

Abstract
Tissue engineered and bioactive scaffolds with different degradation rates are required for the regeneration of diverse tissues/organs. To optimize tissue regeneration in different tissues, it is desirable that the degradation rate of scaffolds can be manipulated to comply with various stages of tissue regeneration. Unfortunately, the degradation of most degradable polymers rely solely on passive controlled degradation mechanisms. To overcome this challenge, we report a new family of reduction-sensitive biodegradable elastomeric polyurethanes containing various amounts of disulfide bonds (PU-SS), in which degradation can be initiated and accelerated with the supplement of a biological product - antioxidant-glutathione (GSH). The polyurethanes can be processed into films and electrospun fibrous scaffolds. Synthesized materials exhibited robust mechanical properties and high elasticity. Accelerated degradation of the materials was observed in the presence of GSH, and the rate of such degradation depends on the amount of disulfide present in the polymer backbone. The polymers and their degradation products exhibited no apparent cell toxicity while the electrospun scaffolds supported fibroblast growth in vitro. The in vivo subcutaneous implantation model showed that the polymers prompt minimal inflammatory responses, and, as anticipated, the polymer with the higher disulfide bond amount had a faster degradation in vivo. This new family of polyurethanes offers tremendous potential of directed scaffold degradation to promote maximal tissue regeneration.
Keywords: Reduction-sensitive, Triggerable, Biodegradation, Polyurethane, Scaffolds, Tissue engineering
Introduction
Personalized medicine has gained increasing attention in the field of tissue engineering.1 To facilitate tissue repair and regeneration, it is critical that the scaffolds be customized to meet needs of an individual patient. For example, children, who exhibit high regenerative ability, will benefit from faster degrading scaffolds that allow for rapid tissue development. On the other hand, a scaffold that degrades more slowly may suit those patients who exhibit low regenerative ability, such as older adults and people with poor health. As another example, at the early stage of tissue regeneration, tissue scaffold with slow degradation rate is needed to provide sufficient mechanical support while permitting cell infiltration and growth. Fast and complete scaffold degradation is required to minimize foreign body reactions at the later stage of the tissue regeneration. Unfortunately, this pattern of material degradation behavior cannot be reproduced using the existing polymers in which degradation rate can only be controlled passively via hydrolysis or enzymatic processes. The goal of this work is to fabricate a new group of polymers in which the polymer degradation can be initiated and facilitated with the presence of a biological trigger.
Several strategies have been implemented in the development of polymers with the active-induced degradation property. A common method is to conjugate functional labile groups into the polymer backbone wherein the polymer can be degraded in the presence of physical or chemical cues, such as light, ultrasound, redox, and enzyme.2-9 Most of these materials are hydrogel or peptide based.10-12 For example, peptide-conjugated polymers have been shown to permit collagenase-mediated degradation.13 However, collagenase and many other enzymes cannot be administered into the patients to trigger degradation on demand without damaging healthy tissues. To overcome these drawbacks, our goal was to fabricate a new material whose degradation can be induced on demand via nonenzymatic processes.
To achieve this goal, we developed a new group of degradable polymers in which degradation can be induced via redox reactions involving thiols and disulfides. For this process, we fabricated a family of biodegradable polyurethane elastomers containing disulfide bonds (SS) that would trigger material degradation in the presence of a biological antioxidant and reducing agent—glutathione (GSH). GSH is the most abundant non-protein thiol compound found in living organisms, including mammalian cells. GSH has been shown to have many beneficial effects on health, including antioxidant and immune-boosting properties.14-15 It has been documented that large quantities of GSH can be administered via oral (up to 3 grams/day), intravenous (up to 2.4 grams/day), intramuscular (up to 0.6 grams/day) and aerosol (up to 1.2 grams/day) routes with no sign of side effect.16-21 These advantageous properties make this degradation strategy suitable for tissue-engineering applications. As a result, studies have examined the chemical structure as well as mechanical and degradation properties of synthesized polymer films. The toxicity of degradation products and the cellular compatibility of the films were also evaluated. The polymers were further processed into fibrous scaffolds by electrospinning. The mechanical properties and cytocompatibility of these scaffolds were characterized. The in vivo tissue compatibility of the scaffolds was also investigated using a mouse subcutaneous implantation model.
2. Materials and Methods
2.1. Materials
PCL (average number molecular weight=2000, Sigma) and 1,4-butanediol (BDO, Sigma) were dried in a vacuum oven at 60°C to remove residual water before synthesis. 1,6-Hexamethylene diisocyanate (HDI, Sigma) was purified by distillation before use. Hydroxyethyl disulfide (HDS, Sigma), stannous octoate (Sn(Oct)2, Sigma), anhydrous dimethyl sulfoxide (DMSO, Sigma), 1,1,1,3,3,3-hexafluroisopropanol (HFIP, Oakwood Product) and GSH (Sigma) were used as received.
2.2. Synthesis of polyurethane containing disulfides (PU-SS)
The polyurethanes containing disulfide bonds in the backbone were synthesized from PCL and HDI with a chain extender HDS using a two-step process according to a previous report (Figure 1).22 First, PCL was dissolved in DMSO at 70°C in a 3-neck flask under N2 protection with stirring. HDI was then added to the solution, followed by 1 droplet of catalyst Sn(Oct)2 (0.5%). After 3 h reaction, drops of HDS/DMSO solution were added into the prepolymer solution. The final polymer solution concentration was approximately 4% (w/v). Afterwards, the reaction was kept at 70°C for 5 d. The polymer was precipitated in an excess volume of cool deionized water, rinsed 3 times, and then dried in a vacuum oven at 60°C for 2 d. The molar ratios of PCL/HDI/HDS were set as 1.5:2:0.5, 1:2:1, 0.5:2:1.5 and 0.2:2:1.8, which were referred to as PU-0.5SS, PU-1SS, PU-1.5SS and PU-1.8SS, respectively. Polyurethane with a chain extender BDO (PU-BDO) was used as a control. The PCL: HDI: BDO was fixed at 1:2:1. The yields of all products were above 90%.
Figure 1.

Schematic synthesis of biodegradable polyurethane containing disulfide bonds (PU-SS).
2.3. Fabrication of polymer films and electrospun fibrous scaffolds
For PU-SS film preparation, the polymers were firstly dissolved in HFIP with a concentration of 2% (w/v). All films were solvent casted into a Teflon dish. After HFIP completely evaporated, the films were dried in vacuum oven at 60 °C for 2 d to remove the residual solvent.
For electrospun fibrous scaffold fabrication, a 10% (w/v) polymer solution in HFIP was used for each group. The polymer solution was electrospun under the following conditions: 1) the positive voltage to the steel tip (1.2 mm inner diameter) connected with the polymer solution–loaded syringe was 15 kV, 2) the negative voltage to the collector steel disc was –10 kV, 3) the distance between the tip and the collector was 19 cm, 4) the solution infuse rate was 1 mL/h, and 5) the electrospinning time was fixed at 2 h. The electrospun fibrous scaffolds were removed from the disc and dried in a vacuum oven at room temperature overnight for further use.
2.4. Polymer characterization
The Fourier transform infrared spectrometer (FTIR, Nicolet 6700, Thermo Electron Corporation) was used to verify polymer chemical structure. Glass transition temperature (Tg) and melting temperature (Tm) were determined using a differential scanning calorimeter (DSC, Shimazu T60) from −100 to 200°C at a heating rate of 10 °C/min with nitrogen flow. For water absorption, the films (n=3 per polymer) were weighted (W0) and then incubated in a phosphate buffer solution (PBS, Sigma) at 37°C. After 24 h, the films were weighted (W1) after removing surface water using filter paper. The water absorption ratio was calculated as (W1-W0)/W0×100%.
For uniaxial tensile mechanical properties, the strips (2 width ×20 length mm, n = 6) were cut from the polymer films, and their mechanical properties were measured on a MTS Tytron 250 MicroForce Testing Workstation with a 500 N load cell and a cross-head rate of 10 mm/min according to ASTM D638-03.23 The sample was measured at dry state and at room temperature. The instant strain recovery (n = 4) was measured under the same conditions as described above. The sample was stretched to 10% strain, held for 1 min, and then released. The procedure was repeated 3 times. The original length (L0) and the length after stretching (L1) were measured using a caliper. The instant strain recovery was calculated as (1-(L1 -L0)/L0)×100%.
For cyclic stretch, the strips (2×20 mm, n=3) were stretched to 30% and 300% strains, respectively, and released back to 0% strain, which was repeated 10 times at a rate of 10 mm/min.24
The inherent viscosity (IV) associated with molecular weight was measured using an Ubbelohde viscometer, since polyurethanes tend to stick to GPC columns because of the strong hydrogen bonding.25 Each polymer solution (0.1g/dl) was prepared using HFIP and filtered by a 1.2 μm glassfiber filter. Each sample was tested at 25°C and the IV was calculated as ln(tp/ts)/Cp, where tp is the time for the polymer solution flowing through the capillary, ts is the time for the solvent HFIP, and Cp represents the polymer concentration.
2.5. Scaffold characterization
The morphology of electrospun fibrous scaffolds was observed using a scanning electronic microscope (SEM, HITACHI S-3000N). The fiber diameter was measured using ImageJ (National Institutes of Health, US). The mechanical properties of the fibrous scaffolds at dry state were measured using the same protocol as for the polymer film at room temperature.
2.6. In vitro degradation
The hydrolytic and reductive degradation of polymer films and scaffolds were measured in PBS and GSH/PBS solutions, respectively. For hydrolysis, the weighed sample (W0) was immersed in 10 mL PBS at 37 °C. For reductive degradation, the weighed sample (W0) was placed in 10 mL GSH/PBS solution (10 mM) at 37°C. For controlled degradation, the samples (W0) were first immersed in PBS at 37°C for 2 wk, and then immersed into 10 mM GSH/PBS solution at 37°C for another 2 wk. At a predetermined time point, the sample was rinsed 3 times with deionized water, dried in a freeze-dryer for 3 d, and then weighed (W1). The mass remaining was calculated as W1/W0 × 100%. Three samples were used for each polymer at each time point. The IVs were measured for the remained polymer films after degradation in GSH solution for 7 d and 14 d using a viscometer as described above. The mechanical properties of the films after GSH treatment (n=3) were measured as described above. The scaffold morphologies after degradation were observed under an SEM.
2.7. Cell toxicity of degradation products
The toxicity of the polymers’ degradation products was measured as described earlier.26 The PU-SS film (50 mg) was immersed in 10 mL 1 M NaOH solution at 37°C for 1 wk to achieve complete degradation. The degradation solution was neutralized using 10 M HCl solution to pH = 7.4 and then sterilized by 0.22 μm membrane filter prior to cell culture study. Mouse 3T3 fibroblast (ATCC, Manassas, VA) were seeded on 96-well cell culture plates with a density of 2×103 cells per well in culture medium of Dulbecco’s modified eagle’s medium (DMEM), which was supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin (Sigma). After incubation for 1 d, the neutralized degradation solution (final concentration of 0.1 mg/mL) or DMEM medium (control) was added to each well. The cellular metabolic activity (n = 4) was measured using a mitochondrial activity assay (MTT, Sigma) at days 1, 3, and 5.27 For the 5-day cell toxicity group, a media change was carried out at day 3 with media containing the same type of degradation products (0.1 mg/mL). In brief, 20 μL of MTT solution (5 mg/mL in PBS solution) was added into each well. After 4h incubation, the medium was removed completely, and then 200μL DMSO was added to each well to dissolve formed formazan crystals. The metabolic index was recorded as absorbance intensity at 490 nm on a microplate reader. A live and dead staining (Live: SYTO 10 green fluorescent nucleic acid stain; Dead: ethidium homodimer-1 nucleic acid stain, Life Technologies, Inc.) kit was used to observe cellular morphology.
2.8. In vitro cellular growth on the films
The polymer disks (6 mm diameter) were punched from the films using standard biopsy punches (6 mm, Miltex), sterilized in 70 % ethanol for 30 min, and then rinsed 3 times using PBS prior to the study. The sterilized disks were immersed in the cell medium overnight and then placed into the wells of 96-well cell culture plate. The mouse 3T3 fibroblasts were seeded on the sample surface with a density of 2×103 per well in 96 well plates. The cell medium (DMEM supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin) were exchanged every 3 d. The MTT assay (n = 5) was used to evaluate the cellular viability at days 1, 3, and 5 as described above. Each day, MTT results were verified using a live/dead kit to visualize the fibroblast cells on the films, and the images were taken on a fluorescence microscope. Tissue culture polystyrene (TCPS) was a control.
2.9. In vitro cell growth on the fibrous scaffolds
The 6 mm diameter scaffold disks were placed in 96-well plates, and the 3T3 fibroblasts (2×103 cells per well) were seeded on the scaffold surface. The cell medium (DMEM supplemented with 10% fetal bovine serum, 10% fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin) were replenished every 3 d. The MTT assay was used to evaluate the cell growth on the scaffold at days 1, 3, and 5 (n = 5 for each group). The morphology of 3T3 fibroblasts cultured on the scaffolds was observed using SEM. TCPS was used as a control.
2.10. Mouse subcutaneous implantation
The animal use protocols were reviewed and approved by the Institutional Animal Care and Use Committee of the University of Texas at Arlington. Female Balb/C mice (20–25 grams, from Taconic Farms, Germantown, NY) were used in this investigation. Selected scaffolds (6 mm diameter disks with 300 μm thickness) were implanted in the dorsal subcutaneous region of the mice. After being implanted for 1 to 2 months, the implants and surrounding tissues were isolated for histological analyses. All tissue samples were frozen as sections into an 8-μm thick segment using a Leica Cryostat (CM1850, Leica Microsystem, Wetzlar, Germany) and then stained with hematoxylin-eosin (H&E). Based on the H&E staining, ImageJ was utilized to analyze the thickness of implants at different time points. The material in vivo degradation rate was then calculated by dividing the thickness of 2 month implants with the thickness of 1 month implant to reflect the percentages of thickness reduction per month.
2.11. Statistical analysis
All results are shown by mean ± standard deviation. All data (different time points and groups) except for degradation were analyzed by one-way ANOVA, followed by a post hoc Tukey-Kramer test. Repeated measures ANOVA was used for polymer and scaffold degradation using Statistics Analysis System (SAS). Significant difference was considered at p<0.05.
3. Results
3.1. Polymer characterization
The chemical structure of PU-SS polymers was verified by FTIR spectra (Figure 2). The symmetric and asymmetric stretching vibrations of -CH2- were shown at 2940 cm−1 and 2860 cm−1, respectively. The peak at 1720 cm−1 corresponded to C=O stretching. The peak at 1240-1260 cm−1 belongs to the C-O-C stretching. The existence of urethane groups was verified by peaks at 3320 cm−1 (N-H stretching) and 1540 cm−1 (N-H bending). The S-S bonds were observed at 1000 cm−1 (C-S bending) and 540 cm−1 (S-S dihedral bending). The intensities of the two peaks increased with increased HDS addition.
Figure 2.
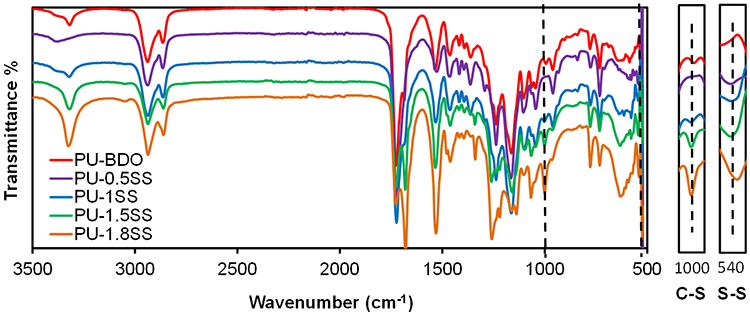
FT-IR spectra of PU-SS polymers.
All polymers had low glass transition temperatures (Tgs) below −55°C, which belonged to the soft segment of PCL in the polyurethane (Table 1 and Supporting Information Figure S1). The Tg increased by reducing the soft segment PCL amount. Single melting temperature was seen for PU-BDO, PU-0.5SS, and PU-1SS, while double melting temperatures (Tms) were observed for PU-1.5SS and PU-1.8SS. The low Tms resulted from the semicrystalline PCL soft segment (from 36 to 16°C).28 The high Tms may be attributed to the HDS hard segment (PU-1.5SS: 110°C, and PU-1.8SS: 126°C). Water absorption of PU-SS films enhanced with increasing HDS content (Table 1). PU-1.8SS has the highest water absorption (76±10%).
Table 1.
Polymer film characterization*
| Samples | Tg (°C) |
Tm1 (°C) |
Tm2 (°C) |
Peak stress (MPa) |
Breaking strain (%) |
Initial modulus (MPa) |
Inherent viscosity (dL/g) |
Water absorption (%) |
Instant recovery (%) |
|---|---|---|---|---|---|---|---|---|---|
| PU-BDO | −58 | 26 | - | 12.3±0.8 | 729±126a | 15±2a | 1.72 | 43±5a | 99±1 |
| PU-0.5SS | −60 | 36 | - | 12.2±1.4 | 1021±274a | 4±1b | 2.43 | 32±16a | 100±1 |
| PU-1SS | −59 | 35 | - | 11.9±0.5 | 1098±231a | 8±1c | 2.32 | 40±7a | 99±1 |
| PU-1.5SS | −59 | 16 | 110 | 12.0±0.6 | 821±116a | 26±4b | 1.72 | 67 ±4b | 99±1 |
| PU-1.8SS | −57 | - | 126 | 11.5±1.4 | 337±45b | 44±3e | 1.63 | 76±10b | 98±1 |
represent significantly different groups for each characteristic.
Mechanical properties for polymer films are summarized in Table 1, and typical stress-strain curves of PU-SS polymers are shown in Supporting Information Figure S2. The tensile strengths and the breaking strains of the polymers ranged from 11.5±1.4 to 12.3±0.8 MPa and from 337 ±45 to 1098 ± 231%, respectively. The tensile strengths and breaking strains of all polymers had insignificant difference. The initial moduli of PU-SS increased with increasing HDS contents (p<0.05). The PU-1.8SS had the highest initial modulus (44±3 MPa), while the PU-0.5SS had the lowest initial modulus (4±1 MPa). The initial modulus of PU-BDO was 15±2 MPa. The instant recoveries for all polymers had no significant difference (p>0.05).
For the resilience of the PU-SS films, the cyclic stretching was performed at a maximum strain of 30% or 300% (Figure 3). All PU-SS polymers and PU-BDO had large hysteresis loops in the first cycle that was not observed in the following nine cycles. With a maximum strain of 30%, all samples showed small irreversible deformations at ~5% (Figure 3A-3D), except for the PU-1.8SS (~18%) (Figure 3E). With a maximum strain of 300%, the irreversible deformations became appreciable for polymer samples (~200%) (Figure 3F-3I). The cyclic stretching test was not done on the PU-1.8SS film due to the fact that its breaking strain (337±45%) is close to 300%.
Figure 3.

Cyclic stretch of PU-SS films at 30% and 300% deformation.
3.2. In vitro degradation of polymer films
The hydrolytic and reductive degradations of the PU-SS films were measured in PBS and GSH solution, respectively (Figure 4A). The degradation rate increased with increasing HDS content in the polymer. For hydrolysis at day 28, the mass remaining of the polymers ranged from 92.8±0.5% to 97.6±0.3%. The degradation rates of PU-1.5SS and PU-1.8SS were significantly higher than those of PU-BDO, PU-0.5SS, and PU-1SS. For reductive degradation, PU-BDO had the lowest degradation rate (97.2±0.9% mass remaining at day 28), while PU-1.8SS had the highest degradation rate (55.3±5.5% mass remaining at day 28) (p<0.05). The degradation rates of PU-BDO in PBS and GSH solution had no significant difference (p>0.05). The degradation rates of PU containing disulfide bonds in GSH solution were statistically higher than those in PBS (p>0.05).
Figure 4.
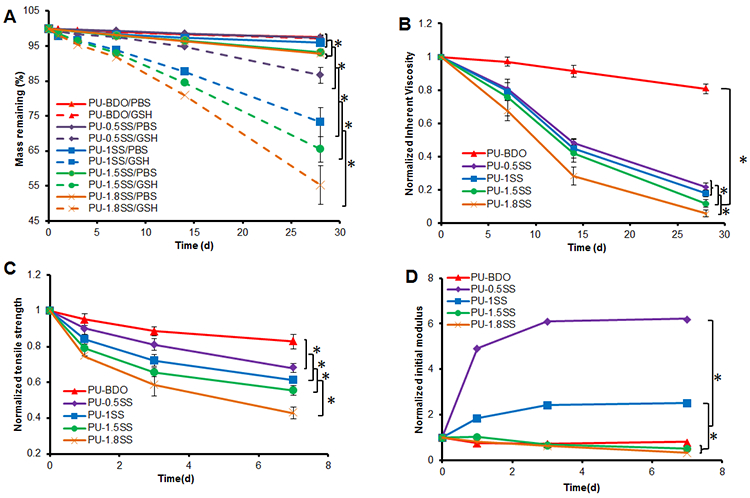
Polymer film degradation. (A) Mass remaining of PU-SS films in PBS and 10 mM GSH at 37 °C. (B) Inherent viscosity, (C) Tensile strength and (D) initial modulus changes of polymer films with degradation in GSH. *represented significant different groups (p<0.05).
The IV changes of the polymer films degraded in GSH solution for 28 days are shown in Figure 4B. The IVs decreased with the incubation time for all polymers. The PU-BDO had the lowest IV reduction (19.2±2.9% at day 28), and the PU-1.8SS had the highest IV reduction (94.4±2.1% at day 28) (p<0.05). PU-1.5SS had a higher IV reduction than PU-1SS and PU-0.5SS (p<0.05).
The mechanical property changes of the PU-SS films degraded in GSH solution were determined at day 7 (Figure 4C-4D), because PU-1.8SS and PU-1.5SS films became brittle at days 14 and 28 in GSH solution and were hard to handle. The tensile strengths of polymer films decreased with increased incubation time. The PU-BDO film had the lowest decrease (17.1±4.1% at day 7), and the PU-1.8SS film had the highest decrease (57.1±3.2% at day 7).
3.3. Cytotoxicity of PU-SS degradation products
The mouse 3T3 fibroblasts were utilized to evaluate the cell toxicity of the degradation products of the polyurethanes (Figure 5A). The cell viabilities in cell culture mediums containing degradation products showed no statistical difference from that of the medium controls (p>0.05). The cell numbers increased from day 1 to day 3 for cell culture medium and culture mediums containing degradation products (p<0.05). In live/dead stained images (Supporting Information Figure S3A), the increase of live cell (green) numbers from day 1 to day 3 was observed without any dead cells (red) as shown in the live/dead cell stained images (Supporting information Figure S3B). These results support that the presence of degradation products have little or no influence on cell growth.
Figure 5.
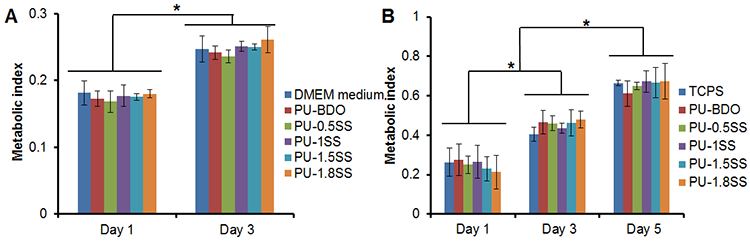
Cytotoxicity of polymer degradation products and cell growth on polymer films. (A) Metabolic index of 3T3 fibroblasts cultured with medium mixed with PU-SS degradation products at 0.1 mg/mL. DMEM culture medium was a control. (B) Metabolic index of 3T3 fibroblast seeded on PU-SS films (TCPS as a control) at days 1, 3 and 5. *represented significant different groups (p<0.05).
3.4. In vitro cellular growth on the films
The 3T3 fibroblasts were seeded on the PU-SS films to assess their in vitro cell compatibility (Figure 5B). The numbers of adherent cells on the polymer films and TCPS increased from day 1 to day 5 (p<0.05). However, there was no significant difference between the polymer films and the control TCPS during 5 days in culture. As anticipated, live/dead stained images show that there are no dead cells found on both PU-1SS films and TCPS (Supporting Information Figure S3B). In addition, with increasing incubation time, adherent cells spread to form cell sheets on PU-1SS forms as well as on TCPS.
3.5. Scaffold characterization
The SEM images of electrospun fibrous scaffolds showed continuous fibers without beads and drops (Figure 6). The fiber diameters of BDO, PU-0.5SS, PU-1SS, PU-1.5SS, and PU-1.8SS scaffolds were 1043 ± 190 nm, 1070 ± 182 nm, 503 ± 258 nm, 1156 ± 414 nm, and 456 ± 21 nm, respectively (Table 2). The peak stresses of the scaffolds ranged from 1.9 ± 0.3 MPa to 2.8±0.7 MPa without significant differences (p>0.05) (Table 2). Raising HDS contents increased the initial moduli of the scaffolds in the following order: PU-BDO ≈ PU-0.5SS ≈ PU-1SS<PU-1.5SS<PU-1.8SS. The strains of PU-1.5SS and PU-1.8SS scaffolds were lower than those of PU-BDO, PU-0.5SS, and PU-1SS scaffolds (p<0.05) (Table 2).
Figure 6.

Electrospun fibrous morphology of PU-SS scaffolds. (A) PU-BDO, (B) PU-0.5SS, (C) PU-1SS, (D) PU-1.5SS, and (E) PU-1.8SS.
Table 2.
Scaffold characterization*
| Samples | Fiber diameter (nm) |
Peak stress (MPa) |
Initial modulus (MPa) |
Breaking strain (%) |
|---|---|---|---|---|
| PU-BDO | 1043±190a | 2.6±0.6 | 1.5±0.3a | 411±50a |
| PU-0.5SS | 1070±182a | 2.6±0.8 | 1.5±0.4a | 531±168a |
| PU-1SS | 503±258b | 2.8±0.7 | 2.0±0.2a | 552±191a |
| PU-1.5SS | 1156±414a | 2.0±0.2 | 3.4±0.5b | 282±35b |
| PU-1.8SS | 456±214b | 1.9±0.3 | 7.8±1.1c | 255±61b |
represent significantly different groups for each characteristic.
The cyclic stretches of scaffolds, at a maximum strain of 30%, are shown in Figure 7. Most of the PU-SS scaffolds showed a larger hysteresis loop in the first cycle than in the additional 9 cycles. Most of the samples showed a very small irreversible deformation (~5%) (Figure 7A-7C), while the irreversible deformation at ~10% exists for PU-1.5SS and PU-1.8SS (Figure 7D-7E).
Figure 7.

Cyclic stretch of PU-SS fibrous scaffolds at 30% deformation.
3.6. In vitro degradation of the scaffolds
The hydrolytic and reductive degradations of the PU-SS scaffolds were measured in PBS and GSH solution, respectively (Figure 8A). The degradation rate increased with increasing the amount of HDS in polyurethanes. For hydrolytic degradation, the mass remaining of the polymers ranged from 92.7±1.1 to 97.2±0.4% within 2 wk. The degradation rates of PU-BDO and PU-0.5SS were significantly lower than those of PU-1SS, PU-1.5SS, and PU-1.8SS. In GSH solution, all polymers containing HDS showed markedly faster degradation than in PBS solution. There was no significant difference between PU-BDO degradation rates in PBS and GSH solutions (p>0.05). At day 14, PU-BDO showed the lowest degradation rate (95.9±0.2% mass remaining), while PU-1.8SS showed the highest degradation rate (42.0±8.3% mass remaining) (p<0.05). Additionally, the degradation rates of PU-SS scaffolds were greater than those of corresponded PU-SS films within 14 d in both PBS and GSH solution.
Figure 8.

Scaffold degradation. (A) Mass remaining of fibrous scaffolds in GSH and PBS at 37°C. (B) Scaffold controllable degradation. Scaffolds were immersed in PBS for 14 d and then in 10 mM GSH for another 14 d. (C) PU-BDO, (D) PU-0.5SS, (E) PU-1SS, (F) PU-1.5SS and (G) PU-1.8SS scaffold morphology after 14 d immersion in 10 mM GSH solution. *represented significant different groups (p<0.05).
The controlled degradation of scaffolds was conducted first in PBS solution for 2 wk and then in GSH solution for another 2 wk (Figure 8B). Within the first 2 wk, scaffold mass remaining in PBS ranged from 92.6±0.6% to 98.0±0.3%. However, after the scaffolds were transferred to the 10mM GSH solution, the scaffold degradation rates increased markedly when compared to their degradation rates in PBS. The degradation rate increased with increasing the amount of HDS in polymers, which is consistent with the mass remaining results of scaffolds. At day 28, the PU-BDO had the lowest degradation rate (95.5±0.2% mass remaining), while the PU-1.8SS had the highest degradation rate (43.6±9.0% mass remaining) (p<0.05).
SEM images were used to further study the degradation behavior of scaffolds in GSH at week 2 (Figure 8C-8G). The morphology of PU-BDO electrospun fibers barely changed (Figure 8C). The fibers of PU-0.5SS and PU-1SS scaffolds exhibited swollen and some fibers broke, but they still maintained fibrous morphology (Figure 8D and 8E). The PU-1.5SS and PU-1.8SS scaffolds completely lost the fibrous morphology (Figure 8F and 8G)
3.7. In vitro cellular growth on the scaffolds
The mouse 3T3 fibroblasts proliferated on all scaffolds from 1d to 5d (Figure 9A). There were no significant differences in cell viability between all of the scaffolds and the TCPS at each time point. Typical cell morphologies on the scaffold (PU-1SS) were visualized (Figure 9B-9D). At day 1, few of cells attached and spread on the fibrous scaffold (Figure 9B). At day 3, cell number obviously increased and partial cell confluence was observed (Figure 9C). At day 5, the complete cell confluence was observed on the scaffold surface (Figure 9D).
Figure 9.

Cell growth on scaffolds. (A) Metabolic index to show the 3T3 fibroblast viability on the scaffold (TCPS as a control). SEM micrographs of 3T3 fibroblasts on the surface of PU-1SS scaffold at (B) 1 d, (C) 3 d, and (D) 5 d. *represented significant different groups (p<0.05).
3.8. In vivo mouse subcutaneous implantation
To determine their in vivo degradation rates and tissue compatibility, PU-BDO, PU-1SS, and PU-1.8SS scaffolds were implanted subcutaneously on the backs of mice for a period of 1 and 2 months. We found very little inflammatory cell accumulation surrounding all implants at both time points (Figure 10A). Intensive cell infiltration was not found in all test materials. The scaffolds exhibited an increased trend of degradation rate with disulfide bond amount (Figure 10B). PU-1.8SS scaffolds showed significantly lower explant thickness than for PU-BDO at 2 month (p<0.05). The degradation rates of PU-1.8SS (34%) and PU-1SS (30%) were higher than that of PU-BDO (13%).
Figure 10.

Histological evaluation of explanted scaffolds in a mouse subcutaneous model. (A) H&E staining were carried out on the scaffolds implanted in mice for 1 and 2 months. Red arrows indicate location of inflammatory cells at the tissue:implant interfaces. (B) Explanted scaffold thicknesses were measured after 1 and 2 month implantation. *: p<0.05, PU-1.8SS compared with other groups at 1 month and 2 month.
4. Discussion
The bioreducible polyurethanes have been fabricated for gene delivery and drug control release.29-32 For example, a targeting-clickable and tumor-cleavable polyurethane micelle has also been reported for multifunctional delivery of antitumor drugs. Those micelles contained redox-responsive disulfide bonds, which triggered the intracellular drug release under the reductive environment of tumor tissue.33 Furthermore, degradable cationic polyurethane micelles bearing redox-responsive disulfide linkages throughout the backbone were developed for magnetic resonance imaging (MRI) and drug delivery.34 Finally, disulfide cross-linked micelles were fabricated by cross-linking of poly(ethylene glycol)/polyurethane block copolymers containing cyclic disulfide moieties via a thiol-disulfide exchange reaction to load anticancer drug for cancer therapy.35 However, it is rarely reported that the polyurethanes containing disulfide bonds serve as scaffolds to provide temporary mechanical support for personalized tissue engineering. Recently, a reduction-sensitive polyurethane was synthesized from poly(ethylene glycol) (PEG) or PCL and HDI with cystine chain extender,36-37 but no further characterization was reported for tissue engineering scaffold use. In this project, elastomeric biodegradable polyurethanes containing disulfide bonds were synthesized by introducing HDS as a chain extender, and were further processed into fibrous scaffolds for in vitro and in vivo assessment.
The PCL, used as a soft segment for all synthesized polyurethanes, is a semicrystalline polymer with a Tg of −60° C and Tm of 59-64°C.38 The low Tgs (<−50°C) of the polyurethanes were attributed to PCL soft segment, while the Tms were attributed to PCL crystalline and the hard segment. The interaction between the PCL and hard segment reduces the PCL crystalline degree and hard segment phase, and then leads to the decrease of Tms.24 Thus, the polyurethanes (PU-BDO, PU-0.5SS and PU-1SS) with high PCL contents only present a single Tm at 30-40°C. With PCL content decrease and HDS content increase, two Tms (PU-1.5SS) were observed and then a single high Tm (PU-1.8SS) was seen.
The initial modulus of the PU-SS films obviously increased with the increase of HDS content. The stiff hard segment increase is majorly attributed to HDS content increase, which leads to initial modulus increase. The tensile strengths of all polymers have no significant difference because the mechanical strength is dependent on molecular weight and phase separation as well as the hard/soft segment ratio. The elasticity of polyurethane is a result of the recoiling of the soft segment, while the permanent deformation is attributed to the plastic deformation of the hard segment phase, according to the strain-softening theory.39 Since PU-1.8SS has the highest hard segment content, it exhibited higher irreversible deformation for 30% cyclic stretching than other polyurethanes with lower disulfide contents. Once the strain reached 300%, all hard segment phases might be deformed to produce the permanent deformation, which results in a large deformation of polyurethanes (~200%) without significant difference for all polyurethanes. The electrospun fibrous scaffolds showed consistent results with the films, but the mechanical strengths decreased due to the porous structure.40
The degradation of PU-SS polymers and scaffolds can be induced in a reductive environment. Their degradation rate can also be adjusted by altering the amount of the disulfide bond, which would compromise the mechanical properties of PU-SS polymers. The PCL is slowly degradable polyester,41 and it would provide a relatively slow degradation before triggering fast degradation. Thus, in PBS, all materials showed slow degradation rates. The PUs with higher disulfide bond amounts exhibited faster degradation. It may be resulted from an increase in polymer hydrophilicity with an increase of disulfide bond amounts. When placed in GSH solution, all polymers and scaffolds with disulfide bonds distinctly increased degradation rates, which verified the reductively triggered degradation of the PU-SS material. It is obvious that higher SS bond amounts in polymer result in higher sensitivity of polymer degradation to the reductive agent. Other reductive agents, such as dithiothreitol (DTT) and cysteine, are also workable to trigger the polymer degradation.42-44 DTT has stronger reductive activity than GSH, and it induced much faster degradation of PU-SS material (Data not shown). However, DTT is toxic to the cells and tissues,44 thus it is not suitable for our future biomedical use. GSH and cysteine are natural products and are compatible with cells and tissues, and have been administered into human via oral intake, inhalation, intravenous and intramuscular injection with no known toxicity and side-effect.16, 45 Thus, high dosage of GSH could be used as a safe on-demand trigger via intramuscular injection to induce the fast degradation of the implant in vivo.
The synthesized PU-SS materials exhibited good cellular compatibility and tissue compatibility from the in vitro and in vivo assessments. The selected chemical components for PU-SS synthesis, including PCL, HDI, and BDO, have been applied for the materials of FDA-approved devices. The final degradation products also lacked cellular toxicity. The degradation products containing the thiol group (-SH) are non-toxic and have no effect on the pH value of the surrounding tissue.46 The PU-SS films and scaffolds supported 3T3 fibroblast cells proliferation. In vivo subcutaneous implantation showed all implants had good biocompatibility with minor inflammation and without severe immune response, which further confirmed their safety for future use. The conventional electrospun scaffold is very dense, which cannot allow a good cellular infiltration in vitro and in vivo.47 The fast degradation of the electrospun scaffolds can accelerate the cell infiltration.48 The PU-1.8SS scaffold had the fastest degradation, which may contribute to the enhanced cell infiltration in vivo as compared to PU-BDO and PU-1SS scaffolds. On the other hand, the reduction-induced degradation of the PU-SS polymer is not sensitive to the low GSH level in the tissue. The intracellular concentrations of GSH range from 0.5-10 mM,14 while the GSH concentrations in body fluids and extracellular matrices are relatively low (e.g., 2-20 μM in plasma).49-50 It was reported that the GSH concentration in rat subcutaneous tissue is ~4 μM, which is much lower than the concentration (10 mM) used for in vitro degradation tests.51 Thus, SS amount in PU-SS scaffolds may not be high enough to respond to this low GSH concentration in subcutaneous tissue, which led to insignificantly different degradation rates between PU-BDO and PU-SS scaffolds in 1 month. However, the SS sensitivity of the PU-SS may be enhanced with time in vivo. In the 2nd month, the PU-1.8SS scaffolds with high amounts of SS obviously exhibited fast degradation in vivo compared with PU-BDO scaffolds.
The developed polyurethane family in this study showed good elasticity, robust mechanical properties, good biocompatibility, and tunable degradation behavior with sensitivity to GSH. It can be processed into nanofibrous scaffolds. As it is a linear thermoplastic polymer, the PU-SS would also be processed into porous scaffold using phase separation, salt leaching, and other technologies.28, 47, 52-54 PU-based scaffolds have been widely used for soft tissue engineering, such as myocardium,55-56 blood vessels,57-58 tender,59 skin,60-61 corneal,62 abdominal wall,47 cartilage,63 and hard tissue engineering, such as bone.64 Scaffolds incorporating disulfide linkages are of particular interest when compared with the hydrolysis process, as the reductive degradation process can be performed relatively faster under physiological conditions by nontoxic reducing agents such as glutathione or cysteine.65 Generally, in vivo degradation of scaffolds tends to weaken the tissue/scaffold system during tissue growth. However, it is necessary to provide sufficient mechanical support at least at the early stage of tissue growth. Thus, it is desirable to develop a slow degradation at an early stage and then an on-demand fast degradation process upon the end of tissue growth, which can be performed by disulfide-bond cleavage.66 Thus, the PU-SS would be promising to find multiple opportunities for tissue repair and regeneration applications.
There are some limitations that should be mentioned. First, we only used 3T3 fibroblast cells for biological evaluation, and we had no specific application focus. In the future, for specific biomedical application, we will use the corresponding cell type to further evaluate the polymer. For example, for myocardium regeneration, cardiomyocytes or stem cells will be used. For blood vessel use, endothelial cells and vascular smooth muscle cells will be used, along with blood compatibility evaluation using human whole blood. Second, to accelerate scaffold degradation, GSH may be administered via direct injection to the implantation sites and/or surrounding tissue/muscles in vivo. However, the potential influence of GSH injection on wound healing process has yet to be determined. Third, there are many disulfide bonds-containing proteins in the blood and tissue. The influence of these disulfide bonds-containing proteins on polymer degradation remains to be evaluated. Finally, it is possible that different degradation rates are needed for different phases of tissue regeneration. Further studies are needed to determine the optimal degradation scheme to achieve the best tissue regeneration outcomes.
5. Conclusion
A family of elastic polyurethanes with active inducible biodegradation property was synthesized by introducing disulfide bonds into the polymer backbone. Both PU-SS films and scaffolds had good elasticity and attractive mechanical properties. Their in vitro fast degradation can be actively trigged by GSH. The polymer degradation rate can be tuned by altering disulfide bond amounts in PU-SS backbone. The polymer films and scaffolds had good cellular compatibility to support cell growth without apparent cytotoxicity. The polymer scaffolds showed good tissue compatibility with minimal immune response in mouse subcutaneous models. These polyurethane elastomers possessed attractive mechanical properties, controllable degradation, and good biocompatibility, which would find opportunities to be applied as 3D scaffolds for tissue repair and regeneration.
Supplementary Material
ACKNOWLEDGMENT
We greatly appreciate the supports from Research Enhanced Program (Y.H.) of the University of Texas at Arlington, Beginning Grant-in-Aid #14BGIA20510066 (Y.H.) from the American Heart Association, and Congressionally Directed Medical Research Program W81XWH-14-0459 (L.T.) from the Department of Defense, the United States of America.
Footnotes
Supporting Information
DSC curves and stress-strain curves of polymers, and live/dead stained cell images.
References
- (1).Hamburg MA; Collins FS The Path to Personalized Medicine. N. Engl. J. Med 2010, 363, 301–304. [DOI] [PubMed] [Google Scholar]
- (2).Wu G; Mikhailovsky A; Khant HA; Fu C; Chiu W; Zasadzinski JA Remotely Triggered Liposome Release by Near-Infrared Light Absorption via Hollow Gold Nanoshells. J. Am. Chem. Soc 2008, 130, 8175–8177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Nazemi A; Schon TB; Gillies ER Synthesis and Degradation of Backbone Photodegradable Polyester Dendrimers. Org. Lett 2013, 15, 1830–1833. [DOI] [PubMed] [Google Scholar]
- (4).Epstein-Barash H; Orbey G; Polat BE; Ewoldt RH; Feshitan J; Langer R; Borden MA; Kohane DS A Microcomposite Hydrogel for Repeated On-Demand Ultrasound-Triggered Drug Delivery. Biomaterials 2010, 31, 5208–5217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Kost J; Leong K; Langer R Ultrasound-Enhanced Polymer Degradation and Release of Incorporated Substances. Proc. Natl. Acad. Sci. U.S.A 1989, 86, 7663–7666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).El-Sayed ME; Hoffman AS; Stayton PS Rational Design of Composition and Activity Correlations for pH-Responsive and Glutathione-Reactive Polymer Therapeutics. J. Controlled. Release 2005, 104, 417–427. [DOI] [PubMed] [Google Scholar]
- (7).Li C; Madsen J; Armes SP; Lewis AL A New Class of Biochemically Degradable, Stimulus-Responsive Triblock Copolymer Gelators. Angew. Chem. Int. Ed 2006, 45, 3510–3513. [DOI] [PubMed] [Google Scholar]
- (8).Gunawan ST; Kempe K; Such GK; Cui J; Liang K; Richardson JJ; Johnston AP; Caruso F Tuning Particle Biodegradation through Polymer–Peptide Blend Composition. Biomacromolecules 2014, 15, 4429–4438. [DOI] [PubMed] [Google Scholar]
- (9).de la Rica R; Aili D; Stevens MM Enzyme-Responsive Nanoparticles for Drug Release and Diagnostics. Adv. Drug. Deliv. Rev 2012, 64, 967–978. [DOI] [PubMed] [Google Scholar]
- (10).Kratz K; Habermann R; Becker T; Richau K; Lendlein A Shape-Memory Properties and Degradation Behavior of Multifunctional Electro-Spun Scaffolds. Int. J. Artif. Organs 2011, 34, 225–230. [DOI] [PubMed] [Google Scholar]
- (11).Martin RA; Yue S; Hanna JV; Lee PD; Newport RJ; Smith ME; Jones JR Characterizing the Hierarchical Structures of Bioactive Sol-Gel Silicate Glass and Hybrid Scaffolds for Bone Regeneration. Philos. Trans. A Math. Phys. Eng. Sci 2012, 370, 1422–1443. [DOI] [PubMed] [Google Scholar]
- (12).Hakimi O; Mouthuy P; Carr A Synthetic and Degradable Patches: An Emerging Solution for Rotator Cuff Repair. Int. J. Exp. Pathol 2013, 94, 287–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Fu H; Hong Y; Little SR; Wagner WR Collagenase-Labile Polyurethane Urea Synthesis and Processing into Hollow Fiber Membranes. Biomacromolecules 2014, 15, 2924–2932. [DOI] [PubMed] [Google Scholar]
- (14).Wu G; Fang YZ; Yang S; Lupton JR; Turner ND Glutathione Metabolism and its Implications for Health. J. Nutr 2004, 134, 489–492. [DOI] [PubMed] [Google Scholar]
- (15).Cai J; Chen Y; Seth S; Furukawa S; Compans RW; Jones DP Inhibition of Influenza Infection by Glutathione. Free Radic. Biol. Med 2003, 34, 928–936. [DOI] [PubMed] [Google Scholar]
- (16).Lomaestro BM; Malone M Glutathione in Health and Disease: Pharmacotherapeutic issues. Ann. Pharmacother 1995, 29, 1263–1273. [DOI] [PubMed] [Google Scholar]
- (17).Arjinpathana N; Asawanonda P Glutathione as an Oral Whitening Agent: a Randomized, Double-Blind, Placebo-Controlled Study. J. Dermatol. Treat 2012, 23, 97–102. [DOI] [PubMed] [Google Scholar]
- (18).Witschi A; Reddy S; Stofer B; Lauterburg BH The Systemic Availability of Oral Glutathione. Eur J Clin Pharmacol. 1992, 43, 667–669. [DOI] [PubMed] [Google Scholar]
- (19).Bardellini E; Bindi P; Borzone S; Caglieris S; Dagnino F; Testa R The Effect of High Doses of Reduced Glutathione on Hepatic Clearances and Fibrogenetic Activity in Patients with Chronic Alcoholic Liver Disease. Adv. Ther 1992, 9, 116–122. [Google Scholar]
- (20).Lenzi A; Culasso F; Gandini L; Lombardo F; Dondero F Placebo-Controlled, Double-Blind, Cross-Over Trial of Glutathione Therapy in Male Infertility. Hum. Reprod 1993, 8, 1657–1662. [DOI] [PubMed] [Google Scholar]
- (21).Borok Z, Buhl R, Grimes GJ, Bokser AD, Hubbard RC, Holroyd KJ, Roum JH, Czerski DB, Cantin AM, Crystal RG. Effect of Glutathione Aerosol on Oxidant-Antioxidant Imbalance in Idiopathic Pulmonary Fibrosis. Lancet, 1991, 338, 215–216. [DOI] [PubMed] [Google Scholar]
- (22).Guan J; Sacks MS; Beckman EJ; Wagner WR Synthesis, Characterization, and Cytocompatibility of Elastomeric, Biodegradable Poly(ester-urethane)ureas Based on Poly(caprolactone) and Putrescine. J. Biomed. Mater. Res 2002, 61, 493–503. [DOI] [PubMed] [Google Scholar]
- (23).Chuang T; Masters KS Regulation of Polyurethane Hemocompatibility and Endothelialization by Tethered Hyaluronic Acid Oligosaccharides. Biomaterials 2009, 30, 5341–5351. [DOI] [PubMed] [Google Scholar]
- (24).Ma Z; Hong Y; Nelson DM; Pichamuthu JE; Leeson CE; Wagner WR Biodegradable Polyurethane Ureas with Variable Polyester or Polycarbonate Soft Segments: Effects of Crystallinity, Molecular Weight, and Composition on Mechanical Properties. Biomacromolecules 2011, 12, 3265–3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Asplund JB; Bowden T; Mathisen T; Hilborn J Synthesis of Highly Elastic Biodegradable Poly(urethane urea). Biomacromolecules 2007, 8, 905–911. [DOI] [PubMed] [Google Scholar]
- (26).Guan J; Sacks MS; Beckman EJ; Wagner WR Biodegradable Poly(ether ester urethane)urea Elastomers Based on Poly(ether ester) Triblock Copolymers and Putrescine: Synthesis, Characterization and Cytocompatibility. Biomaterials 2004, 25, 85–96. [DOI] [PubMed] [Google Scholar]
- (27).Twentyman PR; Luscombe M A Study of Some Variables in a Tetrazolium Dye (MTT) Based Assay for Cell Growth and Chemosensitivity. Br. J. Cancer 1987, 56, 279–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Hong Y; Guan J; Fujimoto KL; Hashizume R; Pelinescu AL, Wagner WR Tailoring the Degradation Kinetics of Poly(ester carbonate urethane)urea Thermoplastic Elastomers for Tissue Engineering Scaffolds. Biomaterials 2010, 31, 4249–4258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Saito G; Swanson JA; Lee K Drug Delivery Strategy Utilizing Conjugation via Reversible Disulfide Linkages: Role and Site of Cellular Reducing Activities. Adv. Drug Deliv. Rev 2003, 55, 199–215. [DOI] [PubMed] [Google Scholar]
- (30).Choh S; Cross D; Wang C Facile Synthesis and Characterization of Disulfide-Cross-Linked Hyaluronic Acid Hydrogels for Protein Delivery and Cell Encapsulation. Biomacromolecules 2011, 12, 1126–1136. [DOI] [PubMed] [Google Scholar]
- (31).Blacklock J; Handa H; Manickam DS; Mao G; Mukhopadhyay A; Oupický D Disassembly of Layer-by-Layer Films of Plasmid DNA and Reducible TAT Polypeptide. Biomaterials 2007, 28, 117–124. [DOI] [PubMed] [Google Scholar]
- (32).Blacklock J; You Y; Zhou Q; Mao G; Oupický D Gene Delivery in Vitro and in Vivo from Bioreducible Multilayered Polyelectrolyte Films of Plasmid DNA. Biomaterials 2009, 30, 939–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Song N; Ding M; Pan Z; Li J; Zhou L; Tan H; Fu Q Construction of Targeting-clickable and Tumor-Cleavable Polyurethane Nanomicelles for Multifunctional Intracellular Drug Delivery. Biomacromolecules 2013, 14, 4407–4419. [DOI] [PubMed] [Google Scholar]
- (34).Ding M; Zeng X; He X; Li J; Tan H; Fu Q Cell Internalizable and Intracellularly Degradable Cationic Polyurethane Micelles as a Potential Platform for Efficient Imaging and Drug delivery. Biomacromolecules 2014, 15, 2896–2906. [DOI] [PubMed] [Google Scholar]
- (35).Yu S; Ding J; He C; Cao Y; Xu W; Chen X. Disulfide Cross-Linked Polyurethane Micelles as a Reduction-Triggered Drug Delivery System for Cancer Therapy. Adv. Healthc. Mater 2014, 3, 752–760. [DOI] [PubMed] [Google Scholar]
- (36).Lu H; Sun P; Zheng Z; Yao X; Wang X; Chang F Reduction-Sensitive Rapid Degradable Poly(urethane-urea)s Based on Cystine. Polym. Degrad. Stab 2012, 97, 661–669. [Google Scholar]
- (37).Wang J; Zheng Z; Chen L; Tu X; Wang X Glutathione-Responsive Biodegradable Poly(urea-urethane)s Containing L-Cystine-based Chain Extender. J. Biomater. Sci. Polym. Ed 2013, 24, 831–848. [DOI] [PubMed] [Google Scholar]
- (38).Woodruff MA; Hutmacher DW The Return of a Forgotten Polymer-Polycaprolactone in the 21st Century. Prog. Polym. Sci 2010, 35, 1217–1256. [Google Scholar]
- (39).Asplund JB; Bowden T; Mathisen T; Hilborn J Synthesis of Highly Elastic Biodegradable Poly(urethane urea). Biomacromolecules 2007, 8, 905–911. [DOI] [PubMed] [Google Scholar]
- (40).Charriere E; Lemaitre J; Zysset P Hydroxyapatite Cement Scaffolds with Controlled Macroporosity: Fabrication Protocol and Mechanical Properties. Biomaterials 2003, 24, 809–817. [DOI] [PubMed] [Google Scholar]
- (41).Vert M. Degradable and Bioresorbable Polymers in Surgery and in Pharmacology: Beliefs and Facts. J. Mater. Sci. Mater. Med 2009, 20, 437–446. [DOI] [PubMed] [Google Scholar]
- (42).Kim T; Lee M; Kim SW A Guanidinylated Bioreducible Polymer with High Nuclear Localization Ability for Gene Delivery Systems. Biomaterials 2010, 31, 1798–1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Li C; Wang H; Cao J; Zhang J; Yu X Bioreducible Cross-Linked Polymers Based on G1 Peptide Dendrimer as Potential Gene Delivery Vectors. Eur. J. Med. Chem 2014, 87, 413–420. [DOI] [PubMed] [Google Scholar]
- (44).Matsusaki M; Yoshida H; Akashi M The Construction of 3D-Engineered Tissues Composed of Cells and Extracellular Matrices by Hydrogel Template Approach. Biomaterials 2007, 28, 2729–2737. [DOI] [PubMed] [Google Scholar]
- (45).GO YM; Jones DP Cysteine/Cystine Redox Signaling in Cardiovascular Disease. Free Radic Biol Med. 2011, 50, 495–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Yoshida H; Klinkhammer K; Matsusaki M; Möller M; Klee D; Akashi M Disulfide-Crosslinked Electrospun Poly(γ-glutamic acid) Nonwovens as Reduction-Responsive Scaffolds. Macromol. Biosci 2009, 9, 568–574. [DOI] [PubMed] [Google Scholar]
- (47).Hong Y; Takanari K; Amoroso NJ; Hashizume R; Brennan-Pierce EP; Freund JM; Badylak SF; Wagner WR An Elastomeric Patch Electrospun from a Blended Solution of Dermal Extracellular Matrix and Biodegradable Polyurethane for Rat Abdominal wall Repair. Tissue Eng. Part C Methods 2011, 18, 122–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Stankus JJ; Freytes DO; Badylak SF,; Wagner WR Hybrid Nanofibrous Scaffolds from Electrospinning of a Synthetic Biodegradable Elastomer and Urinary Bladder Matrix. J. Biomater. Sci. Polym. Ed 2008, 19, 635–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Jones DP Redox Potential of GSH/GSSG Couple: Assay and Biological Significance. Methods Enzymol. 2002, 348, 93–112. [DOI] [PubMed] [Google Scholar]
- (50).Griffith OW Biologic and Pharmacologic Regulation of Mammalian glutathione Synthesis. Free Radic. Biol. Med 1999, 27, 922–935. [DOI] [PubMed] [Google Scholar]
- (51).Wan HL; Tsai PJ; Chiang MJ; Wu JP; Liu L; Tsai WJ; Kuo JS; Yang CS Effect of Acute Skin Thermal Injury on Subcutaneous Glutathione, Ascorbic Acid and Hydroxyl Radical Concentrations in Anesthetized Rats. Redox Rep. 1996, 2, 267–272. [DOI] [PubMed] [Google Scholar]
- (52).Liu SQ; Kodama M Porous Polyurethane Vascular Prostheses with Variable Compliances. J. Biomed. Mater. Res 1992, 26, 1489–1502. [DOI] [PubMed] [Google Scholar]
- (53).Guan J; Fujimoto KL; Sacks MS; Wagner WR Preparation and Characterization of Highly Porous, Biodegradable Polyurethane Scaffolds for Soft Tissue Applications. Biomaterials 2005, 26, 3961–3971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Fromstein J; Woodhouse K Elastomeric Biodegradable Polyurethane Blends for Soft Tissue Applications. J. Biomater. Sci. Polym. Ed 2002, 13, 391–406. [DOI] [PubMed] [Google Scholar]
- (55).Siepe M; Giraud M; Liljensten E; Nydegger U; Menasche P; Carrel T; Tevaearai HT Construction of Skeletal Myoblast-Based Polyurethane Scaffolds for Myocardial Repair. Artif. Organs 2007, 31, 425–433. [DOI] [PubMed] [Google Scholar]
- (56).Fromstein JD; Zandstra PW; Alperin C; Rockwood D; Rabolt JF; Woodhouse KA Seeding Bioreactor-Produced Embryonic Stem Cell-Derived Cardiomyocytes on Different Porous, Degradable, Polyurethane Scaffolds Reveals the Effect of Scaffold Architecture on Cell Morphology. Tissue Eng. Part A 2008, 14, 369–378. [DOI] [PubMed] [Google Scholar]
- (57).Fujimoto K; Minato M; Miyamoto S; Kaneko T; Kikuchi H; Sakai K; Okada M; Ikada Y Porous Polyurethane Tubes as Vascular Graft. J. Appl. Biomater 1993, 4, 347–354. [DOI] [PubMed] [Google Scholar]
- (58).Kowligi RR; von Maltzahn WW; Eberhart RC Fabrication and Characterization of Small-Diameter Vascular Prostheses. J. Biomed. Mater. Res 1988, 22, 245–256. [DOI] [PubMed] [Google Scholar]
- (59).Ruzzini L; Longo UG; Campi S; Maffulli N; Muda AO; Denaro V Adhesion and Collagen Production of Human Tenocytes Seeded on Degradable Poly(urethane urea). Knee Surg. Sports Traumatol. Arthrosc 2013, 21, 1834–1840. [DOI] [PubMed] [Google Scholar]
- (60).Adolph EJ; Pollins AC; Cardwell NL; Davidson JM; Guelcher SA; Nanney LB Biodegradable Lysine-Derived Polyurethane Scaffolds Promote Healing in a Porcine Full-Thickness Excisional Wound Model. J. Biomater. Sci. Polym. Ed 2014, 25, 1973–1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Heo DN; Yang DH; Lee JB; Bae MS; Kim JH; Moon SH; Chun HJ; Kim CH; Lim HN; Kwon IK Burn-Wound Healing Effect of Gelatin/Polyurethane Nanofiber Scaffold Containing Silver-Sulfadiazine. J. Biomed. Nanotechnol 2013, 9, 511–515. [DOI] [PubMed] [Google Scholar]
- (62).Chirila TV First Development of a Polyurethane Keratoprosthesis and its Australian Connection: An Unbeknown Episode in the History of Artificial Cornea. Clin. Experiment Ophthalmol 2006, 34, 485–488. [DOI] [PubMed] [Google Scholar]
- (63).Reyes R; Delgado A; Solis R; Sanchez E; Hernandez A; Roman JS; Evora C Cartilage Repair by Local Delivery of Transforming Growth Factor-β1 or Bone Morphogenetic Protein-2 from a Novel, Segmented Polyurethane/Polylactic-co-Glycolic Bilayered Scaffold. J. Biomed. Mater. Res., Part A 2014, 102, 1110–1120. [DOI] [PubMed] [Google Scholar]
- (64).Mi H; Palumbo S; Jing X; Turng L; Li W; Peng X Thermoplastic Polyurethane/Hydroxyapatite electrospun Scaffolds for Bone Tissue Engineering: Effects of Polymer Properties and Particle Size. J. Biomed. Mater. Res., Part B 2014, 102, 1434–1444. [DOI] [PubMed] [Google Scholar]
- (65).Wang L; Li C; Ryan AJ; Armes SP Synthesis and Peptide-Induced Degradation of Biocompatible Fibers Based on Highly Branched Poly(2-hydroxyethyl methacrylate). Adv. Mater 2006, 18, 1566–1570. [Google Scholar]
- (66).Dispinar T; Van Camp W; De Cock LJ; De Geest BG; Du Prez FE Redox-Responsive Degradable PEG Cryogels as Potential Cell Scaffolds in Tissue Engineering. Macromol. Biosci 2012, 12, 383–394. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.