

Abstract
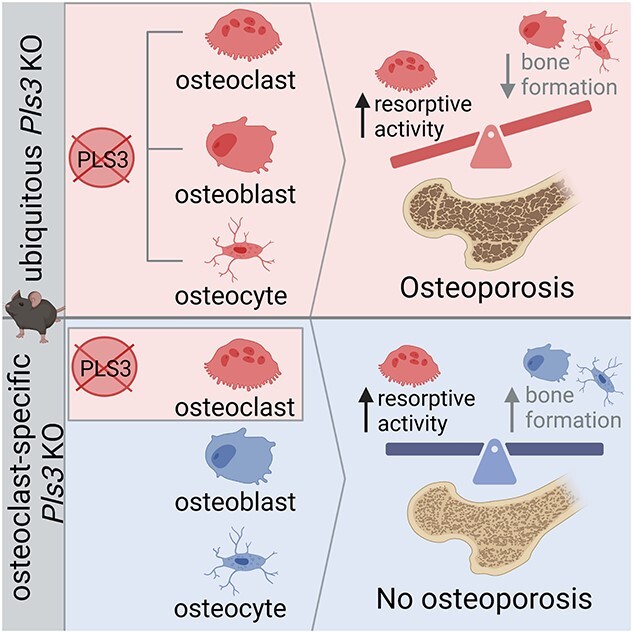
PLS3 loss-of-function mutations in humans and mice cause X-linked primary osteoporosis. However, it remains largely unknown how PLS3 mutations cause osteoporosis and which function PLS3 plays in bone homeostasis. A recent study showed that ubiquitous Pls3 KO in mice results in osteoporosis. Mainly osteoclasts were impacted in their function However, it has not been proven if osteoclasts are the major cell type affected and responsible for osteoporosis development in ubiquitous Pls3 KO mice. Here, we generated osteoclast-specific Pls3 KO mice. Additionally, we developed a novel polyclonal PLS3 antibody that showed specific PLS3 loss in immunofluorescence staining of osteoclasts in contrast to previously available antibodies against PLS3, which failed to show PLS3 specificity in mouse cells. Moreover, we demonstrate that osteoclast-specific Pls3 KO causes dramatic increase in resorptive activity of osteoclasts in vitro. Despite these findings, osteoclast-specific Pls3 KO in vivo failed to cause any osteoporotic phenotype in mice as proven by micro-CT and three-point bending test. This demonstrates that the pathomechanism of PLS3-associated osteoporosis is highly complex and cannot be reproduced in a system singularly focused on one cell type. Thus, the loss of PLS3 in alternative bone cell types might contributes to the osteoporosis phenotype in ubiquitous Pls3 KO mice.
Keywords: genetic animal model, osteoclasts, osteoporosis, Plastin 3, bone resorption
Mutations in the PLS3 gene that result in loss of function of the gene product are known to be responsible for the development of early-onset primary osteoporosis in humans and mice. Osteoporosis is a disease characterized by weakening, brittleness and loss of bones, as well as increased bone fragility and an increased risk of bone fractures. However, it is not clear how the mutations in PLS3 cause osteoporosis and what function PLS3 plays in bone. Recently, it was shown that whole body deletion of Pls3 in mice (ubiquitous Pls3 knockout mice) leads to osteoporosis, characterized by increased bone thickness and stiffness in these mice. This revealed abnormalities in the bone cells that break down bone, called osteoclasts. In particular, their osteoclasts removed significantly more bone than osteoclasts from wild-type mice. Furthermore, osteoclast formation and activity, which are controlled by an important signaling pathway called the NF-κB pathway, were found to be abnormally regulated in the mice’s osteoclasts by whole-body deletion of Pls3. However, it remains unclear whether osteoclasts are the most affected cell type and are responsible for the development of osteoporosis in the mice with whole-body deletion of Pls3.
To find out whether osteoclast function was primarily affected by loss of Pls3, we specifically deleted the Pls3 gene only in osteoclasts and generated so-called osteoclast-specific Pls3 knockout KO mice. These mice only lack Pls3 in their osteoclasts, while Pls3 is still present and functional in the remaining cells and tissues. Remarkably, osteoclasts from these mice removed more bone than osteoclasts from normal mice. However, their bones did not show osteoporosis development in vivo in mice at 12, 24, and 48 wk of age, such as reduced bone strength and stiffness. These results suggest that deletion of Pls3 from osteoclasts alone is not sufficient for the development of osteoporosis. Therefore, additional cells from the bones, such as the bone-forming cells, so-called osteoblasts, or the cells that control the activity of osteoclasts and osteoblasts, so-called osteocytes, could be further impaired in their function by the whole-body Pls3 mice. Therefore, loss of Pls3 from these additional bone cells could also contribute to the cause of osteoporosis. Furthermore, we successfully developed a first antibody against PLS3 to specifically visualize the presence of PLS3 in cells and tissues by immunofluorescence staining.
Graphical Abstract
Graphical Abstract.
Introduction
Osteoporosis is one of the most common skeletal disorders, affecting more than 200 million people worldwide.1 Due to the rising age of the common population, a high number of people, especially above the age of 50, is affected by osteoporosis.2 Thus, osteoporosis is associated with a high economic burden and is considered a public health issue.3,4 This systemic disease, affecting mostly postmenopausal women, Caucasians, and elderly people, is characterized by low BMD, reduced bone mass, and deteriorated bone microarchitecture, causing bone fragility and frequent fractures in the femoral neck, upper limbs, and body of the vertebrae with a T score (BMD score) of ≤ −2.5.5–7 Due to high treatment costs, disease prevention and therapy developments are of great importance.8
Osteoporosis results from the disequilibrium between bone-forming osteoblasts and bone-resorbing osteoclasts, where bone resorption exceeds bone formation.1,9 In this context, it has been shown that mutations in Plastin 3 (PLS3) are causative for BMD quantitative trait locus 18, BMND18; (MIM 300910), a monogenic X-linked, early-onset osteoporosis development in men and postmenopausal women, where the number of PLS3 mutations identified is continuously rising.10,11
The gene encoding the highly conserved PLS3 protein is located on the X-chromosome.10,12–14 PLS3 is an F-actin–binding and F-actin–bundling protein and is ubiquitously expressed in all solid tissues.12,15 Interestingly, in 5% of healthy individuals, it is additionally detected in blood.16,17 PLS3 is expressed in almost all cell types of the bone, including osteoblasts, osteoclasts, osteocytes, and chondrocytes.18–21 Due to its important role in F-actin dynamics, it is essential for actin-dependent cellular processes including vesicle trafficking such as endocytosis and mechanotransduction. Furthermore, as mutations in PLS3 resulted in reduced BMD, it is also suspected to play an essential role in bone metabolism.10,17,22 Moreover, it is speculated that PLS3 is important for Ca2+-dependent mechanotransduction in bone.23
However, it remains elusive, how PLS3 mutations cause osteoporosis and which cellular pathways and cell systems are mainly involved. To elucidate the role of PLS3 in bone diseases, a ubiquitous Pls3 KO mouse model has been generated to study the osteoporotic phenotype.20 Ubiquitous Pls3 KO mice show an osteoporotic phenotype with decreased cortical and trabecular bone parameters and reduced strength and stiffness of the femur and vertebrae. Specifically, the osteoclasts are strongly affected by the loss of PLS3, exhibiting an increased resorptive activity, impaired podosome formation, as well as a disturbed NF-κB pathway, which has been hypothesized to be causative for the enhanced osteoclastogenesis in Pls3 KO osteoclasts.20 Demonstrating these strong PLS3-dependent effects of osteoclast function, here we aim to unravel if the osteoclasts are the main cell type affected by Pls3 loss and if a cell type–specific Pls3 KO in osteoclasts is sufficient to cause a similar osteoporotic phenotype as detected in the ubiquitous Pls3 KO mice. We generated an osteoclast-specific Pls3 KO mouse model driven by LysMCre, which is expressed in the myeloid lineage, from which osteoclasts differentiate.24 This study aims to comprehensively characterize the osteoclasts and the bone phenotype of the generated mouse model and compare these findings to the ubiquitous Pls3 KO mice to gain a deeper understanding of the role of PLS3 in the osteoporosis pathomechanism.
Material and methods
Mouse model—Ethical approval
All mice were housed in the animal facility of the CMMC (Centre for Molecular Medicine Cologne) under a 12-h light/dark cycle with access to food and water ad libitum. Breeding, housing, and experimental use of animals were performed in a specific pathogen-free environment. All animal experiments were approved by the Landesamt für Natur, Umwelt und Verbraucherschutz Nordrhein-Westfalen (LANUV) under the application number: 81-02.04.2020.A196.
Pls3 KO mouse model
Ubiquitous Pls3 KO mice were generated as described in Neugebauer et al.20 Briefly, the B6NTac;B6N-Atm1BrdPls3tm1a (EUCOMM)Wtsi/Wtsi (EMMA-ID: EM:06997, Wellcome Trust Sanger Institute) mouse line were purchased from the Wellcome Trust Sanger Institute and imported into the mouse facility through embryo transfer. Breedings, primers, allele names, and PCR conditions were applied as suggested by the manufacturer. To generate mice with a flipped out mutant allele (tm1c), where exon B of Pls3 gene is flanked by loxP sites, the preconditional mice, containing the complete selection cassette tm1a, were crossbred with FLP-deleter mice.25 To generate ubiquitous Pls3 KO mice, tm1c-expressing mice were then crossbred with ubiquitously expressing CMV-Cre-deleter mice26 to achieve excision of exon 2 of the Pls3 gene (tm1d). Moreover, homo- and heterozygosity for females or males were checked by genotyping for either the WT or the mutant cassette. Genotyping for tm1a, tm1c, tm1d, and Pls3 was performed as described by Neugebauer et al.20. Congenic WT C57BL/6 N mice were used as controls. All mice were held on a congenic C57BL/6 N background.
Study design
The experimenter was blinded regarding genotypes of the mice at all steps until final statistical analysis. To perform micro-computed tomography (microCT) and three-point bending tests (3-PBTs), three different cohorts of animals including each genotype of males and females were sacrificed at 12, 24, and 48 wk of age, respectively. The exact number of animals used for each genotype, sex, and age is given in each figure legend and represented as dots in each graph. Experimenter was blinded regarding cell line genotypes during image acquisition, recordings, and statistical analysis. We conducted all experiments at least in triplicates. To avoid pseudo replication, the mean value per animal and mean value of technical replicates was applied for statistical analysis.
Generation of osteoclast-specific Pls3 KO (Pls3fl/fl;LysMCretg/0) mice
For the generation of osteoclast-specific Pls3 KO (Pls3fl/fl;LysMCretg/0) mice, the Cre-lox system was used. For this, conditional Pls3 KO (Pls3fl) mice, carrying exon 2 of the Pls3 allele flanked by loxP sites (tm1c), were crossbred with LysMCre (LysMCre+/−) mice.24 As LysM expression is restricted to cells of the myeloid lineage, Pls3 will be specifically knocked-out in myeloid progenitor cells such as macrophages and subsequently osteoclasts. The cross-breeding resulted in the generation of Pls3fl/fl;LysMCre+/− (osteoclast-specific Pls3 KO) mice as well as their littermates Pls3fl/fl;LysMCre−/−. Genotyping of WT or mutant cassette was performed, and the following primers and PCR setups were used: Pls3_F: TACGCCATTACTCCCCATCC; Pls3_R: TTTCACACACTCGCCAAACAC; and Cas_R: TCGTGGTATCGTTATGCGCC. For the WT allele, the expected amplicon size was 581 bp, and for the mutant allele, the size was 311 bp. PCR conditions were 5 min 94 °C, 30 s 94 °C, 30 s 58 °C, 45 s 72 °C (repeat 34 cycles), and 5 min 72 °C. To check for the presence of heterozygous LysMCre, following primer setup and PCR conditions were applied: LysM_F: CTCTAGTCAGCCAGCAGCTG and LysM_R: ATGTTTAGCTGGCCCAAATGT. The expected amplicon size was 350 bp, and PCR conditions were similar to conditions described before. The Pls3fl/fl;LysMCre−/− (called Pls3fl/fl) littermates were used as controls. All mice were held on a congenic C57BL/6 N background.
Preparation of murine femora and spines
Preparation of femora and spines was performed as previously described.20 Briefly, mice were sacrificed, and the skin was removed around the hind limbs. To expose the hip joint, hind limb muscles and fascia were removed. With the forceps, the neck of the femur was grasped, and the hip joint was carefully dislocated by applying upward force, and attached ligaments were cut. The hip with femur and tibia was placed into DPBS, and the remaining skin and ligaments were carefully removed. Then the tibia was cut at the knee from the femur. For the spine isolation, the ribs were removed and the spine was carefully cleaned. The femur and the spine were wrapped in gauze tape, which was soaked in DPBS, and stored at −20 °C until analyzed.
Micro-computed tomography
For nondestructive three-dimensional evaluation of bone microstructure in the femur, as well as in the spine, microCT measurements were performed as previously described using a Scanco μCT35 scanner (Scanco Medical AG, Bassersdorf, Switzerland).20,27 Briefly, scans of femora in 0.9% NaCl including protease inhibitors (Sigma-Aldrich, Steinheim, Germany) were captured at a voxel size of 7 × 7 × 7 μm, 70-kVp tube voltage, 114-μA tube current, and 400-ms integration time. To extract the trabecular bone, a global threshold of 22.0% was set. For the cortical bone, a threshold of 26.0% was applied. Trabecular bone was analyzed in the distal metaphysis in a volume situated 2000 –1000 μm proximal of the growth plate, which included only secondary spongiosa. For assessment of cortical bone, a 750 μm long volume was evaluated at the femoral midshaft. Trabecular parameters included bone volume fraction (BV/TV, %), trabecular thickness (Tb.Th, mm), trabecular number (Tb.N, 1/mm), trabecular separation (Tb.Sp, mm), and connectivity density (Conn.D, 1/mm3). At the diaphysis, cortical area (Ct.Ar, mm2), tissue area (Tt.Ar, mm2), cortical area fraction (Ct.Ar/Tt.Ar, %), cortical thickness (Ct.Th, mm), and marrow area (Ma.Ar, mm2) were assessed.
The microCT measurement at the spine was performed on the fourth, and fifth lumbar vertebrae (L4 and L5), as well as on the 12th and 13th thoracic vertebrae (T12 and T13). Scans of the spines in 0.9% NaCl were performed at a voxel size of 12 × 12 × 12 μm, 70-kVp tube voltage, 114-μA tube current, and 400-ms integration time. For the spine, 50% of the central body was assessed (± 25% up and down from the mid slice), segmenting only the trabecular structure, and the data were globally thresholded with 22.5%. We determined the same parameter as for trabecular bone at the femur. In addition, BMD (mgHA/cm3), cross-sectional area vertebral foramen (mm2), vertebral body height (mm), and the diameter of the vertebral foramen in anterior–posterior (A/P, mm) as well as medial-lateral (M/L, mm) direction were evaluated.
Three-point bending test
Subsequently, femora were subjected to 3-PBT using a material testing machine (Z2.5/TN1S; Zwick; Germany) as previously described.28 Briefly, femora were loaded at mid-diaphysis in anterior–posterior direction using a 100-N load cell. The distance between support points, which had a diameter of 1.5 mm, was 5 mm. After preloading at 0.1 N, 0.05 mm/s, a loading rate of 1 mm/min was applied until failure. Breaking force (N) and deformation (mm), stiffness (N/mm), and energy (mJ) were measured from the load-deformation curve. Ultimate stress (MPa) and strain, elastic modulus (MPa), as well as energy density (mJ/mm3) were calculated based on the area moments of inertia obtained from microCT measurements.
Radiographic studies and kyphosis index determination
Mice were sacrificed and placed on a radiographic cassette with a radiographic film. The exposure was done at 55 kV for 30 s. The radiographs were used to determine the kyphosis index (KI). For this, a line was drawn between the caudal margin of the last cervical vertebra to the caudal margin of the sixth lumbar vertebra, which corresponded to the cranial border of the wing of ilium. Then a line, placed perpendicular to this line, was drawn from the dorsal edge of the vertebra, where the curvature showed to be greatest. The KI was calculated by dividing the distance of the line between the vertebra by the distance of the line from this line to the curvature.29
Osteoclast differentiation from spleen
Preparation of primary osteoclasts from spleen was performed as described by Boraschi-Diaz and Komarova.30 Briefly, spleens of 4-wk-old mice were aseptically removed, compressed using a plunger, and filtered through a strainer. After centrifugation, the pellet was dissolved in Red Cell Lysis Buffer, followed by two washing steps of the pellet by two centrifugation step in fresh medium. The cell pellet was resuspended in RPMI-1640 medium (ThermoFisher, #A10491091) + 10% FCS + penicillin/streptomycin. Differentiation of cells was initiated by the addition of 500 ng/mL M-CSF. On the next day, the culture medium was collected and centrifuged, and the pellet was resuspended in culture medium supplemented with M-CSF (250 ng/mL) (PeproTech, #315–02) and RANK-L (500 ng/mL) (PeproTech, #315–11) and added to the original culture flask. Cultures were maintained for 8 more days at 37 °C in an atmosphere of 5% CO2/air, and the medium, supplemented with M-CSF and RANKL, was replaced every 2–3 d.
Primary cell cultures using murine embryonic fibroblasts
Primary murine embryonic fibroblasts (MEFs) were isolated and cultured as described.31
Western blot analysis
Proteins from cell cultures were collected and lysed in RIPA buffer containing cOmplete mini protease inhibitor cocktail (Sigma-Aldrich, #11836153001). Protein concentrations were measured using Bradford reagent. Western blot analysis was performed according to standard protocols. For analysis, the following primary antibodies were used: anti-PLS3 (Eurogentec [17; 1238], anti-N-terminal PLS3 [this manuscript; 3772], anti-PLS3 [Human Protein Atlas, HPA020433], and anti-beta-actin HRP-conjugated (Proteintech Cat# HRP-60008). Signal was detected with rabbit-HRP-conjugated secondary antibody (Cell Signaling Technology Cat# 7074). ACTB was used as internal standard, and the chemiluminescence reagent was used to develop the immunosignals (Thermo Scientific).
Development of an immunofluorescence staining–competent and specific PLS3 antibody
To generate a PLS3-specific antibody, we expressed an 87 AA stretch with the lowest homologies to plastin-1 (64% identities) and to plastin-2 (54% identities). This region (Mus musculus; NP_001159925; AA4..AA90) was expressed with a BM40 signal peptide, a Twin-Strep-tag®, and a carrier protein. Prior to the immunization, the Twin-Strep-tag® was removed by thrombin digestion. Recombinant PLS3 in PBS was used to immunize two rabbits (Davids Biotechnologie GmbH). After 2 mo, sera were collected, and the antibodies affinity purified via a PLS3 affinity column. Tag-free PLS3 was coupled to CNBr-activated Sepharose (GE Healthcare), and after applying the sera to the column, the specific antibodies were eluted with 150 mM NaCl, 0.1 M glycine–HCl, pH 2.5, and immediately neutralized with 1 M Tris–HCl, pH 8, then dialyzed against PBS. The two antibodies were tested and validated by Western blot analysis and immunohistochemistry.
Immunohistochemistry
For immunofluorescence staining, differentiated osteoclasts and MEFs were fixed with 4% PFA and incubated with primary antibody at 4 °C O/N. Incubation of the secondary antibody was performed for 2 h at RT in the dark. The following antibodies were used: anti-N-terminal PLS3 1: 300; Phalloidin, AlexaFluor 647 1:400 (Thermo Fisher Scientific, #A22287); anti-PLS3 1:200 (17 Eurogentec, 1238); anti-PLS3 1:200 (Human Protein Atlas, HPA020433); Hoechst (NucBlue) 1 drop/500 μL (Thermo Fisher Scientific, #R37605); and rabbit AlexaFluor 488 1:300 (Thermo Fisher Scientific, #A21206). Cells were mounted in Mowiol (Sigma, #81381).
Image acquisition and analysis
Fluorescence images and immunofluorescence stainings were acquired with Zeiss microscope (AxioImager.M2) supplied with the Apotome.2 system to mimic a confocal microscope. Images were acquired as Z-stacks 40× oil objective.
Resorption pit assay
Resorption pit assay was performed as previously described.20 Osteoclast precursor cells from spleen-derived macrophages were differentiated into mature osteoclasts for 10 d in RPMI-1640 medium with the supplementation of 250 ng/μm M-CSF and 500 ng/μL RANK-L. Afterward, 250 000 osteoclasts/mL were transferred onto bovine bone slice (boneslices.com), placed in 96-well plates, and allowed to resorb bone for 7 d with the regular exchange of medium supplemented with M-CSF and RANK-L. For the staining of resorption pits, cells were removed by ultrasonication, followed by toluidine blue (Sigma-Aldrich, #T3260) staining. The quantification of the resorption pit area was conducted with the ZEN software (RRID:SCR_013672) (Zeiss).
Statistics
Statistical analyses were performed using the software programs GraphPad Prism 9 (RRID:SCR_002798) and Excel2013 (RRID:SCR_016137) (Microsoft). For equal variances, two-tailed Student t-tests was used; for unequal variances of two independent groups, the Mann–Whitney U-test was applied. The logarithmic resorbed area was evaluated by two-way ANOVA with factors genotype and animal nested within genotype. To guard against type-I-error inflation in pairwise comparisons, Tukey’s HSD method was applied. Notable, the corresponding inference space is narrow,32 i.e. inferences pertain to the specific sample of animals. Specific tests, sample size, data representation, and P-values are indicated in figure legends. Significance thresholds were: P < .05*, P < .01**, P < .001***, and ns = not significant. As indicated, the data are presented as mean ± SEM or as mean ± SD. The box plots show all individual data points with the median as a line, interquartile range (25th–75th percentile), and min to max as whiskers.
Results
Generation of osteoclast-specific Pls3 KO mice
We previously demonstrated that the ubiquitous Pls3 KO in mice results in osteoporosis and strongly impairs the osteoclast function.20 To reveal if the osteoclasts are the major and solely cell system affected by the loss of Pls3 and are responsible for the development of osteoporosis, we generated an osteoclast-specific Pls3 KO mouse line. To achieve osteoclast-specific Pls3 KO, conditional Pls3-floxed mice (Pls3fl/fl) were crossbred with LysMCretg/0 mice, resulting in the specific deletion of Pls3 in cells of the myeloid lineage, which give rise to osteoclasts during development.24 Additional crossbreedings for three generations were conducted to result in the generation of 50% female Pls3fl/fl;LysMCretg/0 or male Pls3fl;LysMCretg/0 mice, defined as osteoclast-specific Pls3 KO mice, and 50% female Pls3fl/fl or male Pls3fl mice, defined as Pls3-floxed mice. Genotyping was used to differentiate Pls3fl/fl;LysMCretg/0 or Pls3fl;LysMCretg/0 from Pls3fl/fl or Pls3fl mice and confirmed the successful osteoclast-specific Pls3 KO for the deleted and the Pls3-floxed allele (Supplemental Figure S1A).
The osteoclast-specific Pls3 KO mice were viable and did not show weight differences compared to Pls3-floxed mice (data not shown). To assess bone-specific morphological changes of the spine, the KI29 was determined in 12-wk-old female and male osteoclast-specific Pls3 KO mice in comparison to Pls3-floxed littermates and WT mice. For both newly generated mouse groups, no kyphosis phenotype was identified in comparison to WT mice (Supplemental Figure S1B). Since both female and male Pls3-floxed mice were unaffected compared to WT mice, these were used as control littermates for further studies.
Characterization of osteoclasts
We hypothesize that PLS3 plays an important role in osteoclast differentiation and bone resorption due to the previously identified effect on the osteoclasts’ resorptive activity, podosome formation, and NF-κB pathway in ubiquitous Pls3 KO mice.20
To demonstrate that Pls3 was specifically and successfully deleted in osteoclasts of osteoclast-specific Pls3 KO mice, spleen-derived primary differentiated osteoclasts were cultured and protein lysates were analyzed by Western blotting. PLS3 protein was absent in the differentiated osteoclast-specific Pls3 KO osteoclasts (Figure 1A).
Figure 1.
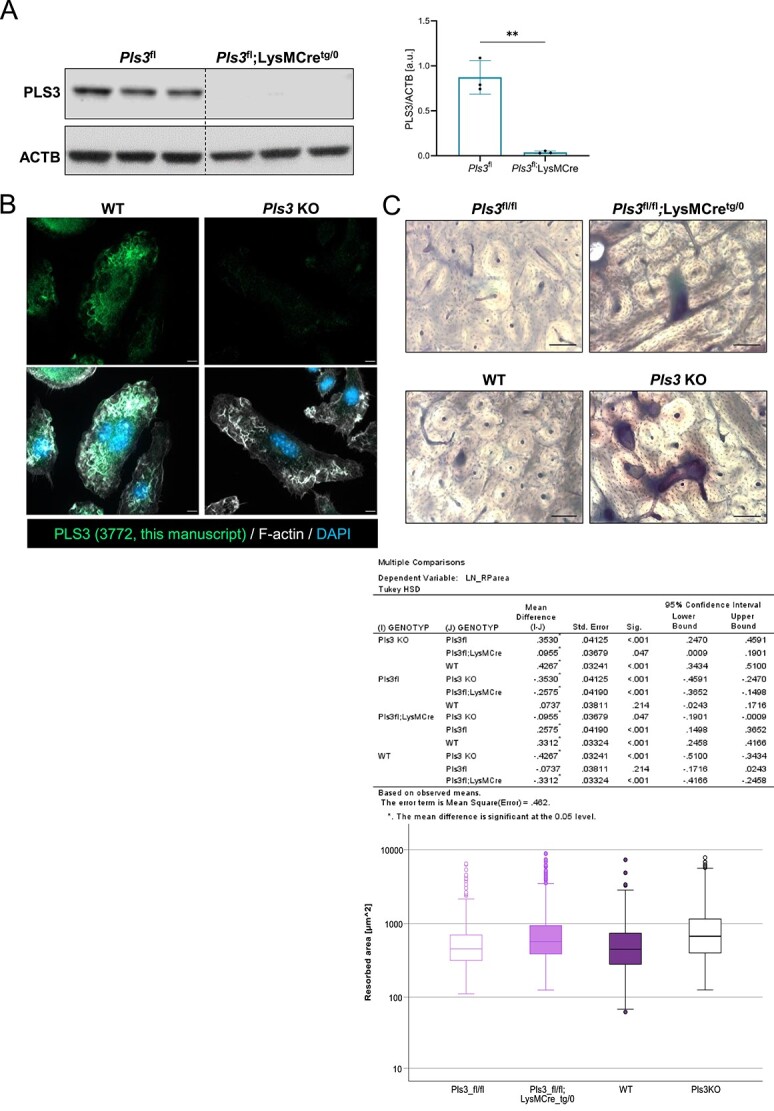
Characterization of Pls3fl/fl;LysMCretg/0 osteoclasts and validation of the specificity of a newly developed polyclonal PLS3 antibody. (A) Protein levels of PLS3 and the housekeeper ACTB in male Pls3fl and Pls3fl;LysMCretg/0 osteoclasts. Quantification of PLS3 protein levels relative to ACTB confirmed the osteoclast-specific Pls3 deletion in the osteoclasts of these mice. N = 3. Results are shown as mean ± SD. Statistical test: Unpaired two-tailed Student’s t-test **p < 0.01 (B) immunofluorescence staining of differentiated WT control and Pls3 KO osteoclasts with a newly developed PLS3 antibody (this manuscript, 3772) confirmed antibody specificity. PLS3 staining resulted in filamentous structures in WT control osteoclasts, whereas no signal was detected in Pls3 KO osteoclasts. The actin cytoskeleton was stained with F-actin and nuclei were stained with DAPI. (C) Resorption pit assay of primary spleen-derived osteoclasts from 1-mo-old mice cultivated on bovine bone slices for 7 d and stained with toluidine blue. Resorbed areas of Pls3fl/fl, Pls3fl/fl;LysMCretg/0, WT and, Pls3 KO were compared to each other. Scale bar: 10 μm. N = 3-7 animals per genotype, n > 90 resorption pits per animal. Results of multiple comparison analyses are shown in the table below and in the graphical representation beneath. Statistical test: Log resorbed area was evaluated by two-way ANOVA with factors genotype and animal nested within genotype. To guard against type-I-error inflation in pairwise comparisons, Tukey‘s HSD method was applied. Notable, the corresponding inference space is narrow,32 i.e. inferences pertain to the specific sample of animals.
Besides, we demonstrated that PLS3 abundancy was equally absent or present in osteoclasts derived from bone marrow of 12-wk-old osteoclast-specific Pls3 KO mice and their Pls3-floxed controls, compared to spleen-derived osteoclasts from ubiquitous Pls3 KO or osteoclast-specific Pls3 KO mice as compared to WT mice (Figure 1A, Supplemental Figure S1C–D).
Based on these data and because the number of osteoclasts derived from precursor cells from spleen was higher than from bone marrow, we were not only able to reduce the number of animals but also to conduct our experiments much faster and to lower costs.
Moreover, we performed immunofluorescence staining of osteoclasts with a newly developed polyclonal PLS3 antibody, which showed no signals in the Pls3 deleted but in WT control osteoclasts (Figure 1B). Importantly to note, our old polyclonal PLS3 antibody developed by Eurogentec (#1238),17 as well as other antibodies available on the market (anti-PLS3 Antibody from Human Protein Atlas [HPA020433]), failed to show PLS3-specific signals in MEFs in contrast to the newly developed PLS3 antibody (this manuscript, 3772) (Supplemental Figure S2A–C). Nonetheless, the former PLS3 antibody from Eurogentec and the newly developed one, but not from Human Protein Atlas, are specific for Western blots (Supplemental Figure S2D–F).
As resorptive activity of osteoclasts from ubiquitous Pls3 KO mice was strongly affected,20 we conducted resorption pit assay with osteoclast-specific Pls3 KO osteoclasts. In addition, we included osteoclasts from WT and ubiquitous Pls3 KO mice to directly compare the results to the osteoclast-specific Pls3 KO mice.
Osteoclasts from osteoclast-specific Pls3 KO mice produced abundant and notably increased resorption pit areas, exhibiting a 49% increase (1.5-fold increase) in comparison to their control littermates (Figure 1C). In line with previous results,20 ubiquitous Pls3 KO osteoclasts showed a 65% increase (1.7-fold increase) in resorption pit area and thus an enhanced resorptive activity compared to the WT controls. The strong increase in resorptive activity of osteoclast-specific Pls3 KO and ubiquitous Pls3 KO osteoclasts occurred likewise when comparing to Pls3-floxed or WT osteoclasts; there was no difference between Pls3-floxed and WT osteoclasts resorptive pit areas (Figure 1C). These results demonstrate the successful generation of osteoclast-specific Pls3 KO mice and confirm that PLS3 regulates the resorptive activity in osteoclasts.
Absence of Pls3 in osteoclasts does not cause osteoporosis in the femur
Increased bone resorptive activity of osteoclasts is known to directly influence the bone quality, leading to a weakening of the bone microarchitecture and decreasing bone strength and stiffness, and might subsequently result in osteoporosis.33,34
To evaluate the influence of the osteoclast-specific Pls3 KO on bone morphology, the trabecular and cortical bone microstructure of femora and spines were analyzed by microCT, and the mechanical properties of the femora were examined by 3-PBT. To this end, the same bone morphometric measurements previously performed in 12-wk-old ubiquitous Pls3 KO mice were applied on osteoclast-specific Pls3 KO mice and their control littermates, and the measurements were compared.20 Female and male mice were analyzed separately, to detect possible sex differences characteristic for osteoporosis.
No marked changes in trabecular parameters of 12-wk-old female and male osteoclast-specific Pls3 KO mice were measured, except of a significant decrease in the trabecular thickness (Tb.Th) (18.3%, P < .01) in female osteoclast-specific Pls3 KO mice (Figure 2A, Supplemental Figure S3A, Table 1). In contrast, for female and male ubiquitous Pls3 KO mice, the Tb.Th was unaffected, but the trabecular number (Tb.N) and connectivity density (Conn.D) were significantly decreased, and the trabecular separation (Tb.Sp) significantly increased (Table 1, first column).20
Figure 2.

Selected microCT data of the femora from 12- and 24-wk-old female Pls3fl/fl;LysMCretg/0 mice in comparison to their Pls3fl/fl littermates. Shown are bone volume fraction (BV/TV, %), trabecular thickness (Tb.Th, mm), trabecular number (Tb.N, 1/mm), trabecular separation (Tb.Sp, mm), cortical area (Ct.Ar, mm2), ratio of cortical area to tissue area (Ct.Ar/Tt.Ar, %), and cortical thickness (Ct.Th, mm) for (A) 12-wk-old female mice and (B) for 24-wk-old female mice. N = 8 of 12-wk-old female Pls3fl/fl;LysMCretg/0 mice, N = 11 of 12-wk-old female Pls3fl/fl mice; N = 10 of 24-wk-old female Pls3fl/fl;LysMCretg/0 mice; N = 13 of 24-wk-old female Pls3fl/fl mice. All results are shown as box plots, representing individual data points with median as a line, interquartile range (25th–75th percentile), and min to max as whiskers. *P < .05, **P < .01, ***P < .001, ns = not significant. Statistical test: Mann–Whitney U test.
Table 1.
microCT data of the femora from ubiquitous Pls3 KO mice compared to osteoclast-specific Pls3 KO mice.
| microCT of femur | 12 wk | 24 wk | 48 wk | ||
|---|---|---|---|---|---|
|
Pls3 KO vs WT |
Pls3 fl/fl;LysMCre vs Pls3fl/fl |
Pls3 fl/fl;LysMCre vs Pls3fl/fl |
Pls3 fl/fl;LysMCre vs Pls3fl/fl |
||
| Neugebauer et al.20 | This study | ||||
| BV/TV (%) | Female | * * decrease | ns | * increase | ns |
| Male | ns | ns | ns | ns | |
| Tb.Th (mm) | Female | ns | * * decrease | ns | ns |
| Male | ns | ns | ns | ns | |
| Tb.N (1/mm) | Female | * * * decrease | ns | ns | ns |
| Male | * * * decrease | ns | ns | ns | |
| Tb.Sp (mm) | Female | * * * increase | ns | ns | ns |
| Male | * * * increase | ns | ns | ns | |
| Conn.D (1/mm3) | Female | * decrease | ns | * increase | ns |
| Male | * decrease | ns | ns | ns | |
| Ct.Ar (mm2) | Female | * * * decrease | ns | * increase | ns |
| Male | * decrease | ns | ns | ns | |
| Tt.Ar (mm2) | Female | * increase | ns | ns | ns |
| Male | ns | ns | ns | ns | |
| Ct.Ar/Tt.Ar (%) | Female | * * * decrease | ns | * increase | ns |
| Male | * * * decrease | ns | ns | ns | |
| Ct.Th (mm) | Female | * * * decrease | ns | * * increase | ns |
| Male | * * * decrease | ns | ns | ns | |
| Ma.Ar (mm2) | Female | * * increase | ns | ns | ns |
| Male | * * increase | ns | ns | ns | |
Previously, trabecular and cortical bone parameters, measured by microCT, were determined in 12-wk-old female and male ubiquitous Pls3 KO mice and compared to WT mice.20 In this study, similar parameters were determined for femora of 12-, 24-, and 48-wk-old osteoclast-specific Pls3 KO female and male mice in comparison to their Pls3-floxed control littermates. Shown are ratio of bone volume fraction (BV/TV, %), trabecular thickness (Tb.Th, mm), trabecular number (Tb.N, 1/mm), trabecular separation (Tb.Sp, mm), connectivity density (Conn.D, 1/mm3), cortical area (Ct.Ar, mm2), total area (Tt.Ar, mm2), ratio of Ct.Area to tissue area (Ct.Ar/Tt.Ar, %) ratio, cortical thickness (Ct.Th, mm), and marrow area (Ma.Ar, mm2). N = 10 of 12-wk-old ubiquitous Pls3 KO mice; N = 10 of 12-wk-old WT mice; N = 8 of 12-wk-old female Pls3fl/fl;LysMCretg/0 mice; N = 11 of 12-wk-old female Pls3fl/fl mice; N = 12 of 12-wk-old male Pls3fl;LysMCretg/0 mice; N = 11 of 12-wk-old male Pls3fl mice; N = 10 of 24-wk-old female Pls3fl/fl;LysMCretg/0 mice; N = 13 of 24-wk-old female Pls3fl/fl mice. N = 13 of 24-wk-old male Pls3fl;LysMCretg/0 mice; N = 9–11 of 24-wk-old male Pls3fl mice; N = 5 of 48-wk-old female Pls3fl/fl;LysMCretg/0 mice; N = 3–8 of 48-wk-old female Pls3fl/fl mice; N = 9 of 48-wk-old male Pls3fl;LysMCretg/0 mice; N = 9 of 48-wk-old male Pls3fl mice. All results are shown as mean ± SEM. *P < .05, **P < .01, ***P < .001, ns = not significant. An increase or decrease in comparison to the control group is indicated. Statistical test: Mann–Whitney U test.
Similarly, all cortical parameters measured in femora of 12-wk-old female and male osteoclast-specific Pls3 KO mice remained unaffected compared to their control littermates (Figure 2A, Supplemental Figures S3 and S4A, Table 1). These findings did not resemble the osteoporotic phenotype, detected in the femora of aged- and sex-matched ubiquitous Pls3 KO mice, which showed significant changes in cortical parameters compared to the WT controls (Table 1, first column).20
Consequently, we included 24- and 48-wk-old female and male osteoclast-specific Pls3 KO and control littermates in our study to test whether phenotypic changes develop at a later time-point. Similar to the results in the 12-wk-old osteoclast-specific Pls3 KO mice, a bone phenotype was absent in 24-wk-old female and male mice (Figure 2B, Supplemental Figure S4B, Table 1). Opposing to the results obtained in 12-wk-old ubiquitous Pls3 KO mice, an increase in the BV/TV (103.1%, P < .05), Conn.D (116.3%, P < .05), Ct.Ar (4.5%, P < .05), Ct.Ar/Tt.Ar (4.5%, P < .05), and Ct.Th (5.4%, P < .01) was detected for female 24-wk-old osteoclast-specific Pls3 KO mice (Figure 2B, Supplemental Figure S4B, Table 1). Unexpectedly, these changes were not detected in 48-wk-old female and male osteoclast-specific Pls3 KO mice compared to control littermates (Supplemental Figures S3 and S4C, Table 1).
Absence of Pls3 in osteoclasts does not cause osteoporosis in the spine
Additionally, Pls3-dependent changes in the spine were investigated by microCT measurements in 12-wk-old female and male osteoclast-specific Pls3 KO mice, focusing on the trabecular bone of the lumbar regions L4 and L5, and the thoracic regions T12 and T13 (Supplemental Figures S5–12, Tables 2 and 3).
Table 2.
microCT data of the vertebrae L4 in 12-, 24-, and 48-wk-old female and male osteoclast-specific Pls3 KO mice in comparison to their Pls3-floxed control littermates.
| microCT of spine L4 | 12 wk | 24 wk | 48 wk | |
|---|---|---|---|---|
|
Pls3 fl/fl;LysMCre vs Pls3fl/fl |
Pls3 fl/fl;LysMCre vs Pls3fl/fl |
Pls3 fl/fl;LysMCre vs Pls3fl/fl |
||
| BV/TV (%) | Female | ns | ns | ns |
| Male | ns | ns | ns | |
| BMD (mgHA/cm3) | Female | ns | ns | ns |
| Male | ns | ns | ns | |
| Tb.N (1/mm) | Female | ns | ns | ns |
| Male | ns | ns | ns | |
| Tb.Sp (mm) | Female | ns | ns | ns |
| Male | ns | ns | ns | |
| Tb.Th (mm) | Female | ns | * increase | ns |
| Male | ns | ns | ns | |
| Conn.D (1/mm3) | Female | ns | ns | ns |
| Male | ns | ns | ns | |
| Vertebral foramen A/P (mm) | Female | ns | * increase | ns |
| Male | ns | ns | ns | |
| Vertebral foramen M/L (mm) | Female | ns | ns | ns |
| Male | ns | ns | ns | |
| Cross-sectional area vertebral foramen (mm2) | Female | ns | ns | ns |
| Male | ns | ns | ns | |
| Vertebral body height (mm) | Female | ns | ns | ns |
| Male | ns | ns | ns | |
Shown are bone volume fraction (BV/TV, %), BMD (mmHA/cm3), trabecular number (Tb.N, 1/mm), trabecular separation (Tb.Sp, mm), trabecular thickness (Tb.Th, mm), and connectivity density (Conn.D, 1/mm3), vertebral foramen A/P (mm), vertebral foramen M/L (mm), cross-sectional area of the vertebral foramen (mm2), and vertebral body height (mm). N = 8 of 12-wk-old female Pls3fl/fl;LysMCretg/0 mice; N = 12 of 12-wk-old female Pls3fl/fl mice; N = 12 of 12-wk-old male Pls3fl;LysMCretg/0 mice; N = 11 of 12-wk-old male Pls3fl mice; N = 11 of 24-wk-old female Pls3fl/fl;LysMCretg/0 mice; N = 13 of 24-wk-old female Pls3fl/fl mice; N = 13 of 24-wk-old male Pls3fl;LysMCretg/0 mice; N = 11 of 24-wk-old male Pls3fl mice; N = 3 of 48-wk-old female Pls3fl/fl;LysMCretg/0 mice; N = 5 of 48-wk-old female Pls3fl/fl mice; N = 5 of 48-wk-old male Pls3fl;LysMCretg/0 mice; N = 4 of 48-wk-old male Pls3fl mice. All results are shown as mean ± SEM. *P < .05, **P < .01, ***P < .001, ns = not significant. An increase or decrease in comparison to the control group is indicated. Statistical test: Mann–Whitney U test.
MicroCT analysis of vertebrae L4 and L5 did not reveal any significant differences in the microstructure for female and male osteoclast-specific Pls3 KO mice, except of a significant increase in the Tb.N (6.0%, P < .01) and a decrease in the Tb.Sp (6.9%, P < .05) in male osteoclast-specific Pls3 KO mice for L5 (Supplemental Figures S5–S8, Tables 2 and 3).
Table 3.
microCT data of the vertebrae L5 in 12-, 24-, and 48-wk-old female and male osteoclast-specific Pls3 KO mice in comparison to their Pls3-floxed control littermates.
| microCT of spine L5 | 12 wk | 24 wk | 48 wk | |
|---|---|---|---|---|
|
Pls3 fl/fl;LysMCre vs Pls3fl/fl |
Pls3 fl/fl;LysMCre vs Pls3fl/fl |
Pls3 fl/fl;LysMCre vs Pls3fl/fl |
||
| BV/TV (%) | Female | ns | ns | ns |
| Male | ns | ns | ns | |
| BMD (mgHA/cm3) | Female | ns | ns | ns |
| Male | ns | ns | ns | |
| Tb.N (1/mm) | Female | ns | ns | ns |
| Male | * * increase | ns | ns | |
| Tb.Sp (mm) | Female | ns | ns | ns |
| Male | * decrease | ns | ns | |
| Tb.Th (mm) | Female | ns | ns | ns |
| Male | ns | ns | ns | |
| Conn.D (1/mm3) | Female | ns | ns | ns |
| Male | ns | ns | ns | |
| Vertebral foramen A/P (mm) | Female | ns | ns | ns |
| Male | ns | ns | ns | |
| Vertebral foramen M/L (mm) | Female | ns | ns | ns |
| Male | ns | ns | ns | |
| Cross-sectional area vertebral foramen (mm2) | Female | ns | ns | ns |
| Male | ns | ns | ns | |
| Vertebral body height (mm) | Female | ns | ns | ns |
| Male | ns | ns | ns | |
Shown are bone volume fraction (BV/TV, %), BMD (mgHA/cm3), trabecular number (Tb.N, 1/mm), trabecular separation (Tb.Sp, mm), trabecular thickness (Tb.Th, mm), and connectivity density (Conn.D, 1/mm3), vertebral foramen A/P (mm), vertebral foramen M/L (mm), cross-sectional area of the vertebral foramen (mm2), and vertebral body height (mm). N = 8 of 12-wk-old female Pls3fl/fl;LysMCretg/0 mice; N = 12 of 12-wk-old female Pls3fl/fl mice; N = 12 of 12-wk-old male Pls3fl;LysMCretg/0 mice; N = 11 of 12-wk-old male Pls3fl mice; N = 11 of 24-wk-old female Pls3fl/fl;LysMCretg/0 mice; N = 13 of 24-wk-old female Pls3fl/fl mice; N = 13 of 24-wk-old male Pls3fl;LysMCretg/0 mice; N = 11 of 24-wk-old male Pls3fl mice; N = 3 of 48-wk-old female Pls3fl/fl;LysMCretg/0 mice; N = 5 of 48-wk-old female Pls3fl/fl mice; N = 5 of 48-wk-old male Pls3fl;LysMCretg/0 mice; N = 4 of 48-wk-old male Pls3fl mice. All results are shown as mean ± SEM. *P < .05, **P < .01, ***P < .001, ns = not significant. An increase or decrease in comparison to the control group is indicated. Statistical test: Mann–Whitney U test.
For the vertebrae T12, the vertebral foramen M/L (3.5%, P < .01) and cross-sectional area of the vertebral foramen (4.8%, P < .05) were markedly increased in male osteoclast-specific Pls3 KO mice (Supplemental Figure S10A, Table 4). Moreover, for the vertebrae T13, an increase in the vertebral foramen M/L (2.1%, P < .05) and vertebral body height (2.8%, P < .05) was measured in 12-wk-old male osteoclast-specific Pls3 KO mice (Supplemental Figure S12A, Table 5). Remaining parameters were unaffected (Supplemental Figures S9–12A, Tables 4 and 5).
Table 4.
microCT data of the vertebrae T12 in 12-, 24-, and 48-wk-old female and male osteoclast-specific Pls3 KO mice in comparison to their Pls3-floxed control littermates.
| microCT of spine T12 | 12 wk | 24 wk | 48 wk | |
|---|---|---|---|---|
|
Pls3 fl/fl;LysMCre vs Pls3fl/fl |
Pls3 fl/fl;LysMCre vs Pls3fl/fl |
Pls3 fl/fl;LysMCre vs Pls3fl/fl |
||
| BV/TV (%) | Female | ns | ns | ns |
| Male | ns | ns | ns | |
| BMD (mgHA/cm3) | Female | ns | ns | ns |
| Male | ns | ns | ns | |
| Tb.N (1/mm) | Female | ns | ns | ns |
| Male | ns | ns | ns | |
| Tb.Sp (mm) | Female | ns | ns | ns |
| Male | ns | ns | ns | |
| Tb.Th (mm) | Female | ns | ns | ns |
| Male | ns | ns | ns | |
| Conn.D (1/mm3) | Female | ns | ns | * increase |
| Male | ns | ns | ns | |
| Vertebral foramen A/P (mm) | Female | ns | ns | ns |
| Male | ns | ns | ns | |
| Vertebral foramen M/L (mm) | Female | ns | ns | ns |
| Male | * * increase | ns | ns | |
| Cross-sectional area vertebral foramen (mm2) | Female | ns | ns | ns |
| Male | * increase | ns | ns | |
| Vertebral body height (mm) | Female | ns | ns | ns |
| Male | ns | ns | ns | |
Shown are bone volume fraction (BV/TV, %), BMD (mgHA/cm3), trabecular number (Tb.N, 1/mm), trabecular separation (Tb.Sp, mm), trabecular thickness (Tb.Th, mm), and connectivity density (Conn.D, 1/mm3), vertebral foramen A/P (mm), vertebral foramen M/L (mm), cross-sectional area of the vertebral foramen (mm2), and vertebral body height (mm). N = 8 of 12-wk-old female Pls3fl/fl;LysMCretg/0 mice; N = 12 of 12-wk-old female Pls3fl/fl mice; N = 12 of 12-wk-old male Pls3fl;LysMCretg/0 mice; N = 11 of 12-wk-old male Pls3fl mice; N = 11 of 24-wk-old female Pls3fl/fl;LysMCretg/0 mice; N = 13 of 24-wk-old female Pls3fl/fl mice; N = 13 of 24-wk-old male Pls3fl;LysMCretg/0 mice; N = 11 of 24-wk-old male Pls3fl mice; N = 3 of 48-wk-old female Pls3fl/fl;LysMCretg/0 mice; N = 5 of 48-wk-old female Pls3fl/fl mice; N = 5 of 48-wk-old male Pls3fl;LysMCretg/0 mice; N = 4 of 48-wk-old male Pls3fl mice. All results are shown as mean ± SEM. *P < .05, **P < .01, ***P < .001, ns = not significant. An increase or decrease in comparison to the control group is indicated. Statistical test: Mann–Whitney U test.
Table 5.
microCT data of the vertebrae T13 in 12-, 24-, and 48-wk-old female and male osteoclast-specific Pls3 KO mice in comparison to their Pls3-floxed control littermates.
| microCT of spine T13 | 12 wk | 24 wk | 48 wk | |
|---|---|---|---|---|
|
Pls3 fl/fl;LysMCre vs Pls3fl/fl |
Pls3 fl/fl;LysMCre vs Pls3fl/fl |
Pls3 fl/fl;LysMCre vs Pls3fl/fl |
||
| BV/TV (%) | Female | ns | ns | ns |
| Male | ns | ns | ns | |
| BMD (mgHA/cm3) | Female | ns | ns | ns |
| Male | ns | ns | ns | |
| Tb.N (1/mm) | Female | ns | ns | ns |
| Male | ns | ns | ns | |
| Tb.Sp (mm) | Female | ns | ns | ns |
| Male | ns | ns | ns | |
| Tb.Th (mm) | Female | ns | ns | ns |
| Male | ns | ns | ns | |
| Conn.D (1/mm3) | Female | ns | ns | ns |
| Male | ns | ns | ns | |
| Vertebral foramen A/P (mm) | Female | ns | ns | ns |
| Male | ns | ns | ns | |
| Vertebral foramen M/L (mm) | Female | ns | ns | ns |
| Male | * increase | ns | ns | |
| Cross-sectional area vertebral foramen (mm2) | Female | ns | ns | ns |
| Male | ns | ns | ns | |
| Vertebral body height (mm) | Female | ns | ns | ns |
| Male | * increase | ns | ns | |
Shown are bone volume fraction (BV/TV [%]),BMD (mgHA/cm3), trabecular number (Tb.N, 1/mm), trabecular separation (Tb.Sp, mm), trabecular thickness (Tb.Th, mm), and connectivity density (Conn.D, 1/mm3); vertebral foramen A/P (mm), vertebral foramen M/L (mm), cross-sectional area of the vertebral foramen (mm2), and vertebral body height (mm). N = 8 of 12-wk-old female Pls3fl/fl;LysMCretg/0 mice; N = 12 of 12-wk-old female Pls3fl/fl mice; N = 12 of 12-wk-old male Pls3fl;LysMCretg/0 mice; N = 11 of 12-wk-old male Pls3fl mice; N = 11 of 24-wk-old female Pls3fl/fl;LysMCretg/0 mice; N = 13 of 24-wk-old female Pls3fl/fl mice; N = 13 of 24-wk-old male Pls3fl;LysMCretg/0 mice; N = 11 of 24-wk-old male Pls3fl mice; N = 3 of 48-wk-old female Pls3fl/fl;LysMCretg/0 mice; N = 5 of 48-wk-old female Pls3fl/fl mice; N = 5 of 48-wk-old male Pls3fl;LysMCretg/0 mice; N = 4 of 48-wk-old male Pls3fl mice. All results are shown as mean ± SEM. *P < .05, **P < .01, ***P < .001, ns = not significant. An increase or decrease in comparison to the control group is indicated. Statistical test: Mann–Whitney U test.
Furthermore, in 24-wk-old female osteoclast-specific Pls3 KO mice, there was a significant increase in Tb.Th (6.5%, P < .05) and vertebral foramen A/P (3.7%, P < .05) measured for L4 (Supplemental Figure S5B, Table 2). In contrast, the remaining measured parameters of L4 and L5 were unaffected in 24-wk-old female and male osteoclast-specific Pls3 KO mice (Supplemental Figures S5–S8, Tables 4 and 5). Likewise, no changes were detected for L4 and L5 of 48-wk-old mice (Supplemental Figures S5–S8C, Tables 2–5).
Similarly, most parameters of the thoracic regions T12 and T13 were unaffected in 24- and 48-wk-old female and male osteoclast-specific Pls3 KO mice (Supplemental Figures S9–12B and C, Tables 4 and 5). Only the Conn.D (56.6%, P < .05) showed a significant increase in 48-wk-old female osteoclast-specific Pls3 KO mice for T12 (Supplemental Figure S9C, Table 4).
Absence of Pls3 in osteoclasts does not cause osteoporosis in the femur as measured by 3-PBT
To analyze the influence of PLS3 on the bone strength, 3-PBT of the femora in 12-wk-old female and male osteoclast-specific Pls3 KO mice was performed and compared to sex-specific ubiquitous Pls3 KO mice (Table 6).20 In osteoclast-specific Pls3 KO females and males, all parameters were unaffected (Figure 3A, Supplemental Figure S13A, Table 6).20 In contrast, ubiquitous Pls3 KO females and males showed a significant decrease in the ultimate stress. In addition, ubiquitous Pls3 KO males showed reduced stiffness (Figure 3A, Table 6). Similar to the results in 12-wk-old osteoclast-specific Pls3 KO mice, no changes were detected in 24- and 48-wk-old mice (Figure 3B, Supplemental Figure S13, Table 6). Overall, female and male osteoclast-specific Pls3 KO mice did not exhibit an osteoporotic phenotype and did not resemble the phenotype of the ubiquitous Pls3 KO mice.20
Table 6.
3-PBT data of the femurs in ubiquitous Pls3 KO mice compared to osteoclast-specific Pls3 KO mice.
| 3-PBT | 12 wk | 24 wk | 48 wk | ||
|---|---|---|---|---|---|
|
Pls3 KO vs WT |
Pls3 fl/fl;LysMCre vs Pls3fl/fl |
Pls3 fl/fl;LysMCre vs Pls3fl/fl |
Pls3 fl/fl;LysMCre vs Pls3fl/fl | ||
| Neugebauer et al.20 | This study | ||||
| Breaking force (N) | Female | ns | ns | ns | ns |
| Male | * * * decrease | ns | ns | ns | |
| Ultimate stress (MPa) | Female | * * decrease | ns | ns | ns |
| Male | * * * decrease | ns | ns | ns | |
| Stiffness (N/mm) | Female | ns | ns | ns | ns |
| Male | * * * decrease | ns | ns | ns | |
| Deformation (mm) | Female | ns | ns | ns | ns |
| Male | ns | ns | ns | ns | |
| E-modulus (MPa) | Female | ns | ns | ns | ns |
| Male | ns | ns | ns | ns | |
Previously, 3-PBT measurements were performed in 12-wk-old female and male ubiquitous Pls3 KO mice and compared to WT mice.20 In this study, similar parameters were determined for femurs of 12-, 24-, and 48-wk-old female and male osteoclast-specific Pls3 KO mice in comparison to their Pls3-floxed control littermates. Shown are breaking force (N), ultimate stress (MPa), stiffness (N/mm), deformation (mm), and elastic modulus (E-modulus, MPa). N = 10 of 12-wk-old Pls3 KO mice; N = 10 of 12-wk-old WT mice; N = 8 of 12-wk-old female Pls3fl/fl;LysMCretg/0 mice; N = 8 of 12-wk-old female Pls3fl/fl mice; N = 8 of 12-wk-old male Pls3fl;LysMCretg/0 mice; N = 4 of 12-wk-old male Pls3fl mice; N = 4 of 24-wk-old female Pls3fl/fl;LysMCretg/0 mice; N = 5 of 24-wk-old female Pls3fl/fl mice; N = 6 of 24-wk-old male Pls3fl;LysMCretg/0 mice; N = 6 of 24-wk-old male Pls3fl mice; N = 5 of 48-wk-old female Pls3fl/fl;LysMCretg/0 mice; N = 8 of 48-wk-old female Pls3fl/fl mice; N = 8 of 48-wk-old male Pls3fl;LysMCretg/0 mice; N = 11 of 48-wk-old male Pls3fl mice. All results are shown as mean ± SEM. *P < .05, **P < .01, ***P < .001, ns = not significant. An increase or decrease in comparison to the control group is indicated. Statistical test: Mann–Whitney U test.
Figure 3.

3-PBT of 12- and 24-wk-old female and male Pls3fl/fl;LysMCretg/0 mice in comparison to their Pls3fl/fl littermates. (A) 12-wk-old and (B) 24-wk-old. Shown are breaking force (N), ultimate stress (MPa), stiffness (N/mm), deformation (mm), and elastic modulus (E-modulus, MPa). N = 8 of 12-wk-old female Pls3fl/fl;LysMCretg/0 mice; N = 8 of 12-wk-old female Pls3fl/fl mice; N = 8 of 12-wk-old male Pls3fl;LysMCretg/0 mice; N = 4 of 12-wk-old male Pls3fl mice; N = 4 of 24-wk-old female Pls3fl/fl;LysMCretg/0 mice; N = 5 of 24-wk-old female Pls3fl/fl mice; N = 6 of 24-wk-old male Pls3fl;LysMCretg/0 mice; N = 6 of 24-wk-old male Pls3fl mice. All results are shown as box plots, representing individual data points with median as a line, interquartile range (25th to 75th percentile), and min to max as whiskers. *P < .05, **P < .01, ***P < .001, ns = not significant. Statistical test: Mann–Whitney U test.
Discussion
To unravel the influence of PLS3 on bone morphology and osteoporosis, recently, a ubiquitous Pls3 KO mouse model has been intensively studied with focus on bone morphometry.20 These findings suggest that ubiquitous Pls3 KO mice, apart from decreased cortical and trabecular bone parameters and impaired strength and stiffness of bones, exhibit disturbed osteoclast formation, activity, and function with increased resorptive activity.20 However, these investigations did not allow the conclusion that osteoclasts are solely responsible for the observed osteoporotic phenotype, and thus, if mainly osteoclasts are affected in their functionality by aberrant PLS3 levels. In the present study, we aimed to unravel if, indeed, the osteoclasts are the main cell type affected in osteoporosis caused by the loss of PLS3. To address this question and to elucidate the role of PLS3 in osteoclast function and bone resorption during postnatal bone remodeling, we successfully generated an osteoclast-specific Pls3 KO mouse model (Figure 1A). This allowed to investigate the isolated effect of Pls3 loss in osteoclasts solely.
Differentiated osteoclasts from osteoclast-specific Pls3 KO mice showed a strong resorptive capacity, which produced large resorptive areas when cultivated on bone slices. The resorptive efficiency was almost as strong as the activity of osteoclasts from ubiquitous Pls3 KO mice (Figure 1C). These observations reflect an increased resorptive function caused by the loss of Pls3 in osteoclasts, irrespective of the PLS3 level in other bone cells. Thus, we can conclude that the specific depletion of PLS3 in osteoclasts is sufficient to cause a hyperresorptive activity in vitro as is observed in osteoporosis in vivo.
Unexpectedly, the osteoclast-specific Pls3 KO mice did not exhibit an osteoporotic phenotype as measured by microCT and 3-PBT at three different ages of the mice (12-, 24-, and 48-wk-old). Due to the strong resorptive activity of the osteoclasts, we expected a similar strong osteoporotic phenotype as observed in the ubiquitous Pls3 KO mice.20 These contrasting outcomes may indicate that the observed osteoporotic phenotype in the ubiquitous Pls3 KO mice was not solely caused by PLS3 level-dependent dysregulation of osteoclast function, but that other cell types of the bone were also affected by the loss of Pls3.20
Indeed, PLS3 is known to be expressed in various bone cell types, such as osteoblasts and osteocytes. In the osteoclast-specific Pls3 KO mice, these cells might potentially compensate for the increased bone resorptive function of osteoclasts to maintain bone homeostasis.18–21 Importantly, the crosstalk and coupling between osteoblasts and osteoclasts regulate bone remodeling, increasing bone formation upon the increase of bone resorption.35–38 Thereby, the hyperactivity of osteoclasts in the osteoclast-specific Pls3 KO might be balanced by the osteoblasts, preserving the bone architecture.
PLS3 is known to regulate the osteoblast mineralization and differentiation through its control of the osteoblasts’ Ca2+ concentrations.13,18,39 Although these crucial functions are unaffected in the osteoblasts of osteoclast-specific Pls3 KO mice, it is likely that the ubiquitous Pls3 KO might directly affect the osteoblasts’ function, being responsible for the contradictory results between the two mouse models. In fact, it was assumed previously that cortical thinning and osteoporosis development in human and mice with PLS3 mutations or Pls3 loss resulted from PLS3-dependent disturbances of the osteoblast-regulated bone mass acquisition and mineralization.20,22,40,41 Additionally, decreased osteoblast differentiation and bone formation, potentially caused by the impairment of Ca2+ regulation of PLS3 in osteoblasts, have been associated with PLS3 mutations or Pls3 loss, concomitantly increasing bone resorption.13,40,42 Likewise, mineralization defects and reduced osteoblast numbers were reported in human and rats with PLS3 mutations, leading to osteoporosis.11,41,43–45 Based on these studies, and due to the absence of an osteoporotic phenotype in the osteoclast-specific Pls3 KO mice, it is very likely that the ubiquitous Pls3 KO equally affects the function of osteoblasts and osteoclasts, therefore interfering with the balance between osteoblast-mediated bone formation and osteoclast-mediated bone resorption, where bone resorption exceeds bone formation.
Likewise, PLS3, through its function in cytoskeleton modification and suggested involvement in dendrite formation, is assumed to be indispensable for osteocytes’ activity and function, thereby regulating the sensing of mechanical stimuli and mechanotransduction of osteocytes. In this way, PLS3 is involved in the regulation of communication and networking between osteocytes, osteoblasts, and osteoclasts, and subsequently controls bone homeostasis.11,46–49 Hence, osteocytes of the osteoclast-specific Pls3 KO mice could be able to respond to the abnormal increased osteoclast function by regulating the osteoblast bone formation. However, this regulatory function might be disturbed upon ubiquitous Pls3 KO, rendering osteocytes incapable to compensate for the increased bone resorptive function of osteoclasts in these mice. In line with this, PLS3 mutations showed to alter osteocyte mechanotransduction, causing the misbalancing of bone homeostasis.11,44 Further studies on PLS3 mutations reported abnormal osteoid accumulations in focal and perilacunar areas of osteocytes, accompanied by an increased osteocyte apoptosis rate, potentially resulting in osteoclast recruitment and osteoclastogenesis to initiate bone resorption.50–52 Thus, dysregulations of the PLS3 level in osteocytes could even augment the effect of the increased osteoclast bone resorption in the ubiquitous Pls3 KO mice, uncoupling bone resorption from bone formation, which contributes loss of trabecular and cortical bone.53–55
Thus, to identify the cell system, which is mostly affected by the loss of Pls3, we believe it is crucial to deeply investigate alternative cell types of the bone, especially osteoblasts and osteocytes, as well as to study the crosstalk between osteoblasts and osteoclasts in the ubiquitous Pls3 KO mice. In this regard, the generation of combinatorial cell–specific Pls3 KO mouse lines will be needed to understand the dependence of specific cells on PLS3 and unveil the pathomechanisms involved.
There are limitations to consider in the present study. No dynamic histomorphometric changes could be analyzed since calcein labeling was not performed to determine the rate of bone formation and mineralization. Additionally, in vivo measurements of the resorptive activity of osteoclasts from osteoclast-specific Pls3 KO mice are lacking in the present study. Thus, in future studies, the coupling between osteoclasts and osteoblasts should further be investigated by in vivo methods such as CTX-1 ELISA to reveal if alternative cell types of the bone, such as osteoblasts are involved in the bone phenotype.
In conclusion, we confirm that PLS3 influences the osteoclast activity by increasing their resorptive function. However, the specific loss of Pls3 in osteoclasts is not sufficient to cause the osteoporotic phenotype in vivo as observed in the ubiquitous Pls3 KO mice.20
Moreover, our results highlight the strong dependence of the whole organism and specifically bone cells on regulated PLS3 levels. We conclude that the development of the PLS3-associated osteoporotic phenotype is highly complex and cannot be reproduced in a system singularly focused on one cell type.
In addition, we were able to develop a novel polyclonal PLS3 antibody, which specifically visualized filamentous structures at the cell edge, colocalizing with F-actin, in WT control osteoclasts and MEFs (Figure 1B, Supplemental Figure 2C). This specific localization of PLS3 to F-actin rich structures at the cell edge, focal adhesions, and stress fibers is in line with previous findings in HEK293 cells, fibroblasts, and U2OS osteoblasts, confirming the specificity of the antibody.13,56
Supplementary Material
Acknowledgments
We thank Jutta Knifka (Department of Orthopedic and Trauma Surgery, University Hospital Cologne, Cologne, Germany and Department of Anatomy I, Medical Faculty, University of Cologne, Cologne, Germany) for conducting the X-ray analysis of the mice. We would like to thank Prof. Martin Hellmich from the Institute of Medical Statistics and Computational Biology, University of Cologne, for conducting the statistical analysis of the data presented in (Figure 1C).
Contributor Information
Ilka Maus, Institute of Human Genetics, University of Cologne, University Hospital of Cologne, 50931 Cologne, Germany; Center for Molecular Medicine Cologne, University of Cologne, 50931 Cologne, Germany.
Maren Dreiner, Institute of Biomechanics and Orthopaedics, German Sport University Cologne, 50933 Cologne, Germany.
Sebastian Zetzsche, Institute of Human Genetics, University of Cologne, University Hospital of Cologne, 50931 Cologne, Germany; Center for Molecular Medicine Cologne, University of Cologne, 50931 Cologne, Germany.
Fabian Metzen, Medical Faculty, Institute for Dental Research and Oral Musculoskeletal Biology, University of Cologne, 50931 Cologne, Germany; Medical Faculty, Center for Biochemistry, University of Cologne, 50931 Cologne, Germany.
Bryony C Ross, Institute of Human Genetics, University of Cologne, University Hospital of Cologne, 50931 Cologne, Germany; Center for Molecular Medicine Cologne, University of Cologne, 50931 Cologne, Germany.
Daniela Mählich, Institute of Biomechanics and Orthopaedics, German Sport University Cologne, 50933 Cologne, Germany.
Manuel Koch, Center for Molecular Medicine Cologne, University of Cologne, 50931 Cologne, Germany; Medical Faculty, Institute for Dental Research and Oral Musculoskeletal Biology, University of Cologne, 50931 Cologne, Germany; Medical Faculty, Center for Biochemistry, University of Cologne, 50931 Cologne, Germany.
Anja Niehoff, Institute of Biomechanics and Orthopaedics, German Sport University Cologne, 50933 Cologne, Germany; Faculty of Medicine, Cologne Center for Musculoskeletal Biomechanics (CCMB), University of Cologne, 50931 Cologne, Germany.
Brunhilde Wirth, Institute of Human Genetics, University of Cologne, University Hospital of Cologne, 50931 Cologne, Germany; Center for Molecular Medicine Cologne, University of Cologne, 50931 Cologne, Germany; Center for Rare Diseases, University of Cologne, University Hospital of Cologne, 50931 Cologne, Germany.
Author contributions
Ilka Maus (Study design, study conduct, data collection, data analysis, data interpretation, drafting manuscript, and review and editing), Maren Dreiner (Study conduct, data collection, data analysis, review and editing), Sebastian Zetzsche (Study conduct, data analysis, review, and editing), Fabian Metzen (Antibody generation, review and editing), Bryony C. Ross (Study conduct, data collection, data analysis, review and editing), Daniela Mählich (Data analysis, review and editing), Manuel Koch (Antibody generation, review and editing), Anja Niehoff (Study design, data collection, data analysis, data interpretation, drafting manuscript, review and editing, supervision), and Brunhilde Wirth (Study design, data interpretation, drafting manuscript, review and editing, supervision)
Supplementary material
Supplementary material is available at JBMR Plus online.
Funding
This project was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation, FOR2722 to B.W. and A.N. (project ID 407176282 and 384170921).
Conflicts of interest
There are no related or potential conflicts of interest.
Data availability
The data that support the findings of this study are available from the corresponding authors upon reasonable request.
References
- 1. Sozen T, Ozisik L, Basaran NC. An overview and management of osteoporosis. Eur J Rheumatol. 2017;4(1):46–56. Epub 20161230. 10.5152/eurjrheum.2016.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. de Villiers TJ, Goldstein SR. Bone health 2022: an update. Climacteric. 2022;25(1):1–3. 10.1080/13697137.2021.1965408. [DOI] [PubMed] [Google Scholar]
- 3. Kaushal N, Vohora D, Jalali RK, Jha S. Prevalence of osteoporosis and osteopenia in an apparently healthy Indian population – a cross-sectional retrospective study. Osteoporos Sarcopenia. 2018;4(2):53–60. 10.1016/j.afos.2018.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Clynes MA, Harvey NC, Curtis EM, Fuggle NR, Dennison EM, Cooper C. The epidemiology of osteoporosis. Brit Med Bull. 2020;133(1):105–117. 10.1093/bmb/ldaa005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Alswat KA. Gender disparities in osteoporosis. J Clin Med Res. 2017;9(5):382–387. 10.14740/jocmr2970w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Harvey N, Dennison E, Cooper C. Osteoporosis: impact on health and economics. Nat Rev Rheumatol. 2010;6(2):99–105. 10.1038/nrrheum.2009.260. [DOI] [PubMed] [Google Scholar]
- 7. Kanis JA. Diagnosis of osteoporosis and assessment of fracture risk. Lancet. 2002;359(9321):1929–1936. 10.1016/S0140-6736(02)08761-5. [DOI] [PubMed] [Google Scholar]
- 8. Svedbom A, Hadji P, Hernlund E, et al. Cost-effectiveness of pharmacological fracture prevention for osteoporosis as prescribed in clinical practice in France, Germany, Italy, Spain, and the United Kingdom. Osteoporosis Int. 2019;30(9):1745–1754. 10.1007/s00198-019-05064-w. [DOI] [PubMed] [Google Scholar]
- 9. Raisz LG. Pathogenesis of osteoporosis: concepts, conflicts, and prospects. J Clin Invest. 2005;115(12):3318–3325. 10.1172/JCI27071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wolff L, Strathmann EA, Muller I, et al. Plastin 3 in health and disease: a matter of balance. Cell Mol Life Sci. 2021;78(13):5275–5301. Epub 20210523. 10.1007/s00018-021-03843-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. van Dijk FS, Zillikens MC, Micha D, et al. PLS3 mutations in X-linked osteoporosis with fractures. N Engl J Med. 2013;369(16):1529–1536. 10.1056/NEJMoa1308223. [DOI] [PubMed] [Google Scholar]
- 12. Lin CS, Chen ZP, Park T, Ghosh K, Leavitt J. Characterization of the human L-plastin gene promoter in normal and neoplastic cells. J Biol Chem. 1993;268(4):2793–2801. 10.1016/S0021-9258(18)53843-6. [DOI] [PubMed] [Google Scholar]
- 13. Schwebach CL, Kudryashova E, Zheng W, et al. Osteogenesis imperfecta mutations in plastin 3 lead to impaired calcium regulation of actin bundling. Bone Res. 2020;8(1):21. 10.1038/s41413-020-0095-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shinomiya H. Plastin family of actin-bundling proteins: its functions in leukocytes, neurons, intestines, and cancer. Int J Cell Biol. 2012;2012:213492 Epub 20120104. 10.1155/2012/213492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Karpova TS, Tatchell K, Cooper JA. Actin-filaments in yeast are unstable in the absence of capping protein or fimbrin. J Cell Biol. 1995;131(6):1483–1493. 10.1083/jcb.131.6.1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lin CS, Lau A, Huynh T, Lue TF. Differential regulation of human T-plastin gene in leukocytes and non-leukocytes: identification of the promoter, enhancer, and CpG island. DNA Cell Biol. 1999;18(1):27–37. 10.1089/104454999315592. [DOI] [PubMed] [Google Scholar]
- 17. Oprea GE, Krober S, McWhorter ML, et al. Plastin 3 is a protective modifier of autosomal recessive spinal muscular atrophy. Science. 2008;320(5875):524–527. 10.1126/science.1155085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fahiminiya S, Majewski J, Al-Jallad H, et al. Osteoporosis caused by mutations in PLS3: clinical and bone tissue characteristics. J Bone Miner Res. 2014;29(8):1805–1814. 10.1002/jbmr.2208. [DOI] [PubMed] [Google Scholar]
- 19. Kamioka H, Sugawara Y, Honjo T, Yamashiro T, Takano-Yamamoto T. Terminal differentiation of osteoblasts to osteocytes is accompanied by dramatic changes in the distribution of actin-binding proteins. J Bone Miner Res. 2004;19(3):471–478. 10.1359/JBMR.040128. [DOI] [PubMed] [Google Scholar]
- 20. Neugebauer J, Heilig J, Hosseinibarkooie S, et al. Plastin 3 influences bone homeostasis through regulation of osteoclast activity. Hum Mol Genet. 2018;27(24):4249–4262. 10.1093/hmg/ddy318. [DOI] [PubMed] [Google Scholar]
- 21. Treurniet S, Eekhoff EMW, Schmidt FN, Micha D, Busse B, Bravenboer N. A clinical perspective on advanced developments in bone biopsy assessment in rare bone disorders. Front Endocri. 2020;11:399. Epub 20200623. 10.3389/fendo.2020.00399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Balasubramanian M, Fratzl-Zelman N, O'Sullivan R, et al. Novel PLS3 variants in X-linked osteoporosis: exploring bone material properties. Am J Med Genet A. 2018;176(7):1578–1586. 10.1002/ajmg.a.38830. [DOI] [PubMed] [Google Scholar]
- 23. Zhong W, Pathak JL, Liang Y, et al. The intricate mechanism of PLS3 in bone homeostasis and disease. Front Endocrinol. 2023;14:1168306. Epub 20230707. 10.3389/fendo.2023.1168306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Clausen BE, Burkhardt C, Reith W, Renkawitz R, Forster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 1999;8(4):265–277. 10.1023/A:1008942828960. [DOI] [PubMed] [Google Scholar]
- 25. Rodriguez CI, Buchholz F, Galloway J, et al. High-efficiency deleter mice show that FLPe is an alternative to Cre-loxP. Nat Genet. 2000;25(2):139–140. 10.1038/75973. [DOI] [PubMed] [Google Scholar]
- 26. Schwenk F, Baron U, Rajewsky K. A cre-transgenic mouse strain for the ubiquitous deletion of loxP-flanked gene segments including deletion in germ cells. Nucleic Acids Res. 995. 23(24):5080–5081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bouxsein ML, Boyd SK, Christiansen BA, Guldberg RE, Jepsen KJ, Muller R. Guidelines for assessment of bone microstructure in rodents using micro-computed tomography. J Bone Miner Res. 2010;25(7):1468–1486. 10.1002/jbmr.141. [DOI] [PubMed] [Google Scholar]
- 28. Blumbach K, Niehoff A, Belgardt BF, et al. Dwarfism in mice lacking collagen-binding integrins alpha2beta1 and alpha11beta1 is caused by severely diminished IGF-1 levels. J Biol Chem. 2012;287(9):6431–6440. Epub 20111230. 10.1074/jbc.M111.283119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Laws N, Hoey A. Progression of kyphosis in mdx mice. J Appl Physiol. 1985;97(5):1970–1977 Epub 20040702. [DOI] [PubMed] [Google Scholar]
- 30. Boraschi-Diaz I, Komarova SV. The protocol for the isolation and cryopreservation of osteoclast precursors from mouse bone marrow and spleen. Cytotechnology. 2016;68(1):105–114. 10.1007/s10616-014-9759-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ackermann B, Krober S, Torres-Benito L, et al. Plastin 3 ameliorates spinal muscular atrophy via delayed axon pruning and improves neuromuscular junction functionality. Hum Mol Genet. 2013;22(7):1328–1347. 10.1093/hmg/dds540. [DOI] [PubMed] [Google Scholar]
- 32. McLean RA, Sanders WL, Stroup WW. A unified approach to mixed linear models. Am Stat. 1991;45(1):54–64 JSTOR. 10.1080/00031305.1991.10475767 Accessed 4 Mar. 2024. [DOI] [Google Scholar]
- 33. Compston J. Bone quality: what is it and how is it measured? Arq Bras Endocrinol Metabol. 2006;50(4):579–585. 10.1590/S0004-27302006000400003. [DOI] [PubMed] [Google Scholar]
- 34. Idris AI, van't Hof RJ, Greig IR, et al. Regulation of bone mass, bone loss and osteoclast activity by cannabinoid receptors. Nat Med. 2005;11(7):774–779. Epub 20050522. 10.1038/nm1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chen X, Wang ZQ, Duan N, Zhu GY, Schwarz EM, Xie C. Osteoblast-osteoclast interactions. Connect Tissue Res. 2018;59(2):99–107. 10.1080/03008207.2017.1290085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pfeilschifter J, Mundy GR. Modulation of type beta transforming growth factor activity in bone cultures by osteotropic hormones. Proc Natl Acad Sci U S A. 1987;84(7):2024–2028. 10.1073/pnas.84.7.2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. O'Brien CA, Plotkin LI, Galli C, et al. Control of bone mass and remodeling by PTH receptor signaling in osteocytes. PLoS One. 2008;3(8):e2942. 10.1371/journal.pone.0002942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Parfitt AM. Bone remodeling. Henry Ford Hosp Med J. 1988;36(3):143–144. [PubMed] [Google Scholar]
- 39. Maeno S, Niki Y, Matsumoto H, et al. The effect of calcium ion concentration on osteoblast viability, proliferation and differentiation in monolayer and 3D culture. Biomaterials. 2005;26(23):4847–4855. 10.1016/j.biomaterials.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 40. Yorgan TA, Sari H, Rolvien T, et al. Mice lacking plastin-3 display a specific defect of cortical bone acquisition. Bone. 2020;130:115062. Epub 20191031. 10.1016/j.bone.2019.115062. [DOI] [PubMed] [Google Scholar]
- 41. Kampe AJ, Costantini A, Levy-Shraga Y, et al. PLS3 deletions lead to severe spinal osteoporosis and disturbed bone matrix mineralization. J Bone Miner Res. 2017;32(12):2394–2404. Epub 20170906. 10.1002/jbmr.3233. [DOI] [PubMed] [Google Scholar]
- 42. Wang L, Zhai Q, Zhao P, et al. Functional analysis of p.Ala253_Leu254insAsn mutation in PLS3 responsible for X-linked osteoporosis. Clin Genet. 2018;93(1):178–181. 10.1111/cge.13081. [DOI] [PubMed] [Google Scholar]
- 43. Hu J, Zhou B, Lin X, et al. Impaired bone strength and bone microstructure in a novel early-onset osteoporotic rat model with a clinically relevant PLS3 mutation. Elife. 2023;12:12. Epub 20230421. 10.7554/eLife.80365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Laine CM, Wessman M, Toiviainen-Salo S, et al. A novel splice mutation in PLS3 causes X-linked early onset low-turnover osteoporosis. J Bone Miner Res. 2015;30(3):510–518. 10.1002/jbmr.2355. [DOI] [PubMed] [Google Scholar]
- 45. Makitie RE, Kampe A, Costantini A, Alm JJ, Magnusson P, Makitie O. Biomarkers in WNT1 and PLS3 osteoporosis: altered concentrations of DKK1 and FGF23. J Bone Miner Res. 2020;35(5):901–912. 10.1002/jbmr.3959. [DOI] [PubMed] [Google Scholar]
- 46. Lv F, Ma M, Liu W, et al. A novel large fragment deletion in PLS3 causes rare X-linked early-onset osteoporosis and response to zoledronic acid. Osteoporos Int. 2017;28(9):2691–2700. 10.1007/s00198-017-4094-0. [DOI] [PubMed] [Google Scholar]
- 47. Qin X, Jiang Q, Nagano K, et al. Runx2 is essential for the transdifferentiation of chondrocytes into osteoblasts. PLoS Genet. 2020;16(11):e1009169. 10.1371/journal.pgen.1009169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pathak JL, Bravenboer N, Klein-Nulend J. The osteocyte as the new discovery of therapeutic options in rare bone diseases. Front Endocrinol. 2020;11:405. 10.3389/fendo.2020.00405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Weinbaum S, Duan Y, Thi MM, You L. An integrative review of mechanotransduction in endothelial, epithelial (renal) and dendritic cells (osteocytes). Cell Mol Bioeng. 2011;4(4):510–537. 10.1007/s12195-011-0179-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wesseling-Perry K, Makitie RE, Valimaki VV, et al. Osteocyte protein expression is altered in low-turnover osteoporosis caused by mutations in WNT1 and PLS3. J Clin Endocrinol Metab. 2017;102(7):2340–2348. 10.1210/jc.2017-00099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Plotkin LI. Apoptotic osteocytes and the control of targeted bone resorption. Curr Osteoporos Rep. 2014;12(1):121–126. 10.1007/s11914-014-0194-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ru JY, Wang YF. Osteocyte apoptosis: the roles and key molecular mechanisms in resorption-related bone diseases. Cell Death Dis. 2020;11(10):846. 10.1038/s41419-020-03059-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Aguirre JI, Plotkin LI, Stewart SA, et al. Osteocyte apoptosis is induced by weightlessness in mice and precedes osteoclast recruitment and bone loss. J Bone Miner Res. 2006;21(4):605–615. Epub 20060405. 10.1359/jbmr.060107. [DOI] [PubMed] [Google Scholar]
- 54. Jilka RL, Noble B, Weinstein RS. Osteocyte apoptosis. Bone. 2013;54(2):264–271. 10.1016/j.bone.2012.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Morse LR, Xu Y, Solomon B, et al. Severe spinal cord injury causes immediate multi-cellular dysfunction at the chondro-osseous junction. Transl Stroke Res. 2011;2(4):643–650. 10.1007/s12975-011-0118-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lyon AN, Pineda RH, Hao le T, Kudryashova E, Kudryashov DS, Beattie CE. Calcium binding is essential for plastin 3 function in Smn-deficient motoneurons. Hum Mol Genet. 2014;23(8):1990–2004. Epub 20131123. 10.1093/hmg/ddt595. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding authors upon reasonable request.