

An E. coli platform for systematic engineering of artificial metalloenzymes that catalyze new-to-nature reactions is described.
Abstract
Artificial metalloenzymes (ArMs) catalyzing new-to-nature reactions could play an important role in transitioning toward a sustainable economy. While ArMs have been created for various transformations, attempts at their genetic optimization have been case specific and resulted mostly in modest improvements. To realize their full potential, methods to rapidly discover active ArM variants for ideally any reaction of interest are required. Here, we introduce a reaction-independent, automation-compatible platform, which relies on periplasmic compartmentalization in Escherichia coli to rapidly and reliably engineer ArMs based on the biotin-streptavidin technology. We systematically assess 400 ArM mutants for five bioorthogonal transformations involving different metals, reaction mechanisms, and reactants, which include novel ArMs for gold-catalyzed hydroamination and hydroarylation. Activity enhancements up to 15-fold highlight the potential of the systematic approach. Furthermore, we suggest smart screening strategies and build machine learning models that accurately predict ArM activity from sequence, which has crucial implications for future ArM development.
INTRODUCTION
Artificial metalloenzymes (ArMs) combine the broad reaction scope of organometallic catalysis with the exceptional catalytic performance, selectivity, and mild reaction conditions of enzymes (1, 2). Therefore, they have a great potential to enable sustainable synthetic routes to various compounds of interest (3, 4) and, if functional in a cellular environment, open up new possibilities for metabolic engineering and synthetic biology (5). ArMs have been constructed by repurposing natural metalloenzymes (6, 7), designing binding sites for metal ions (8–11), and incorporating organometallic cofactors into protein scaffolds (12–20). Moreover, unnatural amino acids (21) and photoexcitation (22, 23) have been used to unlock new reactivity in enzymes. Among these efforts to create new-to-nature biocatalysts, one of the most versatile approaches relies on the biotin-streptavidin technology. This strategy uses the high affinity of the homotetrameric protein streptavidin (Sav) for the vitamin d-biotin to noncovalently anchor biotinylated metal complexes (referred to as cofactor hereafter) within the Sav protein. Using this approach, artificial enzymes have been created for multiple reactions, including hydrogenation, sulfoxidation, C─H activation, and olefin metathesis (24–26). While in some cases wild-type Sav imparts some selectivity or rate acceleration on the reaction, protein engineering is usually required to obtain proficient biocatalysts (27–31). Initial studies in this direction relied on screening (semi-)purified Sav mutants (32, 33), but more recently, a trend toward whole-cell screening with Escherichia coli has emerged for Sav-based ArMs (34, 35) and other artificial enzymes (8, 36, 37). This allows substantially increased throughput and is the method of choice if variants that are functional under in vivo conditions are desired, which is an important prerequisite for synthetic biology applications.
However, a number of challenges arise for cell-based ArM assays, most notably insufficient cofactor uptake into the cell and cofactor poisoning by cellular components such as reduced glutathione (38). To circumvent these challenges, Sav has been exported to the periplasmic space (34) or the cell surface (35). While these studies have established the possibility of engineering ArMs using whole-cell screenings, they did so using case-specific engineering strategies and with a focus on reactions affording fluorescent products. Consequently, applicability of these methods across a wider range of reactions remains to be demonstrated. To generalize the development of ArMs, broadly applicable engineering strategies that enable the rapid identification of highly active variants for ideally any reaction of interest are needed. This requires a robust screening protocol that is compatible with various reaction conditions and analytical readouts. Moreover, it imposes a demand for Sav mutant libraries that embody a high potential to contain highly active variants for different reactions while maintaining a comparably small and thus screenable library size. Such a combination of a versatile, reaction-independent screening method with a “concise” library could serve as a universal starting point for various ArM engineering campaigns and would render tedious case-by-case method and library development obsolete. Here, we present a screening platform that meets the aforementioned requirements as a first systematic approach to ArM engineering. We establish a well plate–based screening protocol that can be easily adapted to new reaction conditions or analytical methods and show that it is amenable to lab automation as an important prerequisite for streamlined ArM engineering. Furthermore, we create a full-factorial, sequence-defined Sav library that is rich in variants with high activity by simultaneously diversifying two crucial amino acid positions in close vicinity to the catalytic metal. To demonstrate the versatility of this platform, we selected five bioorthogonal reactions requiring different cofactors, reaction conditions, and analytical readouts. The platform enabled the identification of substantially improved ArMs for all tested reactions with fast turnaround times. Moreover, the systematic characterization of the local sequence-activity landscape enabled us to identify smart library designs to further enhance the development of ArMs and to construct machine learning models that predict the activity of ArMs with high precision. This study represents the first systematic approach to ArM engineering and thus constitutes an important step toward a streamlined optimization of these promising biocatalysts.
RESULTS
Construction and characterization of a sequence-defined Sav library
To establish a generalizable first engineering step for ArMs, we sought to generate a library of Sav variants that offers a high likelihood of identifying active variants at a minimized screening effort. On the basis of previous experience with Sav expression and whole-cell ArM catalysis (34, 35), we selected a periplasmic compartmentalization strategy for this library. Secretion to the periplasm and subsequent ArM assembly represent a good trade-off between accessibility to the cofactor, expression levels, and compatibility of the reaction conditions (39). To facilitate periplasmic export in E. coli, the signal peptide of the outer membrane protein A (OmpA) was N-terminally fused to T7-tagged mature Sav (referred to as wild type hereafter) as previously reported (34). The holoenzyme (i.e., full ArM with cofactor) can then be assembled in the periplasm by incubating cells in a buffer containing a biotinylated metal cofactor (Fig. 1A). We selected the amino acid residues 112 and 121 in Sav as randomization targets, which correspond to serine and lysine in wild-type Sav. These residues are in close proximity to the biotinylated cofactor (32) (Fig. 1B) and have repeatedly been found to have a substantial impact on the activity of ArMs for diverse cofactors and reactions (33–35). More specifically, we created a combinatorial Sav 112X 121X library (X representing all 20 canonical amino acids) consisting of all 400 possible amino acid combinations for these two positions. Such a full-factorial library makes it possible to identify improved mutants that are the result of synergistic interactions between the two positions. We applied a three-step cloning and sequencing strategy (see Methods and figs. S1 to S3) to produce an arrayed, sequence-verified set of all 400 possible Sav 112X 121X mutants. This minimizes the screening effort by avoiding the requirement for oversampling.
Fig. 1. Systematic screening of ArMs.

(A) Sav is secreted to the periplasm, where it binds an externally added biotinylated cofactor, which consists of a catalytic metal M and ligands L and L′, to afford an ArM. OmpA, periplasmic export signal from outer membrane protein A. (B) Left: Biotinylated metathesis cofactor embedded in the biotin-binding vestibule of homotetrameric Sav (Protein Data Bank 5IRA). Symmetry-related residues S112/S′112 (blue) and K121/K′121 (purple) are in the immediate vicinity of the cofactor and were mutated (right) to afford an Sav library of 400 amino acid combinations. (C) Expression level of periplasmic Sav mutants as determined in cell lysate using a fluorescence quenching assay (Methods) (41). Means of biological triplicates (n = 3) normalized to the OD600 of the cultures are displayed (see fig. S4 for SD). (D) Estimated percentage of Sav mutants with unoccupied biotin-binding sites as a function of the cofactor concentration added to the cell suspension (assuming 50% uptake into the periplasm). Ten micromolars was selected as maximum-permitted cofactor concentration for further experiments to ensure an excess of binding sites for >90% of Sav variants (dashed lines). (E) Overview of the screening workflow (see Results and Methods). Circular arrows represent centrifugation steps for buffer exchange.
Even single mutations can substantially alter the expression level of proteins (40), which complicates the identification of variants with increased specific activity. For this reason, we aimed to screen at cofactor concentrations that do not fully saturate the available biotin-binding sites even for low-expressing variants (i.e., excess of binding sites). Consequently, we determined the expression level of all 400 Sav mutants from the 112X 121X library, relying on a quenching assay with biotinylated fluorescent probes (Fig. 1C and Methods) (41). The majority of variants showed high expression levels ranging from 17 to 536 mg liter−1, which is equivalent to concentrations of 1 to 33 μM biotin-binding sites. When normalized by the density of the cell suspensions, these values amount to 0.2 to 3.2 μM binding sites in a culture with an optical density at 600 nm (OD600) of 1. While the expression of variants harboring a cysteine at position 112 or a tryptophan at either of the two positions appeared to be reduced, 87% of Sav mutants showed an expression level greater than 1 μM binding sites per OD600. On the basis of these measurements, we determined a maximum permitted cofactor concentration (Fig. 1D). This critical experimental parameter should, on the one hand, be high enough to result in well-detectable product concentrations and, on the other hand, remain below the concentration of available biotin-binding sites for the majority of the library. The latter requirement is important to keep the concentration of assembled ArMs constant, irrespective of the expression level of Sav. In light of previous studies (34) and our own observations (fig. S5), we conservatively assumed that less than half of externally added cofactor enters the periplasm. As a consequence, we set the maximum permitted cofactor concentration at the incubation step to 10 μM, because this ensures that an excess of binding sites is maintained for more than 90% of variants (Fig. 1D), thus largely eliminating the expression level dependence of the screening results.
Systematic screening of ArMs for bioorthogonal reactions
To facilitate screening of improved ArMs for diverse reactions, we relied on a 96-well plate–based assay (34). In brief, periplasmic expression cultures are spun down, and cells are resuspended in an incubation buffer containing the cofactor of interest to assemble the ArM in the periplasm. After incubation, cells are washed once to remove unbound cofactor and resuspended in a reaction-specific buffer containing the substrate. Following overnight incubation, the product concentration is determined using a suitable analytical method (Fig. 1E). This screening procedure is compatible with various reaction conditions and analytical methods and, consequently, not restricted to model reactions, which, for example, produce a fluorescent product. Furthermore, it is amenable to lab automation (see below). Relying on this protocol, we sought to systematically test the full-factorial 400-mutant Sav library for various ArM reactions of interest (Fig. 2A). To this end, we selected three reactions based on previously reported ArMs: a ring-closing metathesis (RCM, I) of diallyl-sulfonamide 1 to 2,5-dihydro-pyrrole 2 (34) and two deallylation reactions (II and III) of allyl carbamate–protected substrates 3 and 5, affording amino coumarin 4 and indole 6, respectively. The corresponding cofactors for these reactions are a biotinylated second-generation Hoveyda-Grubbs catalyst (Biot-NHC)Ru (reaction I) and a biotinylated ruthenium cyclopentadienyl complex (Biot-HQ)CpRu (42, 43) (reactions II and III), respectively (Fig. 2B). Furthermore, with the aim of extending the scope of ArM-catalyzed reactions toward biocompatible nucleophilic cyclizations of alkynes, we developed two novel gold-containing ArM cofactors, (Biot-NHC)Au1 and (Biot-NHC′)Au2 (Fig. 2B), to create ArMs for hydroamination (IV) and hydroarylation (44) (V) reactions. None of these reactions are known to be catalyzed by natural enzymes, and therefore, they have potential for applications ranging from the design of novel metabolic pathways to prodrug activation and in vivo labeling (5, 45).
Fig. 2. Systematic screening for ArMs catalyzing diverse reactions.

(A) ArM-catalyzed reactions: I, ring-closing metathesis (RCM) with a diallyl-sulfonamide 1 yielding a 2,5-dihydro-pyrrole 2. II, Deallylation of allylcarbamate-protected coumarin 3 to the corresponding amino coumarin 4. III, Deallylation of allyl carbamate indole 5 to indole 6. IV, Hydroamination of 2-ethynylaniline 7 to indole 6. V, Hydroarylation of profluorophore 8 to afford amino coumarin 9. (B) Biotinylated cofactors used in this study. Biot: d-biotin. (C) Cell-specific activity of 400 ArMs mutated at Sav positions 112 and 121 normalized to the activity of wild-type Sav (S112 K121). The displayed activities are product concentrations after 20 hours of reaction (mean of biological duplicates; for SDs, refer to fig. S8). Note that the screenings for reactions II and V were performed using robotics. (D) Activity distribution in the Sav mutant library for the five ArM reactions. Violins comprise 400 double mutants with the 10 most active ArMs depicted as circles. (E) Validation of hits from the 400 mutant screens. Bars are mean activity of eight biological replicates with SD (error bars) and individual replicates (circles). Mutants are designated by the amino acids in positions 112 and 121.
While preliminary experiments on the RCM I and deallylations II and III confirmed the compatibility of these reactions with the periplasmic screening platform, reactions IV and V showed only little conversion in the whole-cell assay. On the basis of previous experience with ArMs (34, 38), we hypothesized that cellular thiols might inhibit these gold-catalyzed reactions, which we could confirm by in vitro experiments with purified Sav. These revealed a marked inhibitory effect of glutathione and cysteine (fig. S6A). Previously, we had reported that thiol inhibition can be overcome in vitro by adding the oxidizing agent diamide (38). We observed that diamide also neutralizes the detrimental effect of thiols in whole-cell experiments, rendering periplasmic gold catalysis feasible (fig. S6B).
With a functional periplasmic screening assay for the five reactions at hand, we tested all 400 mutants for each reaction at least in biological duplicates relying on the aforementioned workflow in 96–deep well plates. We automated the steps required for incubation with cofactor, washing, and substrate addition and applied this automated protocol for reactions II and V. This substantially reduces the manual labor and the consumption of consumables. The robotic platform can handle up to eight 96-well plates concurrently. Accordingly, the 400-member library can be processed in 1 day, and an entire screening (including Sav expression and analytics) can be performed within 1 week.
Relying on the periplasmic assay, we recorded a local sequence-activity landscape for each ArM (Fig. 2C). The activity patterns varied substantially between reactions, which points to the existence of specific interactions between protein, cofactor, and substrate, as opposed to unspecific effects, for instance, as a result of varying expression levels. In line with this, we observed no correlation between expression level and activity for any of the reactions (fig. S7). Hence, the concentration of cofactor, and not the number of available biotin-binding sites, is limiting, which confirms the validity of the determined maximum cofactor concentration (see above). Similar activity patterns were only found between the two deallylation reactions (II and III), suggesting that, in these cases, the substrates are sufficiently similar or that the observed activity is mainly the result of interactions between protein and cofactor.
For all reactions, the wild-type variant (S112 K121) had a comparably low activity that was only slightly above the background observed for cells lacking Sav. Note that this background activity results from residual cofactor that is unspecifically bound and thus incompletely removed in the washing step. Compared with wild-type Sav, we identified significantly more active mutants for all five ArM reactions (Fig. 2D). To validate these results, we measured the activity of the most promising variants of each ArM again in eight replicates, which confirmed the observed enhancements (Fig. 2E). The best mutants reached fold improvements over the wild type varying between 6- and 15-fold. Notably, we also identified substantially improved variants of the novel, gold-based ArMs for hydroamination (IV) and hydroarylation (V), reaching fold improvements over wild type of about 7- and 10-fold, respectively.
While the screenings were designed toward increased ArM activity in a cellular environment, we sought to investigate whether the identified variants also display increased activity in vitro. We therefore purified the three most active mutants for each reaction and performed ArM reactions with these (Fig. 3). Notably, all mutants significantly outperformed the wild-type ArM and the corresponding free cofactor, as reflected by markedly higher turnover numbers (TONs). Depending on the reaction, the best variants reached between 5- and 20-fold higher TONs than the free cofactor, demonstrating the benefit of embedding these cofactors within an engineered protein scaffold. Note that, in some cases, the relative ranking of variants changed compared with the whole-cell biotransformations, which is likely due to the different reaction conditions and the absence of cellular components in vitro. Nevertheless, the in vitro experiments confirmed that the identified variants have a significantly increased specific activity and further corroborated the potential of the periplasmic screening platform for the rapid and straightforward discovery of different ArMs with improved catalytic properties.
Fig. 3. In vitro turnover number of ArM variants identified in the periplasmic screening.
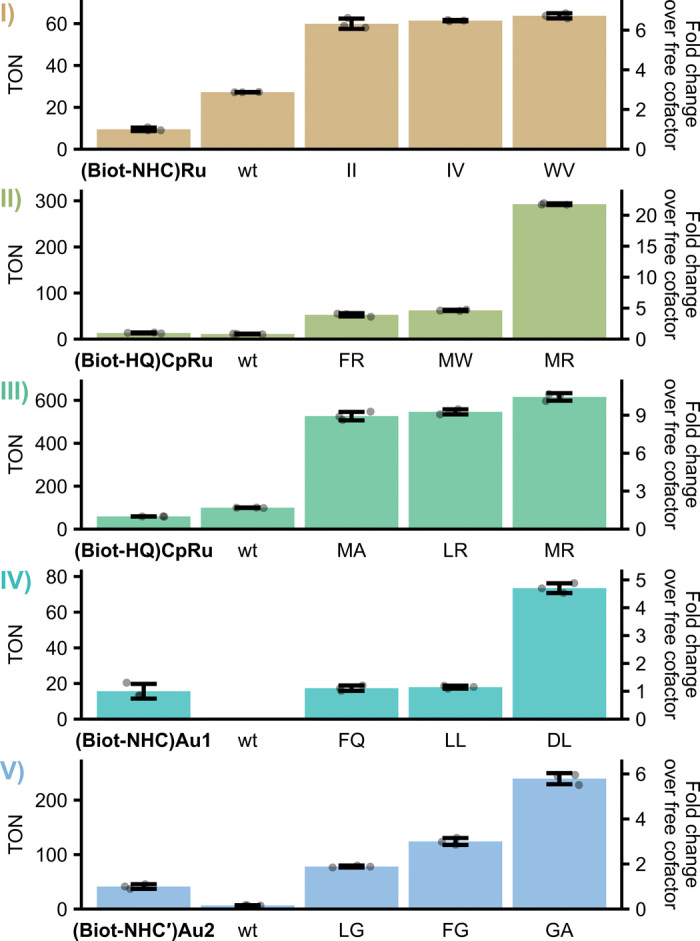
For each reaction, the three most active variants identified in the whole-cell screening were purified, and their TON was determined. Reactions were carried out at 37°C and 200 rpm for 20 hours. Bars represent mean TONs of technical triplicate reactions with SD as error bars and individual replicates as circles. For comparison, the free cofactor and wild-type Sav variant (wt) were included. Mutants are designated by the amino acids in positions 112 and 121.
Analysis of sequence-activity landscapes
The recording of sequence-activity data as performed here provides the basis for additional insight beyond the mere identification of active mutants. To this end, we first sought to understand which biophysical properties result in the observed activity changes. Therefore, we analyzed the effect of individual amino acids on the activity of the different ArMs (Fig. 4A): First, we scaled the activity measurements (Fig. 2C) such that the average activity across the 400 mutant library equals zero and the corresponding SD equals 1. Next, we averaged the scaled activities of all 20 variants that carry the same amino acid in one position. This was performed for all 20 canonical amino acids and either of the two diversified positions. As a result, amino acid– and position-specific averages greater or smaller than zero represent a positive or negative effect on ArM catalysis, respectively. While effects of amino acids varied between positions and reactions (Fig. 4A), an overall positive effect of hydrophobic amino acids on the activity of the corresponding ArMs could be observed, whereas charged amino acids and proline tend toward a negative impact. In addition, we used hierarchical clustering to analyze which amino acids have similar effects on activity. In line with the previous observation, hydrophobic amino acids clustered clearly separated from hydrophilic ones, indicating that this property is crucial in terms of catalytic activity (Fig. 4B). Furthermore, amino acids with similar chemical properties showed a strong tendency to cluster closely together (note, for instance, L-I-V, D-E, H-K, and S-T clusters). This points to an anticipated large potential of library strategies that use reduced amino acid alphabets to maintain a large chemical diversity while significantly reducing the screening effort (see below).
Fig. 4. Amino acid effects and implications for future ArM engineering.

(A) Effect of amino acids on ArM activity. Points outside and inside the dark gray circles indicate a positive and negative effect, respectively. Values were standardized by subtracting the mean activity of all 400 mutants and dividing by the corresponding SD. The mean across all 20 variants harboring the respective amino acid is shown. (B) Hierarchical clustering of amino acids across both positions and all reactions (Methods). NDT-encoded amino acids are highlighted (blue). (C) Comparison of enzyme engineering strategies. Strategies are compared to the full-factorial approach (left to right): screening single mutants (both positions), combining the best single mutations at both positions, ISM, and combinatorial screening with reduced amino acid sets (NDT/NRT codons). Bars are the mean across hits identified by the respective strategy. (D) Predictive machine learning models based on SVM, gradient boosting, and neural networks (Methods). Models were trained on 320 randomly selected data points and evaluated on the remaining 80 variants. Mean and SD of five training runs are displayed for gradient boosting and neural networks. (E) Performance of gradient boosting model for reaction III. Predictions are plotted against experimental measurements for 80 held-out variants (predicted top 10% highlighted in blue).
Next, we compared the full-factorial screening approach as pursued here with other common enzyme engineering strategies. To this end, we used our screening results to analyze which variants would have been identified using such heuristics, and how their activity compares to the most active variant identified in our exhaustive screening. As highlighted in Figure 4C, screening only single mutants (S112X K121 or S112 K121X) would have led to “hits” with activities ranging from 26 to 65% relative to the most active double mutant for the respective ArM reaction. Subsequent combination of the most beneficial single mutations would have led to an activity increase in two cases but a decrease in three cases. This suggests that there are interactions between the two amino acid positions 112 and 121 that lead to nonadditive effects on the overall activity (46). Further analysis revealed that between 24 and 37% of the observed variance can be attributed to interactions between the two positions (table S1). Notably, the activity of the best mutants identified in our screening can only be explained by considering nonadditive effects (fig. S9).
Iterative saturation mutagenesis (ISM) relies on the sequential randomization of positions while using the best variant from the previous round as a starting point for the next (47). In this way, it can leverage nonadditive interactions to some extent while keeping the experimental effort limited. This strategy would have been more successful than the simple combination of single mutations and would have led to the discovery of mutants at or near the local activity optimum for two of the five reactions. Nonetheless, this demonstrates that a full-factorial library enables the discovery of mutants that would likely be missed using other strategies, as they are the result of nontrivial interactions. In addition, note that while sequential strategies require the analysis of fewer variants, they do not provide universally applicable libraries, which would be a substantial disadvantage for a generalizable, reaction-independent ArM screening platform as proposed here.
Unfortunately, full-factorial screening quickly becomes intractable if more positions are to be diversified. An established strategy to decrease library size is to use reduced amino acid sets, such as the 12 amino acids encoded by NDT “(where N = A, C, G or T and D = A, G or T)” codons, which still cover all major classes of amino acid chemistries and lack redundant or stop codons (48). Our analysis shows that this strategy would have been more successful than those discussed above, reaching consistently high activities for all ArM reactions. This observation is in line with the fact that these 12 amino acids cover the clusters identified in our previous analysis well (Fig. 4B). For this reason, it seems promising to screen combinatorial libraries of Sav mutants based on such a reduced amino acid set. Further reductions in the amino acid set are possible [for example, eight amino acids encoded by NRT “(where N = A, C, G or T and R = A or G)’’], but at the cost of a reduced probability of finding highly active mutants.
The recording of sequence-activity data as performed here may also enable data-driven approaches to ArM engineering that efficiently explore the available sequence space. In recent years, machine learning techniques have attracted great interest in the context of enzyme engineering, as these could predict the activity of hitherto untested enzyme variants directly from their sequence, provided that a suitable set of training data is available (49). We therefore assessed the utility of different machine learning techniques for the engineering of ArMs relying on the sequence-activity data obtained in this study. Specifically, we chose three commonly used approaches—support vector machine (SVM) regression (50), gradient boosting (51), and neural networks (52)—and evaluated their performance for all five reactions. After choosing the best sets of hyperparameters using fourfold cross-validation, we trained the models on 320 data points and evaluated their predictions for the remaining 80 variants (Fig. 4D). Overall, predictions correlated well with the experimentally determined activities (coefficient of determination, R2 between 0.34 and 0.71). On average, gradient boosting achieved the best predictive performance and appeared to be most robust across different reactions (R2 of 0.68, 0.68, 0.71, 0.46, and 0.55 for reactions I, II, III, IV, and V, respectively). Last, we selected the best-performing model for each reaction to assess whether the predictive power would be sufficient to facilitate machine learning–guided ArM engineering. We found that the models could be used to design highly active mutants with high confidence (Fig. 4E and fig. S10). This highlights the potential of machine learning in the context of ArMs, and similar approaches may be used to guide their engineering in the future.
DISCUSSION
The results presented in this study highlight that Sav-based ArMs can be tailored to catalyze various new-to-nature reactions. The activity landscapes of ArMs are distinct for each reaction, underscoring the need for flexible enzyme engineering strategies that can be easily adapted to new transformations. The Sav 112X 121X library along with the automation-compatible, periplasmic screening workflow represent the first platform for systematic ArM engineering to this end. Using this approach, we readily identified improved ArMs for five biorthogonal reactions. The activity enhancements compare favorably to previous efforts of optimizing Sav-based ArMs (34, 35). Similarly, the hits appear to be more active than the variants we identified previously for the same reactions. For instance, several studies had previously described allylic deallylases that uncage allylcarbamate-protected substrates. Of these, the most extensive engineering study was a screening of 80 surface-displayed variants with mutations at positions 112 and 121, yielding the double mutants 112M 121A and 112Y 121S as the most active variants for the uncaging of allylcarbamate-protected amino coumarin 3 (II) (35). While both of these mutants proved to be highly active in our periplasmic screen, several others displayed even higher activities, reaching an improvement over wild type of up to 15-fold, compared with 9- and 10-fold for the previously identified variants.
The recorded data on ArM activity and cellular Sav levels indicate that differences in the expression level of Sav mutants do not affect the screening performance of our platform, as an excess of biotin-binding sites is consistently ensured. This notion is corroborated by the observation that the best ArM mutants displayed an increased specific activity in vitro. At the same time, the large excess of binding sites points to a substantial potential to increase the cell-specific ArM activity for applications in biocatalysis, which could be achieved by increasing the concentration of cofactor and/or improving its uptake.
All reactions tested in the context of this study were found to be compatible with whole-cell biotransformations under mild reaction conditions, which is an important prerequisite for advanced applications in the context of synthetic biology (5). In this regard, the indole-producing ArMs (reactions III and IV) are of particular interest, as a variety of applications can be imagined on the basis of this metabolic intermediate and signaling molecule.
The combinatorial library focused on two crucial residues proved to be a powerful tool for the engineering of ArMs, particularly when pursuing multiple catalytic activities in parallel from a common starting point. The combination of such libraries with the screening workflow presented here enables the rapid discovery of active ArMs for any new reaction, potentially as a universal initial step followed by more extensive engineering campaigns. For the latter, the results from this study provide valuable implications such as the indicated high potential of reduced amino acid sets in the context of ArMs. Combined with lab automation, such amino acid sets render larger screening campaigns involving more amino acid positions feasible. For much larger numbers of targeted positions in Sav, ISM, potentially in combination with reduced amino acid alphabets, appears to be a promising strategy, as it offers a good trade-off between experimental effort and the probability of finding highly active mutants. Last, the screening platform could be used to perform undersampling of libraries with a much larger theoretical diversity. Subsequently, the obtained data may be used to train machine learning models, which can guide additional screening rounds and thus streamline ArM engineering (49). Our analyses in this regard (Fig. 4, D and E) highlight the power of such approaches specifically in the context of ArMs. Moreover, our observations indicate that various machine learning techniques can achieve sufficient accuracy to efficiently guide the directed evolution of ArMs (53). Note that the datasets used for the model training here are relatively small and only allow predictions for the two randomized amino acid positions. Nonetheless, we anticipate that future screening campaigns with larger libraries are likely to profit substantially from such predictive models trained on an initial smaller sample of variants.
METHODS
Chemicals and reagents
Unless stated otherwise, chemicals were obtained from Sigma-Aldrich, Acros, or Fluorochem. Primers were synthesized by Sigma-Aldrich, and enzymes for molecular cloning were obtained from New England Biolabs.
Cloning of Sav library
The Sav library was created on the basis of a previously described expression plasmid that contains a T7-tagged Sav gene with an N-terminal ompA signal peptide for export to the periplasm under control of the T7 promoter in a pET30b vector (Addgene, #138589) (34). Positions 112 and 121 were randomized using the codon set described by Tang et al. (54). The plasmid was amplified in two fragments using either primer 1 and a mix of primers 2 to 5 (molar ratio of 12:6:1:1) or primer 10 and a mix of primers 6 to 9 (molar ratio of 12:6:1:1; see table S2 for primer sequences). Polymerase chain reactions (PCRs) were carried out using Q5 polymerase. Template plasmid was digested using DpnI, and the PCR products were purified using a PCR purification kit (Sigma-Aldrich). Subsequently, the fragments were joined by Gibson assembly and used to transform chemically competent BL21-Gold(DE3) cells (Agilent Technologies). Individual clones were sequence verified by Sanger Sequencing (Microsynth AG). Having identified 250 of the 400 possible distinct double mutants this way, a second pool containing only the 150 missing variants was cloned. To this end, sequence-verified plasmids from the first library generation step were used as PCR templates, and 40 fragments, each containing a single amino acid exchange at position 112 or 121, were obtained by amplification with primers 1 and 12 or 10 and 11 for positions 112 and 121, respectively. Following DpnI digest and purification, these fragments were added to Gibson assembly reactions in combinations suitable for obtaining pools of missing variants. More specifically, this was achieved by adding one fragment with a desired mutation at position 112 per reaction, along with multiple fragments for position 121. Again, individual clones were sequenced after transformation. Eventually, 36 remaining variants were cloned individually by assembling the corresponding fragments. Refer to fig. S1 for an overview of the library generation.
Sav expression in 96-well plates
Ninety-six deep-well plates were filled with 500 μl of LB (+ 50 mg liter−1 kanamycin) per well. Cultures were inoculated from glycerol stocks and grown overnight at 37°C and 300 revolutions per minute (rpm) in a Kuhner LT-X shaker (50-mm shaking diameter). Twenty microliters per culture was used to inoculate expression cultures in 1 ml of LB with kanamycin. These cultures were grown at 37°C and 300 rpm for 1.5 hours. At this point, the plates were placed at room temperature for 20 min, and subsequently, Sav expression was induced by addition of 50 μM isopropyl β-d-1-thiogalactopyranoside (IPTG). Expression was carried out at 20°C and 300 rpm for an additional 20 hours.
Quantification of biotin-binding sites
For measurement of Sav expression levels, the OD600 of the cultures was determined in a plate reader (Infinite M1000 PRO, Tecan Group AG) using 50 μl of samples diluted with an equal volume of phosphate-buffered saline (PBS). The remaining cultures were then centrifuged [3220 relative centrifugal force (rcf), 20°C, 10 min], and the pellets were resuspended in 250 μl of lysis buffer [50 mM tris, 150 mM NaCl, 5 mM EDTA, and 1 g liter−1 lysozyme (pH 7.4)]. After 30 min of incubation at room temperature, the cell suspensions were subjected to three freeze-thaw cycles. Afterward, 150 μl of deoxyribonuclease I (DNaseI) buffer [50 mM tris, 150 mM NaCl, and 2 U ml−1 DNaseI (pH 7.4)] was added, and plates were incubated at 37°C for 45 min before centrifugation (4800 rcf, 20°C, 20 min). Subsequently, the concentration of biotin-binding sites in the supernatant was determined using a modified version of the assay described by Kada et al. (41), which relies on the quenching of the fluorescence of a biotinylated fluorophore upon binding to Sav. Specifically, 190 μl of the binding site buffer [1 μM biotin-4-fluorescein, bovine serum albumin (0.1 g liter−1) in PBS] was mixed with 10 μl of supernatant or purified Sav standard. After incubation at room temperature for 90 min, the fluorescence intensity was measured (excitation at 485 nm, emission at 525 nm), and a calibration curve produced with purified Sav was used to calculate the concentration of biotin-binding sites in each sample.
Synthesis of cofactors and substrates
Substrates 1 (34), 3 (35), 5 (55), and cofactors (Biot-NHC)Ru (56) and (Biot-HQ)CpRu (35) were prepared according to reported procedures. Substrate 7 was obtained from Sigma-Aldrich. A detailed description of the synthesis of (Biot-NHC)Au1, (Biot-NHC′)Au2, substrate 8, and product 9 is available in the Supplementary Materials.
Whole-cell screening
Following the expression of Sav mutants in deep-well plates, the OD600 of the cultures was determined in a plate reader using 50 μl of samples diluted with an equal volume of PBS. Afterward, the plates were centrifuged (3220 rcf, 15°C, 10 min), the supernatant was discarded, and the pellets were resuspended in 400 μl of incubation buffer containing the respective reaction-specific cofactor. The composition of the incubation buffer varied for metathesis [2 μM (Biot-NHC)Ru in 50 mM tris, 0.9% (w/v) NaCl (pH 7.4)], deallylation [5 μM (Biot-HQ)CpRu in 50 mM MES, 0.9% NaCl (pH 6.1)], hydroamination [10 μM (Biot-NHC)Au1 in (2-(N-morpholino)ethanesulfonic acid) 50 mM MES, 0.9% NaCl, 5 mM diamide (pH 6.1)], and hydroarylation [10 μM (Biot-NHC′)Au2 in 50 mM MES, 0.9% NaCl, 5 mM diamide (pH 5)] reactions. The compositions of different buffers are summarized in table S3. Cells were incubated with the cofactor for 1 hour at 15°C and 300 rpm. Afterward, plates were centrifuged (2000 rcf, 15°C, 10 min), the supernatant was discarded, and the pellets were resuspended in 500 μl of the respective incubation buffer lacking cofactor to remove unbound cofactor. Following another centrifugation step, cell pellets were resuspended in 200 μl of reaction buffer containing the respective substrate. The composition of this reaction buffer varied for metathesis [5 mM 1 in 100 mM sodium acetate, 0.5 M MgCl2 (pH 4)], deallylation [500 μM 3 or 5 in 50 mM MES, 0.9% NaCl (pH 6.1)], hydroamination [5 mM 7 in 50 mM MES, 0.9% NaCl, 5 mM diamide (pH 6.1)], and hydroarylation [5 mM 9 in 50 mM MES, 0.9% NaCl, 5 mM diamide (pH 5)] reactions (table S3). Reactions were performed at 37°C and 300 rpm for 20 hours before determining the product concentration. Each 96-well plate contained 80 mutants to be tested along with four replicates of cells expressing wild-type Sav, Sav SL, Sav MA, and cells lacking Sav as controls. To account for differences in cell density and plate-to-plate variations, the product concentrations were divided by the OD600 of the culture and normalized to the mean of the cell-specific product concentrations measured for the Sav SL (metathesis, hydroamination, and hydroarylation) or Sav MA (deallylation) controls on the respective plate. These mutants had been identified as active in preliminary experiments. All variants were tested at least in biological duplicates.
Lab automation
The steps required for incubation of cells with cofactor, washing, substrate addition, and OD600 measurement were implemented using an automation platform featuring two Tecan EVO 200 (Tecan Group AG) robotic platforms coupled to each other. Both platforms were controlled using the EVOware standard software (Tecan Group AG). For shaking, incubation, and resuspension of cultures, the platform was equipped with Kuhner ES-X shaking platform (Adolf Kühner AG) running at 300 rpm at 50-mm shaking radius. The shaking platform was surrounded by a custom-made box made of aluminum plastic composite panels (Tecan Group AG). The temperature inside the box was maintained at 15°C using an “Icecube” (Life imaging services) heater/cooler device. Centrifugation of the samples was performed using the integrated Rotanta 46 RSC Robotic centrifuge (Hettich AG). All buffer exchanges during sample preparation were performed using the integrated liquid-displacement pipetting system equipped with eight 2500-μl dilutors and fixed stainless steel needles. OD600 measurements were performed using a Tecan Infinite M200 PRO plate reader. The automation method files are available upon request. The automated protocol was used for the deallylation with allyl carbamate coumarin (II) and the gold-catalyzed hydroarylation (V), while the screenings for the other reactions were performed manually.
Product quantification by ultraperformance liquid chromatography–mass spectrometry
To quantify the metathesis product 2, an extraction was performed by adding 775 μl of methanol and 25 μl of internal standard (100 μM in methanol) to each 200-μl sample. The samples were incubated for 1 hour at 20°C and 300 rpm, followed by centrifugation at 3220 rcf and 20°C for 10 min. Subsequently, 50 μl of the supernatant was mixed with 200 μl of water and analyzed by ultraperformance liquid chromatography–mass spectrometry (UPLC-MS). UPLC analysis was performed using a Waters H-Class Bio using a BEH C18 1.7-μM column and a flow rate of 0.6 ml min−1 (eluent A, 0.1% formic acid in water; eluent B, 0.1% formic acid in acetonitrile; gradient at 0 min: 90% A, 10% B; at 0.5 min: 90% A, 10% B; at 2.5 min: 10% A, 90% B; at 3.5 min: 90% A, 10% B; and at 4.5 min: 90% A, 10% B). Peak integration for single ion recording was used for quantification, and concentrations of the metathesis product (retention time of 1.0 ± 0.25 min) were determined on the basis of a standard curve with the ring-closed product in the presence of a fixed concentration of the nonadeuteraded product (retention time of 1.0 ± 0.25 min).
Fluorescence measurements
The fluorescent product 4 was quantified by measuring the fluorescence intensity at an excitation of 394 nm and an emission of 460 nm. Product 9 was excited at 390 nm and measured at 488 nm. Measurements were carried out in black 96-well plates in an Infinite M1000 PRO plate reader.
Kovac’s assay
Indole was quantified using the photometric Kovac’s assay [adapted from Piñero-Fernandez et al. (57)]. For measurements in culture supernatant, plates were centrifuged (3220 rcf, 20°C, 10 min), and 110 μl of supernatant was mixed with 165 μl of Kovac’s reagent [4-(dimethylamino)benzaldehyde (50 g liter−1), isoamylic alcohol (710 g liter−1), and hydrochloric acid (240 g liter−1)] in a separate plate. After 5 min of incubation, these plates were centrifuged (3220 rcf, 20°C, 10 min). Subsequently, 75 μl of the upper phase was transferred to a new transparent plate, and the absorbance at 540 nm was measured in a plate reader (Infinite M1000 PRO). The same procedure (omitting the first centrifugation step) was applied to measure indole after in vitro experiments. A standard curve was then used to determine indole concentrations.
Sav expression for purification
A single colony of BL21-Gold(DE3) cells harboring a plasmid from the previously described library for periplasmic expression of the desired Sav variant was used to inoculate a starter culture [4 ml of LB with kanamycin (50 mg liter−1)], which was grown overnight at 37°C and 200 rpm. On the following day, 100 ml of LB with kanamycin in a 500-ml flask was inoculated to an OD600 of 0.01. The culture was grown at 37°C and 200 rpm until it reached an OD600 of 0.5. At this point, the flask was placed at room temperature for 20 min, and 50 μM IPTG was added to induce Sav expression. Expression was performed at 20°C and 200 rpm overnight, and cells were harvested by centrifugation (3220 rcf, 4°C, 15 min). Pellets were stored at −20°C until purification.
Sav purification
Cell pellets were resuspended in lysis buffer [50 mM tris, 150 mM NaCl, and lysozyme (1 g liter−1) (pH 7.4)] so as to reach a final OD600 of 10. After 30 min of incubation at room temperature, cell suspensions were subjected to three freeze-thaw cycles. Subsequently, nucleic acids were digested by addition of 1 μl of Benzonase (Merck KGaA) and 10 μl of 1 M MgSO4, followed by incubation at room temperature for 10 min. After centrifugation, the supernatant was transferred to a new tube and mixed with carbonate buffer [50 mM ammonium bicarbonate, 500 mM NaCl (pH 11)] in a ratio of 2:3. Iminobiotin beads (500 μl) were added, and the tubes were incubated at room temperature with shaking (120 rpm) for 1 hour. Afterward, the beads were washed twice with 15 ml of carbonate buffer and resuspended in 2 ml of acetate buffer [50 mM ammonium acetate and 500 mM NaCl (pH 4)]. After 20 min of incubation at room temperature with shaking, the tubes were centrifuged, and the supernatant was dialyzed (SnakeSkin Dialysis tubing with 3.5-kDa molecular weight cutoff; Thermo Fisher Scientific) against 1 liter of the desired buffer (table S4) for 24 hours at room temperature with one buffer exchange.
In vitro catalysis
In vitro reactions were performed with 2.5 μM purified Sav (tetrameric; corresponding to 10 μM biotin-binding sites) in a volume of 200 μl in glass vials. Cofactor and substrate concentrations and buffer conditions varied between reactions and are listed in table S4. Reactions were carried out at 37°C and 200 rpm for 20 hours.
Data analysis
Data were analyzed using R 4.0.2 (58). For clustering and calculating amino acid effects, activity values were standardized by subtracting the mean and dividing by the SD of all activity values for the respective reaction. Hierarchical clustering was done by calculating the Euclidian distances between all standardized activity values of variants harboring the respective amino acid at position 112 or 121 (20 values per position and reaction, amounting to 200 values in total) and by clustering these using the hclust function (58) with complete linkage. The activity of all 400 mutants was further analyzed by two-way analysis of variance (ANOVA). To this end, positions 112 and 121 were treated as explanatory variables (with 20 levels corresponding to the canonical amino acids). For analyzing the variance explained by individual factors, an interaction term was included, whereas it was omitted to generate a purely additive model.
Machine learning
Machine learning was implemented in R using the packages XGBoost (59), Keras (60), and e1071 (61). The 400 activity values for each reaction were randomly split into a training set comprising 320 data points and a test set consisting of the remaining 80 variants. The amino acids at positions 112 and 121 were vectorized to obtain different candidate descriptors using one-hot encoding, PCscores (62), zScales (63), VHSE (64), or physical descriptors (65). The following hyperparameters were optimized using grid search and fourfold cross-validation on the training dataset: descriptor and cost for SVM regression; descriptor, learning rate, maximum depth, subsample ratio of the training instance, subsample ratio of columns when constructing each tree, and number of trees for gradient boosting; descriptor, model architecture (two to four hidden layers, with number of nodes per layer between 50 and 200), batch size, and number of epochs for neural networks. Refer to table S5 for an overview of settings explored during hyperparameter tuning. The best combination of hyperparameters was used to train a final model on all 320 data points, which was then evaluated on the test set.
Acknowledgments
We thank V. Sabatino, F. Schwizer, Y. Cotelle, and J. Vallapurackal for help with substrate and cofactor synthesis, and G. Schmidt for technical support regarding lab automation. Funding: This work was supported by the NCCR “Molecular Systems Engineering” and the European Commission (project: Madonna; grant no. 766975). T.R.W. thanks the SNF (grant no. 200020_182046) for generous support. S.P. and T.V. acknowledge support by the SNF (grant no. 31003A_179521). Author contributions: M.J. and T.R.W. conceived and supervised the study. S.P. advised the project. T.V. carried out the biological and screening experiments, analyzed the data, and performed machine learning. F.C. and M.M.P. developed the gold-catalyzed reactions in vitro and synthesized the corresponding substrates and cofactors. T.V., M.J., S.P., and T.R.W. discussed the data. T.V. and M.J. wrote the manuscript with input from all authors. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data and code required to reproduce the figures and analyses presented in the main text are made available under https://github.com/JeschekLab/Systematic-engineering-of-ArMs. All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/4/eabe4208/DC1
REFERENCES AND NOTES
- 1.Lewis J. C., Artificial metalloenzymes and metallopeptide catalysts for organic synthesis. ACS Catal. 3, 2954–2975 (2013). [Google Scholar]
- 2.Rosati F., Roelfes G., Artificial metalloenzymes. ChemCatChem 2, 916–927 (2010). [Google Scholar]
- 3.Bornscheuer U. T., The fourth wave of biocatalysis is approaching. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 376, 20170063 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Hammer S. C., Knight A. M., Arnold F. H., Design and evolution of enzymes for non-natural chemistry. Curr. Opin. Green Sustain. Chem. 7, 23–30 (2017). [Google Scholar]
- 5.Vornholt T., Jeschek M., The quest for xenobiotic enzymes: From new enzymes for chemistry to a novel chemistry of life. Chembiochem 21, 2241–2249 (2020). [DOI] [PubMed] [Google Scholar]
- 6.Kan S. B. J., Lewis R. D., Chen K., Arnold F. H., Directed evolution of cytochrome c for carbon–silicon bond formation: Bringing silicon to life. Science 354, 1048–1051 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tinoco A., Steck V., Tyagi V., Fasan R., Highly diastereo- and enantioselective synthesis of trifluoromethyl-substituted cyclopropanes via myoglobin-catalyzed transfer of trifluoromethylcarbene. J. Am. Chem. Soc. 139, 5293–5296 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Song W. J., Tezcan F. A., A designed supramolecular protein assembly with in vivo enzymatic activity. Science 346, 1525–1528 (2014). [DOI] [PubMed] [Google Scholar]
- 9.Studer S., Hansen D. A., Pianowski Z. L., Mittl P. R. E., Debon A., Guffy S. L., Der B. S., Kuhlman B., Hilvert D., Evolution of a highly active and enantiospecific metalloenzyme from short peptides. Science 362, 1285–1288 (2018). [DOI] [PubMed] [Google Scholar]
- 10.Drienovská I., Alonso-Cotchico L., Vidossich P., Lledós A., Maréchal J.-D., Roelfes G., Design of an enantioselective artificial metallo-hydratase enzyme containing an unnatural metal-binding amino acid. Chem. Sci. 8, 7228–7235 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cangelosi V. M., Deb A., Penner-Hahn J. E., Pecoraro V. L., A de novo designed metalloenzyme for the hydration of CO2. Angew. Chem. Int. Ed. 53, 7900–7903 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Key H. M., Dydio P., Clark D. S., Hartwig J. F., Abiological catalysis by artificial haem proteins containing noble metals in place of iron. Nature 534, 534–537 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang H., Swartz A. M., Park H. J., Srivastava P., Ellis-Guardiola K., Upp D. M., Lee G., Belsare K., Gu Y., Zhang C., Moellering R. E., Lewis J. C., Evolving artificial metalloenzymes via random mutagenesis. Nat. Chem. 10, 318–324 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mirts E. N., Petrik I. D., Hosseinzadeh P., Nilges M. J., Lu Y., A designed heme-[4Fe-4S] metalloenzyme catalyzes sulfite reduction like the native enzyme. Science 361, 1098–1101 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Raines D. J., Clarke J. E., Blagova E. V., Dodson E. J., Wilson K. S., Duhme-Klair A. K., Redox-switchable siderophore anchor enables reversible artificial metalloenzyme assembly. Nat. Catal. 1, 680–688 (2018). [Google Scholar]
- 16.Eda S., Nasibullin I., Vong K., Kudo N., Yoshida M., Kurbangalieva A., Tanaka K., Biocompatibility and therapeutic potential of glycosylated albumin artificial metalloenzymes. Nat. Catal. 2, 780–792 (2019). [Google Scholar]
- 17.Dydio P., Key H. M., Nazarenko A., Rha J. Y.-E., Seyedkazemi V., Clark D. S., Hartwig J. F., An artificial metalloenzyme with the kinetics of native enzymes. Science 354, 102–106 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oohora K., Kihira Y., Mizohata E., Inoue T., Hayashi T., C(sp3)-H bond hydroxylation catalyzed by myoglobin reconstituted with manganese porphycene. J. Am. Chem. Soc. 135, 17282–17285 (2013). [DOI] [PubMed] [Google Scholar]
- 19.Chevalley A., Cherrier M. V., Fontecilla-Camps J. C., Ghasemi M., Salmain M., Artificial metalloenzymes derived from bovine β-lactoglobulin for the asymmetric transfer hydrogenation of an aryl ketone – synthesis, characterization and catalytic activity. Dalt. Trans. 43, 5482–5489 (2014). [DOI] [PubMed] [Google Scholar]
- 20.Lopez S., Mayes D. M., Crouzy S., Cavazza C., Leprêtre C., Moreau Y., Burzlaff N., Marchi-Delapierre C., Ménage S., A mechanistic rationale approach revealed the unexpected chemoselectivity of an artificial Ru-dependent oxidase: A dual experimental/theoretical approach. ACS Catal. 10, 5631–5645 (2020). [Google Scholar]
- 21.Burke A. J., Lovelock S. L., Frese A., Crawshaw R., Ortmayer M., Dunstan M., Levy C., Green A. P., Design and evolution of an enzyme with a non-canonical organocatalytic mechanism. Nature 570, 219–223 (2019). [DOI] [PubMed] [Google Scholar]
- 22.Emmanuel M. A., Greenberg N. R., Oblinsky D. G., Hyster T. K., Accessing non-natural reactivity by irradiating nicotinamide-dependent enzymes with light. Nature 540, 414–417 (2016). [DOI] [PubMed] [Google Scholar]
- 23.Biegasiewicz K. F., Cooper S. J., Gao X., Oblinsky D. G., Kim J. H., Garfinkle S. E., Joyce L. A., Sandoval B. A., Scholes G. D., Hyster T. K., Photoexcitation of flavoenzymes enables a stereoselective radical cyclization. Science 364, 1166–1169 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heinisch T., Ward T. R., Artificial metalloenzymes based on the biotin–streptavidin technology: Challenges and opportunities. Acc. Chem. Res. 49, 1711–1721 (2016). [DOI] [PubMed] [Google Scholar]
- 25.Nödling A. R., Świderek K., Castillo R., Hall J. W., Angelastro A., Morrill L. C., Jin Y., Tsai Y.-H., Moliner V., Luk L. Y. P., Reactivity and selectivity of iminium organocatalysis improved by a protein host. Angew. Chem. Int. Ed. 57, 12478–12482 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hassan I. S., Ta A. N., Danneman M. W., Semakul N., Burns M., Basch C. H., Dippon V. N., McNaughton B. R., Rovis T., Asymmetric δ-lactam synthesis with a monomeric streptavidin artificial metalloenzyme. J. Am. Chem. Soc. 141, 4815–4819 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zeymer C., Hilvert D., Directed evolution of protein catalysts. Annu. Rev. Biochem. 87, 131–157 (2018). [DOI] [PubMed] [Google Scholar]
- 28.R. K. Zhang, D. K. Romney, S. B. J. Kan, F. H. Arnold, in Artificial Metalloenzymes and MetalloDNAzymes in Catalysis (Wiley-VCH Verlag GmbH & Co. KGaA, 2018), pp. 137–170; http://doi.wiley.com/10.1002/9783527804085.ch5.
- 29.Reetz M. T., Directed evolution of artificial metalloenzymes: A universal means to tune the selectivity of transition metal catalysts? Acc. Chem. Res. 52, 336–344 (2019). [DOI] [PubMed] [Google Scholar]
- 30.Markel U., Sauer D. F., Schiffels J., Okuda J., Schwaneberg U., Towards the evolution of artificial metalloenzymes—A protein engineer’s perspective. Angew. Chem. Int. Ed. 58, 4454–4464 (2019). [DOI] [PubMed] [Google Scholar]
- 31.Turner N. J., Directed evolution of enzymes for applied biocatalysis. Trends Biotechnol. 21, 474–478 (2003). [DOI] [PubMed] [Google Scholar]
- 32.Creus M., Pordea A., Rossel T., Sardo A., Letondor C., Ivanova A., LeTrong I., Stenkamp R. E., Ward T. R., X-ray structure and designed evolution of an artificial transfer hydrogenase. Angew. Chem. Int. Ed. 47, 1400–1404 (2008). [DOI] [PubMed] [Google Scholar]
- 33.Hyster T. K., Knorr L., Ward T. R., Rovis T., Biotinylated Rh(III) complexes in engineered streptavidin for accelerated asymmetric C─H activation. Science 338, 500–503 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jeschek M., Reuter R., Heinisch T., Trindler C., Klehr J., Panke S., Ward T. R., Directed evolution of artificial metalloenzymes for in vivo metathesis. Nature 537, 661–665 (2016). [DOI] [PubMed] [Google Scholar]
- 35.Heinisch T., Schwizer F., Garabedian B., Csibra E., Jeschek M., Vallapurackal J., Pinheiro V. B., Marlière P., Panke S., Ward T. R., E. coli surface display of streptavidin for directed evolution of an allylic deallylase. Chem. Sci. 9, 5383–5388 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grimm A. R., Sauer D. F., Polen T., Zhu L., Hayashi T., Okuda J., Schwaneberg U., A whole cell E. coli display platform for artificial metalloenzymes: Poly(phenylacetylene) production with a rhodium–nitrobindin metalloprotein. ACS Catal. 8, 2611–2614 (2018). [Google Scholar]
- 37.Donnelly A. E., Murphy G. S., Digianantonio K. M., Hecht M. H., A de novo enzyme catalyzes a life-sustaining reaction in Escherichia coli. Nat. Chem. Biol. 14, 253–255 (2018). [DOI] [PubMed] [Google Scholar]
- 38.Wilson Y. M., Dürrenberger M., Nogueira E. S., Ward T. R., Neutralizing the detrimental effect of glutathione on precious metal catalysts. J. Am. Chem. Soc. 136, 8928–8932 (2014). [DOI] [PubMed] [Google Scholar]
- 39.M. Jeschek, S. Panke, T. R. Ward, in Methods in Enzymology (Elsevier Inc., ed. 1, 2016), vol. 580, pp. 539–556; 10.1016/bs.mie.2016.05.037. [DOI] [PubMed]
- 40.Tokuriki N., Tawfik D. S., Stability effects of mutations and protein evolvability. Curr. Opin. Struct. Biol. 19, 596–604 (2009). [DOI] [PubMed] [Google Scholar]
- 41.Kada G., Kaiser K., Falk H., Gruber H. J., Rapid estimation of avidin and streptavidin by fluorescence quenching or fluorescence polarization. Biochim. Biophys. Acta 1427, 44–48 (1999). [DOI] [PubMed] [Google Scholar]
- 42.Völker T., Dempwolff F., Graumann P. L., Meggers E., Progress towards bioorthogonal catalysis with organometallic compounds. Angew. Chem. Int. Ed. 53, 10536–10540 (2014). [DOI] [PubMed] [Google Scholar]
- 43.Völker T., Meggers E., Chemical activation in blood serum and human cell culture: Improved ruthenium complex for catalytic uncaging of alloc-protected amines. Chembiochem 18, 1083–1086 (2017). [DOI] [PubMed] [Google Scholar]
- 44.Do J. H., Kim H. N., Yoon J., Kim J. S., Kim H. J., A rationally designed fluorescence turn-on probe for the gold(III) ion. Org. Lett. 12, 932–934 (2010). [DOI] [PubMed] [Google Scholar]
- 45.Sasmal P. K., Streu C. N., Meggers E., Metal complex catalysis in living biological systems. Chem. Commun. 49, 1581–1587 (2013). [DOI] [PubMed] [Google Scholar]
- 46.Reetz M. T., The importance of additive and non-additive mutational effects in protein engineering. Angew. Chem. Int. Ed. 52, 2658–2666 (2013). [DOI] [PubMed] [Google Scholar]
- 47.Reetz M. T., Carballeira J. D., Iterative saturation mutagenesis (ISM) for rapid directed evolution of functional enzymes. Nat. Protoc. 2, 891–903 (2007). [DOI] [PubMed] [Google Scholar]
- 48.Reetz M. T., Kahakeaw D., Lohmer R., Addressing the numbers problem in directed evolution. Chembiochem 9, 1797–1804 (2008). [DOI] [PubMed] [Google Scholar]
- 49.Yang K. K., Wu Z., Arnold F. H., Machine-learning-guided directed evolution for protein engineering. Nat. Methods 16, 687–694 (2019). [DOI] [PubMed] [Google Scholar]
- 50.Drucker H., Burges C. J. C., Kaufman L., Smola A., Vapnik V., Support vector regression machines. Adv Neural Inf. Process Syst. 28, 779–784 (1997). [Google Scholar]
- 51.Friedman J. H., Stochastic gradient boosting. Comput. Stat. Data Anal. 38, 367–378 (2002). [Google Scholar]
- 52.Schmidhuber J., Deep learning in neural networks: An overview. Neural Netw. 61, 85–117 (2015). [DOI] [PubMed] [Google Scholar]
- 53.Wu Z., Kan S. B. J., Lewis R. D., Wittmann B. J., Arnold F. H., Machine-learning-assisted directed protein evolution with combinatorial libraries. Proc. Natl. Acad. Sci. 116, 8852–8858 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tang L., Gao H., Zhu X., Wang X., Zhou M., Jiang R., Construction of “small-intelligent” focused mutagenesis libraries using well-designed combinatorial degenerate primers. Biotechniques 52, 149–158 (2012). [DOI] [PubMed] [Google Scholar]
- 55.Jacquemard U., Bénéteau V., Lefoix M., Routier S., Mérour J. Y., Coudert G., Mild and selective deprotection of carbamates with Bu4NF. Tetrahedron 60, 10039–10047 (2004). [Google Scholar]
- 56.Kajetanowicz A., Chatterjee A., Reuter R., Ward T. R., Biotinylated metathesis catalysts: Synthesis and performance in ring closing metathesis. Catal. Letters. 144, 373–379 (2014). [Google Scholar]
- 57.Piñero-Fernandez S., Chimerel C., Keyser U. F., Summers D. K., Indole transport across Escherichia coli membranes. J. Bacteriol. 193, 1793–1798 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.R Core Team, R: A language and environment for statistical computing, R Found. Stat. Comput. (2019); https://www.r-project.org/.
- 59.T. Chen, T. He, M. Benesty, V. Khotilovich, Y. Tang, H. Cho, K. Chen, R. Mitchell, I. Cano, T. Zhou, M. Li, J. Xie, M. Lin, Y. Geng, Y. Li, xgboost: Extreme Gradient Boosting, R package version 1.2.0.1. (2020); https://cran.r-project.org/package=xgboost.
- 60.J. Allaire, F. Chollet, keras: R Interface to “Keras,” R package version 2.3.0.0. (2020); https://cran.r-project.org/package=keras.
- 61.D. Meyer, E. Dimitriadou, K. Hornik, A. Weingessel, F. Leisch, e1071: Misc Functions of the Department of Statistics, Probability Theory Group (Formerly: E1071), TU Wien, R package version 1.7–3. (2019); https://cran.r-project.org/package=e1071.
- 62.Xu Y., Verma D., Sheridan R. P., Liaw A., Ma J., Marshall N. M., McIntosh J., Sherer E. C., Svetnik V., Johnston J. M., Deep dive into machine learning models for protein engineering. J. Chem. Inf. Model. 60, 2773–2790 (2020). [DOI] [PubMed] [Google Scholar]
- 63.Sandberg M., Eriksson L., Jonsson J., Sjöström M., Wold S., New chemical descriptors relevant for the design of biologically active peptides. A multivariate characterization of 87 amino acids. J. Med. Chem. 41, 2481–2491 (1998). [DOI] [PubMed] [Google Scholar]
- 64.Mei H., Liao Z. H., Zhou Y., Li S. Z., A new set of amino acid descriptors and its application in peptide QSARs. Biopolymers 80, 775–786 (2005). [DOI] [PubMed] [Google Scholar]
- 65.Barley M. H., Turner N. J., Goodacre R., Improved descriptors for the quantitative structure–activity relationship modeling of peptides and proteins. J. Chem. Inf. Model. 58, 234–243 (2018). [DOI] [PubMed] [Google Scholar]
- 66.Jordan J. P., Grubbs R. H., Small-molecule N-heterocyclic-carbene-containing olefin-metathesis catalysts for use in water. Angew. Chem. Int. Ed. 46, 5152–5155 (2007). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/4/eabe4208/DC1