

Abstract
The 34-kDa protein encoded by the I3 gene of vaccinia virus is expressed at early and intermediate times postinfection and is phosphorylated on serine residues. Recombinant I3 has been expressed in Escherichia coli and purified to near homogeneity, as has the protein from infected cells. Both recombinant and endogenous I3 protein demonstrate a striking affinity for single-stranded, but not for double-stranded, DNA. The interaction with DNA is resistant to salt, exhibits low cooperativity, and appears to involve a binding site of approximately 10 nucleotides. Electrophoretic mobility shift assays indicate that numerous I3 molecules can bind to a template, reflecting the stoichiometric interaction of I3 with DNA. Sequence analysis reveals that a pattern of aromatic and charged amino acids common to many replicative single-stranded DNA binding proteins (SSBs) is conserved in I3. The inability to isolate viable virus containing an interrupted I3 allele provides strong evidence that the I3 protein plays an essential role in the viral life cycle. A likely role for I3 as an SSB involved in DNA replication and/or repair is discussed.
Vaccinia virus, the prototypic member of the poxvirus family, replicates in the cytoplasm of the host cell and exhibits a remarkable degree of genetic autonomy. Compartmentalization of the infectious cycle within the cytoplasm implies that the virus must supply all essential components of the transcription and replication machinery. Indeed, vaccinia virus is known to encode many DNA replicative functions, including a DNA polymerase with intrinsic 5′-3′ polymerization and 3′-5′ exonuclease activity, a processivity factor, a DNA ligase, a thymidine kinase, a thymidylate kinase, and a ribonucleotide reductase (46). Phenotypic analyses of temperature-sensitive (ts) DNA− mutants has also revealed essential roles for the B1 kinase and the D5 nucleoside triphosphatase in DNA replication. Finally, a virally encoded uracil DNA glycosylase (UDG) appears to participate in viral replication (5, 26, 44), with a viral dUTPase playing a supportive role in maintaining the integrity of the genomic DNA.
Our understanding of the minimal functions required for vaccinia virus DNA replication remains incomplete, with several functions postulated by analogy with other systems as yet unidentified. The viral and cellular processes of DNA replication, recombination, and repair share a general requirement for single-stranded DNA (ssDNA) binding proteins (SSBs). These accessory proteins form specific complexes with ssDNA which render it resistant to nucleases and remove barriers of intramolecular secondary structure. This helix-destabilizing activity, as well as the establishment of specific protein-protein interactions, serves to stimulate several replication enzymes. For example, both human replication protein A (hRPA) and bacteriophage T4 gene 32 protein have been found to interact with and stimulate their cognate DNA polymerases (15, 17).
At the onset of these studies, no vaccinia virus SSB had been identified biochemically, genetically or by sequence analysis of the predicted open reading frames (ORFs) encoded by the genome. Nevertheless, the inability of the purified viral DNA polymerase to pass through barriers of DNA secondary structure in vitro (3, 24) suggested that an SSB would be essential for viral replication. Our search for a candidate SSB was guided by early reports in the literature of various abundant DNA binding proteins found within virosomes, the cytoplasmic foci of vaccinia replication. Initial efforts were directed at identifying the gene encoding a 34-kDa phosphoprotein referred to variously as polypeptide B (29, 32) or FP11 (30, 33). Fortuitously, these experiments led us to the analysis of a distinct 34-kDa polypeptide encoded by the vaccinia I3 gene. Here we describe the characterization of this protein and demonstrate that it is the best candidate for a vaccinia virus-encoded SSB.
MATERIALS AND METHODS
Materials.
Restriction endonucleases and DNA-modifying enzymes were obtained from Boehringer Mannheim Biochemicals (Indianapolis, Ind.) or New England Biolabs, Inc. (Beverly, Mass.). [35S]methionine, 32Pi, and 32P-labeled nucleoside triphosphates were obtained from Dupont/New England Nuclear Corp. (Boston, Mass.). Proteins (unlabeled, prestained, and 14C labeled) for use as molecular weight (MW) standards were obtained from GIBCO/Bethesda Research Laboratories, Inc. (Gaithersburg, Md.). HindIII-digested lambda phage DNA and HaeIII-digested phage φX174 DNA size markers were from New England Biolabs. Protein A-Sepharose and antibiotics were purchased from Sigma Chemical Co. (St. Louis, Mo.); nitrocellulose was obtained from Schleicher & Schuell (Keene, N.H.). Immunoblots were developed with reagents obtained from Bio-Rad (Richmond, Calif.). Denatured DNA-cellulose was obtained from Sigma. Phosphocellulose resin was obtained from Whatman BioSystems, Ltd. (Kent, England).
Cells and virus.
The WR strain of vaccinia virus and various derivatives were propagated in BSC40 cells maintained in Dulbecco’s minimal essential medium (DMEM) supplemented with 5% fetal calf serum (FCS) (GIBCO Laboratories, Grand Island, N.Y.). For the purification of I3 protein, HeLa cells grown in suspension culture (Joklik-modified DMEM, 2.5% calf serum, 2.5% horse serum) were infected with vaccinia virus.
Preparation of a bacterial overexpression clone.
A 1,511-bp segment of the vaccinia virus HindIII I genome fragment was subcloned into pUC19, using HindIII and BamHI sites in both the insert and vector. This sequence included about 500 bp of additional sequence 5′ of the I3 ORF and about 300 bp of additional sequence at the 3′ end. The entire I3 ORF, from a BspHI site at the 5′ end of the gene (nucleotide [nt] −3) to an RsaI site at the 3′ end of the gene (nt +865), was excised from this parental pUC-HindIII-BamHI vector, resolved by Tris acetate-EDTA-agarose gel electrophoresis, and purified on glass beads (49). This fragment was cloned into a glass-purified pET 3c backbone (45) prepared by restriction with NdeI followed by blunting with the Klenow fragment of Escherichia coli DNA polymerase I. This construct was used to direct the synthesis of the authentic I3 protein after transformation into competent E. coli HMS174.
Overexpression of the I3 34-kDa protein.
HMS174 cells transformed with the pET-I3 clone were grown to a density of 4 × 108 cells per ml in the presence of maltose (0.2%) and ampicillin (50 μg/ml). Cultures were then induced by infection with λ CE6 at a multiplicity of infection (MOI) of 10 PFU per cell; 20 min of adsorption at room temperature was followed by incubation at 37°C with agitation for 25 min. Cultures were then shaken at room temperature for 3 h. Uninduced cultures were grown under the same conditions but were not infected with λ CE6. Some cultures were metabolically labeled for 5 min at the end of the induction period; labeling was performed in M9 minimal medium supplemented with [35S]methionine (10 μCi/ml) and the remaining unlabeled amino acids. Bacteria were collected by centrifugation at 5,000 × g for 5 min. Pellets were solubilized by freeze-thawing in lysis buffer (1 ml/10 ml of culture) (50 mM Tris [pH 7.5], 1.0 M NaCl, 10 mM EDTA, 1 mM dithiothreitol [DTT], 10% sucrose) containing lysozyme (0.2 mg/ml) followed by incubation on ice for 30 min. After the addition of Triton X-100 to 0.1%, the extract was clarified by centrifugation, and the supernatant was retained and stored at −20°C.
Preparation of a polyclonal anti-I3 serum.
Recombinant I3, overexpressed in E. coli from the pET-I3 vector described above, was excised from a sodium dodecyl sulfate (SDS)-polyacrylamide gel and used as an antigen for the generation of a rabbit polyclonal antiserum. The serum was effective in both immunoprecipitation and immunoblot (used at a dilution of 1:500) assays.
Metabolic labeling of infected cells.
The temporal profile of I3 protein synthesis during viral infections was assessed by immunoprecipitation of extracts prepared from metabolically labeled cells. BSC-40 cells (107) were infected with wild-type (wt) virus at an MOI of 15 PFU per cell. After 30 min of adsorption, the inoculum was removed and the cells were rinsed and fed with DMEM-FCS. At selected times, the infected cultures were pulse labeled for 30 min with [35S]methionine (0.1 mCi/ml) in methionine-free DMEM and subsequently harvested. Cultures were rinsed with ice-cold phosphate-buffered saline (PBS; 140 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.5 mM KH2PO4 [pH 7.4]) and disrupted in phospholysis buffer (100 mM NaPO4 [pH 7.4], 100 mM NaCl, 1% Triton X-100, 0.1% SDS, 0.5% sodium deoxycholate). Lysates were clarified by microcentrifugation at 4°C and stored at −20°C. I3 phosphorylation was examined by immunoprecipitation of extracts prepared from cells metabolically labeled with 32Pi (0.1 mCi/ml) for 2 h in phosphate-free DMEM.
Immunoprecipitation.
Thawed lysates representing 1.2 × 106 cells were clarified and incubated with 4 μl of preimmune or anti-I3 serum on ice for 4 h. Immune complexes were recovered with protein A-Sepharose; bound antigen was liberated from the complex by boiling in protein sample buffer (1% SDS, 1% 2-mercaptoethanol, 50 mM Tris [pH 6.8], 10% glycerol) and then resolved by electrophoresis through SDS–12% polyacrylamide gels (19). Dried gels were visualized by fluorography (2).
Immunofluorescence.
BSC40 cells were grown to confluence in Labtek eight-chamber slides (Nunc, Inc., Naperville, Ill.) and either left uninfected or infected with wt vaccinia virus at an MOI of 15 in the presence or absence of cytosine arabinoside (20 μg/ml). At designated times postinfection, slides were chilled on ice and monolayers were rinsed with cold PBS and then fixed with 4% paraformaldehyde in PBS for 15 min at 4°C. Cells were then rinsed two times with cold PBS, permeabilized with 0.1% Triton X-100 in PBS for 5 min, and rinsed again. Clarified preimmune or anti-I3 immune serum was then applied for 1 h at a dilution of 1:50. Cells were washed three times with cold PBS and then incubated with fluorescein isothiocyanate-conjugated goat anti-rabbit immunoglobulin G (Jackson ImmunoResearch Laboratories, Inc., West Grove, Pa.) at a dilution of 1:100. When used in parallel to detect nucleic acids. Hoechst 33258 (Polysciences, Inc., Warrington, Pa.) was applied at a concentration of 0.5 μg/ml in PBS. Monolayers were rinsed four times with cold PBS, and then glass coverslips were mounted over Aquamount (Lerner Labs, Pittsburgh, Pa.). Immunofluorescence was detected with a Microphot EPI FL fluorescence microscope (Nikon, Inc., Garden City, N.Y.). Photographic slides of these images were then scanned using a Nikon Coolscan II scanner and Adobe Photoshop software (Adobe Systems, Inc., San Jose, Calif.). The figures were labeled by using Canvas software (Deneba Systems, Inc.), and final images were obtained with a Kodak dye sublimation printer.
One-dimensional phosphoamino acid analysis.
32P-labeled vaccinia virus-infected cell extracts were subjected to immunoprecipitation as described above. Protein was eluted from protein A-Sepharose by boiling for 5 min in protein sample buffer without dye. Proteins were precipitated by the addition of trichloroacetic acid to 10% followed by incubation on ice for 60 min. Precipitates were collected by microcentrifugation at 4°C for 15 min, rinsed with 100% ethanol (−20°C), and air dried. Samples were resuspended in 50 μl of constant-boil HCl (Pierce, Rockford, Ill.) and heated at 110°C for 1 h; hydrolysates were collected by lyophilization and resuspended in distilled H2O. Hydrolyzed samples and phosphoamino acid markers (20 nmol) were spotted on thin-layer cellulose F chromatography plates (EM Separations, Gibbstown, N.J.). Electrophoresis was performed in pyridine-glacial acetic acid-water (1:10:189) at 2,000 V for 15 min. The plates were dried, sprayed with ninhydrin (Sigma), developed at 65°C for 20 min, and exposed for autoradiography.
Purification of recombinant I3.
Induced pET-I3 bacterial pellets from 2 liters of culture were frozen, thawed, resuspended in 200 ml of pET lysis buffer containing 1 M NaCl and 0.2 mg of lysozyme per ml, and incubated for 30 min on ice. Triton X-100 was added to 0.1%; lysates were subjected to vigorous vortexing and clarified by centrifugation at 14,000 × g for 30 min. Lysates were then adjusted to contain 50 mM sodium acetate, 12 mM MgCl2, and 4 mM CaCl2 and then treated with 20 μl of DNase I (0.1 μg/ml) for 30 min at 25°C. The reaction was stopped by adding EDTA to 6 mM and EGTA to 3 mM. The lysate was then dialyzed against 2 liters of buffer A (40 mM Tris [pH 7.4], 50 mM NaCl, 6 mM EDTA, 3 mM EGTA, 1 mM β-mercaptoethanol, 10% glycerol). All purification steps were performed on a Pharmacia FPLC apparatus at 4°C. Gradient fractions (and I3 purification profiles) were typically monitored by immunoblot and silver-stained SDS-polyacrylamide gel analysis. Protein concentrations were determined by using the Bradford method and bovine serum albumin (BSA) as a standard. The dialysate (fraction I, 200 ml) was subjected to ammonium sulfate precipitation. The 40 to 60% pellet fraction was resuspended in 20 ml of buffer A and applied to a 15-ml ssDNA-cellulose column that had been equilibrated with buffer A. The column was developed with an 80-ml linear gradient containing 50 mM to 2.5 M NaCl. Peak I3 fractions were pooled and concentrated by reverse dialysis using polyethylene glycol (fraction II). Fraction II was subsequently dialyzed against buffer B (50 mM Tris [pH 7.4], 50 mM NaCl, 1 mM EDTA, 2 mM β-mercaptoethanol, 5% glycerol).
Purification of I3 from vaccinia virus-infected cells.
HeLa cells (5 × 109) in suspension culture were infected with wt vaccinia virus at an MOI of 15 in the presence of hydroxyurea (10 mM) and harvested at 5 h postinfection (hpi). Cytoplasmic extracts were prepared by hypotonic lysis as described previously (25), and fractionated by chromatography on a 25-ml DEAE-cellulose column equilibrated in DEAE buffer (50 mM Tris [pH 7.4], 1 mM DTT, 1 mM EDTA, 10% glycerol)–50 mM NaCl, and developed with a stepwise elution of NaCl in DEAE buffer. I3 was recovered in the flowthrough and applied to a 10-ml ssDNA-cellulose column (equilibrated in DEAE buffer–50 mM NaCl) that was then developed with a linear gradient of 50 mM to 2.5 M NaCl in DEAE buffer. Peak I3 fractions were pooled, dialyzed against buffer B, and concentrated by polyethylene glycol reverse dialysis. The purification yielded approximately 0.5 mg of pure I3 from 5 × 109 infected cells.
Gel filtration analysis of purified endogenous I3.
A 200-μl sample containing 25 μg of I3 in 200 mM NaCl–20 mM Tris (pH 7.4)–1 mM DTT–1 mM EDTA–5% glycerol was injected onto a 24-ml Superdex 75 column. The column was equilibrated and developed in the same buffer, and chromatography was performed at 0.25 ml/min on a Pharmacia FPLC apparatus at 4°C. Fractions of 0.5 ml were collected. The profile of I3 within the fractions was determined by SDS-polyacrylamide gel electrophoresis (PAGE) and immunoblot analysis. Chromatography was also performed with the following protein standards of known Stokes radii in order to calibrate the column: BSA, 36.1 Å; chicken ovalbumin, 27.5 Å; and myoglobin, 19 Å. Dextran blue and l-tryptophan were used to determine the column’s void and total volumes, respectively. The Stokes radii and molecular weights of the I3 species observed were calculated by the method of Siegel and Monty (43).
Electrophoretic mobility shift assays (EMSAs).
Oligonucleotides were prepared with an Applied Biosystems (Foster City, Calif.) model 391 DNA synthesizer. Full-length oligonucleotides were purified by using Poly-Pak cartridges (Glen Research, Sterling, Va.). The 5′ terminus was radiolabeled with [γ-32P]ATP and T4 polynucleotide kinase. For the preparation of double-stranded oligonucleotides, complementary 5′-labeled oligonucleotides were hybridized as described previously (27). DNA probes and protein samples were mixed in reaction mixtures containing 250 μg of BSA per ml, 20 mM Tris (pH 7.4), 50 mM NaCl, 1 mM EDTA, and 5% glycerol. Then 15-μl reaction mixtures were incubated at 30°C for 15 min, added to 1 μl of 5× loading buffer, and analyzed by low-ionic-strength, nondenaturing, 15% polyacrylamide gel electrophoresis. Standard vertical gels were cast and run in 0.25× TBE (25 mM Tris, 21 mM boric acid, 0.25 mM EDTA); electrophoresis was performed at 200 V for 4 h at 4°C. Gels were then dried and exposed for autoradiography. Bands were quantitated with a phosphorimager.
Denatured DNA-cellulose chromatography of vaccinia virus-infected cell extracts.
BSC-40 cells (1.4 × 108) were infected with wt vaccinia virus at an MOI of 15 PFU per cell. Adsorption was carried out for 30 min, at which time monolayers were rinsed with either methionine-free DME in the case of [35S]methionine labeling or phosphate-free DME in the case of 32Pi labeling. Cells were then immediately labeled with medium containing either [35S]methionine (0.1 mCi/ml) or 32Pi (0.1 mCi/ml) for 5 h. Cytoplasmic extracts were prepared by hypotonic lysis (25) of labeled cells, and lysates were treated with DNase I as described above for recombinant protein purification. Each lysate was then dialyzed against buffer A and applied to a 5-ml ssDNA-cellulose column which had been equilibrated with buffer A. The columns were developed with a 60-ml linear gradient of 50 mM–2.5 M NaCl. Resulting fractions were analyzed by SDS-PAGE (12% gel) and subjected to autoradiography, silver staining, and immunoblot analysis.
Construction of an I3 null allele by replacing I3 with the Neor gene.
The pUC HindIII-BamHI plasmid containing 1,511 bp of the HindIII I fragment was cleaved with XbaI and BglII to release a 383-bp fragment representing 46.5% of the I3 gene. The backbone was then treated with phosphatase, made blunt with the Klenow fragment of DNA polymerase I, resolved by Tris acetate-EDTA-agarose electrophoresis, and purified on glass beads. The neomycin resistance (Neor) gene under the direction of the vaccinia virus early/late p7.5 promoter was excised from pVVNEO (8) by digestion with SalI and XbaI; the termini were then made blunt with the Klenow fragment of DNA polymerase I. The Neor cassette was inserted into the XbaI-BglII site of the pUC HindIII-BamHI plasmid in place of the I3 sequence. Two pI3:NEO constructs, in which the Neor cassette was oriented in either direction relative to the flanking sequences, were selected. These plasmids retained 752 bp of viral sequence upstream of, and 376 bp downstream of, the Neor sequence to facilitate recombination into the vaccinia virus genome. BSC-40 cells in 35-mm-diameter dishes were infected with wt vaccinia virus at an MOI of 0.03 PFU per cell; at 3 hpi the replicate dishes were independently transfected with 3.5 μg of a calcium phosphate precipitate of either supercoiled or linearized pI3:NEO DNA. G418 (2 μg/ml) was added to the medium at 15 hpi; cultures were maintained at 37°C in the presence of drug until 3 days postinfection. Monolayers were harvested and subjected to two cycles of freeze-thawing to release cell-associated virus. Viral yields were then titrated, and each stock was independently passaged twice through BSC-40 cells at 37°C in the presence of G418 to enrich for recombinant progeny. Two rounds of plaque purification were then performed to prepare Neor isolates from each of 16 original infections/transfections. Eleven of these had been transfected with supercoiled pI3:NEO (five in one orientation and six in the other); five had received linearized pI3:NEO (two in one orientation and three in the other). Viral expansions were then prepared from these isolates, and portions of the stocks were applied to Zetaprobe (Bio-Rad) with a dot blot apparatus (Bio-Rad). Duplicate filters were then hybridized with one of four radioactive probes prepared by nick translation (38): (i) an 8.1-kb fragment derived from the vaccinia virus HindIII D genomic fragment, (ii) the excised Neor gene (without any vaccinia virus sequences), (iii) the 353-bp internal fragment deleted from the I3 gene during the preparation of pI3:NEO (a probe for the wild-type I3 allele), and (iv) pUC19 plasmid DNA.
RESULTS
With the aim of identifying a viral SSB, we investigated the possibility that a protein previously described in the vaccinia virus literature and designated polypeptide B (29, 32) or FP11 (30, 33) might serve this function. This protein had been described as an abundant DNA binding phosphoprotein of 34 kDa which was a major component of virosomes (replication factories).
We set out to map the gene encoding this protein by using the technique of hybrid selection (7, 35, 37, 48). HindIII restriction fragments spanning the genome were used to select early viral mRNAs which were in turn translated in vitro; radiolabeled protein products were resolved on SDS–12% polyacrylamide gels (data not shown) and compared to 35S- and 32P-labeled extracts prepared from infected cells. An abundant 34-kDa protein whose mRNA was selected by the HindIII I fragment was chosen for further analysis and shown to be encoded by the I3 ORF.
The I3 gene of vaccinia virus is predicted to encode a 267-amino-acid protein which exhibits no significant homology with other protein sequences (9, 16, 39). Transcriptional analysis has shown that I3 mRNA is synthesized at both early and intermediate times postinfection. Indeed, the sequences upstream of the I3 gene contain characteristic promoter elements for both early and intermediate vaccinia virus gene expression (14). From the outset we wished to characterize those properties of the I3 protein which might bear on its potential as an SSB. We began by cloning the authentic ORF into expression vectors which would enable us to obtain large quantities of recombinant protein for both antibody production and biochemical characterization.
Characterization of the temporal expression pattern of I3 during vaccinia virus infection.
Authentic I3 protein was expressed in E. coli through the use of the pET vector system (45). Upon induction of the T7 promoter by infection with a bacteriophage carrying the T7 RNA polymerase gene, the I3 protein accumulated to significant levels (Fig. 1A, lanes 1 and 2). This protein was used in the preparation of a polyclonal anti-I3 serum as described in Materials and Methods. This antiserum was used to examine the temporal regulation of I3 protein synthesis and to look at I3 protein localization within vaccinia virus-infected cells by immunofluorescence.
FIG. 1.

Analysis of I3 expression in E. coli and in vaccinia virus-infected cells. (A) Synthesis of recombinant I3 in E. coli transformants containing plasmid pET-I3. Extracts from uninduced (U) or induced (I) bacteria are shown in lanes 1 and 2, respectively; the 34-kDa I3 protein is apparent in induced extracts. Cultures were metabolically labeled with [35S]methionine as described in Materials and Methods prior to being harvested and analyzed by SDS-PAGE and fluorography. (B) Temporal profile of I3 synthesis. Parallel cultures were left uninfected (M), infected with wt vaccinia virus (MOI of 15), and harvested at 2, 3, 4, 6, or 8 hpi or were infected with wt vaccinia virus (MOI of 15) in the presence of cytosine arabinoside and harvested at 4 hpi (C). Cells were metabolically labeled with [35S]methionine for 45 min prior to being harvested. Aliquots of total cellular lysates (lanes 1 to 7) as well as the species recovered after immunoprecipitation with anti-I3 serum (lanes 8 to 14) were analyzed by SDS-PAGE and fluorography. (C) I3 is phosphorylated in vivo. Cells infected with wt vaccinia virus (MOI of 15) were metabolically labeled with 32Pi from 1 to 3 or 3 to 5 hpi and harvested at the end of the labeling period. The time of harvest is shown above the lanes. Lysates were subjected to immunoprecipitation with preimmune serum (P.I.) or anti-I3 serum and subjected to SDS-PAGE and fluorography. 14C-labeled protein standards are shown at the far left in the lane marked MW, with their molecular masses indicated. The arrow at the far right shows the position of electrophoretic migration of both recombinant and authentic I3 protein.
To determine the temporal profile of I3 synthesis, BSC-40 cells were infected with wt virus at an MOI of 15 PFU per cell at 37°C, radiolabeled with [35S]methionine for 30 min at various times postinfection, and harvested immediately. Extracts were subjected to immunoprecipitation analysis with the anti-I3 serum; the recovered immune complexes were fractionated by SDS-PAGE and visualized by fluorography. The fluorographs (Fig. 1B) reveal that the temporal pattern of I3 expression is that of both an early and an intermediate vaccinia virus protein. Synthesis of I3 in the presence of cytosine arabinoside verifies that it is indeed an early gene (Fig. 1B, lane 14). That it is also an intermediate gene is demonstrated by the observation that although I3 synthesis is initiated by 2 hpi (lane 9), it reaches maximal levels at 3 to 4 hpi (lanes 10 and 11). That I3 is not a late gene is evident by the fact that synthesis begins to decline by 6 hpi and is minimal by 8 hpi (lanes 12 and 13). This profile of protein synthesis is mirrored in immunoblot analyses of I3 accumulation (data not shown), which show that I3 levels have reached a plateau by 8 hpi. This temporal profile would ensure that I3 was present at the onset of DNA replication and throughout its duration but suggests that I3 is unlikely to be a major component of newly assembled virions.
Immunoprecipitation analysis was also used to assess the phosphorylation of I3 in vaccinia virus-infected cell extracts (Fig. 1C). BSC-40 cells were infected with wt vaccinia virus at an MOI of 15 PFU per cell at 37°C and radiolabeled with 32Pi at various times postinfection. Extracts were subjected to immunoprecipitation with anti-I3 serum. Substantial levels of I3 phosphorylation were seen in extracts labeled from 1 to 3 or 3 to 5 hpi (Fig. 1C, lanes 2 and 3). No such radiolabeled species was recovered when preimmune serum was used instead (Fig. 1C, lane 1).
I3 is phosphorylated on serine residues.
32P-labeled I3 immunoprecipitates were used as substrates for phosphoamino acid analysis. Briefly, liberated I3 immunocomplexes were precipitated, resuspended in concentrated HCl, and subjected to partial hydrolysis by heating at 110°C for 1 h. Lyophilized amino acids were then analyzed by one-dimensional high-voltage thin-layer electrophoresis. As shown in Fig. 2, in which identical samples were mixed with unlabeled phosphoamino acid standards individually (lanes 1 to 3) or together (lane 4), I3 is phosphorylated exclusively on serine residues. Since similar studies on polypeptide B have shown it to be phosphorylated on threonine residues (29), our data suggest that I3 and polypeptide B are not the same protein.
FIG. 2.

I3 is phosphorylated on serine residues. Cells were infected with wt virus (MOI of 15) and metabolically labeled with 32Pi. Lysates were prepared and subjected to immunoprecipitation with anti-I3 serum. Liberated immune complexes were subjected to trichloroacetic acid precipitation and then to hydrolysis with HCl. The hydrolysates were mixed with phosphoamino acid markers either individually (lanes 1 through 3) or as a mixture (lane 4). Samples were then applied to thin-layer cellulose plates and resolved by high-voltage electrophoresis. Markers were visualized by color development with ninhydrin; radiolabeled phosphoamino acids derived from I3 were visualized by autoradiography. The dotted wickets indicate the positions of migration of the phosphoserine (pSerine), phosphothreonine (pThreonine), and phosphotyrosine (pTyrosine) markers. The position of migration of free phosphate is indicated, as is the application origin.
Localization of I3 within vaccinia virus-infected cells by immunofluorescence.
A presumed role for I3 as a viral SSB would place it in the virosome during infection, perhaps with this localization dependent on ongoing DNA synthesis. We used immunofluorescence microscopy to address the issue of intracellular localization of I3 during vaccinia virus infection. As shown in Fig. 3A, BSC cells infected with vaccinia virus and fixed at 4 hpi display large discrete cytoplasmic foci of fluorescence only when incubated with anti-I3 immune serum. Mock-infected cells incubated with immune serum (Fig. 3F) or infected cells incubated with preimmune serum (Fig. 3G) exhibited no fluorescence. To control for the possibility that the observed foci represented nonspecific aggregates of I3, monolayers were infected in the presence of the DNA replication inhibitor cytosine arabinoside. These infections showed a more diffuse pattern of cytoplasmic staining with some small punctate foci, as seen in Fig. 3B and C. This result indicates that I3 localization is in fact dependent on DNA replication and accumulation within virosomal structures. Indeed, analysis of infected cells doubly labeled with anti-I3 serum and Hoechst 33258 show an equivalence between cytoplasmic nucleic acid (virosomal) staining (Fig. 3D) and I3 staining (Fig. 3E).
FIG. 3.
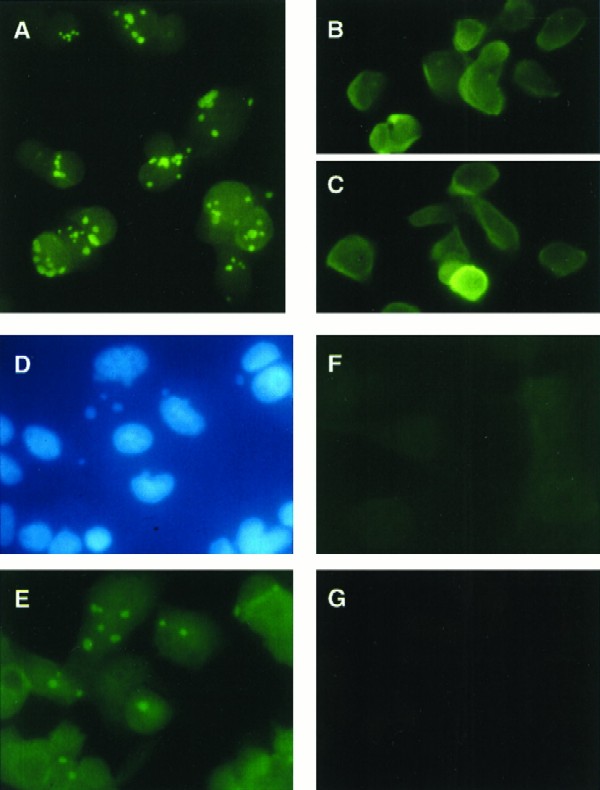
I3 protein localizes to discrete cytoplasmic foci that are coincident with virosomes. Mock- or virus-infected BSC-40 cells were processed for immunofluorescence as described in Materials and Methods. The panels represent the following treatments: (A) vaccinia virus infected, anti-I3 serum; (B and C) vaccinia virus infected in the presence of cytosine arabinoside (20 μg/ml), anti-I3 serum; (D and E) vaccinia virus infected, anti-I3 serum plus Hoechst 33258; (F) mock infected, anti-I3 serum; (G) vaccinia virus infected, preimmune serum. Panels D and E represent the same field.
Purification of recombinant and endogenous I3.
To begin analyses of the biochemical properties of the I3 protein, we purified both the recombinant protein from E. coli and the endogenous protein from vaccinia virus-infected cells. Although several chromatographic strategies were attempted, the following protocols were especially effective and capitalized on the obvious affinity of I3 for ssDNA.
(i) Recombinant I3.
Recombinant I3 protein was purified to near homogeneity from soluble extracts generated by induction of bacteria containing plasmid pET-I3 with λ CE6 at 25°C. Cell lysis was performed under high-salt conditions to maximize protein solubility and to dissociate protein-DNA complexes. Lysates were then treated with DNase I to digest endogenous DNA which might compete with immobilized ssDNA-cellulose for I3 binding. Lysates were subjected to ammonium sulfate precipitation, and the 40 to 60% pellet fraction was applied to an ssDNA-cellulose column which was developed with increasing concentrations of NaCl. I3 eluted in a broad peak between 1.0 and 2.0 M NaCl and was purified to near homogeneity by this procedure, as assessed by SDS-PAGE and silver stain (Fig. 4A) and immunoblot (not shown) analyses. This source of I3 is especially useful because it is free of other viral proteins and is presumably not phosphorylated.
FIG. 4.

Purification of recombinant and authentic I3. (A) Purification of I3 from E. coli transformants containing pET-I3. Expression of I3 was induced as described in Materials and Methods, and I3 was purified by ammonium sulfate precipitation and chromatography on ssDNA-cellulose. Aliquots of uninduced cultures (lane 2), of induced cultures (17 μg of protein; lane 3), and of each purification step (lanes 4 [29 μg of protein] and 5 [0.6 μg of protein] are shown. Positions of MW markers (lane 1) are shown in kilodaltons at the left. Proteins were resolved by SDS-PAGE and visualized by silver staining. (B) Purification of endogenous I3. I3 was purified from cells infected with wt vaccinia virus as described in Materials and Methods. Aliquots of the unfractionated lysate and peak fractions from the DEAE-cellulose and ssDNA-cellulose columns were analyzed by SDS-PAGE and silver staining. Amounts of protein analyzed: lane 2, 50 μg; lane 3, 20 μg; lane 4, 1.2 μg. Protein MW standards were resolved in lane 1; the corresponding molecular masses are indicated in kilodaltons at the left.
(ii) I3 from vaccinia virus-infected cells.
Eight liters of a suspension culture of HeLa cells was infected with vaccinia virus at a high MOI in the presence of hydroxyurea. Cytoplasmic extracts were subjected to DEAE-cellulose chromatography, and the protein pool eluting with 50 mM NaCl was applied to an ssDNA-cellulose column. The column was developed with a linear gradient of 50 mM to 2.75 M NaCl. A broad peak of I3 protein eluted between 1.0 and 2.0 M NaCl and was shown to be purified to near homogeneity, as assessed after SDS-PAGE by silver staining (Fig. 4B) and immunoblot (not shown) analyses. This purification yielded 0.5 mg of pure protein from 5 × 109 infected cells. Compared with the purification of 0.2 mg of overexpressed D5 (6) and 0.3 mg of overexpressed DNA polymerase (25) from 2 × 108 and 1 × 109 infected BSC-40 cells, respectively, this yield indicates that I3 is expressed at high levels during the early phase of wt infections.
Determination of the native MW of the I3 protein.
Gel filtration chromatography was performed on both recombinant and endogenous forms of I3 to examine their native MW in solution. Elution profiles of I3 from a Superdex 75 FPLC column were compared to those of known standards; I3 eluted just after the peak of ovalbumin (MW, 44,000) (not shown). A Stokes radius of 24.7 Å was calculated by the method of Siegel and Monty (43). An MW of 34,000 was extrapolated for I3 based on a plot of MW versus Stokes radius for protein standards, indicating that I3 exists as a monomer in solution.
Recombinant I3 binds specifically to single-stranded oligonucleotides.
The chromatographic properties of I3 suggested that it did indeed have a high affinity for ssDNA (Fig. 4A). To explore the DNA binding character of I3 protein in more detail, we performed EMSAs. As shown in Fig. 5A, purified recombinant I3 formed a specific protein-DNA complex with a 5′-labeled single-stranded 24-nt probe in a concentration-dependent manner. In contrast, virtually no DNA binding was seen if a double-stranded oligonucleotide of the same sequence was used as the probe (Fig. 5B). The plot of these data shown in Fig. 5C illustrates the striking specificity of I3 for ssDNA.
FIG. 5.

EMSAs show that I3 binds specifically to ssDNA. (A) Analysis of complex formation between purified, recombinant I3 and ssDNA oligonucleotides. Reaction mixtures contained 0.2 pmol of a single-stranded, terminally 32P-labeled 24-mer oligonucleotide and various amounts of I3. Amounts of I3 used: lane 1, 0 pmol; lane 2, 0.3 pmol; lane 3, 0.6 pmol; lane 4, 1.3 pmol; lane 5, 2.0 pmol; lane 6, 2.6 pmol; lane 7, 3.2 pmol; lane 8, 3.9 pmol; lane 9, 4.6 pmol; lane 10, 5.2 pmol. After incubation for 15 min at 30°C, samples were fractionated on a 15% nondenaturing polyacrylamide gel at 4°C and visualized by autoradiography. (B) Analysis of complex formation between purified, recombinant I3 and double-stranded DNA oligonucleotides. Reactions were carried out and analyzed as for panel A except that protein samples were mixed with 0.2 pmol of a terminally radiolabeled double-stranded 24-mer oligonucleotide identical in sequence to that used for panel A. (C) Graphic representation of I3-DNA complex formation. The data in panels A and B were quantitated by phosphorimaging and plotted as shown. The ordinate represents the picomoles of DNA bound, as determined by measuring the decrease in the levels of free probe remaining; the abscissa represents the amount of I3 in the reaction. ds, double-stranded.
I3-ssDNA complexes are highly salt resistant.
The elution profile of I3 from ssDNA-cellulose had suggested to us that the I3-DNA interaction was a strong one. Those experiments monitored the salt sensitivity of I3-DNA interactions in the presence of multiple other proteins. To examine the salt sensitivity of the binding of purified recombinant I3 to DNA, EMSAs were performed in the presence of various concentrations of NaCl. As shown in Fig. 6A, no diminution of complex formation was seen until the NaCl concentration reached 0.5 M, and significant binding remained even in the presence of 1 M NaCl. The observation that the addition of increasing amounts of unlabeled single-stranded 24-mer midway through a 30-min incubation could fully compete for I3 binding (Fig. 6B), displacing it from the radiolabeled probe, confirms that our assays are detecting a classic, noncovalent equilibrium interaction between protein and DNA.
FIG. 6.

I3-DNA interaction: NaCl titration and competition analyses. (A) The interaction of I3 with ssDNA is salt resistant. Reaction mixtures containing 3.3 pmol of recombinant I3 and 0.2 pmol of radiolabeled single-stranded 24-mer were incubated at 30°C for 15 min in buffer containing the indicated concentration of NaCl. Samples were fractionated on a 15% nondenaturing polyacrylamide gel at 4°C and visualized by autoradiography. (B) The interaction of I3 with ssDNA can be reversed by the addition of unlabeled competitor DNA. Reaction mixtures containing 3.3 pmol of recombinant I3 and 0.2 pmol radiolabeled single-stranded 24-mer were incubated at 30°C for 15 min. Unlabeled single-stranded 24-mer was then added to the levels indicated, and reaction mixtures were incubated for an additional 15 min at 30°C. Samples were then fractionated on a 15% nondenaturing polyacrylamide gel at 4°C and visualized by autoradiography.
I3 forms distinct complexes on longer oligonucleotides.
To assess the topology of I3’s interaction with DNA, EMSAs were performed with single-stranded oligonucleotides of increasing length. I3 was shown to form a ladder of discrete complexes on longer probes. As shown in Fig. 7 in an autoradiograph of a 5 to 20% gradient acrylamide gel, the number of complexes seen increases as probe length is increased. This is seen most clearly at limiting protein concentrations, since complexes are chased to saturated, lower-mobility species at high I3 concentrations. From the number of complexes seen with each oligonucleotide used, we can estimate the binding site size to be approximately 10 nt per I3 molecule (seven complexes are seen with the 73-mer, five are seen with the 53-mer, three are seen with the 34-mer etc.). However, we find that I3 will not bind to a 12-mer in this assay (not shown), indicating that the minimum length for binding is between 13 and 18 nt. The ability to detect the full array of different complexes under conditions in which most of the probe remains free strongly suggests that I3 binds to DNA in a noncooperative manner.
FIG. 7.

Analysis of the binding site size of I3 on ssDNA. Reaction mixtures contained either 10.5, 1.5, or 0 pmol of purified recombinant I3 and 0.2 pmol of a terminally radiolabeled oligonucleotide of 18, 24, 34, 53, or 73 nt in length. After 15 min of incubation at 30°C, samples were fractionated on a nondenaturing, 5 to 20% polyacrylamide gradient gel at 4°C and visualized by autoradiography.
Endogenous I3 also shows a strong preference for ssDNA.
A comparison of the DNA binding properties of recombinant and endogenous I3 is shown in Fig. 8. The two proteins show comparable binding to the radiolabeled 24-nt probe, although the mobilities of the complexes formed are somewhat different in this particular experiment (Fig. 8A). Recombinant I3 appears to form two complexes, whereas endogenous I3 forms only the more slowly migrating of these two species. As shown in Fig. 8B, the endogenous I3 also has a very strong preference for ssDNA.
FIG. 8.

Endogenous I3 binds specifically to ssDNA. (A) Comparison of the binding of endogenous and recombinant I3 to ssDNA. Reaction mixtures containing 0.2 pmol of radiolabeled single-stranded 24-mer oligonucleotide and the indicated amounts of either endogenous I3 (VV I3) (lanes 1 to 5) or recombinant I3 (Rec I3) (lanes 6 to 11) were incubated at 30°C for 15 min, fractionated on a 15% nondenaturing polyacrylamide gel at 4°C, and visualized by autoradiography. (B) Comparison of the binding of endogenous I3 to single-stranded and double-stranded DNA. Reaction mixtures contained 0.2 pmol of radiolabeled single-stranded 24-mer oligonucleotide (lanes 1 to 3) or 0.2 pmol radiolabeled double-stranded (ds) 24-mer oligonucleotide (of identical sequence) (lanes 4 to 6) and the indicated amounts of purified endogenous I3. After incubation for 15 min at 30°C, samples were fractionated on a 15% nondenaturing polyacrylamide gel at 4°C and visualized by autoradiography.
I3 saturates M13 ssDNA with low cooperativity.
The issue of whether recombinant and/or endogenous I3 bind ssDNA in a cooperative fashion was also addressed in assays using single-stranded, circular M13 DNA and increasing concentrations of I3 (23). Under conditions of DNA excess, highly cooperative binding would result in two bands after electrophoretic fraction through agarose: one representing fully saturated DNA, and one representing free DNA. As protein concentration increased, the intensity of the saturated DNA band would increase. In the absence of cooperativity, one would expect to see a random distribution of protein-DNA complexes, manifested in a diffuse band whose mobility would decrease as protein concentration increased. I3 binding to the 7.2-kbp M13 ssDNA clearly progressed through the formation of a random population of protein-DNA complexes which were of decreasing mobility with increasing protein concentration (Fig. 9). Equivalent titrations are presented for recombinant I3 and for endogenous I3; again, although the profiles are similar, the mobilities of the complexes seen are somewhat different. Elevation of the NaCl concentration in these reactions to 100 or 250 mM had no effect on the distribution of the shifted species (data not shown). Therefore, we do not observe a qualitative salt-dependent change in the cooperativity of DNA binding.
FIG. 9.

I3 binds to ssDNA with low cooperativity. Reaction mixtures containing 0.316 pmol of M13mp18 single-stranded circular DNA (7,249 bases) and various amounts (0, 20, 40, 60, 80, 100, and 120 pmol) of either recombinant (lanes 1 to 7) or endogenous (lanes 8 to 14) I3 protein were incubated at 30°C for 15 min. Reaction mixtures were then fractionated by electrophoresis through a 0.5% agarose gel; the gel was stained in 2 M NaCl–ethidium bromide, and DNA bands were visualized by UV illumination.
Chromatographic profile of I3 on ssDNA cellulose: phosphorylation does not appear to affect affinity.
In our comparisons of the interactions of recombinant and endogenous I3 with DNA, we found similarities in preference for ssDNA (Fig. 5 and 8), stoichiometry of binding (Fig. 8), and lack of cooperativity (Fig. 9). These results suggested that the phosphorylation of endogenous I3 probably had little effect on the DNA binding properties of the protein. During the course of these studies, however, data from another laboratory suggested that this might not be so. Davis and Mathews (4) identified I3 as a protein likely to interact with the vaccinia virus-encoded ribonucleotide reductase by screening vaccinia virus expression libraries with an anti-idiotype antibody raised against anti-ribonucleotide reductase (small subunit). Their studies suggested that whereas 35S-labeled I3 could be coprecipitated with nascent viral DNA metabolically labeled with bromodeoxyuridine, 32P-labeled I3 could not.
We addressed this issue further by looking at the association of I3 with ssDNA-cellulose either [35S]methionine-labeled (Fig. 10A) or 32Pi-labeled (Fig. 10B) cytoplasmic extracts. Fractions eluted from the column were analyzed by SDS-PAGE (12% gel) and visualized by silver staining (data not shown), immunoblot analysis, and autoradiography. As assessed by silver staining and immunoblot analysis, I3 elutes from ssDNA-cellulose as a broad peak from 0.5 to 2 M NaCl. These gels also show that I3 is by far the most abundant (silver stain analysis) and highly synthesized (autoradiography) protein retained on the ssDNA matrix under stringent wash conditions. 32P-labeled I3 also elutes as a broad peak that extends until the highest salt concentrations, indicating that phosphorylated I3 does not have a reduced affinity for ssDNA. (The highly radiolabeled species of approximately 34 kDa that appears to elute prior to immunoreactive I3 does not comigrate precisely with I3 and probably represents the H5 protein.) In support of this conclusion, we have also shown that recombinant I3, which is almost certainly not phosphorylated, exhibits the same affinity for ssDNA-cellulose seen with endogenous I3.
FIG. 10.

Chromatographic profile of [35S]methionine-labeled and 32Pi-labeled I3 on denatured DNA-cellulose. (A) BSC-40 cells were infected for 5 h with wt virus (MOI of 15) at 37°C and metabolically labeled with [35S]methionine throughout. Cytoplasmic extracts were prepared as described in Materials and Methods and applied to a 5-ml denatured DNA-cellulose column. The column was developed with a 60-ml linear gradient of 50 mM to 2.5 M NaCl. Fractions were analyzed by SDS-PAGE (12% gel), and the gel was subjected to autoradiography. Lanes: M, 14C-labeled protein standards (molecular masses are shown in kilodaltons at the left); Load, total cytoplasmic lysate; FT, flowthrough; Wash, material eluting in a 10-ml 50 mM NaCl wash. The remaining lanes contain the samples eluting during development with the NaCl gradient. The majority of the fractions were also subjected to immunoblot analysis using the I3 antiserum; the relevant portion of the blot is positioned below the autoradiograph. (B) BSC-40 cells were infected for 5 h with wt virus (MOI of 15) at 37°C and metabolically labeled with 32Pi throughout. Cytoplasmic extracts were prepared and analyzed as described above for panel A.
Attempted generation of an I3 null mutant virus.
The data presented above demonstrate that I3 is the best candidate for a viral SSB—it is one of the most abundant viral proteins synthesized prior to and during DNA replication and exhibits the strongest and most specific affinity for ssDNA. A role as an SSB would almost certainly require that I3 be an essential viral function. To determine whether I3 was in fact essential for viral replication, we attempted to isolate a viral recombinant in which the I3 allele had been inactivated by insertion of a selectable marker. BSC-40 cells infected with wt vaccinia virus were transfected with a plasmid construct containing modified HindIII I sequences; 46.5% of the I3 gene was deleted and replaced with the Neor gene under the control of a constitutive vaccinia virus promoter. Infected cells were transfected with either supercoiled or linearized plasmid DNA and maintained at 37°C under G418 selection until 3 days postinfection. Under these conditions, recombination between homologous sequences on the plasmid and viral genome should occur. Homologous recombination between the linearized plasmid and the genome should replace the endogenous allele with the interrupted allele containing the neor cassette. If the I3 allele is essential, then acquisition of Neor will come only from illegitimate, nonhomologous recombination events. When circular plasmid is used in the transfections, homologous recombination with the genome will generate two tandem copies of the I3 gene, one of which is intact and one of which is internally deleted and contains the Neor gene. These intermediate viruses are then subjected to two rounds of plaque purification in the absence of G418. It has been shown previously that tandemly repeated sequences are unstable within the vaccinia virus genome; if the I3 gene is not essential, recombination between the I3 alleles will lead to excision of the Neor gene with the concomitant retention of only a deleted I3 allele. When the final viral stocks were analyzed, isolates obtained from all 16 independent infections/transfections retained the Neor gene and an intact I3 gene. These data provide compelling evidence (21, 35, 42) that the I3 gene product plays an essential role during the vaccinia virus infectious cycle.
DISCUSSION
Our study has identified an essential vaccinia virus gene which encodes a protein with virtually all of the characteristics of replicative SSBs. These include abundant expression before and during DNA replication, localization to sites of viral DNA replication, and sequence-nonspecific, high-affinity binding to ssDNA. Our observation that I3 binds approximately 10 nt is also comparable to results obtained for several other SSBs (15, 17). The binding of I3 to ssDNA appears to lack significant cooperativity; the cooperativity of SSBs from different organisms has been reported to differ substantially (17, 22). Low cooperativity may in fact be an intrinsic feature of I3, but it is also possible that our experimental conditions (for example, the lack of an appropriate cation or cofactor) are not optimized for cooperative binding or that cooperativity requires an additional protein partner.
Although SSBs do not have a high degree of primary sequence similarity, there are common sequence motifs that have emerged from comparative analyses. A domain containing conserved aromatic and charged residues thought to mediate DNA binding is present in many SSBs and appears to be present in the I3 protein as well (11, 34, 51) (Fig. 11A). Support for the importance of this loosely defined motif has come from the direct demonstration that for several of these proteins, these aromatic residues are important for DNA binding. Interestingly, this motif is retained in the I3 homolog of molluscum contagiosum virus (MCV), a poxvirus whose genomic sequence differs significantly from that of vaccinia virus (40, 41) (Fig. 11B). Overall, the identity of the MCV protein to the vaccinia protein is only 39.1%, suggesting that those regions that are conserved are likely to be of functional importance in the context of viral infection.
FIG. 11.

Conservation of sequence motifs within SSBs. (A) A common motif is found in diverse SSBs. Partial protein sequences from various SSBs were aligned with that portion of the gene 5 protein (fd bacteriophage) involved in DNA binding (modified from references 11, 34, and 51). This analysis yielded a region in all of the sequences which consists of conserved aromatic and basic amino acids (indicated by single-letter abbreviations) separated by variable numbers of unrelated residues. These spacer regions are designated by X, with a subscript indicating the number of residues in each case. The proteins shown are the I3 protein (39) of vaccinia virus strain WR (VV), gene 5 protein of bacteriophage fd (GP5), gene 32 protein of bacteriophage T4 (GP32), SSB of E. coli (SSB), and DNA binding proteins from bacteriophage ike (PIKE), herpes simplex virus type 1 (HSV-1), varicella-zoster virus (VZV), Epstein-Barr virus (EBV), and adenovirus type 5 (AD-5). Numbers indicate the residue numbers for each sequence shown in the alignment. (B) Sequence comparison of the vaccinia virus I3 protein and the homolog encoded by MCV. The sequence of the I3 protein encoded by the WR strain of vaccinia virus protein was aligned with the sequence of the MCV homolog (ORF MC046L, accession no. U60315). Residues common to both proteins are shaded and boxed; circles are above the residues corresponding to the motif shown in panel A.
We have also shown that the I3 protein is phosphorylated on a serine residue(s). This property suggests that I3 is distinct from the phosphoprotein described earlier in the literature as a possible SSB (polypeptide B or FP11), which was shown to be phosphorylated on threonine residues. I3 also shows a far stronger and more specific affinity for ssDNA than was described for this protein, which is instead more likely to be the recently described product of the H5 gene (1), a 25-kDa protein which migrates anomolously in SDS-PAGE with an apparent MW of 35,000 (1, 10, 18, 38e)).
Our investigation of the elution profiles of 35S- or 32P-labeled extracts from ssDNA-cellulose revealed a single binding mode for phosphorylated and total I3, suggesting that phosphorylation does not alter the ssDNA binding properties on the protein under these conditions. This interpretation, of course, rests on the assumption that both phosphorylated and unphosphorylated I3 are present intracellularly. In support of our conclusion, we also see no appreciable difference in the DNA binding properties of endogenous I3 and recombinant I3 purified from E. coli. Nevertheless, Davis and Mathews previously reported that antibromodeoxyuridine antibody coprecipitated 35S-labeled but not 32P-labeled I3 (4). Our data suggest that their observations may reflect technical considerations (such as differences in the levels of the phosphorylated and unphosphorylated species) or may indicate that phosphorylation of I3 does modulate its interaction with DNA in vivo. We would propose that this modulation is indirect, perhaps by regulating I3’s interactions with other proteins, rather than through a direct effect on its inherent DNA binding properties.
The role of I3’s phosphorylation, therefore, warrants further study, especially since several eukaryotic SSBs are known to be phosphorylated (15, 52). The 34-kDa middle subunit of hRPA is phosphorylated in a cell cycle-specific manner, suggesting that hRPA-mediated events, including DNA replication, may be regulated through modulation of hRPA activity. The major SSB of adenovirus, DBP, is phosphorylated on several serine and threonine residues in its N-terminal domain, which is not its DNA binding domain. The significance of this phosphorylation is also unknown. Studies from our laboratory suggest that recombinant I3 cannot be phosphorylated in vitro by either purified B1 kinase (35, 38a) or F10 kinase (38b, 47). In addition, the phosphorylation of I3 is not compromised in vivo during nonpermissive infections performed with mutants carrying lesions in the B1 (ts2 or ts25) (35, 36, 38c) or F10 (ts28) (38d, 47, 50) kinase. It appears, therefore, that I3 is phosphorylated by cellular kinases. If phosphorylation of I3 is meaningful and not fortuitous, we suggest again that the most obvious hypothesis is that phosphorylation mediates protein-protein interactions central to I3’s role(s) in vivo.
Our characterization of I3’s DNA binding properties prompted us to test whether purified recombinant I3 could substitute for E. coli SSB in permitting recombinant vaccinia virus DNA polymerase to replicate a singly primed M13 template in vitro (24). It could not; moreover, addition of I3 to reactions containing E. coli SSB inhibited RFII formation in a dose-dependent manner (not shown). As more I3 was added, the length of the products formed decreased in a uniform manner, suggesting that I3 was slowing the apparent rate of polymerase elongation. These data could be explained by the presence of an inhibitor of the polymerase in our preparations; the observation that some, but not all, methods of purifying the herpes simplex virus-encoded SSB gave results similar to ours provides a precedent for this interpretation (13, 31). Alternatively, our data might suggest that the function of I3 as an SSB relies on appropriate phosphorylation and/or on interactions with molecules other than the catalytic DNA polymerase. Candidates for such partners could include an additional component of an SSB multimer, a processivity factor, or a replicative helicase. Moreover, the earlier studies of Davis and Matthews indicated that an anti-idiotypic antibody raised against the ribonucleotide reductase bound to the I3 protein; this finding, as well as their supporting data, suggests strongly that I3 could interact with the nucleotide synthetic machinery at the replication fork (4). By analogy to the cellular RPA, which interacts with components of the repair machinery as well as the replication machinery (12, 20, 28), I3 might also associate with such viral enzymes as the UDG. Identification of such a complex might clarify the surprising observation that the vaccinia virus UDG is essential for DNA replication (5, 26, 44). Davis and Mathews noted that both I3 and the SSB of bacteriophage of T4 (gene 32 protein) have a highly acidic C terminus (4). This region of the gene 32 protein has been shown to mediate interactions with other proteins. Assessing the impact of mutations within this and other regions of I3 on the viral life cycle, as well as pursuing biochemical and genetic strategies aimed at identifying I3-interacting proteins, should provide insight into the regulation of viral DNA metabolism.
ACKNOWLEDGMENTS
We thank B. Peabody for technical support and Lori Van Houten for invaluable help with the photography. We also thank Nancy Klemperer, Ke Liu, Bill McDonald, and Joel Pardee for many helpful discussions.
This work was supported by a grant to P.T. from the NIH (AI 21758) and by a generous group of donors from the Dorothy Rodbell Cohen Foundation.
REFERENCES
- 1.Beaud G, Beaud R, Leader D P. Vaccinia virus gene H5R encodes a protein that is phosphorylated by the multisubstrate vaccinia virus B1R protein kinase. J Virol. 1995;69:1819–1826. doi: 10.1128/jvi.69.3.1819-1826.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bonner W M, Laskey R A. A film detection method for tritium-labeled proteins and nucleic acids in polyacrylamide gels. Eur J Biochem. 1974;46:83–88. doi: 10.1111/j.1432-1033.1974.tb03599.x. [DOI] [PubMed] [Google Scholar]
- 3.Challberg M D, Englund P T. The effect of template secondary structure on vaccinia DNA polymerase. J Biol Chem. 1979;254:7820–7826. [PubMed] [Google Scholar]
- 4.Davis R E, Mathews C K. Acidic C terminus of vaccinia virus DNA binding protein interacts with ribonucleotide reductase. Proc Natl Acad Sci USA. 1993;90:745–749. doi: 10.1073/pnas.90.2.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ellison K S, Peng W, McFadden G. Mutations in active-site residues of the uracil-DNA glycosylase encoded by vaccinia virus are incompatible with virus viability. J Virol. 1996;70:7965–7973. doi: 10.1128/jvi.70.11.7965-7973.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Evans E, Klemperer N, Ghosh R, Traktman P. The essential replication protein encoded by the vaccinia virus D5 gene is a nucleic acid-independent NTPase. J Virol. 1995;69:5353–5361. doi: 10.1128/jvi.69.9.5353-5361.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Evans E, Traktman P. Molecular genetic analysis of a vaccinia virus gene with an essential role in DNA replication. J Virol. 1987;61:3152–3162. doi: 10.1128/jvi.61.10.3152-3162.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Franke C A, Rice C M, Strauss J H, Hruby D E. Neomycin resistance as a dominant selectable marker for selection and isolation of vaccinia virus recombinants. Mol Cell Biol. 1985;5:1918–1924. doi: 10.1128/mcb.5.8.1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goebel S J, Johnson G P, Perkus M E, David S W, Winslow J P, Paoletti E. The complete DNA sequence of vaccinia virus. Virology. 1990;179:247–266. doi: 10.1016/0042-6822(90)90294-2. [DOI] [PubMed] [Google Scholar]
- 10.Gordon J, Kovala T, Dales S. Molecular characterization of a prominent antigen of the vaccinia virus envelope. Virology. 1988;167:361–369. [PubMed] [Google Scholar]
- 11.Gutierrez C, Martin G, Sogo J M, Salas M. Mechanism of stimulation of DNA replication by bacteriophage Phi 29 single-stranded DNA-binding protein p5. J Biol Chem. 1991;266:2104–2111. [PubMed] [Google Scholar]
- 12.He Z, Henricksen L A, Wold M S, Ingles C J. RPA involvement in the damage-recognition and incision steps of nucleotide excision repair. Nature. 1995;374:566–569. doi: 10.1038/374566a0. [DOI] [PubMed] [Google Scholar]
- 13.Hernandez T R, Lehman I R. Functional interaction between the herpes simplex-1 DNA polymerase and UL42 protein. J Biol Chem. 1990;265:11227–11232. [PubMed] [Google Scholar]
- 14.Hirschmann P, Vos J C, Stunnenberg H G. Mutational analysis of a vaccinia virus intermediate promoter in vivo and in vitro. J Virol. 1990;64:6063–6069. doi: 10.1128/jvi.64.12.6063-6069.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hubscher U, Maga G, Podust V N. DNA replication accessory proteins. In: DePamphilis M L, editor. DNA replication in eukaryotic cells. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1996. pp. 525–544. [Google Scholar]
- 16.Johnson G P, Goebel S J, Paoletti E. An update on the vaccinia virus genome. Virology. 1993;196:381–401. doi: 10.1006/viro.1993.1494. [DOI] [PubMed] [Google Scholar]
- 17.Kornberg A, Baker T. DNA replication. W. H. New York, N.Y: Freeman and Co.; 1992. [Google Scholar]
- 18.Kovacs G R, Moss B. The vaccinia virus H5R gene encodes late gene transcription factor 4: purification, cloning, and overexpression. J Virol. 1996;70:6796–6802. doi: 10.1128/jvi.70.10.6796-6802.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Laemmli U K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 20.Li L, Lu X, Peterson C A, Legerski R J. An interaction between the DNA repair factor XPA and replication protein A appears essential for nucleotide excision repair. Mol Cell Biol. 1995;15:5396–5402. doi: 10.1128/mcb.15.10.5396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu K, Lemon B, Traktman P. The dual specificity phosphatase encoded by vaccinia virus, VH1, is essential for viral transcription in vivo and in vitro. J Virol. 1995;69:7823–7834. doi: 10.1128/jvi.69.12.7823-7834.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lohman T M, Ferrari M E. Escherichia coli single-stranded DNA-binding protein: multiple DNA-binding modes and cooperativities. Annu Rev Biochem. 1994;63:527–570. doi: 10.1146/annurev.bi.63.070194.002523. [DOI] [PubMed] [Google Scholar]
- 23.Lohman T M, Overman L B. Salt-dependent changes in the DNA binding cooperativity of Escherichia coli single strand binding protein. J Mol Biol. 1986;187:603–615. doi: 10.1016/0022-2836(86)90338-4. [DOI] [PubMed] [Google Scholar]
- 24.McDonald W F, Traktman P. Vaccinia virus DNA polymerase: In vitro analysis of parameters affecting processivity. J Biol Chem. 1994;269:31190–31197. [PubMed] [Google Scholar]
- 25.McDonald W F, Traktman P. Overexpression and purification of the vaccinia virus DNA polymerase. Protein Expression Purif. 1994;5:409–421. doi: 10.1006/prep.1994.1059. [DOI] [PubMed] [Google Scholar]
- 26.Millns A K, Carpenter M S, DeLange A M. The vaccinia virus-encoded uracil DNA glycosylase has an essential role in viral DNA replication. Virology. 1994;198:504–513. doi: 10.1006/viro.1994.1061. [DOI] [PubMed] [Google Scholar]
- 27.Morham S G, Shuman S. Covalent and noncovalent DNA binding by mutants of vaccinia DNA topoisomerase I. J Biol Chem. 1992;267:15984–15992. [PubMed] [Google Scholar]
- 28.Nagelhus T A, Haug T, Singh K K, Keshav K F, Skorpen F, Otterlei M, Bharati S, Lindmo T, Benichou S, Benarous R, Krokan H E. A sequence in the N-terminal region of human uracil-DNA glycosylase with homology to XPA interacts with the C-terminal part of the 34-kDa subunit of replication protein A. J Biol Chem. 1997;272:6561–6566. doi: 10.1074/jbc.272.10.6561. [DOI] [PubMed] [Google Scholar]
- 29.Nowakowski M, Bauer W, Kates J. Characterization of a DNA-binding phosphoprotein from vaccinia virus replication complex. Virology. 1978;86:217–225. doi: 10.1016/0042-6822(78)90022-3. [DOI] [PubMed] [Google Scholar]
- 30.Nowakowski M, Kates J, Bauer W. Isolation of two DNA-binding proteins from the intracellular replication complex of vaccinia virus. Virology. 1978;84:260–267. doi: 10.1016/0042-6822(78)90246-5. [DOI] [PubMed] [Google Scholar]
- 31.O’Donnell M E, Elias P, Funnell B E, Lehman I R. Interaction between the DNA polymerase and single-stranded DNA-binding protein (infected cell protein 8) of herpes simplex virus 1. J Biol Chem. 1987;262:4260–4266. [PubMed] [Google Scholar]
- 32.Polisky B, Kates J. Vaccinia virus intracellular DNA-protein complex: biochemical characteristics of associated protein. Virology. 1972;49:168–179. doi: 10.1016/s0042-6822(72)80018-7. [DOI] [PubMed] [Google Scholar]
- 33.Polisky B, Kates J. Viral-specific polypeptides associated with newly replicated vaccinia DNA. Virology. 1975;66:128–139. doi: 10.1016/0042-6822(75)90184-1. [DOI] [PubMed] [Google Scholar]
- 34.Prasad B V V, Chiu W. Sequence comparison of single-stranded DNA binding proteins and its structural implications. J Mol Biol. 1987;193:579–584. doi: 10.1016/0022-2836(87)90268-3. [DOI] [PubMed] [Google Scholar]
- 35.Rempel R, Traktman P. Vaccinia virus B1 kinase: phenotypic analysis of temperature-sensitive mutants and enzymatic characterization of recombinant proteins. J Virol. 1992;66:4413–4426. doi: 10.1128/jvi.66.7.4413-4426.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rempel R E, Anderson M K, Evans E, Traktman P. Temperature-sensitive vaccinia virus mutants identify a gene with an essential role in viral replication. J Virol. 1990;64:574–583. doi: 10.1128/jvi.64.2.574-583.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ricciardi R P, Miller J S, Roberts B E. Purification and mapping of specific mRNAs by hybridization-selection and cell-free translation. Proc Natl Acad Sci USA. 1979;76:4927–4931. doi: 10.1073/pnas.76.10.4927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rigby P W J, Dieckmann M, Rhodes C, Berg P. Labeling deoxyribonucleic acid to high specific activity in vitro by nick translation with DNA polymerase I. J Mol Biol. 1977;113:237–251. doi: 10.1016/0022-2836(77)90052-3. [DOI] [PubMed] [Google Scholar]
- 38a.Rochester, C. R., and U. Sankar. Unpublished data.
- 38b.Rochester, C. R., and S. Jesty. Unpublished data.
- 38c.Sankar, U. Personal communication.
- 38d.Sankar, U., and P. Traktman. Unpublished data.
- 38e.Sankar, U., M. Derrien, and P. Traktman. Unpublished data.
- 39.Schmitt J F C, Stunnenberg H G. Sequence and transcriptional analysis of the vaccinia virus HindIII I fragment. J Virol. 1988;62:1889–1897. doi: 10.1128/jvi.62.6.1889-1897.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Senkevich T G, Bugert J J, Sisler J R, Koonin E V, Darai G, Moss B. Genome sequence of a human tumorigenic poxvirus: Prediction of specific host responses—evasion genes. Science. 1996;273:813–816. doi: 10.1126/science.273.5276.813. [DOI] [PubMed] [Google Scholar]
- 41.Senkevich T G, Koonin E V, Bugert J J, Darai G, Moss B. The genome of molluscum contagiosum virus: analysis and comparison with other poxviruses. Virology. 1997;233:19–42. doi: 10.1006/viro.1997.8607. [DOI] [PubMed] [Google Scholar]
- 42.Shuman S, Golder M, Moss B. Insertional mutagenesis of the vaccinia virus gene encoding a type I topoisomerase: evidence that the gene is essential for virus growth. Virology. 1989;170:302–306. doi: 10.1016/0042-6822(89)90384-x. [DOI] [PubMed] [Google Scholar]
- 43.Siegel L, Monty K. Determination of molecular weights and frictional ratios of proteins in impure systems by the use of gel filtration and density gradient centrifugation. Biochim Biophys Acta. 1966;112:346–362. doi: 10.1016/0926-6585(66)90333-5. [DOI] [PubMed] [Google Scholar]
- 44.Stuart D T, Upton C, Higman M A, Niles E G, McFadden G. A poxvirus-encoded uracil DNA glycosylase is essential for virus viability. J Virol. 1993;67:2503–2512. doi: 10.1128/jvi.67.5.2503-2512.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Studier F W, Rosenberg A L, Dunn J J, Dubendorff J W. Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol. 1990;185:60–89. doi: 10.1016/0076-6879(90)85008-c. [DOI] [PubMed] [Google Scholar]
- 46.Traktman P. Poxvirus DNA replication. In: DePamphilis M, editor. DNA replication in eukaryotic cells. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1996. pp. 775–798. [Google Scholar]
- 47.Traktman P, Caligiuri A, Jesty S A, Liu K, Sankar U. Temperature-sensitive mutants with lesions in the vaccinia virus F10 kinase undergo arrest at the earliest stage of virion morphogenesis. J Virol. 1995;69:6581–6587. doi: 10.1128/jvi.69.10.6581-6587.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Traktman P, Sridhar P, Condit R C, Roberts B E. Transcriptional mapping of the DNA polymerase gene of vaccinia virus. J Virol. 1984;49:125–131. doi: 10.1128/jvi.49.1.125-131.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vogelstein B, Gillespie D. Preparative and analytical purification of DNA from agarose. Proc Natl Acad Sci USA. 1979;76:615–619. doi: 10.1073/pnas.76.2.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang S, Shuman S. Vaccinia virus morphogenesis is blocked by temperature-sensitive mutations in the F10 gene, which encodes protein kinase-2. J Virol. 1995;69:6376–6388. doi: 10.1128/jvi.69.10.6376-6388.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang Y, Hall J D. Characterization of a major DNA-binding domain in the herpes simplex virus type 1 DNA-binding protein (ICP8) J Virol. 1990;64:2082–2089. doi: 10.1128/jvi.64.5.2082-2089.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weisshart K, Fanning E. Role of phosphorylation in DNA replication. In: DePamphilis M L, editor. DNA replication in eukaryotic cells. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1996. pp. 295–330. [Google Scholar]