

Delivery of antibodies using smart frameworks affords sustained and targeted activation of T cells for improved immunotherapy.
Abstract
The major impediments to the implementation of cancer immunotherapies are the sustained immune effect and the targeted delivery of these therapeutics, as they have life-threatening adverse effects. In this work, biomimetic metal-organic frameworks [zeolitic imidazolate frameworks (ZIFs)] are used for the controlled delivery of nivolumab (NV), a monoclonal antibody checkpoint inhibitor that was U.S. Food and Drug Administration–approved back in 2014. The sustained release behavior of NV-ZIF has shown a higher efficacy than the naked NV to activate T cells in hematological malignancies. The system was further modified by coating NV-ZIF with cancer cell membrane to enable tumor-specific targeted delivery while treating solid tumors. We envisage that such a biocompatible and biodegradable immunotherapeutic delivery system may promote the development and the translation of hybrid superstructures into smart and personalized delivery platforms.
INTRODUCTION
Compared to the conventional chemotherapy and radiation therapy that are more broad in their function and kill both healthy and cancer cells (CCs), immunotherapy can more specifically attack CCs via modulation of the functions of specific immune cells with, in most cases, tolerable side effects (1). Immune checkpoint blockade (ICB) therapy, including inhibition of programmed cell death 1 (PD-1) or PD ligand 1 (PD-L1) and cytotoxic T lymphocyte antigen–4 (CTLA-4), is one of the main strategies in cancer immunotherapy as it targets and inhibits tumor-mediated immunosuppression. Tumors rely on taking advantage of the immune checkpoint pathways to escape from the host immune response. As receptor/ligand interactions can be disrupted by antibodies, inhibitory agents have been designed and synthesized to target and block immune checkpoints, overcoming tumor immune resistance. Despite these achievements, a primary problem facing ICB therapy in clinical trials is the extremely low response rate (2). The therapeutic response to ICB is highly variable, not only between different cancers but also between patients with the same cancer type (3). The biological mechanisms underlying these differences in response are incompletely understood (2). Even in melanoma, one of the most immunogenic types of cancer, only 20 to 50% of patients benefit from ICB treatment (2). A hallmark of immunotherapy is the durability of responses, most likely due to the memory of the adaptive immune system, which translates into long-term survival for a subset of patients (3). Researchers anticipated that immunotherapy would induce long-lasting effects against CCs. However, the main challenge to the broad implementation of immunotherapies remains in their sustained release and efficient delivery to boost or activate the immune system to attack cancer without the adverse side effects such as autoimmunity and nonspecific inflammation (4). To circumvent these broader approaches, biomaterial carriers of various immunotherapies could enable a more controlled therapeutic delivery to CCs directly, avoiding off-target side effects (5). Liposome-based immunotherapies have paved the way for synthetic vehicles to be efficiently used to modulate the immune response (6, 7). Polymeric nanoparticles and gels were also successful in the immune engineering of certain formulations for combined immunotherapy (8, 9). Although extremely biocompatible and leading the field of controlled delivery (10), liposomal and polymeric delivery suffer from limited stability, poor drug loading capacity (LC), and a narrow circulation window; these obstacles drastically influence their ability to sustain the release of therapeutics needed to achieve the required activation of the immune system (11–13). Other organic and inorganic platforms such as carbon nanotubes and silica nanoparticles have been used in immunotherapy, but their implementation is restricted by their limited degradability and possible cytotoxicity (12, 13). Tracking and imaging of cancer-specific T cells were also successfully reported using gold nanoparticles (14, 15). Furthermore, peptide conjugation of immune checkpoint inhibitors was recently used to control the release but was limited to certain sequences (16).
Over the past decade, metal-organic frameworks (MOFs) have been actively used as intricately engineered platforms for biomedical applications (17–19). Zeolitic imidazolate frameworks (ZIF-8)—a subclass of MOFs—are crystalline solids based on Zn2+ ion subunits coordinated to organic 2-methylimidazole (mIM) ligands, resulting in the formation of highly porous structures (20). ZIF-8 has recently emerged as a potential candidate for on-demand drug delivery applications due to its biocompatibility, remarkable LC, superior stability under physiological conditions (no premature drug release), pH responsiveness, and tunable drug release properties (21, 22). In comparison to other delivery vehicles, ZIFs can encapsulate different size and charge therapeutic hosts with a high LC reproducibly and deliver them on demand. Therefore, they were used in the delivery of current breakthrough proteins such as small interfering RNA (23), CRISPR-Cas9 ribonucleoprotein (24, 25), and catalytic enzymes (26, 27). Here, we developed an efficient strategy for the sustained release and high loading of the PD-1 inhibitor, nivolumab (NV), using ZIF-8 (NV-ZIF) with the capability of working on both hematological malignancies and solid tumors (Fig. 1). Targeted delivery, in the case of solid tumors, was achieved by using cell-specific membrane coating (CC), as this technique has proved viable for enhancing targeted cancer therapy (25, 28, 29).
Fig. 1. Schematic illustration of the mode of action of NV-ZIF.
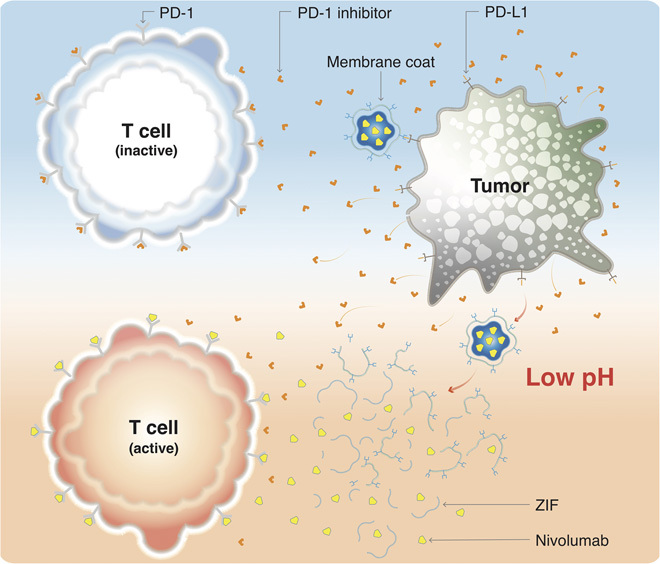
Targeted delivery of NV, a PD-1 antibody, to activate T cells in the slightly acidic tumor environment.
RESULTS
Design and characterization of NV-ZIF
In a typical experiment for the biomimetically mineralized growth of ZIF-8, an aqueous solution containing mIM (2.5 M, 0.9 ml) and NV (1 mg·ml−1) was mixed with a separate aqueous solution of zinc (Zn) nitrate (0.5 M, 0.1 ml) at room temperature for 20 min. The solution immediately turned opaque, indicating crystal formation. Cryogenic transmission electron microscopy (cryo-TEM) and TEM micrographs clearly illustrated octahedral crystals with an average diameter ranging between 102 and 160 nm (Fig. 2A). The energy-dispersive x-ray spectroscopy elemental mapping revealed a uniform distribution of ZIF-8–associated elements, Zn, nitrogen (N), and carbon (C); and NV-associated elements, oxygen (O) and sulfur (S) (Fig. 2B).
Fig. 2. Characterization of NV-ZIF.
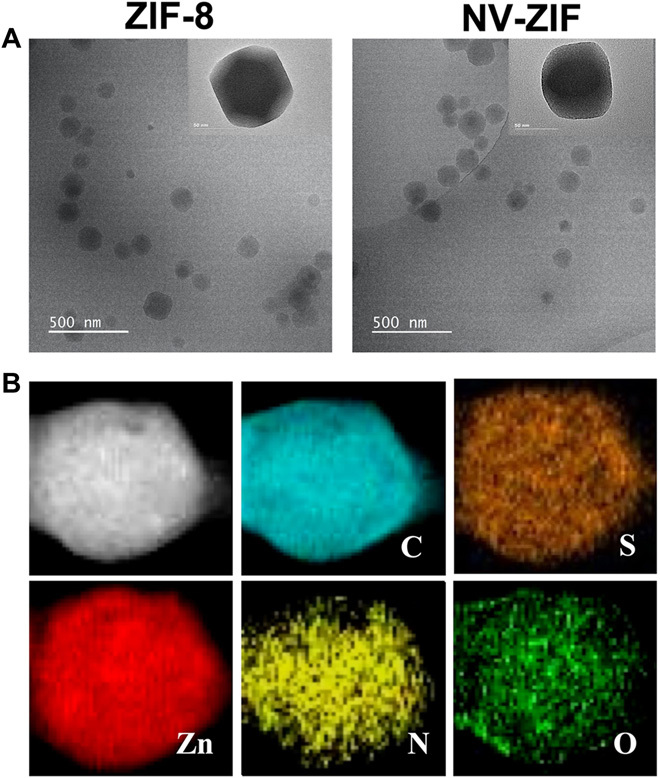
(A) Cryo-TEM and TEM micrographs of ZIF-8 and NV-ZIF. (B) TEM elemental mappings (C, S, Zn, N, and O) of NV-ZIF.
Our powder x-ray diffraction (PXRD) results demonstrate that the embedded NV did not result in any change in ZIF-8 crystallinity (Fig. 3A), which is consistent with other reported MOF-based protein carriers (18, 26, 30). The LC and loading efficiency (LE) were then evaluated using the Bradford assay. The LC and LE were found to be 5.07 ± 1% and 80 ± 3%, respectively. NV content in NV-ZIF was also estimated by thermogravimetric analysis (TGA), and the results were comparable to those obtained by the Bradford assay (fig. S1, A and B). Comparing the thermogram of NV-ZIF with that of ZIF-8 gives information about the formulation’s composition. The TGA spectrum of the NV-ZIF showed a weight loss of 6.1% between 10° and 150°C due to the loss of the adsorbed moisture. The 23% loss observed between 250° and 444.6°C can be attributed to the pyrolysis of the carboxyl or hydroxyl groups, which most probably originated from the NV decomposition. The final range of temperature from 320° to 600°C resulted in an obvious mass loss of 52.9%, which we assigned to the removal of the organic linker molecules and the collapse of ZIF-8. The interaction between ZIF-8 and NV delayed the pyrolysis process of NV that is coved by the in situ growth of ZIF-8. Unlike NV-ZIF, ZIF-8 showed a weight loss of 3.7% between 10° and 150°C, 21.6% between 250° and 444.6°C, and 47.89% from 320° to 600°C. The 23% weight loss between 250° and 444°C supports the presence of the NV. On the basis of TGA analysis, we carried out a calcination process at 320°C for 2 hours to confirm that NV is majorly embedded at the surface of the framework, as previously reported for protein-embedded MOFs (31). The TEM image of NV-ZIF after calcination supports the existence of small cavities (fig. S1C) resulting from the removal of NV molecules and their aggregates. The ultraviolet–visible–near-infrared (IR) (UV-Vis-NIR) spectrum of NV-ZIF clearly showed the absorbance band of NV at 280 nm. The embedded NV resulted in reducing its symmetry; therefore, a red shift was observed (fig. S1D) as previously reported (32). The embedded NV was also confirmed by Fourier transform IR (FT-IR) spectrum through the absence of the vibrational band at 1660 cm−1, characteristic of COO− group of NV (fig. S1E). We further investigated the possible coordination of NV and Zn2+. Therefore, different ratios of NV and Zn2+ (0.1:1 and 1:1) were stirred for 20 min at room temperature. Zn nitrate showed an absorbance band at 300 nm according to our UV spectrum (fig. S2A). When NV was mixed with Zn at different ratios, an absorbance peak at 280 nm appeared for NV only, indicating that no coordination occurred between Zn2+ and NV. The increase in the absorbance of NV is attributed to the increase in NV concentration. The FT-IR analysis showed no shift in the vibrational band at 1700 cm−1 for the COOH and 3400 cm−1 for the N─H, supporting no coordination with Zn2+ and validating the importance of mIM in the NV-ZIF formation (fig. S2B).
Fig. 3. Characterization of NV-ZIF.
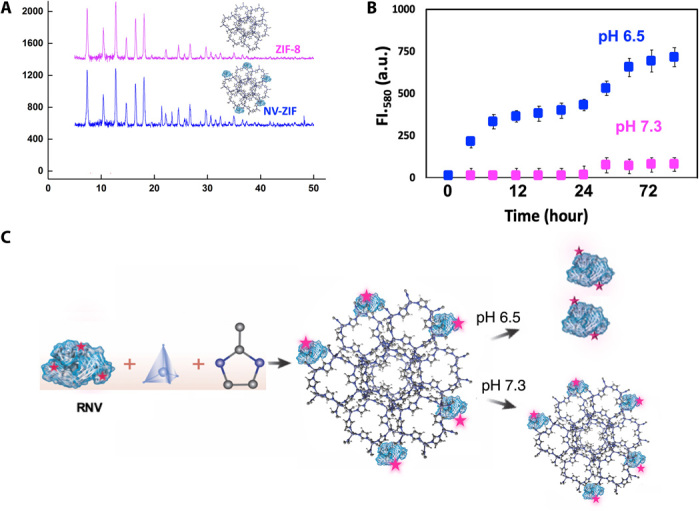
(A) PXRD patterns of ZIF-8 and NV-ZIF. (B) FI.580, Fluorescence intensity at 580. Cumulative release of RNV from NV-ZIF at physiological (pH 7.3) and acidic (pH 6.5) pH. a.u., arbitrary units. (C) Schematic representation of the controlled release of RNV from NV-ZIF under physiological and acidic conditions. NV will be released at slightly acidic pH and will remain embedded in the framework at physiological pH.
To monitor the loading and release, we labeled NV with rhodamine B, Rh (RNV) (fig. S3). By measuring the Rh fluorescence intensity, the majority of NV was embedded in ZIF-8 (fig. S3, A and B). Moreover, the sustained release of RNV from NV-ZIF at varying pH values in phosphate-buffered saline (PBS) was monitored by fluorescence spectroscopy (Fig. 3, B and C). NV-ZIF exhibited a slow sustained release at acidic pH (6.5), and approximately 50% of RNV was released within 12 hours. More stable release of small dosages of encapsulated RNV was observed after 24 hours, reaching more than 70% of RNV release from ZIF-8 (Fig. 3B and fig. S3D). Scanning electron microscopy images of the release process show a gradual dissociation of NV-ZIF at acidic conditions over time. NV-ZIF morphology remarkably changed after 12 hours, extrapolating the 50% release of RNV from ZIF-8 (fig. S3E). In contrast, less than 25% of RNV was released over 3 days under physiological conditions at pH 7.3, and the system exhibited an excellent colloidal stability for over 6 months (Fig. 3B and fig. S3C). Such slow and controlled release behavior is intended to improve treatment outcomes. Next, we evaluated the in vitro cytotoxicity of NV, ZIF-8, and NV-ZIF by cell counting kit-8 (CCK-8) against human embryonic kidney (HEK), HeLa, and Michigan Cancer Foundation-7 (MCF-7). As expected, no obvious cytotoxicity was observed, supporting NV-ZIF biocompatibility (fig. S4, A to C). Our system exhibited excellent NV sustained release performances and triggered pH responsiveness, which is consistent with previously reported drug-loaded ZIF-8 delivery systems (30, 32).
Expression of PD-1 and in vitro pH-responsive NV-ZIF
The expression of PD-1 was first examined on Jurkat T cells that were either activated with anti-CD3/anti-CD28 antibodies or left unactivated. Flow cytometric analysis demonstrated that the expression of PD-1 on activated Jurkat T cells (aTCs; >80%, P < 0.005) was significantly higher than that on unactivated Jurkat T cells (10%, P < 0.005) (fig. S5, A and B). To assess NV activity and integrity after loading, we extracted NV from ZIF-8 by adding EDTA to dissociate the ZIF-8 crystals by breaking the coordination bonds between Zn2+ and 2-mIM. The released NV was then incubated with aTC for 30 min and labeled with a secondary phycoerythrin (PE)–labeled anti-human antibody. The fluorescence intensity of the extracted NV shifted to the right; the same shift was observed with free NV–treated aTC that was used as a control (fig. S5C). The same result was obtained when the residual NV in the supernatant was used, confirming that embedding NV in the framework did not affect the activity and the integrity of this antibody. Subsequently, the release of NV from NV-ZIF was tested in vitro by incubating NV-ZIF with aTC for 6 and 12 hours at slightly acidic (pH 6.5) and physiological (pH 7.3) conditions. The PD-1/anti–PD-1 (NV) interaction was detected by staining aTC against PE-labeled anti-human antibody. No obvious difference was detected when aTC was incubated with NV-ZIF for 6 hours (17.3%). Twelve hours post incubation induced the release of NV (fig. S6). A marked increase was observed at pH 6.5 (66.3%) (fig. S6B). On the other hand, treating aTC with free NV for 30 min showed a rapid binding of anti–PD-1 (NV) with PD-1 on aTC (47.7%) (fig. S6A). Such treatment profile is consistent with the one currently used in the clinics, which is associated with increased levels of toxicity (33). Administering the same dose but at a slower rate would help to avoid adverse reactions. Hence, the slow and sustained release behavior of NV-ZIF will help in reducing immune-related life-threatening events associated with free NV delivery.
Efficacy of NV sustained release on hematological malignancies
Lymphocytes of patients with acute myeloid leukemia (AML) and chronic lymphoid leukemia (CLL) are known to express high levels of PD-1 (34, 35), while PD-L1 was shown to be up-regulated on cancerous cells and antigen-presenting cells (APCs) from these patients (36–38). Peripheral blood mononuclear cells (PBMCs) isolated from patients with AML and CLL were initially treated with ZIF-8 to test its effect on T cell proliferation (CD8+ and CD4+) using Ki-67 as a marker for cell proliferation. Our data demonstrated that CD8+ and CD4+ T cells treated with phytohemagglutinin (PHA) resulted in high Ki-67 expression compared to ZIF-8 and the control (fig. S7, A to C), indicating that ZIF-8 has no effect on lymphocyte proliferation. Next, we treated CD8+ T cells with NV-ZIF for 6 and 12 hours. Compared to free NV, NV-ZIF boosted the activity of PHA-stimulated T cells over time. As shown in Figs. 4 and 5, contrary to ZIF-8, NV-ZIF enhanced the activation of CD8+ T cells compared to nontreated cells as evidenced by the higher levels of CD8+ interferon-γ (IFN-γ) and CD8+ tumor necrosis factor–α (TNF-α) T cells. NV-ZIF increased the level of CD8+ IFN-γ and CD8+ TNF-α T cells 12 hours following treatment (Figs. 4 and 5). This is mostly due to the sustained release of NV from the NV-ZIF over time. The level of activation of those cells at 12 hours was either slightly higher or comparable to cells treated with free NV (Figs. 4 and 5). Although we expected that there is higher cytokine release at 12 hours compared to 6 hours, the comparably high levels of cytokines observed for cells treated with NV-ZIF and NV suggest that there was a sufficient release of NV from the NV-ZIF.
Fig. 4. Anticancer activity of NV-ZIF in PBMCs isolated from CLL patient samples.
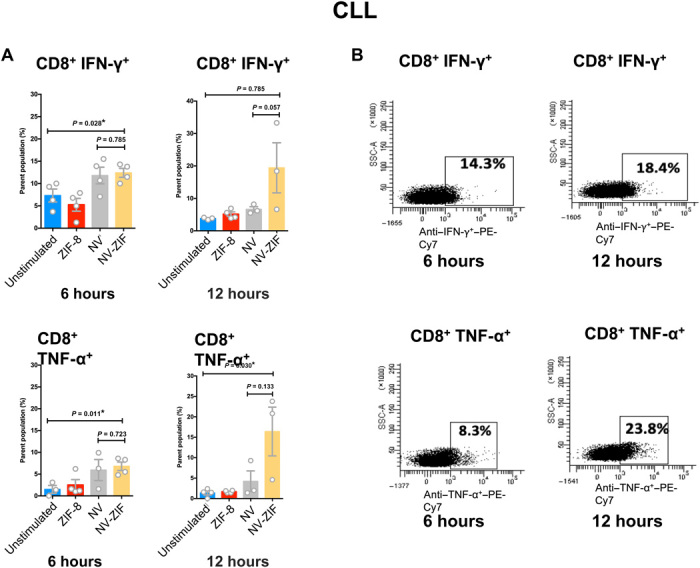
(A) Quantitative analysis of CD8+ IFN-γ+ and CD8+ TNF-α+ T cells in serum in response to various treatments after 6 and 12 hours. Error bars are based on SE (n = 5). (B) Representative flow cytometry plots of lymphocytes of CLL treated with NV-ZIF. SSC-A, side scattered area. Statistical significance was calculated by one- or two-way analysis of variance (ANOVA) and Tukey’s multiple comparisons test: *P < 0.05.
Fig. 5. Anticancer activity of NV-ZIF in PBMCs isolated from AML patient samples.
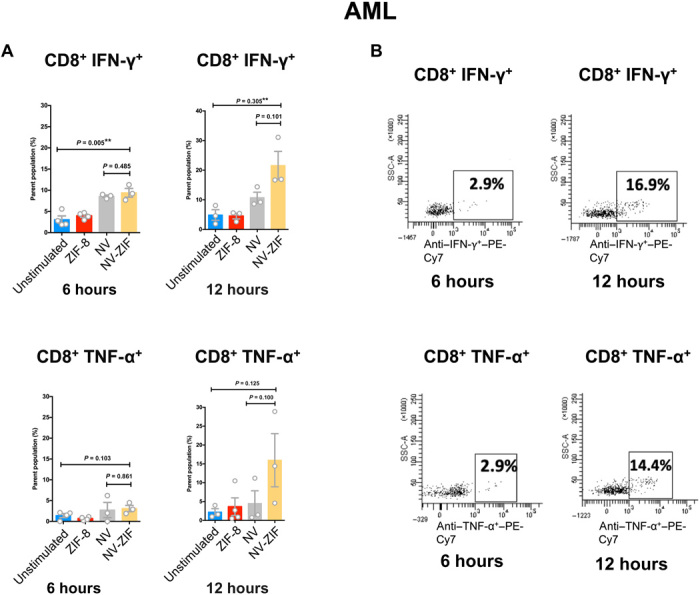
(A) Quantitative analysis of CD8+ IFN-γ+ and CD8+ TNF-α+ T cells in serum in response to various treatments after 6 and 12 hours. Error bars are based on SE (n = 5). (B) Representative flow cytometry plots of lymphocytes of AML treated with NV-ZIF. Statistical significance was calculated by one- or two-way ANOVA and Tukey’s multiple comparisons test: *P < 0.05.
Cell type–specific delivery of NV
Attending to the tumor microenvironment (TME) is crucial when developing therapies for solid tumors. We modified the NV-ZIF to specifically deliver and release NV into the TME, enabling local activation of tumor-specific immune responses and reducing systemic toxicity associated with NV administration. CC membrane was used as a targeting agent in our study, which provided a personalized tumor-specific PD-1 blockade therapy. We previously validated that coating ZIF-8 with CC resulted in preferential accumulation of coated ZIF-8 within CCs from which the membrane was extracted (25). The same protocol was followed for coating NV-ZIF with MCF-7 membrane. TEM micrographs of MCF membrane–coated NV-ZIF (NV-ZIFMCF) showed an octagonal crystal with an average size of 166 nm, and the negative staining of NV-ZIFMCF exhibited rough surface after coating (fig. S8A). The PXRD patterns and intensity of NV-ZIFMCF are similar to those of the NV-ZIF and ZIF-8, which supports that the ZIF-8 crystallinity was maintained after coating (fig. S8B). Zeta potential measurements validated the complexation with CC, as the charge of the NV-ZIF dropped from +11 to −21 mV (fig. S8C). The successful functionalization was further confirmed by SDS–polyacrylamide gel electrophoresis (PAGE), followed by protein staining (fig. S9). The protein profile of the purified CC matches that of the NV-ZIFMCF (fig. S9A), indicating a good retention of the characteristic proteins inherited from the CC. Surface adhesion molecules such as CD44, E-cadherin, and CD49e were detected on CC and NV-ZIFMCF by Western blot (fig. S9B). Biocompatibility of NV-ZIFMCF was tested at different concentrations by incubation with MCF-7 for 24 hours using CCK-8. Compared to the native MCF-7, concentrations below 100 μg·ml−1 were completely safe, whereas high concentrations (100 μg·ml−1) led to a low level of cytotoxicity (fig. S10). To verify the cancer-targeting ability of NV-ZIFMCF, we incubated NV-ZIFMCF with HeLa, HEK, and MCF-7. The results indicated that NV-ZIFMCF accumulated in MCF-7 tumors and that the accumulation steadily increased with longer incubation times (fig. S11, A to C), which is consistent with our previous study (25). The targeted delivery and preferential accumulation of NV-ZIFMCF were further evaluated using homologous 4T1 cancer-bearing mice in vivo. XenoLight DiR (1,1′-dioctadecyl-3,3,3′,3′-tetramethylindotricarbocyanine iodide) was loaded with NV-ZIF for in vivo imaging purposes. Mice-bearing 4T1 tumors were imaged at different time points (3 and 24 hours) following injection of ZIF particles using In Vivo Imaging System (IVIS). Our data revealed that NV-ZIFMCF exhibited a high accumulation in tumors within 3 hours with prolonged tumor retention. Unlike NV-ZIFMCF, the accumulation of NV-ZIF (uncoated) was low and detected after 24 hours, supporting the efficiency of this targeting strategy (Fig. 6A). Measuring Zn2+ content of the tumor by inductively coupled plasma mass spectrometry (ICP-MS) showed a significant increase in the accumulation of NV-ZIFMCF at tumor site compared to that of NV-ZIF (Fig. 6B). Unlike most developed delivery systems that targeted superficial TME, such as melanoma (39, 40), our engineered NV-ZIFMCF efficiently targets TME inside the body. This highly specific cancer recognition ability of NV-ZIFMCF can extensively enhance the therapeutic effect of NV, as the platform showed negligible toxicity to the animals as verified by the control samples.
Fig. 6. Cancer-targeting and retention behavior of NV-ZIFMCF.
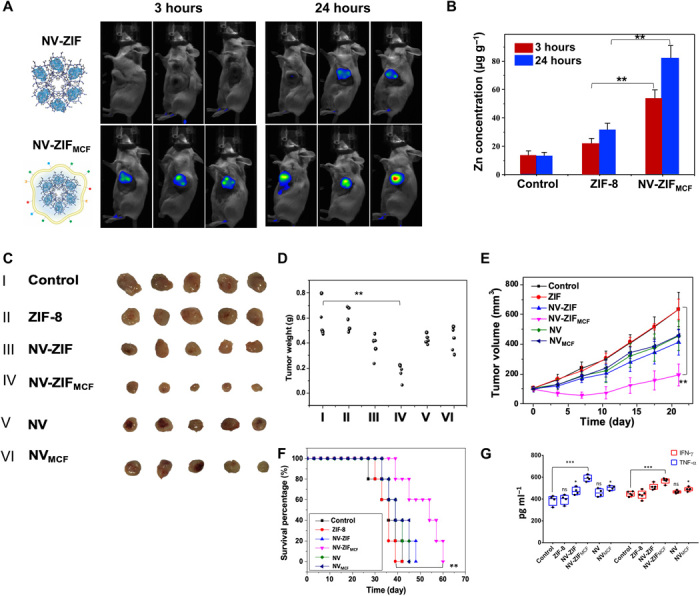
(A) The in vivo fluorescence images of 4T1 cancer-bearing mice after intravenous injection of NV-ZIF and NV-ZIFMCF. Images were taken at 3 hours (left) and 24 hours (right) post-injection. (B) ICP-MS analysis of Zn in tumors of mice injected with PBS (control), NV-ZIF, or NV-ZIFMCF. Error bars are based on SE (n = 5). (C) Representative images of tumors isolated from mice at the end of various treatments (21 days post-injection). (D and E) Tumor weight and tumor growth curves of different groups of 4T1 cancer-bearing mice after various treatments, respectively. Error bars are based on SE (n = 5). (F) Kaplan-Meier survival curve images of 4T1 cancer bearing mice after various treatments. (G) Quantitative analysis of TNF-α and IFN-γ in the serum collected 48 hours post-treatment (n = 4 to 5). Statistical significance was calculated by one- or two-way ANOVA and Tukey’s multiple comparisons test: *P < 0.05, **P < 0.01, and ***P < 0.001, ns, not significant.
Efficacy of NV sustained release on solid tumors
We injected mice with either NV-ZIFMCF, NV-ZIF, CC-NV, or NV (3 mg·kg−1 per mouse for each injection) on days 3, 6, 9, and 12. The mice were then sacrificed on day 21, and tumor sizes were measured. Results in Fig. 6 (C to F) indicate that the NV-ZIFMCF treatment significantly inhibited tumor growth compared to NV-ZIF, CC-NV, or NV treatment alone. To further test the treatment effect, we observed tumor development over 21 days after various treatments and found that the antitumor activity and survival rate were significantly extended (tumor volume maintained <200 mm3, P < 0.01) after the NV-ZIFMCF treatment (Fig. 6, C to F). The survival time was slightly extended from 42 days for untreated mice to 45 and 49 days for NV-ZIF– and NVMCF-treated mice, respectively (Fig. 6F). Free NV did not show superior tumor survival compared to NV-ZIF and NVMCF, indicating the necessity of efficient NV delivery for effective tumor inhibition. In contrast, NV-ZIFMCF significantly prolonged animal survival (Fig. 6F). Mice treated with NV-ZIFMCF also showed a significantly higher production of IFN-γ and TNF-α (P < 0.001) compared to other treated groups that showed comparable levels of production (Fig. 6G). We measured the mice body weight, and as expected, experimental and control mice did not show an obvious difference in body weight (fig. S12).
Next, we characterized CC proliferation by histological assays. CC damage was detected by hematoxylin and eosin (H&E) staining upon treatment with NV-ZIFMCF (Fig. 7A). The Ki-67 staining results revealed marked reduction in Ki-67 levels after NV-ZIFMCF treatment, resulting in significant inhibition of the proliferation of tumor cells compared to other treatments (Fig. 7B). Of a particular note, functionalizing NV with the targeting agent (CC-NV) did not result in equivalent antitumor activity to that of NV-ZIFMCF. Likewise, NV-ZIF did not exhibit antitumor activity similar to that of NV-ZIFMCF, which supports insufficient NV delivery to TME in both cases. The cancer inhibition ratio reached about 73% after treatment with NV-ZIFMCF, indicating that this strategy made TME more sensitive to immunotherapy. To gain a better understanding of the effects of each treatment regimen on lymphocytes present in the TME, we analyzed the population of FoxP3+ regulatory T cells. These FoxP3+ regulatory T cells act to suppress immune responses and, in this case, antitumor immune responses. T cells from tumor tissues were harvested and analyzed by flow cytometry to determine the percentage of regulatory T cells within the total T cell population in the tumor. The percentage of FoxP3+ cells significantly decreased in NV-ZIFMCF–treated groups compared to the control and ZIF-8, whereas NV-, CC-NV–, and NV-ZIF–treated groups showed some level of reduction (Fig. 7C), supporting the enhanced production of IFN-γ and TNF-α in groups treated with NV-ZIFMCF. These results strongly confirm the anticancer efficacy of NV-ZIFMCF enabled by the inhibition of the regulatory T cells. Collectively, the antitumor activity of CC-NV was comparable to that of NV-ZIF, indicating that sufficient delivery of NV to TME is the key for enhanced NV-ZIFMCF antitumor activity. An extrapolation of these results suggests that tumor-specific delivery of NV results in (i) enriching NV within TME, leading to local inhibition of PD-1; (ii) enhancing the sensitivity of TME to anti–PD-1 blockade therapy; and (iii) systemically activating specific antitumor immune response enabled by the local inhibition of the regulatory T cells.
Fig. 7. Immunohistochemical analysis of the tumor sections.
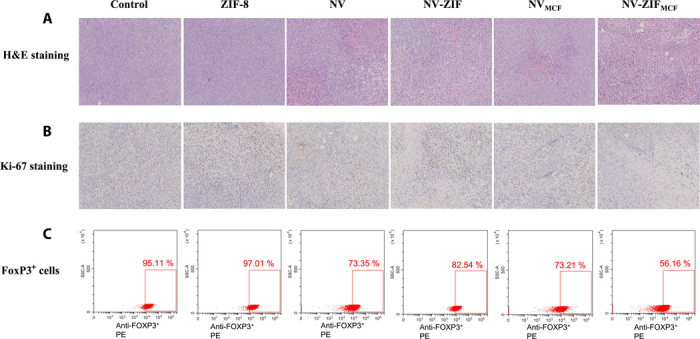
(A) H&E staining and (B) Ki-67 staining of the sacrificed cancer tissues after various treatments for 21 days. (C) Representative flow cytometry plots showing FoxP3+ regulatory T cells.
DISCUSSION
ICB therapy has shown encouraging preclinical and clinical results to treat different types of tumors. Current delivery methods, however, are not antigen specific and result in the systemic blocking of regulatory pathways, leading to systemic activation of immune cells and limiting therapeutic benefits in many patients. Consequently, there is a tremendous need to improve the safety and efficacy of such treatments. Here, we demonstrated the therapeutic potential of the sustained release and targeted delivery of NV by NV-ZIF and NV-ZIFMCF on both hematological malignancies and solid tumors, respectively.
Hematological malignancies, such as leukemia, involve continuous and systemic contact between the tumor clone and the immune system. Our NV-ZIF release behavior showed a slow release of small doses of NV over time that resulted in improving the antitumor activity with longer incubation by inducing T cell activation to a level comparable to that of free NV. This kind of release behavior is expected to reduce immune-related toxicity and increase patient compliance.
On the other hand, solid tumors are characterized by confining infiltrating lymphocytes in localized tissue. The suppressive nature of TME induces the irresponsiveness to PD-1 blockade therapy. Thus, we coated our NV-ZIF with CC to enable tumor-specific recognition, reducing off-target delivery and immune-related side effects, improving the sensitivity of TME to NV, extending the retention of NV-ZIF within tumor, and eliciting tumor-specific immunity. We used the challenging 4T1 mouse breast cancer model to demonstrate the therapeutic potential of NV-ZIFMCF. Our results showed the superior antitumor activity of NV-ZIFMCF over NV, CC-NV, and NV-ZIF. Of a particular note, functionalizing NV with the targeting agent (CC-NV) did not result in equivalent antitumor activity to that of NV-ZIFMCF. Likewise, NV-ZIF did not exhibit antitumor activity similar to that of NV-ZIFMCF, which supports insufficient NV delivery to TME in both cases. The cancer inhibition ratio reached about 73% after treatment with NV-ZIFMCF, indicating that this treatment regimen made TME more sensitive to immunotherapy. Such treatment could be followed by chemotherapy or radiotherapy to completely eradicate tumor. CC coating could be used as a promising strategy to develop a personalized tumor-specific immune response as shown in previous studies (28). Our strategy has shown that local delivery of NV-ZIFMCF leads to systemic and durable activation of antitumor immune response that has the potential to reduce the risk of metastasis. Unlike most developed delivery systems that targeted superficial TME, such as melanoma (39, 40), our developed NV-ZIFMCF was efficient in targeting TME inside the body. Our strategy shows a great clinical translation potential in patients with both hematological malignancies and solid tumors because all the materials used in this system are biocompatible, and it would be a step toward developing personalized immune therapeutics treatment plans.
In summary, we have successfully loaded NV in ZIF-8 and demonstrated the potential utility of the sustained NV release in hematological malignancies and solid tumors. The sustained release behavior of NV-ZIF has shown its efficacy in activating T cells in AML and CLL. The system was further modified to enable tumor-specific targeted delivery while treating solid tumors by coating NV-ZIF with specific CC membrane. NV-ZIFMCF displayed an enhanced antitumor activity due to the preferential accumulation and prolonged retention of NV-ZIFMCF within TME that resulted in efficient NV delivery. Collectively, this work demonstrates that tackling the sustained and targeted delivery is the way forward for the broader impact of ICB therapy in the fight against cancer.
MATERIALS AND METHODS
Fabrication of ZIF-8, NV-ZIF, and NV-ZIFMCF
NV-ZIF was synthesized by stirring NV (1 mg·ml−1) and 2-mIM (2.5 M, 0.9 ml) for 30 min. Zinc nitrate solution (0.5 M, 0.1 ml) was slowly added under mechanical agitation for 20 min. The resulting product was collected by centrifugation and washed three times with deionized water to remove any residues. ZIF-8 was synthesized by slowly adding zinc nitrate solution (0.5 M, 0.1 ml) to 2-mIM (2.5 M, 0.9 ml). The solution was agitated for 20 min. The supernatant of NV-ZIF was collected to calculate the LC and LE by Bradford assay. NV-ZIFMCF was fabricated by mixing 1:1 weight ratio of NV-ZIF and extracted CC membrane in deionized water. The mixture was then transferred into a syringe and successively extruded through 1.0-μm and 800.0- and 450.0-nm polycarbonate membrane. The obtained NV-ZIFMCF in solution was further purified by centrifugation to remove the free CC membrane. The zeta potential of NV-ZIF was performed using a Malvern Zetasizer Nano ZS at 25°C at pH 7.3 in aqueous solutions. PXRD measurements were performed using a Panalytical X’Pert Pro X-ray powder diffractometer using the Cu Kα radiation (40 V, 40 mA, λ = 1.54056 Å) in a θ – θ mode from 20° to 90° (2θ). TEM images were obtained using FEI Tecnai 12 microscope operating at 120 kV. For visualization by TEM, samples were prepared by dropping the solution on a copper grid 300 mesh (Electron Microscopy Sciences, LC 300-Cu). Fluorescence measurements were performed on a Cary Eclipse fluorescence spectrophotometer (Varian). The slits for excitation and emission were set at 10 nm. NV was labeled with rhodamine B (Rh) by N-hydroxysuccinimide (NHS) chemistry. Briefly, 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) (5 mg) and NHS (2.5 mg) were mixed with NV solution (10 mg·ml−1, 1 ml), and the mixed solution was stirred for 2 hours. Rh (63 μg) was then dissolved in dimethyl sulfoxide (100 μl), and the whole solution was stirred overnight at 4°C in the dark. The dialysis technique was used to remove unreacted EDC, NHS, and Rh.
Preparation of CC membrane
Human breast adenocarcinoma cell (MCF-7) cells were incubated in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS) and 1% antibiotics (penicillin-streptomycin). Cells were grown in T-175 culture flasks to full confluency and then detached and washed in PBS three times by centrifuging at 500g. Then, they were suspended in a hypotonic lysing buffer consisting of 20 mM tris-HCl (pH 7.5), 10 mM KCl, 2 mM MgCl2, and 1 EDTA-free mini protease inhibitor tablet per 10 ml of solution and disrupted using a Dounce homogenizer with a tight-fitting pestle. The entire solution was subjected to 20 passes before spinning down at 3200g for 5 min. The supernatant was saved, while the pellet was resuspended in hypotonic lysing buffer and subjected to another 20 passes and spun down again. The supernatants were pooled and centrifuged at 20,000g for 20 min, after which the pellet was discarded, and the supernatant was centrifuged again at 100,000g. The pellet containing the plasma membrane material was then washed again in 10 mM tris-HCl (pH 7.5) and 1 mM EDTA. The final pellet was collected and used as a purified CC membrane.
CC membrane protein characterization
Protein characterization was carried out using the SDS-PAGE method. The cracked CC membrane samples were suspended in lithium dodecyl sulfate loading buffer (Invitrogen). Samples were heated to 90°C for 10 min, and 20 μl of sample was loaded into each well of a NuPAGE Novex 4 to 12% bis-tris minigel, using Mops SDS as the running buffer (Invitrogen) in an XCell SureLock Electrophoresis System based on the manufacturer’s instructions. Protein staining was accomplished using Coomassie Blue (Invitrogen) and destained in water overnight before imaging. For Western blot analysis, the protein was transferred to Protran nitrocellulose membranes (Whatman) using an XCell II Blot Module (Invitrogen) in NuPAGE transfer buffer (Invitrogen) per the manufacturer’s instructions. Membranes were probed using antibodies against CD44 (clone 515; BD Biosciences), E-cadherin (clone 36; BD Biosciences), and CD49e (BD Biosciences), followed by horseradish peroxidase–conjugated anti-mouse immunoglobulin G (Cell Signaling Technology) as the secondary antibody.
LC and LE NV-ZIF
The LC and LE of NV in ZIF-8 nanoparticles were measured with the Bradford method. First, a standard curve of NV at 595 nm was generated. Then, LE and LC of NV in ZIF-8 were obtained by analyzing residual NV in supernatants, which was collected after washing. The LE was calculated as follows
Release of Rh-labeled NV via pH trigger
To evaluate the release of Rh-labeled NV from ZIF-8, the fluorescence signal of Rh-labeled NV was measured by using the microplate spectrophotometer. Aliquots of hydrochloric acid were added to Rh-labeled NV-ZIF (600 μg·ml−1) in PBS to reach a pH of 6.5 at 37°C. PBS only was added to the sample of pH 7.3. The supernatant of the mixture solution was obtained through centrifugation at different time points. The fluorescence of released Rh-labeled NV was monitored by fluorescent spectroscopy (excitation/emission wavelength: 540 nm/625 nm).
In vitro release and Jurkat activation
Anti-CD3 (mouse anti-human CD3, clone: OKT3; eBioscience) was added in a 24-well plate at a final concentration of 5 μg·ml−1 prepared in PBS (300 μl per well) and incubated for 3 hours in a 37°C incubator supplied with 5% CO2. After incubation, antibody solution was removed from each well. In the same well, 106 cells·ml−1 of Jurkat cells (acute T cell leukemia human, Jurkat, clone E6-1) were resuspended in 2 ml of RPMI 1640 (Gibco) supplemented with 10% FBS (Gibco) and 1% streptomycin (Hyclone). Anti-CD28 (purified NA/LE as described in BD mouse anti-human CD28, BD) was added at a final concentration of 1 μg·ml−1 to each well. Last, interleukin-2 was added at a final concentration of 100 U·ml−1. The cells were then incubated in a 37°C humidified incubator supplied with 5% CO2 for 3 days.
Cell viability
CCK-8 assay was performed according to the manufacturer’s protocol. Briefly, MCF-7, HEK, and HeLa cells (5 × 103 cells per well) were seeded onto a 96-well plate. After 12 hours, the culture medium was changed, and the cells were incubated with different concentrations (100, 50, 25, 12, 6, and 3 μg·ml−1) of NV, NV-ZIF, and ZIF-8 in 200 μl of DMEM at 37°C for 24 hours. The media was then discarded, and the prepared culture medium containing 10% CCK-8 solution was added into each well, including a negative control of culture media alone. After 3 hours of incubation, the absorbance was measured at 450 nm using a microplate spectrophotometer (xMark Microplate Absorbance Spectrophotometer).
Specific targeting studies
Flow cytometric assay was used to investigate the specific targeting ability of NV-ZIFMCF. Rh–NV–ZIFMCF was used to track the uptake of the NV-ZIFMCF. Cells were seeded in six-well plates at a density of 5 × 105 cells per single well and cultured for 12 hours in 2 ml of DMEM containing 10% FBS and 1% antibiotics (penicillin-streptomycin). After NV-ZIFMCF (100 μg·ml−1) was co-incubated with the cells for 1, 3, 6, 12, and 24 hours, the cells were washed three times with PBS, detached by trypsin, and lastly collected by centrifugation at 1000 rpm for 5 min. The bottom cells were washed three times with PBS, and then the suspended cells were analyzed by BD LSR II Flow Cytometer equipped with BD FACSDiva (BD Biosciences) software.
Treating stimulated inflammatory cells with NV, ZIF-8, and NV-ZIF
PBMCs were isolated from blood samples of two patients, one with AML and another with CLL, using a Ficoll gradient (Axis Shield, Norway). Cells were collected in complete RPMI 1640 medium (pH 6.5). PBMCs (106 cells) were then divided into five sets for each treatment and incubated with either ZIF-8 (5 μg·ml−1), NV (10 μg·ml−1), or NV-ZIF (5 μg·ml−1) for 1 hour. Cells were then stimulated with PHA (100 ng·ml−1) for 6 or 12 hours (the last 2 hours in the presence of brefeldin A). Intracellular cytokine staining was performed to determine the ability of CD8+ cells to express cytokines. The cells were surface stained with CD3+ APC (0.2 μg·μl−1; R&D Systems, Minneapolis, MN, USA). They were then fixed in 4% paraformaldehyde, resuspended in 0.25% saponin (S4521; Sigma-Aldrich, Germany), and stained with anti–IFN-γ PE-Cy7 [PE-Cy7 mouse anti-human IFN-γ (BD Biosciences), 0.2 μg·μl−1] and anti–TNF-α–PE-Cy7 [PE-Cy7 mouse anti-human TNF-α (BD Biosciences), 0.2 μg·μl−1] antibodies. Samples were analyzed using a BD LSR II flow cytometer equipped with BD FACSDiva (BD Biosciences) software.
Animals and tumor models
All animal experiments were carried out in accordance with the Institute of Laboratory Animal Resources guidelines. Ethical approval was granted by the Institutional Animal Care and Use Committee of Zhejiang Academy of Medical Sciences, China.
Female BALB/c mice (4 weeks old, ~20-g body weight) were purchased from the Zhejiang Academy of Medical Sciences and maintained in a pathogen-free environment under controlled temperature (24°C). A total of 0.1 ml of 4T1 cells (5 × 105) was injected into the breast fat pad of the mice. The tumors were allowed to grow to ~100 mm3 before experimentation.
The tumor volume was calculated as (tumor length) × (tumor width)2/2.
For tumor accumulation studies, the mice were randomly divided into two groups (n = 3) and intravenously injected with XenoLight DiR–NV–ZIF or Rh–NV–ZIFMCF [corresponding to NV (3.0 mg·kg−1)]. The fluorescent images were obtained under an IVIS (CRi USA, IVIS: 710 excitation/760 emission). The tumor samples were then collected at the desired time after injection and were digested using concentrated nitric acid. The amount of Zn2+ in the tumors was measured using ICP-MS.
For antitumor activity study, 4T1 tumor-bearing mice were randomly divided into six groups (n = 5) and intravenously injected with (i) 200 μl of physiological saline, (ii) 200 μl of ZIF-8 solution, (iii) 200 μl of NV-ZIF solution, (iv) 200 μl of NV-ZIFMCF solution, (v) 200 μl of NV solution, and (vi) 200 μl of NVMCF solution, respectively. The dosage of NV is 3.0 mg·kg−1. Mice received treatment four times every 3 days. Physiological saline that is used for in vivo application is 1× PBS (0.01 M). Tumor volume and body weight were measured every 3 days. In the histological assay, the tumor tissues were fixed in 4% paraformaldehyde for 24 hours. The specimens were dehydrated in graded ethanol, embedded in paraffin, and cut into 5-mm-thick sections. The fixed sections were deparaffinized and hydrated according to a standard protocol and stained with H&E for microscopic observation. Tumor sections were also stained with antibody against Ki-67 (Abcam, USA) to visualize viable CCs.
Blood samples (0.1 ml) were taken from retro-orbital sinus to isolate serum for analysis, 48 hours after single injection. TNF-α (MTA00B; R&D systems) and IFN-γ (MIF00; R&D systems) were analyzed with enzyme-linked immunosorbent assay kits according to the vendors’ protocols.
To study the immune cells inside tumors, tumors were harvested from mice in different groups and cut into small pieces. After being ground by the rubber end of a syringe in cell strainers, tumor tissues were treated with 0.25% trypsin-EDTA solution for 5 min at 37°C. Then, cells were filtered through nylon mesh filters with a size of 70 μm and washed with PBS. The single-cell suspension was then incubated with anti-CD16/32 (BD Pharmingen; catalog: 553141) to reduce nonspecific binding to the fragment crystallizable region (Fc receptor). Cells were further stained with anti-mouse FOXP3 (eBioscience; catalog: 12-5773-82) antibodies according to the manufacturer’s protocols. Last, flow cytometry was used for cell sorting.
Statistical analysis
Data are reported as means ± SD. The differences among groups were determined using one- or two-way analysis of variance (ANOVA) analysis. Statistical significance was calculated by one- or two-way ANOVA and Tukey’s multiple comparisons test: *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Acknowledgments
We thank R. Langer, Institute Professor, MIT, for feedback and comments. We acknowledge H. Alrabiah, Associate Professor at the Department of Pharmaceutical Chemistry, College of Pharmacy, King Saud University and H. I. Aljohar, Assistant Professor at the Department of Pharmaceutical Chemistry, College of Pharmacy, King Saud University for providing nivolumab. Funding: This work was funded by the King Abdulaziz City for Science and Technology (KACST) through the MERS-CoV research grant program (number 20-0004), which is a part of the Targeted Research Program (TRP). Author contributions: N.M.K. conceived the idea. S.K.A. carried out synthesis and characterization of NV-ZIF and NV-ZIFMCF and in vitro studies. S.S.Q., S.S., A.A., R.H., W.B., M.A., MR.A., and J.M. helped in characterization and in vitro studies. Z.M. designed and performed in vivo studies. S.K.A. and N.M.K. designed the experiments and wrote the paper. All authors discussed the results and commented on the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/4/eabe7174/DC1
REFERENCES AND NOTES
- 1.Khalil D. N., Smith E. L., Brentjens R. J., Wolchok J. D., Erratum: The future of cancer treatment: Immunomodulation, CARs and combination immunotherapy. Nat. Rev. Clin. Oncol. 13, 394 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ribas A., Wolchok J. D., Cancer immunotherapy using checkpoint blockade. Science 359, 1350–1355 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sharma P., Hu-Lieskovan S., Wargo J. A., Ribas A., Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 168, 707–723 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Riley R. S., June C. H., Langer R., Mitchell M. J., Delivery technologies for cancer immunotherapy. Nat. Rev. Drug Discov. 18, 175–196 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mi Y., Hagan C. T. IV, Vincent B. G., Wang A. Z., Emerging nano−/microapproaches for cancer immunotherapy. Adv. Sci. 6, 1801847 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang H., Mooney D. J., Biomaterial-assisted targeted modulation of immune cells in cancer treatment. Nat. Mater. 17, 761–772 (2018). [DOI] [PubMed] [Google Scholar]
- 7.Kuai R., Ochyl L. J., Bahjat K. S., Schwendeman A., Moon J. J., Designer vaccine nanodiscs for personalized cancer immunotherapy. Nat. Mater. 16, 489–496 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Park C. G., Hartl C. A., Schmid D., Carmona E. M., Kim H.-J., Goldberg M. S., Extended release of perioperative immunotherapy prevents tumor recurrence and eliminates metastases. Sci. Transl. Med. 10, eaar1916 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Moynihan K. D., Opel C. F., Szeto G. L., Tzeng A., Zhu E. F., Engreitz J. M., Williams R. T., Rakhra K., Zhang M. H., Rothschilds A. M., Kumari S., Kelly R. L., Kwan B. H., Abraham W., Hu K., Mehta N. K., Kauke M. J., Suh H., Cochran J. R., Lauffenburger D. A., Wittrup K. D., Irvine D. J., Eradication of large established tumors in mice by combination immunotherapy that engages innate and adaptive immune responses. Nat. Med. 22, 1402–1410 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Silva J. M., Videira M., Gaspar R., Préat V., Florindo H. F., Immune system targeting by biodegradable nanoparticles for cancer vaccines. J. Controll. Release 168, 179–199 (2013). [DOI] [PubMed] [Google Scholar]
- 11.Irvine D. J., Hanson M. C., Rakhra K., Tokatlian T., Synthetic nanoparticles for vaccines and immunotherapy. Chem. Rev. 115, 11109–11146 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Francis D. M., Thomas S. N., Progress and opportunities for enhancing the delivery and efficacy of checkpoint inhibitors for cancer immunotherapy. Adv. Drug Deliv. Rev. 114, 33–42 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen Q., Wang C., Chen G., Hu Q., Gu Z., Delivery strategies for immune checkpoint blockade. Adv. Healthc. Mater. 7, e1800424 (2018). [DOI] [PubMed] [Google Scholar]
- 14.Meir R., Shamalov K., Betzer O., Motiei M., Horovitz-Fried M., Yehuda R., Popovtzer A., Popovtzer R., Cohen C. J., Nanomedicine for cancer immunotherapy: tracking cancer-specific T-cells in vivo with gold nanoparticles and CT imaging. ACS Nano 9, 6363–6372 (2015). [DOI] [PubMed] [Google Scholar]
- 15.Meir R., Shamalov K., Sadan T., Motiei M., Yaari G., Cohen C. J., Popovtzer R., Fast image-guided stratification using anti-programmed death ligand 1 gold nanoparticles for cancer immunotherapy. ACS Nano 11, 11127–11134 (2017). [DOI] [PubMed] [Google Scholar]
- 16.Ishihara J., Fukunaga K., Ishihara A., Larsson H. M., Potin L., Hosseinchi P., Galliverti G., Swartz M. A., Hubbell J. A., Matrix-binding checkpoint immunotherapies enhance antitumor efficacy and reduce adverse events. Sci. Transl. Med. 9, eaan0401 (2017). [DOI] [PubMed] [Google Scholar]
- 17.Horcajada P., Gref R., Baati T., Allan P. K., Maurin G., Couvreur P., Férey G., Morris R. E., Serre C., Metal–organic frameworks in biomedicine. Chem. Rev. 112, 1232–1268 (2012). [DOI] [PubMed] [Google Scholar]
- 18.Li P., Modica J. A., Howarth A. J., Vargas E. L., Moghadam P. Z., Snurr R. Q., Mrksich M., Hupp J. T., Farha O. K., Toward design rules for enzyme immobilization in hierarchical mesoporous metal–organic frameworks. Chem 1, 154–169 (2016). [Google Scholar]
- 19.He C., Lu K., Liu D., Lin W., Nanoscale metal–organic frameworks for the co-delivery of cisplatin and pooled siRNAs to enhance therapeutic efficacy in drug-resistant ovarian cancer cells. J. Am. Chem. Soc. 136, 5181–5184 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li H., Eddaoudi M., O’Keeffe M., Yaghi O. M., Design and synthesis of an exceptionally stable and highly porous metal-organic framework. Nature 402, 276–279 (1999). [Google Scholar]
- 21.Liang K., Ricco R., Doherty C. M., Styles M. J., Bell S., Kirby N., Mudie S., Haylock D., Hill A. J., Doonan C. J., Falcaro P., Biomimetic mineralization of metal–organic frameworks as protective coatings for biomacromolecules. Nat. Commun. 6, 7240 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen T.-T., Yi J.-T., Zhao Y.-Y., Chu X., Biomineralized metal–organic framework nanoparticles enable intracellular delivery and endo-lysosomal release of native active proteins. J. Am. Chem. Soc. 140, 9912–9920 (2018). [DOI] [PubMed] [Google Scholar]
- 23.Yang X., Tang Q., Jiang Y., Zhang M., Wang M., Mao L., Nanoscale ATP-responsive zeolitic imidazole framework-90 as a general platform for cytosolic protein delivery and genome editing. J. Am. Chem. Soc. 141, 3782–3786 (2019). [DOI] [PubMed] [Google Scholar]
- 24.Alsaiari S. K., Patil S., Alyami M., Alamoudi K. O., Aleisa F. A., Merzaban J. S., Li M., Khashab N. M., Endosomal escape and delivery of CRISPR/Cas9 genome editing machinery enabled by nanoscale zeolitic imidazolate framework. J. Am. Chem. Soc. 140, 143–146 (2018). [DOI] [PubMed] [Google Scholar]
- 25.Alyami M. Z., Alsaiari S. K., Li Y., Qutub S. S., Aleisa F. A., Sougrat R., Merzaban J. S., Khashab N. M., Cell-type-specific CRISPR/Cas9 delivery by biomimetic metal organic frameworks. J. Am. Chem. Soc. 142, 1715–1720 (2020). [DOI] [PubMed] [Google Scholar]
- 26.Liao F. S., Lo W. S., Hsu Y. S., Wu C. C., Wang S. C., Shieh F. K., Morabito J. V., Chou L. Y., Wu K. C. W., Tsung C. K., Shielding against unfolding by embedding enzymes in metal-organic frameworks via a de Novo approach. J. Am. Chem. Soc. 139, 6530–6533 (2017). [DOI] [PubMed] [Google Scholar]
- 27.Liang W., Xu H., Carraro F., Maddigan N. K., Li Q., Bell S. G., Huang D. M., Tarzia A., Solomon M. B., Amenitsch H., Vaccari L., Sumby C. J., Falcaro P., Doonan C. J., Enhanced activity of enzymes encapsulated in hydrophilic metal–organic frameworks. J. Am. Chem. Soc. 141, 2348–2355 (2019). [DOI] [PubMed] [Google Scholar]
- 28.Liu W.-L., Zou M. Z., Liu T., Zeng J. Y., Li X., Yu W. Y., Li C. X., Ye J. J., Song W., Feng J., Zhang X. Z., Cytomembrane nanovaccines show therapeutic effects by mimicking tumor cells and antigen presenting cells. Nat. Commun. 10, 3199 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhuang J., Gong H., Zhou J., Zhang Q., Gao W., Fang R. H., Zhang L., Targeted gene silencing in vivo by platelet membrane–coated metal–organic framework nanoparticles. Sci. Adv. 6, eaaz6108 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zheng H. Q., Zhang Y., Liu L., Wan W., Guo P., Nyström A. M., Zou X., One-pot synthesis of metal–organic frameworks with encapsulated target molecules and their applications for controlled drug delivery. J. Am. Chem. Soc. 138, 962–968 (2016). [DOI] [PubMed] [Google Scholar]
- 31.Lyu F., Zhang Y., Zare R. N., Ge J., Liu Z., One-pot synthesis of protein-embedded metal-organic frameworks with enhanced biological activities. Nano Lett. 14, 5761–5765 (2014). [DOI] [PubMed] [Google Scholar]
- 32.Duan F., Feng X., Yang X., Sun W., Jin Y., Liu H., Ge K., Li Z., Zhang J., A simple and powerful co-delivery system based on pH-responsive metal-organic frameworks for enhanced cancer immunotherapy. Biomaterials 122, 23–33 (2017). [DOI] [PubMed] [Google Scholar]
- 33.Larkin J., Chiarion-Sileni V., Gonzalez R., Grob J. J., Cowey C. L., Lao C. D., Schadendorf D., Dummer R., Smylie M., Rutkowski P., Ferrucci P. F., Hill A., Wagstaff J., Carlino M. S., Haanen J. B., Maio M., Marquez-Rodas I., McArthur G. A., Ascierto P. A., Long G. V., Callahan M. K., Postow M. A., Grossmann K., Sznol M., Dreno B., Bastholt L., Yang A., Rollin L. M., Horak C., Hodi F. S., Wolchok J. D., Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N. Engl. J. Med. 373, 23–34 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grzywnowicz M., Karabon L., Karczmarczyk A., Zajac M., Skorka K., Zaleska J., Wlasiuk P., Chocholska S., Tomczak W., Bojarska-Junak A., Dmoszynska A., Frydecka I., Giannopoulos K., The function of a novel immunophenotype candidate molecule PD-1 in chronic lymphocytic leukemia. Leuk. Lymphoma 56, 2908–2913 (2015). [DOI] [PubMed] [Google Scholar]
- 35.Jia B., Wang L., Claxton D. F., Ehmann W. C., Rybka W. B., Mineishi S., Rizvi S., Shike H., Bayerl M., Schell T. D., Hohl R. J., Zheng H., Bone marrow CD8 T cells express high frequency of PD-1 and exhibit reduced anti-leukemia response in newly diagnosed AML patients. Blood Cancer J. 8, 34 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grzywnowicz M., Zaleska J., Mertens D., Tomczak W., Wlasiuk P., Kosior K., Piechnik A., Bojarska-Junak A., Dmoszynska A., Giannopoulos K., Programmed death-1 and its ligand are novel immunotolerant molecules expressed on leukemic B cells in chronic lymphocytic leukemia. PLOS ONE 7, e35178 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Williams P., Basu S., Garcia-Manero G., Hourigan C. S., Oetjen K. A., Cortes J. E., Ravandi F., Jabbour E. J., al-Hamal Z., Konopleva M., Ning J., Xiao L., Hidalgo Lopez J., Kornblau S. M., Andreeff M., Flores W., Bueso-Ramos C., Blando J., Galera P., Calvo K. R., al-Atrash G., Allison J. P., Kantarjian H. M., Sharma P., Daver N. G., The distribution of T-cell subsets and the expression of immune checkpoint receptors and ligands in patients with newly diagnosed and relapsed acute myeloid leukemia. Cancer 125, 1470–1481 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Annibali O., Crescenzi A., Tomarchio V., Pagano A., Bianchi A., Grifoni A., Avvisati G., PD-1 /PD-L1 checkpoint in hematological malignancies. Leuk. Res. 67, 45–55 (2018). [DOI] [PubMed] [Google Scholar]
- 39.Zhang Y., Fang C., Wang R. E., Wang Y., Guo H., Guo C., Zhao L., Li S., Li X., Schultz P. G., Cao Y. J., Wang F., A tumor-targeted immune checkpoint blocker. Proc. Natl. Acad. Sci. U.S.A. 116, 15889–15894 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang C., Ye Y., Hochu G. M., Sadeghifar H., Gu Z., Enhanced cancer immunotherapy by microneedle patch-assisted delivery of anti-PD1 antibody. Nano Lett. 16, 2334–2340 (2016). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/4/eabe7174/DC1