

Abstract
Objectives
Thymidine kinase 2 deficiency (TK2d) is a rare autosomal recessive disorder that stems from a perturbation of the mitochondrial DNA maintenance. Nucleoside treatment has recently shown promise as a disease-modifying therapy. TK2d was initially associated with rapidly progressive fatal myopathy in children featuring mitochondrial DNA depletion. Subsequently, less severe variants of the disease were described, with onset of symptoms during adolescence or adulthood and associated with the presence of multiple mtDNA deletions. These less severe phenotypes have been reported in only 15% of the approximately 120 patients described worldwide. However, some reports suggest that these juvenile and adult-onset presentations may be more common. The objective of this study was to describe the clinical phenotype in a sample of patients from Spain.
Methods
This study includes 53 patients harboring biallelic TK2 pathogenic variants, compiling data retrospectively from 7 Spanish centers. We analyzed allele frequency, investigated the most recent common ancestor of core haplotypes, and used the Runs of Homozygosity approach to investigate variant coalescence.
Results
Symptom onset distribution revealed that 32 patients (60%) experienced symptoms beyond 12 years of age. Approximately 30% of patients died of respiratory insufficiency, while 56% of surviving patients needed mechanical ventilation. Genetic analysis identified 16 distinct variants in TK2. Two variants, p.Lys202del and p.Thr108Met, exhibited significantly higher prevalence in the Spanish population than that reported in gnomAD database (86-fold and 13-fold, respectively). These variants are estimated to have originated approximately 16.8 generations ago for p.Thr108Met and 95.2 generations ago for p.Lys202del within the Spanish population, with the increase in frequency attributed to various forms of inbreeding. In late-onset cases, 46.9% carried the p.Lys202del variant.
Discussion
The higher frequency of TK2d in Spain can be partially attributed to the increased prevalence of 2 variants and consanguinity. Notably, in 60% of the cohort, the disease was late-onset, emphasizing the potential underdiagnosis of this subgroup of patients in other regions. Raising awareness of this potentially treatable disorder is of utmost importance because early interventions can significantly affect the quality of life and survival of affected individuals.
Introduction
Thymidine kinase 2 deficiency (TK2d) is a very rare autosomal recessive mitochondrial disorder that causes a myopathic form of mitochondrial DNA (mtDNA) depletion/multiple deletion syndrome (MIM # 609560).1 The TK2 enzyme plays a crucial role in the pyrimidine deoxynucleotide salvage pathway by phosphorylating deoxythymidine (dThd) and deoxycytidine (dCtd). When TK2 is deficient, the balance of mitochondrial deoxynucleoside triphosphates is disrupted, causing impairments in mtDNA replication.2,3 TK2d can manifest as either a mtDNA quantitative defect (mtDNA depletion),4,5 a qualitative defect characterized by errors during mtDNA replication that lead to multiple mtDNA deletions, or a combination of both.6 Disease severity is closely related to the number of mtDNA copies in muscle tissue, resulting in a continuous spectrum of clinical involvement.7,8 This spectrum spans from an infantile presentation with rapid progression and ultimately fatal outcomes within a few months to late-onset cases characterized by less severe phenotypes and variable rates of disease progression.7,8
Pathogenic variants in the TK2 gene were first identified in 20019 in 4 children who exhibited severe myopathy. The symptoms appeared 6–36 months after an unremarkable early development but rapidly progressed, leading to mechanical ventilation (MV) or death within the first 4 years of life. In these cases, the muscle's mtDNA/nuclear DNA ratio was markedly reduced to 16%–22% of the average value observed in controls. In subsequent years, numerous patients with a similar phenotype were reported, all characterized by mtDNA depletion.10-13 Since 2010, less severe phenotypes have been documented in patients experiencing symptom onset during adolescence or adulthood. Patients exhibiting this less severe phenotype are characterized by multiple mtDNA deletions in the muscle without a significant reduction in mtDNA copy number.10,14-18 A comprehensive literature review conducted in 2018, including 2 studies and a total of 107 individuals, revealed that only 17 of the reported patients experienced an onset of the symptoms after 12 years of age.7,8 However, recent cumulative reports of cases diagnosed in adulthood, mainly from Spain, suggest that the frequency of juvenile and adult presentations may have previously been underestimated.19-22 In this study, we analyze epidemiologic data and delve into the possible genetic factors that may contribute to the remarkably high number of patients diagnosed with this disorder in Spain, in contrast to its rarity in other countries sharing similar characteristics. In natural populations, rare autosomal recessive variants' prevalence deviates from Hardy-Weinberg equilibrium due to increased homozygosity from inbreeding. Consanguineous marriages, more common in lower-prevalence diseases, contribute to this phenomenon.23-25 Increased homozygosity arises from systematic (FIS) and panmictic (FST) inbreeding, each with unique evolutionary implications.26 FIS results from consanguineous practices, causing deviations from Hardy-Weinberg proportions. FST stems from endogamy and reduced effective population size, leading to irreversible genetic diversity loss without migration. Runs of homozygosity (ROH) analysis is crucial for understanding variant coalescence in a population. ROH represents continuous stretches of identical-by-descent homozygous alleles.27 This study uses ROH to explore the coalescent process of pathogenic variants in 53 patients with TK2d in Spain, comparing them with a Spanish control population. In addition, we also extensively analyzed the clinical data of our series to enhance our understanding of the clinical spectrum of TK2d.
Methods
Patients
This is a multicenter historical cohort study of 53 Spanish patients diagnosed harboring biallelic TK2 pathogenic variants between 2003 and 2022, conducted at “Hospital 12 de Octubre” in Madrid, “Hospital de la Santa Creu i Sant Pau,” “Hospital San Joan de Déu,” “Hospital de Bellvitge,” and “Hospital Vall d'Hebron” in Barcelona, “Hospital La Fe” in Valencia, and “Hospital Virgen del Rocío” in Seville, all the National Reference Centers for Rare Neuromuscular Diseases existing in Spain. Medical records were reviewed for demographic data, family history, genetic results, age at disease onset, clinical manifestations, and treatment. Partial clinical data from several patients were previously published elsewhere.16,19,21,28-31
Genetic Studies
The identification of variants in the TK2 gene was performed by direct Sanger sequencing, with a customized next-generation sequencing (NGS) panel associated with mtDNA maintenance defects or by whole-exome sequencing (WES). Variants were classified following the American College of Medical Genetics (ACMG) criteria.32
Mitochondrial DNA Studies
Skeletal muscle DNA was isolated with the DNA Mini Kit (Qiagen, Hilden, Germany). Multiple mtDNA deletions were analyzed by Southern blot using mtDNA probes marked with digoxigenin (Roche Diagnostics) and/or by long-range PCR (LR-PCR) of mtDNA using LA Taq DNA Polymerase (Takara Bio, Kusatsu, Japan). Mitochondrial DNA content (mtDNA/nuclear DNA ratio expressed as a percentage relative to an age-matched control) was assessed by quantitative PCR (qPCR).
Allele Frequency
We analyzed the allele frequency of the 2 most frequent variants identified in the cohort in a total of 21,846 clinical exomes studied in 5 hospital genetic diagnostic centers located in different regions of the country (2 in Madrid -Centre-, and one in Valencia -Eastern-, Catalonia -North-eastern-, and Galicia -North-western-, respectively). We compared it with the allelic frequency described for these 2 variants in the European population in the gnomAD database.
Age of the Most Recent Ancestor: Haplotype Analysis
A core haplotype was defined based on sharing among affected individuals. Haplotype dating was performed using a published method33 and R package. Through a gamma distribution, this method estimates the age of the most recent common ancestor (MRCA) by the maximum likelihood from whom the core haplotype was inherited. This method can also be used for individuals with shared “extended haplotypes,” who are likely to have a more recent MRCA than that for the whole group. The model can assume independent or correlated genealogies, i.e., a “tree-like genealogy where subsets of the sample have common ancestry earlier than the MRCA for the entire sample. This approach has been widely used in the bibliography.34-36
ROH Analysis
Additional explanation for ROH analysis is included in the eMethods.
Control Population
To contextualize patients in the Spanish population, 1,014 individuals from the BNADN data set (507 women and 507 men) were added. DNA samples were genotyped at CeGen-FPGMX, Santiago de Compostela, Spain, using the Axiom Spain Biobank Array with 757,836 variants for genomic, disease, and functional allele coverage, capturing Spain's regional diversity. Genomic DNA (200 ng) was amplified, fragmented, purified, and transferred to the GeneTitan Multichannel Instrument for automated processing. CEL files were processed using the Axiom GT1 algorithm and Axiom Analysis Suite v4.0.3.3. The population comprised individuals with all 4 grandparents born in Spain. PAC analysis demonstrated a lack of structure among patients and controls.
ROH Calling
ROH longer than 300 kb will be identified using PLINK v1.9 software37 with specific parameters: homozyg-snp 30 (minimum number of variants required), —homozyg-kb 300 (sliding window length in kb), —homozyg-density 30 (minimum density for considering an ROH), —homozyg-window-snp 30 (number of SNPs in the sliding window), —homozyg-gap 1000 (length between 2 SNPs in different segments), —homozyg-window-het 1 (allowed heterozygous SNPs in a window), —homozyg-window-missing 5 (allowed missing calls in a window), and —homozyg-window-threshold 0.05 (proportion of overlapping window for defining an SNP as “homozygous”). No linkage disequilibrium pruning will occur. SNPs with minor allele frequencies <0.01 and those deviating from H-W proportions (p < 0.001) will be filtered. These conditions, validated in published studies, identify ROH corresponding to autozygous segments where all SNPs are homozygous-by-descent, even those that do not present on the array.
Estimating Inbreeding and Its Origin
Inbreeding can stem from a departure from panmixia, either through systematic inbreeding (consanguinity or FIS) or genetic isolation and a small effective population size (genetic drift or FST), leading to panmictic inbreeding.26,38 The total inbreeding coefficient FIT is the probability of an individual receiving 2 alleles identical-by-descent: (1-FIT) = (1-FIS) (1-FST).39,40 Traditionally assessed through extensive genealogical records, we utilized the genomic inbreeding coefficient (FROH) as a proxy for FIT, while estimating FIS using single nucleotide variants data. FIS represents the average single nucleotide variants homozygosity within an individual compared to the expected homozygosity of alleles randomly selected from the population. PLINK uses the following expression:
Observed Hom represents the observed homozygous SNPs, Expected Hom is the expected homozygous SNPs considering Hardy-Weinberg proportions, and N is the total number of nonmissing genotyped SNPs. FIS gauges inbreeding in the current generation, with FIS = 0 indicating random mating, FIS >0 indicating consanguinity, and FIS <0 indicating inbreeding avoidance. FROH measures the proportion of the autosomal genome that is autozygous beyond a specified minimum length ROH threshold. Analyzing ROH>1.5 Mb, FROH strongly correlates (r = 0.86) with inbreeding coefficients obtained from six-generation pedigrees.
Genomic Distribution of ROH: ROH Islands (ROHi)
ROH distribution in a population is nonrandom, with certain genome regions exhibiting either a high prevalence or complete absence of ROH.27,41,42 These regions, enriched with protein-coding genes, suggest directional selection.43,44 In this study, we focused on chromosome 16, specifically the TK2 gene, examining shared IBD haplotypes. ROHi, regions with a higher-than-expected proportion of individuals with ROH, were identified using a 100-kb sliding window. A binomial test (p < 2 × 10−7) with Bonferroni correction for 2,500 windows determined significant ROH enrichment across the population in specific genomic windows.
Statistical Analysis
The Fisher exact test was used to assess the potential association between the common variants p.Lys202del and p.Thr108Met when they are in the homozygous state (total of 18 patients) and several clinical parameters: age at disease onset, ptosis, chronic progressive external ophthalmoplegia (CPEO), dysphagia, and onset of MV. A 2-sided p value <0.01 was considered significant.
Ethics Statement
All participants or their caregivers gave informed consent for muscle biopsy and genetic analysis and for the use of anonymized data for scientific purposes in accordance with the ethical standards of the 1964 Declaration of Helsinki. Study approval was obtained from ‘Hospital 12 de Octubre’ ethics committee (project number 16/070).
Results
Clinical Characteristics
Clinical and genetic characteristics are summarized in Table 1 and Table 2 and presented in Figure 1. Of the 53 patients, 27 were women (50,9%). Four patients (7.5%) experienced symptom onset within the first year of life, 11 patients (20.8%) between 12 and 36 months of life, 6 patients (11.3%) between 36 months and 12 years, and 32 patients beyond 12 years of age (60.3%), with 14 of them starting to experience symptoms after the age of 40 years (26.4%), Figure 2.
Table 1.
Main Clinical and Genetic Characteristics of Patients With TK2 Deficiency
| ID | Age at onset | Ambulant | Age at Mv onset | Muscle biopsy | Muscle mtDNA levels | Multiple deletions | Genetics (allele 1) | Genetics (allele 2) | Ns | Exitus (age) |
| 1 | <12 mo | — | — | 1 | 9% | ND | p.Arg130Trp | p.Ile168ThrfsTer2 | 0 | 1 |
| 2 | <12 mo | — | — | 1 | ND | ND | p.Thr108Met | p.Leu233Pro | 0 | 1 (4 y) |
| 3 | <12 mo | — | — | 1 | ND | ND | p.Arg254Ter | p.Arg254Ter | 0 | 1 |
| 4 | <12 mo | 1 | 0 | 1 | 18% | 1 | p.Thr108Met | p.Thr108Met | 1 | 0 |
| 5 | 12–36 mo | 1 | 0 | 1 | 42% | 1 | p.Thr108Met | p.Arg192Lys | 0 | — |
| 6 | 12–36 mo | — | 27 y | 1 | ND | 1 | p.Tyr208Cys | p.Tyr208Cys | 0 | 1 |
| 7 | 12–36 mo | 0 | 17 mo | 1 | 17% | 0 | p.His121Asn | p.Arg192Lys | 1 | 0 |
| 8 | 12–36 mo | 0 | 29 mo | 1 | 15% | 0 | p.Tyr208Cys | p.Arg130Trp | 1 | 0 |
| 9 | 12–36 mo | 1 | 0 | 1 | 17% | 0 | p.Thr108Met | p.Arg192Lys | 1 | 0 |
| 10 | 12–36 mo | 1 | 0 | 1 | 25% | 0 | p.Asp177Tyr | p.Lys202del | 1 | 0 |
| 11 | 12–36 mo | 0 | 21 y | 1 | 45% | 1 | p.Thr108Met | p.Thr108Met | 1 | 0 |
| 12 | 12–36 mo | 1 | 0 | 0 | ND | ND | p.Thr108Met | p.Thr108Met | 1 | 0 |
| 13 | 12–36 mo | 1 | 0 | 0 | ND | ND | p.Lys202del | p.Gln125Ter | 1 | 0 |
| 14 | 12–36 mo | 0 | 6 y | 1 | ND | ND | p.Lys202del | p.Ser51Ilefs*99 | 0 | 1 (8 y) |
| 15 | 12–36 mo | 1 | 0 | 1 | 8% | 0 | p.Thr108Met | p.Thr108Met | 1 | 0 |
| 16 | 3–12 y | 1 | 0 | 1 | ND | ND | p.Thr108Met | p.Pro227Serfs*9 | 1 | 0 |
| 17 | 3–12 y | 0 | 14 y | 1 | <10% | 1 | p.Arg183Gly | p.Lys202del | 1 | 0 |
| 18 | 3–12 y | 1 | 0 | 1 | 57% | 1 | p.Thr108Met | p.Thr108Met | 0 | — |
| 19 | 3–12 y | 1 | 35 y | 1 | 29% | 1 | p.Thr108Met | p.Thr108Met | 1 | 0 |
| 20 | 3–12 y | — | 0 | 1 | ND | ND | p.Lys202del | p.Lys202del | 0 | 1 (29 y) |
| 21 | 3–12 y | 1 | 42 y | 1 | 48% | 1 | p.Tyr208Cys | p.Lys202del | 0 | 0 |
| 22 | 12–40 y | 0 | 38 y | 1 | ND | 1 | p.Thr108Met | p.Thr108Met | 0 | 1 (49 y) |
| 23 | 12–40 y | 1 | 56 y | 1 | ND | 1 | p.Thr108Met | p.Thr108Met | 1 | 0 |
| 24 | 12–40 y | 1 | 32 y | 1 | 17% | 1 | p.Thr108Met | p.Thr108Met | 1 | 0 |
| 25 | 12–40 y | 1 | 0 | 1 | 48% | 1 | p.Thr108Met | p.Thr108Met | 0 | 0 |
| 26 | 12–40 y | 1 | 0 | 1 | ND | 1 | p.Tyr208Cys | p.Tyr208Cys | 1 | 0 |
| 27 | 12–40 y | 1 | 0 | 1 | ND | 1 | p.Lys202del | p.Lys202del | 0 | 0 |
| 28 | 12–40 y | 1 | 38 y | 1 | ND | 1 | p.Thr108Met | p.Thr108Met | 0 | 0 |
| 29 | 12–40 y | 1 | 28 y | 1 | 39% | 1 | p.Thr108Met | p.Thr108Met | 1 | 1 (28 y) |
| 30 | 12–40 y | — | 40 y | 1 | ND | 1 | p.Ser51Ilefs*99 | p.Ala139Thr | 0 | 0 |
| 31 | 12–40 y | — | — | 1 | ND | ND | p.Lys202del | p.Lys202del | 0 | 1 |
| 32 | 12–40 y | 1 | 30 y | 1 | ND | ND | p.Thr108Met | p.Thr108Met | 0 | 1 (40 y) |
| 33 | 12–40 y | 1 | 0 | 1 | ND | ND | p.Tyr208Cys | p.Tyr208Cys | 1 | 0 |
| 34 | 12–40 y | — | — | 1 | ND | ND | p.Lys202del | p.Lys202del | 0 | 1 (42 y) |
| 35 | 12–40 y | 1 | 45 y | 1 | ND | 1 | p.Thr108Met | p.Thr108Met | 1 | 0 |
| 36 | 12–40 y | 1 | 0 | 1 | 53% | 1 | p.Lys202del | p.Lys202del | 0 | 0 |
| 37 | 12–40 y | 1 | 61 y | 1 | ND | ND | p.Lys202del | p.Lys202del | 0 | 0 |
| 38 | 12–40 y | 1 | 0 | 1 | ND | 1 | p.Lys202del | p.Lys202del | 0 | 1 |
| 39 | 12–40 y | 1 | 61 y | 1 | 60% | 1 | p.Lys202del | p.Lys202del | 1 | 0 |
Abbreviations: ID = identification number; MV = mechanical ventilation; ND = not done; UNK = unknown; 0 = No; 1 = yes; — = data not available. Patients with onset of symptoms before 1 year of age, between 1 and 3 years of age, between 3 and 12 years of age, and 12 and 40 years of age.
Table 2.
Main Clinical and Genetic Characteristics of Patients With TK2 Deficiency
| ID | Age at onset | Ambulant | Age at mv onset | Muscle biopsy | Muscle mtDNA levels | Multiple deletions | Genetics (allele 1) | Genetics (allele 2) | Ns | Exitus (age) |
| 41 | >40 y | 1 | 42 y | 1 | ND | ND | p.Gln125Ter | p.Ala139Thr | 0 | 0 |
| 42 | >40 y | 0 | 49 y | 1 | 33% | 1 | p.Lys202del | p.Lys202del | 0 | 1 (68 y) |
| 43 | >40 y | 1 | 50 y | 1 | 66% | 1 | p.Lys202del | p.Lys202del | 1 | 0 |
| 44 | >40 y | 1 | 51 y | 1 | ND | ND | p.Ala139Thr | p.Ser14Argfs*40 | 0 | 0 |
| 45 | >40 y | 1 | 65 y | 1 | ND | ND | p.Lys202del | p.Lys202del | 1 | 0 |
| 46 | >40 y | 1 | 55 y | 1 | ND | 1 | p.His121Asn | p.Lys202del | 0 | 0 |
| 47 | >40 y | 1 | 0 | 1 | ND | 1 | p.Lys202del | p.Lys202del | 0 | 0 |
| 48 | >40 y | — | UNK | 1 | ND | 1 | p.Ala139Thr | p.His121Asn | 0 | 1 (>70 y) |
| 49 | >40 y | 1 | 58 y | 1 | ND | 1 | p.Arg130Trp | p.Ala139Thr | 0 | 1 |
| 50 | >40 y | — | 72 y | 1 | ND | 1 | p.Ala139Thr | p.His121Asn | 0 | 1 (>70 y) |
| 51 | >40 y | 1 | 0 | 1 | ND | 1 | p.Lys202del | p.Tyr208Cys | 0 | 0 |
| 52 | >40 y | 1 | 53 y | 1 | ND | 1 | p.Tyr208Cys | p.Tyr208Cys | 0 | 0 |
| 53 | >40 y | 1 | 73 y | 0 | ND | ND | p.Lys202del | p.Lys202del | 1 | 0 |
Abbreviations: ID = identification number; MV = mechanical ventilation; ND = not done; UNK = unknown; 0 = No; 1 = yes; — = data not available. Patients with onset of symptoms after the age of 40 years.
Figure 1. Main Clinical Characteristics by Age at Onset.

Relevant clinical characteristics in different group of ages are represented; ptosis, progressive external ophthalmoplegia (PEO), facial weakness, cervical weakness, dysphagia, and mechanical ventilation.
Figure 2. Distribution of Patients According to the Age at Which the First Symptoms Appeared.
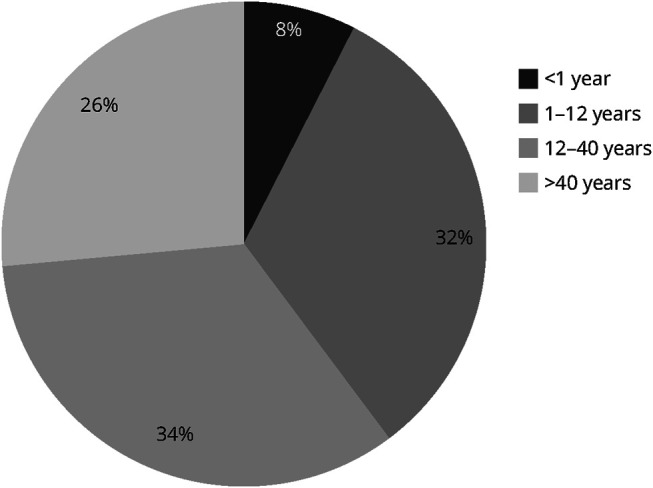
Ages less than 1 year, between 1 and 12 years, between 12 and 40 years, and more than 40 years.
We collected detailed clinical data from 44 patients within the cohort, 16 with symptom onset before 12 years of age, 15 with symptom onset between 12 and 40 years of age, and 13 with a very late onset of the disease, that is, patients who remained asymptomatic until their forties. Only 18.8% (3/16) of patients with symptom onset before the age of 12 years exhibited ptosis and CPEO, other 18.8% presented solely with ptosis (3/16), while the majority, 62.5% (10/16), had no ptosis nor CPEO. Among patients with symptom onset after the age of 12 years but before 40 years of age, 40% (6/15) displayed ptosis and CPEO, 46.7% (7/15) had only ptosis, and merely 13.3% (2/16) presented without ptosis or CPEO. By contrast, all patients with the onset of symptoms after the age of 40 years showed ptosis (13/13), and 85% (11/13) had ptosis and CPEO. Most of the patients (80%) exhibited cervical and facial weakness, irrespective of the age at which symptoms appeared. While we lack specific details regarding the severity of muscle weakness, we collected the ambulatory status of 43 patients of the cohort, and only 7 had lost the ability to walk independently during data collection (16.8%). In addition, 40% of patients experienced some degree of dysphagia, although specific data on its severity were unavailable. Notably, 3 patients (6.8%) only exhibited exercise intolerance, characterized by episodes of exertion-induced rhabdomyolysis, without significant muscle weakness. Ptosis and ophthalmoplegia were absent in these cases, leading to initial suspicion of a metabolic muscle disorder as the primary diagnosis. Of the 34 patients with documented data who remain alive, 19 (55.8%) required MV while maintaining ambulatory capacity.
In 50 of 53 cases (94.3%), a muscle biopsy was performed, and in all these cases, the diagnosis of mitochondrial myopathy could be established based on the presence of signs of mitochondrial proliferation and dysfunction. All muscle biopsies were reported as pathologic. We were able to review 30 in more detail. Samples were taken from different muscles (deltoid, biceps, or quadriceps) and showed varying degrees of ragged-red fibers (2%–18% of total fibers) and cytochrome C oxidase–negative fibers (4%–40% of the total fibers). Furthermore, as previously described,16 we found myopathic changes in 93% of cases (necrotic fibers in 48%, fiber size variability in 93%, nuclear internalization in 50%, and fibrosis in 52%). The performance of electron microscopy was anecdotal and did not provide additional relevant information.
Treatment With Oral Deoxynucleosides
Regarding treatment, 23 of 53 patients (43.4%) received treatment with deoxynucleosides (dThd and dCtd). The administration of deoxynucleosides was facilitated either through compassionate use29 or within a clinical trial (ClinicalTrials.gov ID NCT03845712). Specifically, in the group with symptom onset before the age of 12 years, 12 patients (57.2%) underwent treatment, whereas in the group with symptom onset after the age of 12 years, 11 patients (34.4%) received treatment. The comprehensive analysis of treatment response is beyond the scope of this study. For further insights into the treatment outcomes, interested readers may refer to the compassionate use results, available in ref 29.
By the end of this study (September 2023), 16 patients (30%) from the cohort have died. Among patients with symptom onset before 12 months, 3 of 4 (75%) died before the availability of the treatment, while the 1 still alive is currently receiving treatment. In the subset with onset of symptoms between the ages of 12 and 36 months, we do not have data for 1 case, while 2 of the 10 patients (20%) have died, none of whom had received treatment. There were 7 patients with symptom onset before the age of 12 years who had never received treatment. All except one of them have died (85.7%). The only patient alive did not have muscle mtDNA depletion. However, it is worth noting that all patients with symptom onset before the age of 12 years who initiated treatment remain alive (12 patients). Among the patients with symptom onset after the age of 12 years, 10 have died, and only one of them received treatment. All patients, regardless of the age at onset, died due to respiratory failure.
Genetic Results
In 40 of 53 patients (75.4%), genetic diagnosis was achieved through NGS of a panel of nuclear genes associated with mtDNA maintenance defects. Thirty-one of these patients were included in the panel after the presence of multiple mtDNA deletions in muscle tissue was detected. In 9 patients (17%), the diagnosis was reached through direct Sanger sequencing of the TK2 gene, and in 4 patients (7.5%), the diagnosis was obtained through WES.
Among the 53 patients, 16 distinct variants in the TK2 gene were identified, all of which were present in a biallelic form (either homozygous or compound heterozygous). Additional data are listed in eTable 1. The most frequent pathogenic variants in our series were c.604_606del (NM_004614; p.Lys202del), found in 21 patients (35 alleles), and c.323C>T (p.Thr108Met), identified in 18 patients (32 alleles). Among the 32 late-onset cases, 15 patients harbored the (p.Lys202del) variant in at least 1 allele, accounting for 46.9% in this subgroup.
In addition to these variants, we also identified 2 other recurrent TK2 pathogenic variants: c.623A>G (p.Tyr208Cys) and c.415G>A (p.Ala139Thr), which both were found in 6 patients, encompassing 11 and 6 alleles, respectively.
Pathogenic variants were identified across the entire TK2 gene, except for exons 3 and 4. It is noteworthy the clustering of missense variants within the protein region spanning amino acids 100–141, which plays a crucial role in Mg2+ and ATP binding, essential for the proper functioning of TK2.45 Furthermore, a variety of variants, including predicting in-frame, frameshift, and stop-gain alterations, were observed throughout the entire protein (Figure 3).
Figure 3. TK2 Gene Variants Distribution.

TK2 representation showing the distribution of the variants identified in our cohort and the number of alleles identified for each variant (red background for >20 alleles, yellow background for 5–20 alleles, and blue background for ≤4 alleles).
The only variant that had not been previously reported as pathogenic was c.503del (p.Ile168ThrfsTer2). It is predicted to be pathogenic according to ACMG classification because it fulfils 3 criteria of very strong and moderate pathogenicity: (1) it is a null variant (frameshift) in a gene where loss of function (LOF) is a known mechanism of disease (the gene has several documented pathogenic LOF variants) (PVS1); (2) it is absent in population databases such as the Genome Aggregation Database (gnomAD), Exome Sequencing Project, and 1000 Genomes Project and (PM2); and (3) it was detected in trans with another pathogenic variant in an autosomal recessive disorder. Of note, the variants (p.Asp177Tyr), (p.Pro227Serfs*9), and (p.Leu14Argfs*41) had been previously first reported by Spanish research groups in patients included in this cohort.19,21
Frequency of the p.Thr108Met and p.Lys202del Alleles in the Spanish Population
The frequency of the p.Lys202del variant in the Spanish genetic databases analyzed was 0.034% compared with 0.0004% described in gnomAD (86-fold higher). Within gnomAD, the subpopulation with the highest frequency was the Latino population, exhibiting a frequency of 0.0029%, with the Spanish frequency also being 12 times higher than this.
The frequency of p.Thr108Met, which is the most frequent pathogenic variant reported in TK2d,8 was 0.062% in the Spanish databases and 0.0046% in gnomAD (13-fold difference). The frequency was higher in the Northern Spain area (Galicia), reaching 0.176%, which accounts for 38-fold the frequency registered in gnomAD. This variant is also more frequent in the gnomAD Latino subpopulation, being 0.022%.
Haplotype Analysis
We used genetic lengths of haplotypes containing p.Thr108Met and p.Lys202del and a model-free Gamma method to estimate the most common recent ancestor (MCRA). Assuming a dependent genealogy, as is shown in the eFigure 1, MCRA of the individuals with p.Thr108Met variant lived 425 (125-1,550) years ago and the MCRA of the individuals with p.Lys202del lived 2,375 (1,212 to 3,532) years ago (additional data are listed in eTable 2).
ROH Analysis
Figure 4 summarizes key findings on ROH distribution and origin in affected individuals and the general population. In (A), there are significant differences in ROH longer than 8 Mb between affected individuals and the general population (ANOVA p value = 5.78E-11). (B) and (C) analyze the origin of ROH. In (B), we can discern a rightward shift in individuals from families practicing consanguinity. Specifically, 3 individuals carrying the p.Thr108Met variant exhibit a pronounced right shift. (C) displays a plot of systematic inbreeding coefficient (FIS) against FROH, revealing 3 regions related to inbreeding: individuals near the diagonal line, like p.Thr108Met-affected individuals, exhibit systematic inbreeding (FIS), indicating that the overall inbreeding coefficient (FIT) in this population mainly stems from consanguinity. Those near FIS = 0 experience panmictic inbreeding due to genetic drift and consanguinity. Negative FIS values signal low effective population size, isolation, and significant genetic drift, contributing to FIT and strong FST. Finally, in Figure 4, (C), we observe the physical positioning of ROH on chromosome 16 in affected individuals, with a concentration of ROH evident in the region where the TK2 is located.
Figure 4. ROH Exploratory Analysis.

Different runs of homozygosity (ROH) exploratory analyses are presented in this figure. (A) ROH size distribution by variant. Average total sums are presented over 6 classes of ROH lengths: 0.3 ≤ ROH > 0.5 Mb, 0.5 ≤ ROH > 1 Mb, 1 ≤ ROH > 2 Mb, 2 ≤ ROH > 4 Mb, 4 ≤ ROH > Mb, and ROH ≥8 Mb. (B) Average number and cumulative length of ROH (Runs of Homozygosity) exceeding 1.5 Mb for each individual. The diagonal line on the graph is generated through regression analysis, plotting the number of ROH against the total ROH length. Data for this analysis come from ASW and ACB populations in the 1000 Genomes Project, which are representative of admixed and relatively outbred populations.47 Consanguinity practices in preceding generations manifest as a rightward shift. Simulated data for the number and total length of ROH (ROH >1.5 Mb) in the offspring resulting from various consanguineous matings are also displayed. Points of varying colors represent offspring from different consanguineous relationships: green for second cousins, yellow for first cousins, orange for avuncular (including uncle-niece, aunt-nephew, and double first cousins), and red for incestuous relationships (such as brother-sister and parent-offspring). Each type of consanguineous mating is depicted through 5,000 simulations. Simulations do not account for genetic drift. However, the extent of the rightward shift can be extrapolated to scenarios where there is some degree of autozygosity due to genetic drift. (C) Population analysis and components of the inbreeding coefficient. Systematic inbreeding coefficient (FIS) vs the inbreeding coefficient obtained from ROH (FROH). In this context, FIS is the average SNP homozygosity within an individual relative to the expected homozygosity of alleles randomly drawn from the population. The diagonal broken line represents FIS = FROH. The horizontal broken line represents FIS = 0. (D) Representation of the ROH physical distribution in the Chr 16 of affected individuals. The broken line shows the TK2 gene physical location.
ROH islands in the TK2 gene region on chromosome 16, including shared haplotypes from affected individuals used in MRCA analysis, are summarized in eTable 3. When considering unaffected individuals, an ROHi of 2.3 Mb (Chr16: 66480000–68850000) is found in 42% of all individuals. Notably, 100% of the p.Thr108Met-affected individuals exhibit an ROHi of 0.6 Mb in (Chr16: 66120000–66810000); no statistically significant ROHi in this region was observed in p.Lys202del-affected individuals. By contrast, shared haplotypes are longer for affected individuals: 6.4 Mb for p.Thr108Met and 0.3 Mb in p.Lys202del.
Association of Phenotypes With the Variants p.Lys202del and p.Thr108Met
A statistically significant association (p < 0.01) was identified between the frequent variants p.Lys202del and p.Thr108Met and the age of initiation of MV. Specifically, the 6 patients who were homozygous for the p.Lys202del variant started MV after the age of 40 years. Conversely, among the 9 patients homozygous for the p.Thr108Met variant, 78% started MV within the 12- to 40-year age range, while only 22% required MV after the age of 40 years.
Furthermore, all 7 homozygous patients for the p.Lys202del variant displayed ptosis, while only 64% of homozygous carriers (11 patients) of the p.Thr108Met variant showed ptosis. The age at disease onset also exhibited interesting patterns, with all individuals homozygous for the p.Lys202del variant experiencing onset during the 12- to 40-year-old period or later, in comparison with homozygous carriers of the p.Thr108Met variant, where onset occurred during the 12- to 40-year-old period or earlier.
Discussion
In this study, we conducted a comprehensive analysis of both genetic and clinical characteristics of TK2d, using data retrospectively collected from a cohort of 53 Spanish patients with biallelic TK2 variants. Notably, this compilation stands as the most extensive series documented on this disorder in a single country. Our objective was to elucidate the factors contributing to the high number of patients diagnosed in Spain and to refine our understanding of the clinical spectrum of this deficiency.
In previous studies that compiled all published cases with TK2d, it was reported that the proportion of patients with symptom onset after the age of 12 years accounted for only approximately 15% of the total cases.7,8 However, in our series, which encompasses more than half of all reported cases worldwide, 60% of patients had not presented symptoms before the age of 12 years, and 26% had remained asymptomatic until the fifth decade of life. These findings suggest a potential underdiagnosis of adult patients with this disorder, although the high frequency of the milder variant p.Lys202del in Spain also might contribute to this phenomenon.
Notably, we found that 33% of the identified alleles carried the p.Lys202del variant, which had been exclusively identified in patients of Spanish or Hispanic origin19 and associated with later-onset cases, presenting with a less severe phenotype.7,16 Our results also support these conclusions because homozygous individuals for this variant in our cohort showed a later onset and required MV from the age of 40 years. In addition, approximately 30% of the mutated alleles were attributed to the p.Thr108Met variant, the most prevalent variant reported worldwide. Of interest clinical correlations have not been established for this variant because it is found in clinical forms spanning the entire spectrum of severity. The remaining one-third of cases were associated with 14 different variants, many of which were private and found in isolated cases.
We conducted an analysis to assess the prevalence of the 2 most recurrent variants of the Spanish population, p.Lys202del and p.Thr108Met. Noteworthy, the gnomAD database indicates that these 2 variants are more common in the Latino population subset. Notably, the p.Lys202del variant was found to be 86 times more frequent in the Spanish population and the p.Thr108Met 13 times more frequent than their reported frequency in gnomAD. Remarkably, this later variant was even more prevalent in the northern region of Spain, where it was observed to be 38 times more frequent than in gnomAD. The higher frequency in the northern area could be attributed to the higher consanguinity index observed in this region, which arises from its specific geographical characteristics. It is important to highlight that our analyses were based on exome variant databases obtained from patients with very heterogeneous clinical indications for genetic diagnostic purposes, rather than being collected from a general population. This distinction is important to consider because it may influence the prevalence rates and cannot be directly compared with data derived from healthy individuals.
The high frequency of 2 variants prompted a haplotype study to estimate the MRCA. While literature often associates MRCA estimation with founder events, it is crucial to distinguish this from classical genetics' founder effect. Founder effects result from a small founding population or significant subdivision with reproductive isolation. Genetic drift, not selection, predominates, leading to variant frequency increase.46 Attributing differences to a founder effect requires detailed population structure analysis, and conclusions may be speculative without pedigree records. Moreover, analysis requires additional information about the population's demographic history to determine its founder origin or even the variant's age.47 Clear founder effects are evident in studies of Ashkenazi and other isolated populations globally.48 In this study, we used the ROH approach to explore the coalescent process of analyzed variants. Our analysis revealed that affected individuals have a significantly higher number of larger ROH, correlated with their recent origin within the last 5 to 10 generations. The origin of these ROH varies among affected individuals, with consanguinity and genetic drift playing roles, particularly for the p.Thr108Met variant. Inbreeding, regardless of origin, appears to underlie the elevated prevalence of both variants in affected individuals. Analysis of ROH islands highlighted a shorter island across the TK2 gene for p.Thr108Met. Both variants likely emerged in the Spanish population approximately 16.8 and 95.2 generations ago for p.Thr108Met and p.Lys202del, respectively, with subsequent frequency increase through various forms of inbreeding.
The identification of 14 additional distinct variants suggests that a high diagnostic capacity might also have contributed to the elevated number of identified cases with TK2d, particularly among those with an onset in the juvenile or adult population. In this regard, performing a muscle biopsy at the early stages of the diagnostic process facilitates the use of targeted ancillary tests and expedites the overall diagnostic journey. In our series, a muscle biopsy was performed in 94% of the patients except for a recently diagnosed pediatric case where a WES was prioritized. Because our series includes patients diagnosed over 20 years, it remains to be determined whether this high number of biopsies will be maintained, especially among pediatric cases.
From the analysis of the available clinical information within this cohort, we can infer a higher frequency of ptosis and CPEO in cases presenting in adulthood (Figure 1), without any other distinct clinical features. Notably, respiratory failure is consistently the cause of mortality in all cases, confirming our previous emphasis on the early and prominent involvement of the diaphragm in this condition.16,28 This aspect is highly relevant for diagnosis and monitoring, even in patients with milder phenotypes, such as those with apparent isolated CPEO.14,18,49 Of interest, besides the classical phenotype, this cohort includes 3 patients with isolated exercise intolerance and rhabdomyolysis episodes, one of them previously reported in detail.15 This manifestation could represent the milder end of the severity spectrum and a diagnostic challenge, because mitochondrial disorders rarely present with isolated rhabdomyolysis.50 In these cases, the muscle biopsy was critical to reach the final diagnosis.
TK2d is a severe disease with high mortality in the absence of treatment. Among the 28 untreated patients whose vital status were available, 7 presented with symptoms before the age of 12 years, and 86% died. Among the 21 untreated patients who developed symptoms after the age of 12 years, 6 died (29%) and 67% required MV. However, it is noteworthy that all patients receiving treatment (23), except for 1, are still alive regardless of the age at disease onset.
In conclusion, the elevated prevalence of this disorder in Spain can be attributed to several factors, including the higher frequency of 2 specific variants (p.Thr108Met and p.Lys202del), as well as consanguinity. Notably, the p.Lys202del variant is associated with a less severe phenotype characterized by later onset and slower disease progression. Nonetheless, it is essential to recognize that a third of the cases within this extensive cohort are attributed to other genetic variants, indicating that a high diagnostic capability is also a contributing factor. In addition, a noteworthy proportion of this cohort (60%) comprises patients with late-onset TK2d, a subgroup that could be underdiagnosed in other regions. This observation emphasizes the critical need to increase awareness of this potentially treatable disease within this specific population.
The data sets generated and analyzed during this study are available from the corresponding author on reasonable request.
Acknowledgment
C.D.-G., M.O., N.M., R.J., C.P., and A.N. are members of the European Reference Network for Neuromuscular Disorders (ERN-NMD).
Glossary
- CPEO
chronic progressive external ophthalmoplegia
- NGS
next-generation sequencing
- dCtd
deoxycytidine
- dThd
deoxythymidine
- mtDNA
mitochondrial DNA
- MRCA
most recent common ancestor
- MV
mechanical ventilation
- LR-PCR
long-range PCR
- qPCR
quantitative PCR
- ROH
runs of homozygosity
- TK2d
thymidine kinase 2 deficiency
- WES
whole-exome sequencing
Appendix 1. Authors
| Name | Location | Contribution |
| Francisco Ceballos, PhD | Spanish Network for Biomedical Research in Rare Diseases (CIBERER), Madrid, Spain | Drafting/revision of the article for content, including medical writing for content; major role in the acquisition of data; and analysis or interpretation of data |
| Pablo Serrano-Lorenzo, PhD | Spanish Network for Biomedical Research in Rare Diseases (CIBERER); Mitochondrial and Neuromuscular Research Group ‘12 de Octubre’, Hospital Research Institute (imas12), Madrid, Spain | Drafting/revision of the article for content, including medical writing for content; major role in the acquisition of data; and analysis or interpretation of data |
| Laura Bermejo-Guerrero, MD | Neurology Department, Neuromuscular Disorders Unit, Hospital 12 de Octubre, Madrid, Spain | Drafting/revision of the article for content, including medical writing for content; major role in the acquisition of data; and analysis or interpretation of data |
| Alberto Blázquez, BSc | Spanish Network for Biomedical Research in Rare Diseases (CIBERER); Mitochondrial and Neuromuscular Research Group ‘12 de Octubre’, Hospital Research Institute (imas12), Madrid, Spain | Drafting/revision of the article for content, including medical writing for content; analysis or interpretation of data |
| Juan F. Quesada-Espinosa, PhD | Genetics Department, Hospital Universitario 12 de Octubre, Madrid, Spain | Drafting/revision of the article for content, including medical writing for content; analysis or interpretation of data |
| Jorge Amigo, PhD | Spanish Network for Biomedical Research in Rare Diseases (CIBERER), Madrid; Fundación Pública Galega de Medicina Xenómica (FPGMX); Genetic’s Group, Santiago de Compostela Research Institute (IDIS); Medicine Xenómica’s Group, Research Center for Molecular Medicine and Chronic Diseases (CIMUS), Santiago de Compostela University (USC), Spain | Drafting/revision of the article for content, including medical writing for content; study concept or design; and analysis or interpretation of data |
| Pablo Minguez, PhD | Spanish Network for Biomedical Research in Rare Diseases (CIBERER); Department of Genetics and Genomics, Instituto de Investigación Sanitaria-Fundación Jiménez Díaz University Hospital; Bioinformatics Unit, Health Research Institute-Fundación Jiménez Díaz University Hospital, Universidad Autónoma de Madrid (IIS-FJD, UAM), Spain | Drafting/revision of the article for content, including medical writing for content; analysis or interpretation of data |
| Carmen Ayuso, MD, PhD | Spanish Network for Biomedical Research in Rare Diseases (CIBERER); Department of Genetics and Genomics, Instituto de Investigación Sanitaria-Fundación Jiménez Díaz University Hospital, Universidad Autónoma de Madrid (IIS-FJD, UAM), Spain | Drafting/revision of the article for content, including medical writing for content; analysis or interpretation of data |
| Elena García-Arumí, MD, PhD | Spanish Network for Biomedical Research in Rare Diseases (CIBERER), Madrid; Department of Clinical and Molecular Genetics, Valld’Hebron University Hospital; Research Group on Neuromuscular and Mitochondrial Disorders, Vall d’Hebron Research Institut (VHIR), Universitat Autónoma de Barcelona, Spain | Drafting/revision of the article for content, including medical writing for content; analysis or interpretation of data |
| Nuria Muelas, MD, PhD | Spanish Network for Biomedical Research in Rare Diseases (CIBERER), Madrid; Neuromuscular Unit, Department of Neurology, Hospital Universitari I Politècnic La Fe, Neuromuscular and Ataxias Research Group, Instituto de Investigación Sanitaria La Fe, Valencia, Spain | Drafting/revision of the article for content, including medical writing for content; major role in the acquisition of data; and analysis or interpretation of data |
| Teresa Jaijo, PhD | Spanish Network for Biomedical Research in Rare Diseases (CIBERER), Madrid; Department of Genetics, Hospital Universitari I Politècnic la Fe de Valencia, Spain | Drafting/revision of the article for content, including medical writing for content; major role in the acquisition of data; and analysis or interpretation of data |
| Andres Nascimento, MD | Spanish Network for Biomedical Research in Rare Diseases (CIBERER), Madrid; Neuromuscular Unit, Neurology Department, Sant Joan de Déu Research Institute, Sant Joan de Déu Hospital, Barcelona, Spain | Drafting/revision of the article for content, including medical writing for content; major role in the acquisition of data; and analysis or interpretation of data |
| Beatriz Galán-Rodriguez, MD | Neurology Department, Neuromuscular Disorders Unit, Instituto de Biomedicina de Sevilla, Hospital U. Virgen del Rocío, Spain | Drafting/revision of the article for content, including medical writing for content; major role in the acquisition of data; and analysis or interpretation of data |
| Carmen Paradas, MD, PhD | Neurology Department, Neuromuscular Disorders Unit, Instituto de Biomedicina de Sevilla, Hospital U. Virgen del Rocío; Spanish Network for Biomedical Research in Neurodegenerative Diseases (CIBERNED), Madrid, Spain | Drafting/revision of the article for content, including medical writing for content; major role in the acquisition of data; and analysis or interpretation of data |
| Joaquín Arenas, PhD | Spanish Network for Biomedical Research in Rare Diseases (CIBERER); Mitochondrial and Neuromuscular Research Group ‘12 de Octubre’, Hospital Research Institute (imas12), Madrid, Spain | Drafting/revision of the article for content, including medical writing for content; major role in the acquisition of data; and analysis or interpretation of data |
| Angel Carracedo, MD; PhD | Spanish Network for Biomedical Research in Rare Diseases (CIBERER), Madrid; Fundación Pública Galega de Medicina Xenómica (FPGMX); Genetic’s Group, Santiago de Compostela Research Institute (IDIS); Medicine Xenómica’s Group, Research Center for Molecular Medicine and Chronic Diseases (CIMUS), Santiago de Compostela University (USC), Spain | Drafting/revision of the article for content, including medical writing for content; analysis or interpretation of data |
| Ramon Martí, PhD | Spanish Network for Biomedical Research in Rare Diseases (CIBERER), Madrid; Research Group on Neuromuscular and Mitochondrial Disorders, Vall d’Hebron Research Institut (VHIR), Universitat Autónoma de Barcelona, Spain | Drafting/revision of the article for content, including medical writing for content; study concept or design; and analysis or interpretation of data |
| Miguel A. Martín, PhD | Spanish Network for Biomedical Research in Rare Diseases (CIBERER); Mitochondrial and Neuromuscular Research Group ‘12 de Octubre’, Hospital Research Institute (imas12); Genetics Department, Hospital Universitario 12 de Octubre, Madrid, Spain | Drafting/revision of the article for content, including medical writing for content; major role in the acquisition of data; study concept or design; and analysis or interpretation of data |
| Cristina Domínguez-González, MD, PhD | Spanish Network for Biomedical Research in Rare Diseases (CIBERER); Mitochondrial and Neuromuscular Research Group ‘12 de Octubre’, Hospital Research Institute (imas12), Madrid, Spain | Drafting/revision of the article for content, including medical writing for content; major role in the acquisition of data; study concept or design; and analysis or interpretation of data |
Appendix 2. Coinvestigators
| Name | Location | Role | Contribution |
| Carlos de Fuenmayor Fernández-de la Hoz | Hospital 12 de Octubre | Neurologist | Collect relevant clinical information |
| Paloma Martin Jimenez | Hospital 12 de Octubre | Neurologist | Collect relevant clinical information |
| Aurelio Hernández-Laín | Hospital 12 de Octubre | Neuropathologist | Collect relevant clinical information |
| Ana Hernández-Voth | Hospital 12 de Octubre | Pulmonologist | Collect relevant clinical information |
| Javier Sayas | Hospital 12 de Octubre | Pulmonologist | Collect relevant clinical information |
| Germán Moris | Hospital Central de Asturias | Neurologist | Collect relevant clinical information |
| Jorge García-García | Hospital General de Alicante | Neurologist | Collect relevant clinical information |
| María Rabasa Pérez | Hospital de Fuenlabrada | Neurologist | Collect relevant clinical information |
| Montse Olivé | Hospital de la Santa Creu i Sant Pau | Neurologist | Collect relevant clinical information |
| Daniel Sánchez-Tejerina | Hospital Vall d’Hebron | Neurologist | Collect relevant clinical information |
| Raúl Juntas | Hospital Vall d’Hebron | Neurologist | Collect relevant clinical information |
| Juan Carlos León | Hospital La Candelaria | Neurologist | Collect relevant clinical information |
Study Funding
This research was funded by the Instituto de Salud Carlos III, (ISCIII) and the Ministerio de Ciencia e Innovación (Madrid, Spain; cofunded by European Regional Development Fund “A way to make Europe”), grant number PI22/01587, to C.D.-G., PI19/01772 to E.G.-A. and PI21/00381 to M.A.M.
Disclosure
F. Ceballos reports no disclosures relevant to the manuscript, P. Serrano-Lorenzo reports no disclosures relevant to the manuscript, L. Bermejo-Guerrero reports no disclosures relevant to the manuscript, A. Blázquez reports no disclosures relevant to the manuscript, J.F. Quesada-Espinosa reports no disclosures relevant to the manuscript, J. Amigo reports no disclosures relevant to the manuscript, P. Minguez reports no disclosures relevant to the manuscript, C. Ayuso reports no disclosures relevant to the manuscript, E. García-Arumí reports no disclosures relevant to the manuscript, N. Muelas reports no disclosures relevant to the manuscript, T. Jaijo reports no disclosures relevant to the manuscript; A. Nascimento serves on the advisory board of UCB Pharma; B. Galán-Rodriguez reports no disclosures relevant to the manuscript; C. Paradas serves on the advisory board of UCB Pharma; TK2d Spanish-Group report no disclosures relevant to the manuscript; J. Arenas reports no disclosures relevant to the manuscript; A. Carracedo reports no disclosures relevant to the manuscript; R. Martí serves on the advisory board of UCB Pharma; MAM serves on the advisory board of UCB Pharma; and C. Domínguez-González serves on the advisory board of UCB Pharma. Go to Neurology.org/NG for full disclosures.
References
- 1.El-Hattab AW, Scaglia F. Mitochondrial DNA depletion syndromes: review and updates of genetic basis, manifestations, and therapeutic options. Neurother J Am Soc Exp Neurother. 2013;10(2):186-198. doi: 10.1007/s13311-013-0177-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang L, Munch-Petersen B, Herrström Sjöberg A, et al. Human thymidine kinase 2: molecular cloning and characterisation of the enzyme activity with antiviral and cytostatic nucleoside substrates. FEBS Lett. 1999;443(2):170-174. doi: 10.1016/s0014-5793(98)01711-6 [DOI] [PubMed] [Google Scholar]
- 3.Ashley N, Adams S, Slama A, et al. Defects in maintenance of mitochondrial DNA are associated with intramitochondrial nucleotide imbalances. Hum Mol Genet. 2007;16(12):1400-1411. doi: 10.1093/hmg/ddm090 [DOI] [PubMed] [Google Scholar]
- 4.Rahman S, Poulton J. Diagnosis of mitochondrial DNA depletion syndromes. Arch Dis Child. 2009;94(1):3-5. doi: 10.1136/adc.2008.147983 [DOI] [PubMed] [Google Scholar]
- 5.Moraes CT, Shanske S, Tritschler HJ, et al. mtDNA depletion with variable tissue expression: a novel genetic abnormality in mitochondrial diseases. Am J Hum Genet. 1991;48(3):492-501. [PMC free article] [PubMed] [Google Scholar]
- 6.Viscomi C, Zeviani M. MtDNA-maintenance defects: syndromes and genes. J Inherit Metab Dis. 2017;40(4):587-599. doi: 10.1007/s10545-017-0027-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang J, Kim E, Dai H, et al. Clinical and molecular spectrum of thymidine kinase 2-related mtDNA maintenance defect. Mol Genet Metab. 2018;124(2):124-130. doi: 10.1016/j.ymgme.2018.04.012 [DOI] [PubMed] [Google Scholar]
- 8.Garone C, Taylor RW, Nascimento A, et al. Retrospective natural history of thymidine kinase 2 deficiency. J Med Genet. 2018;55(8):515-521. doi: 10.1136/jmedgenet-2017-105012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saada A, Shaag A, Mandel H, Nevo Y, Eriksson S, Elpeleg O. Mutant mitochondrial thymidine kinase in mitochondrial DNA depletion myopathy. Nat Genet. 2001;29(3):342-344. doi: 10.1038/ng751 [DOI] [PubMed] [Google Scholar]
- 10.Chanprasert S, Wang J, Weng SW, et al. Molecular and clinical characterization of the myopathic form of mitochondrial DNA depletion syndrome caused by mutations in the thymidine kinase (TK2) gene. Mol Genet Metab. 2013;110(1-2):153-161. doi: 10.1016/j.ymgme.2013.07.009 [DOI] [PubMed] [Google Scholar]
- 11.Oskoui M, Davidzon G, Pascual J, et al. Clinical spectrum of mitochondrial DNA depletion due to mutations in the thymidine kinase 2 gene. Arch Neurol. 2006;63(8):1122-1126. doi: 10.1001/archneur.63.8.1122 [DOI] [PubMed] [Google Scholar]
- 12.Mancuso M, Salviati L, Sacconi S, et al. Mitochondrial DNA depletion: mutations in thymidine kinase gene with myopathy and SMA. Neurology. 2002;59(8):1197-1202. doi: 10.1212/01.wnl.0000028689.93049.9a [DOI] [PubMed] [Google Scholar]
- 13.Tulinius M, Moslemi AR, Darin N, Holme E, Oldfors A. Novel mutations in the thymidine kinase 2 gene (TK2) associated with fatal mitochondrial myopathy and mitochondrial DNA depletion. Neuromuscul Disord. 2005;15(6):412-415. doi: 10.1016/j.nmd.2005.03.010 [DOI] [PubMed] [Google Scholar]
- 14.Behin A, Jardel C, Claeys KG, et al. Adult cases of mitochondrial DNA depletion due to TK2 defect: an expanding spectrum. Neurology. 2012;78(9):644-648. doi: 10.1212/WNL.0b013e318248df2b [DOI] [PubMed] [Google Scholar]
- 15.de Fuenmayor-Fernández de la Hoz CP, Morís G, Jiménez-Mallebrera C, et al. Recurrent rhabdomyolysis and exercise intolerance: a new phenotype of late-onset thymidine kinase 2 deficiency. Mol Genet Metab Rep. 2021;26:100701. doi: 10.1016/j.ymgmr.2020.100701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Domínguez-González C, Hernández-Laín A, Rivas E, et al. Late-onset thymidine kinase 2 deficiency: a review of 18 cases. Orphanet J Rare Dis. 2019;14(1):100. doi: 10.1186/s13023-019-1071-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Paradas C, Gutiérrez Ríos P, Rivas E, Carbonell P, Hirano M, DiMauro S. TK2 mutation presenting as indolent myopathy. Neurology. 2013;80(5):504-506. doi: 10.1212/WNL.0b013e31827f0ff7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alston CL, Schaefer AM, Raman P, et al. Late-onset respiratory failure due to TK2 mutations causing multiple mtDNA deletions. Neurology. 2013;81(23):2051-2053. doi: 10.1212/01.wnl.0000436931.94291.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Domínguez-González C, Madruga-Garrido M, Hirano M, et al. Collaborative model for diagnosis and treatment of very rare diseases: experience in Spain with thymidine kinase 2 deficiency. Orphanet J Rare Dis. 2021;16(1):407. doi: 10.1186/s13023-021-02030-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bychkov IO, Itkis YS, Tsygankova PG, et al. Mitochondrial DNA maintenance disorders in 102 patients from different parts of Russia: mutational spectrum and phenotypes. Mitochondrion. 2021;57:205-212. doi: 10.1016/j.mito.2021.01.004 [DOI] [PubMed] [Google Scholar]
- 21.Domínguez-González C, Hernández-Voth A, de Fuenmayor-Fernández de la Hoz CP, et al. Metrics of progression and prognosis in untreated adults with thymidine kinase 2 deficiency: an observational study. Neuromuscul Disord. 2022;32(9):728-735. doi: 10.1016/j.nmd.2022.07.399 [DOI] [PubMed] [Google Scholar]
- 22.Rodríguez-López C, García-Cárdaba LM, Blázquez A, et al. Clinical, pathological and genetic spectrum in 89 cases of mitochondrial progressive external ophthalmoplegia. J Med Genet. 2020;57(9):643-646. doi: 10.1136/jmedgenet-2019-106649 [DOI] [PubMed] [Google Scholar]
- 23.Alkuraya FS. Autozygome decoded. Genet Med. 2010;12(12):765-771. doi: 10.1097/GIM.0b013e3181fbfcc4 [DOI] [PubMed] [Google Scholar]
- 24.Salman DO, Mahfouz R, Bitar ER, Samaha J, Karam PE. Challenges of genetic diagnosis of inborn errors of metabolism in a major tertiary care center in Lebanon. Front Genet. 2022;13:1029947. doi: 10.3389/fgene.2022.1029947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abouelhoda M, Faquih T, El-Kalioby M, Alkuraya FS. Revisiting the morbid genome of Mendelian disorders. Genome Biol. 2016;17(1):235. doi: 10.1186/s13059-016-1102-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Templeton AR, Read B. Inbreeding: one word, several meanings, much confusion. In: Loeschcke V, Jain SK, Tomiuk J, ed. Conservation Genetics. Birkhäuser Basel; 1994:91-105. doi: 10.1007/978-3-0348-8510-2_9 [DOI] [PubMed] [Google Scholar]
- 27.Ceballos FC, Joshi PK, Clark DW, Ramsay M, Wilson JF. Runs of homozygosity: windows into population history and trait architecture. Nat Rev Genet. 2018;19(4):220-234. doi: 10.1038/nrg.2017.109 [DOI] [PubMed] [Google Scholar]
- 28.Laine-Menéndez S, Domínguez-González C, Blázquez A, et al. Preferent diaphragmatic involvement in TK2 deficiency: an autopsy case study. Int J Mol Sci. 2021;22(11):5598. doi: 10.3390/ijms22115598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Domínguez‐González C, Madruga‐Garrido M, Mavillard F, et al. Deoxynucleoside therapy for thymidine kinase 2–deficient myopathy. Ann Neurol. 2019;86(2):293-303. doi: 10.1002/ana.25506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dominguez-Gonzalez C, Badosa C, Madruga-Garrido M, et al. Growth Differentiation Factor 15 is a potential biomarker of therapeutic response for TK2 deficient myopathy. Sci Rep. 2020;10(1):10111. doi: 10.1038/s41598-020-66940-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Domínguez-González C, Fernández-Torrón R, Moore U, et al. Muscle MRI characteristic pattern for late-onset TK2 deficiency diagnosis. J Neurol. 2022;269(7):3550-3562. doi: 10.1007/s00415-021-10957-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical genetics and genomics and the association for molecular Pathology. Genet Med. 2015;17(5):405-424. doi: 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gandolfo LC, Bahlo M, Speed TP. Dating rare mutations from small samples with dense marker data. Genetics. 2014;197(4):1315-1327. doi: 10.1534/genetics.114.164616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pagnamenta AT, Kaiyrzhanov R, Zou Y, et al. An ancestral 10-bp repeat expansion in VWA1 causes recessive hereditary motor neuropathy. Brain. 2021;144(2):584-600. doi: 10.1093/brain/awaa420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Park YH, Remmers EF, Lee W, et al. Ancient familial Mediterranean fever mutations in human pyrin and resistance to Yersinia pestis. Nat Immunol. 2020;21(8):857-867. doi: 10.1038/s41590-020-0705-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Florian RT, Kraft F, Leitão E, et al. Unstable TTTTA/TTTCA expansions in MARCH6 are associated with familial adult myoclonic epilepsy type 3. Nat Commun. 2019;10(1):4919. doi: 10.1038/s41467-019-12763-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559-575. doi: 10.1086/519795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jacquard A. Inbreeding: one word, several meanings. Theor Popul Biol. 1975;7(3):338-363. doi: 10.1016/0040-5809(75)90024-6 [DOI] [PubMed] [Google Scholar]
- 39.Weir BS. Estimating F -statistics: a historical view. Philos Sci. 2012;79(5):637-643. doi: 10.1086/667904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wills C. Principles of population genetics, 4th edition. J Hered. 2007;98(4):382-382. doi: 10.1093/jhered/esm035 [DOI] [Google Scholar]
- 41.Nothnagel M, Lu TT, Kayser M, Krawczak M. Genomic and geographic distribution of SNP-defined runs of homozygosity in Europeans. Hum Mol Genet. 2010;19(15):2927-2935. doi: 10.1093/hmg/ddq198 [DOI] [PubMed] [Google Scholar]
- 42.Palamara PF, Lencz T, Darvasi A, Pe’er I. Length Distributions of Identity by descent reveal fine-scale demographic history. Am J Hum Genet. 2012;91(5):809-822. doi: 10.1016/j.ajhg.2012.08.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ceballos FC, Virseda-Berdices A, Resino S, et al. Metabolic profiling at COVID-19 onset shows disease severity and sex-specific dysregulation. Front Immunol. 2022;13:925558. doi: 10.3389/fimmu.2022.925558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ceballos FC, Hazelhurst S, Ramsay M. Runs of homozygosity in sub-Saharan African populations provide insights into complex demographic histories. Hum Genet. 2019;138(10):1123-1142. doi: 10.1007/s00439-019-02045-1 [DOI] [PubMed] [Google Scholar]
- 45.Wang L, Eriksson S. Mutational analyses of human thymidine kinase 2 reveal key residues in ATP-Mg2+ binding and catalysis. Nucleosides Nucleotides Nucleic Acids. 2022;41(3):264-272. doi: 10.1080/15257770.2021.2001005 [DOI] [PubMed] [Google Scholar]
- 46.Provine WB. Ernst Mayr: genetics and speciation. Genetics. 2004;167(3):1041-1046. doi: 10.1093/genetics/167.3.1041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Slatkin M. A population-genetic test of founder effects and implications for Ashkenazi Jewish diseases. Am J Hum Genet. 2004;75(2):282-293. doi: 10.1086/423146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bray SM, Mulle JG, Dodd AF, Pulver AE, Wooding S, Warren ST. Signatures of founder effects, admixture, and selection in the Ashkenazi Jewish population. Proc Natl Acad Sci USA. 2010;107(37):16222-16227. doi: 10.1073/pnas.1004381107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tyynismaa H, Sun R, Ahola-Erkkila S, et al. Thymidine kinase 2 mutations in autosomal recessive progressive external ophthalmoplegia with multiple mitochondrial DNA deletions. Hum Mol Genet. 2012;21(1):66-75. doi: 10.1093/hmg/ddr438 [DOI] [PubMed] [Google Scholar]
- 50.Urtizberea JA, Severa G, Malfatti E. Metabolic myopathies in the era of next-generation sequencing. Genes. 2023;14(5):954. doi: 10.3390/genes14050954 [DOI] [PMC free article] [PubMed] [Google Scholar]