

Abstract

An efficient methodology for the synthesis of halogenated benzodioxepinones and benzoxazecinones has been developed via tandem oxidation and iodolactonization reaction of 2-O/N-tethered alkenyl benzaldehydes mediated by CuI and tertiarybutylhydro-peroxide in acetonitrile at 70 °C in moderate to good yields. The reaction involves initial oxidation of aldehyde to acid followed by iodolactonization. Terminal propargyl ether resulted in a mixture of mono- and diiodido-3-methylene-1,4-dioxepin-5-ones. The post-synthetic modification of the reaction products leads to the formation of corresponding thiocyanate, azide, thioether, and triazole derivatives.
Introduction
Lactones are important structural motifs in organic chemistry and found in many biologically active molecules and natural products.1 For example, octalactin A,2 an eight-membered lactone, has cytotoxic activity against melanoma and colon tumor cells; (+)-penicillide and (+)-purpactin A are natural products isolated from Penicillium simplicissimum, which have inhibitory activity against acyl-CoA-cholestrol acyltransferase (Figure 1).3 Seven-membered lactones such as 2,3-dihydro-5H-1,4-benzodioxepin-5-ones (2,3-DHB) are the starting material for the synthesis of polyester (Figure 1).4 Therefore, several methods have been developed for the synthesis of lactones via classical ring closure of alcohols and acids.5 The classical methods require either activation of acid or alcohol functionality to achieve good yield, chemoselectivity, and to avoid side products. In contrast, transition metal-catalyzed intramolecular ketone hydroacylation reactions,6 allylic oxidation/lactonization,7 and N-heterocyclic carbene catalyzed oxidative lactonization8 have also been used.
Figure 1.

Biologically active lactones.
Synthesis of medium-ring lactones is challenging as the cyclization suffers from low reactivity due to transannular strain and high degree of conformational flexibility.9 Recently, alkene halolactonization of unsaturated acids has received special attention in preparing medium-ring lactones (Scheme 1).10 Gataullin reported the synthesis of benzoxazocinones via halolactonization of N-acyl-N-(2-cyclohex-1-en-1-yl-6-methylphenyl) (Scheme 1a).10a Xiang has reported the catalytic asymmetric halocyclization procedure for the synthesis of benzoxazepinones and benzoxazecinones using (DHQ)2PHAL as the catalyst (Scheme 1b).10b Although halolactonization of intramolecular bifunctional alkenes and acids is reported in the literature (Scheme 1c),11 halolactonization of intramolecular bifunctional alkenes and aldehydes is not reported so far. In this article, we have disclosed a new methodology for the synthesis of benzodioxepinones and benzoxazepinones via tandem oxidation and iodolactonization of 2-O/N-tethered alkenyl benzaldehydes mediated by the CuI/TBHP system for the first time (Scheme 1d).
Scheme 1. Halolactonization of Alkenes.

Results and Discussion
In our study, compound 1a was considered as standard starting material and treated with 5.0 equiv of tertiarybutylhydro-peroxide (TBHP) and 1.0 equiv of CuI in acetonitrile at room temperature for 6 h. To our dismay, no product was observed from the reaction (Table 1, entry 1). However, when the reaction was performed at 80 °C compound 2a was obtained in 67% yield (Table 1, entry 6). While decreasing the temperature from 80 to 70 and 60 °C, it provided 69 and 61% yields, respectively (Table 1, entries 7–8). Subsequent increase in TBHP to 6.0 equiv and CuI to 1.2 equiv at 70 °C resulted in 75% yield (Table 1, entry 9). Mixed solvents like acetonitrile and water gave 73% yield (Table 1, entry 10). Increasing the amount of CuI to 1.5 equiv resulted in same yield of the corresponding product (Table 1, entry 11). Other variants like oxidants H2O2, K2S2O8,and molecular oxygen (Table 1, entries 13–15); halogen sources like I2, NIS, NaI, CuCl2, CuBr could not provide better yields (Table 1, entries 16–21). Solvents such as DCM, DCE, 1,4-dioxane, and toluene were proved to be inefficient for this transformation (Table 1, entries 2–5). Therefore, 6.0 equiv of TBHP and 1.2 equiv of CuI in CH3CN at 70 °C for 6 h are the optimal conditions for the reaction.
Table 1. Optimization of the Reactiona.

| entry | oxidant (equiv) | halogen precursor (equiv) | solvent | temp. (°C) | yieldb (%) |
|---|---|---|---|---|---|
| 1 | TBHP (5.0) | Cul (1.0) | CH3CN | rt | c |
| 2 | TBHP (5.0) | Cul (1.0) | DCM | 40 | 55 |
| 3 | TBHP (5.0) | Cul (1.0) | DCE | 80 | c |
| 4 | TBHP (5.0) | Cul (1.0) | 1,4-dioxane | 100 | 35 |
| 5 | TBHP (5.0) | Cul (1.0) | toluene | 110 | c |
| 6 | TBHP (5.0) | Cul (1.0) | CH3CN | 80 | 67 |
| 7 | TBHP (5.0) | Cul (1.0) | CH3CN | 70 | 69 |
| 8 | TBHP (6.0) | Cul (1.2) | CH3CN | 60 | 61 |
| 9 | TBHP (6.0) | Cul (1.2) | CH3CN | 70 | 75 |
| 10 | TBHP (6.0) | Cul (1.5) | CH3CN:H2O (3:1) | 70 | 73 |
| 11 | TBHP (8.0) | Cul (1.5) | CH3CN | 70 | 75 |
| 12 | H202 (3.0) | Cul (1.2) | CH3CN | 70 | 70 |
| 13 | K2S208 (3.0) | Cul (1.2) | CH3CN | 70 | c |
| 14 | O2 | Cul (1.2) | CH3CN | 70 | c |
| 15 | TBHP (6.0) | NIS (2.0) | CH3CN | 70 | c |
| 16 | TBHP (6.0) | Nal (2.0) | CH3CN | 70 | 35 |
| 17 | TBHP (6.0) | KI (2.0) | CH3CN | 70 | 45 |
| 18 | TBHP (6.0) | 12 (2.0) | CH3CN | 70 | 50 |
| 19 | TBHP (6.0) | CuCl2 (1.2) | CH3CN | 70 | c |
| 20 | TBHP (6.0) | CuBr (1.2) | CH3CN | 70 | c |
| 21 | 70 | c |
Reaction conditions: 1a (0.57 mmol, 1.0 equiv), oxidant, halogen precursor, solvent 4.0 mL, N2 atmosphere, 6 h.
lsolated yield.
No reaction.
With this optimal condition in hand, the scope of the reaction was investigated with a variety of substrates as depicted in Scheme 2. It was observed from Scheme 2 that both substituted and unsubstituted allyl/homoallyl ethers work well to provide the seven-membered iodolactones 2a–2p and eight-membered lactones 2q–2s in moderate to good yields. Similarly, electron-withdraing and electron-donating groups in the aromatic ring are also compatible under the reaction conditions. However, the yield of the eight-membered iodolactones 2q–2s is lower than the seven-membered rings. The methodology is applicable to the synthesis of benzoxazepinones 2t–2y and benzooxazocinone 2z. The reaction is also compatible with the substrate without a heteroatom (X = −CH2−) giving 3-(iodomethyl)-4,5-dihydrobenzo[c]oxepin-1(3H)-one 2a′a in 68% yield. However, the reaction with internal alkene 1b′ resulted in the formation of benzodioxinone 2b′b in 10% yield (Scheme 3).
Scheme 2. Synthesis of 3-Iodomethyl-1,4-dioxepin-5-one and 3-Iodomethyl-1,4-oxazepin-5-one Derivatives.

Reaction conditions: 1(0.57 mmol, 1.0 equiv), CH3CN 4.0 mL, N2 atmosphere.
Scheme 3. Reaction with Internal Alkene.

The scope of the reaction was extended to propargyl ethers. The reaction with terminal propargyl ether resulted in a mixture of mono- and di-iodosubstituted products (Scheme 4), whereas silyl substituted propargyl ether gave single diiodide product 4a in 38% yield. The structure of all the compounds was determined by 1H, 13C{1H} NMR, IR spectroscopy, high-resolution mass spectrometry (HRMS), and finally by X-ray crystallographic analysis of the compounds 2a, 4a, and 4a′.
Scheme 4. Synthesis of 3-(Diiodomethylene)-1,4-dioxepin-5-one and 3-(Iodomethylene)-1,4-dioxepin-5-one Derivatives.

Reaction conditions: 1(0.63 mmol, 1.0 equiv), CH3CN 4.0 mL, N2 atmosphere.
A comparison of the present reaction conditions for the conversion of carboxylic acid to lactone with an earlier reaction was carried out considering acid C, as shown in Table 2. It was observed that although time required for conversion is decreased from 5 to 2.5 h in the present system, the yield decreased from 63 to 43%. In the case of a source of halogen in both cases 1.2 equiv was used. The major advantage of the earlier reaction was the usage of a catalytic amount of the reagent, which yielded a chiral product with 32% ee. On the other hand, present reaction conditions provided more reactive iodo derivatives for further transformations.
Table 2. Comparison of Present Reaction Conditions with the Reported Reaction.

| entry | catalyst/oxidant (equiv) | halogen source (equiv) | time (h) | yield (%) |
|---|---|---|---|---|
| 1 | (DHQ)2PHAL (0.05) | NBS (1.2) | 5 | 63 |
| 2 | TBHP (6.0) | CuI (1.2) | 2.5 | 43 |
In order to establish the reaction pathway some control experiments were undertaken (Scheme 5). The compound 1a was subjected to react with TEMPO (3.0 equiv) and BHT (3.0 equiv) under standard reaction conditions (Scheme 5a). The product 2a was obtained in 21% with TEMPO and trace amount in the case of BHT. The intermediate 2,2,6,6-tetramethylpiperidin-1-yl 2-((2- methylallyl)oxy)benzoate 5 was detected by the HRMS experiment. This indicates that the reaction proceeds via a radical mechanism and aldehyde is oxidized to acid in situ. On the other hand, the reaction with corresponding benzoic acid 5a with CuI in the absence of TBHP resulted in 2a with 45% yield, indicating the formation of benzoic acid as reaction intermediate (Scheme 5b). Interestingly, the yield of the reaction decreased by 30% (Scheme 5b). However, the reaction of 1a with TBHP in the absence of CuI resulted in the formation of carboxylic acid in trace amount (Scheme 5c). Therefore, CuI is important in converting aldehyde to corresponding carboxylic acid.
Scheme 5. Controlled Experiments.

From the above experiments and literature evidence,12 a plausible mechanism is proposed (Scheme 6). Reaction of CuI and TBHP generates tert-butoxide and tert-butylhydroperoxide radicals of which tert-butoxide radical abstracts hydrogen from aldehyde to form radical intermediate A. The intermediate A then reacts with tert-butylhydroperoxide radical to form peroxy ester B, which after hydrolysis gives acid C. Again, the reaction between CuI and tBuOOH produced reactive species tBuOI, which reacts with olefin to form intermediate E via D. The nucleophilic attack of carboxylate ion on iodonium ion forms the final compound 2. It is evident from the mechanism that TBHP is involved in oxidizing Cu(I) to Cu(II) and back to Cu(I); transforming CuI to tBuOI for the iodolactonization reaction. Therefore, an excess amount of TBHP is required for this reaction.
Scheme 6. Plausible Mechanism.

The applicability of the synthesized iodolactone was demonstrated by converting the iodofunctionality to its thiocyanate 6 and azide 7 by reacting with ammonium thiocyanate and sodium azide, respectively (Scheme 7). The azide can conveniently be converted into its triazole derivative 8 with the reaction of phenylacetylene in 89% yield. The iodofunctionality can also be converted to its thioether 9 by reacting with thiophenol in 51% yield (Scheme 7).
Scheme 7. Post-Synthetic Applications.

In order to check the scalability of the reaction a gram-scale synthesis of the product 2a was carried out from starting compound 1a (1.00g) under standard reaction conditions. This resulted product 2a in 61% yield (1.10 g) (Scheme 8).
Scheme 8. Gram-Scale Synthesis.

In conclusion, we have demonstrated an efficient methodology for the synthesis of benzodioxepinones and benzoxazepinones via tandem oxidation and iodolactonization of 2-O/N-tethered alkenyl benzaldehyde mediated by CuI/TBHP in moderate to good yields. The reaction is compatible with a variety of functional groups. The synthetic utility of the methodology is extended to the synthesis of its azide, thiocyanide, and thioether. The azide can also be converted to its 1,2,3-triazole derivative.
Experimental Section
General Information
All the reagents were of reagent grade (AR grade) and were used as purchased without further purification. Silica gel (60–120 mesh size) was used for column chromatography. Reactions were monitored by thin-layer chromatography (TLC) on silica gel GF254 (0.25 mm). Melting points were recorded in an open capillary tube and are uncorrected. Fourier transform-infrared spectra were recorded as neat liquid or KBr pellets. NMR spectra were recorded in CDCl3 with tetramethylsilane as the internal standard for 1H (500 and 400 MHz) or 13C{1H} (100 and 125 MHz) NMR. Chemical shifts (δ) are reported in ppm and spin–spin coupling constants (J) are given in Hertz. HRMS spectra were recorded using Q-TOF mass spectrometer.
The starting material 2-((2-methylallyl)oxy)benzaldehyde
derivatives (1a–1b, 1d–1f, 1i–1k, 1n–1q), N-(2-formylphenyl)-4-methyl-N-(2-methylallyl)benzenesulfonamide derivative (1y),
1a′, 1b′, and 2-(prop-2-yn-1-yloxy)benzaldehyde
derivatives (3a, 3b, and 3c) were prepared
by following the previous work literature.13 The substrates 1a′(14)1b′(15) were prepared
from the literature procedure. The spectroscopic data of the above
compounds are in good agreement with the literature one. The experimental
procedure and the characterization data of the remaining starting
material 2-((2-methylallyl)oxy)benzaldehyde derivatives (1c, 1g, 1h, 1l, 1m, 1r, and 1s), N-(2-formylphenyl)-4-methyl-N-(2-methylallyl)benzenesulfonamide derivatives (1t–1x and 1z) and 2-((3-(trimethylsilyl)prop-2-yn-1-yl)oxy)benzaldehyde
are given as follows:
General Experimental Procedure for the Synthesis of 1c, 1g, 1h, 1l, and 1m
To a suspension of K2CO3 (7.38 mmol, 3 equiv) in DMF under the N2 atmosphere was added substituted 2-hydroxybenzaldehyde (2.46 mmol, 1.0 equiv) and 3-chloro-2-methylprop-1-ene (3.69 mmol, 1.5 equiv). The reaction mixture was then allowed to stir at room temperature. The progress of the reaction was monitored by TLC analysis. After completion of the reaction (12–20 h), the combined organic layer was washed with brine and ice water and further extracted with ethyl acetate (3 × 15 mL) followed by drying over anhydrous Na2SO4. The organic phase was concentrated in a rotary evaporator to give the crude product, which was then subjected to column chromatography over silica gel to provide the desired product 1.
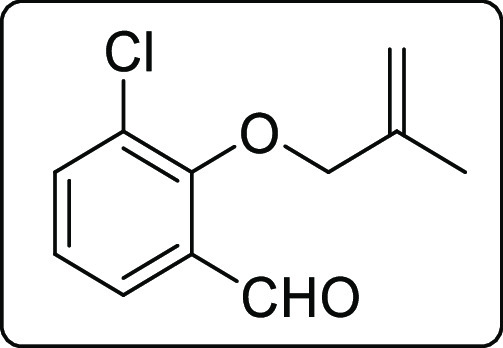
3-Chloro-2-((2-methylallyl)oxy)benzaldehyde (1c)
 Yellow liquid; Rf (hexane/EtOAc,
9:1) 0.55; yield 403 mg, 78%; 1H NMR (400 MHz, CDCl3) δ 10.29 (s, 1 H), 7.69 (dd, J = 8.0,
2.0 Hz, 1 H), 7.57 (dd, J = 8.0, 1.6 Hz, 1 H), 7.12
(t, J = 8.0 Hz, 1 H), 5.09 (s, 1 H), 4.99 (s, 1 H),
4.45 (s, 2 H), 1.87 (s, 3 H); 13C{1H} NMR (100
MHz, CDCl3) δ 189.1, 158.1, 140.1, 136.4, 131.3,
129.0, 126.9, 125.2, 114.5, 79.6, 19.8. IR (KBr, neat) 3081, 2862,
1690, 1586, 1443, 1379, 1243, 1136, 956, 908, 785, 728, 620 cm–1; HRMS (ESI) calcd. for C11H12ClO2 (M + H)+ 211.0520, found 211.0520.
Yellow liquid; Rf (hexane/EtOAc,
9:1) 0.55; yield 403 mg, 78%; 1H NMR (400 MHz, CDCl3) δ 10.29 (s, 1 H), 7.69 (dd, J = 8.0,
2.0 Hz, 1 H), 7.57 (dd, J = 8.0, 1.6 Hz, 1 H), 7.12
(t, J = 8.0 Hz, 1 H), 5.09 (s, 1 H), 4.99 (s, 1 H),
4.45 (s, 2 H), 1.87 (s, 3 H); 13C{1H} NMR (100
MHz, CDCl3) δ 189.1, 158.1, 140.1, 136.4, 131.3,
129.0, 126.9, 125.2, 114.5, 79.6, 19.8. IR (KBr, neat) 3081, 2862,
1690, 1586, 1443, 1379, 1243, 1136, 956, 908, 785, 728, 620 cm–1; HRMS (ESI) calcd. for C11H12ClO2 (M + H)+ 211.0520, found 211.0520.
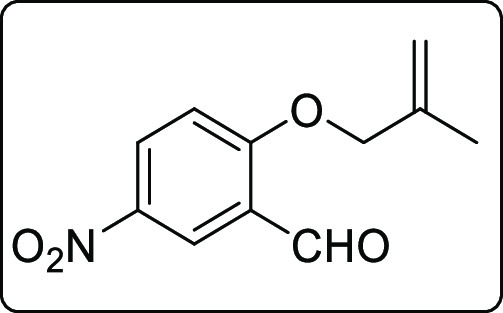
2-((2-Methylallyl)oxy)-5-nitrobenzaldehyde (1g)
Brown solid; Rf (hexane/EtOAc, 4:1) 0.55; mp 73–75 °C; yield 163 mg, 30%; 1H NMR (400 MHz, CDCl3) δ 10.48 (s, 1 H), 8.65 (d, J = 3.2 Hz, 1 H), 8.37 (dd, J = 9.2, 3.2 Hz, 1 H), 7.11 (d, J = 9.2 Hz, 1 H), 5.13 (s, 1 H), 5.09 (s, 1 H), 4.69 (s, 2 H), 1.86 (s, 3 H); 13C{1H} NMR (100 MHz, CDCl3) δ 187.6, 165.0, 141.8, 138.9, 130.7, 124.9, 124.7, 114.6, 113.6, 73.3, 19.4. IR (KBr, neat) 2958, 2919, 1691, 1608, 1522, 1488, 1344, 1272, 1078, 997, 823, 749, 664 cm–1; anal. calcd. for C11H11NO4: C, 59.73; H, 5.01; N, 6.33. Found: C, 60.39; H, 5.19; N, 6.25.
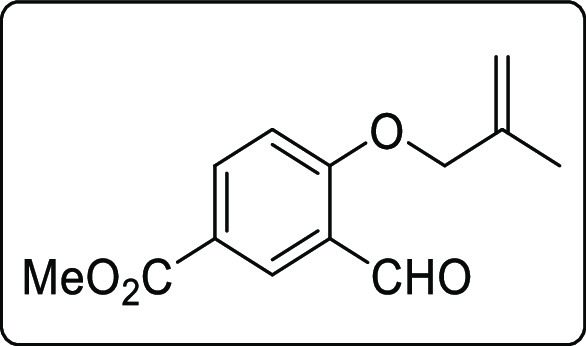
Methyl-3-formyl-4-((2-methylallyl)oxy)benzoate (1h)
 White solid; Rf (hexane/EtOAc,
9:1) 0.50; mp 49–51 °C; yield 351 mg, 61%; 1H NMR (400 MHz, CDCl3) δ 10.43 (s, 1 H), 8.40 (d, J = 2.4 Hz, 1 H), 8.12 (dd, J = 8.8, 2.4
Hz, 1 H), 6.96 (d, J = 8.8 Hz, 1 H), 5.07 (s, 1 H),
5.00 (s, 1 H), 4.55 (s, 2 H), 3.83 (s, 3 H), 1.80 (s, 3 H); 13C{1H} NMR (100 MHz, CDCl3) δ 188.7, 166.0,
164.0, 139.4, 137.0, 130.5, 124.7, 123.0, 113.8, 112.8, 72.5, 52.2,
19.4. IR (KBr, neat) 2955, 2923, 1722, 1688, 1607, 1437, 1265, 1126,
1000, 766, 654 cm–1; HRMS (ESI) calcd. for C13H15O4 (M + H)+ 235.0965,
found 235.0977.
White solid; Rf (hexane/EtOAc,
9:1) 0.50; mp 49–51 °C; yield 351 mg, 61%; 1H NMR (400 MHz, CDCl3) δ 10.43 (s, 1 H), 8.40 (d, J = 2.4 Hz, 1 H), 8.12 (dd, J = 8.8, 2.4
Hz, 1 H), 6.96 (d, J = 8.8 Hz, 1 H), 5.07 (s, 1 H),
5.00 (s, 1 H), 4.55 (s, 2 H), 3.83 (s, 3 H), 1.80 (s, 3 H); 13C{1H} NMR (100 MHz, CDCl3) δ 188.7, 166.0,
164.0, 139.4, 137.0, 130.5, 124.7, 123.0, 113.8, 112.8, 72.5, 52.2,
19.4. IR (KBr, neat) 2955, 2923, 1722, 1688, 1607, 1437, 1265, 1126,
1000, 766, 654 cm–1; HRMS (ESI) calcd. for C13H15O4 (M + H)+ 235.0965,
found 235.0977.
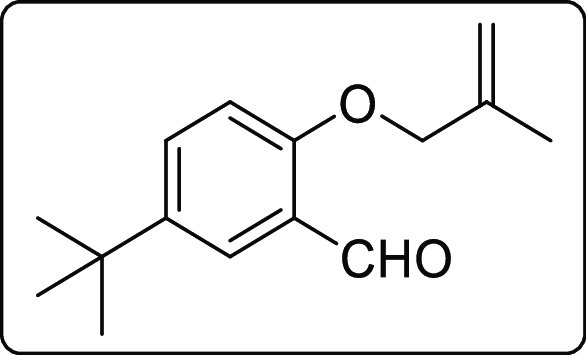
5-(tert-Butyl)-2-((2-methylallyl)oxy)benzaldehyde (1l)
 Pale yellow liquid; Rf (hexane/EtOAc,
9:1) 0.55; yield 434 mg, 76%; 1H NMR (400 MHz, CDCl3) δ 10.54 (s, 1 H), 7.86 (d, J = 2.8
Hz, 1 H), 7.55 (dd, J = 8.4, 2.4 Hz, 1 H), 6.91 (d, J = 8.8 Hz, 1 H), 5.11 (s, 1 H), 5.02 (s, 1 H), 4.52 (s,
2 H), 1.84 (s, 3 H), 1.30 (s, 9 H); 13C{1H}
NMR (100 MHz, CDCl3) δ 190.2, 159.4, 143.9, 140.5,
133.3, 125.0, 124.6, 113.3, 112.8, 72.4, 34.4, 31.5, 19.6; IR (KBr,
neat) 2962, 2866, 1684, 1608, 1494, 1264, 1188, 1010, 818, 646 cm–1; HRMS (ESI) calcd. for C15H21O2 (M + H)+ 233.1536, found 233.1534.
Pale yellow liquid; Rf (hexane/EtOAc,
9:1) 0.55; yield 434 mg, 76%; 1H NMR (400 MHz, CDCl3) δ 10.54 (s, 1 H), 7.86 (d, J = 2.8
Hz, 1 H), 7.55 (dd, J = 8.4, 2.4 Hz, 1 H), 6.91 (d, J = 8.8 Hz, 1 H), 5.11 (s, 1 H), 5.02 (s, 1 H), 4.52 (s,
2 H), 1.84 (s, 3 H), 1.30 (s, 9 H); 13C{1H}
NMR (100 MHz, CDCl3) δ 190.2, 159.4, 143.9, 140.5,
133.3, 125.0, 124.6, 113.3, 112.8, 72.4, 34.4, 31.5, 19.6; IR (KBr,
neat) 2962, 2866, 1684, 1608, 1494, 1264, 1188, 1010, 818, 646 cm–1; HRMS (ESI) calcd. for C15H21O2 (M + H)+ 233.1536, found 233.1534.
2-((2-Methylallyl)oxy)-1-naphthaldehyde (1m)
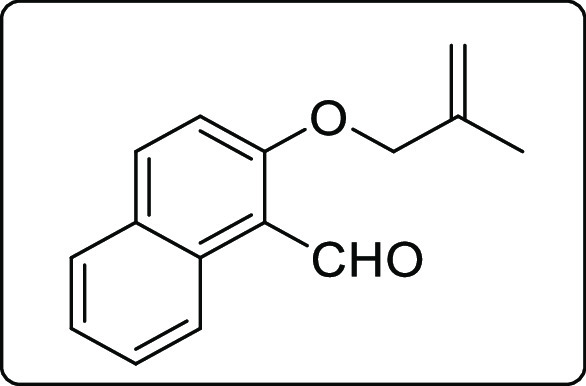 Brown solid; Rf (hexane/EtOAc,
9:1) 0.50; mp 72–74 °C; yield 367 mg, 66%; 1H NMR (500 MHz, CDCl3) 1H NMR δ 10.99
(s, 1 H), 9.31 (d, J = 9 Hz, 1 H), 8.04 (d, J = 9.0 Hz, 1 H), 7.79 (d, J = 8.0 Hz,
1 H), 7.64 (t, J = 7.0 Hz, 1 H), 7.44 (t, J = 7.5 Hz, 1 H), 7.27 (d, J = 9.0 Hz,
1 H), 5.18 (s, 1 H), 5.09 (s, 1 H), 4.70 (s, 2 H), 1.91 (s, 3 H); 13C{1H} NMR (125 MHz, CDCl3) δ
192.2, 163.5, 140.2, 137.6, 131.8, 130.0, 128.8, 128.4, 125.2, 125.0,
117.2, 114.0, 113.8, 73.2, 19.6; IR (KBr, neat) 2955, 2918, 1670,
1591, 1511, 1436, 1242, 1155, 1022, 811, 753, 710 cm–1; HRMS (ESI) calcd. for C15H15O2 (M + H)+ 227.1067, found 227.1066.
Brown solid; Rf (hexane/EtOAc,
9:1) 0.50; mp 72–74 °C; yield 367 mg, 66%; 1H NMR (500 MHz, CDCl3) 1H NMR δ 10.99
(s, 1 H), 9.31 (d, J = 9 Hz, 1 H), 8.04 (d, J = 9.0 Hz, 1 H), 7.79 (d, J = 8.0 Hz,
1 H), 7.64 (t, J = 7.0 Hz, 1 H), 7.44 (t, J = 7.5 Hz, 1 H), 7.27 (d, J = 9.0 Hz,
1 H), 5.18 (s, 1 H), 5.09 (s, 1 H), 4.70 (s, 2 H), 1.91 (s, 3 H); 13C{1H} NMR (125 MHz, CDCl3) δ
192.2, 163.5, 140.2, 137.6, 131.8, 130.0, 128.8, 128.4, 125.2, 125.0,
117.2, 114.0, 113.8, 73.2, 19.6; IR (KBr, neat) 2955, 2918, 1670,
1591, 1511, 1436, 1242, 1155, 1022, 811, 753, 710 cm–1; HRMS (ESI) calcd. for C15H15O2 (M + H)+ 227.1067, found 227.1066.
General Experimental Procedure for the Synthesis of 1r and 1s
To a suspension of K2CO3 (7.38 mmol, 3 equiv) in DMF under the N2 atmosphere was added substituted 2-hydroxybenzaldehyde (2.46 mmol, 1.0 equiv) and 4-bromobut-1-ene (3.69 mmol, 1.5 equiv). The reaction mixture was then allowed to stir at 50 °C and the progress of the reaction was monitored by TLC analysis. After completion of the reaction (12 h), the combined organic layer was washed with brine and ice water and further extracted with ethyl acetate (3 × 15 mL) followed by drying over anhydrous Na2SO4. The organic phase was concentrated in a rotary evaporator to give the crude product, which was then subjected to column chromatography over silica gel to provide the desired product 1r and 1s.
2-(But-3-en-1-yloxy)-4-methoxybenzaldehyde (1r)
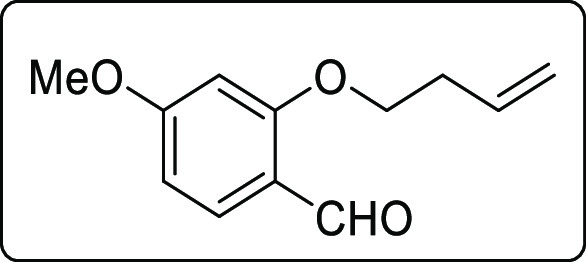 White liquid; Rf (hexane/EtOAc,
9:1) 0.50; yield 233 mg, 46%; 1H NMR (500 MHz, CDCl3) δ 10.30 (s, 1 H), 7.79 (d, J = 8.5
Hz, 1 H), 6.52 (dd, J = 9.0, 2.5 Hz, 1 H), 6.41 (d, J = 2.5 Hz, 1 H), 5.92–5.84 (m, 1 H), 5.19–5.15
(m, 1 H), 5.11 (d, J = 10.0 Hz, 1 H), 4.08 (t, J = 6.5 Hz, 2 H), 3.84 (s, 3 H), 2.60–2.56 (m, 2
H); 13C{1H} NMR (125 MHz, CDCl3)
δ 188.5, 166.3, 163.3, 134.1, 130.4, 119.4, 117.8, 106.2, 98.8,
67.9, 55.8, 33.6; IR (KBr, neat) 2928, 2845, 1674, 1596, 1443, 1257,
1198, 1112, 1028, 815, 549, 406 cm–1; HRMS (ESI)
calcd. for C12H15O3 (M + H)+ 207.1016, found 207.1022.
White liquid; Rf (hexane/EtOAc,
9:1) 0.50; yield 233 mg, 46%; 1H NMR (500 MHz, CDCl3) δ 10.30 (s, 1 H), 7.79 (d, J = 8.5
Hz, 1 H), 6.52 (dd, J = 9.0, 2.5 Hz, 1 H), 6.41 (d, J = 2.5 Hz, 1 H), 5.92–5.84 (m, 1 H), 5.19–5.15
(m, 1 H), 5.11 (d, J = 10.0 Hz, 1 H), 4.08 (t, J = 6.5 Hz, 2 H), 3.84 (s, 3 H), 2.60–2.56 (m, 2
H); 13C{1H} NMR (125 MHz, CDCl3)
δ 188.5, 166.3, 163.3, 134.1, 130.4, 119.4, 117.8, 106.2, 98.8,
67.9, 55.8, 33.6; IR (KBr, neat) 2928, 2845, 1674, 1596, 1443, 1257,
1198, 1112, 1028, 815, 549, 406 cm–1; HRMS (ESI)
calcd. for C12H15O3 (M + H)+ 207.1016, found 207.1022.
4-Bromo-2-(but-3-en-1-yloxy)benzaldehyde (1s)
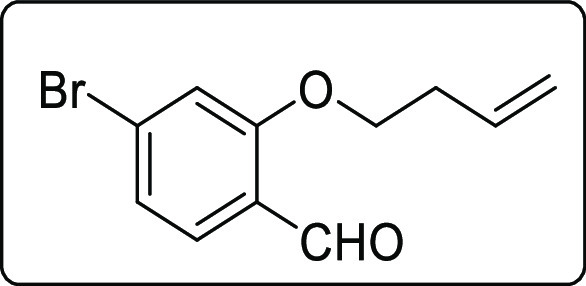 White solid; Rf (hexane/EtOAc,
9:1) 0.55; mp 70–72 °C; yield 358 mg, 57%; 1H NMR (500 MHz, CDCl3) δ 10.36 (d, J = 4.0 Hz, 1 H), 7.61 (q, J = 6.0 Hz 1 H), 7.12–7.08
(m, 2 H), 5.89–5.80 (m, 1 H), 5.18–5.09 (m, 2 H), 4.09–4.05
(m, 2 H), 2.58–2.54 (m, 2 H); 13C{1H}
NMR (125 MHz, CDCl3) δ 188.8, 161.5, 133.7, 130.5,
129.5, 124.3, 123.9, 117.9, 116.3, 68.2, 33.5; IR (KBr, neat) 2926,
2867, 1684, 1585, 1381, 1236, 1020, 915, 840, 808 cm–1; HRMS (ESI) calcd. for C11H12BrO2 (M + H)+ 255.0015, found 255.0020.
White solid; Rf (hexane/EtOAc,
9:1) 0.55; mp 70–72 °C; yield 358 mg, 57%; 1H NMR (500 MHz, CDCl3) δ 10.36 (d, J = 4.0 Hz, 1 H), 7.61 (q, J = 6.0 Hz 1 H), 7.12–7.08
(m, 2 H), 5.89–5.80 (m, 1 H), 5.18–5.09 (m, 2 H), 4.09–4.05
(m, 2 H), 2.58–2.54 (m, 2 H); 13C{1H}
NMR (125 MHz, CDCl3) δ 188.8, 161.5, 133.7, 130.5,
129.5, 124.3, 123.9, 117.9, 116.3, 68.2, 33.5; IR (KBr, neat) 2926,
2867, 1684, 1585, 1381, 1236, 1020, 915, 840, 808 cm–1; HRMS (ESI) calcd. for C11H12BrO2 (M + H)+ 255.0015, found 255.0020.
General Experimental Procedure for the Synthesis of 1t–1x and 1z
To a suspension of substituted 2-aminobenzaldehyde (4.13 mmol, 1.0 equiv) and p-TsCl (4.54 mmol, 1.1 equiv) in DCM under the N2 atmosphere was added pyridine (9.09 mmol, 2.2 equiv) dropwise at 0 °C. The reaction mixture was then allowed to stir at 0 °C for 5 min, which was then allowed to stir at room temperature. The progress of the reaction was monitored by TLC. After completion of the reaction (12 h), the combined organic layer was washed with brine and further extracted with DCM (3 × 15 mL) followed by drying over anhydrous Na2SO4. The organic phase was concentrated in a rotary evaporator to give the crude product, which was then subjected to column chromatography over silica gel to provide the desired N-(2-formylphenyl)-4-methylbenzenesulfonamide derivative A.
To a suspension of K2CO3 (3.27 mmol, 3 equiv) in DMF under the N2 atmosphere was added N-(2-formylphenyl)-4-methylbenzenesulfonamide derivative A (1.09 mmol, 1.0 equiv) and B (1.64 mmol, 1.5 equiv). Then, the reaction mixture was allowed to stir at 50 °C, and the reaction time was monitored by TLC. After completion of the reaction (4 h), the combined organic layer was washed with brine and ice water and further extracted with ethyl acetate (3 × 15 mL) followed by drying over anhydrous Na2SO4. The organic phase was concentrated in a rotary evaporator to give the crude product, which was then subjected to column chromatography over silica gel to provide the desired product 1t–1x and 1z.
N-(2-Formylphenyl)-4-methyl-N-(2-methylallyl)benzenesulfonamide (1t)
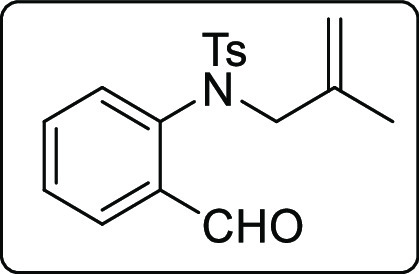 Yellow solid; Rf (hexane/EtOAc,
4:1) 0.50; mp 120–122 °C; yield 326 mg, 91%; 1H NMR (500 MHz, CDCl3) δ 10.44 (s, 1 H), 8.00 (dd, J = 7.0, 2.5 Hz, 1 H), 7.45–7.41 (m, 4 H), 7.28 (s,
1 H), 7.26 (d, J = Hz, 1 H), 6.69 (d, J = 7.5 Hz, 1 H), 4.75 (s, 1 H), 4.62 (s, 1 H), 4.46 (s, 1 H), 3.80
(s, 1 H), 2.44 (s, 3 H), 1.75 (s, 3 H); 13C{1H} NMR (125 MHz, CDCl3) δ 190.1, 144.5, 141.4, 139.0,
136.3, 134.1, 133.9, 129.9, 128.7, 128.6, 128.3, 127.5, 117.2, 57.8,
21.8, 20.6; IR (KBr, neat) 2917, 1691, 1597, 1348, 1163, 1089, 819,
661, 575 cm–1; HRMS (ESI) calcd. for C18H20NO3S (M + H)+ 330.1158, found
330.1155.
Yellow solid; Rf (hexane/EtOAc,
4:1) 0.50; mp 120–122 °C; yield 326 mg, 91%; 1H NMR (500 MHz, CDCl3) δ 10.44 (s, 1 H), 8.00 (dd, J = 7.0, 2.5 Hz, 1 H), 7.45–7.41 (m, 4 H), 7.28 (s,
1 H), 7.26 (d, J = Hz, 1 H), 6.69 (d, J = 7.5 Hz, 1 H), 4.75 (s, 1 H), 4.62 (s, 1 H), 4.46 (s, 1 H), 3.80
(s, 1 H), 2.44 (s, 3 H), 1.75 (s, 3 H); 13C{1H} NMR (125 MHz, CDCl3) δ 190.1, 144.5, 141.4, 139.0,
136.3, 134.1, 133.9, 129.9, 128.7, 128.6, 128.3, 127.5, 117.2, 57.8,
21.8, 20.6; IR (KBr, neat) 2917, 1691, 1597, 1348, 1163, 1089, 819,
661, 575 cm–1; HRMS (ESI) calcd. for C18H20NO3S (M + H)+ 330.1158, found
330.1155.
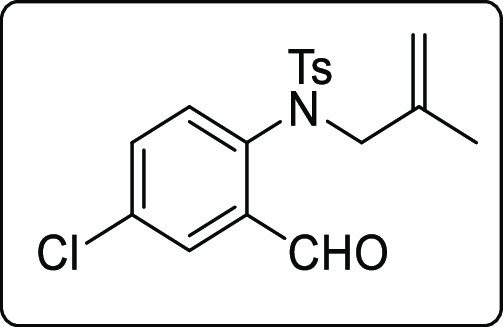
N-(4-Chloro-2-formylphenyl)-4-methyl-N-(2-methylallyl)benzenesulfonamide (1u)
 Pale yellow solid; Rf (hexane/EtOAc,
4:1) 0.50; mp 105–107 °C; yield 298 mg, 75%; 1H NMR (400 MHz, CDCl3) δ 10.34 (s, 1 H), 7.90 (d, J = 2.8 Hz, 1 H), 7.40–7.33 (m, 3 H), 7.26 (s, 1
H), 7.24 (d, J = 4.0 Hz, 1 H), 6.57 (d, J = 8.4 Hz, 1 H), 4.73 (s, 1 H), 4.59 (s, 1 H), 4.43 (s, 1 H), 3.69
(s, 1 H), 2.40 (s, 3 H), 1.68 (s, 3 H); 13C{1H} NMR (125 MHz, CDCl3) δ 188.6, 144.8, 139.6, 138.6,
137.3, 134.8, 133.8, 133.5, 130.0, 128.7, 128.5, 128.2, 117.5, 57.5,
21.8, 20.5; IR (KBr, neat) 2955, 2919, 1694, 1593, 1474, 1348, 1161,
1090, 1025, 862, 725, 666, 583, 553, 493 cm–1; HRMS
(ESI) calcd. for C18H19ClNO3S (M
+ H)+ 364.0769, found 364.0785.
Pale yellow solid; Rf (hexane/EtOAc,
4:1) 0.50; mp 105–107 °C; yield 298 mg, 75%; 1H NMR (400 MHz, CDCl3) δ 10.34 (s, 1 H), 7.90 (d, J = 2.8 Hz, 1 H), 7.40–7.33 (m, 3 H), 7.26 (s, 1
H), 7.24 (d, J = 4.0 Hz, 1 H), 6.57 (d, J = 8.4 Hz, 1 H), 4.73 (s, 1 H), 4.59 (s, 1 H), 4.43 (s, 1 H), 3.69
(s, 1 H), 2.40 (s, 3 H), 1.68 (s, 3 H); 13C{1H} NMR (125 MHz, CDCl3) δ 188.6, 144.8, 139.6, 138.6,
137.3, 134.8, 133.8, 133.5, 130.0, 128.7, 128.5, 128.2, 117.5, 57.5,
21.8, 20.5; IR (KBr, neat) 2955, 2919, 1694, 1593, 1474, 1348, 1161,
1090, 1025, 862, 725, 666, 583, 553, 493 cm–1; HRMS
(ESI) calcd. for C18H19ClNO3S (M
+ H)+ 364.0769, found 364.0785.
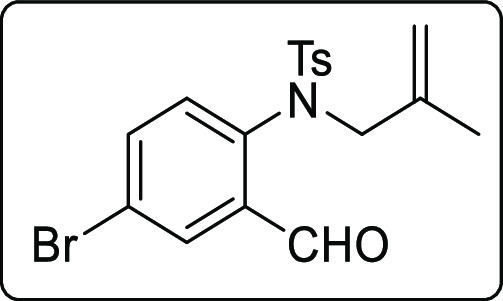
N-(4-Bromo-2-formylphenyl)-4-methyl-N-(2-methylallyl)benzenesulfonamide (1v)
 Brown solid; Rf (hexane/EtOAc,
4:1) 0.50; mp 91–93 °C; yield 342 mg, 77%; 1H NMR (500 MHz, CDCl3) δ 10.36 (s, 1 H), 8.08 (d, J = 2.5 Hz, 1 H), 7.52 (dd, J = 8.5, 2.5
Hz, 1 H), 7.41 (d, J = 8.0 Hz, 2 H), 7.28 (d, J = 8.0 Hz, 2 H), 6.53 (d, J = 8.5 Hz,
1 H), 4.76 (s, 1 H), 4.62 (s, 1 H), 4.46 (s, 1 H), 3.71 (s, 1 H),
2.43 (s, 3 H), 1.71 (s, 3 H); 13C{1H} NMR (125
MHz, CDCl3) δ 188.5, 144.8, 140.1, 138.5, 137.5,
136.7, 133.5, 131.5, 130.0, 128.9, 128.2, 122.7, 117.5, 57.5, 21.8,
20.5; IR (KBr, neat) 2958, 2922, 1692, 1596, 1474, 1349, 1162, 1091,
860, 723, 664, 581, 551, 419 cm–1; HRMS (ESI) calcd.
for C18H19BrNO3S (M + H)+ 408.0264, found 408.0284.
Brown solid; Rf (hexane/EtOAc,
4:1) 0.50; mp 91–93 °C; yield 342 mg, 77%; 1H NMR (500 MHz, CDCl3) δ 10.36 (s, 1 H), 8.08 (d, J = 2.5 Hz, 1 H), 7.52 (dd, J = 8.5, 2.5
Hz, 1 H), 7.41 (d, J = 8.0 Hz, 2 H), 7.28 (d, J = 8.0 Hz, 2 H), 6.53 (d, J = 8.5 Hz,
1 H), 4.76 (s, 1 H), 4.62 (s, 1 H), 4.46 (s, 1 H), 3.71 (s, 1 H),
2.43 (s, 3 H), 1.71 (s, 3 H); 13C{1H} NMR (125
MHz, CDCl3) δ 188.5, 144.8, 140.1, 138.5, 137.5,
136.7, 133.5, 131.5, 130.0, 128.9, 128.2, 122.7, 117.5, 57.5, 21.8,
20.5; IR (KBr, neat) 2958, 2922, 1692, 1596, 1474, 1349, 1162, 1091,
860, 723, 664, 581, 551, 419 cm–1; HRMS (ESI) calcd.
for C18H19BrNO3S (M + H)+ 408.0264, found 408.0284.
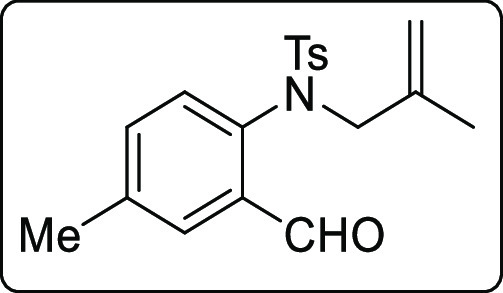
N-(2-Formyl-4-methylphenyl)-4-methyl-N-(2-methylallyl)benzenesulfonamide (1w)
 Red solid; Rf (hexane/EtOAc,
4:1) 0.50; mp 102–104 °C; yield 187 mg, 50%; 1H NMR (500 MHz, CDCl3) δ 10.40 (s, 1 H), 7.78 (s,
1 H), 7.44 (d, J = 8.5 Hz, 2 H), 7.27 (d, J = 8.0 Hz, 2 H), 7.23 (d, J = 8.5 Hz,
1 H), 6.57 (d, J = 8.0 Hz, 1 H), 4.74 (s, 1 H), 4.61
(s, 1 H), 4.43 (s, 1 H), 3.75 (s, 1 H), 2.43 (s, 3 H), 2.37 (s, 3
H), 1.73 (s, 3 H); 13C{1H} NMR (125 MHz, CDCl3) δ 190.4, 144.4, 139.1, 138.9, 138.7, 135.8, 134.8,
134.2, 129.8, 128.9, 128.3, 127.3, 117.0, 57.8, 21.8, 21.2, 20.6;
IR (KBr, neat) 2923, 2866, 1692, 1492, 1346, 1163, 864, 682, 658,
589, cm–1; HRMS (ESI) calcd. for C19H22NO3S (M + H)+ 344.1315, found 344.1320.
Red solid; Rf (hexane/EtOAc,
4:1) 0.50; mp 102–104 °C; yield 187 mg, 50%; 1H NMR (500 MHz, CDCl3) δ 10.40 (s, 1 H), 7.78 (s,
1 H), 7.44 (d, J = 8.5 Hz, 2 H), 7.27 (d, J = 8.0 Hz, 2 H), 7.23 (d, J = 8.5 Hz,
1 H), 6.57 (d, J = 8.0 Hz, 1 H), 4.74 (s, 1 H), 4.61
(s, 1 H), 4.43 (s, 1 H), 3.75 (s, 1 H), 2.43 (s, 3 H), 2.37 (s, 3
H), 1.73 (s, 3 H); 13C{1H} NMR (125 MHz, CDCl3) δ 190.4, 144.4, 139.1, 138.9, 138.7, 135.8, 134.8,
134.2, 129.8, 128.9, 128.3, 127.3, 117.0, 57.8, 21.8, 21.2, 20.6;
IR (KBr, neat) 2923, 2866, 1692, 1492, 1346, 1163, 864, 682, 658,
589, cm–1; HRMS (ESI) calcd. for C19H22NO3S (M + H)+ 344.1315, found 344.1320.
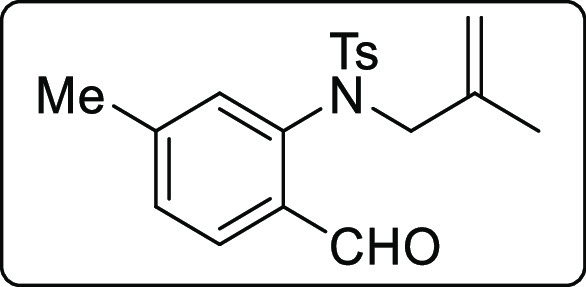
N-(2-Formyl-5-methylphenyl)-4-methyl-N-(2-methylallyl)benzenesulfonamide (1x)
 White solid; Rf (hexane/EtOAc,
4:1) 0.50; mp 100–102 °C; yield 206 mg, 55%; 1H NMR (500 MHz, CDCl3) δ 10.32 (s, 1 H), 7.88 (d, J = 8.0 Hz, 1 H), 7.45 (d, J = 8.5 Hz,
2 H), 7.28 (d, J = 8.0 Hz, 2 H), 7.21 (d, J = 8.0 Hz, 1 H), 6.47 (s, 1 H), 4.75 (s, 1 H), 4.62 (s,
1 H), 4.41 (s, 1 H), 3.77 (s, 1 H), 2.44 (s, 3 H), 2.26 (s, 3 H),
1.74 (s, 3 H); 13C{1H} NMR (125 MHz, CDCl3) δ 190.0, 145.2, 144.4, 141.5, 139.1, 134.2, 133.8,
129.7, 129.5, 128.6, 128.3, 128.2, 116.9, 58.0, 21.8, 21.8, 20.6;
IR (KBr, neat) 2955, 2917, 1689, 1604, 1345, 1162, 1090, 816, 687,
657, 577, 545 cm–1; HRMS (ESI) calcd. for C19H22NO3S (M + H)+ 344.1315,
found 344.1320.
White solid; Rf (hexane/EtOAc,
4:1) 0.50; mp 100–102 °C; yield 206 mg, 55%; 1H NMR (500 MHz, CDCl3) δ 10.32 (s, 1 H), 7.88 (d, J = 8.0 Hz, 1 H), 7.45 (d, J = 8.5 Hz,
2 H), 7.28 (d, J = 8.0 Hz, 2 H), 7.21 (d, J = 8.0 Hz, 1 H), 6.47 (s, 1 H), 4.75 (s, 1 H), 4.62 (s,
1 H), 4.41 (s, 1 H), 3.77 (s, 1 H), 2.44 (s, 3 H), 2.26 (s, 3 H),
1.74 (s, 3 H); 13C{1H} NMR (125 MHz, CDCl3) δ 190.0, 145.2, 144.4, 141.5, 139.1, 134.2, 133.8,
129.7, 129.5, 128.6, 128.3, 128.2, 116.9, 58.0, 21.8, 21.8, 20.6;
IR (KBr, neat) 2955, 2917, 1689, 1604, 1345, 1162, 1090, 816, 687,
657, 577, 545 cm–1; HRMS (ESI) calcd. for C19H22NO3S (M + H)+ 344.1315,
found 344.1320.
N-(But-3-en-1-yl)-N-(2-formylphenyl)-4-methylbenzenesulfonamide (1z)
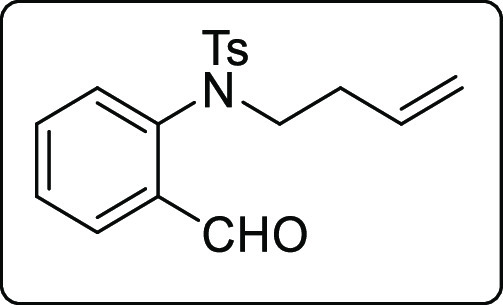 White solid; Rf (hexane/EtOAc,
4:1) 0.50; mp 118–120 °C; yield 233 mg, 65%; 1H NMR (500 MHz, CDCl3) δ 10.43 (s, 1 H), 8.01 (dd, J = 7.0, 2.0 Hz, 1 H), 7.48–7.44 (m, 3 H), 7.43 (s,
1 H), 7.27 (d, J = 8.0 Hz, 2 H), 6.69 (d, J = 7.0
Hz, 1 H), 5.73–5.67 (m, 1 H), 5.03 (d, J =
10.0 Hz, 1 H), 4.97 (d, J = 17.0 Hz, 1 H), 4.04 (s,
1 H), 3.33 (s, 1 H), 2.43 (s, 3 H), 2.21 (q, J =
7.0 Hz, 2 H); 13C{1H} NMR (125 MHz, CDCl3) δ 190.4, 144.4, 141.6, 136.5, 134.4, 134.2, 134.2,
129.8, 128.8, 128.7, 128.2, 127.3, 118.0, 50.6, 32.8, 21.8; IR (KBr,
neat) 2924, 2867, 1692, 1595, 1346, 1162, 1061, 719, 661, 574 cm–1; HRMS (ESI) calcd. for C18H20NO3S (M + H)+330.1158, found 330.1155.
White solid; Rf (hexane/EtOAc,
4:1) 0.50; mp 118–120 °C; yield 233 mg, 65%; 1H NMR (500 MHz, CDCl3) δ 10.43 (s, 1 H), 8.01 (dd, J = 7.0, 2.0 Hz, 1 H), 7.48–7.44 (m, 3 H), 7.43 (s,
1 H), 7.27 (d, J = 8.0 Hz, 2 H), 6.69 (d, J = 7.0
Hz, 1 H), 5.73–5.67 (m, 1 H), 5.03 (d, J =
10.0 Hz, 1 H), 4.97 (d, J = 17.0 Hz, 1 H), 4.04 (s,
1 H), 3.33 (s, 1 H), 2.43 (s, 3 H), 2.21 (q, J =
7.0 Hz, 2 H); 13C{1H} NMR (125 MHz, CDCl3) δ 190.4, 144.4, 141.6, 136.5, 134.4, 134.2, 134.2,
129.8, 128.8, 128.7, 128.2, 127.3, 118.0, 50.6, 32.8, 21.8; IR (KBr,
neat) 2924, 2867, 1692, 1595, 1346, 1162, 1061, 719, 661, 574 cm–1; HRMS (ESI) calcd. for C18H20NO3S (M + H)+330.1158, found 330.1155.
General Experimental Procedure for the Synthesis of 3d

To a suspension of K2CO3 (4.92 mmol, 2 equiv) in DMF under the N2 atmosphere was added 2-hydroxybenzaldehyde (2.46 mmol, 1.0 equiv) and (3-bromoprop-1-yn-1-yl)trimethylsilane (4.92 mmol, 2 equiv). The reaction mixture was then allowed to stir at room temperature. The progress of the reaction was monitored by TLC analysis. After completion of the reaction (12 h), the combined organic layer was washed with brine and ice water and further extracted with ethyl acetate (3 × 15 mL) followed by drying over anhydrous Na2SO4. The organic phase was concentrated in a rotary evaporator to give the crude product, which was then subjected to column chromatography over silica gel to provide the desired product 3d.
2-((3-(Trimethylsilyl)prop-2-yn-1-yl)oxy)benzaldehyde (3d)
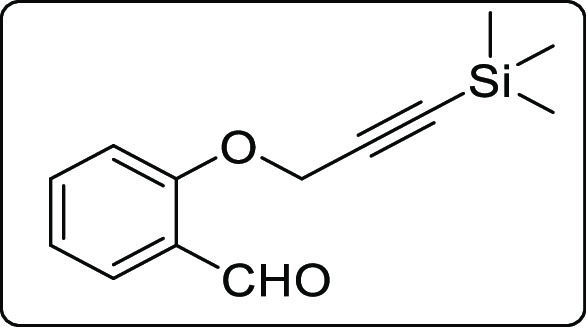 White liquid; Rf (hexane/EtOAc,
9:1) 0.55; yield 143 mg, 25%; 1H NMR (500 MHz, CDCl3) δ 10.49 (s, 1 H), 7.85 (d, J = 6.0
Hz, 1 H), 7.55 (t, J = 8.5 Hz, 1 H), 7.11 (d, J = 8.5 Hz, 1 H), 7.07 (t, J = 7.5 Hz,
1 H), 4.80 (s, 2 H), 0.16 (s, 9 H); 13C{1H}
NMR (125 MHz, CDCl3) δ 189.9, 160.3, 135.8, 128.6,
125.8, 121.8, 113.8, 99.3, 94.2, 57.7, −0.2; IR (KBr, neat)
2960, 2864, 2180, 1688, 1595, 1481, 1459, 1250, 1216, 1029, 837, 755,
638 cm–1; HRMS (ESI) calcd. for C13H17O2Si (M + H)+ 233.0992, found 233.1014.
White liquid; Rf (hexane/EtOAc,
9:1) 0.55; yield 143 mg, 25%; 1H NMR (500 MHz, CDCl3) δ 10.49 (s, 1 H), 7.85 (d, J = 6.0
Hz, 1 H), 7.55 (t, J = 8.5 Hz, 1 H), 7.11 (d, J = 8.5 Hz, 1 H), 7.07 (t, J = 7.5 Hz,
1 H), 4.80 (s, 2 H), 0.16 (s, 9 H); 13C{1H}
NMR (125 MHz, CDCl3) δ 189.9, 160.3, 135.8, 128.6,
125.8, 121.8, 113.8, 99.3, 94.2, 57.7, −0.2; IR (KBr, neat)
2960, 2864, 2180, 1688, 1595, 1481, 1459, 1250, 1216, 1029, 837, 755,
638 cm–1; HRMS (ESI) calcd. for C13H17O2Si (M + H)+ 233.0992, found 233.1014.
General Experimental Procedure for the Synthesis of 2a–2s, 2b′b

To a stirred solution of 1 (0.57 mmol, 1.0 equiv) and CuI (0.68 mmol, 1.2 equiv) in CH3CN (4 mL) was added 70% aq. solution of TBHP (3.42 mmol, 6.0 equiv,) dropwise at room temperature. Then, the reaction mixture was allowed to stir at 70 °C, and the reaction time was monitored by TLC. After completion of the reaction (6–12 h), the reaction mixture was brought to room temperature. The solvent was removed under vacuo in a rotary evaporator and extracted with ethyl acetate (3 × 15 mL) and washed with aqueous solution of Na2S2O3, NH4Cl, and saturated brine solution. The combined organic extracts were dried over Na2SO4 and concentrated in a rotary evaporator. The crude was subjected to column chromatography over silica gel to give the corresponding product 2a–2s.
3-(Iodomethyl)-3-methyl-2,3-dihydro-5H-benzo[e][1,4]dioxepin-5-one (2a)
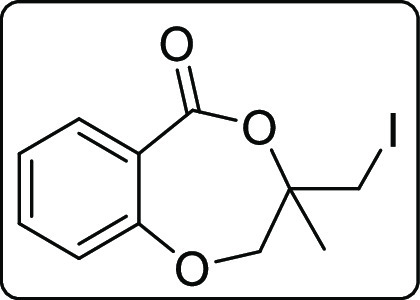 Pale yellow solid; Rf (hexane/EtOAc,
4:1) 0.50; mp 85–87 °C; yield 136 mg, 75%; 1H NMR (400 MHz, CDCl3) δ 8.19 (d, J = 8.0 Hz, 1 H), 7.51–7.46 (m, 1 H), 7.10–7.03 (m,
2 H), 4.53 (d, J = 13.6 Hz, 1 H), 4.38 (d, J = 13.6 Hz, 1 H), 3.42 (d, J = 10.8 Hz,
1 H), 3.36 (d, J = 10.8 Hz, 1 H), 1.62 (s, 3 H); 13C{1H} NMR (100 MHz, CDCl3) δ
165.3, 157.5, 136.1, 135.3, 122.3, 120.0, 116.5, 80.3, 75.1, 23.2,
8.1; IR (KBr, neat) 2969, 1738, 1696, 1480, 1277, 1261, 1118, 753,
cm–1; HRMS (ESI) calcd. for C11H12IO3 (M + H)+ 318.9826, found 318.982.
Pale yellow solid; Rf (hexane/EtOAc,
4:1) 0.50; mp 85–87 °C; yield 136 mg, 75%; 1H NMR (400 MHz, CDCl3) δ 8.19 (d, J = 8.0 Hz, 1 H), 7.51–7.46 (m, 1 H), 7.10–7.03 (m,
2 H), 4.53 (d, J = 13.6 Hz, 1 H), 4.38 (d, J = 13.6 Hz, 1 H), 3.42 (d, J = 10.8 Hz,
1 H), 3.36 (d, J = 10.8 Hz, 1 H), 1.62 (s, 3 H); 13C{1H} NMR (100 MHz, CDCl3) δ
165.3, 157.5, 136.1, 135.3, 122.3, 120.0, 116.5, 80.3, 75.1, 23.2,
8.1; IR (KBr, neat) 2969, 1738, 1696, 1480, 1277, 1261, 1118, 753,
cm–1; HRMS (ESI) calcd. for C11H12IO3 (M + H)+ 318.9826, found 318.982.
7-Chloro-3-(iodomethyl)-3-methyl-2,3-dihydro-5H-benzo[e][1,4]dioxepin-5-one (2b)
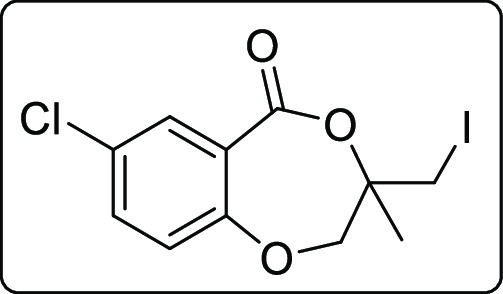 White solid; Rf (hexane/EtOAc,
4:1) 0.50; mp 82–84 °C; yield 127 mg, 63%; 1H NMR (500 MHz, CDCl3) δ 8.16 (d, J = 2.5 Hz, 1 H), 7.42 (dd, J = 9.0, 2.5 Hz, 1 H),
7.00 (d, J = 8.5 Hz, 1 H), 4.52 (d, J = 13.5 Hz, 1 H), 4.38 (d, J = 14 Hz, 1 H), 3.40
(d, J = 11.0 Hz, 1 H), 3.36 (d, J = 10.5 Hz, 1 H), 1.61 (s, 3 H).; 13C{1H} NMR
(125 MHz, CDCl3) δ 164.03, 156.06, 135.34, 134.99,
127.67, 121.74, 117.69, 80.59, 75.41, 23.13, 7.74; IR (KBr, neat)
2955, 2911, 1696, 1477, 1389, 1276, 1134, 824, 723 cm–1; HRMS (ESI) calcd. for C11H11ClIO3 (M + H)+ 352.9436, found 352.9432
White solid; Rf (hexane/EtOAc,
4:1) 0.50; mp 82–84 °C; yield 127 mg, 63%; 1H NMR (500 MHz, CDCl3) δ 8.16 (d, J = 2.5 Hz, 1 H), 7.42 (dd, J = 9.0, 2.5 Hz, 1 H),
7.00 (d, J = 8.5 Hz, 1 H), 4.52 (d, J = 13.5 Hz, 1 H), 4.38 (d, J = 14 Hz, 1 H), 3.40
(d, J = 11.0 Hz, 1 H), 3.36 (d, J = 10.5 Hz, 1 H), 1.61 (s, 3 H).; 13C{1H} NMR
(125 MHz, CDCl3) δ 164.03, 156.06, 135.34, 134.99,
127.67, 121.74, 117.69, 80.59, 75.41, 23.13, 7.74; IR (KBr, neat)
2955, 2911, 1696, 1477, 1389, 1276, 1134, 824, 723 cm–1; HRMS (ESI) calcd. for C11H11ClIO3 (M + H)+ 352.9436, found 352.9432
9-Chloro-3-(iodomethyl)-3-methyl-2,3-dihydro-5H-benzo[e][1,4]dioxepin-5-one (2c)
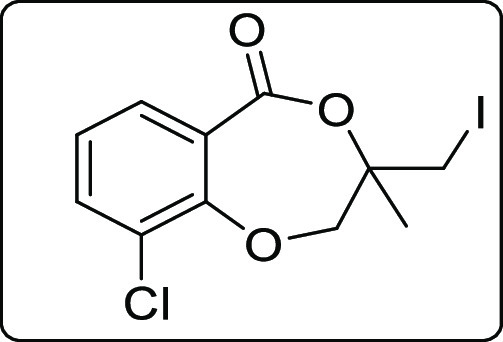 Brown solid; Rf (hexane/EtOAc,
4:1) 0.50; mp 128–130 °C; yield 143 mg, 71%; 1H NMR (500 MHz, CDCl3) δ 8.10 (dd, J = 8.5, 2.0 Hz, 1 H), 7.59 (dd, J = 8.0, 2.0 Hz,
1 H), 7.02 (t, J = 8.0 Hz, 1 H), 4.61 (d, J = 13.5 Hz, 1 H), 4.51 (d, J = 14 Hz,
1 H), 3.42 (d, J = 11.0 Hz, 1 H), 3.38 (d, J = 11.0 Hz, 1 H), 1.63 (s, 3 H); 13C{1H} NMR (125 MHz, CDCl3) δ 164.3, 153.1, 135.7, 134.7,
124.8, 122.3, 118.5, 80.4, 76.0, 23.1, 8.1; IR (KBr, neat) 2922, 1698,
1465, 1282, 1184, 1080, 748 cm–1; HRMS (ESI) calcd.
for C11H11ClIO3 (M + H)+ 352.9436, found 352.9440.
Brown solid; Rf (hexane/EtOAc,
4:1) 0.50; mp 128–130 °C; yield 143 mg, 71%; 1H NMR (500 MHz, CDCl3) δ 8.10 (dd, J = 8.5, 2.0 Hz, 1 H), 7.59 (dd, J = 8.0, 2.0 Hz,
1 H), 7.02 (t, J = 8.0 Hz, 1 H), 4.61 (d, J = 13.5 Hz, 1 H), 4.51 (d, J = 14 Hz,
1 H), 3.42 (d, J = 11.0 Hz, 1 H), 3.38 (d, J = 11.0 Hz, 1 H), 1.63 (s, 3 H); 13C{1H} NMR (125 MHz, CDCl3) δ 164.3, 153.1, 135.7, 134.7,
124.8, 122.3, 118.5, 80.4, 76.0, 23.1, 8.1; IR (KBr, neat) 2922, 1698,
1465, 1282, 1184, 1080, 748 cm–1; HRMS (ESI) calcd.
for C11H11ClIO3 (M + H)+ 352.9436, found 352.9440.
7-Bromo-3-(iodomethyl)-3-methyl-2,3-dihydro-5H-benzo[e][1,4]dioxepin-5-one (2d)
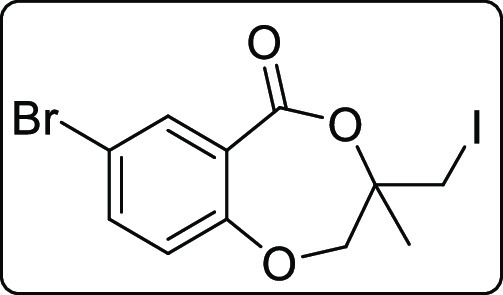 Brown solid; Rf (hexane/EtOAc,
4:1) 0.50; mp 105–107 °C; yield 149 mg, 66%; 1H NMR (400 MHz, CDCl3) δ 8.33 (d, J = 2.8 Hz, 1 H), 7.56 (dd, J = 8.8, 2.4 Hz, 1 H),
6.95 (d, J = 8.8 Hz, 1 H), 4.53 (d, J = 13.6 Hz, 1 H), 4.38 (d, J = 14 Hz, 1 H), 3.41
(d, J = 10.8 Hz, 1 H), 3.36 (d, J = 10.4 Hz, 1 H), 1.62 (s, 3 H); 13C{1H} NMR
(100 MHz, CDCl3) δ 163.90, 156.53, 138.11, 137.94,
122.00, 118.12, 114.68, 80.52, 75.40, 23.10, 7.77; IR (KBr, neat)
2958, 2924, 1695, 1474, 1386, 1275, 1135, 823, 764 cm–1; HRMS (ESI) calcd. for C11H11BrIO3 (M + H)+ 396.8931, found 396.8928.
Brown solid; Rf (hexane/EtOAc,
4:1) 0.50; mp 105–107 °C; yield 149 mg, 66%; 1H NMR (400 MHz, CDCl3) δ 8.33 (d, J = 2.8 Hz, 1 H), 7.56 (dd, J = 8.8, 2.4 Hz, 1 H),
6.95 (d, J = 8.8 Hz, 1 H), 4.53 (d, J = 13.6 Hz, 1 H), 4.38 (d, J = 14 Hz, 1 H), 3.41
(d, J = 10.8 Hz, 1 H), 3.36 (d, J = 10.4 Hz, 1 H), 1.62 (s, 3 H); 13C{1H} NMR
(100 MHz, CDCl3) δ 163.90, 156.53, 138.11, 137.94,
122.00, 118.12, 114.68, 80.52, 75.40, 23.10, 7.77; IR (KBr, neat)
2958, 2924, 1695, 1474, 1386, 1275, 1135, 823, 764 cm–1; HRMS (ESI) calcd. for C11H11BrIO3 (M + H)+ 396.8931, found 396.8928.
8-Bromo-3-(iodomethyl)-3-methyl-2,3-dihydro-5H-benzo[e][1,4]dioxepin-5-one (2e)
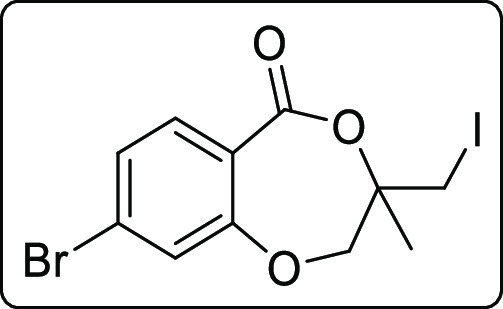 White solid; Rf (hexane/EtOAc,
4:1) 0.50; mp 107–109 °C; yield 163 mg, 72%; 1H NMR (400 MHz, CDCl3) δ 8.06 (d, J = 8.4 Hz, 1 H), 7.27–7.26 (m, 1 H), 7.22 (dd, J = 8.8, 2.0 Hz, 1 H), 4.54 (d, J = 14 Hz, 1 H),
4.39 (d, J = 13.6 Hz, 1 H), 3.41 (d, J = 10.8 Hz, 1 H), 3.37 (d, J = 10.8 Hz, 1 H), 1.62
(s, 3 H); 13C{1H} NMR (100 MHz, CDCl3) δ 164.54, 157.65, 137.31, 129.75, 125.93, 123.02, 115.58,
80.30, 75.39, 23.11, 7.71; IR (KBr, neat) 2924, 1696, 1593, 1410,
1277, 1105, 864, 755 cm–1; HRMS (ESI) calcd. for
C11H11BrIO3 (M + H)+ 396.8931,
found 396.8934.
White solid; Rf (hexane/EtOAc,
4:1) 0.50; mp 107–109 °C; yield 163 mg, 72%; 1H NMR (400 MHz, CDCl3) δ 8.06 (d, J = 8.4 Hz, 1 H), 7.27–7.26 (m, 1 H), 7.22 (dd, J = 8.8, 2.0 Hz, 1 H), 4.54 (d, J = 14 Hz, 1 H),
4.39 (d, J = 13.6 Hz, 1 H), 3.41 (d, J = 10.8 Hz, 1 H), 3.37 (d, J = 10.8 Hz, 1 H), 1.62
(s, 3 H); 13C{1H} NMR (100 MHz, CDCl3) δ 164.54, 157.65, 137.31, 129.75, 125.93, 123.02, 115.58,
80.30, 75.39, 23.11, 7.71; IR (KBr, neat) 2924, 1696, 1593, 1410,
1277, 1105, 864, 755 cm–1; HRMS (ESI) calcd. for
C11H11BrIO3 (M + H)+ 396.8931,
found 396.8934.
7-Fluoro-3-(iodomethyl)-3-methyl-2,3-dihydro-5H-benzo[e][1,4]dioxepin-5-one (2f)
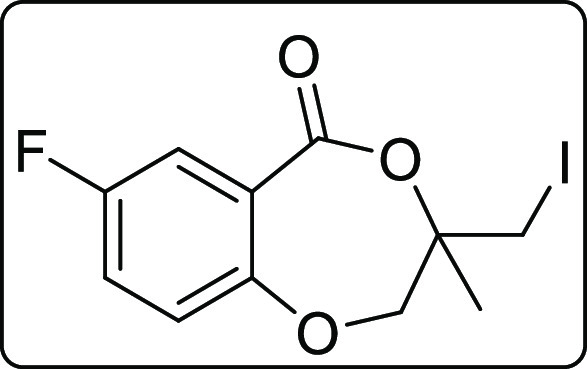 Red solid; Rf (hexane/EtOAc,
4:1) 0.50; mp 117–119 °C; yield 117 mg, 61%; 1H NMR (500 MHz, CDCl3) δ 7.89–7.86 (m, 1
H), 7.24–7.19 (m, 1 H), 7.04–7.01 (m, 1 H), 4.51 (dd, J = 14.0, 2.0 Hz, 1H), 4.36 (d, J = 13.5
Hz, 1 H), 3.42 (d, J = 10.5 Hz, 1 H), 3.37 (d, J = 10.5 Hz, 1 H), 1.62 (s, 3 H); 13C{1H} NMR (125 MHz, CDCl3) δ 164.0, 164.0, 157.5 (d, J = 240.4 Hz), 153.9, 153.8, 123.3, 123.1, 121.8, 121.8,
120.80.(d, J = 25.25 Hz), 117.5, 117.4, 80.8, 75.4,
23.2, 7.9; 19F NMR (470 MHz, C6F6/CDCl3) δ 40.41 (s, -F); IR (KBr, neat) 2980, 2927,
1695, 1488, 1411, 1275, 1167, 827, 751 cm–1; HRMS
(ESI) calcd. for C11H11FIO3 (M +
H)+ 336.9731, found 336.9736.
Red solid; Rf (hexane/EtOAc,
4:1) 0.50; mp 117–119 °C; yield 117 mg, 61%; 1H NMR (500 MHz, CDCl3) δ 7.89–7.86 (m, 1
H), 7.24–7.19 (m, 1 H), 7.04–7.01 (m, 1 H), 4.51 (dd, J = 14.0, 2.0 Hz, 1H), 4.36 (d, J = 13.5
Hz, 1 H), 3.42 (d, J = 10.5 Hz, 1 H), 3.37 (d, J = 10.5 Hz, 1 H), 1.62 (s, 3 H); 13C{1H} NMR (125 MHz, CDCl3) δ 164.0, 164.0, 157.5 (d, J = 240.4 Hz), 153.9, 153.8, 123.3, 123.1, 121.8, 121.8,
120.80.(d, J = 25.25 Hz), 117.5, 117.4, 80.8, 75.4,
23.2, 7.9; 19F NMR (470 MHz, C6F6/CDCl3) δ 40.41 (s, -F); IR (KBr, neat) 2980, 2927,
1695, 1488, 1411, 1275, 1167, 827, 751 cm–1; HRMS
(ESI) calcd. for C11H11FIO3 (M +
H)+ 336.9731, found 336.9736.
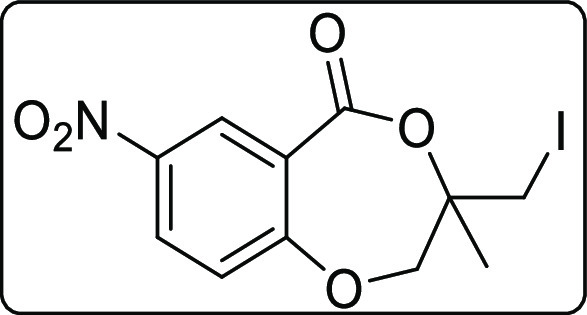
3-(Iodomethyl)-3-methyl-7-nitro-2,3-dihydro-5H-benzo[e][1,4]dioxepin-5-one (2g)
 Pale yellow solid; Rf (hexane/EtOAc,
4:1) 0.40; mp 142–144 °C; yield 124 mg, 60%; 1H NMR (500 MHz, CDCl3) δ 9.13 (d, J = 3.0 Hz, 1 H), 8.32 (dd, J = 9.5, 3.0 Hz, 1 H),
7.20 (d, J = 9.0 Hz, 1 H), 4.66 (d, J = 13.5 Hz, 1 H), 4.53 (d, J = 14.0 Hz, 1 H), 3.41
(d, J = 10.5 Hz, 1 H), 3.39 (d, J = 11.0 Hz, 1 H), 1.66 (s, 3 H); 13C{1H} NMR
(125 MHz, CDCl3) δ 163.0, 161.4, 142.8, 132.8, 129.7,
121.6, 121.6, 117.0, 80.5, 76.1, 23.0, 7.2; IR (KBr, neat) 2958, 2919,
1702, 1524, 1337, 1276, 1132, 841, 750 cm–1; HRMS
(ESI) calcd. for C11H11INO5 (M +
H)+ 363.9676, found 363.9674.
Pale yellow solid; Rf (hexane/EtOAc,
4:1) 0.40; mp 142–144 °C; yield 124 mg, 60%; 1H NMR (500 MHz, CDCl3) δ 9.13 (d, J = 3.0 Hz, 1 H), 8.32 (dd, J = 9.5, 3.0 Hz, 1 H),
7.20 (d, J = 9.0 Hz, 1 H), 4.66 (d, J = 13.5 Hz, 1 H), 4.53 (d, J = 14.0 Hz, 1 H), 3.41
(d, J = 10.5 Hz, 1 H), 3.39 (d, J = 11.0 Hz, 1 H), 1.66 (s, 3 H); 13C{1H} NMR
(125 MHz, CDCl3) δ 163.0, 161.4, 142.8, 132.8, 129.7,
121.6, 121.6, 117.0, 80.5, 76.1, 23.0, 7.2; IR (KBr, neat) 2958, 2919,
1702, 1524, 1337, 1276, 1132, 841, 750 cm–1; HRMS
(ESI) calcd. for C11H11INO5 (M +
H)+ 363.9676, found 363.9674.
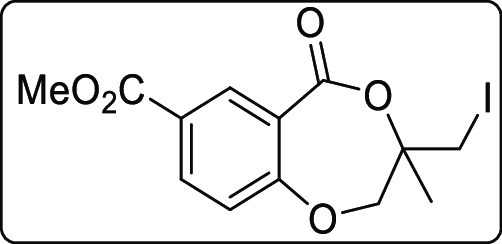
Methyl-3-(iodomethyl)-3-methyl-5-oxo-2,3-dihydro-5H-benzo[e][1,4]dioxepine-7-carboxylate (2h)
 White solid; Rf (hexane/EtOAc,
3:1) 0.50; mp 166–168 °C; yield 150 mg, 70%; 1H NMR (500 MHz, CDCl3) δ 8.89 (d, J = 2.5 Hz, 1 H), 8.12 (dd, J = 8.5, 2.0 Hz, 1 H),
7.10 (d, J = 8.5 Hz, 1 H), 4.59 (d, J = 14.0 Hz, 1 H), 4.46 (d, J = 14.0 Hz, 1 H), 3.91 (s, 3 H), 3.41
(d, J = 11.0, 1 H), 3.37 (d, J =
10.5, 1 H), 1.63 (s, 3 H); 13C{1H} NMR (125
MHz, CDCl3) 13C NMR δ 165.8, 164.3, 160.4, 138.6,
135.9, 124.7, 120.5, 116.3, 80.2, 75.5, 52.4, 23.1, 7.6; IR (KBr,
neat) 2955, 2922, 1686, 1611, 1409, 1259, 1112, 852, 761 cm–1; HRMS (ESI) calcd. for C13H14IO5 (M + H)+ 376.9880, found 376.9884.
White solid; Rf (hexane/EtOAc,
3:1) 0.50; mp 166–168 °C; yield 150 mg, 70%; 1H NMR (500 MHz, CDCl3) δ 8.89 (d, J = 2.5 Hz, 1 H), 8.12 (dd, J = 8.5, 2.0 Hz, 1 H),
7.10 (d, J = 8.5 Hz, 1 H), 4.59 (d, J = 14.0 Hz, 1 H), 4.46 (d, J = 14.0 Hz, 1 H), 3.91 (s, 3 H), 3.41
(d, J = 11.0, 1 H), 3.37 (d, J =
10.5, 1 H), 1.63 (s, 3 H); 13C{1H} NMR (125
MHz, CDCl3) 13C NMR δ 165.8, 164.3, 160.4, 138.6,
135.9, 124.7, 120.5, 116.3, 80.2, 75.5, 52.4, 23.1, 7.6; IR (KBr,
neat) 2955, 2922, 1686, 1611, 1409, 1259, 1112, 852, 761 cm–1; HRMS (ESI) calcd. for C13H14IO5 (M + H)+ 376.9880, found 376.9884.
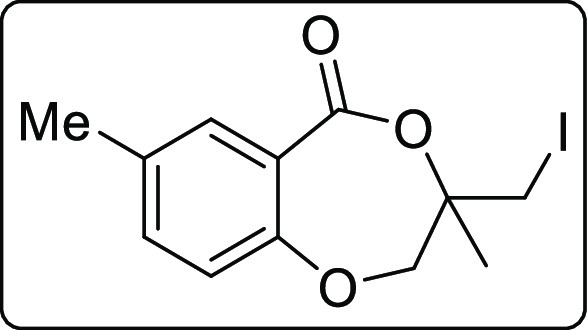
3-(Iodomethyl)-3,7-dimethyl-2,3-dihydro-5H-benzo[e][1,4]dioxepin-5-one (2i)
 Pale yellow solid; Rf (hexane/EtOAc,
4:1) 0.50; mp 89–91 °C; yield 138 mg, 73%; 1H NMR (400 MHz, CDCl3) δ 7.98 (s, 1 H), 7.29 (dd, J = 8.4, 2.4 Hz, 1 H), 6.94 (d, J = 8.4
Hz, 1 H), 4.50 (d, J = 13.6 Hz, 1 H), 4.34 (d, J = 13.6 Hz, 1 H), 3.42 (d, J = 10.8, 1
H), 3.36 (d, J = 10.4, 1 H), 2.31 (s, 3 H), 1.61
(s, 3 H).; 13C{1H} NMR (125 MHz, CDCl3) δ 165.4, 155.4, 136.4, 135.5, 131.7, 119.8, 115.9, 80.3,
75.0, 23.2, 20.4, 8.2. IR (KBr, neat) 2966, 2919, 1694, 1617, 1494,
1398, 1286, 1137, 822, 750 cm–1; HRMS (ESI) calcd.
for C12H14IO3 (M + H)+ 332.9982, found 332.9980.
Pale yellow solid; Rf (hexane/EtOAc,
4:1) 0.50; mp 89–91 °C; yield 138 mg, 73%; 1H NMR (400 MHz, CDCl3) δ 7.98 (s, 1 H), 7.29 (dd, J = 8.4, 2.4 Hz, 1 H), 6.94 (d, J = 8.4
Hz, 1 H), 4.50 (d, J = 13.6 Hz, 1 H), 4.34 (d, J = 13.6 Hz, 1 H), 3.42 (d, J = 10.8, 1
H), 3.36 (d, J = 10.4, 1 H), 2.31 (s, 3 H), 1.61
(s, 3 H).; 13C{1H} NMR (125 MHz, CDCl3) δ 165.4, 155.4, 136.4, 135.5, 131.7, 119.8, 115.9, 80.3,
75.0, 23.2, 20.4, 8.2. IR (KBr, neat) 2966, 2919, 1694, 1617, 1494,
1398, 1286, 1137, 822, 750 cm–1; HRMS (ESI) calcd.
for C12H14IO3 (M + H)+ 332.9982, found 332.9980.
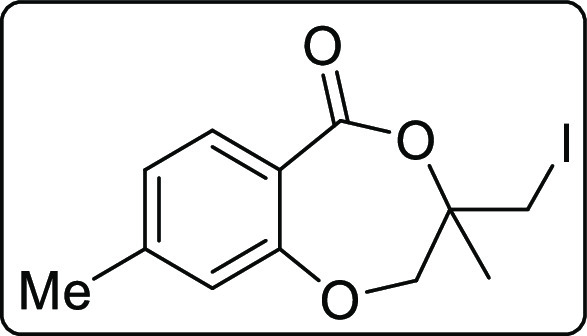
3-(Iodomethyl)-3,8-dimethyl-2,3-dihydro-5H-benzo[e][1,4]dioxepin-5-one (2j)
 White solid; Rf (hexane/EtOAc,
4:1) 0.50; mp 138–140 °C; yield 144 mg, 76%; 1H NMR (400 MHz, CDCl3) δ 8.08 (d, J = 8.4 Hz, 1 H), 6.89 (d, J = 8.0, 2 H)6.85 (s,
1 H), 4.51 (d, J = 13.6 Hz, 1 H), 4.35 (d, J = 13.6 Hz, 1 H), 3.42 (d, J = 10.4, 1
H), 3.35 (d, J = 10.8, 1 H), 2.35 (s, 3 H), 1.61
(s, 3 H); 13C{1H} NMR (125 MHz, CDCl3) δ 165.3, 157.4, 146.7, 136.0, 123.7, 120.0, 113.7, 80.1,
75.0, 23.2, 21.4, 8.1; IR (KBr, neat) 2922, 1692, 1619, 1412, 1283,
1136, 1093, 963, 760 cm–1; HRMS (ESI) calcd. for
C12H14IO3 (M + H)+ 332.9982,
found 332.9991.
White solid; Rf (hexane/EtOAc,
4:1) 0.50; mp 138–140 °C; yield 144 mg, 76%; 1H NMR (400 MHz, CDCl3) δ 8.08 (d, J = 8.4 Hz, 1 H), 6.89 (d, J = 8.0, 2 H)6.85 (s,
1 H), 4.51 (d, J = 13.6 Hz, 1 H), 4.35 (d, J = 13.6 Hz, 1 H), 3.42 (d, J = 10.4, 1
H), 3.35 (d, J = 10.8, 1 H), 2.35 (s, 3 H), 1.61
(s, 3 H); 13C{1H} NMR (125 MHz, CDCl3) δ 165.3, 157.4, 146.7, 136.0, 123.7, 120.0, 113.7, 80.1,
75.0, 23.2, 21.4, 8.1; IR (KBr, neat) 2922, 1692, 1619, 1412, 1283,
1136, 1093, 963, 760 cm–1; HRMS (ESI) calcd. for
C12H14IO3 (M + H)+ 332.9982,
found 332.9991.
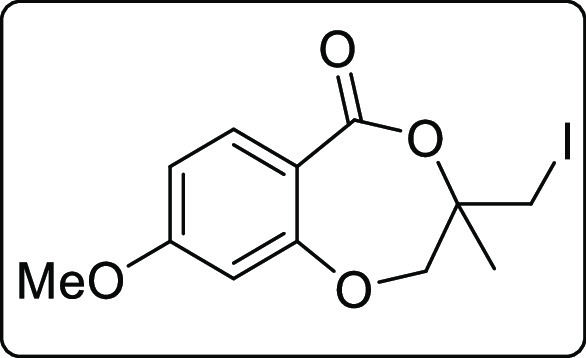
3-(Iodomethyl)-8-methoxy-3-methyl-2,3-dihydro-5H-benzo[e][1,4]dioxepin-5-one (2k)
 Gray solid; Rf (hexane/EtOAc,
4:1) 0.40; mp 108–110 °C; yield 159 mg, 80%; 1H NMR (500 MHz, CDCl3) δ 8.14 (dd, J = 9.5, 3.5 Hz, 1 H), 6.65 (m, 1 H), 6.48 (t, J =
2.5 Hz, 1 H), 4.52 (d, J = 13.5 Hz, 1 H), 4.35 (d, J = 13.5 Hz, 1 H), 3.84 (s, 3 H), 3.42 (d, J = 10.5, 1 H), 3.36 (d, J = 10.5, 1 H), 1.61 (s,
3 H); 13C{1H} NMR (125 MHz, CDCl3) δ 165.3, 164.9, 159.4, 137.9, 110.8, 109.2, 102.9, 79.8,
75.1, 55.9, 23.2, 8.0; IR (KBr, neat) 2960, 2924, 1687, 1612, 1419,
1231, 1120, 1027, 841, 755 cm–1; HRMS (ESI) calcd.
for C12H14IO4 (M + H)+ 348.9931, found 348.9941.
Gray solid; Rf (hexane/EtOAc,
4:1) 0.40; mp 108–110 °C; yield 159 mg, 80%; 1H NMR (500 MHz, CDCl3) δ 8.14 (dd, J = 9.5, 3.5 Hz, 1 H), 6.65 (m, 1 H), 6.48 (t, J =
2.5 Hz, 1 H), 4.52 (d, J = 13.5 Hz, 1 H), 4.35 (d, J = 13.5 Hz, 1 H), 3.84 (s, 3 H), 3.42 (d, J = 10.5, 1 H), 3.36 (d, J = 10.5, 1 H), 1.61 (s,
3 H); 13C{1H} NMR (125 MHz, CDCl3) δ 165.3, 164.9, 159.4, 137.9, 110.8, 109.2, 102.9, 79.8,
75.1, 55.9, 23.2, 8.0; IR (KBr, neat) 2960, 2924, 1687, 1612, 1419,
1231, 1120, 1027, 841, 755 cm–1; HRMS (ESI) calcd.
for C12H14IO4 (M + H)+ 348.9931, found 348.9941.
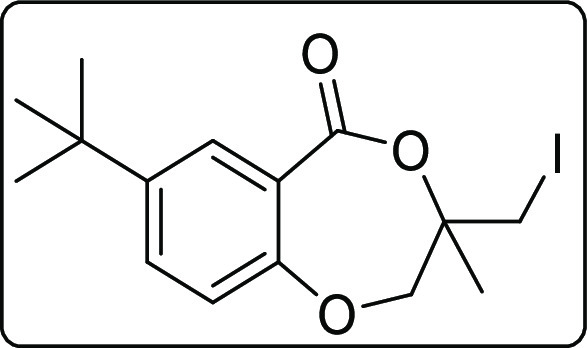
7-(tert-Butyl)-3-(iodomethyl)-3-methyl-2,3-dihydro-5H-benzo[e][1,4]dioxepin-5-one (2l)
 Brown gummy; Rf (hexane/EtOAc,
4:1) 0.55; yield 153 mg, 72%; 1H NMR (400 MHz, CDCl3) δ 8.18 (d, J = 2.4 Hz, 1 H), 7.53
(dd, J = 8.8, 2.8 Hz, 1 H), 6.99 (d, J = 8.4 Hz, 1 H), 4.51 (d, J = 13.6 Hz, 1 H), 4.35
(d, J = 13.6 Hz, 1 H), 3.43 (d, J = 10.8, 1 H), 3.37 (d, J = 10.4, 1 H), 1.62 (s,
3 H), 1.31 (s, 9 H); 13C{1H} NMR (125 MHz, CDCl3) δ 165.7, 155.4, 145.2, 133.1, 132.2, 119.7, 115.6,
80.4, 75.0, 34.6, 31.5, 23.3, 8.1; IR (KBr, neat) 2961, 2873, 1696,
1611, 1494, 1400, 1294, 1251, 1144, 832, 766 cm–1; HRMS (ESI) calcd. for C15H20IO3 (M + H)+ 375.0452, found 375.0464.
Brown gummy; Rf (hexane/EtOAc,
4:1) 0.55; yield 153 mg, 72%; 1H NMR (400 MHz, CDCl3) δ 8.18 (d, J = 2.4 Hz, 1 H), 7.53
(dd, J = 8.8, 2.8 Hz, 1 H), 6.99 (d, J = 8.4 Hz, 1 H), 4.51 (d, J = 13.6 Hz, 1 H), 4.35
(d, J = 13.6 Hz, 1 H), 3.43 (d, J = 10.8, 1 H), 3.37 (d, J = 10.4, 1 H), 1.62 (s,
3 H), 1.31 (s, 9 H); 13C{1H} NMR (125 MHz, CDCl3) δ 165.7, 155.4, 145.2, 133.1, 132.2, 119.7, 115.6,
80.4, 75.0, 34.6, 31.5, 23.3, 8.1; IR (KBr, neat) 2961, 2873, 1696,
1611, 1494, 1400, 1294, 1251, 1144, 832, 766 cm–1; HRMS (ESI) calcd. for C15H20IO3 (M + H)+ 375.0452, found 375.0464.
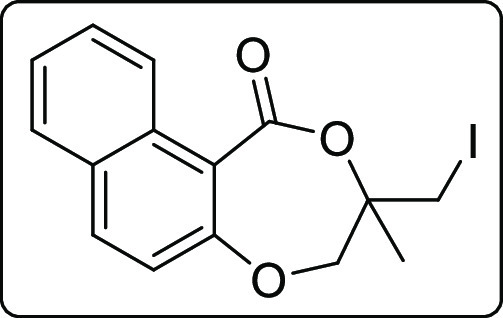
3-(Iodomethyl)-3-methyl-3,4-dihydro-1H-naphtho[2,1-e][1,4]dioxepin-1-one (2m)
 Brown gummy; Rf (hexane/EtOAc,
4:1) 0.55; yield 96 mg, 46%; 1H NMR (400 MHz, CDCl3) δ 8.50 (d, J = 8.8 Hz, 1 H), 7.89
(d, J = 8.8 Hz, 1 H), 7.78 (d, J = 8.0 Hz, 1 H), 7.58 (t, J = 7.6 Hz, 1 H), 7.45
(t, J = 7.6 Hz, 1 H), 7.15 (d, J = 8.8 Hz, 1 H), 4.63 (d, J = 13.2 Hz, 1 H), 4.55
(d, J = 13.6 Hz, 1 H), 3.48 (d, J = 10.8 Hz, 1 H), 3.41 (d, J = 10.8 Hz, 1 H), 1.64
(s, 3 H); 13C{1H} NMR (125 MHz, CDCl3) δ 166.2, 156.5, 135.1, 132.8, 130.1, 128.8, 128.5, 125.9,
125.4, 119.8, 113.2, 81.4, 75.6, 23.7, 8.9; IR (KBr, neat) 2919, 2851,
1710, 1472, 1343, 1275, 1236, 827, 750 cm–1; HRMS
(ESI) calcd. for C15H14IO3 (M + H)+ 368.9982, found 368.9982.
Brown gummy; Rf (hexane/EtOAc,
4:1) 0.55; yield 96 mg, 46%; 1H NMR (400 MHz, CDCl3) δ 8.50 (d, J = 8.8 Hz, 1 H), 7.89
(d, J = 8.8 Hz, 1 H), 7.78 (d, J = 8.0 Hz, 1 H), 7.58 (t, J = 7.6 Hz, 1 H), 7.45
(t, J = 7.6 Hz, 1 H), 7.15 (d, J = 8.8 Hz, 1 H), 4.63 (d, J = 13.2 Hz, 1 H), 4.55
(d, J = 13.6 Hz, 1 H), 3.48 (d, J = 10.8 Hz, 1 H), 3.41 (d, J = 10.8 Hz, 1 H), 1.64
(s, 3 H); 13C{1H} NMR (125 MHz, CDCl3) δ 166.2, 156.5, 135.1, 132.8, 130.1, 128.8, 128.5, 125.9,
125.4, 119.8, 113.2, 81.4, 75.6, 23.7, 8.9; IR (KBr, neat) 2919, 2851,
1710, 1472, 1343, 1275, 1236, 827, 750 cm–1; HRMS
(ESI) calcd. for C15H14IO3 (M + H)+ 368.9982, found 368.9982.
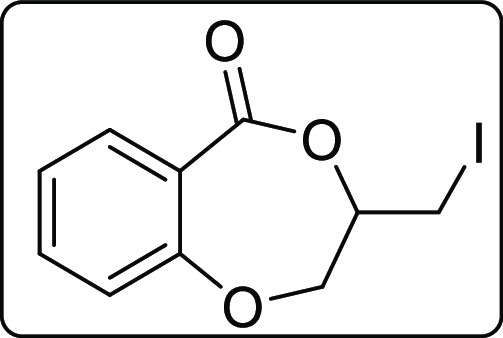
3-(Iodomethyl)-2,3-dihydro-5H-benzo[e][1,4]dioxepin-5-one (2n)
 Brown Solid; Rf (hexane/EtOAc,
4:1) 0.55; mp 90–92 °C; yield 113 mg, 65%; 1H NMR (400 MHz, CDCl3) δ 7.94–7.89 (m, 1
H), 7.52–7.48 (m, 1 H), 7.16–7.12 (m, 1 H), 7.05–7.01
(m, 1 H), 4.65–4.62 (m, 1 H), 4.59–4.54 (m, 1 H), 4.48–4.41
(m, 1 H), 3.41–3.36 (m, 1 H), 3.33–3.29 (m, 1 H), 13C{1H} NMR (125 MHz, CDCl3) δ
167.0, 155.5, 135.3, 134.0, 123.3, 121.1, 119.4, 75.7, 74.3, −0.3;
IR (KBr, neat) 2928, 1716, 1603, 1480, 1444, 1293, 1217, 1115, 751,
443 cm–1; HRMS (ESI) calcd. for C10H10IO3 (M + H)+ 304.9669, found 304.9667.
Brown Solid; Rf (hexane/EtOAc,
4:1) 0.55; mp 90–92 °C; yield 113 mg, 65%; 1H NMR (400 MHz, CDCl3) δ 7.94–7.89 (m, 1
H), 7.52–7.48 (m, 1 H), 7.16–7.12 (m, 1 H), 7.05–7.01
(m, 1 H), 4.65–4.62 (m, 1 H), 4.59–4.54 (m, 1 H), 4.48–4.41
(m, 1 H), 3.41–3.36 (m, 1 H), 3.33–3.29 (m, 1 H), 13C{1H} NMR (125 MHz, CDCl3) δ
167.0, 155.5, 135.3, 134.0, 123.3, 121.1, 119.4, 75.7, 74.3, −0.3;
IR (KBr, neat) 2928, 1716, 1603, 1480, 1444, 1293, 1217, 1115, 751,
443 cm–1; HRMS (ESI) calcd. for C10H10IO3 (M + H)+ 304.9669, found 304.9667.
3-(Iodomethyl)-3-phenyl-2,3-dihydro-5H-benzo[e][1,4]dioxepin-5-one (2o)
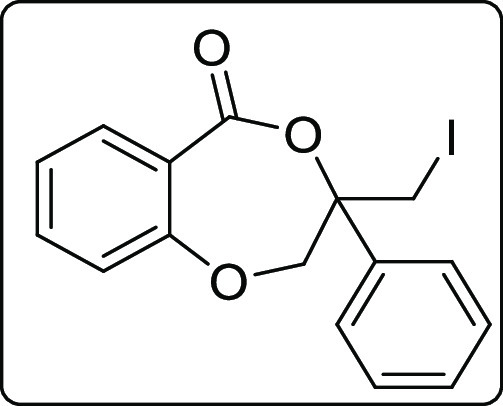 Black solid; Rf (hexane/EtOAc,
4:1) 0.55; mp 90–92 °C; yield 117 mg, 54%; 1H NMR (500 MHz, CDCl3) δ 8.09 (d, J = 7.5 Hz, 1 H), 7.40 (d, J = 7.5 Hz, 2 H), 7.33–7.27
(m, 4 H), 6.95 (t, J = 7.5 Hz, 1 H), 6.79 (d, J = 8.0 Hz, 1 H), 4.92 (d, J = 14.0 Hz,
1 H), 4.79 (d, J = 14.0 Hz, 1 H), 3.60 (d, J = 11.0 Hz, 1 H), 3.56 (d, J = 11.0 Hz,
1 H); 13C{1H} NMR (125 MHz, CDCl3) δ 166.0, 156.5, 137.1, 135.5, 135.1, 128.9, 126.0, 121.9,
119.7, 116.8, 82.2, 76.0, 12.2; IR (KBr, neat) 2969, 1735, 1277, 750,
454 cm–1; HRMS (ESI) calcd. for C16H14IO3 (M + H)+ 380.9982, found 380.9980.
Black solid; Rf (hexane/EtOAc,
4:1) 0.55; mp 90–92 °C; yield 117 mg, 54%; 1H NMR (500 MHz, CDCl3) δ 8.09 (d, J = 7.5 Hz, 1 H), 7.40 (d, J = 7.5 Hz, 2 H), 7.33–7.27
(m, 4 H), 6.95 (t, J = 7.5 Hz, 1 H), 6.79 (d, J = 8.0 Hz, 1 H), 4.92 (d, J = 14.0 Hz,
1 H), 4.79 (d, J = 14.0 Hz, 1 H), 3.60 (d, J = 11.0 Hz, 1 H), 3.56 (d, J = 11.0 Hz,
1 H); 13C{1H} NMR (125 MHz, CDCl3) δ 166.0, 156.5, 137.1, 135.5, 135.1, 128.9, 126.0, 121.9,
119.7, 116.8, 82.2, 76.0, 12.2; IR (KBr, neat) 2969, 1735, 1277, 750,
454 cm–1; HRMS (ESI) calcd. for C16H14IO3 (M + H)+ 380.9982, found 380.9980.
3-(Iodomethyl)-3-(p-tolyl)-2,3-dihydro-5H-benzo[e][1,4]dioxepin-5-one (2p)
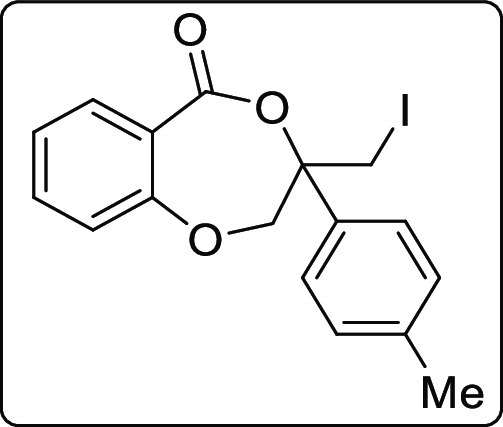 White solid; Rf (hexane/EtOAc,
4:1) 0.55; mp 80–82 °C; yield 119 mg, 53%; 1H NMR (400 MHz, CDCl3) δ 8.10 (d, J = 8.4 Hz, 1 H), 7.35–7.27 (m, 3 H), 7.11 (d, J = 8.0 Hz, 2 H), 6.96 (t, J = 7.6 Hz, 1 H), 6.80
(d, J = 8.4 Hz, 1 H), 4.89 (d, J = 13.6 Hz, 1 H), 4.77 (d, J = 14.0 Hz, 1 H), 3.58
(d, J = 11.2 Hz, 1 H), 3.53 (d, J = 11.2 Hz, 1 H), 2.28 (s, 3 H); 13C{1H} NMR
(125 MHz, CDCl3) δ 166.0, 156.5, 138.8, 135.5, 135.1,
134.1, 129.6, 125.9, 121.8, 119.8, 116.8, 82.2, 76.0, 21.3, 12.5;
IR (KBr, neat) 2955, 2919, 1700, 1290, 1119, 751, 458 cm–1; HRMS (ESI) calcd. for C17H16IO3 (M + H)+ 395.0139, found 395.0136.
White solid; Rf (hexane/EtOAc,
4:1) 0.55; mp 80–82 °C; yield 119 mg, 53%; 1H NMR (400 MHz, CDCl3) δ 8.10 (d, J = 8.4 Hz, 1 H), 7.35–7.27 (m, 3 H), 7.11 (d, J = 8.0 Hz, 2 H), 6.96 (t, J = 7.6 Hz, 1 H), 6.80
(d, J = 8.4 Hz, 1 H), 4.89 (d, J = 13.6 Hz, 1 H), 4.77 (d, J = 14.0 Hz, 1 H), 3.58
(d, J = 11.2 Hz, 1 H), 3.53 (d, J = 11.2 Hz, 1 H), 2.28 (s, 3 H); 13C{1H} NMR
(125 MHz, CDCl3) δ 166.0, 156.5, 138.8, 135.5, 135.1,
134.1, 129.6, 125.9, 121.8, 119.8, 116.8, 82.2, 76.0, 21.3, 12.5;
IR (KBr, neat) 2955, 2919, 1700, 1290, 1119, 751, 458 cm–1; HRMS (ESI) calcd. for C17H16IO3 (M + H)+ 395.0139, found 395.0136.
4-(Iodomethyl)-3,4-dihydro-2H,6H-benzo[b][1,5]dioxocin-6-one (2q)
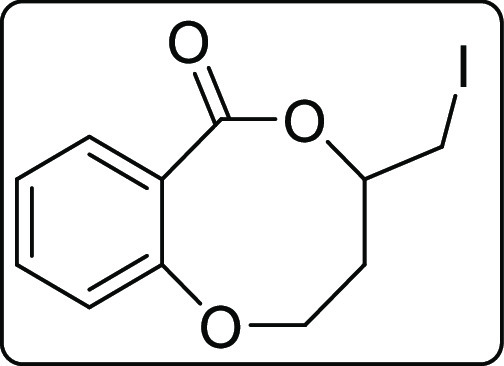 White gummy; Rf (hexane/EtOAc,
4:1) 0.55; yield 87 mg, 48%; 1H NMR (500 MHz, CDCl3) δ 7.54 (dd, J = 7.5, 1.5 Hz, 1 H),
7.42–7.39 (m, 1 H), 7.05 (t, J = 7.5 Hz, 1
H), 6.95 (d, J = 8.5 Hz, 1 H), 4.53–4.48 (m,
1 H), 4.38–4.28 (m, 2 H), 3.37–3.29 (m, 2 H), 2.47–2.40
(m, 1 H), 2.02–1.95 (m, 1 H); 13C{1H}
NMR (125 MHz, CDCl3) δ 169.9, 157.3, 133.6, 133.3,
122.2, 119.9, 116.8, 75.6, 65.6, 36.7, 5.5; IR (KBr, neat) 2957, 2923,
1709, 1603, 1485, 1441, 1289, 1122, 1089, 1054, 752 cm–1; HRMS (ESI) calcd. for C11H12IO3 (M + H)+ 318.9826, found 318.9838.
White gummy; Rf (hexane/EtOAc,
4:1) 0.55; yield 87 mg, 48%; 1H NMR (500 MHz, CDCl3) δ 7.54 (dd, J = 7.5, 1.5 Hz, 1 H),
7.42–7.39 (m, 1 H), 7.05 (t, J = 7.5 Hz, 1
H), 6.95 (d, J = 8.5 Hz, 1 H), 4.53–4.48 (m,
1 H), 4.38–4.28 (m, 2 H), 3.37–3.29 (m, 2 H), 2.47–2.40
(m, 1 H), 2.02–1.95 (m, 1 H); 13C{1H}
NMR (125 MHz, CDCl3) δ 169.9, 157.3, 133.6, 133.3,
122.2, 119.9, 116.8, 75.6, 65.6, 36.7, 5.5; IR (KBr, neat) 2957, 2923,
1709, 1603, 1485, 1441, 1289, 1122, 1089, 1054, 752 cm–1; HRMS (ESI) calcd. for C11H12IO3 (M + H)+ 318.9826, found 318.9838.
4-(Iodomethyl)-9-methoxy-3,4-dihydro-2H,6H-benzo[b][1,5]dioxocin-6-one (2r)
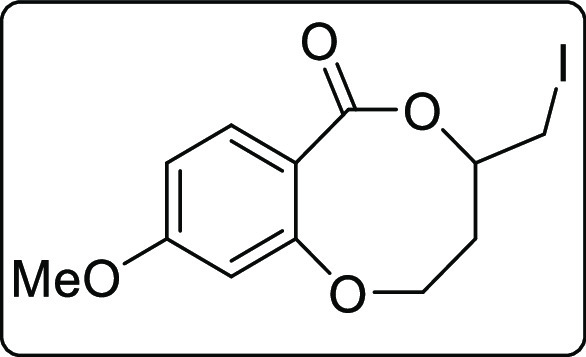 White gummy; Rf (hexane/EtOAc,
4:1) 0.50; yield 105 mg, 53%; 1H NMR (500 MHz, CDCl3) δ 7.52 (d, J = 9.5 Hz, 1 H), 6.63–6.60 (m,
1 H), 6.42 (s, 1 H), 4.52–4.50 (m, 1 H), 4.38–4.35 (m,
1 H), 4.29–4.24 (m, 1 H), 3.81 (s, 3 H), 3.38–3.28 (m,
2 H), 2.46–2.42 (m, 1 H), 1.96–1.90 (m, 1 H); 13C{1H} NMR (125 MHz, CDCl3) δ 168.7, 162.9,
158.3, 134.4, 108.7, 107.9, 102.4, 64.6, 54.8, 35.5, 4.5; IR (KBr,
neat) 2960, 2917, 1707, 1613, 1258, 1010, 1161, 789, cm–1; HRMS (ESI) calcd. for C12H14IO4 (M + H)+ 348.9931, found 348.9931.
White gummy; Rf (hexane/EtOAc,
4:1) 0.50; yield 105 mg, 53%; 1H NMR (500 MHz, CDCl3) δ 7.52 (d, J = 9.5 Hz, 1 H), 6.63–6.60 (m,
1 H), 6.42 (s, 1 H), 4.52–4.50 (m, 1 H), 4.38–4.35 (m,
1 H), 4.29–4.24 (m, 1 H), 3.81 (s, 3 H), 3.38–3.28 (m,
2 H), 2.46–2.42 (m, 1 H), 1.96–1.90 (m, 1 H); 13C{1H} NMR (125 MHz, CDCl3) δ 168.7, 162.9,
158.3, 134.4, 108.7, 107.9, 102.4, 64.6, 54.8, 35.5, 4.5; IR (KBr,
neat) 2960, 2917, 1707, 1613, 1258, 1010, 1161, 789, cm–1; HRMS (ESI) calcd. for C12H14IO4 (M + H)+ 348.9931, found 348.9931.
9-Bromo-4-(iodomethyl)-3,4-dihydro-2H,6H-benzo[b][1,5]dioxocin-6-one (2s)
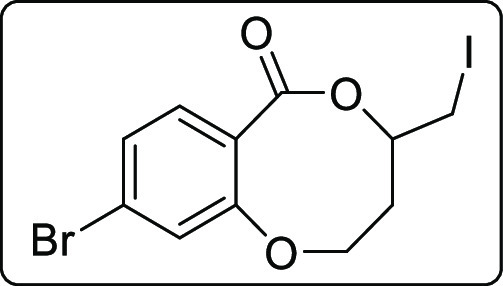 White gummy; Rf (hexane/EtOAc,
4:1) 0.55; yield 93 mg, 41%; 1H NMR (400 MHz, CDCl3) δ 7.41 (d, J = 8.4 Hz, 1 H), 7.19–7.15
(m, 2 H), 4.51–4.44 (m, 1 H), 4.41–4.36 (m, 1 H), 4.32–4.25
(m, 1 H), 3.41–3.28 (m, 2 H), 2.50–2.41(m, 1 H), 2.00–1.92
(m, 1 H); 13C{1H} NMR (125 MHz, CDCl3) δ 169.0, 157.7, 134.6, 127.5, 125.5, 122.8, 115.5, 75.7,
65.7, 36.5, 5.0; IR (KBr, neat) 2955, 2917, 1713, 1592, 1405, 1365,
1272, 1131, 1044, 932 cm–1; HRMS (ESI) calcd. for
C11H11BrIO3 (M + H)+ 396.8931,
found 396.8931.
White gummy; Rf (hexane/EtOAc,
4:1) 0.55; yield 93 mg, 41%; 1H NMR (400 MHz, CDCl3) δ 7.41 (d, J = 8.4 Hz, 1 H), 7.19–7.15
(m, 2 H), 4.51–4.44 (m, 1 H), 4.41–4.36 (m, 1 H), 4.32–4.25
(m, 1 H), 3.41–3.28 (m, 2 H), 2.50–2.41(m, 1 H), 2.00–1.92
(m, 1 H); 13C{1H} NMR (125 MHz, CDCl3) δ 169.0, 157.7, 134.6, 127.5, 125.5, 122.8, 115.5, 75.7,
65.7, 36.5, 5.0; IR (KBr, neat) 2955, 2917, 1713, 1592, 1405, 1365,
1272, 1131, 1044, 932 cm–1; HRMS (ESI) calcd. for
C11H11BrIO3 (M + H)+ 396.8931,
found 396.8931.
General Experimental Procedure for the Synthesis of 2t–2z
 To a stirred solution of 1 (0.30 mmol, 1.0
equiv) and CuI (0.36 mmol, 1.2 equiv) in CH3CN (4 mL) was
added 70% aq. solution of TBHP (1.8 mmol, 6.0 equiv) dropwise at room
temperature. Then, the reaction mixture was allowed to stir at 70
°C, and the reaction time was monitored by TLC. After completion
of the reaction, the reaction mixture was brought to room temperature.
The solvent was removed under vacuo in a rotary evaporator, extracted
with ethyl acetate, and washed with aqueous solution of Na2S2O3, NH4Cl, and saturated brine
solution. The combined organic extracts were dried over Na2SO4 and concentrated in a rotary evaporator. The crude
was subjected to column chromatography over silica gel to give the
corresponding product 2t–2z.
To a stirred solution of 1 (0.30 mmol, 1.0
equiv) and CuI (0.36 mmol, 1.2 equiv) in CH3CN (4 mL) was
added 70% aq. solution of TBHP (1.8 mmol, 6.0 equiv) dropwise at room
temperature. Then, the reaction mixture was allowed to stir at 70
°C, and the reaction time was monitored by TLC. After completion
of the reaction, the reaction mixture was brought to room temperature.
The solvent was removed under vacuo in a rotary evaporator, extracted
with ethyl acetate, and washed with aqueous solution of Na2S2O3, NH4Cl, and saturated brine
solution. The combined organic extracts were dried over Na2SO4 and concentrated in a rotary evaporator. The crude
was subjected to column chromatography over silica gel to give the
corresponding product 2t–2z.
3-(Iodomethyl)-3-methyl-1-tosyl-2,3-dihydrobenzo[e][1,4]oxazepin-5(1H)-one (2t)
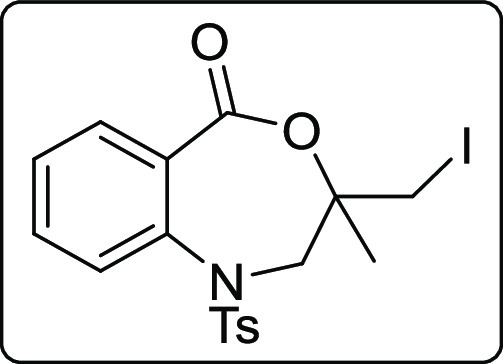 Brown solid; Rf (hexane/EtOAc,
3:1) 0.50; mp 153–155 °C; yield 96 mg, 68%; 1H NMR (400 MHz, CDCl3) δ 7.72–7.61 (m, 3
H), 7.52–7.47 (m, 3 H), 7.26 (t, J = 7.6 Hz,
2 H), 4.35 (d, J = 14.8 Hz, 1 H), 4.16 (d, J = 14.8 Hz, 1 H), 3.20 (d, J = 10.8 Hz,
1 H), 3.08 (d, J = 10.8 Hz, 1 H), 2.43 (s, 3 H),
1.37 (s, 3 H); 13C{1H} NMR (125 MHz, CDCl3) δ 165.8, 144.8, 136.0, 135.5, 134.3, 131.9, 131.8,
131.5, 130.1, 129.9, 127.7, 78.5, 57.4, 27.6, 21.9, 13.9; IR (KBr,
neat) 2955, 2922, 1727, 1595, 1455, 1348, 1291, 1161, 1085, 712, 660,
575, 545 cm–1; HRMS (ESI) calcd. for C18H19INO4S (M + H)+ 472.0074, found
472.0076.
Brown solid; Rf (hexane/EtOAc,
3:1) 0.50; mp 153–155 °C; yield 96 mg, 68%; 1H NMR (400 MHz, CDCl3) δ 7.72–7.61 (m, 3
H), 7.52–7.47 (m, 3 H), 7.26 (t, J = 7.6 Hz,
2 H), 4.35 (d, J = 14.8 Hz, 1 H), 4.16 (d, J = 14.8 Hz, 1 H), 3.20 (d, J = 10.8 Hz,
1 H), 3.08 (d, J = 10.8 Hz, 1 H), 2.43 (s, 3 H),
1.37 (s, 3 H); 13C{1H} NMR (125 MHz, CDCl3) δ 165.8, 144.8, 136.0, 135.5, 134.3, 131.9, 131.8,
131.5, 130.1, 129.9, 127.7, 78.5, 57.4, 27.6, 21.9, 13.9; IR (KBr,
neat) 2955, 2922, 1727, 1595, 1455, 1348, 1291, 1161, 1085, 712, 660,
575, 545 cm–1; HRMS (ESI) calcd. for C18H19INO4S (M + H)+ 472.0074, found
472.0076.
7-Chloro-3-(iodomethyl)-3-methyl-1-tosyl-2,3-dihydrobenzo[e][1,4]oxazepin-5(1H)-one (2u)
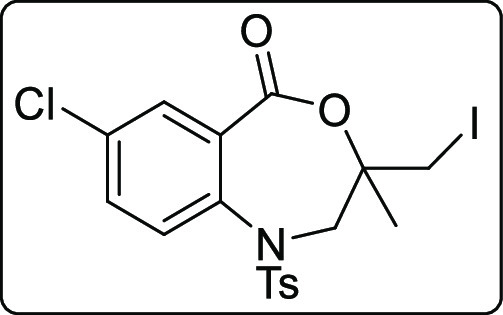 Pale yellow solid; Rf (hexane/EtOAc,
3:1) 0.50; mp 135–137 °C; yield 83 mg, 55%; 1H NMR (500 MHz, CDCl3) δ 7.68 (d, J = 2.5 Hz, 1 H), 7.62–7.55 (m, 2 H), 7.47 (d, J = 8.0 Hz, 2 H), 7.26 (t, J = 3.5 Hz 2 H), 4.36
(d, J = 14.5 Hz, 1 H), 4.11 (d, J = 14.5 Hz, 1 H), 3.22 (d, J = 11.0 Hz, 1 H), 3.12
(d, J = 11.0 Hz, 1 H), 2.42 (s, 3 H), 1.38 (s, 3
H); 13C{1H} NMR (125 MHz, CDCl3)
δ 164.4, 145.0, 136.0, 135.2, 134.5, 134.3, 133.1, 132.8, 131.6,
130.2, 127.7, 78.8, 57.4, 27.6, 21.9, 13.7; IR (KBr, neat) 2956, 2922,
1725, 1596, 1478, 1350, 1161, 1087, 707, 660, 589, 546 cm–1; HRMS (ESI) calcd. for C18H18ClINO4S (M + H)+ 505.9684, found 505.9689.
Pale yellow solid; Rf (hexane/EtOAc,
3:1) 0.50; mp 135–137 °C; yield 83 mg, 55%; 1H NMR (500 MHz, CDCl3) δ 7.68 (d, J = 2.5 Hz, 1 H), 7.62–7.55 (m, 2 H), 7.47 (d, J = 8.0 Hz, 2 H), 7.26 (t, J = 3.5 Hz 2 H), 4.36
(d, J = 14.5 Hz, 1 H), 4.11 (d, J = 14.5 Hz, 1 H), 3.22 (d, J = 11.0 Hz, 1 H), 3.12
(d, J = 11.0 Hz, 1 H), 2.42 (s, 3 H), 1.38 (s, 3
H); 13C{1H} NMR (125 MHz, CDCl3)
δ 164.4, 145.0, 136.0, 135.2, 134.5, 134.3, 133.1, 132.8, 131.6,
130.2, 127.7, 78.8, 57.4, 27.6, 21.9, 13.7; IR (KBr, neat) 2956, 2922,
1725, 1596, 1478, 1350, 1161, 1087, 707, 660, 589, 546 cm–1; HRMS (ESI) calcd. for C18H18ClINO4S (M + H)+ 505.9684, found 505.9689.
7-Bromo-3-(iodomethyl)-3-methyl-1-tosyl-2,3-dihydrobenzo[e][1,4]oxazepin-5(1H)-one (2v)
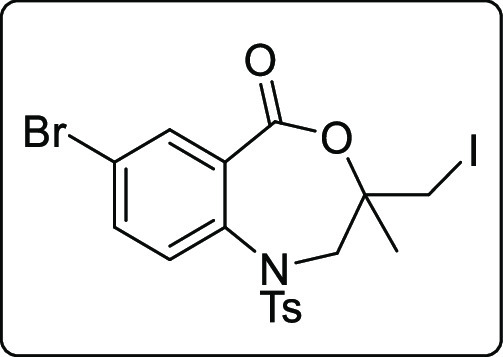 Brown solid; Rf (Hexane/EtOAc,
3:1) 0.50; mp 153–155 °C; yield 99 mg, 60%; 1H NMR (500 MHz, CDCl3) δ 7.82 (d, J = 2.5 Hz, 1H), 7.75 (dd, J = 8.5, 2.5 Hz, 1 H),
7.47 (t, J = 9.0 Hz, 3 H), 7.24 (d, J = 8.0 Hz, 2 H), 4.35 (d, J = 14.5 Hz, 1 H), 4.09
(d, J = 14.5 Hz, 1 H), 3.21 (d, J = 11.0 Hz, 1 H), 3.11 (d, J = 11.0 Hz, 1 H), 2.41
(s, 3 H), 1.37 (s, 3 H); 13C{1H} NMR (125 MHz,
CDCl3) δ 164.3, 145.0, 137.3, 135.1, 135.0, 134.5,
133.2, 132.9, 130.2, 127.7, 123.7, 78.8, 57.3, 27.6, 21.9, 13.7; IR
(KBr, neat) 2956, 2917, 1729, 1456, 1401, 1352, 1162, 1085, 737, 663,
647, 588 cm–1; HRMS (ESI) calcd. for C18H18BrINO4S (M + H)+ 549.9179, found
549.9180.
Brown solid; Rf (Hexane/EtOAc,
3:1) 0.50; mp 153–155 °C; yield 99 mg, 60%; 1H NMR (500 MHz, CDCl3) δ 7.82 (d, J = 2.5 Hz, 1H), 7.75 (dd, J = 8.5, 2.5 Hz, 1 H),
7.47 (t, J = 9.0 Hz, 3 H), 7.24 (d, J = 8.0 Hz, 2 H), 4.35 (d, J = 14.5 Hz, 1 H), 4.09
(d, J = 14.5 Hz, 1 H), 3.21 (d, J = 11.0 Hz, 1 H), 3.11 (d, J = 11.0 Hz, 1 H), 2.41
(s, 3 H), 1.37 (s, 3 H); 13C{1H} NMR (125 MHz,
CDCl3) δ 164.3, 145.0, 137.3, 135.1, 135.0, 134.5,
133.2, 132.9, 130.2, 127.7, 123.7, 78.8, 57.3, 27.6, 21.9, 13.7; IR
(KBr, neat) 2956, 2917, 1729, 1456, 1401, 1352, 1162, 1085, 737, 663,
647, 588 cm–1; HRMS (ESI) calcd. for C18H18BrINO4S (M + H)+ 549.9179, found
549.9180.
3-(Iodomethyl)-3,7-dimethyl-1-tosyl-2,3-dihydrobenzo[e][1,4]oxazepin-5(1H)-one (2w)
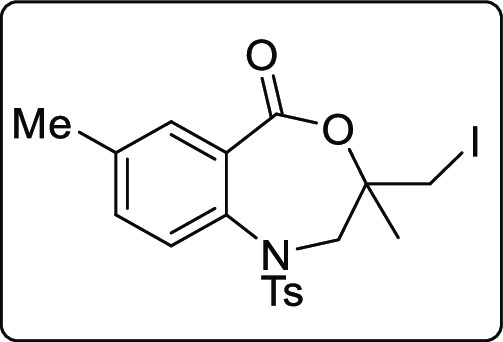 Brown solid; Rf (hexane/EtOAc,
3:1) 0.50; mp 140–142 °C; yield 93 mg, 64%; 1H NMR (500 MHz, CDCl3) δ 7.48–7.43 (s, 1
H), 7.45 (m, 4 H), 7.22 (d, J = 8.0 Hz, 2 H), 4.31
(d, J = 14.5 Hz, 1 H), 4.12 (d, J = 14.5 Hz, 1 H), 3.17 (d, J = 11.0 Hz, 1 H), 3.07
(d, J = 11.0 Hz, 1 H), 2.40 (s, 6 H), 1.34 (s, 3
H); 13C{1H} NMR (125 MHz, CDCl3)
δ 166.0, 144.6, 140.4, 135.5, 135.1, 133.2, 132.0, 131.7, 131.4,
130.1, 127.7, 78.5, 57.3, 27.7, 21.9, 21.2, 14.0; IR (KBr, neat) 2924,
1725, 1493, 1349, 1161, 1089, 814, 713, 662, 598, 547 cm–1; HRMS (ESI) calcd. for C19H21INO4S (M + H)+ 486.0230, found 486.0234.
Brown solid; Rf (hexane/EtOAc,
3:1) 0.50; mp 140–142 °C; yield 93 mg, 64%; 1H NMR (500 MHz, CDCl3) δ 7.48–7.43 (s, 1
H), 7.45 (m, 4 H), 7.22 (d, J = 8.0 Hz, 2 H), 4.31
(d, J = 14.5 Hz, 1 H), 4.12 (d, J = 14.5 Hz, 1 H), 3.17 (d, J = 11.0 Hz, 1 H), 3.07
(d, J = 11.0 Hz, 1 H), 2.40 (s, 6 H), 1.34 (s, 3
H); 13C{1H} NMR (125 MHz, CDCl3)
δ 166.0, 144.6, 140.4, 135.5, 135.1, 133.2, 132.0, 131.7, 131.4,
130.1, 127.7, 78.5, 57.3, 27.7, 21.9, 21.2, 14.0; IR (KBr, neat) 2924,
1725, 1493, 1349, 1161, 1089, 814, 713, 662, 598, 547 cm–1; HRMS (ESI) calcd. for C19H21INO4S (M + H)+ 486.0230, found 486.0234.
3-(Iodomethyl)-3,8-dimethyl-1-tosyl-2,3-dihydrobenzo[e][1,4]oxazepin-5(1H)-one (2x)
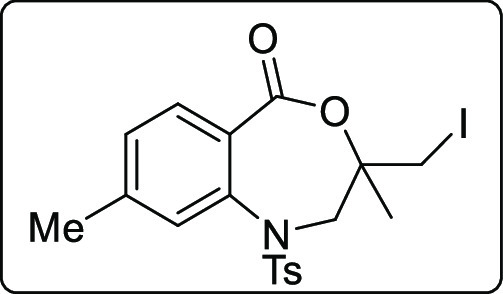 White solid; Rf (hexane/EtOAc,
3:1) 0.50; mp 133–135 °C; yield 95 mg, 65%; 1H NMR (400 MHz, CDCl3) δ 7.58 (d, J = 8.0 Hz, 1 H), 7.46 (d, J = 8.4 Hz, 2 H), 7.42
(s, 1 H), 7.28 (d, J = 8.0 Hz, 1 H), 7.23 (d, J = 8.0 Hz, 2 H), 4.30 (d, J = 14.8 Hz,
1 H), 4.13 (d, J = 14.4 Hz, 1 H), 3.16 (d, J = 11.2 Hz, 1 H), 3.06 (d, J = 11.2 Hz,
1 H), 2.46 (s, 3 H), 2.41 (s, 3 H), 1.34 (s, 3 H); 13C{1H} NMR (125 MHz, CDCl3) δ 165.9, 145.6, 144.7,
135.9, 135.5, 132.1, 131.7, 130.7, 130.1, 128.9, 127.7, 78.4, 57.4,
27.7, 21.9, 21.9, 14.0; IR (KBr, neat) 2917, 1728, 1374, 1349, 1162,
1090, 800, 707, 661, 577 cm–1; HRMS (ESI) calcd.
for C19H21INO4S (M + H)+ 486.0230, found 486.0256.
White solid; Rf (hexane/EtOAc,
3:1) 0.50; mp 133–135 °C; yield 95 mg, 65%; 1H NMR (400 MHz, CDCl3) δ 7.58 (d, J = 8.0 Hz, 1 H), 7.46 (d, J = 8.4 Hz, 2 H), 7.42
(s, 1 H), 7.28 (d, J = 8.0 Hz, 1 H), 7.23 (d, J = 8.0 Hz, 2 H), 4.30 (d, J = 14.8 Hz,
1 H), 4.13 (d, J = 14.4 Hz, 1 H), 3.16 (d, J = 11.2 Hz, 1 H), 3.06 (d, J = 11.2 Hz,
1 H), 2.46 (s, 3 H), 2.41 (s, 3 H), 1.34 (s, 3 H); 13C{1H} NMR (125 MHz, CDCl3) δ 165.9, 145.6, 144.7,
135.9, 135.5, 132.1, 131.7, 130.7, 130.1, 128.9, 127.7, 78.4, 57.4,
27.7, 21.9, 21.9, 14.0; IR (KBr, neat) 2917, 1728, 1374, 1349, 1162,
1090, 800, 707, 661, 577 cm–1; HRMS (ESI) calcd.
for C19H21INO4S (M + H)+ 486.0230, found 486.0256.
3-(Iodomethyl)-1-tosyl-2,3-dihydrobenzo[e][1,4]oxazepin-5(1H)-one (2y)
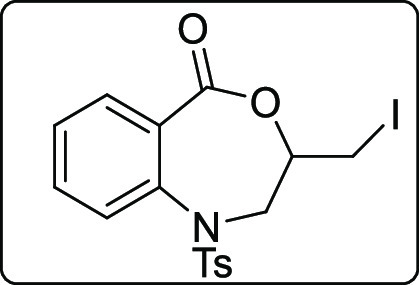 Pale yellow solid; Rf (hexane/EtOAc,3:1)
0.50; mp 163–165 °C; yield 102 mg, 62%; 1H
NMR (500 MHz, CDCl3) δ 7.69–7.61 (m, 3 H),
7.51–7.48 (m, 1 H), 7.41 (d, J = 8.0 Hz, 2
H), 7.23 (d, J = 8.5 Hz, 2 H), 4.31 (q, J = 11.5 Hz, 1 H), 4.10–4.06 (m, 1 H), 3.94 (dd, J = 13.5, 3.5 Hz, 1 H), 3.27 (d, J = 5.5 Hz, 2 H),
2.41 (s, 3 H); 13C{1H} NMR (125 MHz, CDCl3) δ 166.5, 144.8, 135.0, 134.9, 134.2, 132.5, 131.7,
130.2, 129.8, 127.6, 75.3, 55.1, 21.9, 0.5; IR (KBr, neat) 2958, 2928,
1738, 1595, 1455, 1351, 1296, 1161, 1115, 926, 750, 663, 580, 545
cm–1; HRMS (ESI) calcd. for C17H17INO4S (M + H)+ 547.9917, found 547.9917.
Pale yellow solid; Rf (hexane/EtOAc,3:1)
0.50; mp 163–165 °C; yield 102 mg, 62%; 1H
NMR (500 MHz, CDCl3) δ 7.69–7.61 (m, 3 H),
7.51–7.48 (m, 1 H), 7.41 (d, J = 8.0 Hz, 2
H), 7.23 (d, J = 8.5 Hz, 2 H), 4.31 (q, J = 11.5 Hz, 1 H), 4.10–4.06 (m, 1 H), 3.94 (dd, J = 13.5, 3.5 Hz, 1 H), 3.27 (d, J = 5.5 Hz, 2 H),
2.41 (s, 3 H); 13C{1H} NMR (125 MHz, CDCl3) δ 166.5, 144.8, 135.0, 134.9, 134.2, 132.5, 131.7,
130.2, 129.8, 127.6, 75.3, 55.1, 21.9, 0.5; IR (KBr, neat) 2958, 2928,
1738, 1595, 1455, 1351, 1296, 1161, 1115, 926, 750, 663, 580, 545
cm–1; HRMS (ESI) calcd. for C17H17INO4S (M + H)+ 547.9917, found 547.9917.
4-(Iodomethyl)-1-tosyl-1,2,3,4-tetrahydro-6H-benzo[c][1,5]oxazocin-6-one (2z)
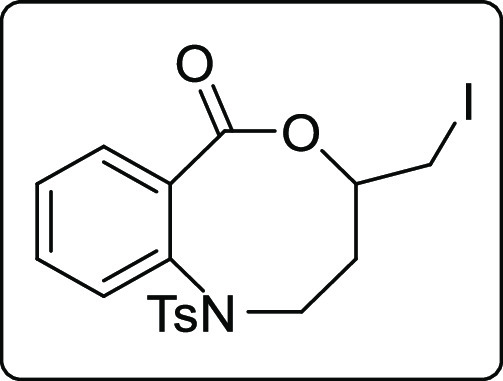 White solid; Rf (hexane/EtOAc,
3:2) 0.50; mp 163–165 °C; yield 85 mg, 60%; 1H NMR (500 MHz, CDCl3) δ 7.63–7.61 (m, 1
H), 7.57 (d, J = 8.0 Hz, 2 H), 7.48–7.46 (m,
2 H), 7.27 (s, 1 H), 7.25 (s, 1 H), 7.01–6.99 (m, 1 H), 4.39–4.35
(m, 1 H), 4.30–4.25 (m, 1 H), 3.32–3.29 (m, 1 H), 3.20–3.13
(m, 2 H), 2.41 (s, 3 H), 2.34–2.25 (m, 1 H), 1.98–1.94
(m, 1 H); 13C{1H} NMR (125 MHz, CDCl3) δ 167.8, 144.2, 138.1, 136.8, 134.2, 133.3, 130.6, 130.1,
130.0, 129.9, 127.7, 79.3, 49.7, 35.7, 21.8, 8.4.; IR (KBr, neat)
2956, 2923, 1727, 1596, 1344, 1287, 1241, 1158, 1091, 818, 684, 569
cm–1; HRMS (ESI) calcd. for C18H19INO4S (M + H)+ 472.0074, found 472.0076.
White solid; Rf (hexane/EtOAc,
3:2) 0.50; mp 163–165 °C; yield 85 mg, 60%; 1H NMR (500 MHz, CDCl3) δ 7.63–7.61 (m, 1
H), 7.57 (d, J = 8.0 Hz, 2 H), 7.48–7.46 (m,
2 H), 7.27 (s, 1 H), 7.25 (s, 1 H), 7.01–6.99 (m, 1 H), 4.39–4.35
(m, 1 H), 4.30–4.25 (m, 1 H), 3.32–3.29 (m, 1 H), 3.20–3.13
(m, 2 H), 2.41 (s, 3 H), 2.34–2.25 (m, 1 H), 1.98–1.94
(m, 1 H); 13C{1H} NMR (125 MHz, CDCl3) δ 167.8, 144.2, 138.1, 136.8, 134.2, 133.3, 130.6, 130.1,
130.0, 129.9, 127.7, 79.3, 49.7, 35.7, 21.8, 8.4.; IR (KBr, neat)
2956, 2923, 1727, 1596, 1344, 1287, 1241, 1158, 1091, 818, 684, 569
cm–1; HRMS (ESI) calcd. for C18H19INO4S (M + H)+ 472.0074, found 472.0076.
Experimental Procedure for the Synthesis of 2a′a
 To a stirred solution of 1 (0.63 mmol, 1.0
equiv) and CuI (0.76 mmol, 1.2 equiv) in CH3CN (4 mL) was
added 70% aq solution of TBHP (3.78 mmol, 6.0 equiv, 70% aq solution)
dropwise at room temperature. Then, the reaction mixture was allowed
to stir at 70 °C, and the reaction time was monitored by TLC.
After completion of the reaction (5 h), the reaction mixture was brought
to room temperature and CH3CN was evaporated in a rotary
evaporator. Then, the reaction mixture was extracted with ethyl acetate
(3 × 15 mL) and washed with saturated Na2S2O3, NH4Cl, and brine solution. The combined
organic extracts were dried over Na2SO4 and
concentrated in a rotary evaporator. The crude was subjected to column
chromatography over silica gel to give the corresponding product 2a′a in 68% yield.
To a stirred solution of 1 (0.63 mmol, 1.0
equiv) and CuI (0.76 mmol, 1.2 equiv) in CH3CN (4 mL) was
added 70% aq solution of TBHP (3.78 mmol, 6.0 equiv, 70% aq solution)
dropwise at room temperature. Then, the reaction mixture was allowed
to stir at 70 °C, and the reaction time was monitored by TLC.
After completion of the reaction (5 h), the reaction mixture was brought
to room temperature and CH3CN was evaporated in a rotary
evaporator. Then, the reaction mixture was extracted with ethyl acetate
(3 × 15 mL) and washed with saturated Na2S2O3, NH4Cl, and brine solution. The combined
organic extracts were dried over Na2SO4 and
concentrated in a rotary evaporator. The crude was subjected to column
chromatography over silica gel to give the corresponding product 2a′a in 68% yield.
3-(Iodomethyl)-4,5-dihydrobenzo[c]oxepin-1(3H)-one (2a′a)
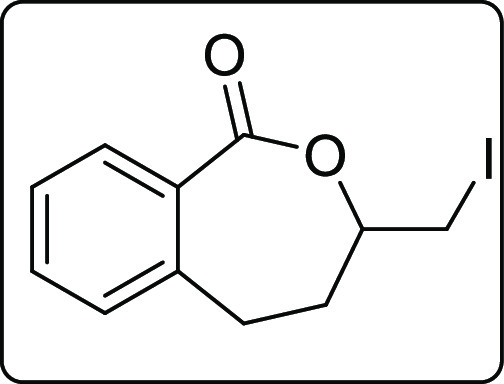 White solid; Rf (hexane/EtOAc,
9:1) 0.50; mp 86–88 °C; yield 129 mg, 68%; 1H NMR (400
MHz, CDCl3) δ 7.73 (dd, J = 7.2,
1.2 Hz, 1 H), 7.51–7.47 (m, 1 H), 7.40–7.36 (m, 1 H),
7.22 (d, J = 7.6 Hz, 1 H), 4.16–4.09 (m, 1
H), 3.39 (dd, J = 10.4, 6.0 Hz, 1 H), 3.33 (dd, J = 10.4, 6.0 Hz, 1 H), 3.06–2.98 (m, 1 H), 2.83–2.77
(m, 1 H), 2.31–2.22 (m, 1 H), 2.16–2.08 (m, 1 H); 13C{1H} NMR (125 MHz, CDCl3) δ 137.9, 133.1,
131.4, 130.5, 129.0, 127.8, 77.5, 34.2, 29.8, 5.5; IR (KBr, neat)
2953, 2924, 1719, 1453, 1293, 1257, 1085, 756, 705, 593 cm–1; HRMS (ESI) calcd. for C11H12IO2 (M + H)+ 302.9876, found 302.9891.
White solid; Rf (hexane/EtOAc,
9:1) 0.50; mp 86–88 °C; yield 129 mg, 68%; 1H NMR (400
MHz, CDCl3) δ 7.73 (dd, J = 7.2,
1.2 Hz, 1 H), 7.51–7.47 (m, 1 H), 7.40–7.36 (m, 1 H),
7.22 (d, J = 7.6 Hz, 1 H), 4.16–4.09 (m, 1
H), 3.39 (dd, J = 10.4, 6.0 Hz, 1 H), 3.33 (dd, J = 10.4, 6.0 Hz, 1 H), 3.06–2.98 (m, 1 H), 2.83–2.77
(m, 1 H), 2.31–2.22 (m, 1 H), 2.16–2.08 (m, 1 H); 13C{1H} NMR (125 MHz, CDCl3) δ 137.9, 133.1,
131.4, 130.5, 129.0, 127.8, 77.5, 34.2, 29.8, 5.5; IR (KBr, neat)
2953, 2924, 1719, 1453, 1293, 1257, 1085, 756, 705, 593 cm–1; HRMS (ESI) calcd. for C11H12IO2 (M + H)+ 302.9876, found 302.9891.
(E)-2-Styryl-4H-benzo[d][1,3]dioxin-4-one (2b′b)
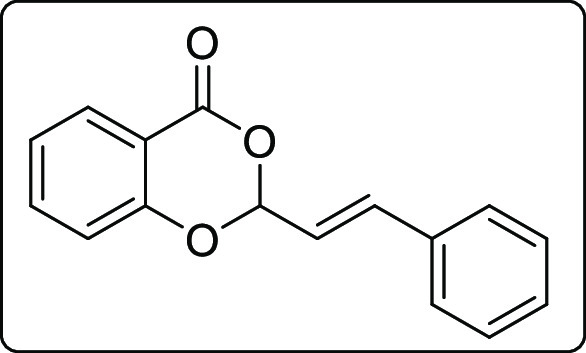 White solid; Rf (hexane/EtOAc,
4:1) 0.50; mp 82–84 °C; yield 14 mg, 10%; 1H NMR (500 MHz, CDCl3) δ 8.03 (d, J = 8.0 Hz, 1 H), 7.60 (t, J = 7.0 Hz, 1 H), 7.48
(d, J = 7.0 Hz, 2 H), 7.38 (t, J = 7.0 Hz, 2 H), 7.35 (d, J = 7.0 Hz, 1 H), 7.21
(t, J = 7.5 Hz, 1 H), 7.10 (d, J = 8.5 Hz, 1 H), 7.04 (d, J = 16.0 Hz, 1 H), 6.42
(dd, J = 16.0, 5.0 Hz, 1 H), 6.19 (d, J = 5.5 Hz, 1 H); 13C{1H} NMR (125 MHz, CDCl3) δ 162.1, 158.3, 137.1, 136.6, 135.1, 130.5, 129.4,
129.0, 127.4, 123.8, 121.2, 117.1, 114.9, 100.4; IR (KBr, neat) 2920,
1743, 1613, 1469, 1301, 1236, 954, 759, 691, 588 cm–1; HRMS (ESI) calcd. for C16H13O3 (M + H)+ 253.0859, found 253.0879.
White solid; Rf (hexane/EtOAc,
4:1) 0.50; mp 82–84 °C; yield 14 mg, 10%; 1H NMR (500 MHz, CDCl3) δ 8.03 (d, J = 8.0 Hz, 1 H), 7.60 (t, J = 7.0 Hz, 1 H), 7.48
(d, J = 7.0 Hz, 2 H), 7.38 (t, J = 7.0 Hz, 2 H), 7.35 (d, J = 7.0 Hz, 1 H), 7.21
(t, J = 7.5 Hz, 1 H), 7.10 (d, J = 8.5 Hz, 1 H), 7.04 (d, J = 16.0 Hz, 1 H), 6.42
(dd, J = 16.0, 5.0 Hz, 1 H), 6.19 (d, J = 5.5 Hz, 1 H); 13C{1H} NMR (125 MHz, CDCl3) δ 162.1, 158.3, 137.1, 136.6, 135.1, 130.5, 129.4,
129.0, 127.4, 123.8, 121.2, 117.1, 114.9, 100.4; IR (KBr, neat) 2920,
1743, 1613, 1469, 1301, 1236, 954, 759, 691, 588 cm–1; HRMS (ESI) calcd. for C16H13O3 (M + H)+ 253.0859, found 253.0879.
General Experimental Procedure for the Synthesis of 4a–4c and 4a′–4c′
 To a stirred solution of 2-(prop-2-yn-1-yloxy)benzaldehyde
derivative (0.63 mmol, 1.0 equiv) and CuI (0.76 mmol, 1.2 equiv) in
CH3CN (4 mL) was added 70% aq solution of TBHP (3.78 mmol,
6.0 equiv) dropwise at room temperature. Then, the reaction mixture
was allowed to stir at 70 °C, and the reaction time was monitored
by TLC. After completion of the reaction (6–12 h), the reaction
mixture was brought to room temperature. The solvent was removed under
vacuo in a rotary evaporator, extracted with ethyl acetate (3 ×
15 mL), and washed with Na2S2O3, NH4Cl, and saturated brine solution. The combined organic extract
was dried over Na2SO4 and concentrated in a
rotary evaporator. The crude was subjected to column chromatography
over silica gel to give the corresponding product 4 and 4′.
To a stirred solution of 2-(prop-2-yn-1-yloxy)benzaldehyde
derivative (0.63 mmol, 1.0 equiv) and CuI (0.76 mmol, 1.2 equiv) in
CH3CN (4 mL) was added 70% aq solution of TBHP (3.78 mmol,
6.0 equiv) dropwise at room temperature. Then, the reaction mixture
was allowed to stir at 70 °C, and the reaction time was monitored
by TLC. After completion of the reaction (6–12 h), the reaction
mixture was brought to room temperature. The solvent was removed under
vacuo in a rotary evaporator, extracted with ethyl acetate (3 ×
15 mL), and washed with Na2S2O3, NH4Cl, and saturated brine solution. The combined organic extract
was dried over Na2SO4 and concentrated in a
rotary evaporator. The crude was subjected to column chromatography
over silica gel to give the corresponding product 4 and 4′.
3-(Diiodomethylene)-2,3-dihydro-5H-benzo[e][1,4]dioxepin-5-one (4a)
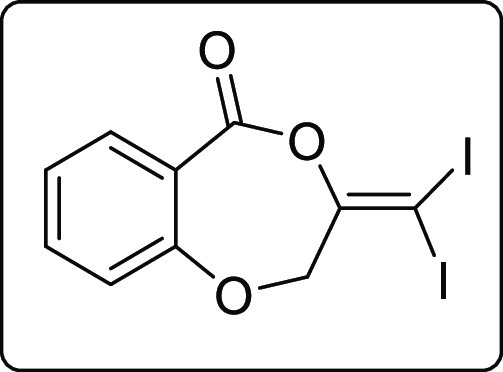 Pale yellow solid; Rf (hexane/EtOAc,
4:1) 0.55; mp 153–155 °C; yield 140 mg, 52%; 1H NMR (500 MHz, CDCl3) δ 7.92 (dd, J = 8.0, 2.0 Hz, 1 H), 7.56–7.53 (m, 1 H), 7.20–7.16
(m, 1 H), 7.09 (dd, J = 8.0, 1.0 Hz, 1 H), 5.06 (s,
2 H); 13C{1H} NMR (125 MHz, CDCl3) δ 163.4, 156.8, 150.4, 135.7, 134.1, 123.8, 121.1, 118.5,
71.2, 11.3; IR (KBr, neat) 3745, 2955, 2922, 1735, 1601, 1477, 1274,
1033, 750 cm–1; HRMS (ESI) calcd. for C10H7I2O3 (M + H)+ 428.8479,
found 428.8470.
Pale yellow solid; Rf (hexane/EtOAc,
4:1) 0.55; mp 153–155 °C; yield 140 mg, 52%; 1H NMR (500 MHz, CDCl3) δ 7.92 (dd, J = 8.0, 2.0 Hz, 1 H), 7.56–7.53 (m, 1 H), 7.20–7.16
(m, 1 H), 7.09 (dd, J = 8.0, 1.0 Hz, 1 H), 5.06 (s,
2 H); 13C{1H} NMR (125 MHz, CDCl3) δ 163.4, 156.8, 150.4, 135.7, 134.1, 123.8, 121.1, 118.5,
71.2, 11.3; IR (KBr, neat) 3745, 2955, 2922, 1735, 1601, 1477, 1274,
1033, 750 cm–1; HRMS (ESI) calcd. for C10H7I2O3 (M + H)+ 428.8479,
found 428.8470.
(Z)-3-(Iodomethylene)-2,3-dihydro-5H-benzo[e][1,4]dioxepin-5-one (4a′)
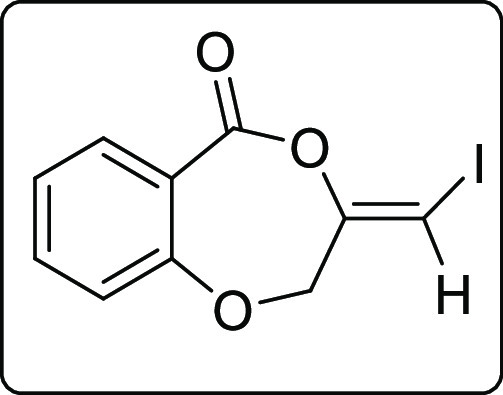 Brown solid; Rf (hexane/EtOAc,
4:1) 0.50; mp 104–106 °C; yield 76 mg, 40%; 1H NMR (500 MHz, CDCl3) δ 7.97–7.94 (m, 1
H), 7.53–7.49 (m, 1 H), 7.16–7.13 (m, 1 H), 7.03 (d, J = 8.0 Hz, 1 H), 6.14 (s, 1 H), 4.83 (s, 2 H); 13C{1H} NMR (125 MHz, CDCl3) δ 164.2, 156.7,
152.1, 135.5, 134.2, 123.4, 120.9, 118.5, 71.1, 70.3; IR (KBr, neat)
3081, 2960, 2922, 1736, 1635, 1604, 1478, 1281, 1226, 1116, 1037,
753 cm–1; HRMS (ESI) calcd. for C10H8IO3 (M + H)+ 302.9513, found 302.9509.
Brown solid; Rf (hexane/EtOAc,
4:1) 0.50; mp 104–106 °C; yield 76 mg, 40%; 1H NMR (500 MHz, CDCl3) δ 7.97–7.94 (m, 1
H), 7.53–7.49 (m, 1 H), 7.16–7.13 (m, 1 H), 7.03 (d, J = 8.0 Hz, 1 H), 6.14 (s, 1 H), 4.83 (s, 2 H); 13C{1H} NMR (125 MHz, CDCl3) δ 164.2, 156.7,
152.1, 135.5, 134.2, 123.4, 120.9, 118.5, 71.1, 70.3; IR (KBr, neat)
3081, 2960, 2922, 1736, 1635, 1604, 1478, 1281, 1226, 1116, 1037,
753 cm–1; HRMS (ESI) calcd. for C10H8IO3 (M + H)+ 302.9513, found 302.9509.
7-Chloro-3-(diiodomethylene)-2,3-dihydro-5H-benzo[e][1,4]dioxepin-5-one (4b)
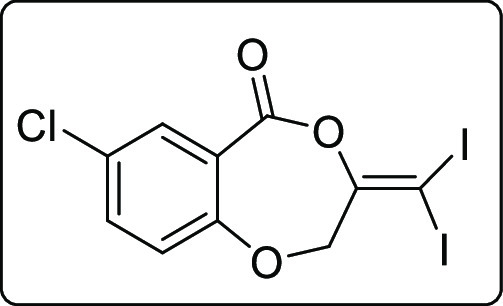 Pale yellow solid; Rf (hexane/EtOAc,
4:1) 0.55; mp 208–210 °C; yield 146 mg, 50%; 1H NMR (400 MHz, CDCl3) δ 7.89 (s, 1 H), 7.48 (d, J = 7.6 Hz, 1 H), 7.04 (d, J = 8.8 Hz,
1 H), 5.05 (s, 2 H); 13C{1H} NMR (125 MHz, CDCl3) δ 162.2, 155.3, 149.9, 135.7, 133.2, 129.1, 122.7,
119.6, 71.3, 12.5; IR (KBr, neat) 3745, 2960, 2919, 1714, 1606, 1471,
1397, 1266, 1164, 1130, 1024, 763, 750 cm–1; HRMS
(ESI) calcd. for C10H6ClI2O3 (M + H)+ 462.8089, found 462.8086.
Pale yellow solid; Rf (hexane/EtOAc,
4:1) 0.55; mp 208–210 °C; yield 146 mg, 50%; 1H NMR (400 MHz, CDCl3) δ 7.89 (s, 1 H), 7.48 (d, J = 7.6 Hz, 1 H), 7.04 (d, J = 8.8 Hz,
1 H), 5.05 (s, 2 H); 13C{1H} NMR (125 MHz, CDCl3) δ 162.2, 155.3, 149.9, 135.7, 133.2, 129.1, 122.7,
119.6, 71.3, 12.5; IR (KBr, neat) 3745, 2960, 2919, 1714, 1606, 1471,
1397, 1266, 1164, 1130, 1024, 763, 750 cm–1; HRMS
(ESI) calcd. for C10H6ClI2O3 (M + H)+ 462.8089, found 462.8086.
(Z)-7-Chloro-3-(iodomethylene)-2,3-dihydro-5H-benzo[e][1,4]dioxepin-5-one (4b′)
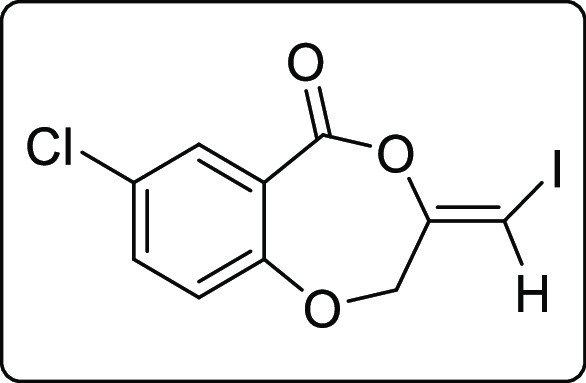 Brown gummy; Rf (hexane/EtOAc,
4:1) 0.50; yield 74 mg, 35%; 1H NMR (400 MHz, CDCl3) δ 7.88 (d, J = 2.8 Hz, 1 H), 7.47
(dd, J = 8.8, 2.4 Hz, 1 H), 7.04 (d, J = 8.8 Hz, 1 H), 6.51 (s, 1 H), 5.01 (s, 2 H); 13C{1H} NMR (125 MHz, CDCl3) δ 163.3, 155.4, 148.9,
135.5, 133.3, 128.9, 122.8, 120.2, 72.2, 71.6; IR (KBr, neat) 3073,
2955, 2920, 1729, 1633, 1602, 1474, 1398, 1269, 1222, 1130, 1026,
826 cm–1; HRMS (ESI) calcd. for C10H7ClIO3 (M + H)+ 336.9123, found 336.9120.
Brown gummy; Rf (hexane/EtOAc,
4:1) 0.50; yield 74 mg, 35%; 1H NMR (400 MHz, CDCl3) δ 7.88 (d, J = 2.8 Hz, 1 H), 7.47
(dd, J = 8.8, 2.4 Hz, 1 H), 7.04 (d, J = 8.8 Hz, 1 H), 6.51 (s, 1 H), 5.01 (s, 2 H); 13C{1H} NMR (125 MHz, CDCl3) δ 163.3, 155.4, 148.9,
135.5, 133.3, 128.9, 122.8, 120.2, 72.2, 71.6; IR (KBr, neat) 3073,
2955, 2920, 1729, 1633, 1602, 1474, 1398, 1269, 1222, 1130, 1026,
826 cm–1; HRMS (ESI) calcd. for C10H7ClIO3 (M + H)+ 336.9123, found 336.9120.
3-(Diiodomethylene)-8-methoxy-2,3-dihydro-5H-benzo[e][1,4]dioxepin-5-one (4c)
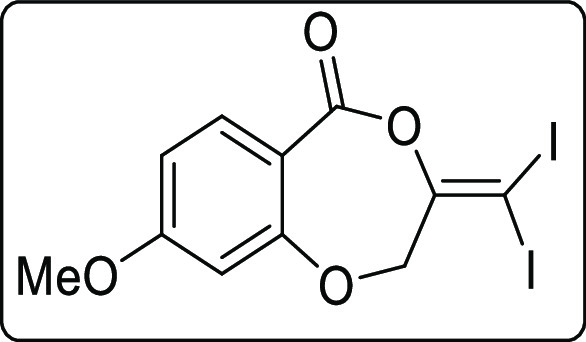 White solid; Rf (hexane/EtOAc,
3:1) 0.55; mp 148–150 °C; yield 164 mg, 57%; 1H NMR (500 MHz, CDCl3) δ 7.91 (d, J = 9.0 Hz, 1 H), 6.70 (dd, J = 9.0, 2.5 Hz, 1 H),
6.51 (d, J = 2.5 Hz, 1 H), 5.04 (s, 2 H), 3.85 (s,
3 H); 13C{1H} NMR (125 MHz, CDCl3) δ 165.6, 162.8, 159.1, 150.5, 136.4, 111.3, 109.5, 104.0,
70.3, 56.0, 10.6; IR (KBr, neat) 2955, 2914, 1724, 1609, 1450, 1208,
1115, 1030 cm–1; HRMS (ESI) calcd. for C11H9I2O4 (M + H)+ 458.8585,
found 458.8583.
White solid; Rf (hexane/EtOAc,
3:1) 0.55; mp 148–150 °C; yield 164 mg, 57%; 1H NMR (500 MHz, CDCl3) δ 7.91 (d, J = 9.0 Hz, 1 H), 6.70 (dd, J = 9.0, 2.5 Hz, 1 H),
6.51 (d, J = 2.5 Hz, 1 H), 5.04 (s, 2 H), 3.85 (s,
3 H); 13C{1H} NMR (125 MHz, CDCl3) δ 165.6, 162.8, 159.1, 150.5, 136.4, 111.3, 109.5, 104.0,
70.3, 56.0, 10.6; IR (KBr, neat) 2955, 2914, 1724, 1609, 1450, 1208,
1115, 1030 cm–1; HRMS (ESI) calcd. for C11H9I2O4 (M + H)+ 458.8585,
found 458.8583.
(Z)-3-(Iodomethylene)-8-methoxy-2,3-dihydro-5H-benzo[e][1,4]dioxepin-5-one (4c′)
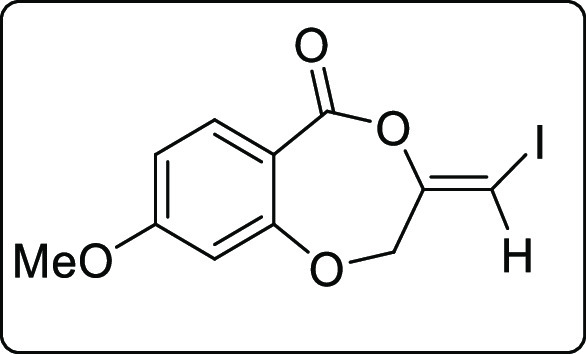 Pale yellow solid; Rf (hexane/EtOAc,
3:1) 0.50; mp 135–137 °C; yield 84 mg, 40%; 1H NMR (400 MHz, CDCl3) δ 7.97 (d, J = 8.8 Hz, 1 H), 6.68 (dd, J = 8.8, 2.4 Hz, 1 H),
6.46 (d, J = 2.4 Hz, 1 H), 6.10 (s, 1 H), 4.79 (s,
2 H), 3.83 (s, 3 H); 13C{1H} NMR (125 MHz, CDCl3) δ 165.5, 163.4, 159.1, 152.1, 136.6, 110.9, 109.8,
103.9, 70.3, 68.8, 55.9; IR (KBr, neat) 3739, 3007, 1716, 1609, 1275,
1261, 1225, 750 cm–1; HRMS (ESI) calcd. for C11H10IO4 (M + H)+ 332.9618,
found 332.961.
Pale yellow solid; Rf (hexane/EtOAc,
3:1) 0.50; mp 135–137 °C; yield 84 mg, 40%; 1H NMR (400 MHz, CDCl3) δ 7.97 (d, J = 8.8 Hz, 1 H), 6.68 (dd, J = 8.8, 2.4 Hz, 1 H),
6.46 (d, J = 2.4 Hz, 1 H), 6.10 (s, 1 H), 4.79 (s,
2 H), 3.83 (s, 3 H); 13C{1H} NMR (125 MHz, CDCl3) δ 165.5, 163.4, 159.1, 152.1, 136.6, 110.9, 109.8,
103.9, 70.3, 68.8, 55.9; IR (KBr, neat) 3739, 3007, 1716, 1609, 1275,
1261, 1225, 750 cm–1; HRMS (ESI) calcd. for C11H10IO4 (M + H)+ 332.9618,
found 332.961.
Experimental Procedure for the Synthesis of 6
 3-(Iodomethyl)-3-methyl-2,3-dihydro-5H-benzo[e][1,4]dioxepin-5-one 2a (0.31 mmol, 1.0 equiv) and NH4SCN (1.55 mmol,
5.0 equiv) in DMF (4 mL) under the N2 atmosphere was allowed
to stir at 80 °C in an oil bath and the reaction time was monitored
by TLC. After completion of the reaction (16 h), the reaction mixture
was brought to room temperature, diluted with ethyl acetate, and saturated
brine solution. The organic phase was extracted with ice water (3
× 10 mL). The combined organic extracts were dried over Na2SO4 and concentrated in a rotary evaporator. The
crude was subjected to column chromatography over silica gel to give
the corresponding product 6.
3-(Iodomethyl)-3-methyl-2,3-dihydro-5H-benzo[e][1,4]dioxepin-5-one 2a (0.31 mmol, 1.0 equiv) and NH4SCN (1.55 mmol,
5.0 equiv) in DMF (4 mL) under the N2 atmosphere was allowed
to stir at 80 °C in an oil bath and the reaction time was monitored
by TLC. After completion of the reaction (16 h), the reaction mixture
was brought to room temperature, diluted with ethyl acetate, and saturated
brine solution. The organic phase was extracted with ice water (3
× 10 mL). The combined organic extracts were dried over Na2SO4 and concentrated in a rotary evaporator. The
crude was subjected to column chromatography over silica gel to give
the corresponding product 6.
3-Methyl-3-(thiocyanatomethyl)-2,3-dihydro-5H-benzo[e][1,4]dioxepin-5-one (6)
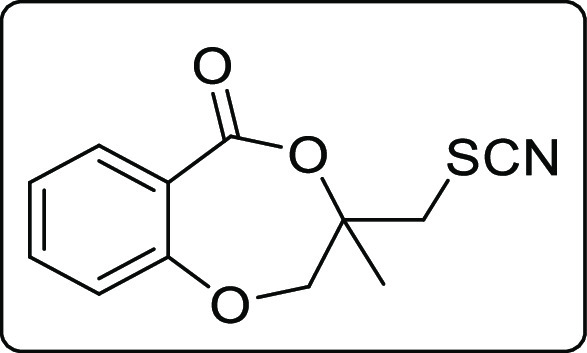 Pale yellow solid; Rf (hexane/EtOAc,
4:1) 0.40; mp 109–111 °C; yield 56 mg, 72%; 1H NMR (500 MHz, CDCl3) δ 8.19 (dd, J = 8.0, 2.0
Hz, 1 H), 7.52–7.49 (m, 1 H), 7.11 (t, J =
7.5 Hz, 1 H), 7.06 (d, J = 8.0 Hz, 1 H), 4.52 (d, J = 13.5 Hz, 1 H), 4.40 (d, J = 13.5 Hz,
1 H), 3.33 (q, J = 14 Hz, 2 H), 1.61 (s, 3 H); 13C{1H} NMR (125 MHz, CDCl3) δ
164.8, 157.1, 136.1, 135.6, 122.7, 120.1, 116.5, 112.1, 81.4, 74.4,
40.6, 21.8; IR (KBr, neat) 2955, 2919, 1695, 1481, 1447, 1280, 1118,
751 cm–1; HRMS (ESI) calcd. for C12H12NO3S (M + H)+ 250.0532, found 250.0525.
Pale yellow solid; Rf (hexane/EtOAc,
4:1) 0.40; mp 109–111 °C; yield 56 mg, 72%; 1H NMR (500 MHz, CDCl3) δ 8.19 (dd, J = 8.0, 2.0
Hz, 1 H), 7.52–7.49 (m, 1 H), 7.11 (t, J =
7.5 Hz, 1 H), 7.06 (d, J = 8.0 Hz, 1 H), 4.52 (d, J = 13.5 Hz, 1 H), 4.40 (d, J = 13.5 Hz,
1 H), 3.33 (q, J = 14 Hz, 2 H), 1.61 (s, 3 H); 13C{1H} NMR (125 MHz, CDCl3) δ
164.8, 157.1, 136.1, 135.6, 122.7, 120.1, 116.5, 112.1, 81.4, 74.4,
40.6, 21.8; IR (KBr, neat) 2955, 2919, 1695, 1481, 1447, 1280, 1118,
751 cm–1; HRMS (ESI) calcd. for C12H12NO3S (M + H)+ 250.0532, found 250.0525.
Experimental Procedure for the Synthesis of 7
 3-(Iodomethyl)-3-methyl-2,3-dihydro-5H-benzo[e][1,4]dioxepin-5-one 2a (0.31 mmol, 1.0 equiv) and NaN3 (0.62 mmol,
2.0 equiv) in DMF (4 mL) under the N2 atmosphere was allowed
to stir at 80 °C in an oil bath and the reaction time was monitored
by TLC. After completion of the reaction (12 h), the reaction mixture
was brought to room temperature, diluted with ethyl acetate, and saturated
brine solution. The organic phase was extracted with ice water (3
× 10 mL). The combined organic extracts were dried over Na2SO4 and concentrated in a rotary evaporator. The
crude was subjected to column chromatography over silica gel to give
the corresponding product 7.
3-(Iodomethyl)-3-methyl-2,3-dihydro-5H-benzo[e][1,4]dioxepin-5-one 2a (0.31 mmol, 1.0 equiv) and NaN3 (0.62 mmol,
2.0 equiv) in DMF (4 mL) under the N2 atmosphere was allowed
to stir at 80 °C in an oil bath and the reaction time was monitored
by TLC. After completion of the reaction (12 h), the reaction mixture
was brought to room temperature, diluted with ethyl acetate, and saturated
brine solution. The organic phase was extracted with ice water (3
× 10 mL). The combined organic extracts were dried over Na2SO4 and concentrated in a rotary evaporator. The
crude was subjected to column chromatography over silica gel to give
the corresponding product 7.
3-(Azidomethyl)-3-methyl-2,3-dihydro-5H-benzo[e][1,4]dioxepin-5-one (7)
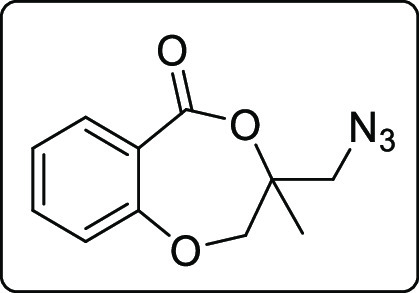 Pale yellow solid; Rf (hexane/EtOAc,
7:3) 0.50; mp 53–55 °C; yield 55 mg, 76%; 1H NMR (500 MHz, CDCl3) δ 8.21 (dd, J = 8.0, 1.5 Hz, 1 H), 7.50–7.47 (m, 1 H), 7.08 (t, J = 8.0 Hz, 1 H), 7.04 (d, J = 8.0 Hz,
1 H), 4.35 (q, J = 14.0 Hz, 2 H), 3.60 (d, J = 13.0 Hz, 1 H), 3.40 (d, J = 12.5 Hz,
1 H), 1.46 (s, 3 H); 13C{1H} NMR (125 MHz, CDCl3) δ 165.4, 157.5, 136.1, 135.3, 122.3, 120.0, 116.7,
81.5, 74.1, 56.4, 20.4; IR (KBr, neat) 2925, 2108, 1696, 1606, 1482,
1443, 1292, 1119, 1069, 945, 752 cm–1; HRMS (ESI)
calcd. for C11H12N3O3 (M
+ H)+ 234.0873, found 234.0872.
Pale yellow solid; Rf (hexane/EtOAc,
7:3) 0.50; mp 53–55 °C; yield 55 mg, 76%; 1H NMR (500 MHz, CDCl3) δ 8.21 (dd, J = 8.0, 1.5 Hz, 1 H), 7.50–7.47 (m, 1 H), 7.08 (t, J = 8.0 Hz, 1 H), 7.04 (d, J = 8.0 Hz,
1 H), 4.35 (q, J = 14.0 Hz, 2 H), 3.60 (d, J = 13.0 Hz, 1 H), 3.40 (d, J = 12.5 Hz,
1 H), 1.46 (s, 3 H); 13C{1H} NMR (125 MHz, CDCl3) δ 165.4, 157.5, 136.1, 135.3, 122.3, 120.0, 116.7,
81.5, 74.1, 56.4, 20.4; IR (KBr, neat) 2925, 2108, 1696, 1606, 1482,
1443, 1292, 1119, 1069, 945, 752 cm–1; HRMS (ESI)
calcd. for C11H12N3O3 (M
+ H)+ 234.0873, found 234.0872.
Experimental Procedure for the Synthesis of 8
 To a stirred solution of 3-(azidomethyl)-3-methyl-2,3-dihydro-5H-benzo[e][1,4]dioxepin-5-one
(0.43 mmol, 1.0 equiv), CuSO4 0.5H2O (0.043 mmol, 0.1 equiv),
and sodium L ascorbate (0.086 mmol, 0.2 equiv) in CHCl3/H2O (3:1) (15 mL) was added phenylacetylene (0.43 mmol,
1.0 equiv) dropwise at 0 °C. Then, the reaction mixture was allowed
to stir at room temperature. After completion of the reaction, the
reaction mixture was diluted with DCM and saturated brine solution.
The combined organic extracts were dried over Na2SO4 and concentrated in rotary evaporator. The crude was subjected
to column chromatography over silica gel to give the corresponding
product 8.
To a stirred solution of 3-(azidomethyl)-3-methyl-2,3-dihydro-5H-benzo[e][1,4]dioxepin-5-one
(0.43 mmol, 1.0 equiv), CuSO4 0.5H2O (0.043 mmol, 0.1 equiv),
and sodium L ascorbate (0.086 mmol, 0.2 equiv) in CHCl3/H2O (3:1) (15 mL) was added phenylacetylene (0.43 mmol,
1.0 equiv) dropwise at 0 °C. Then, the reaction mixture was allowed
to stir at room temperature. After completion of the reaction, the
reaction mixture was diluted with DCM and saturated brine solution.
The combined organic extracts were dried over Na2SO4 and concentrated in rotary evaporator. The crude was subjected
to column chromatography over silica gel to give the corresponding
product 8.
3-Methyl-3-((4-phenyl-1H-1,2,3-triazol-1-yl)methyl)-2,3-dihydro-5H-benzo[e][1,4]-dioxepin-5-one (8)
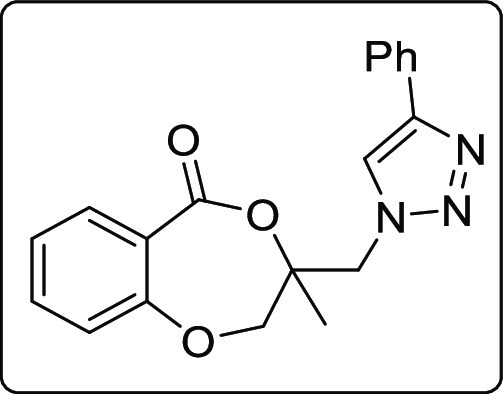 White solid; Rf (hexane/EtOAc,
3:2) 0.50; mp 165–167 °C; yield 89 mg, 89%; 1H NMR (500 MHz, CDCl3) δ 8.18 (dd, J = 8.0, 1.5
Hz, 1 H), 8.10 (s, 1 H), 7.84 (d, J = 8.0 Hz, 2 H),
7.50–7.47 (m, 1 H), 7.41 (t, J = 7.5 Hz, 2
H), 7.35–7.31 (m, 1 H), 7.08 (t, J = 7.0 Hz,
1 H), 7.04 (d, J = 8.0 Hz, 1 H), 4.70 (s, 2 H), 4.57
(d, J = 14.0 Hz, 1 H), 4.02 (d, J = 14.0 Hz, 1 H), 1.47 (s, 3 H); 13C{1H} NMR
(125 MHz, CDCl3) δ 165.2, 157.5, 148.5, 135.9, 135.5,
130.4, 129.1, 128.6, 126.0, 122.5, 121.5, 120.1, 116.4, 81.3, 74.2,
56.4, 20.1; IR (KBr, neat) 2956, 2918, 1694, 1606, 1481, 1290, 1120,
752, 694, cm–1; HRMS (ESI) calcd. for C19H18N3O3 (M + H)+ 336.1343,
found 336.1366.
White solid; Rf (hexane/EtOAc,
3:2) 0.50; mp 165–167 °C; yield 89 mg, 89%; 1H NMR (500 MHz, CDCl3) δ 8.18 (dd, J = 8.0, 1.5
Hz, 1 H), 8.10 (s, 1 H), 7.84 (d, J = 8.0 Hz, 2 H),
7.50–7.47 (m, 1 H), 7.41 (t, J = 7.5 Hz, 2
H), 7.35–7.31 (m, 1 H), 7.08 (t, J = 7.0 Hz,
1 H), 7.04 (d, J = 8.0 Hz, 1 H), 4.70 (s, 2 H), 4.57
(d, J = 14.0 Hz, 1 H), 4.02 (d, J = 14.0 Hz, 1 H), 1.47 (s, 3 H); 13C{1H} NMR
(125 MHz, CDCl3) δ 165.2, 157.5, 148.5, 135.9, 135.5,
130.4, 129.1, 128.6, 126.0, 122.5, 121.5, 120.1, 116.4, 81.3, 74.2,
56.4, 20.1; IR (KBr, neat) 2956, 2918, 1694, 1606, 1481, 1290, 1120,
752, 694, cm–1; HRMS (ESI) calcd. for C19H18N3O3 (M + H)+ 336.1343,
found 336.1366.
Experimental Procedure for the Synthesis of 9
 To a stirred solution of NaH (0.31 mmol, 1.0 equiv) in DMF
(4 mL) under the N2 atmosphere, thiophenol (0.37 mmol,
1.2 equiv) was added dropwise at 0 °C, and the resulting mixture
was allowed to stir for 30 min at room temperature. Then, the substrate
3-(iodomethyl)-3-methyl-2,3-dihydro-5H-benzo[e][1,4]dioxepin-5-one 2a (0.31 mmol, 1.0 equiv)
in DMF was added dropwise to the reaction mixture. The progress of
the reaction was monitored by TLC. After completion of the reaction
(36 h), the reaction mixture was brought to room temperature, diluted
with ethyl acetate, and saturated brine solution. The organic phase
was extracted with ice water (3 × 10 mL). The combined organic
extracts were dried over Na2SO4 and concentrated
in a rotary evaporator. The crude was subjected to column chromatography
over silica gel to give the corresponding product 9.
To a stirred solution of NaH (0.31 mmol, 1.0 equiv) in DMF
(4 mL) under the N2 atmosphere, thiophenol (0.37 mmol,
1.2 equiv) was added dropwise at 0 °C, and the resulting mixture
was allowed to stir for 30 min at room temperature. Then, the substrate
3-(iodomethyl)-3-methyl-2,3-dihydro-5H-benzo[e][1,4]dioxepin-5-one 2a (0.31 mmol, 1.0 equiv)
in DMF was added dropwise to the reaction mixture. The progress of
the reaction was monitored by TLC. After completion of the reaction
(36 h), the reaction mixture was brought to room temperature, diluted
with ethyl acetate, and saturated brine solution. The organic phase
was extracted with ice water (3 × 10 mL). The combined organic
extracts were dried over Na2SO4 and concentrated
in a rotary evaporator. The crude was subjected to column chromatography
over silica gel to give the corresponding product 9.
3-Methyl-3-((phenylthio)methyl)-2,3-dihydro-5H-benzo[e][1,4]dioxepin-5-one (9)
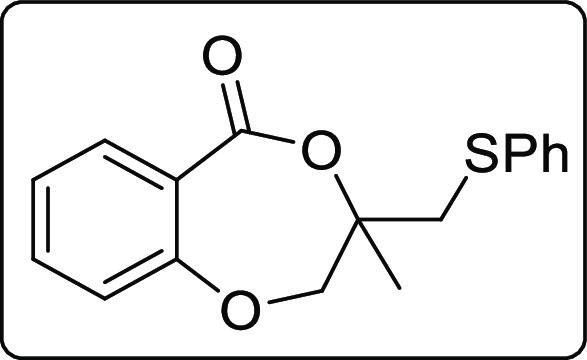 Yellow liquid; Rf (hexane/EtOAc,
4:1) 0.50; yield 47 mg, 51%; 1H NMR (500 MHz, CDCl3) δ 8.22 (dd, J = 8.5, 2.0 Hz, 1 H), 7.52–7.48
(m, 1 H), 7.46 (d, J = 7.0 Hz, 2 H), 7.34 (t, J = 7.0 Hz, 2 H), 7.28 (d, J = 7.0 Hz,
1 H), 7.10 (d, J = 7.5 Hz, 1 H), 7.50 (d, J = 8.5 Hz, 1 H), 4.50 (d, J = 14.0 Hz,
1 H), 4.39 (d, J = 13.5 Hz, 1 H), 3.34 (d, J = 2.0 Hz, 2 H), 1.54 (s, 3 H); 13C{1H} NMR (125 MHz, CDCl3) δ 165.6, 157.4, 135.9, 135.9,
135.0, 130.7, 129.3, 127.2, 122.0, 119.9, 116.7, 83.0, 74.6, 41.7,
22.0; IR (KBr, neat) 2919, 1694, 1605, 1481, 1292, 1119, 750, 691
cm–1; HRMS (ESI) calcd. for C17H20NO3S (M + NH4)+ 318.1158,
found 318.1169.
Yellow liquid; Rf (hexane/EtOAc,
4:1) 0.50; yield 47 mg, 51%; 1H NMR (500 MHz, CDCl3) δ 8.22 (dd, J = 8.5, 2.0 Hz, 1 H), 7.52–7.48
(m, 1 H), 7.46 (d, J = 7.0 Hz, 2 H), 7.34 (t, J = 7.0 Hz, 2 H), 7.28 (d, J = 7.0 Hz,
1 H), 7.10 (d, J = 7.5 Hz, 1 H), 7.50 (d, J = 8.5 Hz, 1 H), 4.50 (d, J = 14.0 Hz,
1 H), 4.39 (d, J = 13.5 Hz, 1 H), 3.34 (d, J = 2.0 Hz, 2 H), 1.54 (s, 3 H); 13C{1H} NMR (125 MHz, CDCl3) δ 165.6, 157.4, 135.9, 135.9,
135.0, 130.7, 129.3, 127.2, 122.0, 119.9, 116.7, 83.0, 74.6, 41.7,
22.0; IR (KBr, neat) 2919, 1694, 1605, 1481, 1292, 1119, 750, 691
cm–1; HRMS (ESI) calcd. for C17H20NO3S (M + NH4)+ 318.1158,
found 318.1169.
Experimental Procedure for Gram-Scale Synthesis of Compound 2a
 To a stirred solution of 2-((2-methylallyl)oxy)benzaldehyde
(5.68 mmol, 1.0 equiv) and CuI (6.82 mmol, 1.2 equiv) in CH3CN (15 mL) was added 70% aq solution of TBHP (34.08 mmol, 6.0 equiv,)
dropwise at room temperature. Then, the reaction mixture was allowed
to stir at 70 °C, and the reaction time was monitored by TLC.
After completion of the reaction (6 h), the reaction mixture was brought
to room temperature, extracted with ethyl acetate (3 × 10 mL),
and washed with Na2S2O3, NH4Cl, and saturated brine solution. The combined organic extract was
dried over Na2SO4 and concentrated in a rotary
evaporator. The crude was subjected to column chromatography over
silica gel to give the corresponding product 2a in 61%
yield (1.10 g).
To a stirred solution of 2-((2-methylallyl)oxy)benzaldehyde
(5.68 mmol, 1.0 equiv) and CuI (6.82 mmol, 1.2 equiv) in CH3CN (15 mL) was added 70% aq solution of TBHP (34.08 mmol, 6.0 equiv,)
dropwise at room temperature. Then, the reaction mixture was allowed
to stir at 70 °C, and the reaction time was monitored by TLC.
After completion of the reaction (6 h), the reaction mixture was brought
to room temperature, extracted with ethyl acetate (3 × 10 mL),
and washed with Na2S2O3, NH4Cl, and saturated brine solution. The combined organic extract was
dried over Na2SO4 and concentrated in a rotary
evaporator. The crude was subjected to column chromatography over
silica gel to give the corresponding product 2a in 61%
yield (1.10 g).
Acknowledgments
A.C. gratefully acknowledges the Ministry of Education (MoE) for a Prime Ministers Research Fellowship (PMRF). The authors are grateful to the DST-FIST program (Grant No. SR/FST/CS-II/2017/23C) for providing analytical facility.
Data Availability Statement
The data underlying this study are available in the published article and its Supporting Information.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.3c09878.
Control experimental procedure and copies of 1H, 13C{1H}, and 19F NMR spectra of all new compounds; X-ray crystallographic data of compound 2a and 4a (PDF)
Accession Codes
CCDC 2299354, 2299353, and 2299355 contain the supplementary crystallographic data for this paper.
The authors declare no competing financial interest.
Due to a production error, this paper was published ASAP on March 12, 2024 with the structures misplaced. The corrected version was reposted on March 13, 2024.
Supplementary Material
References
- a Schulz S.; Hötling S. The use of the lactone motif in chemical communication. Nat. Prod. Rep. 2015, 32, 1042–1066. 10.1039/C5NP00006H. [DOI] [PubMed] [Google Scholar]; b Shiina I. Total synthesis of natural 8-and 9-membered lactones: recent advancements in medium-sized ring formation. Chem. Rev. 2007, 107, 239–273. 10.1021/cr050045o. [DOI] [PubMed] [Google Scholar]; c Sartori S. K.; Diaz M. A. N.; Munoz G. D. Lactones: Classification, synthesis, biological activities, and industrial applications. Tetrahedron 2021, 84, 132001. [Google Scholar]
- Buszek K. R.; Sato N.; Jeong Y. Total Synthesis of Octalactin A and B. J. Am. Chem. Soc. 1994, 116, 5511–5512. 10.1021/ja00091a082. [DOI] [Google Scholar]
- a Komai S.; Hosoe T.; Nozawa T. I. K.; Fukushima T. Y. K.; Kawai K. New penicillide derivatives isolated from Penicillium simplicissimu. J. Nat. Med. 2006, 60, 185–190. [DOI] [PubMed] [Google Scholar]; b Nishida H.; Tomoda H.; Cao J.; Okuda S.; Omura S. Urpactins, New Inhibitors of Acyl-CoA: Cholesterol Acyltransferase Produced by Penicillium purpurogenum. J. Antibiot. 1991, 44, 144–151. 10.7164/antibiotics.44.144. [DOI] [PubMed] [Google Scholar]
- a Lizundia E.; Makwana V. A.; Larrañaga A.; José Luis Vilas J. L.; Shaver M. P. Thermal, structural and degradation properties of an aromatic–aliphatic polyester built through ringopening polymerization. Polym. Chem. 2017, 8, 3530–3538. [Google Scholar]; b MacDonald J. P.; Shaver M. P. An aromatic/aliphatic polyester prepared via ring-opening polymerisation and its remarkably selective and cyclable depolymerisation to monomer. Polym. Chem. 2016, 7, 553–559. 10.1039/C5PY01606A. [DOI] [Google Scholar]
- a Parenty A.; Moreau X.; Campagne J.-M. Macrolactonizations in the Total Synthesis of Natural Products. Chem. Rev. 2006, 106, 911–939. 10.1021/cr0301402. [DOI] [PubMed] [Google Scholar]
- a Phan D. H. T.; Kim B.; Dong V. M. Phthalides by Rhodium-Catalyzed Ketone Hydroacylation. J. Am. Chem. Soc. 2009, 131, 15608–15609. 10.1021/ja907711a. [DOI] [PubMed] [Google Scholar]; b Shen Z.; Khan H. A.; Dong V. M. Rh-Catalyzed Carbonyl Hydroacylation: An Enantioselective Approach to Lactones. J. Am. Chem. Soc. 2008, 130, 2916–2917. 10.1021/ja7109025. [DOI] [PubMed] [Google Scholar]; c Ye Z.; Lv G.; Wang W.; Zhang M.; Cheng J. Rhodium-Catalyzed Cascade Reaction: Aryl Addition/Intramolecular Esterification to Access 3-Aryl and 3-Alkenyl Phthalides. Angew. Chem., Int. Ed. 2010, 49, 3671–3674. 10.1002/anie.201000302. [DOI] [PubMed] [Google Scholar]; d Ye Z.; Qian P.; Lv G.; Luo F.; Cheng J. Palladium-Catalyzed Cascade Aryl Addition/Intramolecular Lactonization of Phthalaldehyde To Access 3-Aryl- and Alkenylphthalides. J. Org. Chem. 2010, 75, 6043–6045. 10.1021/jo101203b. [DOI] [PubMed] [Google Scholar]
- a Fraunhoffer K. J.; Prabagaran N.; Sirois L. E.; White M. C. Macrolactonization via Hydrocarbon Oxidation. J. Am. Chem. Soc. 2006, 128, 9032–9033. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Velasco-Rubio Á.; Varela J. A.; Saá C. Pd-Catalyzed allylic C-H activation to seven-membered N, O-heterocycles. Chem. Commun. 2021, 57, 10915–10918. 10.1039/D1CC04056A. [DOI] [PubMed] [Google Scholar]
- Rose C. A.; Zeitler K. Efficient Catalytic, Oxidative Lactonization for the Synthesis of Benzodioxepinones Using Thiazolium-Derived Carbene Catalysts. Org. Lett. 2010, 12, 4552–4555. 10.1021/ol101854r. [DOI] [PubMed] [Google Scholar]
- a Galli C.; Illuminati G.; Mandolini L.; Tamborra P. Ring-Closure Reactions. 7. Kinetics and Activation Parameters of Lactone Formation in the Range of 3-to 23-Membered Rings. J. Am. Chem. Soc. 1977, 99, 2591–2597. 10.1021/ja00450a031. [DOI] [Google Scholar]; b Illuminati G.; Mandolini L. Ring closure reactions of bifunctional chain molecules. Acc. Chem. Res. 1981, 14, 95–102. 10.1021/ar00064a001. [DOI] [Google Scholar]
- a Gataullin R. R. Halolactonization of N-Acyl-N-(2-cyclohex-1-en-1-yl-6-methylphenyl) glycines: Towards Production of 4,1-Benzoxazoheterocycles. Synthesis 2023, 55, 2053–2060. 10.1055/s-0042-1751427. [DOI] [Google Scholar]; b Jiang X.; Xu X.; Xu W.; Yu P.; Yeung Y.-Y. Catalytic Enantioselective Halocyclizations to Access Benzoxazepinones and Benzoxazecinones. Org. Lett. 2021, 23, 6316–6320. 10.1021/acs.orglett.1c02117. [DOI] [PubMed] [Google Scholar]; c Verma A.; Jana S.; Prasad C. D.; Yadav A.; Kumar S. Organoselenium and DMAP co-catalysis: regioselective synthesis of medium-sized halolactones and bromooxepanes from unactivated alkenes Chem. Commun. 2016, 52, 4179–4182. [DOI] [PubMed] [Google Scholar]; d Payne J. L.; Deng Z.; Flach A. L.; Johnston J. N. Enantioselective iodolactonization to prepare 3-lactone rings using hypervalent iodine. Chem. Sci. 2022, 13, 7318–7324. 10.1039/D2SC01587K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W.; Zheng S.; Liu N.; Werness J. B.; Guzei I. A.; Tang W. Enantioselective Bromolactonization of Conjugated (Z)-Enynes. J. Am. Chem. Soc. 2010, 132, 3664–3665. 10.1021/ja100173w. [DOI] [PubMed] [Google Scholar]
- a Minisci F.; Fontana F.; Araneo S.; Recupero F.; Banfi S.; Quid S. Kharasch and Metalloporphyrin Catalysis in the Functionalization of Alkanes, Alkenes, and Alkylbenzenes by t-BuOOH. Free Radical Mechanisms, Solvent Effect, and Relationship with the Gif Reaction. J. Am. Chem. Soc. 1995, 117, 226–232. [Google Scholar]; b Zhu Y.; Yan H.; Lu L.; Liu D.; Rong G.; Mao J. Copper-Catalyzed Methyl Esterification Reactions via C–C Bond Cleavage. J. Org. Chem. 2013, 78, 9898–9905. 10.1021/jo4016387. [DOI] [PubMed] [Google Scholar]; c Singh J. B.; Mishra K.; Gupta T.; Singh R. M. TBHP-promoted oxidative cyclization of o-alkynylquinoline aldehydes: Metal/additive-free domino synthesis of pyrano[4,3-b]quinolin-1-ones. Tetrahedron Lett. 2018, 59, 1019–1022. 10.1016/j.tetlet.2018.01.083. [DOI] [Google Scholar]
- a Martín-Acosta P.; Feresin G.; Tapia A.; Estévez-Braun A. Microwave-assisted organocatalytic intramolecular Knoevenagel/Hetero Diels–Alder reaction with O-(arylpropynyloxy)-salicylaldehydes: Synthesis of polycyclic embelin derivatives. J. Org. Chem. 2016, 81, 9738–9756. 10.1021/acs.joc.6b01818. [DOI] [PubMed] [Google Scholar]; b Zhang X.; Sivaguru P.; Zanoni G.; Han X.; Tong M.; Bi X. Catalytic asymmetric C (sp3)–H carbene insertion approach to access enantioenriched 3-fluoroalkyl 2, 3-dihydrobenzofurans. ACS Catalysis. 2021, 11, 14293–14301. 10.1021/acscatal.1c04523. [DOI] [Google Scholar]; c Piel I.; Steinmetz M.; Hirano K.; Froehlich R.; Grimme S.; Glorius F. Highly Asymmetric NHC-Catalyzed Hydroacylation of Unactivated Alkenes. Angew. Chem., Int. Ed. 2011, 50, 4983–4987. 10.1002/anie.201008081. [DOI] [PubMed] [Google Scholar]; d Rohlmann R.; Daniliuc C. G.; Mancheño O. G. Highly enantioselective synthesis of chiral 7-ring O-and N-heterocycles by a one-pot nitro-Michael–cyclization tandem reaction. Chem. Comm. 2013, 49, 11665–11667. 10.1039/c3cc47397j. [DOI] [PubMed] [Google Scholar]; e Sharma A.; Gogoi P. A cascade process for the synthesis of ortho-formyl allyl aryl ethers and 2 H-chromen-2-ol derivatives from arynes via trapping of o-quinone methide with an activated alkene. Org. Biomol. Chem. 2019, 17, 333–346. 10.1039/C8OB02507J. [DOI] [PubMed] [Google Scholar]; f Martínez-Gualda A. M.; Domingo-Legarda P.; Rigotti T.; Díaz-Tendero S.; Fraile A.; Alemán J. Asymmetric [2+ 2] photocycloaddition via charge transfer complex for the synthesis of tricyclic chiral ethers. Chem. Commun. 2021, 57, 3046–3049. 10.1039/D1CC00035G. [DOI] [PubMed] [Google Scholar]; g Dreis A. M.; Douglas C. J. Catalytic carbon–carbon σ bond activation: an intramolecular carbo-acylation reaction with acylquinolines. J. Am. Chem. Soc. 2009, 131, 412–413. 10.1021/ja8066308. [DOI] [PubMed] [Google Scholar]; h Sanguinetti M.; Sanfilippo S.; Castagnolo D.; Sanglard D.; Posteraro B.; Donzellini G.; Botta M. Novel macrocyclic amidinoureas: Potent non-azole antifungals active against wild-type and resistant Candida species. ACS Med. Chem. Lett. 2013, 4, 852–857. 10.1021/ml400187w. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Hu H.; Chen X.; Sun K.; Wang J.; Liu Y.; Liu H.; Zhao Y. Silver-catalyzed radical cascade cyclization toward 1, 5-/1, 3-dicarbonyl heterocycles: an atom-/step-economical strategy leading to chromenopyridines and isoxazole-/pyrazole-containing chroman-4-ones. Org. Lett. 2018, 20, 6157–6160. [DOI] [PubMed] [Google Scholar]; j Xuyu Z.; Aijun Z.; Qingqing Z.; Qing’an L.; Jun X. Visible Light-Induced 4-Chromanones Synthesis: Radical Cascade Cyclization of α-Oxocarboxylic Acids with o-(Allyloxy) arylaldehydes Promoted by Phenyliodine (III) Diacetate. Chin. J. Org. Chem. 2022, 42, 2488. [Google Scholar]
- Wu N.; Li R.; Cui F.; Pan Y. Application of Dehydroabietic Acid in Palladium-Catalysed Enyne Cycloisomerisation. Adv. Synth. Catal. 2017, 359, 2442–2447. 10.1002/adsc.201700061. [DOI] [Google Scholar]
- Miege F.; Meyer C.; Cossy J. Rhodium-Catalyzed Cycloisomerization Involving Cyclopropenes: Efficient Stereoselective Synthesis of Medium-Sized Heterocyclic Scaffolds. Angew. Chem., Int. Ed. 2011, 50, 5932–5937. 10.1002/anie.201101220. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this study are available in the published article and its Supporting Information.