

Abstract
A quantitatively‐driven evaluation of existing clinical data and associated knowledge to accelerate drug discovery and development is a highly valuable approach across therapeutic areas, but remains underutilized. This is especially the case for rare diseases for which development is particularly challenging. The current work outlines an organizational framework to support a quantitatively‐based reverse translation approach to clinical development. This approach was applied to characterize predictors of the trajectory of cognition in Hunter syndrome (Mucopolysaccharidosis Type II; MPS‐II), a rare X‐linked lysosomal storage disorder, highly heterogeneous in its course. Specifically, we considered ways to refine target populations based on age, cognitive status, and biomarkers, that is, cerebrospinal fluid glycosaminoglycans (GAG), at trial entry. Data from a total of 138 subjects (age range 2.5 to 10.1 years) from Takeda‐sponsored internal studies and external natural history studies in MPS‐II were included. Quantitative analyses using mixed‐effects models were performed to characterize the relationships between neurocognitive outcomes and potential indicators of disease progression. Results revealed a specific trajectory in cognitive development across age with an initial progressive phase, followed by a plateau between 4 and 8 years and then a variable declining phase. Additionally, results suggest a faster decline in cognition among subjects with lower cognitive scores or with higher cerebrospinal fluid GAG at enrollment. These results support differences in the neurocognitive course of MPS‐II between distinct groups of patients based on age, cognitive function, and biomarker status at enrollment. These differences should be considered when designing future clinical trials.
INTRODUCTION
A quantitatively‐driven approach to the evaluation of all existing data as part of a deliberate reverse translation approach can empower patient‐centric clinical trial design, and ultimately accelerate access to treatments. 1 Taking such an approach, and implementing it early within the drug development process, is particularly critical in the rare disease area. Rare disease trials have inherent challenges in development including, but not limited to, small sample sizes, substantial patient trial burden, a lack of well‐established, predictive correlations between biomarkers and clinical endpoints, and challenges in patient recruitment for clinical development compared to more prevalent diseases.
To overcome these limitations, it is critical to incorporate real‐world evidence, including available external data and lessons learned from previous clinical trials, in early clinical development plans. Such an evidence‐based advancement in clinical development is possible in the context of an organizational scientifically‐driven framework centering quantitative analyses in clinical trial decision making as depicted in Figure 1. This framework can be implemented at any stage of drug development but emphasizes the value of formally incorporating quantitative clinical data in a structured manner. It relies on three ordered steps: (1) identify program‐ and asset‐specific questions and unmet needs, (2) collate relevant internal and external data to address these questions, and (3) execute fit‐for‐purpose quantitative analyses needed for data‐driven decision making and development planning. Herein, we report on the application of this approach in an effort to characterize cognitive aspects of Hunter syndrome (Mucopolysaccharidosis Type II) and advance clinical development of potential treatment candidates.
FIGURE 1.
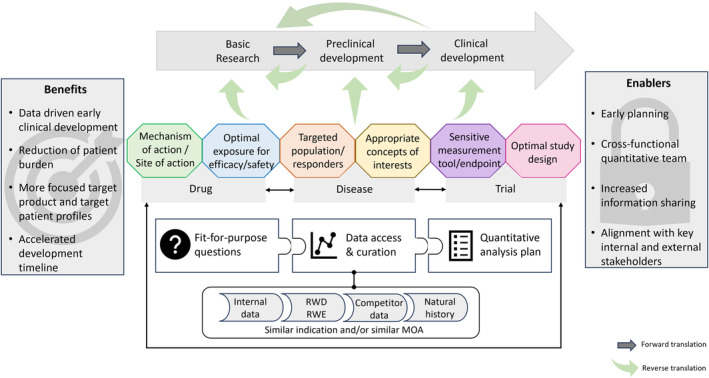
Quantitatively‐driven reverse translation framework. MOA, mechanism of action; RWD, real‐world data; RWE, real‐world evidence.
Hunter Syndrome (Mucopolysaccharidosis Type II; MPS‐II), a rare X‐linked recessive lysosomal storage disorder primarily occurring in males, is due to the deficiency of the enzyme iduronate‐2‐sulfatase that catabolizes several glycosaminoglycans (GAG). 2 The progressive accumulation of GAG, particularly heparan sulfate and dermatan sulfate, within tissues and organs is responsible for the clinical manifestations expressed along a continuum of severity involving both peripheral tissues and central nervous system symptomology. Among patients with neuronopathic MPS‐II, there is considerable heterogeneity in the disease course, and, at present, there is no available CNS‐directed treatment, leaving an unmet medical need. 3 Takeda (formerly Shire) initiated several clinical programs to address the neurological features of MPS‐II that failed to progress due, in part, to the inability to accommodate the heterogeneity of the patient population. However, the data generated from these trials are well suited to support the depicted quantitatively‐driven reverse translation framework to benefit future clinical development.
To address the scientific unmet need in predicting the trajectory of cognitive decline in MPS‐II, our objectives were to better understand the drivers of neurocognitive disease progression, especially across distinct patient groups based on age, cognitive status, and disease burden biomarkers (i.e., cerebrospinal fluid GAG [GAGcsf] levels) at trial entry. External and internal historical trial data were quantitatively leveraged to establish a more accurate reference trajectory against which to evaluate therapeutic outcomes and to refine target populations that would potentially benefit the most from a CNS‐targeted treatment.
METHOD
Data analysis sample
All available relevant data were identified and included: (1) internal data from historical Takeda‐sponsored MPS‐II trials and (2) published natural history studies data. For each study, age, cognitive status at the time of enrollment and change over time, and, when available, GAGcsf levels were retrieved. Internal data included non‐interventional and placebo data from a total of 73 participants from three Takeda‐sponsored studies (NCT00937794, NCT01822184, NCT02055118). 4 , 5 , 6 External data included published natural history data from a total of 65 participants, previously reviewed by Shapiro and Eisengart 3 and digitized from data reported in the original publications. 7 , 8 , 9 Participants from internal clinical trials were all boys, with a median enrollment age of 4.4 years (range 2.5–8.8 years), and a median enrollment Differential Ability Scales‐II (DAS‐II) general conceptual ability (GCA) score of 73 (range 30–122). GAGcsf measurements were available for one trial (NCT02055118) with a median enrollment value of 933.5 ng/mL (range 693–3630). Participants from external studies had a median entry age of 3.7 years (range 0.2–10.1 years) and a median entry development quotient (DQ) score of 53 (range 4.8–109); no biomarker data were available.
Initial efforts required the standardization of reporting cognitive change across disparate data sets. Consistent with Shapiro and Eisengart, 3 standardized age equivalent (AE) scores were used to characterize age‐related cognitive development. For subject‐specific drivers of cognitive disease trajectory, DAS‐II GCA scores were used for internal trial data, and, given variability in assessments used across external studies, DQ scores were considered for external natural history data.
Statistical data analysis
Several analyses were performed to characterize associations between the neurocognitive outcomes and potential predictors of disease progression.
Each outcome (i.e., AE score, GCA score, and DQ score) was modeled based on chronological age using generalized additive mixed‐effects models (GAMMs) assuming a normal distribution of the outcome and subject‐specific random intercept. The use of GAMMs allowed for a non‐linear shape of the mean estimate of the outcome at the group level across chronological age and accounted for multiple observations per subject. To model AE score within natural history data, we fitted a GAMM for each study data separately as well as one GAMM for three studies combined. To model AE score in Takeda‐sponsored Hunter studies, we employed a GAMM with a truncated normal distribution to address left‐ and right‐censoring that occurred within a subset of outcome measurements. After modeling the data at the population level, we then estimated mean curves separately by groups based on characteristics at the time of enrollment: cognitive score (GCA or DQ score ≤70, >70), and biomarker burden (GAGcsf ≤ 1181 or >1181 ng/mL 10 ).
Additionally, we estimated the change in cognition over time using linear mixed‐effects models (LMMs). LMMs assumed subject‐specific random intercept and random slope for change over time. The subject‐specific slopes obtained from LMMs were summarized (boxplot) by groups by characteristics at enrollment to visualize the potential impact of cognitive scores (GCA or DQ score ≤70, >70) and/or biomarker burden (GAGcsf ≤ 1181 or >1181 ng/mL 10 ). Two‐sample t‐tests were performed to compare the means of individuals' slope between groups. All analyses were performed using R version 4.1.2.
RESULTS
Figure 2 depicts cognitive outcome trajectories across age and the impact of covariates at the time of trial entry for internal data (left column) and natural history data (right column). The AE trajectory across age observed with our internal data (Panel a) was consistent with that previously reviewed 3 and replicated within the natural history data, both across studies as well as for each study individually (Panel b). Specifically, cognitive development demonstrates an initial progressive phase at an early age, followed by a plateau in development between 4 and 8 years of age, and then a more variable declining phase. The GCA and DQ score trajectories across age were overall consistent across internal and external data, showing a progressive decline with increasing age (Panels c1, d1, and e1).
FIGURE 2.
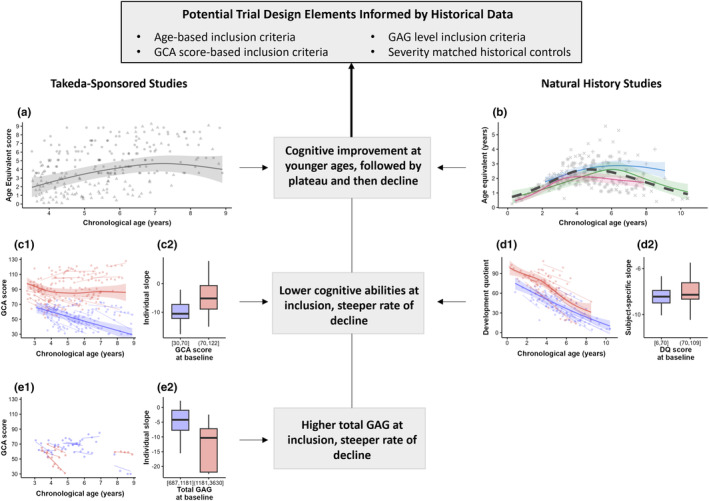
Cognitive abilities across chronological age for Takeda‐sponsored studies (left column) and natural history studies (right column). Panels (a) and (b): Age equivalent scores versus chronological age. Panels (c) and (d): General conceptual ability (GCA) and development quotient (DQ) scores versus chronological age based upon cognitive score at enrollment (blue color for low scores ≤70 and red color for high scores >70). Panel (e): GCA scores versus chronological age based on biomarker burden (GAG) at trial entry (blue color for low burden ≤1181 ng/mL and red color for high burden >1181 ng/mL). In Panels a, b, c1, and d1, solid lines represent mean predicted trends and shaded bands represent 95% confidence intervals for generalized additive mixed‐effects models (GAMM)‐estimated linear predictor. In Panel (a), ball‐shaped points represent observed measurement and triangle‐shaped points represent censored observations simulated from the model to support visualization. In Panel (b): Mean trends are color‐coded for the three natural history studies reviewed in Shapiro & Eisengart (2021) 3 : blue—Young et al. (1983) 8 , red—Seo et al. (2020) 7 , green—Holt et al. (2011) 9 ; distinct point shapes correspond to distinct studies. The black dashed line represents outcome's mean trend for the three studies collectively. In Panels c1, d1, and e1, thin colored lines represent linear mixed‐effects models (LMM)‐predicted subject‐specific slopes. In Panels c2, d2, and e2, LMM‐predicted subject‐specific slopes are summarized by characteristics at the time of enrollment (boxplots).
GAMM (Panels c1, d1, and e1) and LMM (Panels c2, d2, and e2) were leveraged to characterize the impact of cognitive scores and biomarker burden at the time of enrollment. Overall, our results indicated a faster cognitive decline in subjects with lower cognitive scores at trial entry (≤70). In internal studies, substantial differences were observed in GCA score mean trends across groups based on GCA score at entry (Panel c1). Moreover, subjects with high GCA scores at enrollment demonstrated a slower decline (subject‐specific slope median −5.2) compared to those with low GCA trial entry scores (slope median −10.5, p = 0.000007, Panel c2). In contrast, within natural history studies the DQ mean scores were consistently higher for subjects with lower trial entry DQ scores (Panel d1); the individuals' decline was comparable across the two groups (slope median −8.5 and −8.4, respectively, p = 0.96, Panel d2). Finally, results suggest a faster GCA/DQ score decline in subjects with a higher GAGcsf burden at trial entry (Panel e) with a median change slope of −4.2 in subjects with GAG >1181 ng/mL compared to −10.3 in subjects with GAG ≤1181 ng/mL (p = 0.0474).
DISCUSSION
A quantitatively driven approach to the evaluation of both historical external and internal data was applied to address the scientific unmet need in the ability to predict the cognitive disease trajectory in MPS‐II patients. Our analyses support differences in the CNS‐related disease course between distinct groups of MPS‐II patients based on age, cognitive function, and biomarker (GAGcsf) status at enrollment. Specifically, these results suggest that (1) cognitive improvement is more likely when patients are younger at the time of enrollment, (2) lower cognitive abilities at enrollment predict a steeper rate of cognitive decline, and (3) higher GAG burdens predict a steeper rate of decline. These results can be used to guide the development of future clinical studies evaluating CNS‐directed treatments for MPS‐II, as they are directly relevant to patient selection considerations and the selection of endpoints or thresholds required to demonstrate clinically meaningful effects.
This work was performed in the context of an organizational, scientifically‐driven quantitative reverse translational framework (Figure 1). Although reverse translation is often conceptualized within the context of bedside‐to‐benchtop research, it also encompasses the quantitative leveraging of all existing clinical data to inform early clinical drug development with regard to the specific drug, disease, and trial conduct. 11 Indeed, reverse translation is valuable for investigating a range of hurdles across the drug development timeline, including mechanisms of drug action for repurposing, efficacy/safety balance to define the optimal drug exposure to reach at the right site of action, variability observed in drug responsiveness to determine the best population or sub‐population to treat, relevant endpoints to optimize the sensitivity of measurement tools available, and study designs that improve the likelihood of successfully bringing medicines to the right patients faster. Quantitative integration of available relevant data is the sine qua non of reverse translation. Whereas there has been an increased appreciation, in both academic and industrial settings, of the value of quantitatively‐driven reverse translation, it is still underutilized and requires the deliberate allocation of resources.
This is particularly important in the context of rare diseases, for which the totality of evidence must be considered to overcome inherent challenges including the relatively limited data generated across drug development efforts and the heterogeneity of data across studies with regard to measurement tools and endpoints, described in the example of MPS‐II. Scientists will be forced to determine the extent to which data may be pooled (e.g., via standardized summary indices, like AE scores) versus demonstrating cross‐study consistency across disparate assessments of common concepts of interest (e.g., DAS‐II GCA), with the same goal of minimizing the limitations related to sample size or data heterogeneity. Regardless of data manipulation needs and associated decisions, reverse translation efforts are instructive for clinical development and benefit from a formal organizational framework emphasizing cross‐disciplinary collaboration. These efforts must be formalized early enough within the drug discovery and development lifecycle to maximize its value in support of data‐driven decision making. Within the current MPS‐II‐focused example, our results support differences in the CNS‐related disease course between distinct groups of patients based on age, cognitive function, and biomarker (GAG) status at enrollment. These differences should be considered when designing future clinical trials and defining estimands.
The success of this approach also relies on a shared patient‐centric purpose to accelerate access to effective treatments. The scarcity of patients in rare disease populations, and the resulting outsized burden placed on them in clinical development, requires the maximal use of all data generated to ensure clinical development is as data‐driven as that of more prevalent diseases. Rare disease data registries, data curation, and access have been primary efforts of many rare disease advocacy organizations to support therapeutics development. Thus, there is a patient‐driven onus on sponsors to allocate resources and promote collaboration among quantitative scientists across functional lines to appropriately access, analyze, and interpret existing data.
All told, structured quantitatively‐driven reverse translation frameworks are critical for driving innovation to get efficacious treatments to patients with the greatest unmet needs sooner. The success of such efforts will rely on the engagement and strong collaboration among quantitative translational scientists and other key stakeholders of drug development, all of whom share a common set of patient‐centric values.
AUTHOR CONTRIBUTIONS
R.D.L., and O.C. wrote the manuscript. R.D.L., O.C., and M.E.M. designed the research. R.D.L., O.C., M.E.M., M.K., C.J.M., and D.A.H.W. performed the research. R.D.L., O.C., and M.K. analyzed the data.
FUNDING INFORMATION
Funding for this work was provided by Takeda Development Center Americas, Inc.
CONFLICT OF INTEREST STATEMENT
R.D. Latzman, O. Campagne, M. Karas, C.J. Malanga, & D.A.H. Whiteman are employees of Takeda Pharmaceutical Company Limited and stockholders of Takeda Pharmaceutical Company Limited. M.E. Modi was employed by Takeda Development Center Americas, Inc. when this work was completed and is a stockholder of Takeda Pharmaceutical Company Limited.
Latzman RD, Campagne O, Modi ME, Karas M, Malanga CJ, Whiteman DAH. Advancing clinical development for neuronopathic Hunter syndrome through a quantitatively‐driven reverse translation framework. Clin Transl Sci. 2024;17:e13776. doi: 10.1111/cts.13776
REFERENCES
- 1. Heatherington AC, Kasichayanula S, Venkatakrishnan K. How well are we applying quantitative methods to reverse translation to inform early clinical development? Clin Pharmacol Ther. 2018;103(2):174‐176. doi: 10.1002/cpt.948 [DOI] [PubMed] [Google Scholar]
- 2. Whiteman DAH, Kimura A. Development of idursulfase therapy for mucopolysaccharidosis type II (hunters syndrome): the past, the present and the future. Drug Des Devel Ther. 2017;11:2467‐2480. doi: 10.2147/DDDT.S139601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Shapiro EG, Eisengart JB. The natural history of neurocognition in MPS disorders: a review. Mol Genet Metab. 2021;133:8‐34. doi: 10.1016/j.ymgme.2021.03.002 [DOI] [PubMed] [Google Scholar]
- 4. Muenzer J, Hendriksz CJ, Fan Z, et al. A phase I/II study of intrathecal idursulfase‐IT in children with severe mucopolysaccharidosis II. Genet Med. 2016;18:73‐81. doi: 10.1038/gim.2015.36 [DOI] [PubMed] [Google Scholar]
- 5. Muenzer J, Burton BK, Harmatz P, et al. Neurodevelopmental status and adaptive behavior of pediatric patients with hunter syndrome: a longitudinal observational study. Mol Genet Metab. 2019;126(2):S103. doi: 10.1016/j.ymgme.2018.12.260 [DOI] [Google Scholar]
- 6. Muenzer J, Burton BK, Harmatz P, et al. Intrathecal idursulfase‐IT in patients with neuronopathic mucopolysaccharidosis II: results from a phase 2/3 randomized study. Mol Genet Metab. 2022;137(1–2):127‐139. doi: 10.1016/j.ymgme.2022.07.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Seo J‐H, Okuyama T, Shapiro E, Fukuhara Y, Kosuga M. Natural history of cognitive development in neuronopathic mucopolysaccharidosis type II (hunter syndrome): contribution of genotype to cognitive developmental course. Mol Genet Metab Rep. 2020;24:100630. doi: 10.1016/j.ymgmr.2020.100630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Young ID, Harper PS. The natural history of the severe form of Hunter's syndrome: a study based on 52 cases. Dev Med Child Neurol. 1983;25(4):481‐489. doi: 10.1111/j.1469-8749.1983.tb13794.x [DOI] [PubMed] [Google Scholar]
- 9. Holt JB, Poe MD, Escolar ML. Natural progression of neurological disease in mucopolysaccharidosis type II. Pediatrics. 2011;127(5):e1258‐e1265. doi: 10.1542/peds.2010-1274 [DOI] [PubMed] [Google Scholar]
- 10. Hendriksz CJ, Muenzer J, Vanderver A, et al. Levels of glycosaminoglycans in the cerebrospinal fluid of healthy young adults, surrogate‐normal children, and hunter syndrome patients with and without cognitive impairment. Mol Genet Metab Rep. 2015;5:103‐106. doi: 10.1016/j.ymgmr.2015.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shakhnovich V. It's time to reverse our thinking: the reverse translation research paradigm. Clin Transl Sci. 2018;11(2):98‐99. doi: 10.1111/cts.12538 [DOI] [PMC free article] [PubMed] [Google Scholar]