

Abstract
The rabbit hemorrhagic disease virus (RHDV) (isolate AST/89) RNA-dependent RNA-polymerase (3Dpol) coding region was expressed in Escherichia coli by using a glutathione S-transferase-based vector, which allowed milligram purification of a homogeneous enzyme with an expected molecular mass of about 58 kDa. The recombinant polypeptide exhibited rifampin- and actinomycin D-resistant, poly(A)-dependent poly(U) polymerase. The enzyme also showed RNA polymerase activity in in vitro reactions with synthetic RHDV subgenomic RNA in the presence or absence of an oligo(U) primer. Template-size products were synthesized in the oligo(U)-primed reactions, whereas in the absence of added primer, RNA products up to twice the length of the template were made. The double-length RNA products were double stranded and hybridized to both positive- and negative-sense probes.
Rabbit hemorrhagic disease virus (RHDV), a member of the Caliciviridae family (3, 16, 17), consists of a single plus-stranded RNA genome of about 7.4 kb (12) that has a virus-encoded protein, VPg (24), attached covalently to its 5′ end (13) and is polyadenylated at the 3′ end. Viral particles also encapsidate an abundant VPg-linked polyadenylated subgenomic RNA of about 2.2 kb (13), which is coterminal with the 3′ end of the viral genome. The data obtained from the in vitro translation (24) and Escherichia coli expression studies (11) revealed that the viral RNA is translated into a polyprotein that is subsequently cleaved to give rise to mature structural and nonstructural proteins.
Progress on the replication of caliciviruses has been negligible (2) compared with the advances made with the picornaviruses. Because of the lack of a cell culture system for most caliciviruses, such as RHDV, recombinant DNA technology will be important for the production and characterization of viral proteins. Although functional studies have not yet been performed for most of the RHDV nonstructural proteins, the extensive sequence similarities between the RNA-dependent RNA polymerase 3D of picornavirus and the recently described RHDV polyprotein cleavage product p58 (24) indicate that this polypeptide might have a similar role in genome replication. This putative mature RNA-dependent RNA polymerase of RHDV (3Dpol) is generated by cleavage at specific glutamicacid-threonine and glutamic acid-glycine bonds by a viral trypsin-like cysteine proteinase (1, 25).
The expression of enzymatically active 3Dpol from poliovirus (14, 19, 20) and encephalomyocarditis (EMC) virus (22) in E. coli has been reported, and the recombinant enzyme was found to have properties similar to those of the polymerase obtained from virus-infected cells (15). Attempts to isolate the RNA polymerase of RHDV from infected rabbit livers for in vitro studies were unsuccessful. This suggested the alternative approach to produce the mature RHDV 3Dpol in E. coli, using an expression system known to allow the rapid purification of similar recombinant polypeptides under nondenaturing conditions.
MATERIALS AND METHODS
Construction of plasmid pGEX-3D.
The 3Dpol coding region was cloned as a BamHI cassette by PCR amplification of plasmid pRC30 (11). This was done by using oligonucleotide primers 3D5′ (5′ggatccatgacatcaaacttcttctgcg3′) and 3CD3′ (5′ggatcctcactccataacattcacaaactc3′), which also added a BamHI restriction enzyme recognition sequence (underlined residues) at both ends of the amplified region. The resulting fragment was ligated to the pGEM-T vector (Promega). After digestion of the recombinant plasmid with the restriction enzyme BamHI, the 1.5-kb fragment containing the 3Dpol gene was purified and inserted into BamHI-digested, alkaline phosphatase-treated expression vector pGEX-2T (Pharmacia) to generate the recombinant plasmid pGEX-3D (Fig. 1). Cells of E. coli XL1-Blue were subsequently transformed with this vector.
FIG. 1.

Schematic representation of expression plasmid pGEX-3D. The expanded region indicates the junction between GST and 3Dpol coding regions and deduced amino acid sequence. The thrombin cleavage site and the authentic 3Dpol amino terminus are also indicated.
Plasmid pGEX-3D (Fig. 1) directed the synthesis of a fusion protein composed of Schistosoma japonicum glutathione S-transferase (GST) and RHDV 3Dpol.
Expression and purification of RHDV 3Dpol.
Overnight cultures of E. coli transformed with pGEX-3D were diluted 1:250 in 500 ml of fresh 2× Luria-Bertani medium containing ampicillin (50 μg ml−1). The cultures were incubated at 37°C to an optical density at 600 nm of 0.5, and isopropyl-1-thio-β-d-galactopyranoside (IPTG) was added up to 200 μM. After 2 h of growth at 37°C, the cells were harvested by centrifugation and the pellet was suspended in 30 ml of 50 mM Tris-HCl (pH 8.0)–150 mM NaCl–0.25 mM EDTA (buffer 1). After a 30-min incubation with lysozyme, the suspension was sonicated by using five 30-s bursts. The supernatant, recovered after centrifugation at 15,000 rpm for 15 min in a Kontron A8.24 rotor, was loaded onto a 5 ml glutathione-Sepharose 4B column (Pharmacia) equilibrated in buffer 1. The eluate was retained and reapplied to the column. Following the absorption step, the column was washed five times with 5 ml of buffer 1. After the final wash, the gel slurry was suspended in 400 μl of buffer 1 containing 480 ng of thrombin (Pharmacia) and incubated at room temperature for 3 h. The purified RHDV 3Dpol was recovered by collecting the column eluate. The purified RHDV 3Dpol preparation was stored at −20°C after addition of 5% glycerol and 10 mM MgCl2.
Preparation of oligo(U).
Oligo(U) was prepared by alkali hydrolysis of poly(U) as described by Plotch et al. (19). The size of the resulting oligo(U) was determined by end-labeling with [γ-32P]ATP (ICN) and electrophoresis on a 6% polyacrylamide gel.
Preparation of synthetic RHDV subgenomic RNA.
The viral subgenomic RNA was made by in vitro transcription of plasmid pRNA2.2T7R (Fig. 2). Transcription vector pRNA2.2T7R consisted of pEMBL 18 in which a modified T7 RNA polymerase promoter followed by nucleotide (nt) residues 5296 to 7437 of RHDV (isolate AST/89) cDNA, a poly(A) tail of 18 residues, hepatitis delta virus (HDV) antigenome ribozyme (18), and T7 RNA polymerase terminator sequences were cloned (Fig. 2). In vitro transcription of XhoI-digested pRNA2.2T7R with T7 RNA polymerase yielded a 2.2-kb RNA transcript with a nucleotide sequence identical to the authentic subgenomic mRNA of RHDV but lacking the VPg attached to its 5′ terminus and having a shorter poly(A) tail of only 18 residues. A 3′ truncated synthetic subgenomic RNA, lacking 312 nt residues and the poly(A) tail, was also made by runoff transcription of vector pRNA2.2T7R digested with HindIII (Fig. 2).
FIG. 2.

Schematic representation of transcription vector pRNA2.2T7R. The solid triangle and rectangle represent the modified T7 RNA polymerase promoter and T7 RNA polymerase terminator sequences, respectively. The hatched rectangle denotes the HDV antigenome ribozyme sequence. The asterisk represents the poly(A) tail. The numbers indicate RHDV cDNA nucleotide residues. The expanded region shows the modified T7 RNA polymerase promoter (italic boldface) and the RHDV cDNA 5′ region (underlined). The restriction sites XhoI and HindIII, used to linearize the plasmid, and the transcription initiation site (+1) are also indicated.
Gel electrophoresis and protein concentration.
RNA was analyzed on 1.2% formaldehyde-agarose gels (21). Sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis was performed as described elsewhere (9). The protein concentration was determined by the Bio-Rad protein assay.
Enzymatic assays.
The poly(A)-dependent oligo(U)-primed poly(U) polymerase assay was performed as described by Sankar and Porter (22). Briefly, the reaction was carried out at 30°C for 60 min in 50-μl samples containing appropriate concentrations of purified 3Dpol, 50 mM HEPES (pH 8.0), 10 μM UTP, 4 mM dithiothreitol, 3 mM magnesium acetate, 6 μM zinc chloride, 20 μg of rifampin ml−1, 3 nmol of oligo(U) nt−1, 7 nmol of poly(A) nt−1, and 2.5 μmol of [α-32P]UTP (Amersham) (400 Ci/mmol). The in vitro-synthesized product was precipitated with 10% trichloroacetic acid after addition of 100 μg of carrier tRNA in 0.2 M sodium pyrophosphate, collected onto 0.45-μm-pore-size Whatman GF/C filters, and vacuum dried. The radioactivity on the filters was measured by using a scintillation counter.
RNA polymerase RNA-dependent activity was assayed in 50-μl samples containing appropriate concentrations of the purified 3Dpol, 50 mM HEPES (pH 8.0), 10 μM ATP, 10 μM CTP, 10 μM GTP, 5 μM UTP, 4 mM dithiothreitol, 3 mM magnesium acetate, 6 μM zinc chloride, 50 U of ribonuclease inhibitor (Promega), 25 μmol of [α-32P]UTP, and 14 to 28 nM synthetic RHDV subgenomic RNA. After incubation at 30°C for 60 min, the reaction mixture was phenol-chloroform extracted and then ethanol precipitated in the presence of 0.3 M sodium acetate (pH 6.0) and 20 μg of carrier tRNA. The sediments were dissolved in electrophoresis sample buffer, loaded onto 1.2% formaldehyde-agarose gels, and electrophoresed at 60 to 70 V. The gels were then dried, and the in vitro 32P-labeled RNA was detected by autoradiography.
RNase treatment.
The products synthesized by 3Dpol were treated with 14 μg of RNase A ml−1 in 0.5 M or 15 mM NaCl for 15 min at room temperature (8) and analyzed by electrophoresis on 1.2% formaldehyde–agarose gels as described above.
Northern blot hybridization.
Template synthetic RHDV subgenomic RNA and the products synthesized from it by 3Dpol in an enzymatic assay similar to that described above but without [α-32P]UTP were analyzed in parallel tracks of denaturing agarose gels and transferred onto nylon membranes. After blotting, hybridization was done at 42°C for 24 h in the presence of formamide and 32P-labeled RNA transcripts made in vitro with T7 or T3 RNA polymerases to obtain the positive and negative probes, respectively. The recombinant transcription vector contained cDNA sequences representing nt residues 5436 to 7043 from RHDV, cloned into the EcoRI site of pBluescript SK+ (Stratagene). After hybridization the filters were washed at 55°C for 1 h with two changes of 0.1× SSC (150 mM NaCl, 15 mM sodium citrate)–0.1% SDS and autoradiographed.
RESULTS
Cloning and enzyme purification.
To obtain large quantities of 3Dpol, we used the expression vector pGEX-2T, designed for inducible expression of foreign polypeptides in E. coli as fusion proteins with GST. The protein chimera was purified from bacterial lysates by affinity chromatography using the Bulk GST Purification Module (Pharmacia), and the 3Dpol was then released from GST by proteolysis of the fusion protein as described in Materials and Methods.
After induction with IPTG, a major protein band of about 84 kDa was observed in the extracts corresponding to the cells transformed with the recombinant vector pGEX-3D (Fig. 3, lane 2). The molecular mass of this polypeptide was in the range of that expected for the fusion protein GST-3Dpol. The 84-kDa polypeptide was retained on a glutathione-Sepharose column (Fig. 3, lane 3), supporting further the conclusion that this band was indeed the fusion protein. The 3Dpol moiety was released from the carrier protein by in situ proteolysis of the fusion protein adsorbed onto the glutathione-Sepharose column. The released 3Dpol was washed off the gel slurry, showing the expected molecular mass of 58 kDa (Fig. 3, lane 4). The recombinant polypeptide was almost identical in amino acid sequence to the authentic RHDV 3Dpol, except that it contained three extra amino acid residues (Gly-Ser-Met) at the N terminus.
FIG. 3.
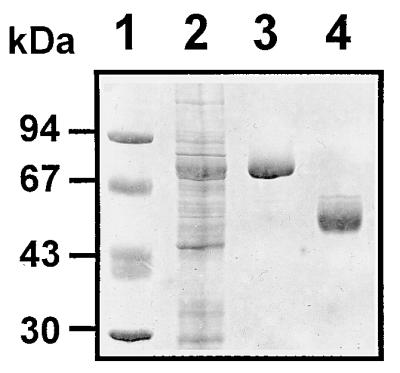
Sodium dodecyl sulfate-polyacrylamide electrophoretic analysis of recombinant RHDV 3Dpol preparations followed by Coomassie Blue staining. Lane 1, molecular weight markers; lane 2, cell-free extract from pGEX-3D-transformed, IPTG-induced E. coli; lane 3, purified GST-3Dpol fusion protein; lane 4, purified recombinant RHDV 3Dpol.
Poly(U) polymerase activity of recombinant RHDV 3Dpol.
As a first step towards establishing an assay system for the RHDV 3Dpol, we used the poly(A)-dependent oligo(U)-primed poly(U) polymerase assay, as described by Sankar and Porter (22). Polymerase activity was quantified as the rate of incorporation of [α-32P]UTP into acid-precipitable material in response to a poly(A) template and an oligo(U) primer. Low concentrations of the purified recombinant 3Dpol exhibited significant poly(U) polymerase activity that was resistant to rifampin at 20 μg ml−1 (Table 1), an antibiotic which is known to block the reinitiation of RNA chains by the E. coli DNA-dependent RNA polymerase holoenzyme. The poly(U) polymerase activity exhibited by 3Dpol was next characterized with respect to template and primer requirements for activity. The results obtained showed that it was completely dependent on the presence of both oligo(U) and poly(A) in the reaction mixture: omission of either one resulted in an almost complete loss of activity (Table 1). The presence of 8 μg of actinomycin D ml−1 did not inhibit the poly(U) polymerase activity (Table 1).
TABLE 1.
Poly(U) polymerase activity of recombinant RHDV 3Dpol
| Reaction mixturea | [α-32P]UMP incorporated (fmol) [%] |
|---|---|
| Poly(A), oligo(U), rifampin (20 μg ml−1) | 151.8 [100] |
| Without rifampin | 147.7 [97.3] |
| Without oligo(U) | 10.1 [6.7] |
| Without poly(A) | 1.8 [1.2] |
| With actinomycin D (8 μg ml−1) | 125.4 [82.6] |
The reaction mixture contained in all cases 2.8 μM purified recombinant enzyme.
Further biochemical characterization of the recombinant 3Dpol was carried out to determine the kinetics of poly(U) polymerase activity (Fig. 4). The amount of product synthesized in a 60-min reaction was found to be a linear function of enzyme concentration, from 0.7 to 3.5 μM 3Dpol (Fig. 4A). Under those assay conditions the poly(U) polymerase activity was linear for 120 min at 30°C (Fig. 4B) and showed oligo(U) saturation kinetics (Fig. 4C).
FIG. 4.

Poly(U) polymerase activity of recombinant RHDV 3Dpol. (A) Effect of enzyme concentration in a 60-min assay carried out at 30°C; (B) 2.4 μM purified recombinant 3Dpol was assayed at 30°C for poly(A)-dependent oligo(U)-primed poly(U) polymerase activity as a function of time; (C) amount of 32P-labeled UMP incorporated in a 60-min assay, using 2.4 μM purified enzyme, as a function of oligo(U) concentration.
RNA-dependent RNA polymerase activity of recombinant RHDV 3Dpol.
To determine whether the recombinant enzyme exhibited any RNA polymerase activity, a synthetic RHDV subgenomic RNA, made by in vitro transcription of XhoI-digested plasmid pRNA2.2T7R (see Materials and Methods), was incubated with the purified enzyme in the presence of all four ribonucleoside triphosphates. The RHDV RNA transcribed in vitro differed somewhat from authentic subgenomic RNA in that it lacked VPg at its 5′ end and its 3′ end had a shorter poly(A) tail of only 18 residues.
The size and quantity of the labeled RNA products synthesized in the absence or presence of increasing amounts of oligo(U) were analyzed by formaldehyde-agarose gel electrophoresis. The major reaction product synthesized in the absence of oligo(U) was an RNA of twice the length (4.4 kb) of the template (Fig. 5, lane 1), indicating that in the presence of an RNA template the enzyme did not have an absolute requirement for oligo(U), in contrast with the results obtained in the poly(U) polymerase assay (Table 1). When increasing amounts of oligo(U) were added to the above reaction mixture, increasing amounts of a template-size product (2.2 kb) were observed together with an equivalent decrease in the 4.4-kb product concentration (Fig. 5, lanes 2 to 6). These data indicate that the replication of RHDV RNA could also be dependent on a preformed primer, as shown for other RNA polymerases (22).
FIG. 5.

Autoradiograph of an agarose-formaldehyde gel showing 32P-labeled RNA products synthesized by recombinant 3Dpol with full-length synthetic RHDV subgenomic RNA as the template, in the presence of 0, 3, 15, 30, 60 or 150 pmol (lanes 1 to 6) of added oligo(U) as the primer nt−1, or with 3′ truncated subgenomic RNA, in the presence of 0, 15, 30, 60 or 150 pmol (lanes 7 to 11) of added oligo(U) as the primer nt−1.
The observation that the largest product RNA was twice the size of the template RNA suggested that the newly synthesized labeled RNA could be covalently linked to the template as a result of a self-priming event. The synthesis of double-length RNA, made in the presence of host factors (26) or oligo(U) (10), has also been shown for the poliovirus replicase.
To further determine whether the dimer-length products are in fact the result of a self-priming event at the extreme 3′ terminus of the template, an RNA transcript of 1.8 kb derived from the same transcription vector but lacking 312 nt of its 3′ region and the complete poly(A) tail was also used. The product RNA synthesized in this reaction was twice the size (3.6 kb) of the template RNA (Fig. 5, lane 7), and this reaction was not inhibited when increasing amounts of oligo(U) were added (Fig. 5, lanes 8 to 11). This result demonstrated that the ability of oligo(U) to inhibit the synthesis of the dimer-length products derived from the full-length RNA is due to a specific interaction at the 3′ end of the template.
These results also showed that the recombinant enzyme exhibited RNA polymerase activity on nonpolyadenylated RNAs.
The RNA polymerase activity in the presence or absence of oligo(U) was dependent on the addition of Mg2+ and all ribonucleoside triphosphates and was inhibited by EDTA (data not shown).
To further analyze the nature of the double-size RNA product synthesized from the full-length template in the absence of oligo(U), a time course experiment was performed. In the oligo(U)-independent reaction the RNA products increased in length from a few hundred residues larger than the template, at short reaction times (5 min), to approximately twice the length of the template RNA after 20 min or longer incubation periods (Fig. 6).
FIG. 6.

Autoradiograph of an agarose-formaldehyde gel showing the time course of oligo(U)-independent RNA synthesis by recombinant RHDV 3Dpol using full-length synthetic RHDV subgenomic RNA as the template. Lanes 1 to 6 correspond to 0, 5, 10, 20, 40, and 60 min of incubation at 30°C, respectively.
This double-length RNA product was not digested by RNase A (14 μg ml−1) in the presence of 0.5 M NaCl but was degraded when the salt concentration was lowered to 0.015 M (Fig. 7). These data supported the hypothesis that most of the 4.4-kb RNA product is double stranded (8) as the result of the self-complementarity of the newly synthesized product and the plus-stranded template, both strands being covalently linked at one end. To investigate further this hypothesis, we have determined the polarity of the 4.4-kb transcript in a Northern blot assay. The results showed that the double-length RNA transcript was recognized by both positive (Fig. 8A, lane 2) and negative (Fig. 8B, lane 2) probes. The template-size RNA recognized by the negative probe in the reaction mixture (Fig. 8B, lane 2) corresponded to an excess of template RNA, and negative polarity products of template length were not detected in the absence of the oligo(U) primer (Fig. 8A, lane 2). These data suggested that under these experimental conditions, the RHDV 3Dpol synthesized double-size transcripts in an oligo(U)-independent reaction which consisted of a newly made minus-strand RNA covalently linked to the plus-strand RNA template.
FIG. 7.
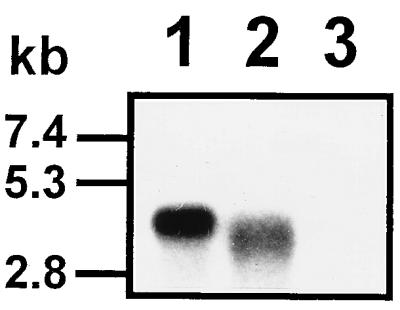
Autoradiograph of an agarose-formaldehyde gel showing RNase A sensitivity of the 32P-labeled RNA product synthesized by recombinant RHDV 3Dpol with full-length synthetic RHDV subgenomic RNA as the template, in the absence of added oligo(U). Lane 1, untreated control; lane 2, 14 μg of RNase A ml−1 in 0.5 M NaCl; lane 3, 14 μg of RNase A ml−1 in 0.015 M NaCl.
FIG. 8.

Northern blot analysis of the RNA product synthesized by recombinant RHDV 3Dpol with full-length synthetic RHDV subgenomic RNA as the template in the absence of added oligo(U). Lanes 1, template RNA; lanes 2, RHDV 3Dpol RNA transcription reaction. (A) Positive probe; (B) negative probe.
In a preliminary study of template specificity, the recombinant enzyme was incubated with purified luciferase mRNA. The product RNA synthesized was also twice the size of the template (data not shown).
DISCUSSION
RNA-dependent RNA polymerases are unique to viruses, and there are no known homologs in animal cells; therefore, this class of enzyme is an interesting target for antiviral chemotherapy. On the other hand, from an evolutionary point of view RNA-dependent RNA polymerases were probably among the first enzymes, and it would be interesting to see how their structure and function have evolved.
Studies on the replication of caliciviruses have lagged behind the advances made with picornaviruses, despite the fact that some of them can be propagated readily in tissue culture cells (2). In the case of RHDV, the lack of a cell system to propagate this pathogen impeded studies of this type and made recombinant DNA technology an essential approach for the characterization of viral functions.
The extensive sequence similarities between the RNA-dependent RNA 3Dpol of picornavirus and the recently described RHDV polyprotein cleavage product p58 (24) suggested that this polypeptide might have a similar role in RHDV genome replication. However, a direct demonstration of polymerase activity associated with this polypeptide has not been made.
The purpose of this work was to develop a system for heterologous expression of the RHDV p58 coding region for functional studies. Using a GST-based vector, we have achieved high-level expression of the fusion protein and purified the recombinant polypeptide to near homogeneity after thrombin cleavage of the fusion protein. The recombinant p58 was designed to be identical in sequence to the RHDV native enzyme; nevertheless, it contained three extra amino acid residues (Gly-Ser-Met) at the N terminus as a consequence of the expression strategy used. The fact that 3Dpol activity was present in a recombinant polypeptide, not generated as the result of cleavage of a polyprotein, suggested that the enzyme did not need to be generated from a precursor to fold into an active conformation. Furthermore, a prokaryotic system was also able to produce a functional enzyme, indicating that eukaryotic posttranslational modifications were not essential for 3Dpol activity. These data agreed with similar results reported for poliovirus and EMC virus replicases (19, 22).
Considering that RHDV minus-strand synthesis might involve poly(U) synthesis as a first step, we assayed the recombinant enzyme for poly(A)-dependent poly(U) polymerase activity. As expected, the recombinant 3Dpol showed a poly(U) polymerase activity, which was dependent on both oligo(U) and poly(A) and which was not inhibited by rifampin or by actinomycin D. The requirements for primer and template ruled out the possibility that the observed activity was due to a terminal transferase-like reaction. These results agreed with those found for other RNA-dependent RNA polymerases of members of the picornavirus superfamily of positive-sense single-stranded RNA viruses: poliovirus (4, 19), EMC virus (22), and potyvirus (7). Furthermore, we have also found that at high concentrations, the oligo(U) primer saturated the enzyme.
In order to investigate the ability of purified recombinant 3Dpol to transcribe RHDV RNA in an in vitro system, we produced a synthetic RNA template from a DNA plasmid which directed the transcription of RHDV subgenomic RNA. This plasmid contained a full-length cDNA of RHDV subgenomic message, inserted between a promoter site for bacteriophage T7 RNA polymerase and a cDNA copy of the self-cleaving HDV antigenome ribozyme (18). On addition of T7 RNA polymerase such plasmid made RNA transcripts which, after autolytic cleavage, yielded a 2.2-kb RNA transcript with a nucleotide sequence identical to that of the authentic subgenomic mRNA of RHDV, lacking the VPg attached to its 5′ terminus and having a shorter poly(A) tail of only 18 residues. It should be stated at this point that this RNA is coterminal to the 3′ end of the RHDV genome and its 5′ untranslated region is very similar to the one found in the virus genome. Considering these structural features, this synthetic RNA template would also mimic a viral genome of reduced size, and the results obtained with this template might be predictably similar to the ones obtained with the authentic genomic RNA. This system has the additional advantages of avoiding experimental animal infections to purify RHDV virions and producing good yields of a highly purified and homogeneous RNA template.
In the presence of the full-length synthetic RHDV subgenomic RNA, the recombinant 3Dpol synthesized template-size products from an oligo(U) primer. Nevertheless, the enzyme produced RNA molecules up to twice the size of the template, in the absence of oligo(U). Several authors have reported the synthesis of double-length product RNA by poliovirus RNA-dependent RNA polymerase in host factor-dependent reactions (5, 26) and in the absence of primer or eukaryotic host factors (19). The omission of ATP, CTP, or GTP from the reaction mixture completely abolished polymerase activity (data not shown). This result was consistent with the activity of a template-dependent RNA polymerase activity and not with a terminal transferase-like reaction. The enzyme activity was also dependent on the addition of Mg2+ and was inhibited by EDTA (data not shown), as described for poliovirus 3Dpol (14) and EMC virus 3Dpol (22).
The observation that the product RNA, in the absence of oligo(U), was twice the size of the template RNA suggested that a covalent linkage existed between the product RNA and the template. The products made in the absence of oligo(U) might derive from internal hairpins, as described in the poliovirus literature, or by priming at the extreme 3′ end of the template, resulting in a self-complementary double-stranded product. Although no sequence analysis was performed to test priming at the 3′ terminus of the template, this can be deduced from the results obtained in the presence of oligo(U) with both types of template RNA (Fig. 5). It can be seen that most of the 4.4-kb product formed from the 2.2-kb template RNA (Fig. 5, lane 1) is replaced by template-length product (Fig. 5, lanes 2 to 6) in the presence of increasing amounts of oligo(U). That this effect of oligo(U) is sequence specific and occurred at the 3′ end can be deduced from the lack of competitive effect of increasing oligo(U) concentrations on the 3′ truncated template (Fig. 5, lanes 8 to 11). Moreover, priming from more internal hairpins should yield products ranging from template length to twice the template length. If a significant amount of these products were formed, then additional bands of intermediate size should be observed.
The results obtained after treatment of the 4.4-kb product with RNase A showed that this RNA was in fact double stranded. Moreover, the Northern blot experiments indicated that this RNA was made of a positive (template sense) strand and a negative strand covalently linked.
The mechanism involved in the generation of these dimer-length products is not known, nor is it known whether such template-primed products represent intermediates of the normal replication reaction or whether they are aberrant products of the in vitro reaction or the recombinant enzyme used.
In poliovirus, nucleolytic activation of poliovirus RNA templates appeared to be responsible for the host factor-mediated synthesis observed. Other authors (6) showed that a host factor preparation that contained a low-level nuclease (and possibly a 3′ phosphatase) was sufficient to activate RNA templates for transcription by 3Dpol to generate dimer-length products. Since the reaction mixture used in this work did not contain added host factors, a putative nuclease activity could also be related to the recombinant 3Dpol.
An alternative explanation for the synthesis of double-length products is that the 3Dpol may stabilize a small hairpin structure at the 3′ end of the template RNA or hold the 3′ end of a second molecule of RNA in position so that it can function as a primer. In this model, it is assumed that the primer nucleotide is held by the recombinant enzyme itself and not by conventional base pairing. This model would also explain why the largest product RNA was twice the size of the template RNA and contained a snapback sequence.
We have also found that recombinant RHDV 3Dpol can also act on nonviral RNA templates, such as the luciferase mRNA, yielding double-length RNA products in the absence of an oligo(U) primer as described for poliovirus replicase (23).
If RHDV RNA synthesized in vivo by the polymerase is covalently linked to the template RNA or to another RNA that was used as a primer, then a mechanism must exist for specifically processing the linkage between the product RNA and the priming RNA. Tobin et al. (23) showed that the VPg linkage reaction in poliovirus was very specific for template-linked minus-strand RNA synthesized by the polymerase and host factor on poliovirion RNA. In their current model the VPg attaches covalently to poliovirus RNA by a transesterification reaction. The nucleophilic attack by VPg appears to take place at the terminal hairpin promoting a breakage of a phosphodiester bond in the RNA and a formation of a new phosphodiester bond between VPg and the product RNA. A similar replication intermediate formed on nonviral polyadenylated RNA was not active in the VPg linkage reaction. It has been suggested that the VPg linkage reaction would serve two functions: the separation of the covalently linked template and product RNAs and the linkage of VPg to minus-strand RNA. A similar role could also be true for RHDV VPg.
Additional work is required to further characterize the structure of the template-linked products synthesized in vitro and the mechanism by which the double-length RNA is made. It must also be determined whether covalently linked products are synthesized on negative-strand RNA templates. This should help us to understand the molecular mechanism involved in the replication of RHDV RNA and the function of the viral and cellular proteins required for this process.
The availability of purified RHDV RNA polymerase will also allow us to map the functional domains of this unique enzyme and determine its relatedness to the DNA-dependent RNA polymerases and reverse transcriptases. Because the GST expression system has allowed the purification of milligram quantities of RHDV 3Dpol, efforts to crystallize this enzyme and solve its structure can also be attempted.
ACKNOWLEDGMENTS
We thank L. A. Ball from the Department of Microbiology, University of Alabama (Birmingham), for providing a plasmid containing the HDV antigenome ribozyme sequence.
This work was supported by grants BIO96-1172-CO2-O2 and PB96-0552-CO2-O1. A.L.V. and R.C. are recipients of fellowships from Fundación Ramón Areces and FICYT, respectively.
REFERENCES
- 1.Boniotti B, Wirblich C, Sibilia M, Meyers G, Thiel H J, Rossi C. Identification and characterization of a 3C-like protease from rabbit hemorrhagic disease virus, a calicivirus. J Virol. 1994;68:6487–6495. doi: 10.1128/jvi.68.10.6487-6495.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Clarke I N, Lambden P R. The molecular biology of caliciviruses. J Gen Virol. 1997;78:291–301. doi: 10.1099/0022-1317-78-2-291. [DOI] [PubMed] [Google Scholar]
- 3.Cubbit, D., D. W. Bradley, M. J. Carter, S. Chiba, M. K. Estes, L. J. Saif, F. L. Schaffer, A. W. Smith, M. J. Studdert, and H. J. Thiel. 1995. Family Caliciviridae. Arch. Virol. 10(Suppl.):359–363.
- 4.Flanegan J B, Baltimore D. Poliovirus-specific primer-dependent RNA polymerase able to copy poly(A) Proc Natl Acad Sci USA. 1977;74:3677–3680. doi: 10.1073/pnas.74.9.3677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hey T D, Richards O C, Ehrenfeld E. Synthesis of plus- and minus-strand RNA from poliovirion RNA template in vitro. J Virol. 1986;58:790–796. doi: 10.1128/jvi.58.3.790-796.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hey T D, Richards O C, Ehrenfeld E. Host factor-induced template modification during synthesis of poliovirus RNA in vitro. J Virol. 1987;61:802–811. doi: 10.1128/jvi.61.3.802-811.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hong H, Hunt A G. RNA polymerase activity catalyzed by a potyvirus-encoded RNA-dependent RNA polymerase. Virology. 1996;226:146–151. doi: 10.1006/viro.1996.0639. [DOI] [PubMed] [Google Scholar]
- 8.Hull R. Purification, biophysical and biochemical characterization of viruses with especial reference to plant viruses. In: Mahy B W J, editor. Virology, a practical approach. Oxford, United Kingdom: IRL Press; 1985. pp. 1–24. [Google Scholar]
- 9.Laemmli U K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature (London) 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 10.Lubinski J M, Ransone L J, Dasgupta A. Primer-dependent synthesis of covalently linked dimeric RNA molecules by poliovirus replicase. J Virol. 1987;61:2997–3003. doi: 10.1128/jvi.61.10.2997-3003.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martín Alonso J M, Casais R, Boga J A, Parra F. Processing of rabbit hemorrhagic disease virus polyprotein. J Virol. 1996;70:1261–1265. doi: 10.1128/jvi.70.2.1261-1265.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meyers G, Wirblich C, Thiel H J. Rabbit hemorrhagic disease virus: molecular cloning and nucleotide sequencing of a calicivirus genome. Virology. 1991;184:664–676. doi: 10.1016/0042-6822(91)90436-f. [DOI] [PubMed] [Google Scholar]
- 13.Meyers G, Wirblich C, Thiel H J. Genomic and subgenomic RNAs of rabbit hemorrhagic disease virus are both protein linked and packaged into particles. Virology. 1991;184:677–686. doi: 10.1016/0042-6822(91)90437-G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morrow C D, Warren B, Lentz M R. Expression of enzymatically active poliovirus RNA-dependent RNA polymerase in Escherichia coli. Proc Natl Acad Sci USA. 1987;84:6050–6054. doi: 10.1073/pnas.84.17.6050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Neufeld K L, Richards O C, Ehrenfeld E. Purification, characterization, and comparison of poliovirus RNA polymerase from native and recombinant sources. J Biol Chem. 1991;266:24212–24219. [PubMed] [Google Scholar]
- 16.Ohlinger V F, Haas B, Meyers G, Weiland F, Thiel H J. Identification and characterization of the virus causing rabbit hemorrhagic disease. J Virol. 1990;64:3331–3336. doi: 10.1128/jvi.64.7.3331-3336.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Parra F, Prieto M. Purification and characterization of a calicivirus as the causative agent of a lethal hemorrhagic disease in rabbits. J Virol. 1990;64:4013–4015. doi: 10.1128/jvi.64.8.4013-4015.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Perrotta A T, Been M D. A pseudoknot-like structure required for efficient self-cleavage of hepatitis delta virus RNA. Nature (London) 1991;350:434–436. doi: 10.1038/350434a0. [DOI] [PubMed] [Google Scholar]
- 19.Plotch S J, Palant O, Gluzman Y. Purification and properties of poliovirus RNA polymerase expressed in Escherichia coli. J Virol. 1989;63:216–225. doi: 10.1128/jvi.63.1.216-225.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rothstein M A, Richards O C, Amin C, Ehrenfeld E. Enzymatic activity of poliovirus RNA polymerase synthesized in Escherichia coli from viral cDNA. Virology. 1988;164:301–308. doi: 10.1016/0042-6822(88)90542-9. [DOI] [PubMed] [Google Scholar]
- 21.Sambrook J, Firtsch E F, Maniatis T. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory; 1989. [Google Scholar]
- 22.Sankar S, Porter A G. Expression, purification, and properties of recombinant encephalomyocarditis virus RNA-dependent RNA polymerase. J Virol. 1991;65:2993–3000. doi: 10.1128/jvi.65.6.2993-3000.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tobin G J, Young D C, Flanegan J B. Self-catalyzed linkage of poliovirus terminal protein VPg to poliovirus RNA. Cell. 1989;59:511–519. doi: 10.1016/0092-8674(89)90034-2. [DOI] [PubMed] [Google Scholar]
- 24.Wirblich C, Thiel H-J, Meyers G. Genetic map of the calicivirus rabbit hemorrhagic disease virus as deduced from in vitro translation studies. J Virol. 1996;70:7974–7983. doi: 10.1128/jvi.70.11.7974-7983.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wirblich C, Sibilia M, Boniotti B, Rossi C, Thiel H J, Meyers G. 3C-like protease of rabbit hemorrhagic disease virus: identification of cleavage sites in the ORF1 polyprotein and analysis of cleavage specificity. J Virol. 1995;69:7159–7168. doi: 10.1128/jvi.69.11.7159-7168.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Young D C, Tuschall D M, Flanegan J B. Poliovirus RNA-dependent RNA polymerase and host cell protein synthesize product RNA twice the size of poliovirion RNA in vitro. J Virol. 1985;54:256–264. doi: 10.1128/jvi.54.2.256-264.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]