

Abstract
INTRODUCTION –
The prevalence and impact of symptoms affecting individuals with pediatric forms of myotonic dystrophy type-1 (DM1) are not well understood.
METHODS –
Patients from United States, Canada, and Sweden completed a survey that investigated 20 themes associated with pediatric-onset DM1. Participants reported the prevalence and importance of each theme affecting their lives. Surveys from participants were matched with surveys from their caregivers for additional analysis.
RESULTS –
The most prevalent symptomatic themes included problems with hands or fingers (79%) and gastrointestinal issues (75%). Problems with urinary/bowel control and gastrointestinal issues were reported to have the greatest impact on patients’ lives. Responses between participants and their caregivers had varying levels of agreement among symptomatic themes.
DISCUSSION –
Many symptoms have meaningful impact on disease burden. The highest levels of agreement between caregivers and individuals with pediatric forms of myotonic dystrophy were found for physical activity themes.
Keywords: Myotonic Dystrophy, Neuromuscular Disease, Symptom Assessment, Health Status Indicators, Caregivers
INTRODUCTION
Myotonic dystrophy type-1 (DM1) is an autosomal dominant, multisystem disorder associated with a CTG repeat expansion in the DMPK gene on chromosome 19q13.3.1–3 It affects approximately 1 in 8000 adults; presenting symptoms vary but can include myotonia, weakness, fatigue, or an early onset of cataracts. CTG repeat length is unstable and can expand from generation to generation, contributing to the phenomenon of genetic anticipation.4 In past studies of pediatric-onset DM1, patients have been classified as having congenital myotonic dystrophy (CDM) when manifestations are present in-utero or at birth and as having childhood myotonic dystrophy (ChDM) when symptoms present before the age of 10, with an uneventful pre- and neonatal history and with normal psychomotor development during the first year of life.5,6 Regardless of the exact classification and timing of onset of symptoms, there is agreement that the impact and spectrum of disease manifestations throughout all of childhood require further study.
The incidence of CDM is approximately 1 in 47,000 live births.7 Diagnosis of CDM can occur prenatally with polyhydramnios and reduced fetal movements.8 At birth, symptoms can include hypotonia, respiratory distress, feeding difficulties, and clubfoot deformity.9 Infants often require an intensive level of care and may need prolonged respiratory support with mechanical ventilation.10 Children with CDM can have profound dysarthria and other gastrointestinal complications and may require gastric tube feedings. Interestingly, individuals that survive the neonatal period show improvement during childhood in many aspects of the disease.11 This contrasts with adult-onset, which has a slowly progressive worsening time course. These differences of disease progression need further investigation.
Intellectual disability is common and CDM is associated with higher rates of social communication and autism spectrum disorder (ASD) as well as attention-deficit-hyperactivity disorder (ADHD).12–14 Children with ChDM have later onset of similar symptoms as children with CDM, but typically these symptoms are not as severe and have a lesser number of manifestations.
Previous studies have explored symptomatic themes in pediatric forms of DM1 through qualitative interviews with patients and caregivers.15 A recent study described the impact of pediatric DM1 disease burden using parent-reported surveys.16 Caregivers reported that individuals with pediatric forms of myotonic dystrophy were most affected by communication issues, problems with hands or fingers, and fatigue. However, this study did not assess disease burden from the patient’s perspective. This study seeks to better understand the impact of DM1 disease burden by assessing patient-reported surveys and comparing them to previously published parent-reported surveys.
METHODS
Participants
Participants included individuals with an onset of myotonic dystrophy before the age of 18 years. In the United States, participants were identified from the National Registry of Myotonic Dystrophy and Facioscapulohumeral Muscular Dystrophy Patients and Family Members.17 In Canada, participants were identified from the Canadian Neuromuscular Disease Registry and from the site investigator’s (CC) neuromuscular clinic.18 In Sweden, participants were identified via rehabilitation center directors. Paper surveys were distributed to all identified individuals. Respondents in the United States and Canada had the option of completing the survey by telephone. Caregivers were asked to assist in completing the survey depending on the age of the participant. All participants were given a 3-month period to complete the survey.
Instrument design
The survey inquired about potential symptomatic themes previously identified through qualitative interviews with individuals with pediatric onset DM1 and their parents.15 Participants identified if they currently experienced the symptom and rated the relative impact using a Likert scale. Participants 5–7 years of age utilized a 3-point Likert while participants aged 8 years of age and older utilized a 5-point Likert scale. Survey question wording was reviewed by a pediatric neuropsychologist for age appropriate comprehension. For example, to query about limitations with mobility and ambulation, participants aged 5–7 were asked to comment on “It is hard for me to walk or run”, participants aged 8–17 were asked to use comment on, “It is hard for me to walk, run, or get around,” and participants aged over the age of 18 years were asked to comment on “Limitations with mobility and walking.” In addition, there was a picture depicting the action for those participants aged 5–7. Demographics, medical history, and developmental milestones were also collected from each participant.
An established systematic translation process was used to create the Swedish version of the surveys.19 A native English speaker fluent in Swedish translated the survey into Swedish. Two bilingual Swedish speaking physicians revised the translation for fluency and syntax. A native Swedish speaker fluent in English then back-translated the survey from Swedish to English. Final corrections were made after the two English versions were compared.
All surveys and methods were approved by local ethics boards (Institutional Review Board). Patients and their caregivers gave consent to the study and to the publication.
Statistical analysis
The prevalence of each theme was determined. Average life impact scores (a score ranging from 0 to 4 that measures the relative importance of a symptom to a participant) were calculated based on the responses from participants who stated they experienced the theme. Numerical values were assigned to each response for participants with an age of 8 or older as follows: 0=‘This never happens to me’; 0=‘I experience, but this does not affect my life’; 1=‘This affects my life a little’; 2=‘This affects my life somewhat’; 3=‘This affects my life very much’; 4=‘This affects my life severely’. To enhance responder comprehension, numerical values were assigned to each response from participants with an age of 7 or younger as follows: 0= ‘This does not happen to me’; 2=‘This is a little problem for me’; 4=‘This is a big problem for me’ (Supplemental Methods 1). The population impact score (also a score ranging from 0 to 4) was calculated as the average life impact score multiplied by the prevalence of the symptom.20
Participant responses were categorized in several ways to identify changes associated with age, CTG repeat length, and age of disease onset. Responses were examined based on the age of the individual with DM1. Age subgroups included: 5 to 7 years, 8 to 11 years, 12 to 17 years, 18–28 years, and older than 28 years. These age subgroups were selected based on developmental age after consultation with a pediatric neuropsychologist. Responses were also categorized based on the CTG repeat length of the participant. CTG repeat length subgroups included: 0 to 500, 501 to 1000, 1001 to 1500, and over 1500. Age of disease onset subgroups included: disease onset less than one year of age and disease onset greater than one year of age. Prevalence, average life impact scores, and population impact scores for each theme were calculated for each subgroup. Fisher’s exact tests were used to compare the prevalence of each theme across all different subgroups. Kruskal-Wallis tests were employed to compare average life impact scores across different subgroups. A two-tailed p value less than or equal to 0.05 was considered significant. Multiple comparison adjustment was not used in this study as it is considered exploratory.21,22
Participant responses were compared to responses from their parents or primary caregivers. If participants over the age of 18 were living independently, their parent or primary care giver was not required to be living with the participant at the time of the survey. Parents and primary caregivers received the same survey as the individuals with pediatric forms of myotonic dystrophy and their responses were reported previously.15 Weighted Cohen’s Kappa Coefficients were utilized to measure agreement between participants and caregivers. Traditional labels to describe the strength of agreement associated with kappa values include: poor (<0); slight (0–0.20); fair (0.21–0.40); moderate (0.41–0.60); substantial (0.61–0.80); and almost perfect (0.81–1.0).23 Caregivers were categorized into subgroups for additional analysis and compared to their child. Affected caregivers were defined as individuals with a diagnosis of myotonic dystrophy. Unaffected caregivers were defined as individuals who do not carry a diagnosis of myotonic dystrophy. Mothers were defined as individuals who were the biological mother of a child who may or may not have a diagnosis of myotonic dystrophy. Fathers were defined as individuals who were the biological father a child who may or may not have a diagnosis of myotonic dystrophy. Caregivers were also categorized into two other subgroups depending on the age of their child at the time of survey completion.
RESULTS
Demographics
One-hundred and twenty-nine participants responded from the 340 surveys distributed. There was an overall response rate of 38% (48% US registry, 34% Canadian, and 26% Swedish). There were five participants that did not have a matching parent or caregiver survey completed and these participants were excluded. 71 participants were from the United States, 21 participants were from Canada, and 32 patients were from Sweden. The number of participants over the age of 18 years at the time of the study was nearly 55%. Demographic data and other participant characteristics are provided in Table 1.
Table 1:
Demographic, developmental, and intellectual characteristics of study participants with pediatric-onset myotonic dystrophy type 1
| Characteristic | Data Field |
|---|---|
| Number of Individuals with DM1 studied | 124 |
| Individuals with DM1, sex (n) | |
| Male | 74 |
| Female | 50 |
| Mean age of participant with DM1, (SD) | 21 years 1 month (10 years 3 months) |
| Age 5–7 years, (%) | 11 (8.9) |
| Age 8–11 years, (%) | 12 (9.7) |
| Age 12–17 years, (%) | 33 (26.6) |
| Age 18–28 years, (%) | 39 (31.4) |
| Age 29+ years, (%) | 29 (23.4) |
| Age of onset less than 1 year, (%) | 54 (44) |
| Mean age of disease onset, (SD) | 4.7 (5.3) |
| Mean CTG repeat length, (SD) | 820.5 (522.7) |
| Race (%) | |
| Asian | 7.9 |
| Caucasian | 88.2 |
| Hispanic | 3.3 |
| Other | 4 |
| Requiring Assistance in School (%) | 61 |
| Requiring Speech Therapy (%) | 49 |
| Characteristic | Data Field |
| Requiring Occupational Therapy (%) | 35 |
| Requiring Physical Therapy (%) | 26 |
| Participated in smaller classroom sizes in school (%) | 35 |
| Obtained test modifications during school (%) | 45 |
| Require assistive devices for ambulation (%) | 65 |
| Require wheelchair for mobility (%) | 14 |
| Diagnosed with scoliosis (%) | 15 |
| Diagnosed with Autism Spectrum Disorders (%) | 17 |
| Diagnosed with Attention Deficit Hyperactivity Disorder (%) | 28 |
| Unemployed >18 years (%) | 54 |
Prevalence and impact of themes
Overall prevalence, average life impact score, and population impact scores for each theme are reported in Supplemental Table 1. The most prevalent themes reported included: problems with hands or fingers; gastrointestinal issues; and fatigue. Themes with the greatest average life impact scores included: problems with urinary or bowel control; gastrointestinal issues; and communication issues. Themes with the greatest population impact scores included: gastrointestinal issues; fatigue; and communication issues. Theme prevalence, average life impact score, and population impact score by age are reported in Figure 1, Supplemental Table 2, and Supplemental Table 4. Theme prevalence, average life impact score, and population impact score by CTG repeat length are reported in Figure 2, Supplemental Table 3, and Supplemental Table 5. CTG repeat length was reported by 57% of participants.
Figure 1.
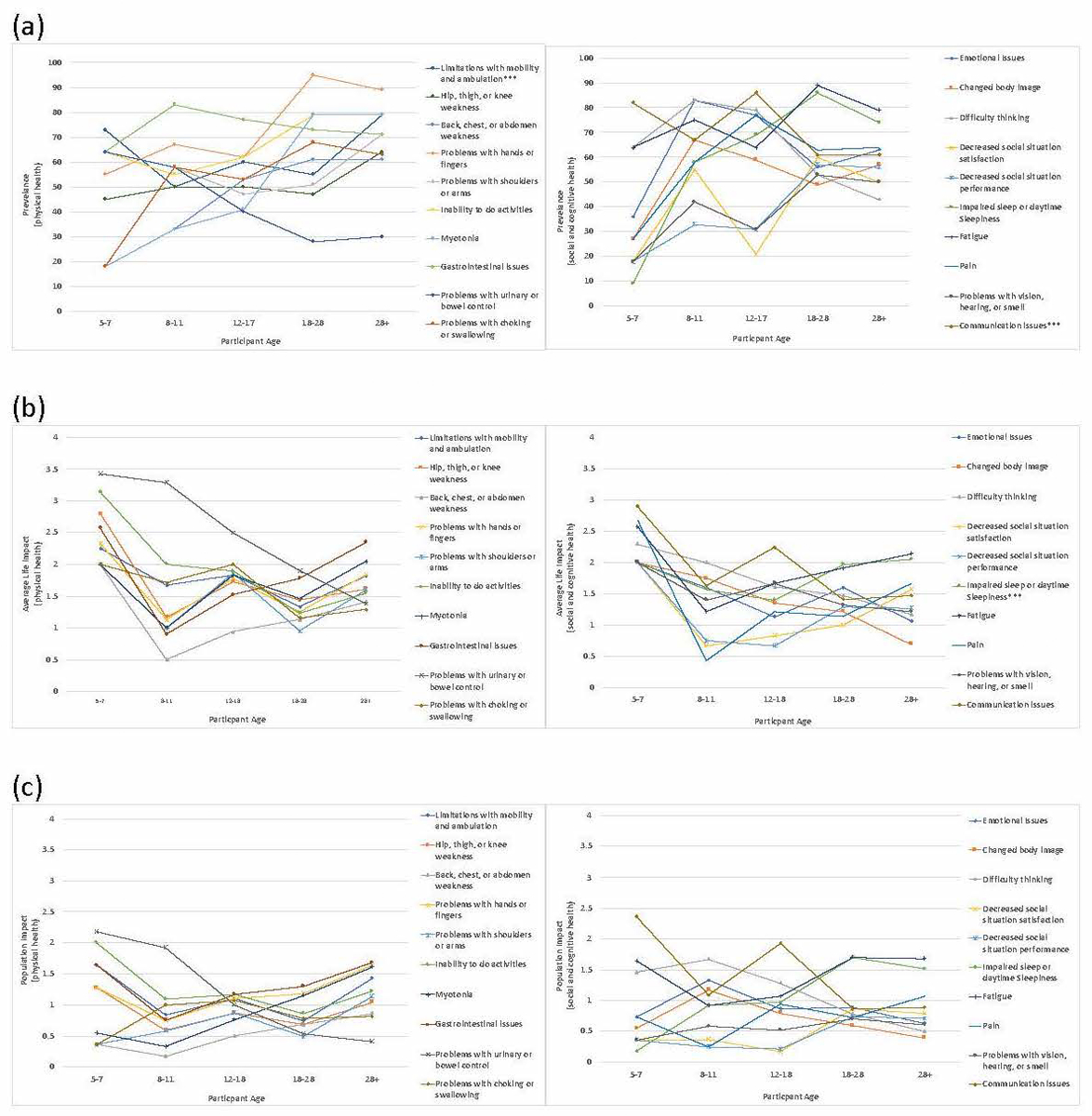
Prevalence, Average Life Impact, and Population Impact by age. (a) Prevalence of themes. (b) Average Life Impact. (c) Population Impact. ***Indicates p<0.05
Figure 2.
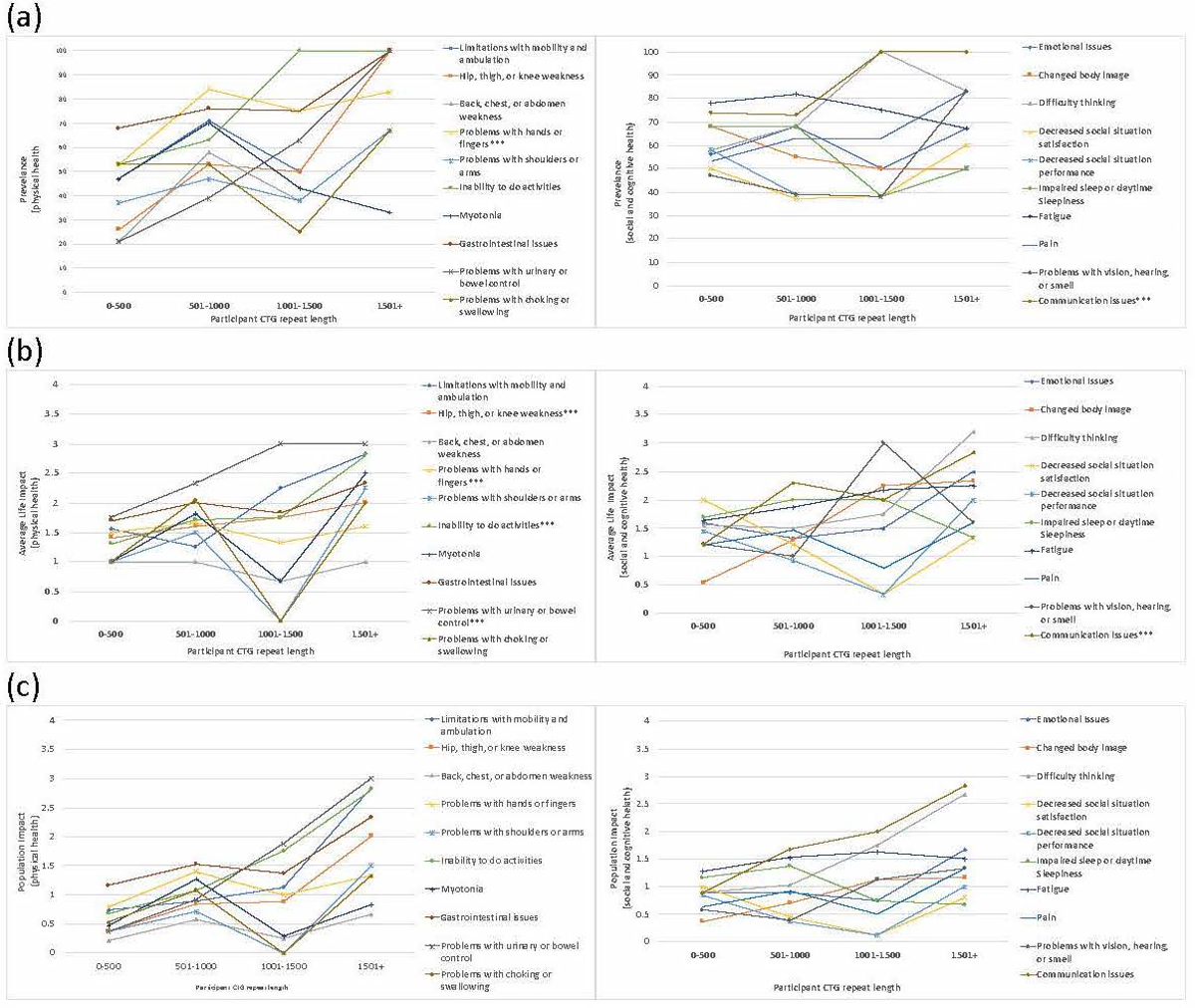
Prevalence, Average Life Impact, and Population Impact by CTG repeat length. (a) Prevalence of themes (b) Average Life Impact. (c) Population Impact. ***Indicates p<0.05
Agreement between caregivers and participants
Weighted kappa values with corresponding confidence intervals are reported in Table 3. When comparing responses from all participants and their caregivers, none of the symptomatic themes had a perfect agreement between patients and caregivers. One theme had moderate agreement, 15 themes had fair agreement, and 4 themes had a slight agreement between the two groups. The themes with the highest weighted kappa values included: problems with urinary or bowel control; limitations with mobility and ambulation; and hip, thigh, and knee weakness. The themes with the lowest weighted kappa values included: problems with vision hearing, or smell; difficulty thinking; and changed body image.
Agreement between participants and their caregivers was also compared in various subgroups. The highest weighted kappa values included: problems with urinary or bowel control when compared between father and child; problems with urinary or bowel control when compared between unaffected caregiver and child; and impaired sleep or daytime sleepiness when compared between father and child. The lowest weighted kappa values included: decreased social situation performance when compared between affected caregiver and child; decreased social situation performance when compared between mother and child; and decreased social situation satisfaction when compared between affected caregiver and child.
DISCUSSION
This study provides patient insight into the wide spectrum of disease burden experienced by individuals with childhood-onset myotonic dystrophy. It addresses a primary question that is commonly asked by clinicians and researchers: what manifestations of a disease matter the most to affected individuals?
CTG repeat length and average life impact score did not significantly correlate among themes. Disease severity does not always relate to CTG repeat length and that there are likely many other genetic and environmental factors contributing to disease severity. 24–26 There was no correlation between symptom theme impact and age, supporting recent evidence suggesting that pediatric-onset myotonic dystrophy is more complex and dynamic than previously understood and that the disease has a nonlinear relationship with age.11 Responses from this population demonstrate that the most prevalent symptoms do not always have the greatest impact on disease burden. For example, problems with urinary or bowel control were prevalent in only 39% of the population but this theme had the greatest average impact in affected individuals. Emphasis should be placed on identifying and treating symptoms that have a great impact on the patient condition even if they are not as prevalent.
Participants reported issues which have not typically been highlighted as problematic symptoms for those clinically managing pediatric-onset myotonic dystrophy. This observation was also seen when surveying individuals with adult myotonic dystrophy.20 Results from both adult and pediatric population groups showed that fatigue and impaired sleep were themes with higher average life impact scores compared to themes related to problems with hands or fingers and limitations with mobility and ambulation.
This study supports prior work in myotonic dystrophy that showed differences exist between the perception of disease burden between individuals with disease manifestations and individuals without disease manifestations.27 Ideally, a patient’s input should be prioritized when discussing their disease burden; however, due to cognitive or age restrictions in certain cases it may be both necessary and reasonable to obtain input from individuals who spend the greatest time with the patient.
Levels of agreement were highest between participants and their fathers than in any other subgroup. Five themes were in ‘substantial’ agreement and three themes were in ‘almost perfect’ agreement. Fathers may be more in agreement with their child given congenital DM1 primarily occurs when the mother is the parent affected by DM1. Two-thirds of responding mothers reported a diagnosis of DM1 while one-third of responding fathers reported a diagnosis of DM1. Parental disease burden may have contributed to caregivers’ responses. Agreement between affected caregivers and child was lower than the agreement between unaffected caregivers and child.
There are other possible factors contributing to the level of agreement among patients and caregivers. Proxy bias, age and development differences between parent and child, and the influence of a child’s disease on a caregiver’s own life may all have contributed to the level of agreement among the two groups. It may be that caregivers relate more to physical or extrinsic issues compared to other issues their child faces. Many of the themes with higher levels of agreement were themes associated with the outward appearance of the disease (urinary or bowel control; limitations of mobility and ambulation; hip, thigh, knee weakness) and many of the themes with lower levels of agreement were themes associated with more intrinsic manifestations of the disease (problems with vision, hearing, smell; decreased social situation performance; difficulty thinking). Other studies comparing parent-proxy surveys and child reported surveys have found similar patterns of discordance with generally good agreement between parents and their child in physical activity domains and poor agreement in domains reflecting social and emotional issues.28
The prevalence and impact of themes affecting individuals with childhood-onset DM1 are distinct from those reported in adult-onset DM1.20 The approach to managing childhood-onset myotonic dystrophy should be different than adult-onset myotonic dystrophy and emphasis should be placed on the symptoms that have the greatest importance to the given population.
Limitations of this study must be considered. Demographic and clinical data was obtained by self or proxy report and not confirmed by a review of medical records. The response rate, though reasonably high for this type of study, was less than 50% for every participating country. There is potential for sampling bias as our study may overrepresent patients with DM1 who have the cognitive capabilities and physical function needed to participate in research and may underrepresent individuals with a more severe disease state. The population group in this study included individuals in the United States, Canada, and Sweden. This patient population probably does not completely represent the world’s population of pediatric-onset DM1 patients. Although individuals in these three countries appeared to provide similar responses, it is unknown how patients outside these countries may have responded to similar survey questions.20 It is plausible that symptoms could have a higher impact on patients residing in less developed countries; further studies would be needed to evaluate this suspicion. National registries were used to identify individuals for the study in United States and Canada. Individuals participate in these registries on a voluntary basis and it is likely that there are individuals with pediatric onset myotonic dystrophy residing in these countries who are not represented. In Sweden, only individuals with pediatric-onset myotonic dystrophy who seek rehabilitation care were represented in this population. This was an exploratory study and subsequent studies should be conducted to confirm the observed associations between symptom impact, age, and CTG repeat length.
This study demonstrates that it is feasible to use a patient-centered approach to identify the most common and most important issues of a pediatric disease even if the disease is rare and has a vast range of clinical manifestations. A systematic approach like this may be beneficial in other disease states. Studies comparing patient-reported severity of symptoms and functional outcome measures is still needed for future clinical trials in pediatric myotonic dystrophy populations.
Supplementary Material
Table 2.
Agreement levels (confidence intervals) between pediatric-onset myotonic dystrophy type 1 patients and their caregiver responses
| Caregiver and Child (n =105) | Affected Caregiver and Child (n = 50) | Unaffected Caregiver and Child (n = 42) | Mother and Child (n = 70) | Father and Child (n = 35) | Caregiver and Child ≥ 18 (n = 51) | Caregiver and Child <18 (n=54) | |
|---|---|---|---|---|---|---|---|
| Limitations with mobility and ambulation | 0.58 (0.44–0.72) | 0.50 (0.22–0.78) | 0.51 (0.23–0.80) | 0.51 (0.29–0.73) | 0.77 (0.52–1.02) | 0.64 (0.46–0.81) | 0.57 (0.39–0.75) |
| Hip, thigh, or knee weakness | 0.49 (0.3–0.68) | 0.40 (0.07–0.73) | 0.36 (−0.03–0.75) | 0.45 (0.20–0.71) | 0.21 (−0.32–0.73) | 0.65 (0.47–0.83) | 0.4 (0.17–0.63) |
| Back, chest, or abdomen weakness | 0.37 (0.14–0.59) | 0.17 (−0.15–0.30) | 0.11 (−0.31–0.54) | 0.16 (−0.10–0.41) | 0.39 (−0.24–1.03) | 0.65 (0.45–0.85) | 0.22 (−0.02–0.45) |
| Problems with hands or fingers | 0.37 (0.14–0.59) | 0.24 (−0.07–0.52) | 0.44 (0.16–0.72) | 0.25 (0.0–0.50) | 0.46 (0.03–0.88) | 0.34 (0.12–0.55) | 0.31 (0.09–0.52) |
| Problems with shoulders or arms | 0.35 (0.14–0.57) | 0.28 (−0.10–0.66) | 0.35 (0.04–0.66) | 0.30 (0.01–0.58) | 0.43 (0.04–0.82) | 0.44 (0.24–0.64) | 0.33 (0.10–0.57) |
| Inability to do activities | 0.30 (0.13–0.47) | 0.49 (0.23–0.75) | 0.47 (0.19–0.74) | 0.47 (0.26–0.69) | 0.71 (0.41–1.0) | 0.41 (0.22–0.61) | 0.54 (0.36–0.72) |
| Myotonia | 0.45 (0.28–0.61) | 0.38 (0.06–0.72) | 0.13 (−0.20–0.45) | 0.28 (0.05–0.51) | 0.24 (−0.32–0.80) | 0.59 (0.41–0.78) | 0.29 (0.08–0.51) |
| Gastrointestinal issues | 0.45 (0.30–0.59) | 0.55 (0.29–0.81) | 0.46 (0.13–0.78) | 0.55 (0.33–0.76) | 0.46 (0.12–0.80) | 0.53 (0.33–0.72) | 0.53 (0.35–0.71) |
| Problems with urinary or bowel control | 0.59 (0.40–0.77) | 0.74 (0.52–0.95) | 0.9 (0.70–1.10) | 0.77 (0.60–0.94) | 1.0 (1.00–1.00) | 0.56 (0.32–0.80) | 0.82 (0.69–0.95) |
| Problems with choking or swallowing | 0.50 (0.33–0.67) | 0.66 (0.43–0.89) | 0.60 (0.30–0.91) | 0.67 (0.49–0.85) | 0.62 (0.22–1.01) | 0.55 (0.37–0.73) | 0.66 (0.50–0.83) |
| Emotional Issues | 0.46 (0.28–0.63) | 0.36 (0.07–0.65) | 0.74 (0.51–0.97) | 0.43 (0.19–0.66) | 0.79 (0.53–1.05) | 0.51 (0.30–0.71) | 0.51 (0.32–0.70) |
| Changed body image | 0.25 (0.01–0.48) | 0.11(−0.12–0.34) | 0.20 (−0.18–0.59) | 0.18 (−0.07–0.43) | 0.46 (0.04–0.87) | 0.24 (0.02–0.47) | 0.25 (0.03–0.47) |
| Difficulty thinking | 0.21 (0–0.41) | 0.34 (0.08–0.59) | 0.16 (−0.23–0.55) | 0.31 (0.08–0.54) | 0.34 (−0.12–0.79) | 0.30 (0.12–0.47) | 0.33 (0.13–0.53) |
| Decreased social situation satisfaction | 0.42 (0.23–0.61) | 0.08 (−0.21–0.37) | 0.24 (0.03–0.45) | 0.16 (−0.03–0.34) | 0.39 (0.05–0.73) | 0.50 (0.30–0.69) | 0.24 (0.07–0.42) |
| Decreased social situation performance | 0.27 (0.21–0.41) | 0.01 (−0.22–0.23) | 0.13 (−0.01–0.28) | 0.07 (−0.10–0.24) | 0.17 (−0.04–0.87) | 0.45 (0.28–0.62) | 0.11 (−0.03–0.25) |
| Impaired sleep or daytime sleepiness | 0.42 (0.27–0.58) | 0.61 (0.37–0.85) | 0.61 (0.35–0.86) | 0.49 (0.28–0.70) | 0.89 (0.78–0.99) | 0.54 (0.37–0.72) | 0.6 (0.43–0.76) |
| Fatigue | 0.34 (0.18–0.50) | 0.42 (0.16–0.68) | 0.52 (0.18–0.87) | 0.49 (0.22–0.67) | 0.74 (0.41–1.07) | 0.49 (0.29–0.68) | 0.52 (0.33–0.71) |
| Pain | 0.39 (0.19–0.59) | 0.51 (0.21–0.82) | 0.56 (0.28–0.84) | 0.39 (0.16–0.63) | 0.83 (0.48–1.17) | 0.63 (0.46–0.80) | 0.49 (0.28–0.69) |
| Problems with vision, hearing, or smell | 0.20 (−0.04–0.44) | 0.29 (−0.04–0.61) | 0.28 (−0.11–0.66) | 0.29 (0.03–0.54) | 0.27 (−0.13–0.67) | 0.38 (0.16–0.60) | 0.25 (0.02–0.48) |
| Communication issues | 0.37 (0.21–0.53) | 0.47 (0.22–0.71) | 0.41 (0.07–0.74) | 0.40 (0.11–0.57) | 0.35 (−0.06–0.77) | 0.38 (0.19–0.57) | 0.44 (0.24–0.63) |
Acknowledgements:
The research is supported by the Myotonic Dystrophy Foundation, The National Institutes of Health (NIAMS K23 AR055947 and K23 NS091511 and NIH NINDS Paul D Wellstone Muscular Dystrophy Cooperative Research Center (MDCRC) Grant). The funding source did not participate in study design, data analysis, or manuscript decisions.
ABBREVIATIONS
- DM1
Myotonic Dystrophy Type 1
- CDM
Congenital Myotonic Dystrophy
- ChDM
Childhood Myotonic Dystrophy
Footnotes
Ethical Publication Statement:
We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Disclosure of Conflicts of Interest:
CC is a site investigator for the AMO Therapeutics clinical trial for myotonic dystrophy and has held an investigator-initiated grant from Valerion Therapeutics. CH is the CEO of the Neuromuscular Institute of Quality-of-Life Studies and Outcome measure Department and provides consultation to the following companies: Cytokinetics, Biogen, Accelron, Ionis, AMO, Regeneron. NEJ reports serving as a consultant or on advisory boards for the following companies: AMO Pharma, FLEX Pharma, AveXis, Cytokinetics, Vertex. The remaining authors have no conflicts of interest.
REFERNCES
- 1.Fu YH, Pizzuti A, Fenwick RG Jr, King J, Rajnarayan S, Dunne PW, et al. An unstable triplet repeat in a gene related to myotonic dystrophy. Science 1992; 255: 1256–58. [DOI] [PubMed] [Google Scholar]
- 2.Mahadevan M, Tsilfidis C, Sabourin L, Shutler G, Amemiya C, Jansen G, et al. Myotonic dystrophy mutation: an unstable CTG repeat in the 3’ untranslated region of the gene. Science 1992; 255: 1253–55. [DOI] [PubMed] [Google Scholar]
- 3.Brook JD, McCurrach ME, Harley HG, Buckler AJ, Church D, Aburatani H, et al. Molecular basis of myotonic dystrophy: expansion of the trinucleotide repeat at the 3’ end of a transcript encoding a protein kinase family member. Cell 1992; 68: 799–808. [DOI] [PubMed] [Google Scholar]
- 4.Harper P Myotonic Dystrophy, 3rd edn. London: W.B. Saunders, 2001. [Google Scholar]
- 5.Koch MC, Grimm T, Harley HG, Harper PS. Genetic risks for children of women with myotonic dystrophy. Am J Hum Genet 1991; 48: 1084–91. [PMC free article] [PubMed] [Google Scholar]
- 6.De Antonio M, Dogan C, Hamroun D, Mati M, Zerrouki S, Eymard B, et al. Unravelling the myotonic dystrophy type 1 clinical spectrum: a systematic registry-based study with implications for disease classification. Rev Neurol (Paris) 2016; 172: 572–580. DOI: [DOI] [PubMed] [Google Scholar]
- 7.Campbell C, Levin S, Siu VM, Venace S, Jacob P. Congenital myotonic dystrophy: Canadian population-based surveillance study. J Pediatr 2013; 163: 120–5. DOI: 10.1016/j.jpeds.2012.12.070. . [DOI] [PubMed] [Google Scholar]
- 8.Zaki M, Boyd PA, Impey L, Roberts A, Chamberlain P. Congenital myotonic dystrophy: prenatal ultrasound findings and pregnancy outcome. Ultrasound Obstet Gynecol 2007; 29: 284–88. DOI: 10.1002/uog.3859. [DOI] [PubMed] [Google Scholar]
- 9.Canavese F, Sussman MD. Orthopaedic manifestations of congential myotonic dystrophy during childhood and adolescence. J Pediatr Orthop 2009: 29:208–13. DOI: 10.1097/BPO.0b013e3181982bf6. [DOI] [PubMed] [Google Scholar]
- 10.Campbell C, Sherlock R, Jacob P. Congenital myotonic dystrophy: assisted ventilation duration and outcome. Pediatrics 2004; 113: 811–16. [DOI] [PubMed] [Google Scholar]
- 11.Johnson NE, Butterfield R, Berggren K, Hung M, Chen W, DiBella D, et al. Disease burden and functional outcomes in congenital myotonic dystrophy. Neurology 2016; 87: 160–67. DOI: 10.1212/WNL.0000000000002845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ekstrom AB, Hakenas-Plate L, Tulinius M, Wentz E. Cognition and adaptive skills in myotonic dystrophy type 1: a study of 55 individuals with congenital and childhood forms. Dev Med Child Neurol 2009; 51: 982–90. DOI: doi: 10.1111/j.1469-8749.2009.03300.x. [DOI] [PubMed] [Google Scholar]
- 13.Angeard N, Huerta E, Jacquette A, Cohen D, Xavier J, Gargiulo M, et al. Childhood-onset form of myotonic dystrophy type 1 and autism spectrum disorder: is there comorbidity. Neuromuscul Disord 2018; 28: 216–21. DOI: 10.1016/j.nmd.2017.12.006. [DOI] [PubMed] [Google Scholar]
- 14.Ekström AB, Hakenäs-Plate L, Samuelsson L, Tulinius M, Wentz E. Autism spectrum conditions in myotonic dystrophy type 1: a study on 57 individuals with congenital and childhood forms. Am J Med Genet B Neuropsychiatr Genet 2008; 147: 918–26. DOI: 10.1002/ajmg.b.30698. [DOI] [PubMed] [Google Scholar]
- 15.Johnson NE, Leubbe E, Eastwood E, Chin N, Moxley RT 3rd, Heatwole CR. The impact of congenital and childhood myotonic dystrophy on quality of life: a qualitative study of associated symptoms. J Child Neurol 2014; 29: 983–86. DOI: 10.1177/0883073813484804. [DOI] [PubMed] [Google Scholar]
- 16.Johnson NE, Ekstrom AB, Campbell C, Hung M, Adams HR, Chen W, et al. Parent-reported multi-national study of the impact of congenital and childhood onset myotonic dystrophy. Dev Med Neurol 2016; 58: 698–705. DOI: 10.1111/dmcn.12948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hilbert JE, Kissel JT, Luebbe EA, Martens WB, McDermott MP, Sanders DB, et al. If you build a rare disease registry, will they enroll and will they use it? Methods and data from the National Registry of Myotonic Dystrophy (DM) and Facioscapulohumeral Muscular Dystrophy (FSHD). Contemp Clin Trials 2012; 33:302–11. DOI: 10.1016/j.cct.2011.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Korngut L, Campbell C, Johnston M, Benstead T, Genge A, Mackenzie A, et al. The CNDR: collaborating to translate new therapies for Canadians. Can J Neurol Sci 2013; 40: 698–704. DOI: 0.1017/S0317167100014943 [DOI] [PubMed] [Google Scholar]
- 19.Terwee CB, Roorda LD, de Vet HC, Dekker J, Westhovens R, van Leeuwen J, et al. Dutch-Flemish translation of 17 item banks from the patient-reported outcomes measurement information system (PROMIS). Qual Life Res 2014; 23: 1733–41. DOI: 10.1007/s11136-013-0611-6. [DOI] [PubMed] [Google Scholar]
- 20.Heatwole C, Bode R, Johnson N, Quinn C, Martens W, McDermott MP, et al. Patient-reported impact of symptoms in myotonic dystrophy type 1 (PRISM-1). Neurology 2012; 79:348–57. DOI: 10.1212/WNL.0b013e318260cbe6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rothman KJ No adjustments are needed for multiple comparisons. Epidemiology. 1990; 1: 43–46 [PubMed] [Google Scholar]
- 22.Althouse AD. Adjust for multiple comparisons, it’s not that simple. The annals of thoracic surgery. 2016; 101(5): 1644–1645 [DOI] [PubMed] [Google Scholar]
- 23.Landis JR, Koch GG. The measurement of observer agreement for categorical data. Biometrics 1977; 33: 159–74. [PubMed] [Google Scholar]
- 24.Gharehbaghi-Schnell EB, Finsterer J, Korschineck I, Mamoli B, Binder BR. Genotype-phenotype correlation in myotonic dystrophy. Clin Genet 1998; 53: 20–26. [DOI] [PubMed] [Google Scholar]
- 25.Gennarelli M, Novelli G, Andreasi Bassi F, Martorell L, Cornet M, Menegazzo E, et al. Prediction of myotonic dystrophy clinical severity based on the number of intragenic [CTG]n trinucleotide repeats. Am J Med Genet 1996: 65: 342–47. [DOI] [PubMed] [Google Scholar]
- 26.Laberge L, Veillette S, Mathieu J, Auclair J, Perron M. The correlation of CTG repeat length with material and social deprivation in myotonic dystrophy. Clin Genet 2007; 71: 59–66. [DOI] [PubMed] [Google Scholar]
- 27.Laberge L, Ptevost C, Perron M, Mathieu J, Auclair J, Gaudreault M, et al. Clinical and genetic knowledge and attitudes of patients with myotonic dystrophy type 1. Public Health Genomics 2010; 13: 424–30. DOI: 10.1159/000316238. [DOI] [PubMed] [Google Scholar]
- 28.Davis E, Nicholas C, Waters E, Cook K, Gibbs L, Gosch A, et al. Parent-proxy and child self-reported health-related quality of life: using qualitative methods to explain the discordance. Qual Life Res 2007; 16: 863–87 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.