

Abstract
Targeted genome editing in hematopoietic stem and progenitor cells (HSPCs) using CRISPR/Cas9 can potentially provide a permanent cure for hematologic diseases. However, the utility of CRISPR/Cas9 systems for therapeutic genome editing can be compromised by their off-target effects. In this chapter, we outline the procedures for CRISPR/Cas9 off-target identification and validation in HSPCs. This method is broadly applicable to diverse CRISPR/Cas9 systems and cell types. Using this protocol, researchers can perform computational prediction and experimental identification of potential off-target sites followed by off-target activity quantification by next-generation sequencing.
Keywords: Genome editing, CRISPR/Cas9, off-target, Next generation sequencing (NGS), Hematopoietic stem and progenitor cells (HSPCs), GUIDE-seq
1. Introduction
Targeted genome editing using CRISPR/Cas9 can potentially provide a permanent cure for genetic disorders through targeted deletion, precise sequence replacement, or site specific insertion of exogenous DNA. The hematologic diseases such as sickle cell disease, β-thalassemia, and primary immunodeficiencies, are ideal candidates for ex vivo hematopoietic stem cell (HSC) gene therapy followed by autologous transplantation. However, the utility of CRISPR-Cas9 systems for therapeutic genome editing may be compromised by their off-target toxicity due to the tolerance for nucleotide mismatches between the genome and the targeting region of the guide RNA (gRNA) [1–3]. The off-target activity of Cas9 nuclease can disrupt normal gene function and induce genome instability via large chromosomal rearrangements between two simultaneous DNA breaks [4], which is of serious concern in human gene therapies, potentially leading to difficult-to-predict side effects. As CRISPR/Cas9 moves towards the clinic, it is reasonable to assume that CRISPR/Cas9 treatments will have some degree of off-target edits that will require careful monitoring over time to ensure that off-target edits in hematopoietic stem cells (HSCs) do not have a proliferative effect in vivo. Therefore, potential off-target effects need to be carefully analyzed, and significant challenges exist with both accurately predicting potential off-target sites and performing genome-wide unbiased searches. Herein, we provide a detailed protocol for the CRISPR/Cas9 off-target identification and validation in CD34+ hematopoietic stem and progenitor cells (HSPCs). Using this protocol, researchers can perform computational off-target site searches using a web-based tool CRISPR Off-target Sites with Mismatches, Insertions, and Deletions (COSMID) [5], and experimental identification of potential off-target sites using Genome-wide, Unbiased Identification of DSBs Enabled by sequencing (GUIDE-Seq) [6]. Identified off-target sites can be validated by PCR amplification, followed by next-generation sequencing with a detection limit of 0.1%. This method allows for multiple sites to be assessed simultaneously with a high degree of sensitivity. Although the CRISPR/Cas9 system derived from Streptococcus pyogenes (SpCas9) is used in the protocol here, our method can be readily adopted for off-target analysis of other CRISPR/Cas systems.
2. Materials
2.1. Expansion and Culture of HSPCs
Reconstitute cytokines in sterile 1x PBS containing 0.1% BSA. Distribute to aliquots and store at −20°C to −80°C (for 3-12 months).
Ficoll-Hypaque
CD34 Microbeads
Serum free medium (or other growth medium) for expansion of hematopoietic cells
Stem cell factor (SCF)
Thrombopoietin (TPO)
Flt3 ligand
Interleukin 3 (IL3)
CD34+ HSPC expansion medium supplemented with 300 ng/mL SCF, 100 ng/mL TPO, 300 ng/mL Flt3 ligand, 60 ng/mL IL3, and 1× Penicillin-Streptomycin. Store at 4 °C.
Fresh or frozen CD34+ HSPCs.
12-well plate.
Laminar flow hood with UV light source.
Cell culture CO2 incubator.
15 mL tubes.
Centrifuge for 15 mL tubes
2.2. HSPC genome-editing
SpCas9 protein, 10 μg/ul stock.
Chemically modified CRISPR sgRNAs. Rehydrate in nuclease-free 1x TE. 2.5 μg/ul in stock. Distribute to aliquots and store at −80°C.
Sterile PCR strip tubes (0.2 mL).
1x phosphate buffered saline (PBS), Ph 7.4, without calcium.
15 mL tubes.
Centrifuge for 15mL tubes.
24-well plate.
P3 Primary Nucleofection Kit
Lonza Nucleofector 4-D.
2.3. Genomic DNA extraction and quantification
Genomic DNA extraction kit.
1X phosphate-buffered saline (PBS), Ph 7.4, without calcium.
56°C heat block.
Molecular biology grade ethanol (200 Proof).
DNase free 1.5 mL microcentrifuge tubes.
Table-top microcentrifuge.
Fluorometric DNA quantification assay kit
Fluorometer for DNA quantification
2.4. GUIDE-seq [6]
-
Oligonucleotides for dsDNA tag. Rehydrate in nuclease-free 1xTE to make a 200 μM stock. Store at −20°C.
Sense oligonucleotide:
5′-/Phos/G*T*TTAATTGAGTTGTCATATGTTAATAACGGT*A*T -3′
Anti-sense oligonucleotide:
5′-/Phos/A*T*ACCGTTATTAACATATGACAACTCAATTAA*A*C -3′
* Indicates a Phosphorothioate Bond Modification.
Thermocycler.
SpCas9 protein.
Chemically modified CRISPR dsODNs. Rehydrate in nuclease free 1xTE and store at −80°C.
P3 Nucleofection kit (Lonza). Store at 4°C.
Lonza Nucleofector 4-D.
CD34+ HSPCs.
CD34+ expansion medium.
24-well plate.
Sterile PCR strip tubes (0.2 mL).
PCR clean-up magnetic beads. Light sensitive, do not freeze, store at 4°C.
Magnetic 96-well plate.
80% Ethanol.
Balance.
Agarose.
1x TAE buffer: Dilute 1:50 from a 50x TAE buffer stock with DI water.
Microwave.
Agarose electrophoresis apparatus.
Nucleic acid gel stain.
Gel loading dye.
DNA ladder.
UV gel box with gel imager.
Covaris S220 microTube.
M220 Covaris.
Illumina Y-adapters (See Table 1 for oligonucleotide sequences).
T4 DNA ligase. Store at −20°C.
dNTP mix. Store at −20°C.
Ligation buffer, 10x. Store at −20°C.
End-repair mix (low)
10x Taq buffer, Mg2+-free
Taq polymerase (non-hot start)
10x Taq buffer, Mg2+-free
25 mM MgCl2+
High fidelity Taq polymerase
-
GSP oligonucleotide primers. Rehydrate in 1x TE to make a 100 μM stock and dilute to a 10 μM working stock. Store at −20°C.
GSP1+: GGATCTCGACGCTCTCCCTATACCGTTATTAACATATGACA
GSP1−: GGATCTCGACGCTCTCCCTGTTTAATTGAGTTGTCATATGTTAATAAC
GSP2+: CCTCTCTATGGGCAGTCGGTGATACATATGACAACTCAATTAAAC
GSP2−: CCTCTCTATGGGCAGTCGGTGATTTGAGTTGTCATATGTTAATAACGGTA
-
P5 oligonucleotide primers. Rehydrate in 1x TE to make a 100 μM stock and dilute to a 10 μM working stock. Store at −20 °C.
P5_1: AATGATACGGCGACCACCGAGATCTA
P5_2: AATGATACGGCGACCACCGAGATCTACAC
P7 oligonucleotide primers (See Table 2 for primer sequences).
Tetramethylammonium chloride (TMAC).
Fluorometric DNA quantification assay kit.
Fluorometer for DNA quantification.
NGS library quantification kits.
MiSeq Reagent Kit v2 (300-cycles) (Illumina).
miSeq (Illumina).
-
GUIDE-seq read and index primers. Rehydrate in 1xTE to make a 100 μM stock and store at −20°C.
GUIDE-seq read2 primer: GTGACTGGAGTCCTCTCTATGGGCAGTCGGTGAT
GUIDE-seq index1 primer: ATCACCGACTGCCCATAGAGAGGACTCCAGTCAC
Table 1.
Y-adapter sequences [6]
| Illumina Y-Adapters | Sequence (5′ → 3′) |
|---|---|
| Miseq Common Adapter | [Phos]GATCGGAAGAGC*C*A |
| A01 | AATGATACGGCGACCACCGAGATCTACACTAGATCGCNNWNNWNNACA CTCTTTCCCTACACGACGCTCTTCCGATC*T |
| A02 | AATGATACGGCGACCACCGAGATCTACACCTCTCTATNNWNNWNNACAC TCTTTCCCTACACGACGCTCTTCCGATC*T |
| A03 | AATGATACGGCGACCACCGAGATCTACACTATCCTCTNNWNNWNNACAC TCTTTCCCTACACGACGCTCTTCCGATC*T |
| A04 | AATGATACGGCGACCACCGAGATCTACACAGAGTAGANNWNNWNNACA CTCTTTCCCTACACGACGCTCTTCCGATC*T |
| A05 | AATGATACGGCGACCACCGAGATCTACACGTAAGGAGNNWNNWNNACA CTCTTTCCCTACACGACGCTCTTCCGATC*T |
| A06 | AATGATACGGCGACCACCGAGATCTACACACTGCATANNWNNWNNACA CTCTTTCCCTACACGACGCTCTTCCGATC*T |
| A07 | AATGATACGGCGACCACCGAGATCTACACAAGGAGTANNWNNWNNACA CTCTTTCCCTACACGACGCTCTTCCGATC*T |
| A08 | AATGATACGGCGACCACCGAGATCTACACCTAAGCCTNNWNNWNNACA CTCTTTCCCTACACGACGCTCTTCCGATC*T |
| A09 | AATGATACGGCGACCACCGAGATCTACACGACATTGTNNWNNWNNACA CTCTTTCCCTACACGACGCTCTTCCGATC*T |
| A10 | AATGATACGGCGACCACCGAGATCTACACACTGATGGNNWNNWNNACA CTCTTTCCCTACACGACGCTCTTCCGATC*T |
| A11 | AATGATACGGCGACCACCGAGATCTACACGTACCTAGNNWNNWNNACA CTCTTTCCCTACACGACGCTCTTCCGATC*T |
| A12 | AATGATACGGCGACCACCGAGATCTACACCAGAGCTANNWNNWNNACA CTCTTTCCCTACACGACGCTCTTCCGATC*T |
| A13 | AATGATACGGCGACCACCGAGATCTACACCATAGTGANNWNNWNNACA CTCTTTCCCTACACGACGCTCTTCCGATC*T |
| A14 | AATGATACGGCGACCACCGAGATCTACACTACCTAGTNNWNNWNNACA CTCTTTCCCTACACGACGCTCTTCCGATC*T |
| A15 | AATGATACGGCGACCACCGAGATCTACACCGCGATATNNWNNWNNACA CTCTTTCCCTACACGACGCTCTTCCGATC*T |
| A16 | AATGATACGGCGACCACCGAGATCTACACTGGATTGTNNWNNWNNACA CTCTTTCCCTACACGACGCTCTTCCGATC*T |
Rehydrate in 1xTE to make a 100 μM stock and store at −20°C.
Table 2.
P7 primer sequences [6]
| P7 Adapters | Sequence (5′ → 3′) |
|---|---|
| P701 | CAAGCAGAAGACGGCATACGAGATTCGCCTTAGTGACTGGAGTCCTCTCTATG GGCAGTCGGTGA |
| P702 | CAAGCAGAAGACGGCATACGAGATCTAGTACGGTGACTGGAGTCCTCTCTATG GGCAGTCGGTGA |
| P703 | CAAGCAGAAGACGGCATACGAGATTTCTGCCTGTGACTGGAGTCCTCTCTATG GGCAGTCGGTGA |
| P704 | CAAGCAGAAGACGGCATACGAGATGCTCAGGAGTGACTGGAGTCCTCTCTATG GGCAGTCGGTGA |
| P705 | CAAGCAGAAGACGGCATACGAGATAGGAGTCCGTGACTGGAGTCCTCTCTATG GGCAGTCGGTGA |
| P706 | CAAGCAGAAGACGGCATACGAGATCATGCCTAGTGACTGGAGTCCTCTCTATG GGCAGTCGGTGA |
| P707 | CAAGCAGAAGACGGCATACGAGATGTAGAGAGGTGACTGGAGTCCTCTCTATG GGCAGTCGGTGA |
| P708 | CAAGCAGAAGACGGCATACGAGATCCTCTCTGGTGACTGGAGTCCTCTCTATG GGCAGTCGGTGA |
Rehydrate in 1xTE to make a 100 μM stock and dilute to a 10 μM working stock. Store at −20 °C.
2.5. Off-target NGS library preparation and sequencing (see Fig. 1)
Figure 1. Schematics of off-target library preparation for NGS.
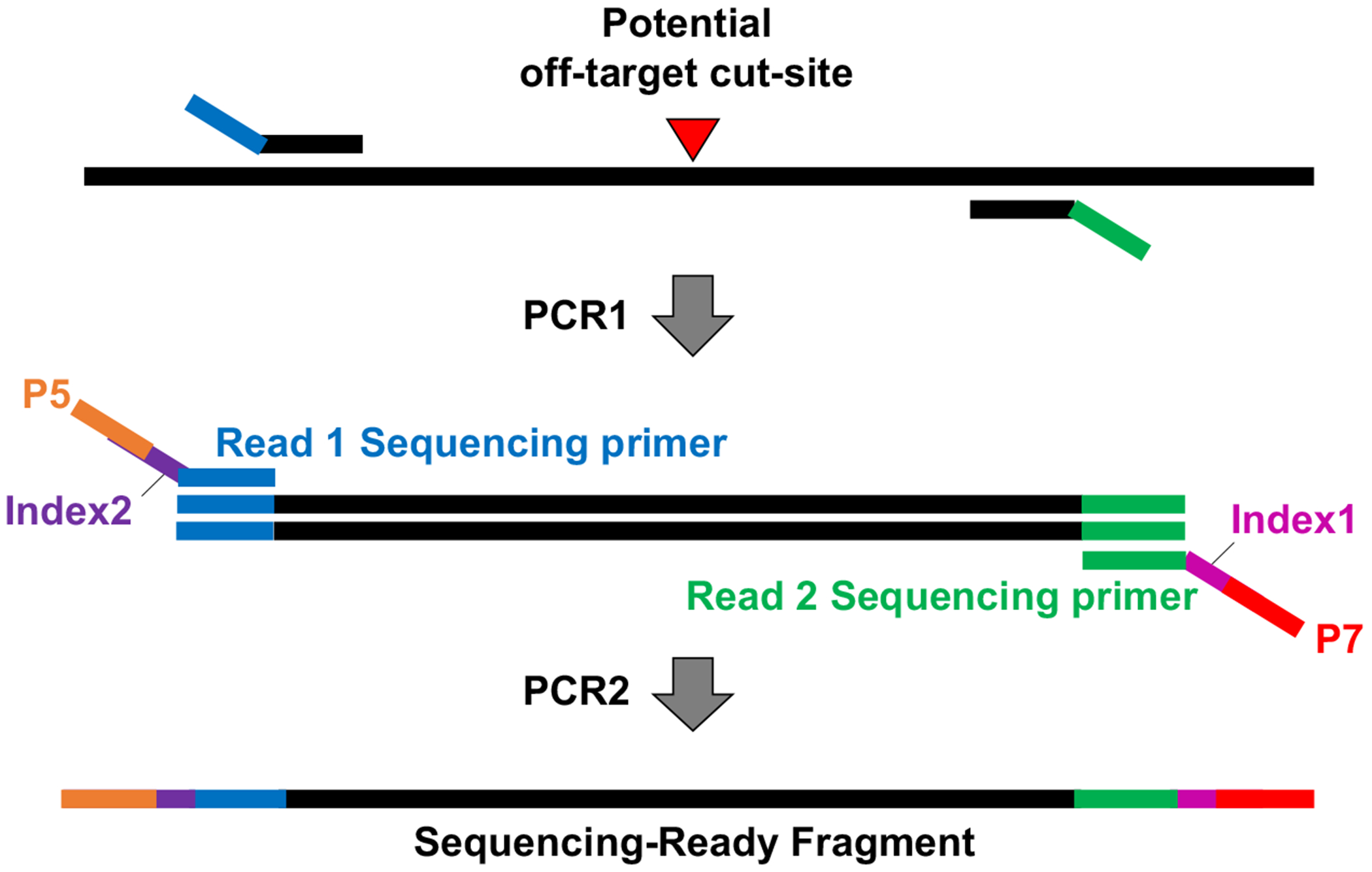
DNA flanking the gRNA off-target sites was amplified using locus-specific primers followed by a second PCR to introduce Illumina sequencing adaptors and sample barcodes.
Thermocycler.
5x OneTaq standard buffer. Store at −20°C.
OneTaq Polymerase. Distribute to aliquots and store at −20°C.
-
Oligonucleotide primers. Rehydrate in 1x TE to make a 100 μM stock and dilute to a 10 μM working stock. Store at −20°C.
PCR1:
Forward primer: 5’-TCTACAGTCCGACGATCA(N)n - 3’ where (N)n denotes target specific primer sequence
Reverse primer: 5’ -GACGTGTGCTCTTCCGATC(N)n - 3’ where (N)n denotes target specific primer sequence
PCR2: (See Table 3 for i7 base sequences and See Table 4 for i5 bases sequences)
Custom Index 1 (i7) adapters
CAAGCAGAAGACGGCATACGAGAT[i7]ATGTGACTGGAGTTCAGACGTGTGCTCTTCCGATC
Custom Index 2 (i5) adapters
AATGATACGGCGACCACCGAGATCTACAC[i5]TGTTCAGAGTTCTACAGTCCGACGATCA
Magnetic 96-well plate.
Magnetic beads. Store at 4°C.
96 well plate.
Reagent reservoirs (for multichannel pipette).
80% Ethanol.
Balance.
Agarose.
1x TAE buffer: Dilute 1:50 from a 50x TAE buffer stock with DI water.
Microwave.
Agarose electrophoresis apparatus.
Nucleic acid gel stain.
Gel loading dye.
DNA ladder.
UV gel box with gel imager.
Nanodrop.
Fluorometric DNA quantification assay kit.
Fluorometer for DNA quantification.
MiSeq Reagent Kit v2 (500-cycles) (Illumina).
MiSeq Instrument (Illumina).
-
Read and index primers. Rehydrate in 1xTE to make a 100 μM stock. Store at −20°C.
Custom read2 primer: 5’ - TGTGACTGGAGTTCAGACGTGTGCTCTTCCGATC - 3′
Custom index primer: 5’ - GATCGGAAGAGCACACGTCTGAACTCCAGTCACAT - 3′
Table 3.
i7 base sequences
| i7 Index Name | Bases in Adapter |
|---|---|
| P7_1 | CACTCACG |
| P7_2 | ACACGATC |
| P7_3 | TATCTGAC |
| P7_4 | CACGTCGT |
| P7_5 | TAGCGACG |
| P7_6 | AGCTCTAG |
| P7_7 | ACTAGAGC |
| P7_8 | CGTACGCA |
| P7_9 | CTACACTA |
| P7_10 | TGCTGCTT |
| P7_11 | TCACGCGT |
| P7_12 | GTAGATCG |
| P7_13 | GTGACGCA |
| P7_14 | CATACTAG |
| P7_15 | AGTGTAGA |
| P7_16 | CGAGAGTT |
| P7_17 | GACATAGT |
| P7_18 | ACGCTACT |
| P7_19 | ACTCACTG |
| P7_20 | TGAGTACG |
| P7_21 | CTGCGTAG |
| P7_22 | TAGTCTCC |
| P7_23 | ACTACGAC |
| P7_24 | GTCTGCTA |
| P7_25 | GTCTATGA |
| P7_26 | CTCGACTT |
| P7_27 | CGAAGTAT |
| P7_28 | TAGCAGCT |
| P7_29 | TCTCTATG |
| P7_30 | GATCTACG |
| P7_31 | GTAACGAG |
| P7_32 | ATAGTACC |
| P7_33 | GCGTATAC |
| P7_34 | TGCTCGTA |
| P7_35 | AACGCTGA |
| P7_36 | CGTAGCGA |
| P7_37 | ATAGCGCT |
| P7_38 | TCTAGACT |
| P7_39 | TCCTCATG |
| P7_40 | CGAGCTAG |
| P7_41 | CTCTAGAG |
| P7_42 | ATGAGCTC |
| P7_43 | AGCATACC |
| P7_44 | CGTCATAC |
| P7_45 | TCAGTCTA |
| P7_46 | CATCGTGA |
| P7_47 | GAGCTCGA |
| P7_48 | TACTAGGT |
| P7_49 | ACGTACGT |
| P7_50 | CGCGATAT |
| P7_51 | CTATCGTG |
| P7_52 | GCGATACG |
| P7_53 | AGTCGCAG |
| P7_54 | GTTACAGC |
| P7_55 | TAACGTCC |
| P7_56 | CTACGACC |
| P7_57 | GAGACTTA |
| P7_58 | ACTGTGTA |
| P7_59 | TGCGTCAA |
Table 4.
i5 base sequences
| i5 Index Name | Bases in Adapter |
|---|---|
| P5_0 | GTTCAGAG |
| P5_1 | CTGATCGT |
| P5_2 | ACTCTCGA |
| P5_3 | TGAGCTAG |
| P5_4 | GAGACGAT |
| P5_5 | CTTGTCGA |
| P5_6 | TTCCAAGG |
| P5_7 | CGCATGAT |
| P5_8 | ACGGAACA |
| P5_9 | CGGCTAAT |
| P5_10 | ATCGATCG |
3. Methods
3.1. COSMID in silico off-target site prediction (See Fig. 2 [7])
Figure 2. COSMID in silico off-target screening.
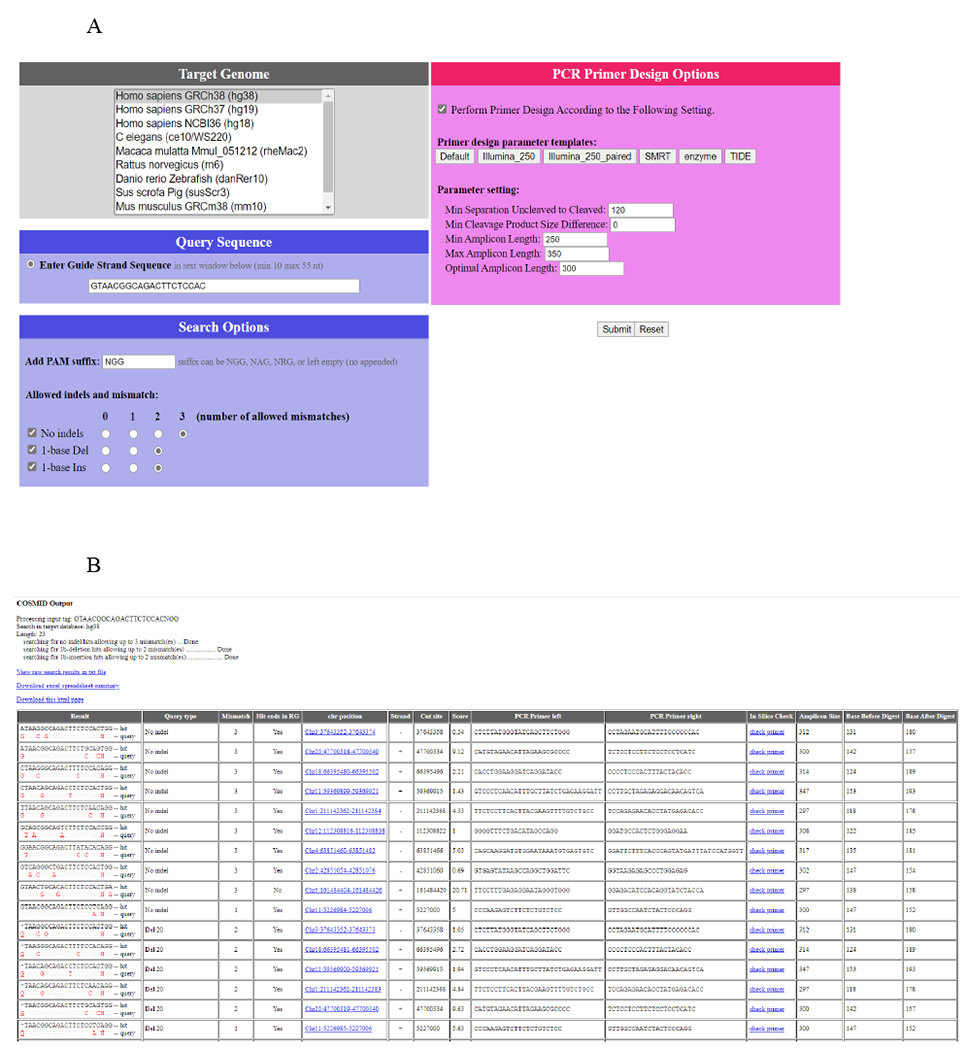
COSMID was performed for R-66 SCD gRNA that targets the HBB sequence with SCD mutation [7]. (a) Bioinformatic prediction of potential off-target sites for the gRNAs was carried out using the COSMID with up to 3 mismatches or with up to 2 mismatches and an insertion or deletion allowed in the 19 PAM proximal bases. The Homo sapiens genome assembly GRCh38/hg38 genome build was used as a reference. (b) Typical COSMID output. Only part of the output is shown.
Select the ‘Target Genome’ for which off-target analysis should be performed (See Note 1).
Enter the gRNA sequence in the ‘Query Sequence’ box.
Enter the PAM sequence of the Cas9 ortholog (NGG for SpCas9) and select the number of allowed indels and mismatches.
COSMID can perform PCR primer design for each off-target site identified. In the ‘PCR Primer Design Options’, check ‘Perform Primer Design According to the Following Setting’ and select ‘Illumina_250_paired’.
Click ‘Download excel spreadsheet summary’ to download the search results (See Note 2).
-
Order oligonucleotide primers for each off-target site identified.
Forward primer requires TCTACAGTCCGACGATCA 5’ of the site-specific primer sequence output by COSMID.
Reverse primer requires GACGTGTGCTCTTCCGATC 5’ of the site-specific primer sequence output by COSMID.
3.2. Experimental identification of off-target sites using GUIDE-seq [6]
3.2.1. Genome editing in CD34+ HSPCs for integration of dsODN tag at Cas9 cut sites (see Note 3 for GUIDE-seq in other cell types)
- Generate the 100 μM dsODN tag by combining the sense oligo-nucleotide (200 μM in 1xTE) and the anti-sense oligonucleotide (200 μM in 1xTE) and run the following annealing program on a thermocycler:
- Heat to 95°C for 2 min.
- Ramp cool to 25°C over 45 min.
- Hold at 12°C.
Aliquot and store annealed dsODN tag at −20°C. Avoid freeze thaw cycles.
Harvest CD34+ HSPCs and prepare 5 x 105 cells for each electroporation reaction. Transfer cells to 15 mL tube and fill the tube to 15 mL with 1xPBS. Pellet the cells by spinning at 300 g for 5 min.
Meanwhile, assemble the RNP complex. Carefully mix 1 μL sgRNA, 0.5 μL SpCas9 and 1 μL dsODN tag (20 μM) in a 0.2 mL PCR tube per electroporation. Incubate at room temperature for 15 min. For the mock-treated control sample, prepare the same amount dsODN tag as treated samples without RNP.
Aspirate as much medium and PBS as possible without disturbing the cell pellet.
Add 22 μL P3 solution per 5 x 105 CD34+ HSPCs for each electroporation and gently resuspend cells.
Transfer 20 μL of cells in P3 solution mixture to the 0.2 mL tube containing the RNP and dsODN tag.
Mix well by gentle flick. Transfer 20 μL of the mixture in one pipet into the center groove of Lonza 4D electroporation cuvette. Avoid bubbles.
Immediately perform electroporation using the CA137 program. Incubate cells for 10 min at room temperature.
Add 80 μL pre-warmed CD34+ HSPCs expansion medium into each cuvette. Transfer all cells from each cuvette to a 24-well plate containing 500 μL pre-warmed culture medium per well.
48 hours after electroporation, add 500 μL fresh pre-warmed medium to the wells.
After 4 days, harvest cells and extract genomic DNA.
3.2.2. Confirmation of dsODN integration at the target site
Design primers that yield amplicon length between 400-500 bp.
Set up PCR using the genomic DNA of cells treated with RNP and from mock-treated cells for use as controls.
Send purified PCR products for Sanger sequencing using the forward or reverse primer used for PCR amplification.
Go to Inference of CRISPR Editing (ICE) [8] web-based tool that determines rates of CRISPR/Cas9 knockout and knock-in editing: https://ice.synthego.com/#/
Enter the Sample Label, 17-23 nt guide RNA sequences without PAM and the Donor Sequence. The donor sequences should contain the 34-bp dsODN Tag sequence inserted at the cut-site in forward or reverse orientation.
Upload a chromatogram file for both a Control File (mock treated cells) and an Experiment File (RNP and dsODN tag treated cells).
Confirm targeted integration of dsODN Tag in forward or reverse orientation
See Fig. 3 for typical ICE output.
Figure 3. dsODN tag knockin rate quantification at on and off-target sites in CD34+ HSPCs by Inference of CRISPR Edits.
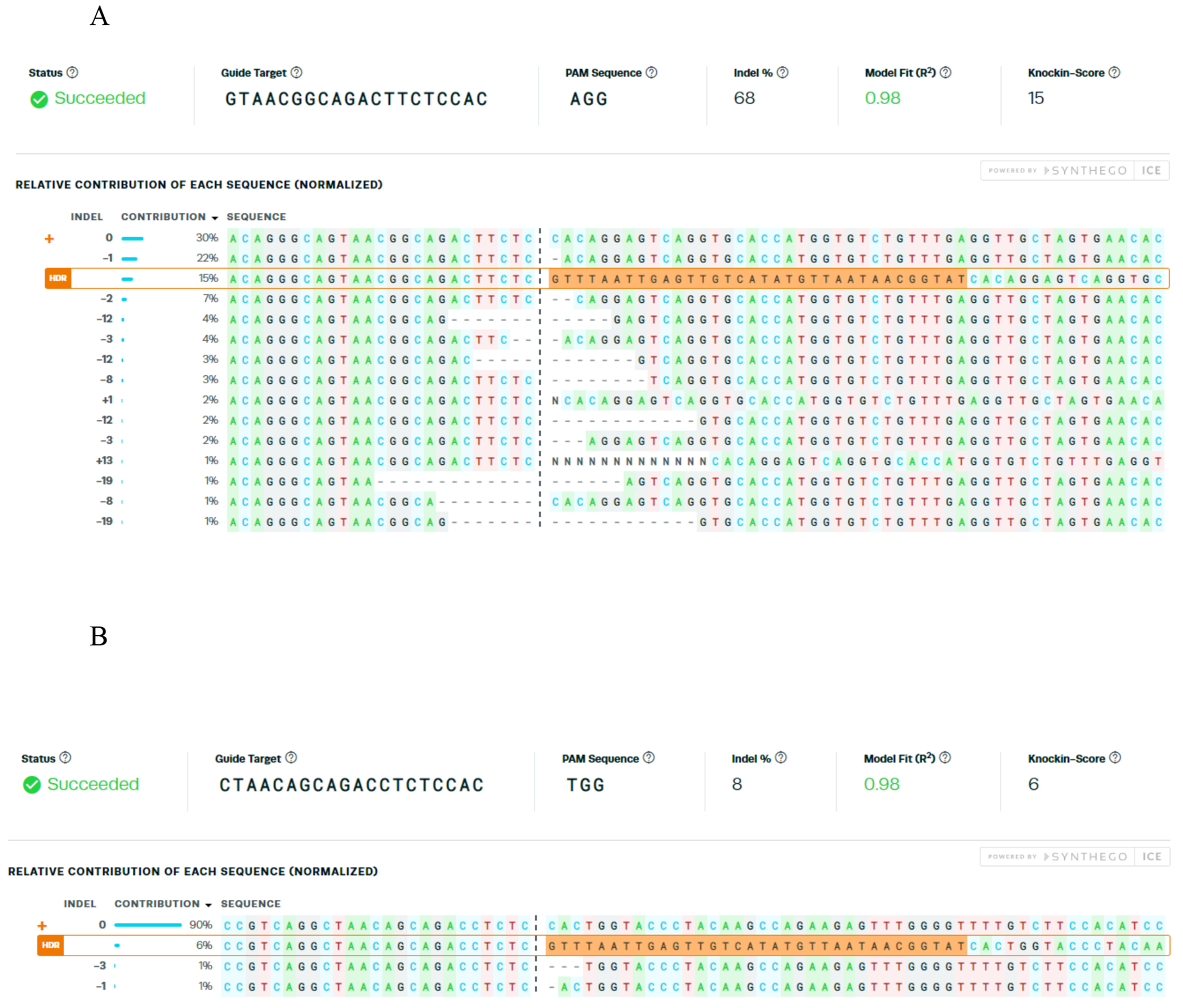
(ICE)[8]. GUIDE-seq was performed in sickle cell disease patient derived CD34+ HSPCs for R-66 SCD gRNA that targets the HBB sequence with sickle mutation [7]. Relative contribution of alleles with dsODN tag knockin in forward orientation at (a) the on-target and (b) the active off-target CRISPR cleavage site.
3.2.3. Quantification and shearing of genomic DNA (See Note 4)
Determine the concentration of extracted genomic DNA by fluorometric DNA quantification assay.
For each genomic DNA sample, prepare two tubes (anti-sense/ sense or +/−) of 1 μg DNA in a total volume of 140 μL 1x TE buffer.
- Place 130 μL of each tube in a Covaris S220 microTube and shear for 25 seconds on a M220 Covaris following manufacturer’s instructions. Run settings should be:
- Average incident power = 10 watts
- Peak incident power = 50 watts
- Duty factor = 20%
- Cycles/ burst = 200 counts
Move samples from microtube to a new plastic tube.
Run 5 μL of sheared DNA on a 1.5% agarose gel and verify that the bulk of the shearing is 500 bps.
Clean up samples with 1x magnetic beads (120 μL) and elute with 15 μL water (See Note 5 for detailed magnetic bead clean up protocol). You may leave the samples with the beads on the magnet and move them when ready to add to tubes with end repair master mix.
3.2.4. Y-adapter preparation
-
Prepare the following annealing reaction by mixing MiSeq Common Adapter with each of the A01 – A16 oligos (see Table 1):
80 μL 1x TE 10 μL 100 μM MiSeq Common Adapter 10 μL 100 μM A## oligo - Run the following annealing program on a thermocycler:
- Heat to 95°C for 2 min.
- Ramp cool to 25°C over 45 min.
- 4°C hold.
3.2.5. End Repair
-
Make the following master mix at 1.2x number of samples and dispense 8.5 μL to each PCR tube or well:
0.5 μL Nuclease-free water 1.0 μL 5 mM dNTP mix 2.5 μL Ligation buffer, 10x 2.0 μL End-repair mix (low) 2.0 μL 10x Taq buffer, Mg2+-free 0.5 μL Taq polymerase (non-hot start) Add 14 μL of water eluted samples to tubes containing the master mix. You should have two PCR tubes for each sample, one sense and one anti-sense.
Mix with pipet set at 22 μL.
- Run the following on a thermocycler with heated lid turned off:
- 12°C for 15 min.
- 37°C for 15 min.
- 72°C for 15 min.
- 4°C hold.
3.2.6. Adapter Ligation
To new PCR tubes, add 1.0 μL of one barcode of annealed A01 – A16 oligos. Use the same barcode for the +/− strands of the same sample.
Add 2.0 μL T4 DNA ligase to each tube.
Add 22.5 μL of the end-repaired sample. Mix by pipetting, vortex, quick spin.
- Run the following on a thermocycler with heated lid turned off:
- 16°C for 30 min.
- 22°C for 30 min.
- 4°C hold.
Clean up samples with 0.9x magnetic beads (22.95 μL). Elute with 22 μL water (See Note 5).
3.2.7. PCR1 – Amplification of dsTag integrated sites
-
Make the following master mix for PCR1. Prepare two PCR1 reactions for each sample using GSP1+ or GSP1− primer. Add sufficient volume of all reagents for the appropriate number of PCR1 reactions. Only one GSP1 primer is used per PCR1 reaction. Dispense 20 μL master mix to each PCR tube:
10.7 μL Nuclease-free water 0.6 μL 10 mM dNTP mix 3.0 μL 10x Taq buffer, Mg2+-free 2.4 μL 25 mM MgCl2+ 0.3 μL Platinum Taq polymerase 1.0 μL 10 μM GSP1 + or − 1.5 μL 0.5 M TMAC 0.5 μL 10 μM P5_1 Add 10 μL of adapter ligated sample prepared in 3.2.5 (f) to 20 μL PCR1 reaction. Make sure to set up two PCR1 reactions (GSP1+ or GSP1−) for each sample. Mix by pipetting, vortex, quick spin.
- Run the following program on a thermocycler with heated lid on:
- 95°C for 5 min.
- (95°C for 30 sec, 70°C (−1°C /cycle) for 2 min., 72°C for 30 sec) 15 cycles
- (95°C for 30 sec, 55°C for 1 min, 72°C for 30 sec) 10 cycles
- 72°C for 5 min
- 4°C hold.
Clean up samples with 1.2x magnetic beads (36 μL) and elute with 15 μL water (See Note 5).
3.2.8. PCR2 – Adaptor Labeling of PCR1 amplicons
-
Make the following master mix for PCR2. Prepare two PCR2 reactions for each sample using GSP2+ or GSP2− primer. Add sufficient volume of all reagents for the appropriate number of PCR1 reactions. Only one GSP2 primer is used per PCR2 reaction:
4.2 μL Nuclease-free water 0.6 μL 10 mM dNTP mix 3.0 μL 10x Taq buffer, Mg2+-free 2.4 μL 25 mM MgCl2+ 0.3 μL Platinum Taq polymerase 1.0 μL 10 μM GSP2 + or − 1.5 μL 0.5 M TMAC 0.5 μL 10 μM P5_2 Dispense 13.5 μL to each PCR tube or well:
Add 1.5 μL of 10 μM P7_# to each tube to make a unique pair with A01 – A16.
Add 15 μL of the PCR1 sample to the PCR2 master mix. Make sure to use GSP2+ primer for the GSP1+ PCR1 mix and GSP2-primer for the GSP1− PCR1 mix. Mix by pipetting, vortex, quick spin.
- Run the following program on a thermocycler with heated lid on:
- 95°C for 5 min.
- (95°C for 30 sec, 70°C (−1°C /cycle) for 2 min., 72°C for 30 sec) 15 cycles
- (95°C for 30 sec, 55°C for 1 min, 72°C for 30 sec) 10 cycles
- 72°C for 5 min.
- 4°C hold.
Clean up samples with 0.7x magnetic beads (36 μL) and elute with 30 μL TE (See Note 5).
3.2.9. Sample quantification and normalization
Quantify each sample using a Fluorometric DNA quantification assay fluorometer.
- Use the following formula to convert from ng/μl to nM:
Choose as high a stock concentration as possible and normalize each sample to the same stock concentration.
Pool the normalized samples (ex. Pool 3 μL of each normalized sample into the one tube).
Quantify the pooled library using a fluorometric DNA quantification assay according to manufacturer instructions.
Dilute an aliquot of the pooled library to 4 nM in nuclease free water.
Make a serial dilution of the pooled library and quantify using NGS library quantification kit as per manufacturer’s instructions.
3.2.10. Illumina Sequencing
Thaw cartridge from MiSeq Reagent Kit v2 (300-cycles) ≥ 1.5 hours prior to use in a room temperature water bath and thaw buffer HT1 at 4°C.
Dilute 5 μL of 4 nM pooled library with equal volume of freshly prepared 0.2N NaOH and vortex briefly. Centrifuge at 280 x g for 1 minute and incubate at room temperature for 5 min.
Dilute denatured library to 20 pM by adding 990 μL of chilled buffer HT1.
Further dilute denatured library to 12.5 pM by mixing 225 μL prechilled HT1 and 375 μL of a 20 pM denatured library.
Invert to mix and store on ice until ready to load onto the MiSeq reagent cartridge.
Dilute each GUIDE-seq read2 and GUIDE-seq index primers to 0.5 μM with HT1.
Prepare MiSeq Sample Sheet according to the MiSeq Sample Sheet Quick Ref Guide (15028392 J).
Pierce the foil seal of position 17, 19 and 20 on the reagent cartridge with a clean 1 mL pipette.
Pipette 600 μL of the 12.5 pM denatured library to the position 17 designated as ‘Load Sample’. Avoid buddle.
Pipette 600 μL of GUIDE-seq index1 primer to position 19 of the reagent cartridge.
Pipette 600 μL of GUIDE-seq read 2 primer to position 20 of the reagent cartridge.
Select ‘Sequence’ on the software interface and follow the instruction on the screen.
Select the sample sheet containing the run specific information.
Rinse the flow cell with Milli-Q water. Dry with lint-free cleaning tissue.
Load the flow cell, cartridge, and PR2.
Sequence.
3.2.11. Bioinformatic analysis of Sequencing Data
- Install bioconda packages required to run GUIDE-seq script.
- conda install -c bioconda bwa
- conda install -c bioconda bedtools
- conda install -c anaconda numpy
- conda install -c dranew bcl2fastq
- git clone --recursive https://github.com/aryeelab/guideseq.git
In the Illumina raw sequencing output folder, run bcl2fastq.
Copy and paste Read1, 2 Index1, 2 files to ‘GuideSeq_WorkingFolder’.
Add yaml file to the same folder.
-
Run guide-seq.
python guideseq/guideseq/guideseq.py all -m ~/GuideSeq_WorkingFolder/sample.yaml
Figure 4. Example of GUIDE-seq results and off-target activity quantification by NGS in CD34+ HSPCs.
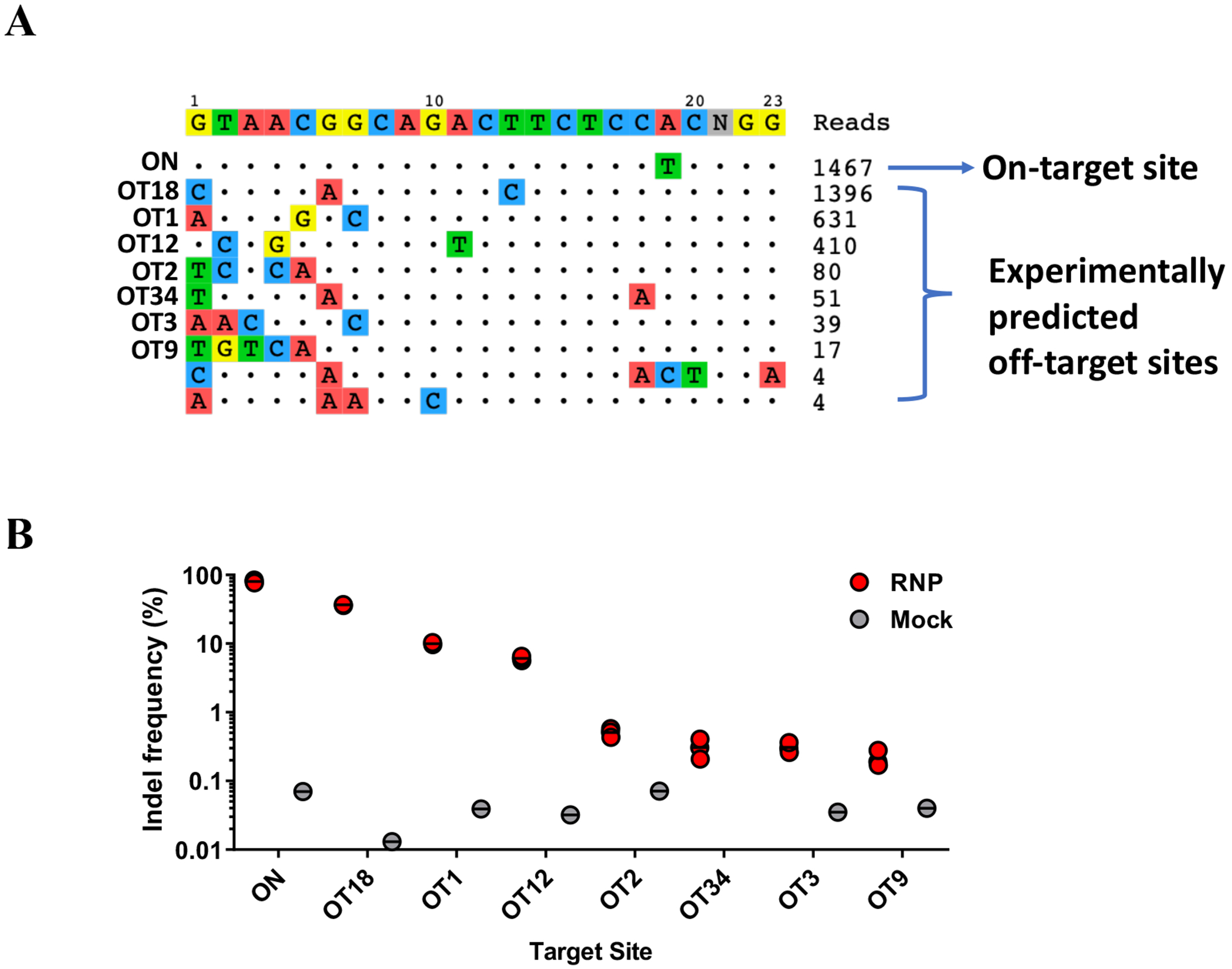
(a) Visualization of GUIDE-seq output. (b) The cleavage activities at the on-target site (ON) and 57 COSMID and GUIDE-seq identified off-target sites (OT) were analyzed by NGS. The dot-plot shows indel frequencies at the on-target site (HBB) and the 7 off-target sites with measurable cleavage activity [7]. GUIDE-seq read counts correlated highly with the indel frequency quantified by NGS, with on-target sites having the highest GUIDE-seq read counts.
3.2.12. Primer design for GUIDE-seq identified off-target sites
In identified.txt GUIDE-seq output file, the chromosome position of the identified off-target site in the target genome can be found in column G (chromosome) and column H (position).
In UCSC genome browser (http://genome.ucsc.edu/cgi-bin/hgGateway), select the relevant human assembly (same as the reference genome used for GUIDE-seq alignment). Search the chromosome position.
Get DNA in ‘Window for the searched position’ using the following ‘Sequence Retrieval Region Options’: Add 175 extra bases upstream (5’) and 175 extra downstream (3’).
Copy and Paste the template sequence in Primer3Plus (https://primer3plus.com/cgibin/dev/primer3plus.cgi) and define ‘Product Size Ranges’ in ‘General Settings’ to 275-325.
-
Order oligonucleotide primers for each off-target site identified with the following 5’ adaptor sequences:
Forward primer requires TCTACAGTCCGACGATCA 5’ of the site-specific primer sequence output by Primer3Plus
Reverse primer requires GACGTGTGCTCTTCCGATC 5’ of the site-specific primer sequence output by Primer3Plus
3.3. Identification of Off-target Activity in CD34+ HSPCs
3.3.1. CD34+ cell culture [9]
Purify CD34+ HSPCs from the peripheral blood. Separate the mononuclear fraction from the peripheral blood of patients with SCD by Ficoll-Hypaque density centrifugation. Extract CD34+ cells from the mononuclear fraction by immunomagnetic separation using the CD34 Microbeads Kit. Assess the purity of CD34+ by flow cytometric analysis.
Extract CD34+ HSPCs from the mononuclear fraction by immunomagnetic separation using the CD34 Microbeads Kit according to the manufacturers’ instructions.
Culture the CD34+ cells using HSPC expansion medium for 2-3 days before electroporation.
3.3.2. RNP delivery using electroporation
Count the cells and prepare 2 x 105 CD34+ cells for each electroporation reaction. Transfer cells to 15 mL tube and fill the tube to 15mL with 1X PBS. Pellet the cells by spinning at 300 g for 5 min.
Meanwhile, assemble the RNP complex by adding5 μg (30.5 pmol) of SpCas9 protein and 2.5 μg (73 pmol) of chemically modified CRISPR sgRNA to a 0.2 mL PCR tube and incubate at room temperature for 10 minutes.
Aspirate as much medium and PBS without disturbing the cell pellet.
Add 22 μL P3 solution per 2 x 105 CD34+ cells for each electroporation reaction.
Transfer 20 μL of cell suspension to each 0.2 mL tube containing RNP complexes. For mock-treated CD34+ cells, electroporate of the same number of cells using the same program as the treated cells, but without RNP.
Mix well by gentle flick. Transfer 20 μL of the mixture in one pipet into the center groove of Lonza 4D electroporation cuvette. One transfection per well. Avoid bubbles.
Immediately perform electroporation using CA137 program. Wait 10 min at room temperature.
Add 80 μL pre-warmed CD34+ expansion medium into each cuvette. Transfer all cells from each cuvette to a 24-well plate with 500 μL pre-warmed expansion medium per well.
Add fresh expansion medium every two days and culture at a density under 1 x 106 live cells/mL.
After 3-5 days, harvest cells and extract genomic DNA using genomic DNA extraction kit according to the manufacturer’s protocol.
3.3.3. Off Target library sample preparation (see Fig. 1).
-
Prepare PCR1 reactions with 50 ng of gDNA for each off-target site as follows:
25 μL reaction Component 5 μL 5x OneTaq standard buffer 0.5 μL 10 mM dNTPs 0.5 μL 10 μM Forward primer 0.5 μL 10 μM Reverse primer 2 μL 50 ng gDNA at 25 ng/μL 0.125 μL OneTaq Polymerase 16.375 μL Nuclease-free water 94°C for 2 min.
(94°C for 30s, 63°C (−1°C/cycle) for 30s, 68°C for 30s) 7 cycles.
(94°C for 30s, 57°C for 30s, 68°C for 25s) 35 cycles.
68°C for 10 min.
4°C hold.
Use 2 μL to verify a single band at the proper size for each amplicon (usually 250 - 350 bps) on a 1.5 % agarose gel (TAE running buffer).
Clean up PCR product with 0.8x magnetic beads. Add 18.4 μL beads to each PCR reaction and elute by adding 25 μL water. Transfer 22 μL to a clean tube or plate (See Note 5).
-
Prepare PCR2 reactions using 10 μL PCR 1 sample with adapters (barcoding) as follows. Use a unique combination of i7 and i5 index adapters for each amplicon.
25 μL reaction Component 5 μL 5x OneTaq standard buffer 0.5 μL 10 mM dNTPs 1 μL 5 μM i5 adapter 1 μL 5 μM i7 adapter 10 μL PCR I sample 0.125 μL OneTaq polymerase 7.375 μL Nuclease-free water 94°C for 2 min.
(94°C for 30s, 60°C for 30s, 68°C for 35s) 30 cycles.
68°C for 5 min.
4°C hold.
Use 2 μL to verify a single band at the proper size (usually 300 bp) on a 1.5 % agarose gel (1x TAE running buffer).
Clean up PCR product with magnetic beads using 1.2x beads to PCR product ratio. Add 30 μL beads/ PCR rxn. Elute by adding 25 μL water. Move 22 μL to a clean tube or plate. (See Note 5).
3.3.4. Quantification and pooling of off-target amplicons
- Quantify purified amplicons by Nanodrop. Use the following formula to convert from ng/μl to nM:
Choose as high a stock concentration as possible and normalize all samples to the same stock concentration. Pool the normalized samples (ex. Pool 3 μL of each normalized sample into the one tube).
See Note 6 for optional sample concentration and See Note 7 for adapter-dimer removal.
Quantify the pool concentration using a Fluorometric DNA quantification assay® dsDNA HS Assay Kits according to manufacturer instructions.
Dilute an aliquot of the pooled library to 4 nM (See Note 8).
3.3.5. Illumina Sequencing
Thaw cartridge from MiSeq Reagent Kit v2 (500-cycles) ≥ 1.5 hours prior to use in a room temperature water bath or overnight at 2 − 8°C.
Ensure HT1 is thawed at 4°C.
Use within 12 hours freshly diluted 0.2N NaOH.
Dilute 5 μL of 4 nM pooled library with equal volume of 0.2N NaOH and vortex briefly.
Centrifuge at 280 x g for 1 minute and incubate at room temperature for 5 minutes.
Add 990 μL chilled HT1. The result is 1000 μL of a 20 pM denatured library.
Dilute denatured library to 12.5 pM by mixing 225 μL prechilled HT1 and 375 μL of a 20 pM denatured library.
Invert to mix. The result is 600 μL of a 12.5 pM denatured library.
Set aside on ice until you are ready to load it onto the reagent cartridge.
Dilute each Custom read2 and Custom index primers to 0.5 μM with HT.
Prepare each Custom read2 and Custom index primers at 100 μM in nuclease free water.
Add 3 μL primer to 597 μL chilled HT1.
Prepare MiSeq Sample Sheet according to the MiSeq Sample Sheet Quick Ref Guide (15028392 J).
Add 3 μL primer to 597 μL chilled HT1.
Pierce the foil seal of position 17, 19 and 20 on the reagent cartridge with a clean 1 mL pipette.
Pipette 600 μL of the 12.5 pM denatured library to the position 17 designated as ‘Load Sample’. Avoid buddle.
Pipette 600 μL of custom index primer to position 19.
Pipette 600 μL of custom read 2 primer to position 20.
Select ‘Sequence’ on the software interface and follow the instruction on the screen.
Select the sample sheet containing the run specific information.
Rinse the flow cell with Milli-Q water. Dry with lint-free cleaning tissue.
Load the flow cell, cartridge, and PR2.
3.3.6. Bioinformatic Analysis of Sequencing Data
CRISPR/Cas9 genome editing outcomes from deep sequencing data were analyzed using a custom pipeline [6]. Details of the custom pipeline can be find https://github.com/piyuranjan/NucleaseIndelActivityScript
- Install bioconda packages required to run nuclease activity script
- -c bioconda perl-sort-naturally
- install -c bioconda perl-list-moreutils
- conda install -c bioconda perl-List-Util
- conda install -c bioconda cutadapt
- conda install -c bioconda trim-galore
- conda install -c bioconda prinseq
- conda install -c bioconda bwa
- conda isntall -c bioconda samtools openssl=1.0
In Illumina run folder, perform ‘bcl2fastq.’ Be sure to use the run specific sample sheet and that it is in this Illumina runs main folder. The Barcodes and samples in this sample sheet will demultiplex your reads into Read1 and Read2 .fastq files, output in ‘Data/Intensities/BaseCalls/.
These demultiplexed files exist as zipped .fastq files (fastq.gz). Unzip the files using the command: gunzip *fastq.gz.
In your working folder, be sure it contains: your unzipped R1 and R2 fastq’s, your reference file (-ref.fa), your cutsites file (Cutsites.txt), the indelQuantificationFromFastqPaired2bp-1.0.1.pl script, and your parameters.config file.
parameter.config file contains information specific to each run. Change the sample name (First Column), the R1 and R2 names (Second and Third Column), the -ref.fa filename (the Eighth Column), and the Cutsite.txt filename (the Ninth Column) to reflect the files in your working folder.
Run the following command: perl indelQuantificationFromFastqPaired2bp-1.0.1.pl -c parameters.config -v. The output gives the level of activity at each off-target site analyzed.
Acknowledgement
This work was supported by the National Institutes of Health (UG3HL151545, R01HL152314 and OT2HL154977 to G.B.) and the Cancer Prevention and Research Institute of Texas (RR140081 to G.B.).
4. Notes
Use the same Target Genome for COSMID and GUIDE-seq to easily identify overlapping sites.
COSMID identifies the same off-target site multiple times when there are multiple possible sequence alignment patterns. Users need to remove duplicated sites from the list manually.
Cell types other than CD34+ HSPCs could be used for GUIDE-seq. To optimize genome editing conditions for GUIDE-seq in the cell type of interest:
- Perform dsODN tag dose optimization using RNP and dsODN Tag. It is recommended to deliver 10 pmol to 100 pmol of dsODN tag.
- Determine the optimal dsODN tag dose that resulted in the highest rate of dsODN tag integration at the on-target cleavage site measured by ICE.
Alternatively, enzymatic fragmentation of genomic DNA, end-repair and adapter ligation can be performed using Lotus™ DNA Library Prep Kit (IDT) according to the manufacturer’s protocol.
Magnetic beads clean up protocol
- Determine the desired volumetric ratio of magnetic beads to sample for the cleanup.
- Vigorously mix stock tube of beads until foamy before use.
- Add beads to sample and mix by pipetting.
- Incubate at room temperature for 5 minutes.
- Place and leave on magnet through the remaining steps until indicated to be removed.
- Room temperature for 10 minutes.
- Remove liquid with a pipet.
- Add 80% ethanol (200 μL for PCR reactions).
- Room temperature for 1 minute.
- Remove liquid with a pipet.
- Repeat steps h – j.
- Use a pipette to remove any residual liquid from the bottom of the tubes.
- Remove samples from the magnet.
- Air dry for 2 minutes.
- Mix sample by pipetting with desired volume and solution for elution. Make sure no air bubbles are present.
- Return to the magnet.
- Incubate for 5 minutes at room temperature.
- Transfer eluate to new tube.
Clean up the pool with 2x magnetic beads and elute with 30 μL water (aimed at concentrating the normalized pool).
If adapter-dimers are present in the library, gel extract the pooled library from a 1.5 % agarose gel. Use DNA gel extraction kit as per manufacturer’s instructions.
If more than one library is being run at once (pool several libraries), determine the number of samples in each pool and pool the 4 nM libraries according to the number of samples. Keep the same ratio or percentage of samples/pool.
References
- 1.Hsu PD, Scott DA, Weinstein JA, et al. , DNA targeting specificity of RNA-guided Cas9 nucleases. Nature biotechnology, 2013. 31(9): p. 827–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cradick TJ, Fine EJ, Antico CJ, et al. , CRISPR/Cas9 systems targeting ϐ-globin and CCR5 genes have substantial off-target activity. Nucleic acids research, 2013. 41(20): p. 9584–9592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fu Y, Foden JA, Khayter C, et al. , High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nature biotechnology, 2013. 31(9): p. 822–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Torres R, Martin MC, Garcia A, et al. , Engineering human tumour-associated chromosomal translocations with the RNA-guided CRISPR-Cas9 system. Nat Commun, 2014. 5: p. 3964. [DOI] [PubMed] [Google Scholar]
- 5.Cradick TJ, Qiu P, Lee CM, et al. , COSMID: A Web-based Tool for Identifying and Validating CRISPR/Cas Off-target Sites. Molecular therapy. Nucleic acids, 2014. 3(12): p. e214–e214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tsai SQ, Zheng Z, Nguyen NT, et al. , GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat Biotechnol, 2015. 33(2): p. 187–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Park SH, Lee CM, Dever DP, et al. , Highly efficient editing of the beta-globin gene in patient-derived hematopoietic stem and progenitor cells to treat sickle cell disease. Nucleic Acids Res, 2019. 47(15): p. 7955–7972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hsiau T, Conant D, Rossi N, et al. , Inference of CRISPR Edits from Sanger Trace Data. bioRxiv, 2019: p. 251082. [DOI] [PubMed] [Google Scholar]
- 9.Ronzoni L, Sonzogni L, Fossati G, et al. , Modulation of gamma globin genes expression by histone deacetylase inhibitors: an in vitro study. Br J Haematol, 2014. 165(5): p. 714–21. [DOI] [PubMed] [Google Scholar]