10 min each and incubated with Alexa Fluor conjugated secondary antibodies and DAPI in 3% BSA in PBST for 1 h at RT. Slides were then washed six times for 10 min each in PBST. Samples were mounted on glass coverslips using ProLong Diamond Antifade Mountant (Thermo Fisher) and cured overnight. Images were acquired using a GE OMX SR system. Signal intensity was measured using ImageJ analysis software (FIJI) and normalized to number of cells in image field (DAPI).
Cycloheximide chase assay
Cells were seeded in a 12-well plate and allowed to recover overnight. The next morning, cycloheximide was added at a final concentration of 50 μM. Cells were harvested in RIPA lysis buffer (1% NP-40 Tergitol, 0.5% deoxycholate, 0.1% SDS, 130 mM NaCl, 20 mM NaF, 2 mM EDTA, and 20 mM Tris pH 7.5 (at 4°C) along with 1 mM AEBSF, 10 mM leupeptin/pepstatin, and 1 mM benzamidine) at 0, 2, 4, 6, and 8 h timepoints. Lysates were centrifuged at 15,000 × g for 10 min and resulting supernatants were analyzed by immunoblotting.
BrdU ELISA assay
Cells were seeded at 10,000 cells/well in a 96-well plate. Each condition was run in triplicate. Assay was optimized for cell type used and otherwise performed according to manufacturer’s protocol. 48 h after plating, BrdU labeling solution was added at 10 μM to each well and cells were returned to incubator. After 4 h, media in each well was replaced with FixDenat solution at 25°C for 30 min. FixDenat solution was then thoroughly removed and replaced by Anti-BrdU POD-conjugated antibody solution for 90 min at 25°C, followed by 3x wash with 200 μL PBS. 100 μL TMB substrate solution was then added to each well and color was allowed to develop over 30 min. Absorbance was read at 370 nm with a reference wavelength of 492 nm.
Etoposide-induced apoptosis assay
Cells were seeded at 200,000 cells/well in a 12-well plate. 48 h after plating, media was replaced with fresh media containing either DMSO or etoposide (50 μM) and cells were returned to 37°C incubator. 72 h later, cells were imaged on a Keyence BZ-X710 microscope, then washed once with DPBS and cells were lysed in RIPA buffer (as described above) for immunoblot analysis.
Cell viability assays
For crystal violet staining, cells were seeded at 50,000 cells/well in a 24-well plate. 48 h after plating, media was replaced with fresh media containing either DMSO, etoposide (50 μM), navitoclax (100 nM), or a combination of the two and cells were returned to 37°C incubator. After 72 h, media was removed and replaced with crystal violet solution (0.25% crystal violet powder and 10% methanol in water) for 20 min. Cells were then washed 3 times with water and plate was allowed to dry for 24 h before imaging. For CellTiter-Glo, cells were seeded at 3,000 cells/well in a 96-well plate. Cells were allowed to recover for 16–24 h, then were treated either DMSO, etoposide (50 μM), navitoclax (100 nM), or a combination of the two and returned to 37°C incubator. After 72 h, CellTiter-Glo reagent was added and plate was placed on a dual-orbital shaker for 2 min to induce cell lysis. Plate was incubated at room temperature for 10 min and luminescence was recorded using a POLARstar Omega microplate reader.
Caspase activity assay
Cells were seeded at 2,000 cells/well in a 96-well white plate (Greiner) and incubated at 37°C and 5% CO2 overnight. 48 h after plating, cells were treated with drug at a final concentration of 50 μM etoposide and/or 5 μM BLU2864 and returned to incubator. After 72 h, cells were incubated with Caspase-Glo 3/7 assay reagents at room temperature for 30 min. The assay was performed as instructed by the manufacturer, and the data was collected using a luminescence plate reader. All experiments were performed with six technical replicates.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data quantification and statistical analyses as indicated in each figure legend were performed with GraphPad Prism 9 for Mac. All data are presented with mean ± SEM unless otherwise noted in the figure legend. Individual figure legends contain specific information on statistical parameters. Experiments involving more than three conditions used one-way ANOVA with subsequent t-tests corrected for multiple comparisons. Specific statistical approaches were determined based on the distributions and parameters for each dataset.
Supplementary Material
Highlights.
The FLC fusion kinase DNAJ-PKAc is not constrained within AKAP signaling islands
Aberrant DNAJ-PKAc associations and catalytic events drive enhanced translation
The co-chaperone BAG2 is recruited to DNAJ-PKAc and is elevated in FLC tumors
Apoptotic resistance conferred by BAG2 can be targeted • using combination therapy
ACKNOWLEDGMENTS
The authors would like to thank K. Collins, L. Wordeman, J. Vicente, K. Riehle, K. Jones, and J. Nelson for technical assistance and helpful discussions. This work was supported by NIH grants T32GM775043 (to S.M.L.) and DK119192 (to J.D.S.), a research grant from the Fibrolamellar Foundation (to J.D.S.), and CA279997 (to J.D.G. and J.D.S.).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2024.113678.
REFERENCES
- 1.Gallicchio L, Daee DL, Rotunno M, Barajas R, Fagan S, Carrick DM, Divi RL, Filipski KK, Freedman AN, Gillanders EM, et al. (2021). Epidemiologic Research of Rare Cancers: Trends, Resources, and Challenges. Cancer Epidemiol. Biomarkers Prev. 30, 1305–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stransky N, Cerami E, Schalm S, Kim JL, and Lengauer C (2014). The landscape of kinase fusions in cancer. Nat. Commun. 5, 4846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Honeyman JN, Simon EP, Robine N, Chiaroni-Clarke R, Darcy DG, Lim IIP, Gleason CE, Murphy JM, Rosenberg BR, Teegan L, et al. (2014). Detection of a recurrent DNAJB1-PRKACA chimeric transcript in fibrolamellar hepatocellular carcinoma. Science 343, 1010–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zack T LK, Maisel SM, Wild J, Yaqubie A, Herman M, Knox JJ, Mayer RJ, Venook AP, Butte A, O’Neill AF, et al. (2023). Defining Incidence and Complications of Fibrolamellar Liver Cancer through Tiered Computational Analysis of Clinical Data. Nature Precision Oncology 7, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eggert T, McGlynn KA, Duffy A, Manns MP, Greten TF, and Altekruse SF (2013). Fibrolamellar hepatocellular carcinoma in the USA, 2000–2010: A detailed report on frequency, treatment and outcome based on the Surveillance, Epidemiology, and End Results database. United European Gastroenterol. J. 1, 351–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dinh TA, Jewell ML, Kanke M, Francisco A, Sritharan R, Turnham RE, Lee S, Kastenhuber ER, Wauthier E, Guy CD, et al. (2019). MicroRNA-375 Suppresses the Growth and Invasion of Fibrolamellar Carcinoma. Cell. Mol. Gastroenterol. Hepatol. 7, 803–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dinh TA, Utria AF, Barry KC, Ma R, Abou-Alfa GK, Gordan JD, Jaffee EM, Scott JD, Zucman-Rossi J, O’Neill AF, et al. (2022). A framework for fibrolamellar carcinoma research and clinical trials. Nat. Rev. Gastroenterol. Hepatol. 19, 328–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alfarouk KO, Stock CM, Taylor S, Walsh M, Muddathir AK, Verduzco D, Bashir AHH, Mohammed OY, Elhassan GO, Harguindey S, et al. (2015). Resistance to cancer chemotherapy: failure in drug response from ADME to P-gp. Cancer Cell Int. 15, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cornella H, Alsinet C, Sayols S, Zhang Z, Hao K, Cabellos L, Hoshida Y, Villanueva A, Thung S, Ward SC, et al. (2015). Unique genomic profile of fibrolamellar hepatocellular carcinoma. Gastroenterology 148, 806–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Graham RP, Lackner C, Terracciano L, González-Cantú Y, Maleszewski JJ, Greipp PT, Simon SM, and Torbenson MS (2018). Fibrolamellar carcinoma in the Carney complex: PRKAR1A loss instead of the classic DNAJB1-PRKACA fusion. Hepatology 68, 1441–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Turnham RE, and Scott JD (2016). Protein kinase A catalytic subunit isoform PRKACA; History, function and physiology. Gene 577, 101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cheung J, Ginter C, Cassidy M, Franklin MC, Rudolph MJ, Robine N, Darnell RB, and Hendrickson WA (2015). Structural insights into mis-regulation of protein kinase A in human tumors. USA 112, 1374–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tomasini MD, Wang Y, Karamafrooz A, Li G, Beuming T, Gao J, Taylor SS, Veglia G, and Simon SM (2018). Conformational Landscape of the PRKACA-DNAJB1 Chimeric Kinase, the Driver for Fibrolamellar Hepatocellular Carcinoma. Sci. Rep. 8, 720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Turnham RE, Smith FD, Kenerson HL, Omar MH, Golkowski M, Garcia I, Bauer R, Lau H-T, Sullivan KM, Langeberg LK, et al. (2019). An acquired scaffolding function of the DNAJ-PKAc fusion contributes to oncogenic signaling in fibrolamellar carcinoma. Elife 8, e44187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fujimoto A, Furuta M, Shiraishi Y, Gotoh K, Kawakami Y, Arihiro K, Nakamura T, Ueno M, Ariizumi S. i., Nguyen HH, et al. (2015). Whole-genome mutational landscape of liver cancers displaying biliary phenotype reveals hepatitis impact and molecular diversity. Nat. Commun. 6, 6120. [DOI] [PubMed] [Google Scholar]
- 16.Wang Z, Jensen MA, and Zenklusen JC (2016). A Practical Guide to The Cancer Genome Atlas (TCGA). In Statistical Genomics: Methods and Protocols, E. Mathé and S. Davis, eds. (Springer; New York: ), pp. 111–141. [DOI] [PubMed] [Google Scholar]
- 17.Vyas M, Hechtman JF, Zhang Y, Benayed R, Yavas A, Askan G, Shia J, Klimstra DS, and Basturk O (2020). DNAJB1-PRKACA fusions occur in oncocytic pancreatic and biliary neoplasms and are not specific for fibrolamellar hepatocellular carcinoma. Mod. Pathol. 33, 648–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Langeberg LK, and Scott JD (2015). Signalling scaffolds and local organization of cellular behaviour. Nat. Rev. Mol. Cell Biol. 16, 232–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith FD, Omar MH, Nygren PJ, Soughayer J, Hoshi N, Lau H-T, Snyder CG, Branon TC, Ghosh D, Langeberg LK, et al. (2018). Single nucleotide polymorphisms alter kinase anchoring and the subcellular targeting of A-kinase anchoring proteins. USA 115, E11465–E11474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gopalan J, Omar MH, Roy A, Cruz NM, Falcone J, Jones KN, Forbush KA, Himmelfarb J, Freedman BS, and Scott JD (2021). Targeting an anchored phosphatase-deacetylase unit restores renal ciliary homeostasis. Elife 10, e67828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hinke SA, Navedo MF, Ulman A, Whiting JL, Nygren PJ, Tian G, Jimenez-Caliani AJ, Langeberg LK, Cirulli V, Tengholm A, et al. (2012). Anchored phosphatases modulate glucose homeostasis. EMBO J. 31, 3991–4004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aggarwal S, Gabrovsek L, Langeberg LK, Golkowski M, Ong SE, Smith FD, and Scott JD (2019). Depletion of dAKAP1-protein kinase A signaling islands from the outer mitochondrial membrane alters breast cancer cell metabolism and motility. J. Biol. Chem. 294, 3152–3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Efendiev R, Bavencoffe A, Hu H, Zhu MX, and Dessauer CW (2013). Scaffolding by A-kinase anchoring protein enhances functional coupling between adenylyl cyclase and TRPV1 channel. J. Biol. Chem. 288, 3929–3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Scott JD, and Pawson T (2009). Cell signaling in space and time: where proteins come together and when they’re apart. Science 326, 1220–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smith FD, Esseltine JL, Nygren PJ, Veesler D, Byrne DP, Vonderach M, Strashnov I, Eyers CE, Eyers PA, Langeberg LK, and Scott JD (2017). Local protein kinase A action proceeds through intact holoenzymes. Science 356, 1288–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Scott JD, Dessauer CW, and Taskén K (2013). Creating order from chaos: cellular regulation by kinase anchoring. Annu. Rev. Pharmacol. Toxicol. 53, 187–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qin L, Guo J, Zheng Q, and Zhang H (2016). BAG2 structure, function and involvement in disease. Cell. Mol. Biol. Lett. 21, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang KM, Bae E, Ahn SG, Pang K, Park Y, Park J, Lee J, Ooshima A, Park B, Kim J, et al. (2017). Co-chaperone BAG2 Determines the Pro-oncogenic Role of Cathepsin B in Triple-Negative Breast Cancer Cells. Cell Rep. 21, 2952–2964. [DOI] [PubMed] [Google Scholar]
- 29.Yue X, Zhao Y, Liu J, Zhang C, Yu H, Wang J, Zheng T, Liu L, Li J, Feng Z, and Hu W (2015). BAG2 promotes tumorigenesis through enhancing mutant p53 protein levels and function. Elife 4, e08401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Branon TC, Bosch JA, Sanchez AD, Udeshi ND, Svinkina T, Carr SA, Feldman JL, Perrimon N, and Ting AY (2018). Efficient proximity labeling in living cells and organisms with TurboID. Nat. Biotechnol. 36, 880–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Omar MH, Lauer SM, Lau HT, Golkowski M, Ong SE, and Scott JD (2023). Proximity biotinylation to define the local environment of the protein kinase A catalytic subunit in adrenal cells. STAR Protoc. 4, 101992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Ce-pas J, Simonovic M, Doncheva NT, Morris JH, Bork P, et al. (2019). STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47, D607–D613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, and Ma’ayan A (2013). Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinf. 14, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, et al. (2016). Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90–W97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xie Z, Bailey A, Kuleshov MV, Clarke DJB, Evangelista JE, Jenkins SL, Lachmann A, Wojciechowicz ML, Kropiwnicki E, Jagodnik KM, et al. (2021). Gene Set Knowledge Discovery with Enrichr. Curr. Protoc. 1, e90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bataller R, and Brenner DA (2005). Liver fibrosis. J. Clin. Invest. 115, 209–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang P, Ma X, Song E, Chen W, Pang H, Ni D, Gao Y, Fan Y, Ding Q, Zhang Y, and Zhang X (2013). Tubulin cofactor A functions as a novel positive regulator of ccRCC progression, invasion and metastasis. Int. J. Cancer 133, 2801–2811. [DOI] [PubMed] [Google Scholar]
- 38.Parker AL, Teo WS, McCarroll JA, and Kavallaris M (2017). An Emerging Role for Tubulin Isotypes in Modulating Cancer Biology and Chemotherapy Resistance. Int. J. Mol. Sci. 18, 1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hennessy F, Nicoll WS, Zimmermann R, Cheetham ME, and Blatch GL (2005). Not all J domains are created equal: implications for the specificity of Hsp40-Hsp70 interactions. Protein Sci. 14, 1697–1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chan GKL, Maisel S, Hwang YC, Pascual BC, Wolber RRB, Vu P, Patra KC, Bouhaddou M, Kenerson HL, Lim HC, et al. (2023). Oncogenic PKA signaling increases c-MYC protein expression through multiple targetable mechanisms. Elife 12, e69521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Penzo M, Montanaro L, Trere D, and Derenzini M (2019). The Ribosome Biogenesis-Cancer Connection. Cells 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zisi A, Bartek J, and Lindström MS (2022). Targeting Ribosome Biogenesis in Cancer: Lessons Learned and Way Forward. Cancers 14, 2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Frankiw L, Baltimore D, and Li G (2019). Alternative mRNA splicing in cancer immunotherapy. Nat. Rev. Immunol. 19, 675–687. [DOI] [PubMed] [Google Scholar]
- 44.Blijlevens M, Li J, and van Beusechem VW (2021). Biology of the mRNA Splicing Machinery and Its Dysregulation in Cancer Providing Therapeutic Opportunities. Int. J. Mol. Sci. 22, 5110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cowen L, Ideker T, Raphael BJ, and Sharan R (2017). Network propagation: a universal amplifier of genetic associations. Nat. Rev. Genet. 18, 551–562. [DOI] [PubMed] [Google Scholar]
- 46.Bordeleau ME, Mori A, Oberer M, Lindqvist L, Chard LS, Higa T, Belsham GJ, Wagner G, Tanaka J, and Pelletier J (2006). Functional characterization of IRESes by an inhibitor of the RNA helicase eIF4A. Nat. Chem. Biol. 2, 213–220. [DOI] [PubMed] [Google Scholar]
- 47.Pelletier J, Graff J, Ruggero D, and Sonenberg N (2015). Targeting the eIF4F translation initiation complex: a critical nexus for cancer development. Cancer Res. 75, 250–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xu Z, Page RC, Gomes MM, Kohli E, Nix JC, Herr AB, Patterson C, and Misra S (2008). Structural basis of nucleotide exchange and client binding by the Hsp70 cochaperone Bag2. Nat. Struct. Mol. Biol. 15, 1309–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Arndt V, Daniel C, Nastainczyk W, Alberti S, and Höhfeld J (2005). BAG-2 acts as an inhibitor of the chaperone-associated ubiquitin ligase CHIP. Mol. Biol. Cell 16, 5891–5900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dai Q, Qian SB, Li HH, McDonough H, Borchers C, Huang D, Takayama S, Younger JM, Ren HY, Cyr DM, and Patterson C (2005). Regulation of the cytoplasmic quality control protein degradation pathway by BAG2. J. Biol. Chem. 280, 38673–38681. [DOI] [PubMed] [Google Scholar]
- 51.Yoon CI, Ahn SG, Cha YJ, Kim D, Bae SJ, Lee JH, Ooshima A, Yang KM, Park SH, Kim SJ, and Jeong J (2021). Metastasis Risk Assessment Using BAG2 Expression by Cancer-Associated Fibroblast and Tumor Cells in Patients with Breast Cancer . Cancers 13, 4654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shabb JB (2001). Physiological substrates of cAMP-dependent protein kinase. Chem. Rev. 101, 2381–2411. [DOI] [PubMed] [Google Scholar]
- 53.Stokoe D, Caudwell B, Cohen PT, and Cohen P (1993). The substrate specificity and structure of mitogen-activated protein (MAP) kinase-activated protein kinase-2. Biochem. J. 296, 843–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ueda K, Kosako H, Fukui Y, and Hattori S (2004). Proteomic identification of Bcl2-associated athanogene 2 as a novel MAPK-activated protein kinase 2 substrate. J. Biol. Chem. 279, 41815–41821. [DOI] [PubMed] [Google Scholar]
- 55.Huang X, Shi D, Zou X, Wu X, Huang S, Kong L, Yang M, Xiao Y, Chen B, Chen X, et al. (2023). BAG2 drives chemoresistance of breast cancer by exacerbating mutant p53 aggregate. Theranostics 13, 339–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang X, Zhang J, Liu Y, Li J, Tan J, and Song Z (2021). Bcl-2 Associated Athanogene 2 (BAG2) is Associated With Progression and Prognosis of Hepatocellular Carcinoma: A Bioinformatics-Based Analysis. Pathol. Oncol. Res. 27, 594649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Francisco AB, Kanke M, Massa AP, Dinh TA, Sritharan R, Vakili K, Bardeesy N, and Sethupathy P (2022). Multiomic analysis of micro-RNA-mediated regulation reveals a proliferative axis involving miR-10b in fibrolamellar carcinoma. JCI insight 7, e154743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Takayama S, Sato T, Krajewski S, Kochel K, Irie S, Millan JA, and Reed JC (1995). Cloning and functional analysis of BAG-1: a novel Bcl-2-binding protein with anti-cell death activity. Cell 80, 279–284. [DOI] [PubMed] [Google Scholar]
- 59.Lee JH, Takahashi T, Yasuhara N, Inazawa J, Kamada S, and Tsujimoto Y (1999). Bis, a Bcl-2-binding protein that synergizes with Bcl-2 in preventing cell death. Oncogene 18, 6183–6190. [DOI] [PubMed] [Google Scholar]
- 60.Shi CS, and Kehrl JH (2019). Bcl-2 regulates pyroptosis and necroptosis by targeting BH3-like domains in GSDMD and MLKL. Cell Death Dis. 5, 151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chaitanya GV, Steven AJ, and Babu PP (2010). PARP-1 cleavage fragments: signatures of cell-death proteases in neurodegeneration. Cell Commun. Signal. 8, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Broecker-Preuss M, Becher-Boveleth N, Müller S, and Mann K (2016). The BH3 mimetic drug ABT-737 induces apoptosis and acts synergistically with chemotherapeutic drugs in thyroid carcinoma cells. Cancer Cell Int. 16, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Townsend PA, Kozhevnikova MV, Cexus ONF, Zamyatnin AA Jr., and Soond SM (2021). BH3-mimetics: recent developments in cancer therapy. J. Exp. Clin. Cancer Res. 40, 355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S, Johnson EF, Marsh KC, Mitten MJ, Nimmer P, et al. (2008). ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 68, 3421–3428. [DOI] [PubMed] [Google Scholar]
- 65.Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, et al. (2013). Mutational landscape and significance across 12 major cancer types. Nature 502, 333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Xu L, Hazard FK, Zmoos AF, Jahchan N, Chaib H, Garfin PM, Rangaswami A, Snyder MP, and Sage J (2015). Genomic analysis of fibrolamellar hepatocellular carcinoma. Hum. Mol. Genet. 24, 50–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Engelholm LH, Riaz A, Serra D, Dagnæs-Hansen F, Johansen JV, Santoni-Rugiu E, Hansen SH, Niola F, and Frödin M (2017). CRISPR/Cas9 Engineering of Adult Mouse Liver Demonstrates That the Dnajb1-Prkaca Gene Fusion Is Sufficient to Induce Tumors Resembling Fibrolamellar Hepatocellular Carcinoma. Gastroenterology 153, 1662–1673.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Oikawa T, Wauthier E, Dinh TA, Selitsky SR, Reyna-Neyra A, Carpino G, Levine R, Cardinale V, Klimstra D, Gaudio E, et al. (2015). Model of fibrolamellar hepatocellular carcinomas reveals striking enrichment in cancer stem cells. Nat. Commun. 6, 8070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kastenhuber ER, Lalazar G, Houlihan SL, Tschaharganeh DF, Baslan T, Chen CC, Requena D, Tian S, Bosbach B, Wilkinson JE, et al. (2017). DNAJB1-PRKACA fusion kinase interacts with beta-catenin and the liver regenerative response to drive fibrolamellar hepatocellular carcinoma. USA 114, 13076–13084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dinh TA, Vitucci ECM, Wauthier E, Graham RP, Pitman WA, Oikawa T, Chen M, Silva GO, Greene KG, Torbenson MS, et al. (2017). Comprehensive analysis of The Cancer Genome Atlas reveals a unique gene and non-coding RNA signature of fibrolamellar carcinoma. Sci. Rep. 7, 44653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Riggle KM, Riehle KJ, Kenerson HL, Turnham R, Homma MK, Kazami M, Samelson B, Bauer R, McKnight GS, Scott JD, and Yeung RS (2016). Enhanced cAMP-stimulated protein kinase A activity in human fibrolamellar hepatocellular carcinoma. Pediatr. Res. 80, 110–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dinh TA, Sritharan R, Smith FD, Francisco AB, Ma RK, Bunaciu RP, Kanke M, Danko CG, Massa AP, Scott JD, and Sethupathy P (2020). Hotspots of Aberrant Enhancer Activity in Fibrolamellar Carcinoma Reveal Candidate Oncogenic Pathways and Therapeutic Vulnerabilities. Cell Rep. 31, 107509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Taskén K, and Aandahl EM (2004). Localized effects of cAMP mediated by distinct routes of protein kinase A. Physiol. Rev. 84, 137–167. [DOI] [PubMed] [Google Scholar]
- 74.Bauman AL, Soughayer J, Nguyen BT, Willoughby D, Carnegie GK, Wong W, Hoshi N, Langeberg LK, Cooper DMF, Dessauer CW, and Scott JD (2006). Dynamic regulation of cAMP synthesis through anchored PKA-adenylyl cyclase V/VI complexes. Mol. Cell 23, 925–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Omar MH, and Scott JD (2020). AKAP Signaling Islands: Venues for Precision Pharmacology. Trends Pharmacol. Sci. 41, 933–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Smith FD, Reichow SL, Esseltine JL, Shi D, Langeberg LK, Scott JD, and Gonen T (2013). Intrinsic disorder within an AKAP-protein kinase A complex guides local substrate phosphorylation. Elife 2, e01319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mayer MP, and Bukau B (2005). Hsp70 chaperones: cellular functions and molecular mechanism. Cell. Mol. Life Sci. 62, 670–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Omar MH, Byrne DP, Jones KN, Lakey TM, Collins KB, Lee KS, Daly LA, Forbush KA, Lau HT, Golkowski M, et al. (2022). Mislocalization of protein kinase A drives pathology in Cushing’s syndrome. Cell Rep. 40, 111073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bathon K, Weigand I, Vanselow JT, Ronchi CL, Sbiera S, Schlosser A, Fassnacht M, and Calebiro D (2019). Alterations in Protein Kinase A Substrate Specificity as a Potential Cause of Cushing Syndrome. Endocrinology 160, 447–459. [DOI] [PubMed] [Google Scholar]
- 80.Lubner JM, Dodge-Kafka KL, Carlson CR, Church GM, Chou MF, and Schwartz D (2017). Cushing’s syndrome mutant PKA(L)(205R) exhibits altered substrate specificity. FEBS Lett. 591, 459–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Walker C, Wang Y, Olivieri C, Karamafrooz A, Casby J, Bathon K, Calebiro D, Gao J, Bernlohr DA, Taylor SS, and Veglia G (2019). Cushing’s syndrome driver mutation disrupts protein kinase A allosteric network, altering both regulation and substrate specificity. Sci. Adv. 5, eaaw9298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Riehle KJ, Yeh MM, Yu JJ, Kenerson HL, Harris WP, Park JO, and Yeung RS (2015). mTORC1 and FGFR1 signaling in fibrolamellar hepatocellular carcinoma. Mod. Pathol. 28, 103–110. [DOI] [PubMed] [Google Scholar]
- 83.Lafaro KJ, and Pawlik TM (2015). Fibrolamellar hepatocellular carcinoma: current clinical perspectives. J. Hepatocell. Carcinoma 2, 151–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Takayama S, and Reed JC (2001). Molecular chaperone targeting and regulation by BAG family proteins. Nat. Cell Biol. 3, E237–E241. [DOI] [PubMed] [Google Scholar]
- 85.Doong H, Vrailas A, and Kohn EC (2002). What’s in the ‘BAG’?–A functional domain analysis of the BAG-family proteins. Cancer Lett. 188, 25–32. [DOI] [PubMed] [Google Scholar]
- 86.Kabbage M, and Dickman MB (2008). The BAG proteins: a ubiquitous family of chaperone regulators. Cell. Mol. Life Sci. 65, 1390–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sherman MY, and Gabai VL (2015). Hsp70 in cancer: back to the future. Oncogene 34, 4153–4161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wang HQ, Zhang HY, Hao FJ, Meng X, Guan Y, and Du ZX (2008). Induction of BAG2 protein during proteasome inhibitor-induced apoptosis in thyroid carcinoma cells. Br. J. Pharmacol. 155, 655–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sun L, Chen G, Sun A, Wang Z, Huang H, Gao Z, Liang W, Liu C, and Li K (2020). BAG2 Promotes Proliferation and Metastasis of Gastric Cancer via ERK1/2 Signaling and Partially Regulated by miR186. Front. Oncol. 10, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yu H, Ding J, Zhu H, Jing Y, Zhou H, Tian H, Tang K, Wang G, and Wang X (2020). LOXL1 confers antiapoptosis and promotes glioma-genesis through stabilizing BAG2. Cell Death Differ. 27, 3021–3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhang X, Dong K, Zhang J, Kuang T, Luo Y, Yu J, Yu J, and Wang W (2023). GNB1 promotes hepatocellular carcinoma progression by targeting BAG2 to activate P38/MAPK signaling. Cancer Sci. 114, 2001–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kudaravalli S, den Hollander P, and Mani SA (2022). Role of p38 MAP kinase in cancer stem cells and metastasis. Oncogene 41, 3177–3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Oltvai ZN, Milliman CL, and Korsmeyer SJ (1993). Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell 74, 609–619. [DOI] [PubMed] [Google Scholar]
- 94.Qian S, Wei Z, Yang W, Huang J, Yang Y, and Wang J (2022). The role of BCL-2 family proteins in regulating apoptosis and cancer therapy. Front. Oncol. 12, 985363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Song Z, Xu S, Song B, and Zhang Q (2015). Bcl-2-associated athanogene 2 prevents the neurotoxicity of MPP+ via interaction with DJ-1. J. Mol. Neurosci. 55, 798–802. [DOI] [PubMed] [Google Scholar]
- 96.Liang S, Song Z, Wu Y, Gao Y, Gao M, Liu F, Wang F, and Zhang Y (2018). MicroRNA-27b Modulates Inflammatory Response and Apoptosis during Mycobacterium tuberculosis Infection. J. Immunol. 200, 3506–3518. [DOI] [PubMed] [Google Scholar]
- 97.Hande KR (1998). Etoposide: four decades of development of a topoisomerase II inhibitor. Eur. J. Cancer 34, 1514–1521. [DOI] [PubMed] [Google Scholar]
- 98.Potluri J, Harb J, Masud AA, and Hutti JE (2020). A Phase 3, Double-Blind, Placebo-Controlled, Randomized Study Evaluating Navitoclax in Combination with Ruxolitinib in Patients with Myelofibrosis (TRANSFORM-1). Blood 136, 4.32614961 [Google Scholar]
- 99.Harrison CN, Garcia JS, Somervaille TCP, Foran JM, Verstovsek S, Jamieson C, Mesa R, Ritchie EK, Tantravahi SK, Vachhani P, et al. (2022). Addition of Navitoclax to Ongoing Ruxolitinib Therapy for Patients With Myelofibrosis With Progression or Suboptimal Response: Phase II Safety and Efficacy. J. Clin. Oncol. 40, 1671–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Rüland L, Andreatta F, Massalini S, Chuva de Sousa Lopes S, Clevers H, Hendriks D, and Artegiani B. (2023). Organoid models of fibrolamellar carcinoma mutations reveal hepatocyte transdifferentiation through cooperative BAP1 and PRKAR2A loss. Nat. Commun. 14, 2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wu JC, Merlino G, and Fausto N (1994). Establishment and characterization of differentiated, nontransformed hepatocyte cell lines derived from mice transgenic for transforming growth factor alpha. USA 91, 674–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Barger CJ, Branick C, Chee L, and Karpf AR (2019). Pan-Cancer Analyses Reveal Genomic Features of FOXM1 Overexpression in Cancer. Cancers 11, 251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Subach FV, Patterson GH, Manley S, Gillette JM, Lippincott-Schwartz J, and Verkhusha VV (2009). Photoactivatable mCherry for high-resolution two-color fluorescence microscopy. Nat. Methods 6, 153–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wang H, Qian WJ, Mottaz HM, Clauss TRW, Anderson DJ, Moore RJ, Camp DG 2nd, Khan AH, Sforza DM, Pallavicini M, et al. (2005). Development and evaluation of a micro- and nanoscale proteomic sample preparation method. J. Proteome Res. 4, 2397–2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Golkowski M, Vidadala VN, Lau HT, Shoemaker A, Shimizu-Albergine M, Beavo J, Maly DJ, and Ong SE (2020). Kinobead/LC-MS Phosphokinome Profiling Enables Rapid Analyses of Kinase-Dependent Cell Signaling Networks. J. Proteome Res. 19, 1235–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Villén J, and Gygi SP (2008). The SCX/IMAC enrichment approach for global phosphorylation analysis by mass spectrometry. Nat. Protoc. 3, 1630–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Rappsilber J, Mann M, and Ishihama Y (2007). Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2, 1896–1906. [DOI] [PubMed] [Google Scholar]
- 108.Fabregat A, Jupe S, Matthews L, Sidiropoulos K, Gillespie M, Garapati P, Haw R, Jassal B, Korninger F, May B, et al. (2018). The Reactome Pathway Knowledgebase. Nucleic Acids Res. 46, D649–D655. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The proximity biotinylation mass spectrometry proteomics and phosphoproteomics data have been deposited to MassIVE with the identifier MSV000092440 (ftp://massive.ucsd.edu/MSV000092440/) and are available as of the date of publication. Accession numbers are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| V5-Tag | Thermo Fisher |
R96025 RRID:AB_2556564 |
| Alexa Fluor™ 488, donkey anti-mouse | Thermo Fisher | A21202 RRID:AB_141607 |
| Pierce™ High Sensitivity NeutrAvidin™-HRP | Thermo Fisher | 31030 |
| PKAcα, mouse | BD Biosciences | 610981 |
| Alexa Fluor™ 647, donkey anti-rabbit | Thermo Fisher |
A31573 RRID:AB_2536183 |
| BAG2, polyclonal rabbit | Invitrogen | PA5-78853 |
| Hsp70, polyclonal rabbit | Proteintech | 10995-1-AP |
| V5-HRP | Invitrogen | 46–0708 |
| Puromycin, clone 12D10 | Millipore | MABE343 |
| RFP | Rockland | 200-101-379 |
| Bcl-2, monoclonal rabbit | Invitrogen | MA5-41210 |
| Cleaved PARP (Asp214) (D6X6X) | Cell Signaling Technology | 94885 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| Stable Competent Cells | NEB | C3040I |
| GC10 Competent Cells | Genesee | 42–659 |
| One Shot™ Stbl3™ Competent Cells | Invitrogen | C737303 |
|
| ||
| Biological samples | ||
|
| ||
| Human liver tissue (FLC and non-tumor liver) | Obtained via R.S.Y., UW Department of Surgery | N/A |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Blasticidin S HCl | Invitrogen | 46–1120 |
| Doxycycline hyclate | Sigma | D9891 |
| Biotin | Sigma | B4501 |
| Polybrene | Santa Cruz | sc-134220 |
| DMEM/F-12 Medium, HEPES, no phenol red | Life Technologies | 11039047 |
| DPBS (PBS) | Thermo Fisher | 14190144 |
| 0.25% Trypsin-EDTA, phenol red | Thermo Fisher | 25200056 |
| Paraformaldehyde, 16% Solution, EM Grade | Electron Microscopy Sciences | 15710 |
| Albumin, Bovine (BSA) | VWR | 0332 |
| Normal Donkey Serum | Jackson ImmunoResearch | 017-000-121 |
| ProLong® Diamond Antifade Mountant | Thermo Fisher | P36962 |
| Triton™ X-100 | Sigma | T9284 |
| Tween® 20 | Sigma | P2287 |
| DMSO | Sigma | D2438 |
| NanoLink® magnetic streptavidin beads | Tri-link Biotechnologies, Vector Labs | M-1002-020 |
| Urea | Fisher Scientific | BP169-10 |
| Tris HCl powder | Fisher Scientific | H5123 |
| TCEP-HCl | Goldbio | TCEP25 |
| 2-Chloroacetamide (CAM) | Sigma | C0267 |
| Triethylammonium bicarbonate buffer, 1 M, pH 8.5 | Sigma | T7408 |
| Endoproteinase LysC | NEB | P8109S |
| Pierce™ Trypsin protease, MS-grade | Thermo Fisher | 90057 |
| Sodium fluoride | Sigma | S7920 |
| Tergitol (NP-40) | Sigma | NP-40 |
| Sodium deoxycholate | Sigma | D6750 |
| Sodium dodecyl sulfate | Fisher | BP166-500 |
| AEBSF | Sigma | A8456 |
| Benzamidine hydrochloride hydrate | Sigma | B6506 |
| Leupeptin | Sigma | L2884 |
| Pepstatin A | MP Biomedicals | 195368 |
| β-Glycerophosphate | Sigma | G6251 |
| 2-mercaptoethanol | Millipore | M3148 |
| Cellstar Easystrainers 40 mm | VWR | 89508–342 |
| DAPI solution (1 mg/mL) | Life Technologies | 62248 |
| TBST (10x) | Cell Signaling Technologies | 9997S |
| Sodium hydroxide | Fisher | S318-500 |
| Hydrochloric acid | Fisher | A144S-500 |
| 0.2 μm sterile filter | Fisher | 09-741-04 |
| GemCell Fetal Bovine Serum | Gemini | 100–500 |
| DMEM | Life Technologies | 11965–118 |
| Insulin, Transferrin, and Selenium (ITS) | Lonza | 17-838Z |
| Mirus Trans-IT LT | Mirus Bio | MIR 2305 |
| Lipofectamine™ 3000 Transfection Reagent | Invitrogen | L3000008 |
| Sodium chloride | Fisher | S671-3 |
| EDTA | Invitrogen | 15576–028 |
| Acetic acid, glacial | Fisher Scientific | 0714 |
| Formic acid | Sigma | F4636 |
| High-Select Fe-NTA Phosphopeptide Enrichment Kit | Thermo Scientific | A32992 |
| Puromycin dihydrochloride from Streptomyces alboniger | Sigma | P7255-25MG |
| Ponceau S solution | Sigma | P7170 |
| Protein G Agarose, Fast Flow | Millipore | 16–266 |
| cOmplete His-Tag Purification Resin | Roche | 08778850001 |
| Re-Blot Plus Strong Solution (10x) | Fisher | 2504 |
| Cycloheximide | Sigma | C1988-1G |
| Etoposide (VP-16) | Santa Cruz | sc-3512B |
| Navitoclax | Selleckchem | S1001 |
| BLU2864 | Med Chem Express | HY-150076 |
| Crystal Violet | Sigma | C3886-26 |
| RPMI | Thermo Fisher | 11875093 |
| Recombinant Human HGF (HEK293 derived) | PeproTech | 100-39H |
| Recombinant Human BAG2 His Protein | Novus Biologicals | NBP1-51025 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Pierce™ BCA Protein Assay Kit | Thermo Fisher | PI23227 |
| Cell Proliferation ELISA, Brdu (colorimetric) | Roche | 11647229001 |
| CellTiter-Glo® Luminescent Cell Viability Assay | Promega | G7572 |
| Caspase-Glo® 3/7 Assay System | Promega | G8091 |
|
| ||
| Deposited data | ||
|
| ||
| Proximity biotinylation mass spectrometry proteomics/phosphoproteomics | This paper | MassIVE: MSV000092440 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| HEK293T cell line | GE Lifesciences | HCL4517 |
| AML12 cell line | ATCC; obtained via K.J.R. by way of Nelson Fausto lab (original depositor; Wu et al.)101 | CRL-2254 RRID:CVCL_0140 |
| AML12DNAJ–PKAc cell line | Generated by R.E.T. (Turnham et al.)14 | N/A |
| FLX1 cell line | Obtained via J.D.G. by way of Bardeesy lab (original source) | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| Cloning Primer SL_AgeI_DP_fwd (TATTATACC GGTCACCATGGGTAAAGACTACTACCAGAC) | This paper | N/A |
| Cloning Primer SL_AgeI_DP_rev (TATTATACC GGTAAACTCAGAAAACTCCTTGCCACACTTC) | This paper | N/A |
| Cloning Primer ds_DP_k72h_fwd (CACATCCT CGACAAACAGAAGGTGG) | This paper | N/A |
| Cloning Primer ds_DP_k72h_rev (CATGGCATAGTGGTTCCCGG) | This paper | N/A |
| Cloning Primer SL_AgeI_mTurboGSG_fwd (TATTATACCGGTGGTTCAGGCGGCAAG CCCATCCCCAAC) |
This paper | N/A |
| Cloning Primer SL_MluI_mTurbo_rev (TATTATACGCGTTCATTCGGCAGACCGC) | This paper | N/A |
| Sequencing Primer mTrb_nTerm_R (CGACCACAGGCAGGACTG) | This paper | N/A |
| Cloning Primer SL_NheI_DP_fwd (ATTATTGCTAGCCACCATGGGTAA AGACTACTACCAGAC) |
This paper | N/A |
| Cloning Primer SL_ggAgeI_DP_rev (TATTATACCGGTCCAAACTCAGAAAA CTCCTTGCCACACTTC) |
This paper | N/A |
| Cloning Primer SL_DP_H33Q_fwd (CAACCGGACAAGAACAAGGAGC) | This paper | N/A |
| Cloning Primer SL_DP_H33Q_rev (GTAGCGCAGCGCCTG) | This paper | N/A |
| Cloning Primer SL_d14PKAc_fwd (GTGAAAGAATTCTTAGCCAAAGCC) | This paper | N/A |
| Cloning Primer SL_d14PKAc_rev (CATGGTGCCAACIIIIIIGTAC) | This paper | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| pCW57-MCS1-P2A-MCS2 (Blast) | (Barger etal.)102 | Adam Karpf; Addgene Plasmid #80921 |
| pMD2.G | Trono Lab (unpublished) | Didier Trono; Addgene Plasmid #12259 |
| psPAX2 | Trono Lab (unpublished) | Didier Trono; Addgene Plasmid #12260 |
| BAG-2 CRISPR Plasmids (m) | Santa Cruz | sc-431882 |
| pEYFP-N1_AKAP79 | Generated by M.H.O. (Omar et al.)78 | N/A |
| piRFP-N1_RIIa | Generated by M.H.O. (Omar et al.)78 | N/A |
| pPAmCherry1-N1 | (Subach et al.)103 | Vladislav Verkhusha; Addgene Plasmid #31928 |
| pPAmCherry-N1 _PKAcWT | This paper | N/A |
| pPAmCherry-N1_DNAJPKAc | This paper | N/A |
| pPAmCherry-N1_ DNAJH33QPKAc | This paper | N/A |
| pPAmCherry-N1 _PKAcΔ14 | This paper | N/A |
| pCW57-PKAcWT-V5-miniTurbo | This paper | N/A |
| pCW57-DNAJPKAc-V5-miniTurbo | This paper | N/A |
| pCW57-DNAJPKAcK72H-V5-miniTurbo | This paper | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| Perseus | https://maxquant.net/perseus/ | N/A |
| ImageJ (FIJI) | https://ImageJ.net/downloads | N/A |
| GraphPad Prism 9 | GraphPad Prism (https://graphpad.com) | N/A |
| MaxQuant | https://www.maxquant.org/ | N/A |
| STRING Database | https://string-db.org/ | N/A |
| The Gene Ontology Resource powered by PANTHER | https://geneontology.org/ | N/A |
| Enrichr | https://maayanlab.doud/Enrichr/ | N/A |
| PhosphoSitePlus® | https://www.phosphosite.org/homeAction.action | N/A |
| Cytoscape | https://cytoscape.org/ | N/A |
|
| ||
| Other | ||
|
| ||
| DynaMag™-2 Magnet | Thermo Fisher | 12321D |
| Thermo Scientific™ Low Protein Binding Collection Tubes, 1.5 mL | Thermo Fisher | 90410 |
| Eppendorf ThermoMixer® F1.5 | Eppendorf | 5384000020 |