

Abstract
Hearing loss is a clinically and genetically heterogeneous disorder, with over 148 genes and 170 loci associated with its pathogenesis. The spectrum and frequency of causal variants vary across different genetic ancestries and are more prevalent in populations that practice consanguineous marriages. Pakistan has a rich history of autosomal recessive gene discovery related to non‐syndromic hearing loss. Since the first linkage analysis with a Pakistani family that led to the mapping of the DFNB1 locus on chromosome 13, 51 genes associated with this disorder have been identified in this population. Among these, 13 of the most prevalent genes, namely CDH23, CIB2, CLDN14, GJB2, HGF, MARVELD2, MYO7A, MYO15A, MSRB3, OTOF, SLC26A4, TMC1 and TMPRSS3, account for more than half of all cases of profound hearing loss, while the prevalence of other genes is less than 2% individually. In this review, we discuss the most common autosomal recessive non‐syndromic hearing loss genes in Pakistani individuals as well as the genetic mapping and sequencing approaches used to discover them. Furthermore, we identified enriched gene ontology terms and common pathways involved in these 51 autosomal recessive non‐syndromic hearing loss genes to gain a better understanding of the underlying mechanisms. Establishing a molecular understanding of the disorder may aid in reducing its future prevalence by enabling timely diagnostics and genetic counselling, leading to more effective clinical management and treatments of hearing loss.
Keywords: genetic counselling, genetic epidemiology, genotype, non‐syndromic hearing loss, Pakistani population, phenotype
1. INTRODUCTION
Pakistan, home to a population of 232 million according to the February 2023 United Nations census, ranks as the fifth most populous country in the world. The country's population is dispersed across multiple provinces, each with unique ethnic and linguistic diversity backgrounds. Consanguineous marriage is prevalent in most communities, making Pakistan one of the countries with the highest consanguinity rates globally. Consequently, homozygous pathogenic alleles steadily accumulate, leading to an ultimate increase in the prevalence of autosomal recessive disorders such as congenital hearing loss (HL). Out of the estimated 14.5 million Pakistanis living with HL, about half have a genetic aetiology. 1 According to the World Health Organization, more than 1.5 billion people around the world are impacted by HL, and roughly 28% of them, or 430 million individuals, have a disabling level of HL. This is concerning because the number of people with disabling HL is projected to exceed 700 million by 2050 (https://www.who.int/health‐topics/hearing‐loss).
Although individuals from the Pakistani community represent a minority of patients tested within the Asian population, 2 their contributions have yielded valuable insights into the molecular genetics and biology of hearing and deafness that hold relevance across diverse global genetic backgrounds. While this review does not aim to provide an exhaustive catalogue of all the variants identified in the Pakistani population, its goal is to highlight the exciting discoveries that have enriched our current understanding of the genetic epidemiology of HL. Although many genes have been implicated in HL (with many more yet to be discovered) in the Pakistani population, a dozen or so genes appear to play a predominant role in its genetic makeup. In this review, we provide a brief overview of the clinical heterogeneity and history of HL gene discovery, analyse gene ontology (GO) terms and pathways associated with HL and expand upon the key genes that explain a significant proportion of HL in the Pakistani population.
2. CHARACTERISTICS AND CLINICAL CLASSIFICATIONS FOR HEARING LOSS
HL, the permanent loss of hearing ability, encompasses several key characteristics. 3 Firstly, the anatomical site of the cochlear defect is used to distinguish between conductive or sensorineural HL (SNHL), with the latter being most prevalent. 4 Conductive HL occurs due to abnormalities in the outer or middle ear that hinder sound conduction, while SNHL results from damage to the inner ear or auditory nerve. When both conductive and sensorineural components are present, mixed HL ensues. Additionally, abnormalities or malfunctions beyond the cochlea, such as in the eighth cranial nerve, auditory brainstem, or cortex, lead to central auditory impairment. 5 The second characteristic pertains to the age of onset relative to speech development. 3 Pre‐lingual HL manifests before the critical period for speech development, including congenital onset or HL already present at birth. Post‐lingual HL, on the other hand, includes individuals with onset following the critical period for speech acquisition, spanning from childhood to advanced age and includes age‐related HL. 3 The third feature relates to the degree of HL and the associated hearing thresholds used to define severity levels, which include mild (hearing threshold 20–40 dB), moderate (41–55 dB), moderately severe (56–70 dB), severe (71–90 dB) and profound (90 dB). 3 HL can affect low (<1000 Hz), mid (1000–2000 Hz) or high (>2000 Hz) frequencies. Classifying HL based on these factors is crucial for determining the most appropriate treatment and management strategies for each individual. The fourth attribute of HL concerns the presence or absence of progression, which is determined by serial audiometry. Although most late‐onset HL typically follows a progressive course, the majority of individuals with congenital HL have a non‐progressive form. HL can affect one or both ears, referred to as unilateral or bilateral, respectively. Asymmetric HL is another possibility where both ears are affected to differing degrees. 6 The sixth feature is whether HL is part of a syndrome that involves concomitant abnormalities in additional organ systems or is the sole presenting feature, as seen with non‐syndromic HL (NSHL), which is observed in the vast majority of congenital cases. 3
Inheritance patterns are another defining characteristic of HL. Most hereditary HL exhibits an autosomal recessive inheritance, accounting for around 75% of congenital cases, 7 while roughly 18%, 1%–3% and 1% of NSHL are caused by autosomal dominant, X‐linked and mitochondrial forms, respectively. 8 Those with autosomal recessive non‐syndromic hearing loss (ARNSHL) inherit either a single heterozygous variant from each asymptomatic parent, as observed for homozygous variants, or receive two distinct variants in the same gene, one from each parent, known as compound heterozygous variants. If both parents carry a single disease allele on the same gene, their offspring have a 25% chance of inheriting two disease alleles to develop HL, a 50% chance of inheriting one disease allele to be an asymptomatic carrier and a 25% chance of inheriting no disease alleles and being unaffected.
3. METHODS USED FOR IDENTIFICATION OF ARNSHL GENES
Over the past three decades, there has been significant progress in identifying genes and loci associated with HL in Pakistani families (see Table 1 and Figure 1), using a range of evolving technologies. One method used to locate Mendelian disease loci involves classic parametric linkage analysis, which utilizes genetic STRP markers or SNP maps along with information about mode of inheritance, penetrance and allele frequencies. 9 This approach is based on the principle that chromosomal regions near genetic variants segregate together during meiosis. By analysing nearby markers, a genomic locus carrying a causative mutation may be statistically linked to the disease. The strength of the linkage evidence (or lack thereof) is assessed using the estimation of a logarithm of the odds (LOD) score. A LOD score greater than 3.0 is considered significant for linkage. 10 Once one or more potentially significant linked intervals have been identified through linkage analysis, the next step involves candidate gene sequencing of all genes in the region. Thirty‐nine ARNSHL genes have been identified in the Pakistani population with the support of linkage analysis. Homozygosity mapping is considered the best method for uncovering chromosomal regions that may harbour a causal variant in consanguineous families. 11 This method involves examining genotypes from multiple affected and unaffected family members to identify the number and size of runs of homozygosity. This information can be used to narrow down the genomic regions that are likely to contain the disease‐causing variant. In genetically heterogeneous disorders, homozygosity mapping can reduce the need to sequence various candidate genes and instead focus on specific genomic regions. 12 Twelve ARNSHL genes have been identified by using homozygosity mapping.
TABLE 1.
Fifty‐one ARNSHL genes were found mutated in the Pakistani population in the last 25 years listed in chronological order.
| Gene symbol | DFN locus | Chr position | Gene name | MIM | Discovery method | Onset and severity | Reference |
|---|---|---|---|---|---|---|---|
| GJB2 | DFNB1 | 13q12.11 | Gap junction protein beta 2 | 220290 | LA | Prelingual, severe to profound | [13] |
| MYO15A | DFNB3 | 17q11.2 | Myosin XVA | 600316 | NGS | Congenital, severe to profound | [14] |
| CLDN14 | DFNB29 | 21q22.1 | Claudin 14 | 614035 | LA | Congenital, profound | [15] |
| TMIE | DFNB6 | 3p21.31 | Transmembrane inner ear | 600971 | LA | Prelingual, severe to profound | [16] |
| TMC1 | DFNB7/11 | 9q21.13 | Transmembrane channel like 1 | 600974 | LA | Prelingual, severe to profound, and vestibular dysfunction | [17] |
| USH1C | DFNB18 | 11p15.1 | Greb1‐like retinoic acid receptor coactivator | 602092 | LA | Prelingual, moderate to severe | [18] |
| PCDH15 | DFNB23 | 10q21.1 | Protocadherin‐related 15 | 609533 | LA | Prelingual, profound | [19] |
| TECTA | DFNB21 | 11q23.3 | Tectorin alpha | 603629 | LA | Prelingual, moderate to severe | [20] |
| SLC26A4 | DFNB4 | 7q22.3 | Solute carrier family 26, member 4 | 600791 | HM, LA | Prelingual, severe to profound | [21] |
| MYO6 | DFNB37 | 6q14.1 | Myosin VI | 607821 | LA | Congenital, profound | [22] |
| TMPRSS3 | DFNB8/10 | 21q22.3 | Transmembrane serine protease 3 | 614861 | LA | Congenital, profound | [23] |
| ESPN | DFNB36 | 1p36.3 | Espin | 609006 | HM, LA | Congenital, profound, with or without vestibular dysfunction | [24] |
| WHRN | DFNB31 | 9q32 | Cask‐interacting protein | 607084 | LA | Congenital, profound | [25] |
| TRIOBP | DFNB28 | 22q13.1 | TRIO and F‐actin‐binding protein | 609823 | LA | Congenital, profound | [26] |
| MARVELD2 | DFNB49 | 5q13.2 | MARVEL domain containing 2 | 610153 | LA | Prelingual, Moderate to severe | [27] |
| LHFPL5 | DFNB67 | 6p21.31 | LHFPL tetraspan subfamily member 5 | 610265 | LA, HM | Prelingual, severe to profound | [28] |
| RDX | DFNB24 | 11q22.3 | Radixin | 611022 | LA | Pre‐lingual, profound | [29] |
| ESRRB | DFNB35 | 14q24.3 | Oestrogen‐related receptor beta | 608565 | LA | Prelingual, severe to profound | [30] |
| LRTOMT | DFNB63 | 11q13.4 | Leucine‐rich transmembrane and O‐methyltransferase domain containing | 611451 | LA | Congenital, profound | [31] |
| OTOF | DFNB9 | 2p23.3 | Otoferlin | 601071 | LA | Prelingual, severe‐to‐profound | [32] |
| HGF | DFNB39 | 7q21.11 | Hepatocyte growth factor | 608265 | LA | Prelingual, severe to profound | [33] |
| BSND | DFNB73 | 1q32.3 | Barttin CLCNK type accessory subunit beta | 602522 | LA | Prelingual, profound | [34] |
| TPRN | DFNB79 | 9q34.3 | Taperin | 613307 | NGS / LA | Prelingual, severe to profound | [35] |
| GRXCR1 | DFNB25 | 4p13 | Glutaredoxin and cysteine‐rich domain containing 1 | 615837 | HM, LA | Congenital, moderate to profound | [36] |
| MSRB3 | DFNB74 | 12q14.3 | Methionine sulfoxide reductase B3 | 613718 | LA | Prelingual, severe to profound | [37] |
| GIPC3 | DFNB15/72/95 | 19p13.3 | GIPC PDZ domain containing family member 3 | 601869 | LA | Prelingual, mild to profound | [38] |
| ILDR1 | DFNB42 | 3q13.33 | Immunoglobulin‐like domain containing receptor 1 | 609646 | LA | Prelingual, profound | [39] |
| PJVK | DFNB59 | 2q31.2 | Pejvakin | 610220 | LA, HM | Prelingual, profound | [40] |
| CIB2 | DFNB48 | 15q25.1 | Calcium‐ and integrin‐binding family member 2 | 609439 | LA | [41] | |
| CDH23 | DFNB12 | 17q12 | Cadherin 23 | 601386 | ES | Congenital, Profound | [42] |
| KARS1 | DFNB89 | 16q23.1 | Lysyl‐tRNA synthetase 1 | 613916 | LA, ES | Prelingual, profound | [43] |
| OTOA | DFNB22 | 16p13.1‐q11.2 | Otoancorin | 607039 | HM | Congenital, moderate to severe | [44] |
| ELMOD3 | DFNB88 | 2p11.2 | ELMO domain containing 3 | 615429 | ES, HM | Prelingual, severe to profound | [45] |
| GRXCR2 | DFNB101 | 5q32 | Glutaredoxin and cysteine‐rich domain containing 2 | 615837 | LA, HM, ES | Congenital, severe to profound | [46] |
| TBC1D24 | DFNB86 | 16p13.3 | TBC1 domain family member 24 | 614617 | ES, LA | Prelingual, profound | [47] |
| ADCY1 | DFNB44 | 7p12.3 | Adenylate cyclase 1 | 610154 | LA | Congenital, Severe to profound | [48] |
| NARS2 | DFNB94 | 11q14.1 | Asparaginyl‐tRNA synthetase 2, mitochondrial | 618434 | ES, LA | Prelingual severe to profound | [49] |
| MET | DFNB97 | 7q31.2 | MET proto‐oncogene, receptor tyrosine kinase | 164860 | LA, HM, ES | Congenital, severe | [50] |
| S1PR2 | DFNB68 | 19p13.2 | Sphingosine‐1‐phosphate receptor 2 | 608565 | ES, LA, HM | Congenital, profound | [51] |
| EPS8L2 | DFNB102 | 11p15.5 | EPS8 like 2 | 617637 | ES, GS | Post lingual, severe | [52] |
| PDZD7 | DFNB57 | 10q24.31 | PDZ domain containing 7 | 618003 | ES | Congenital, severe to profound | [53] |
| PPIP5K2 | DFNB100 | 5q21.1 | Diphosphoinositol pentakisphosphate kinase 2 | 618422 | ES, HM, LA | Prelingual, severe to profound | [54] |
| GAB1 | DFNB26 | 4q31.21 | GRB2‐associated binding protein 1 | 605429 | LA, ES | Congenital, severe to profound | [55] |
| TMEM132E | DFNB99 | 17q12 | Transmembrane protein 132E | 618481 | ES | Prelingual, profound | [56] |
| LOXHD1 | DFNB77 | 18q21.1 | Lipoxygenase homology PLAT domains 1 | 613079 | ES | Congenital, profound | [57] |
| CABP2 | DFNB93 | 11q13.2 | Calcium‐binding protein 2 | 614899 | ES | Prelingual, moderate to severe | [58] |
| SERPINB6 | DFNB91 | 6p25.2 | Serpin family B member 6 | 613453 | ES | Congenital, profound | [59] |
| PTPRQ | DFNB84 | 12q21.31 | Protein tyrosine phosphatase receptor type Q | 613391 | ES, LA | Prelingual, severe to profound | [59] |
| SIX5 | ND | 19q13.32 | SIX homeobox 5 | 610896 | ES | Congenital and post‐lingual, severe to profound | [60] |
| STX4 | ND | 16p11.2 | Syntaxin 4 | 186591 | ES, HM, LA | Congenital, profound | [61] |
| GREB1L | ND | 18q11.1‐q11.2 | GREB1 Like Retinoic Acid Receptor Coactivator | 619274 | ES | Congenital, profound | [62] |
Abbreviations: ES, exome sequencing; GS, genome sequencing; HM, homozygosity mapping; LA, linkage analysis; ND, not defined; NGS, next‐generation sequencing.
FIGURE 1.
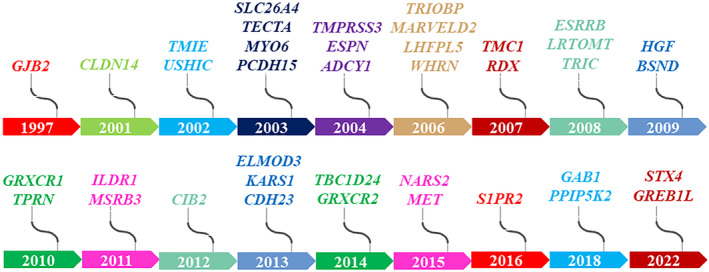
Timeline of 39 ARNSHL genes identified by collaborating with the Pakistani population over the past 25 years.
Gene discovery has been further revolutionized by advances in exome and genome sequencing. Exome sequencing targets the protein‐coding portion of genes, representing only about 1% (30 Mb) of the genome but capturing 85% of the disease‐causing mutations in Mendelian disorders. 63 Consequently, sequencing the entire exome has become a powerful tool for identifying disease‐associated variants, including monogenic disorders like HL as well as predisposing variants in common diseases. This has led to faster identification of such variants. 64 Genome sequencing provides a comprehensive view of an individual's entire genome, including both coding and non‐coding regions, allowing for the identification of both rare and common variants associated with HL. In recent years, genome sequencing has enabled the discovery of new HL genes, particularly when variants reside in non‐coding regions that were previously overlooked. 65 Using exome and genome sequencing, 22 gene variants in novel NSHL‐associated genes in the Pakistani population have been identified. Some of these 22 genes (12 genes) were also identified using other techniques described above such as linkage analysis and homozygosity mapping (as mentioned in Table 1). Moreover, it has facilitated the identification of pathogenic variants in known genes and enables the assessment of structural variations, such as copy number variations and structural rearrangements that can contribute to HL. As sequencing technologies continue to advance and become more cost‐effective, genome sequencing is likely to become an increasingly valuable tool for HL gene discovery and expedite the development of effective therapies.
In the Pakistani population, variants in a total of 51 genes have been implicated in ARNSHL through the approaches described above (Table 1). Owing to the unique genetic background and high consanguinity rate of the Pakistani population, 39 of these 51 genes have been discovered between 1997 and 2022 (Figure 1), underscoring the significant contribution of the Pakistani population to unravelling the genetics of HL.
Although next‐generation sequencing‐based molecular tests are still in their infancy, they have shown clinical value for single‐gene diseases. The full potential of exome and genome sequencing will enrich genomic medicine beyond rare single‐gene disorders. Further developments in next‐generation sequencing technologies and bioinformatics tools will enhance data analysis and clinical extraction. Recent studies justify the effort and expense of incorporating these innovative ideas into molecular diagnostics practice. While advances in next‐generation sequencing have sped up the detection of disease‐causing variants, they cannot overcome universal limitations faced in Mendelian disorders. Exploration of the genetics of ARNSHL is optimal with large pedigrees that ideally support gene mapping approaches and next‐generation sequencing. Furthermore, identification of variants in compound heterozygosity is critical for validating genetic findings. Variants with limited evidence to conclusively assign likely pathogenic or likely benign classifications remain a great challenge in genomic medicine in general. Variable expressivity can affect interpretation as well as outcomes, as not everyone who harbours particular variants will develop the disease due to possible incomplete penetrance. It is critical to use these technologies in collaboration with various patient populations, genetic counsellors, and medical geneticists. This collaborative endeavour will be crucial for increasing our understanding of the genetic basis of HL and discovering novel therapies to treat it.
4. GENE ONTOLOGIES AND PATHWAYS OF RECURRENT ARNSHL GENES IN PAKISTAN
By conducting molecular genetic studies on consanguineous pedigrees, researchers have been able to identify deleterious alleles in 51 out of the 68 genes linked to ARNSHL, thanks in part to participation of Pakistani families with HL. 66 Notably, among the 51 autosomal recessive genes we describe, nine genes (GJB2, TECTA, MYO6, ESPN, TMC1, TPRN, ELMOD3, TBC1D24 and PTPRQ) have been associated with an autosomal‐dominant inheritance pattern as well. While the identification of 51 genes associated with HL may seem daunting, researchers can simplify the complexity by examining the established metabolic and cell signalling pathways through which these genes interact in various organs or by identifying enriched ontology clusters. One of the fundamental presumptions of network analysis and ontology clustering is that there are functional connections between genes whose malfunction leads to disease manifestation. Understanding biological systems, disease states and the mechanisms by which drugs affect them relies on identifying these common pathways and ontology terms. Therefore, by recognizing the shared biological pathways involved in HL and other disorders, researchers can gain insights into the underlying causes of these diseases and develop effective therapeutic interventions.
To gain further insights into the functional roles of the 51 ARNSHL‐associated genes, gene enrichment analysis and MCODE algorithm were employed using Metascape. 67 The annotation, enrichment and genes used to perform the Metascape analysis are shown in Table S1. Enrichment analysis using Metascape identified four areas of enriched protein clusters where proteins are closely related based on similar pathways involved. The first cluster of genes was involved in sensory perception of sound (27 genes, GO:0007605), the second cluster in sensory processing of sound by cochlear inner hair cells (IHCs) (19 genes, R‐HSA‐9662360), the third cluster in actin cytoskeleton organization (7 genes, GO:0030036), and the fourth cluster in retina homeostasis (6 genes, GO:0001895) (Figure 2). To facilitate the visualization of the results, we created a pie chart using Microsoft Excel software to illustrate the count of genes belonging to each category of enriched GO terms.
FIGURE 2.
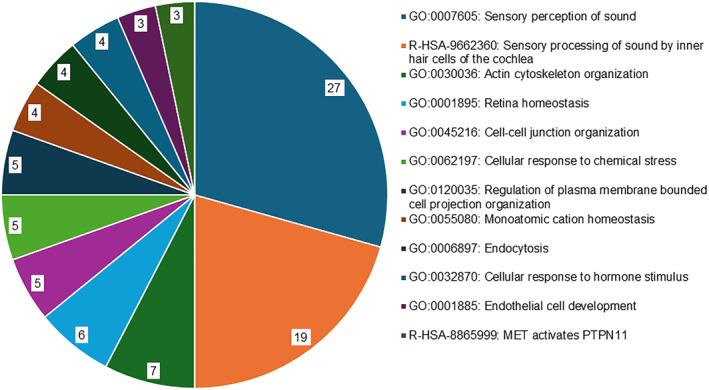
Pie chart showing the results of Metascape analysis of enriched gene ontology (GO) clusters. The chart displays the top 12 clusters and their representative enriched terms (one per cluster). The ‘Count’ refers to the number of genes in our list of 51 genes associated with ARNSHL that are included in the given ontology term. To classify the genes associated with ARNSHL according to their function, we performed pathway and process enrichment and network analysis using Metascape (http://metascape.org). Using this approach, we identified the enrichment of GO terms and genes related to various biological pathways.
Protein clustering also revealed that the 51 genes associated with ARNSHL regulate several common biological pathways. These pathways include cell–cell junction organization (5 genes, GO:0045216), cellular response to chemical stress (5 genes, GO:0062197) and regulation of plasma membrane‐bounded cell projection organization (5 genes, GO:0120035). Other biological processes involved were monoatomic cation homeostasis (4 genes, GO:0055080), endocytosis (4 genes, GO:0006897), cellular response to hormone stimulus (4 genes, GO:0032870), endothelial cell development (3 genes, GO:0001885) and MET activates PTPN11 (3 genes, R‐HSA‐8865999) (Figure 2). ‘MET activates PTPN11’ refers to a signalling pathway that involves the interaction of two proteins of MET and PTPN11. This pathway has been implicated in a variety of cellular processes and diseases, including cancer and developmental disorders.
Overall, our bioinformatics analysis highlights the intricate role of ARNSHL genes in various cellular signalling pathways and biological processes. These findings have important implications for understanding the underlying mechanisms of ARNSHL and for the development of targeted therapies aimed at restoring normal function to disrupted pathways.
5. RECURRENT GENES AND VARIANTS ASSOCIATED WITH HEARING LOSS IN THE PAKISTANI POPULATION
Despite the extensive genetic heterogeneity in ARNSHL, a few genes account for over half of the cases. One large cohort study of Pakistani patients identified variants in six genes (GJB2, HGF, MYO7A, SLC26A4, CIB2 and TMC1) that collectively explained up to 57% of recessively inherited severe to profound HL. 66 Another cohort study found pathogenic or possibly deleterious mutations in GJB2, MYO7A, CDH23 and MYO15A in 13 out of 21 (61.9%) consanguineous Pakistani families with HL. 68 This part of the review aims to shed light on the most prevalent genes and variants in the Pakistani population. By providing valuable insights into the molecular genetics and epidemiology of ARNSHL, this information can be utilized to develop effective genetic screening and counselling programs for individuals and families affected by this condition and also develop personalized treatment strategies for the affected individuals.
5.1. CDH23
CDH23 (DFNB12, USH1D) encodes cadherin 23, a crucial component of the extracellular filaments responsible for formation and function of the mechanosensory hair bundles of the inner ear. 69 The hair bundle consists of stereocilia, hair‐like projections that respond to vibration of the basilar membrane and stimulate hair cells by depolarization. 70 CDH23 is expressed not only in the cochlear hair cells but also in the vestibular hair cells and the photoreceptor layer of the retina. 71 In fact, CDH23 is one of the most highly expressed genes in the vestibular hair cells, where it plays a crucial role in maintaining their mechanical and electrophysiological properties. Different isoforms of CDH23 have tissue‐specific functions. Biallelic variants disrupting CDH23 cause ARNSHL (DFNB12) and Usher syndrome type I (USH1D), characterized by congenital SNHL, vestibular dysfunction and early onset retinitis pigmentosa. 72 Although CDH23‐associated NSHL has traditionally been associated with congenital onset and profound severity, recent studies have revealed a broad phenotypic spectrum that includes individuals with biallelic variants presenting HL in the second to seventh decade of life, expanding the understanding of DFNB12. 73 As one of the most common genetic causes of HL, many CDH23 pathogenic alleles have been identified in affected individuals of Pakistani ancestry, contributing to up to 5% of NSHL and USH1D 1 due to numerous pathogenic alleles of CDH23. 66 The c.2968G>A, p.(Asp990Asn) and c.6133G>A, p.(Asp2045Asn) alleles are major causes of CDH23‐associated deafness in Pakistan but the most commonly occurring variant of these genes is c.6050‐9G>A (splicing). 66 , 74 At least 28 additional CDH23 variants have been identified in different ethnic groups in Pakistan. However, so far no experimental evidence shows an impact on protein function. 66 , 74 , 75 A comprehensive understanding of disease mechanisms in CDH23‐associated disorders may significantly impact the diagnosis and management of HL and deaf‐blindness worldwide.
5.2. CIB2
Calcium‐ and integrin‐binding protein 2 (CIB2) is a crucial component of the mechano‐transduction (MET) process in cochlear hair cells, demonstrated by the complete abolishment of MET currents upon genetic disruption. 76 , 77 MET is the process by which sound vibrations are converted into electrical signals that the brain can interpret as sound. CIB2 is an elongation factor‐hand domain‐containing protein that binds Ca2+ ions and has been associated with various functions, including integrin signalling in platelets and skeletal muscle and autophagy, indicating its considerable functional flexibility. 78 Although its clinical presentation seems to be limited to hearing deficits, typically manifesting as bilateral, profound, pre‐lingual HL, CIB2 has been linked to a wide range of functions beyond hearing physiology. A brief association with Usher syndrome was eventually refuted. 41 , 79 A large Pakistani cohort study has estimated the prevalence of CIB2‐associated HL as 8.6%. 66 The homozygous recurrent c.272T>C, p.Phe91Ser (NM_006383.3) variant is the major cause of NSHL in the Pakistani community and has been identified in 81 families. 41 Two additional rare variants have been identified in affected Pakistani individuals: c.192G>C, p.Glu64Asp and c.297C>G, p.Cys99Trp. 41
5.3. CLDN14
CLDN14, also known as DFNB29, encodes the claudin‐14 protein, an essential membrane protein forming tight junctions in the inner ear. 15 These tight junctions are crucial for compartmentalizing the endolymphatic and perilymphatic fluid compartments, maintaining cell polarity, and regulating intercellular permeability to solutes, ions, and water. 80 CLDN14 is expressed in the inner and outer hair cells, supporting cells, and Reissner's membrane. 81 It plays a crucial role in maintaining ion homeostasis and calcium levels in the endolymph and perilymph fluids, which is essential for the MET process of cochlear hair cells. 82 MET is initiated by the opening of cation channels near the tip of the stereocilia and requires ion homeostasis to maintain ion gradients for preservation of the endocochlear potential. 80 While the endocochlear potential in Cldn14‐null mice has been determined as normal, rapid degeneration of outer hair cells and progressive slower degeneration of IHCs is thought to be due to the compromised tight junction barrier. 83 In Pakistani families, seven different variants of CLDN14 are responsible for 2%–3.3% of cases of profound deafness or moderate to severe HL 66 , 84 with the most prevalent variant being c.254T>A, p.Val85Asp (NM_001146079.2) identified in 21 families. 85 It has been shown that a founder effect contributes to the recurrence of this variant. 85 A modifier of CLDN14‐associated HL has been proposed but remains uncharacterized. The discovery of CLDN14's role in regulating intercellular permeability in the inner ear highlights the importance of genetic testing for CLDN14 in individuals with HL in the moderate range.
5.4. GJB2
Gap junctions play a crucial role in maintaining cochlear potassium homeostasis, which is essential for hearing. GJB2 encodes connexin 26 and is one of the most common causes of HL worldwide. 86 Connexin 26 oligomerizes to form hexameric hemichannels called ‘connexons’ in the plasma membrane. The connexons of neighbouring cells come together to form gap junctions, which facilitate intercellular communication. 87 Gap junctions can be homomeric or heteromeric, depending on whether they are made up of one or many different connexin proteins. This affects their ability to selectively permeate certain molecules and ions. 88 Gap junctions allow ions, nutrients and signalling molecules with molecular weights up to 1200 Da to pass between cells. 89 This intercellular communication is critical for the normal growth, function, and repair of the sensory epithelia of the inner ear. These processes that are disrupted through genetic mutation result in eventual cell death and HL. In the Pakistani population, the prevalence of GJB2‐associated HL ranges from 6.1% to 53% 90 and it is the most common cause of NSHL in South Asia. 66 Patients with biallelic pathogenic variants typically have congenital‐onset HL, ranging from mild to profound severity, depending on the variant, and it can be progressive in roughly half of patients. 86 Many pathogenic variants have been identified in GJB2, with the most common ones in the Pakistani population being c.231G>A, p.Trp77Ter, c.71G>A, p.Trp24Ter and c.35delG, p.Gly12ValfsTer2, (NM_004004.5). 1 , 7 , 68 , 91 Among these variants, individuals homozygous for the c.35delG variant exhibit extreme phenotypic heterogeneity worldwide, ranging from mild HL to profound deafness. A founder effect has been attributed to its high prevalence. The GJB2 c.35delG variant causes a premature stop codon in exon 2 out of 2 total exons, leading to a loss of protein function. 92 In Pakistan, except for one variant associated with moderate–to‐severe HL, all others are associated with severe to profound deafness. 66 To date, 29 pathogenic variants of GJB2 have been identified in the Pakistani population. 66
5.5. HGF
HGF (DFNB39) encodes hepatocyte growth factor (HGF), which plays a crucial role in various biological processes, including cell growth, survival, differentiation and branching morphogenesis, with implications for neuronal survival and differentiation. 93 HGF dosage in the inner ear must be precisely calibrated for normal hearing. Transgenic mouse models of Hgf overexpression and deficiency present deafness resulting from the failure of neural crest cell migration to the intermediate cell layer of the stria vascularis, which causes thinning, reducing the endocochlear potential and hair cell loss. 33 , 93 While HGF‐associated HL is relatively prevalent in the Pakistani population, explaining 6–8% of severe to profound pre‐lingual HL, it is rarely implicated in the diagnosis of individuals with HL in other populations. 66 In the Pakistani population, three HGF variants have been identified, including a synonymous variant that affects splicing and two deletions in a highly conserved region of intron 4, which is part of the 3′ untranslated region of a short HGF isoform. 75 The most common variant in the Pakistani population is c.482+1986_1988delTGA (NM_000601.6) in intron 4. 33 Mice with the 10 bp deletion corresponding to c.482+1991_2000delGATGATGAAA in human intron 4 developed defects in the stria vascularis due to failure of neural crest cell migration during development, resulting in a significantly reduced endocochlear potential. 93 HGF activates the MET receptor‐mediated signalling pathway and mediates diverse downstream pathways, including those involved in the epithelial‐mesenchymal transition and the development of neural crest‐derived lineages. Overall, deleterious variants in HGF can result in impaired cell migration and the development of neural crest‐derived structures in the inner ear, leading to a reduced endocochlear potential and HL.
5.6. MARVELD2
MARVELD2 (DFNB49) encodes a member of the marvel protein family called tricellulin that is concentrated at the tight junctions, forming part of the continuous intercellular barrier between epithelial cells. Tight junctions are multi‐protein complexes preventing leakage of solutes and water, acting as a seal between epithelial cells. 94 Tricellular tight junctions are present in epithelial cells between supporting and hair cells, cochlear supporting cells and marginal cells of the stria vascularis. 27 Although MARVELD2 is ubiquitously expressed in epithelial junctions, only the inner ear appears to be affected by the genetic disruption, suggesting that genetic compensation may be present in other organ systems that are absent in the inner ear. 27 Biallelic variants in the Occludin‐ELL domain of MARVELD2 have been shown to cause HL in multiple Pakistani families. 95 These variants explain between 1.5% and 2.4% of all HL in Pakistan to a moderate to profound degree. The most recurrent variant of MARVELD2 is c.1331+2T>C (NM_001038603.3), and at least seven additional rare disease‐associated variants have also been reported in the Pakistani population. 1 Tricellulin is essential for maintaining the integrity and stability of epithelial cells and their junctions. 27 The occluding‐ELL domain of MARVELD2 may be necessary for the maintenance of tricellular junctions and the proper functioning of the inner ear. 96 Deleterious variants in tricellulin lead to tight junction disorganization, resulting in severe to profound sensorineural HL. 27
5.7. MYO7A
MYO7A (DFNB2) encodes an unconventional myosin that plays a crucial role in maintaining the mechanical stability of the hair bundle. The hair bundle is the sensory structure located on the surface of hair cells in the inner ear, acting as a mechanotransducer that transforms sound waves or orientation information into electrical signals, which the brain interprets (Houdusse & Titus, 2021). 97 This is achieved through the transportation of extracellular stereocilia links along actin filaments 98 at the upper tip‐link density and ankle link region of the stereocilia by MYO7A, which is essential for the cohesion of the hair bundle. 99 MYO7A co‐localizes with several other proteins at the upper tip‐link density and ankle link region of the stereocilia, including CDH23, USH1C, USH1G, ADGRV1 and USH2A, which are integral for the proper functioning of the hair bundle (Houdusse & Titus, 2021). 97 Moreover, independent of molecular trafficking, MYO7A exerts force at the upper tip‐link density region and tensions the MET complex, further highlighting its important role in hearing. 100 In addition to its role in the inner ear, MYO7A is also expressed in retinal pigment epithelial cells where it is required for functional RPE65, a key protein in the retinoid cycle. 101 MYO7A is also associated with Usher syndrome type I, an autosomal recessive condition characterized by deafness, vestibular impairment and retinitis pigmentosa. Biallelic variants in MYO7A account for 29% to 50% of all USH1 cases globally, making it the most common cause of this condition. 102 Moreover, at least 11% of moderate to severe pre‐lingual SNHL in the Pakistani population is due to MYO7A disruption 42 with at least 59 variants reported. 66 Among these variants, c.397dupC, p.His133ProfsTer7 and c.470G>A, p.Ser157Asn (NM_000260.3) are the most prevalent in the Pakistani population. 68 These variants exhibit founder effects that are not seen in other populations. 68
5.8. MYO15A
Myosin XVa, a product of MYO15A, belongs to the unconventional myosin superfamily and is essential for the elongation of stereocilia in cochlear sensory and vestibular hair cells. The growth of the hair bundle is facilitated by the transportation of whirlin and Eps8 to the tip of stereocilia to form the stereocilia tip complex by Myosin XVa, which can help in the conversion of microvilli into fully mature stereocilia. 103 MYO15A is involved in the regulation of actin and the transportation of elongation complexes at the distal stereocilia tip as well as other cargoes for actin. 99 Notably, the motor and tail domains of myosin XVa have been identified as crucial for normal auditory structure and function. 104 In Pakistani families, at least 49 MYO15A variants have been linked to 5–13% of progressive severe to profound bilateral SNHL. 66 The majority of the variants in this gene have been found in affected individuals from one to three Pakistani families, except for two variants, c.6589C>T, p.Gln2197Ter and c.8158G>C, p.Asp2720His (NM_016239.3) found in deaf members of four families each. 59 MYO15A variants significantly impact the motor domain, leading to dysfunction causing shorter stereocilia with an ectopic staircase structure, a condition associated with severe deafness. 105
5.9. MSRB3
MSRB3 (DFNB74) encodes methionine sulfoxide reductase B3 (MSRB3) that plays a critical role in repairing oxidatively damaged proteins by catalysing the stereo‐specific reduction of methionine‐R‐sulfoxides to methionine. 37 Msrb3 localizes to the base of the stereocilia on the apical surface of hair cells. 106 Studies on the Msrb3 −/− mouse model have shown that MSRB3 is essential for the maturation and/or maintenance of stereociliary bundles since these mice exhibit progressive degeneration of the stereociliary bundles and apoptosis of hair cells. 106 Moreover, genetic disruption of Msrb3 is more likely to be due to degeneration rather than abnormal development, since the hair cells of KO mice develop normally and have functional mechanotransduction channels until at least P3. 106 The c.265T>G, p.Cys89Gly and c.55C>T, p.Arg19Ter (NM_001031679.2) MSRB3 variants are associated with deafness in six and two unrelated DFNB74 Pakistani families, respectively. 37 A homozygous missense variant c.20T>G, p.Leu7Arg was identified in 2014 in one Pakistani family, and a homozygous splice variant, c.412‐1G>A was described in another Pakistani family in 2019. 66 These variants are associated with severe to profound HL. Among the four variants reported in individuals with genetic ancestry from Pakistan so far, p.Cys89Gly is the most recurrent variant. This MSRB3 cysteine residue, conserved in orthologs from yeast to humans, is involved in structural zinc binding. In vitro, this non‐synonymous substitution (p.Cys89Gly) reduced zinc binding and MSRB3 enzymatic activity. 37
5.10. OTOF
OTOF (DFNB9) encodes otoferlin, a protein that is essential for normal hearing. Otoferlin plays a crucial role in various functions of synaptic signalling, including sensing of pre‐synaptic Ca+2 for exocytosis following IHC depolarization, priming and replenishment of the synaptic vesicles to maintain fast neurotransmitter release and coupling of exocytosis‐endocytosis. 107 , 108 Most individuals exhibit profound pre‐lingual deafness due to biallelic pathogenic or likely pathogenic OTOF variants. The affected individuals show isolated failure in synaptic transmission, and their otoacoustic emissions are usually initially unaffected, with healthy outer hair cells, particularly in younger individuals. The HL resulting from OTOF‐associated deficits is commonly known as auditory synaptopathy. 109 , 110 OTOF variants account for 3.1%–4% of pre‐lingual moderate or profound HL in Pakistan. The first variant described, a nonsense variant c.4491T>A, p.Tyr1497Ter (NM_001287489.2) in OTOF, was identified in four independent Lebanese families by a candidate gene approach. 111 Since then, at least 23 OTOF variants associated with moderate–to‐severe or profound HL have been identified in hearing‐impaired individuals from Pakistan. 32 , 84 Among these, one of the most common is the c.2122C>T, p.Arg708Ter 66 variant. 112 This variant has been detected in both homozygous and compound heterozygous states in individuals with prelingual NSHL or auditory neuropathy in a large population cohort. 32
5.11. SLC26A4
SLC26A4, also known as pendrin, is a protein that belongs to the solute carrier family 26 and functions as an anion exchanger, transporting negatively charged ions such as chloride, iodide and bicarbonate across cell membranes. This protein is crucial for the development of the cochlea and vestibular duct's bony snail shape structure. 113 Variants in SLC26A4 (DFNB4) have been linked to both ARNSHL and Pendred syndrome. 114 While individuals with isolated HL may experience bilateral, profound SNHL, those with Pendred syndrome may have concomitant HL, enlarged vestibular aqueduct and abnormal iodine organification. 115 Approximately 12.4% of HL cases in the Pakistani community can be attributed to SLC26A4. 66 While the majority of the identified variants are described as non‐syndromic, especially in young individuals, the presence of Pendred syndrome may be frequently overlooked. SLC26A4 has several recurring variations. In the majority of affected individuals, HL is associated with three variants: c.269C>T, p.(Ser90Leu), c.716 T > A, p.(Val239Asp), and c.1337A > G, p.(Gln446Arg) (NM_000441.1). A founder effect has been shown for their recurrence. 84 , 116
5.12. TMC1
Transmembrane channel‐like protein isoform‐1 (TMC1) plays a crucial role in the auditory system as it forms an ion‐conducting pore of the MET channel in auditory hair cells. 117 Over the past 5 years, TMC1 has emerged as a leading contender for the MET channel in auditory hair cells of the inner ear. Hair cells convert acoustic and vestibular stimuli into electrical responses through the activation of MET. 118 TMC1 is thought to have a six‐transmembrane domain structure similar to several other ion‐channel subunits and is transported to the tips of the stereocilia in the sensory hair bundle where the MET channel is located. 119 TMC1 variants associated with human deafness result in loss of typical MET currents and hair cell senescence, causing cell death. Valuable insights into the pathogenesis of TMC1‐associated deafness have been gained from studies of mutant mice. 120 Variants in TMC1 cause both dominant (DFNA36) and recessive (DFNB7/11) forms of NSHL. Both progressive postlingual and pre‐lingual profound HL have been associated with TMC1 variants. In Pakistan, at least 32 different TMC1 variants account for 6.4% of recessively inherited HL cases. 66 The most frequently occurring TMC1 variant in the Pakistani population is c.100C>T, p.Arg34Ter (NM_138691.2), which has been reported in two studies. 84 , 121 This common pathogenic nonsense variant is a likely founder mutation in North African and Middle Eastern populations. 7 , 17
5.13. TMPRSS3
Genetic mutation of type II transmembrane serine protease (TMPRSS3) causes variable HL. TMPRSS3 has four functional domains that include an N‐terminal transmembrane domain, a low‐density lipoprotein receptor A domain, a scavenger receptor cysteine‐rich domain and a C‐terminal serine protease domain. 122 TMPRSS3 is expressed in various components of the developing inner ear, such as the stria vascularis, spiral ganglion neurons, 123 IHCs and cochlear aqueduct of the foetal cochlea and is critical for their normal development and maintenance 124 Additionally, cytoplasmic domains of TMPRSS3 raise the possibility that they may participate in intracellular signal transduction. 122 Biallelic variants in TMPRSS3 are known for causing different types of HL with variable onset. DFNB10 is characterized by congenital or childhood‐onset bilateral profound HL, 125 whereas DFNB8 is associated with a milder postlingual progressive HL, 126 both of which are caused by variants in TMPRSS3 (DFNB8/10). In Pakistan, TMPRSS3 variants are associated with stable, moderate–to‐severe or profound HL and contribute up to 4% of the prevalence of HL. 66 Other frequently occurring variants include c.323‐6G>A (splicing) and c.1219T>C, p.Cys407Arg. 84 The recurring homozygous c.1219T>C, p.Cys407Arg (NM_024022.2) variant has been identified in 20 Pakistani families. 127 Functional studies show that the variant protein has defective protease activity compared to the wild‐type (Lee et al., 2003) as well as a failure to undergo proteolytic cleavage and activate the epithelial sodium channel (ENaC) in vitro. 124 , 128 In silico analysis also supports that this missense variant has a negative effect on protein structure/function. There are 14 other rare variants in this gene that contribute to NSHL in the Pakistani population.
6. CONCLUSION
This review explores the clinical and genetic complexities of HL, defining and describing key characteristics, terminology and genetics. After 25 years of engaging the Pakistani population, researchers have uncovered variants in 51 genes and made significant progress in understanding the most frequently implicated ARNSHL genes. Of these, 39 genes causally related to HL were discovered through the use of gene mapping methodologies and sequencing strategies in consanguineous Pakistani families. This aggregated knowledge has established 13 being the most commonly involved in the molecular diagnosis of Pakistani patients (CDH23, CIB2, CLDN14, GJB2, HGF, MARVELD2, MYO7A, MYO15A, MSRB3, OTOF, SLC26A4, TMC1 and TMPRSS3). We detailed their purpose and highlighted important variants from the Pakistani community's standpoint. For a detailed molecular understanding of these ARNSHL genes, we also categorized enriched GO terms and shared pathways using Metascape.
Engaging the Pakistani community has been fundamental in advancing gene discovery. The high prevalence of consanguinity and congenital HL, a predominantly recessive trait, has provided fundamental insights into the genes and variants underlying HL over several decades. This has amplified the global knowledge base, providing valuable information for the selection of therapeutic targets and improving genetic diagnoses. However, HL continues to impose a significant burden on affected individuals, necessitating the discovery of new strategies for more precise diagnosis, alleviation, and treatment. Much work remains to achieve a comprehensive understanding of all genes and variants causing HL.
AUTHOR CONTRIBUTIONS
Madiha Shadab: Conceptualization (equal); data curation (equal); investigation (equal); methodology (equal); project administration (equal); resources (equal); validation (equal); visualization (equal); writing – original draft (equal); writing – review and editing (equal). Ansar Ahmed Abbasi: Methodology (equal); project administration (equal); supervision (equal); writing – review and editing (equal). Ahsan Ejaz: Data curation (equal); resources (equal). Afif Ben‐Mahmoud: Methodology (equal); writing – review and editing (equal). Vijay Gupta: Formal analysis (equal); writing – review and editing (equal). Hyung‐Goo Kim: Funding acquisition (equal); methodology (equal); supervision (equal); visualization (equal); writing – review and editing (equal). Barbara Vona: Conceptualization (equal); funding acquisition (equal); investigation (equal); methodology (equal); project administration (equal); validation (equal); writing – original draft (equal); writing – review and editing (equal).
FUNDING INFORMATION
Barbara Vona is funded by the German Research Foundation DFG VO 2138/7‐1 grant 469177153. Hyung‐Goo Kim is funded by IGP5 funding from the Qatar Biomedical Research Institute at Hamad Bin Khalifa University.
CONFLICT OF INTEREST STATEMENT
The authors confirm that there are no conflicts of interest.
Supporting information
Table S1.
ACKNOWLEDGMENTS
We acknowledge support by the Open Access Publication Funds of the University of Göttingen. Open Access funding enabled and organized by Projekt DEAL.
Shadab M, Abbasi AA, Ejaz A, et al. Autosomal recessive non‐syndromic hearing loss genes in Pakistan during the previous three decades. J Cell Mol Med. 2024;28:e18119. doi: 10.1111/jcmm.18119
Contributor Information
Madiha Shadab, Email: madiha.shadab@hotmail.com.
Barbara Vona, Email: barbara.vona@med.uni-goettingen.de.
DATA AVAILABILITY STATEMENT
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.
REFERENCES
- 1. Naz S. Molecular genetic landscape of hereditary hearing loss in Pakistan. Hum Genet. 2022;141(3–4):633‐648. doi: 10.1007/s00439-021-02320-0 [DOI] [PubMed] [Google Scholar]
- 2. Rouse SL, Florentine MM, Taketa E, Chan DK. Racial and ethnic disparities in genetic testing for hearing loss: a systematic review and synthesis. Hum Genet. 2022;141(3–4):485‐494. doi: 10.1007/s00439-021-02335-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hoefsloot LH, Feenstra I, Kunst HPM, Kremer H. Genotype phenotype correlations for hearing impairment: approaches to management: genotype phenotype correlations for hearing impairment. Clin Genet. 2014;85(6):514‐523. doi: 10.1111/cge.12339 [DOI] [PubMed] [Google Scholar]
- 4. Stelma F, Bhutta MF. Non‐syndromic hereditary sensorineural hearing loss: review of the genes involved. J Laryngol Otol. 2014;128(1):13‐21. doi: 10.1017/S0022215113003265 [DOI] [PubMed] [Google Scholar]
- 5. Sanders RD, Gillig PM. Cranial nerve VIII: hearing and vestibular functions. Psychiatry. 2010;7(3):17‐22. [PMC free article] [PubMed] [Google Scholar]
- 6. Cai Y, Zhao F, Chen Y, et al. The effect of symmetrical and asymmetrical hearing impairment on music quality perception. Eur Arch Otorhinolaryngol. 2016;273(9):2451‐2459. doi: 10.1007/s00405-015-3838-8 [DOI] [PubMed] [Google Scholar]
- 7. Shafique S, Siddiqi S, Schraders M, et al. Genetic spectrum of autosomal recessive non‐syndromic hearing loss in Pakistani families. PLoS ONE. 2014;9(6):e100146. doi: 10.1371/journal.pone.0100146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bayazit YA, Yılmaz M. An overview of hereditary hearing loss. ORL J Otorhinolaryngol Relat Spec. 2006;68(2):57‐63. doi: 10.1159/000091090 [DOI] [PubMed] [Google Scholar]
- 9. Vink JM, Boomsma DI. Gene finding strategies. Biol Psychol. 2002;61(1–2):53‐71. doi: 10.1016/S0301-0511(02)00052-2 [DOI] [PubMed] [Google Scholar]
- 10. Génin E, Martinez M, Clerget‐Darpoux F. Posterior probability of linkage and maximal lod score. Ann Hum Genet. 1995;59(1):123‐132. doi: 10.1111/j.1469-1809.1995.tb01610.x [DOI] [PubMed] [Google Scholar]
- 11. Lander ES, Botstein D. Homozygosity mapping: a way to map human recessive traits with the DNA of inbred children. Science. 1987;236(4808):1567‐1570. doi: 10.1126/science.2884728 [DOI] [PubMed] [Google Scholar]
- 12. Alkuraya FS. Homozygosity mapping: one more tool in the clinical geneticist's toolbox. Genet Med. 2010;12(4):236‐239. doi: 10.1097/GIM.0b013e3181ceb95d [DOI] [PubMed] [Google Scholar]
- 13. Kelsell DP, Dunlop J, Stevens HP, et al. Connexin 26 mutations in hereditary non‐syndromic sensorineural deafness. Nature. 1997;387(6628):80‐83. doi: 10.1038/387080a0 [DOI] [PubMed] [Google Scholar]
- 14. Liburd N, Ghosh M, Riazuddin S, et al. Novel mutations of MYO15A associated with profound deafness in consanguineous families and moderately severe hearing loss in a patient with Smith‐Magenis syndrome. Hum Genet. 2001;109(5):535‐541. doi: 10.1007/s004390100604 [DOI] [PubMed] [Google Scholar]
- 15. Wilcox ER, Burton QL, Naz S, et al. Mutations in the gene encoding tight junction claudin‐14 cause autosomal recessive deafness DFNB29. Cell. 2001;104(1):165‐172. doi: 10.1016/s0092-8674(01)00200-8 [DOI] [PubMed] [Google Scholar]
- 16. Naz S, Giguere CM, Kohrman DC, et al. Mutations in a novel gene, TMIE, are associated with hearing loss linked to the DFNB6 locus. Am J Hum Genet. 2002;71(3):632‐636. doi: 10.1086/342193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kurima K, Peters LM, Yang Y, et al. Dominant and recessive deafness caused by mutations of a novel gene, TMC1, required for cochlear hair‐cell function. Nat Genet. 2002;30(3):277‐284. doi: 10.1038/ng842 [DOI] [PubMed] [Google Scholar]
- 18. Ahmed ZM, Smith TN, Riazuddin S, et al. Nonsyndromic recessive deafness DFNB18 and usher syndrome type IC are allelic mutations of USHIC. Hum Genet. 2002;110(6):527‐531. doi: 10.1007/s00439-002-0732-4 [DOI] [PubMed] [Google Scholar]
- 19. Ahmed ZM, Riazuddin S, Ahmad J, et al. PCDH15 is expressed in the neurosensory epithelium of the eye and ear and mutant alleles are responsible for both USH1F and DFNB23. Hum Mol Genet. 2003b;12(24):3215‐3223. doi: 10.1093/hmg/ddg358 [DOI] [PubMed] [Google Scholar]
- 20. Naz S. Distinctive audiometric profile associated with DFNB21 alleles of TECTA. J Med Genet. 2003;40(5):360‐363. doi: 10.1136/jmg.40.5.360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Park H‐J. Origins and frequencies of SLC26A4 (PDS) mutations in east and south Asians: global implications for the epidemiology of deafness. J Med Genet. 2003;40(4):242‐248. doi: 10.1136/jmg.40.4.242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ahmed ZM, Morell RJ, Riazuddin S, et al. Mutations of MYO6 are associated with recessive deafness, DFNB37. Am J Hum Genet. 2003a;72(5):1315‐1322. doi: 10.1086/375122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ahmed ZM, Cindy Li X, Powell SD, et al. Characterization of a new full length TMPRSS3 isoform and identification of mutant alleles responsible for nonsyndromic recessive deafness in Newfoundland and Pakistan. BMC Med Genet. 2004;5(1):24. doi: 10.1186/1471-2350-5-24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Naz S. Mutations of ESPN cause autosomal recessive deafness and vestibular dysfunction. J Med Genet. 2004;41(8):591‐595. doi: 10.1136/jmg.2004.018523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Petersen M, Willems P. Non‐syndromic, autosomal‐recessive deafness: autosomal‐recessive deafness. Clin Genet. 2006;69(5):371‐392. doi: 10.1111/j.1399-0004.2006.00613.x [DOI] [PubMed] [Google Scholar]
- 26. Riazuddin S, Khan SN, Ahmed ZM, et al. Mutations in TRIOBP, which encodes a putative cytoskeletal‐organizing protein, are associated with nonsyndromic recessive deafness. Am J Hum Genet. 2006b;78(1):137‐143. doi: 10.1086/499164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Riazuddin S, Ahmed ZM, Fanning AS, et al. Tricellulin is a tight‐junction protein necessary for hearing. Am J Hum Genet. 2006a;79(6):1040‐1051. doi: 10.1086/510022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shabbir MI. Mutations of human TMHS cause recessively inherited non‐syndromic hearing loss. J Med Genet. 2006;43(8):634‐640. doi: 10.1136/jmg.2005.039834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Khan SY, Riazuddin S, Tariq M, et al. Autosomal recessive nonsyndromic deafness locus DFNB63 at chromosome 11q13.2–q13.3. Hum Genet. 2007;120(6):789‐793. doi: 10.1007/s00439-006-0275-1 [DOI] [PubMed] [Google Scholar]
- 30. Collin RWJ, Kalay E, Tariq M, et al. Mutations of ESRRB encoding estrogen‐related receptor Beta cause autosomal‐recessive nonsyndromic hearing impairment DFNB35. Am J Hum Genet. 2008;82(1):125‐138. doi: 10.1016/j.ajhg.2007.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ahmed, ZM , Masmoudi, S , Kalay, E , et al. Mutations ofLRTOMT, a fusion gene with alternative reading frames, cause nonsyndromicdeafness in humans. Nature Genetics. 2008;40(11):1335‐1340. 10.1038/ng.245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Choi B, Ahmed Z, Riazuddin S, et al. Identities and frequencies of mutations of the otoferlin gene (OTOF) causing DFNB9 deafness in Pakistan. Clin Genet. 2009;75(3):237‐243. doi: 10.1111/j.1399-0004.2008.01128.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schultz JM, Khan SN, Ahmed ZM, et al. Noncoding mutations of HGF are associated with nonsyndromic hearing loss, DFNB39. Am J Hum Genet. 2009;85(1):25‐39. doi: 10.1016/j.ajhg.2009.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Riazuddin S, Anwar S, Fischer M, et al. Molecular basis of DFNB73: mutations of BSND can cause nonsyndromic deafness or Bartter syndrome. Am J Hum Genet. 2009;85(2):273‐280. doi: 10.1016/j.ajhg.2009.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rehman AU, Morell RJ, Belyantseva IA, et al. Targeted Capture and Next‐Generation Sequencing Identifies C9orf75, Encoding Taperin, as the Mutated Gene in Nonsyndromic Deafness DFNB79. The American Journal of Human Genetics. 2010;86(3):378–388. 10.1016/j.ajhg.2010.01.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schraders M, Lee K, Oostrik J, et al. Homozygosity mapping reveals mutations of GRXCR1 as a cause of autosomal‐recessive nonsyndromic hearing impairment. Am J Hum Genet. 2010;86(2):138‐147. doi: 10.1016/j.ajhg.2009.12.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ahmed ZM, Yousaf R, Lee BC, et al. Functional null mutations of MSRB3 encoding methionine sulfoxide reductase are associated with human deafness DFNB74. Am J Hum Genet. 2011;88(1):19‐29. doi: 10.1016/j.ajhg.2010.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rehman AU, Gul K, Morell RJ, et al. Mutations of GIPC3 cause nonsyndromic hearing loss DFNB72 but not DFNB81 that also maps to chromosome 19p. Hum Genet. 2011;130(6):759‐765. doi: 10.1007/s00439-011-1018-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Borck G, Rehman AU, Lee K, et al. Loss‐of‐function mutations of ILDR1 cause autosomal‐recessive hearing impairment DFNB42. Am J Hum Genet. 2011;88(2):127‐137. doi: 10.1016/j.ajhg.2010.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mujtaba G, Bukhari I, Fatima A, Naz S. A p.C343S missense mutation in PJVK causes progressive hearing loss. Gene. 2012;504(1):98‐101. doi: 10.1016/j.gene.2012.05.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Riazuddin S, Belyantseva IA, Giese APJ, et al. Alterations of the CIB2 calcium‐ and integrin‐binding protein cause usher syndrome type 1J and nonsyndromic deafness DFNB48. Nat Genet. 2012;44(11):1265‐1271. doi: 10.1038/ng.2426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Shahzad M, Sivakumaran TA, Qaiser TA, et al. Genetic analysis through OtoSeq of Pakistani families segregating Prelingual hearing loss. Otolaryngol Head Neck Surg. 2013;149(3):478‐487. doi: 10.1177/0194599813493075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Santos‐Cortez RLP, Lee K, Azeem Z, et al. Mutations in KARS, encoding Lysyl‐tRNA Synthetase, cause autosomal‐recessive nonsyndromic hearing impairment DFNB89. Am J Hum Genet. 2013;93(1):132‐140. doi: 10.1016/j.ajhg.2013.05.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lee K, Chiu I, Santos‐Cortez R, et al. Novel OTOA mutations causeautosomal recessive non‐syndromic hearing impairment in Pakistani families: Letter to the Editor. Clinical Genetics. 2013;84(3):294–296. 10.1111/cge.12047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jaworek TJ, Richard EM, Ivanova AA, et al. An alteration in ELMOD3, an Arl2 GTPase‐activating protein, is associated with hearing impairment in humans. PLoS Genet. 2013;9(9):e1003774. doi: 10.1371/journal.pgen.1003774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Imtiaz A, Kohrman DC, Naz S. A frameshift mutation in GRXCR2 causes recessively inherited hearing loss. Hum Mutat. 2014;35(5):618‐624. doi: 10.1002/humu.22545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rehman AU, Santos‐Cortez RLP, Morell RJ, et al. Mutations in TBC1D24, a gene associated with epilepsy, also cause nonsyndromic deafness DFNB86. Am J Hum Genet. 2014;94(1):144‐152. doi: 10.1016/j.ajhg.2013.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Santos‐Cortez RLP, Lee K, Giese AP, et al. Adenylate cyclase 1 (ADCY1) mutations cause recessive hearing impairment in humans and defects in hair cell function and hearing in zebrafish. Hum Mol Genet. 2014;23(12):3289‐3298. doi: 10.1093/hmg/ddu042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Simon M, Richard EM, Wang X, et al. Mutations of human NARS2, encoding the mitochondrial Asparaginyl‐tRNA Synthetase, cause nonsyndromic deafness and Leigh syndrome. PLoS Genet. 2015;11(3):e1005097. doi: 10.1371/journal.pgen.1005097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mujtaba G, Schultz JM, Imtiaz A, Morell RJ, Friedman TB, Naz S. A mutation of MET, encoding hepatocyte growth factor receptor, is associated with human DFNB97 hearing loss. J Med Genet. 2015;52(8):548‐552. doi: 10.1136/jmedgenet-2015-103023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Santos‐Cortez RLP, Faridi R, Rehman AU, et al. Autosomal‐recessive hearing impairment due to rare missense variants within S1PR2. Am J Hum Genet. 2016;98(2):331‐338. doi: 10.1016/j.ajhg.2015.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wang R, Han S, Khan A, Zhang X. Molecular analysis of twelve Pakistani families with nonsyndromic or syndromic hearing loss. Genet Test Mol Biomarkers. 2017a;21(5):316‐321. doi: 10.1089/gtmb.2016.0328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Le Quesne Stabej P, James C, Ocaka L, et al. An example of the utility of genomic analysis for fast and accurate clinical diagnosis of complex rare phenotypes. Orphanet J Rare Dis. 2017;12(1):24. doi: 10.1186/s13023-017-0582-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yousaf R, Gu C, Ahmed ZM, et al. Mutations in Diphosphoinositol‐Pentakisphosphate kinase PPIP5K2 are associated with hearing loss in human and mouse. PLoS Genet. 2018b;14(3):e1007297. doi: 10.1371/journal.pgen.1007297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yousaf R, Ahmed ZM, Giese APJ, et al. Modifier variant of METTL13 suppresses human GAB1–associated profound deafness. J Clin Investig. 2018a;128(4):1509‐1522. doi: 10.1172/JCI97350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Liaqat K, Hussain S, Bilal M, et al. Further evidence of involvement of TMEM132E in autosomal recessive nonsyndromic hearing impairment. J Hum Genet. 2020;65(2):187‐192. doi: 10.1038/s10038-019-0691-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhou Y, Tariq M, He S, Abdullah U, Zhang J, Baig SM. Whole exome sequencing identified mutations causing hearing loss in five consanguineous Pakistani families. BMC Med Genet. 2020;21(1):151. doi: 10.1186/s12881-020-01087-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Park HR, Kanwal S, Lim SO, Nam DE, Choi YJ, Chung KW. Homozygous mutations in Pakistani consanguineous families with prelingual nonsyndromic hearing loss. Mol Biol Rep. 2020;47(12):9979‐9985. doi: 10.1007/s11033-020-06037-7 [DOI] [PubMed] [Google Scholar]
- 59. Mahmood U, Bukhari SA, Ali M, Ahmed ZM, Riazuddin S. Identification of hearing loss‐associated variants of PTPRQ, MYO15A, and SERPINB6 in Pakistani families. Biomed Res Int. 2021;2021:1‐6. doi: 10.1155/2021/5584788 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 60. Kakar MU, Akram M, Zubair Mehboob M, et al. Identification of homozygous missense variant in SIX5 gene underlying recessive nonsyndromic hearing impairment. PLoS ONE. 2022;17(6):e0268078. doi: 10.1371/journal.pone.0268078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Schrauwen I, Ghaffar A, Bharadwaj T, et al. Syntaxin 4 is essential for hearing in human and zebrafish. Hum Mol Genet. 2022;ddac257:1184‐1192. doi: 10.1093/hmg/ddac257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Adadey SM, Aboagye ET, Esoh K, et al. A novel autosomal dominant GREB1L variant associated with non‐syndromic hearing impairment in Ghana. BMC Med Genet. 2022;15(1):237. doi: 10.1186/s12920-022-01391-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Rabbani B, Tekin M, Mahdieh N. The promise of whole‐exome sequencing in medical genetics. J Hum Genet. 2014;59(1):5‐15. doi: 10.1038/jhg.2013.114 [DOI] [PubMed] [Google Scholar]
- 64. Vona B, Nanda I, Hofrichter MAH, Shehata‐Dieler W, Haaf T. Non‐syndromic hearing loss gene identification: a brief history and glimpse into the future. Mol Cell Probes. 2015;29(5):260‐270. doi: 10.1016/j.mcp.2015.03.008 [DOI] [PubMed] [Google Scholar]
- 65. Avraham KB, Khalaily L, Noy Y, Kamal L, Koffler‐Brill T, Taiber S. The noncoding genome and hearing loss. Hum Genet. 2022;141(3–4):323‐333. doi: 10.1007/s00439-021-02359-z [DOI] [PubMed] [Google Scholar]
- 66. Richard EM, Santos‐Cortez RLP, Faridi R, et al. Global genetic insight contributed by consanguineous Pakistani families segregating hearing loss. Hum Mutat. 2019;40(1):53‐72. doi: 10.1002/humu.23666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zhou Y, Zhou B, Pache L, et al. Metascape provides a biologist‐oriented resource for the analysis of systems‐level datasets. Nat Commun. 2019;10(1):1523. doi: 10.1038/s41467-019-09234-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Doll J, Vona B, Schnapp L, et al. Genetic Spectrum of syndromic and non‐syndromic hearing loss in Pakistani families. Genes. 2020;11(11):1329. doi: 10.3390/genes11111329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Müller U. Cadherins and mechanotransduction by hair cells. Curr Opin Cell Biol. 2008;20(5):557‐566. doi: 10.1016/j.ceb.2008.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ramzan K, Al‐Numair NS, Al‐Ageel S, et al. Identification of novel CDH23 variants causing moderate to profound progressive nonsyndromic hearing loss. Genes. 2020;11(12):1474. doi: 10.3390/genes11121474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Lagziel A, Overlack N, Bernstein SL, Morell RJ, Wolfrum U, Friedman TB. Expression of cadherin 23 isoforms is not conserved: implications for a mouse model of usher syndrome type 1D. Mol Vis. 2009;15:1843‐1857. [PMC free article] [PubMed] [Google Scholar]
- 72. Bolz H, von Brederlow B, Ramírez A, et al. Mutation of CDH23, encoding a new member of the cadherin gene family, causes usher syndrome type 1D. Nat Genet. 2001;27(1):108‐112. doi: 10.1038/83667 [DOI] [PubMed] [Google Scholar]
- 73. Usami S‐I, Isaka Y, Miyagawa M, Nishio S‐Y. Variants in CDH23 cause a broad spectrum of hearing loss: from non‐syndromic to syndromic hearing loss as well as from congenital to age‐related hearing loss. Hum Genet. 2022;141(3–4):903‐914. doi: 10.1007/s00439-022-02431-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Bork JM, Peters LM, Riazuddin S, et al. Usher syndrome 1D and nonsyndromic autosomal recessive deafness DFNB12 are caused by allelic mutations of the novel cadherin‐like gene CDH23. Am J Hum Genet. 2001;68(1):26‐37. doi: 10.1086/316954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Schultz JM, Bhatti R, Madeo AC, et al. Allelic hierarchy of CDH23 mutations causing non‐syndromic deafness DFNB12 or usher syndrome USH1D in compound heterozygotes. J Med Genet. 2011;48(11):767‐775. doi: 10.1136/jmedgenet-2011-100262 [DOI] [PubMed] [Google Scholar]
- 76. Michel V, Booth KT, Patni P, et al. CIB2, defective in isolated deafness, is key for auditory hair cell mechanotransduction and survival. EMBO Mol Med. 2017;9(12):1711‐1731. doi: 10.15252/emmm.201708087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Wang Y, Li J, Yao X, et al. Loss of CIB2 causes profound hearing loss and abolishes Mechanoelectrical transduction in mice. Front Mol Neurosci. 2017b;10:401. doi: 10.3389/fnmol.2017.00401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Dal Cortivo G, Dell'Orco D. Calcium‐ and integrin‐binding protein 2 (CIB2) in physiology and disease: bright and dark sides. Int J Mol Sci. 2022;23(7):3552. doi: 10.3390/ijms23073552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Booth KT, Kahrizi K, Babanejad M, et al. Variants in CIB2 cause DFNB48 and not USH1J. Clin Genet. 2018;93(4):812‐821. doi: 10.1111/cge.13170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Kitano T, Kitajiri S‐I, Nishio S‐Y, Usami S‐I. Detailed clinical features of deafness caused by a Claudin‐14 variant. Int J Mol Sci. 2019;20(18):4579. doi: 10.3390/ijms20184579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Nishio S‐Y, Hattori M, Moteki H, et al. Gene expression profiles of the cochlea and vestibular endorgans: localization and function of genes causing deafness. Ann Otol Rhinol Laryngol. 2015;124(Suppl 1):6S‐48S. doi: 10.1177/0003489415575549 [DOI] [PubMed] [Google Scholar]
- 82. Kawashima Y, Géléoc GSG, Kurima K, et al. Mechanotransduction in mouse inner ear hair cells requires transmembrane channel‐like genes. J Clin Invest. 2011;121(12):4796‐4809. doi: 10.1172/JCI60405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Ben‐Yosef T, Belyantseva IA, Saunders TL, et al. Claudin 14 knockout mice, a model for autosomal recessive deafness DFNB29, are deaf due to cochlear hair cell degeneration. Hum Mol Genet. 2003;12(16):2049‐2061. doi: 10.1093/hmg/ddg210 [DOI] [PubMed] [Google Scholar]
- 84. Naz S, Imtiaz A, Mujtaba G, et al. Genetic causes of moderate to severe hearing loss point to modifiers: genetic causes of hearing loss point to modifiers. Clin Genet. 2017;91(4):589‐598. doi: 10.1111/cge.12856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Bashir R, Fatima A, Naz S. Mutations in CLDN14 are associated with different hearing thresholds. J Hum Genet. 2010;55(11):767‐770. doi: 10.1038/jhg.2010.104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Kenna MA, Feldman HA, Neault MW, et al. Audiologic phenotype and progression in GJB2 (Connexin 26) hearing loss. Arch Otolaryngol Head Neck Surg. 2010;136(1):81‐87. doi: 10.1001/archoto.2009.202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Denoyelle F, Lina‐Granade G, Plauchu H, et al. Connexin 26 gene linked to a dominant deafness. Nature. 1998;393(6683):319‐320. doi: 10.1038/30639 [DOI] [PubMed] [Google Scholar]
- 88. Anselmi F, Hernandez VH, Crispino G, et al. ATP release through connexin hemichannels and gap junction transfer of second messengers propagate Ca 2+ signals across the inner ear. Proc Natl Acad Sci. 2008;105(48):18770‐18775. doi: 10.1073/pnas.0800793105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Bruzzone R, Giaume C, eds. Connexin Methods and Protocols. Humana Press; 2001. [Google Scholar]
- 90. Santos RLP, Wajid M, Pham TL, et al. Low prevalence of Connexin 26 (GJB2) variants in Pakistani families with autosomal recessive non‐syndromic hearing impairment. Clin Genet. 2005;67(1):61‐68. doi: 10.1111/j.1399-0004.2005.00379.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Kausar N, Haque A, Masoud MS, et al. Disease‐associated variants of gap junction Beta 2 protein (GJB2) in the deaf population of southern Punjab of Pakistan. PLoS ONE. 2021;16(10):e0259083. doi: 10.1371/journal.pone.0259083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Sloan‐Heggen CM, Bierer AO, Shearer AE, et al. Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Hum Genet. 2016;135(4):441‐450. doi: 10.1007/s00439-016-1648-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Morell RJ, Olszewski R, Tona R, et al. Noncoding microdeletion in mouse Hgf disrupts neural crest migration into the Stria Vascularis, reduces the Endocochlear potential, and suggests the neuropathology for human nonsyndromic deafness DFNB39. J Neurosci Off J Soc Neurosci. 2020;40(15):2976‐2992. doi: 10.1523/JNEUROSCI.2278-19.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Anderson JM, Van Itallie CM. Physiology and function of the tight junction. Cold Spring Harb Perspect Biol. 2009;1(2):a002584. doi: 10.1101/cshperspect.a002584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Nayak G, Varga L, Trincot C, et al. Molecular genetics of MARVELD2 and clinical phenotype in Pakistani and Slovak families segregating DFNB49 hearing loss. Hum Genet. 2015;134(4):423‐437. doi: 10.1007/s00439-015-1532-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Nayak G, Lee SI, Yousaf R, et al. Tricellulin deficiency affects tight junction architecture and cochlear hair cells. J Clin Invest. 2013;123(9):4036‐4049. doi: 10.1172/JCI69031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Houdusse A, Titus MA. The many roles of myosins in filopodia, microvilli and stereocilia. Current Biology: CB. 2021;31(10):R586–R602. 10.1016/j.cub.2021.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Sweeney HL, Holzbaur ELF. Motor proteins. Cold Spring Harb Perspect Biol. 2018;10(5):a021931. doi: 10.1101/cshperspect.a021931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Moreland ZG, Bird JE. Myosin motors in sensory hair bundle assembly. Curr Opin Cell Biol. 2022;79:102132. doi: 10.1016/j.ceb.2022.102132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Li S, Mecca A, Kim J, et al. Myosin‐VIIa is expressed in multiple isoforms and essential for tensioning the hair cell mechanotransduction complex. Nat Commun. 2020;11(1):2066. doi: 10.1038/s41467-020-15936-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Lopes VS, Gibbs D, Libby RT, et al. The usher 1B protein, MYO7A, is required for normal localization and function of the visual retinoid cycle enzyme, RPE65. Hum Mol Genet. 2011;20(13):2560‐2570. doi: 10.1093/hmg/ddr155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Jaijo T, Aller E, Oltra S, et al. Mutation profile of the MYO7A gene in Spanish patients with usher syndrome type I. Hum Mutat. 2006;27(3):290‐291. doi: 10.1002/humu.9404 [DOI] [PubMed] [Google Scholar]
- 103. Belyantseva IA, Boger ET, Naz S, et al. Myosin‐XVa is required for tip localization of whirlin and differential elongation of hair‐cell stereocilia. Nat Cell Biol. 2005;7(2):148‐156. doi: 10.1038/ncb1219 [DOI] [PubMed] [Google Scholar]
- 104. Ballesteros A, Yadav M, Cui R, Kurima K, Kachar B. Selective binding and transport of protocadherin 15 isoforms by stereocilia unconventional myosins in a heterologous expression system. Sci Rep. 2022;12(1):13764. doi: 10.1038/s41598-022-17757-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Zhang J, Guan J, Wang H, et al. Genotype‐phenotype correlation analysis of MYO15A variants in autosomal recessive non‐syndromic hearing loss. BMC Med Genet. 2019;20(1):60. doi: 10.1186/s12881-019-0790-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Kwon T‐J, Cho H‐J, Kim U‐K, et al. Methionine sulfoxide reductase B3 deficiency causes hearing loss due to stereocilia degeneration and apoptotic cell death in cochlear hair cells. Hum Mol Genet. 2014;23(6):1591‐1601. doi: 10.1093/hmg/ddt549 [DOI] [PubMed] [Google Scholar]
- 107. Pangrsic T, Lasarow L, Reuter K, et al. Hearing requires otoferlin‐dependent efficient replenishment of synaptic vesicles in hair cells. Nat Neurosci. 2010;13(7):869‐876. doi: 10.1038/nn.2578 [DOI] [PubMed] [Google Scholar]
- 108. Vogl C, Cooper BH, Neef J, et al. Unconventional molecular regulation of synaptic vesicle replenishment in cochlear inner hair cells. J Cell Sci. 2015;128(4):638‐644. doi: 10.1242/jcs.162099 [DOI] [PubMed] [Google Scholar]
- 109. Moser T, Starr A. Auditory neuropathy—neural and synaptic mechanisms. Nat Rev Neurol. 2016;12(3):135‐149. doi: 10.1038/nrneurol.2016.10 [DOI] [PubMed] [Google Scholar]
- 110. Vona B, Rad A, Reisinger E. The many faces of DFNB9: relating OTOF variants to hearing impairment. Genes. 2020;11(12):1411. doi: 10.3390/genes11121411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Yasunaga S, Grati M, Cohen‐Salmon M, et al. A mutation in OTOF, encoding otoferlin, a FER‐1‐like protein, causes DFNB9, a nonsyndromic form of deafness. Nat Genet. 1999;21(4):363‐369. doi: 10.1038/7693 [DOI] [PubMed] [Google Scholar]
- 112. Rodríguez‐Ballesteros M, del Castillo FJ, Martín Y, et al. Auditory neuropathy in patients carrying mutations in the otoferlin gene (OTOF). Hum Mutat. 2003;22(6):451‐456. [DOI] [PubMed] [Google Scholar]
- 113. Ito T, Choi BY, King KA, et al. SLC26A4 genotypes and phenotypes associated with enlargement of the vestibular aqueduct. Cell Physiol Biochem. 2011;28(3):545‐552. doi: 10.1159/000335119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Albert S, Blons H, Jonard L, et al. SLC26A4 gene is frequently involved in nonsyndromic hearing impairment with enlarged vestibular aqueduct in Caucasian populations. Eur J Hum Genet. 2006;14(6):773‐779. doi: 10.1038/sj.ejhg.5201611 [DOI] [PubMed] [Google Scholar]
- 115. Garabet Diramerian L, Ejaz S. Pendred Syndrome. StatPearls. StatPearls Publishing; 2022. http://www.ncbi.nlm.nih.gov/books/NBK549839/ [PubMed] [Google Scholar]
- 116. Anwar S, Riazuddin S, Ahmed ZM, et al. SLC26A4 mutation spectrum associated with DFNB4 deafness and Pendred's syndrome in Pakistanis. J Hum Genet. 2009;54(5):266‐270. doi: 10.1038/jhg.2009.21 [DOI] [PubMed] [Google Scholar]
- 117. Fettiplace R, Furness DN, Beurg M. The conductance and organization of the TMC1‐containing mechanotransducer channel complex in auditory hair cells. Proc Natl Acad Sci. 2022;119(41):e2210849119. doi: 10.1073/pnas.2210849119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Basu A, Lagier S, Vologodskaia M, Fabella BA, Hudspeth A. Direct mechanical stimulation of tip links in hair cells through DNA tethers. eLife. 2016;5:e16041. doi: 10.7554/eLife.16041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Pan B, Akyuz N, Liu X‐P, et al. TMC1 forms the pore of Mechanosensory transduction channels in vertebrate inner ear hair cells. Neuron. 2018;99(4):736‐753.e6. doi: 10.1016/j.neuron.2018.07.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Nakanishi H, Kurima K, Kawashima Y, Griffith AJ. Mutations of TMC1 cause deafness by disrupting mechanoelectrical transduction. Auris Nasus Larynx. 2014;41(5):399‐408. doi: 10.1016/j.anl.2014.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Kitajiri S, McNamara R, Makishima T, et al. Identities, frequencies and origins of TMC1 mutations causing DFNB7/B11 deafness in Pakistan. Clin Genet. 2007;72(6):546‐550. doi: 10.1111/j.1399-0004.2007.00895.x [DOI] [PubMed] [Google Scholar]
- 122. Liu W, Löwenheim H, Santi PA, Glueckert R, Schrott‐Fischer A, Rask‐Andersen H. Expression of trans‐membrane serine protease 3 (TMPRSS3) in the human organ of Corti. Cell Tissue Res. 2018;372(3):445‐456. doi: 10.1007/s00441-018-2793-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Chen Y‐S, Cabrera E, Tucker BJ, et al. TMPRSS3 expression is limited in spiral ganglion neurons: implication for successful cochlear implantation. J Med Genet. 2022;59(12):1219‐1226. doi: 10.1136/jmg-2022-108654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Guipponi M. The transmembrane serine protease (TMPRSS3) mutated in deafness DFNB8/10 activates the epithelial sodium channel (ENaC) in vitro. Hum Mol Genet. 2002;11(23):2829‐2836. doi: 10.1093/hmg/11.23.2829 [DOI] [PubMed] [Google Scholar]
- 125. Molina L, Fasquelle L, Nouvian R, et al. Tmprss3 loss of function impairs cochlear inner hair cell Kcnma1 channel membrane expression. Hum Mol Genet. 2013;22(7):1289‐1299. doi: 10.1093/hmg/dds532 [DOI] [PubMed] [Google Scholar]
- 126. Weegerink NJD, Schraders M, Oostrik J, et al. Genotype–phenotype correlation in DFNB8/10 families with TMPRSS3 mutations. J Assoc Res Otolaryngol. 2011;12(6):753‐766. doi: 10.1007/s10162-011-0282-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Lee K, Khan S, Islam A, et al. Novel TMPRSS3 variants in Pakistani families with autosomal recessive non‐syndromic hearing impairment. Clin Genet. 2012;82(1):56‐63. doi: 10.1111/j.1399-0004.2011.01695.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Lee SJ, Lee S, Han JH, et al. Structural analysis of pathogenic TMPRSS3 variants and theircochlear implantation outcomes of sensorineural hearing loss. Gene. 2023;865:147335. 10.1016/j.gene.2023.147335 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1.
Data Availability Statement
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.