

Abstract
Despite significant advances in developing selective JAK2 inhibitors, JAK2 kinase inhibitor (TKI) therapy is ineffective in suppressing the disease. Reactivation of compensatory MEK-ERK and PI3K survival pathways sustained by inflammatory cytokine signaling causes treatment failure. Concomitant inhibition of MAPK pathway and JAK2 signaling showed improved in vivo efficacy compared to JAK2 inhibition alone but lacked clonal selectivity. We hypothesized that cytokine signaling in JAK2V617F induced MPNs increases the apoptotic threshold that causes TKI persistence or refractoriness. Here, we show that JAK2V617F and cytokine signaling converge to induce MAPK negative regulator, DUSP1. Enhanced DUSP1 expression blocks p38 mediated p53 stabilization. Deletion of Dusp1 increases p53 levels in the context of JAK2V617F signaling that causes synthetic lethality to Jak2V617F expressing cells. However, inhibition of Dusp1 by a small molecule inhibitor (BCI) failed to impart Jak2V617F clonal selectivity due to pErk1/2 rebound caused by off-target inhibition of Dusp6. Ectopic expression of Dusp6 and BCI treatment restored clonal selectively and eradicated the Jak2V617F cells. Our study shows that inflammatory cytokines and JAK2V617F signaling converge to induce DUSP1, which downregulates p53 and establishes a higher apoptotic threshold. These data suggest that selectively targeting DUSP1 may provide a curative response in JAK2V617F-driven MPN.
INTRODUCTION
The clinical success of ABL tyrosine kinase inhibitor (TKI) therapy in treating Ph+ chronic myeloid leukemia (CML) prompted to identify the oncogenic lesions in Ph− MPN for therapeutic intervention. This led to the discovery of activating mutations in JAK2 [1, 2], MPL [3] and CALR [4]. Interestingly, all these mutations converge in activating the JAK-STAT signaling, which provided a strong rationale for targeting JAK2 kinase activity using small molecule kinase inhibitor. However, unlike the cytotoxic effect of ABL inhibitors in CML, JAK2 inhibition imparted a cytostatic response and failed to eradicate the malignant clones or induce molecular remission [5]. Clonal persistence and adaptation to JAK2-tyrosine kinase inhibitors (TKIs) are the primary cause of treatment failure, which is mainly driven by the activation of survival pathways by inflammatory cytokines.
Constitutive cytokine and JAK2 signaling also activate MAPK pathway by activating the PI3 kinase. Consequently, inhibition of MAPK or PI3K with JAK2 inhibitors synergistically suppressed the proliferation of JAK2V617F expressing MPN cells [6, 7]. In preclinical mouse model, combinatorial targeting of JAK2 with MEK/ERK or PI3K in vivo showed modest improvement compared to JAK2 inhibition alone, as most mice exhibited persistent disease [8–11]. Given that both MEK and JAK2 inhibitors exert cytostatic response in vivo, combinatorial targeting of both proteins would least likely exert clonal selectivity. The underlying mechanism driving cytostatic response has yet to be fully understood. Nonetheless, constitutive cytokine signaling induced persistent MAPK activity is suspected for the cytostatic response.
The therapeutic efficacy of TKI was attributed to be a function of oncogene-dependence where acute inhibition of driver-oncogene by TKI results in apoptotic cell death while sparing normal cells [12–14]. While TKI therapy in CML kills most leukemic cells, it is not curative because leukemic stem cells (LSCs) are intrinsically resistant to treatment resulting in minimal residual disease (MRD). Even the most potent TKIs are ineffective in eliminating the LSCs [15]. Although the molecular mechanisms underlying the failure of TKI to eradicate these persistent cells are not fully understood, growth-factor signaling is implicated in rescuing these cells during TKI therapy in leukemia and solid organ tumors [16–20]. We have shown that the growth-factor signaling in the context of oncogenic signaling induces c-Fos and Dusp1 (Dual specificity phosphatase 1) expression, together these genes modulate the AP1 transcriptional network and the activity of p38 kinase, which establishes a higher apoptotic threshold resulting in LSC persistence and TKI refractoriness [21].
Failure of JAK2 TKIs to completely suppress the overproduction of inflammatory cytokines, a cardinal feature of MPN development and transition to AML, drives clonal persistence and TKI refractoriness [22]. We reasoned that, like LSCs in CML where growth-factor signaling increases the apoptotic threshold, inflammatory cytokine signaling in MPNs drives a higher apoptotic threshold. Consequently, these cells are intrinsically resistant to TKI treatment. Here, we show that MPN cells derived from the primary patients exhibit increased expression of DUSP1 but not the c-FOS. Using mouse genetic models, we show that ablation of DUSP1 is synthetic lethal to MPN development. Biochemical analysis revealed that JAK2V617F signaling downregulates p53 expression by DUSP1 mediated inhibition of p38 activity. Phosphorylation of p53 at Serine 15 prevents its interaction with Mdm2. Reduced p53 phosphorylation at Serine 15 due to p38 blockade enhances p53 degradation resulting in reduced expression. Consequently, DUSP1 deletion activates p38 that results in enhanced p53 stabilization and selective demise of Jak2V617F clones. Our study provides evidence that MAPK-negative feedback confers dependence to JAK2V617F-induced MPN, and its targeting may provide a curative response.
METHODS
Available as supplementary material.
RESULTS
To test the effect of growth-factor signaling on TKI response, BaF3 cells stably expressing MPN genes (Jak2V617F, MPLW515L and CALRmut) were examined to determine the efficacy of ruxolitinib in the presence and absence of IL3. Parental BaF3 cells require IL3 for survival, expression of oncogenes renders growth-factor independent growth that allows evaluation of targeted therapy. As expected, ruxolitinib treatment inhibited the proliferation of BaF3-Jak2V617F, BaF3-MPLW515L and BaF3-CALRmut cells with IC50 values of 79 and 46, and 57 nM, respectively (Fig. 1A). The addition of growth factor (IL3) conferred resistance to ruxolitinib resulting in increased IC50 values similar to parental BaF3 cells. Immunoblotting of phospho-JAK2 and phospho-STAT5 revealed complete inhibition of JAK2 and STAT5 in both conditions (Fig. 1B). Thus, providing evidence that resistance to ruxolitinib is not due to growth-factor-mediated JAK2 reactivation. Similarly, primary bone marrow derived c-Kit+ cells expressing Jak2V617F, MPLW515L and CALRmut grown with hematopoietic cytokines (SCF, FLT3 ligand, IL3, IL6 and TPO) exhibited higher IC50 values for ruxolitinib compared to the control, pMSCV-ires-GFP expressing cells (Fig. 1C). Altogether these data provide evidence that growth-factor signaling confers resistance to ruxolitinib.
Fig. 1. Growth factor induced DUSP1 expression confers resistance to ruxolitinib.

A A dose dependent cell proliferation assay of BaF3 cells expressing MPN inducing genes (Jak2V617F, MPLW515L, and CALRmut) showing resistance to ruxolitinib in the presence of growth-factor (IL3) signaling. B Immunoblot analysis of BaF3-Jak2V617F cells showing complete inhibition of Jak2 (increased phospho-JAK2 protein) and phosphoStat5 by ruxolitinib in both IL3+/− conditions. C Bone marrow derived primary Kit+ cells expressing Jak2V617F, MPLW515L, and CALRmut showing resistance to ruxolitinib in the presence of hematopoietic cytokines compared to control vector (pMIG). D Venn diagram showing the commonly expressed genes between Jak2V617F, MPLW515L, and BCR-ABL (treated with IL3 and imatinib). E Heat map showing the expression of 19 commonly deregulated genes in Jak2V617F, MPLW515L, and BCR-ABL cells. F Bar graph showing the relative expression of Dusp1 in LSK cells (Lin−, Sca1+ and Kit+) expressing Jak2V617F, MPLW515L, and CALRmut. G Violin plot showing significant induction of DUSP1 expression in PV and PMF patients but not ET. Expression data (GSE55976) is derived from the CD34+ cells from the MPN patients. H Real-time qPCR analysis of DUSP1 expression in primary MPN patient-derived mononuclear cells. Two PV and two MF patients were analyzed. Data are shown from two independent qPCR analysis ± SD. I A cell proliferation growth curve of Dusp1 knockdown in BaF3 cells expressing Jak2V617F, MPLW515L, and CALRmut showing significantly reduced proliferation compared to the parental BaF3 control. J Dose dependent cell proliferation assays showing Dusp1 depletion abrogated IL3 induced resistance to ruxolitinib in Jak2V617F, MPLW515L, and CALRmut expressing BaF3 cells. K DUSP1 knockdown by shRNA in HEL cells significantly suppressed the proliferation under growth-factor signaling (GF). Extent of DUSP1 knockdown and its expression ± GF conditions are shown in western blot (inset). Please note significant induction of DUSP1 during GF signaling. L Dose dependent cell proliferation assay showing DUSP1 depletion in HEL cells significantly reduced the resistance conferred by GF signaling. M Depletion of DUSP1 by shRNA-mediated knockdown in SET2 cells significantly suppressed the proliferation under growth-factor signaling (GF). Extent of DUSP1 knockdown and its expression in GF ± conditions are shown in western blot (inset). Please note significant induction of DUSP1 during GF signaling. N Dose dependent cell proliferation assay showing DUSP1 depletion in SET2 cells significantly reduced the resistance conferred by GF signaling. Presented data are from two independent experiments shown as the means ± SD. *p < 0.05, **p < 0.01 and ***p < 0.001.
To understand the molecular mechanism driving resistance, the BM-derived Kit+ cells expressing MPLW515L, Jak2V617F, and vector (pMIG) were subjected to the whole genome RNA-seq analysis. A comparative analysis revealed altered expression of 4208 genes in Jak2V617F and MPLW515L expressing cells compared to the vector control (Fig. 1D, Supplementary Fig. 1A). Among these, 2796 genes (2062 induced and 736 downregulated) are commonly modulated between MPLW515L and Jak2V617F, and a significant number of genes were uniquely regulated by MPLW515L (1412; 1038 induced and 374 downregulated) and Jak2V617F (815; 479 induced and 337 downregulated) Supplementary Fig. 1A. A network analysis of genes induced explicitly by JAK2V617F exhibited significant enrichment to pathways previously reported to be activated by JAK2 signaling, such as signal transduction, cellular differentiation, and growth-factor signaling [23]. We observed a more significant enrichment of genes downregulating WNT and JNK pathways in Jak2V617F expressing cells (Supplementary Fig. 1B–D). In contrast, analysis of MPLW515L-induced genes showed enrichment to pathways implicated in interferon β-signaling, platelet activation, TLR signaling and negative regulation of apoptosis (Supplementary Fig. 1E). However, network analysis of commonly induced genes exhibited more significant enrichment in pathways driving chemokine production (one of the hallmarks of MPN), cytokine signaling, and negative regulation of MAPK pathway besides modulating the previously reported MAPK and JAK-STAT pathways (Supplementary Fig. 1F–H and Supplementary Figs. 2 and 3). To identify the genes governing TKI (ruxolitinib) resistance under growth-factor signaling, we compared the expression profile generated from the growth-factor induced TKI resistant CML cells (185 genes) [21]. Comparison of these three data sets identified 19 commonly regulated genes between them (Fig. 1D, E). Among these, expression of Dusp1, Atp6v0d2, Scin, and Ifi27l2a are upregulated in Jak2V617F and MPLW515L cells compared to BCR-ABL expressing cells (Fig. 1E). Using CML as a model, we have shown that FOS, DUSP1, and ZFP36 are functional mediators of growth-factor-induced TKI resistance. Depletion of FOS and DUSP1 alone or together sensitized BaF3-BCR-ABL cells to imatinib, even in the presence of IL3. However, the lack of ZFP36 conferred equal TKI sensitivity to both parental BaF3 and BaF3-BCR-ABL cells, suggesting that ZFP36 is redundant. Given that the oncogenic JAK2 signaling robustly induces MAPK-ERK activation besides activating the JAK-STAT pathway, we reasoned that induced expression of DUSP1 seemingly provides feedback regulation to MAPK signaling to suppress the apoptotic stimulation commonly associated with unrestrained MAPK/ERK activation [24–28]. Therefore, we focused subsequent analyses on DUSP1. DUSP1.
Quantitative expression analysis of bone marrow-derived LSK cells expressing Jak2V617F, MPLW515L and CALRmut exhibited higher Dusp1 expression than the control (Fig. 1F). Likewise, analysis of patient samples revealed significantly increased DUSP1 expression (2–4-fold) in PV and PMF but not in ET compared to normal CD34+ cells (Fig. 1G, H). Next, genetic depletion of Dusp1 by shRNA-mediated knockdown in parental BaF3 cells did not show any adverse effect while its depletion in BaF3-Jak2V617F, BaF3-MPLW515L, BaF3-CALRmut significantly suppressed the proliferation (Fig. 1I). Importantly, DUSP1 depletion abolished cytokine-induced TKI resistance in BaF3- cells expressing Jak2V617F, MPLW515L and CALRmut (Fig. 1J). Likewise, human MPN cells, HEL and SET2, grown with hematopoietic cytokines (IL3, IL6, SCF, FLTLG and GM-CSF) exhibited induced DUSP1 expression and resistance to ruxolitinib (Fig. 1K–N). Depletion of DUSP1 by shRNA-mediated knockdown suppressed their proliferation and restored TKI sensitivity under growth-factor (GF) signaling (Fig. 1K–N). Together, these results suggest that growth-factor induced DUSP1 expression causes TKI refractoriness in MPN cells.
Deletion of Dusp1 is synthetic lethal to JAK2V617F-induced MPN
Next, we examined the role of DUSP1 in MPN development and treatment response. Bone marrow derived c-Kit+ positive cells from the wild-type and Dusp1−/− mice were transduced with Jak2V617F-Ires-GFP, MplW515L-Ires-GFP, and pMSCV-Ires-GFP retroviruses. GFP-positive cells sorted by FACS were used for ex vivo CFU assays and in vivo BM transplantation to monitor disease development and treatment response (Fig. 2A). Deletion of Dusp1 significantly reduced the JAK2V167F (>90%) and MPLW515L (>70%) induced CFUs compared to vector control, cells expressing MSCV-Ires-GFP (Fig. 2B). As reported earlier [3, 29], mice transplanted with wild-type Kit+ cells expressing Jak2V617F developed PV exhibiting higher RBC, Hb and HCT levels with a modest increase in WBC levels while mice recipients of MplW515L expressing Kit cells are more prone to develop leukemia (elevated WBCs) besides inducing the phenotypic characteristics of MPN, elevated RBCs, Hb, and HCT (Fig. 2C, D and Supplementary Fig. 4A–D). Unexpectedly, we observed Dusp1 deficiency abolished Jak2V617F-induced MPN, while MPLW515L-induced leukemogenesis was unaffected (Fig. 2E–H and Supplementary Fig. 4E–H). Strikingly, Jak2V617F expressing cells lacking Dusp1 were gradually removed from the bone marrow compared to control (Dusp1 deficient c-Kit+ cells expressing vector), suggesting that Dusp1 deletion is synthetic lethal to Jak2V617F expression (Fig. 2E, H). To ensure that Jak2V617F expressing cells were completely eradicated and mice were cured of the disease, secondary transplantation was performed. Secondary recipients of wild-type primary BM cells expressing Jak2V617F showed normal engraftment and disease development (increased RBCs, Hb, and HCT) while mice recipients of Jak2V617F primary BM cells lacking Dusp1 exhibited disease-free survival without any trace of Jak2V617F expressing cells determined by quantitation of GFP-positive cells (data not shown). Altogether, these data provide evidence that deletion of Dusp1 is synthetic lethality to Jak2V617F induced MPN.
Fig. 2. Deletion of Dusp1 is synthetic lethal to JAK2V617F induced MPN.

A An schematic showing the experimental design for evaluating the role of DUSP1 in mouse model of MPN. B Percent CFUs from the C57Bl/6-WT and Dusp1−/− Kit+ cells expressing Jak2V617F and MPLW515L. The presented data are the mean colony number from two independent experiments ± SD. C–E MPN development in mice transplanted with wild-type BM-derived Kit+ cells expressing Jak2V617F and MPLW515L. C Peripheral blood smears showing blast cells from the transplanted mice at week six. D WBC levels determined biweekly. E Graph showing GFP+ cells as a surrogate leukemic burden from the peripheral blood. Dotted lines represent normal WBC levels. Representative data are from the two independent transplant experiments. F–H. Mice transplanted with Dusp1 deficient BM-derived Kit+ cells expressing Jak2V617F and MPLW515L. Note, normalized blood count from the mice recipients of Jak2V617F expressing cells. F Peripheral blood smear from the transplanted mice at week six. G Graph showing the normalized and reduced WBC levels in Jak2V617F and MPLW515L transplanted mice, respectively. H Graph showing gradual depletion of GFP+ cells expressing Jak2V617F but not the MPLW515L or pMIG transplanted Kit+ cells. Representative data are from two independent experiments (five mice per group) shown as the means ± SD. *p < 0.05, **p < 0.01 and ***p < 0.001.
Next, we examined the Dusp1 dependency using a Jak2V617F/+ knock-in mouse model to rule out retroviral-mediated bias in cellular transformation. The Dusp1−/−/RosaCreER mice were bred with Jak2floxed/+ mice [30] to generate Dusp1−/−/Jak2floxed/+/RosaCreER. Bone marrow cells were harvested from the Dusp1−/−/RosaCreER, Jak2floxed/+/RosaCreER, and Dusp1−/−/Jak2floxed/+/RosaCreER mice, treated overnight with 4-OHT to generate Jak2V617F/+. One million Jak2V617F/+ (CD45.2) recombined cells were mixed with equal amount of normal BM cells from the BoyJ (CD45.1) mice and transplanted in lethally irradiated BoyJ recipients (Fig. 3A). Mice recipients of Jak2V617F/+ cells developed all features of MPN as described earlier [30] (Fig. 3). In contrast, mice transplanted with Jak2V617F/+/Dusp1−/− cells did not develop the disease. Instead, these cells were gradually depleted and cleared from the bone marrow within thirty weeks, determined by the levels of CD45.2 (Fig. 3B). As reported earlier, the mice transplanted with Dusp1−/−/RosaCreER exhibited persistent engraftment like wild-type cells15. Altogether these results provide clear evidence that Dusp1 confers dependence to Jak2V617F induced MPNs, and its deletion is synthetic lethal.
Fig. 3. Dusp1 deletion eradicates Jak2V617F expressing primary bone marrow cells.

A An schema showing the experimental design for evaluating the role of Dusp1 in Jak2V617F transgenic mouse model. One million Jak2V617F (CD45.2) cells mixed with normal boyJ (CD45.1) BM cells were transplanted in irradiated BoyJ mice. Engraftment and levels of Jak2V617F cells were determined after four weeks of transplantation. Drug treatments were started after week eight and continued for forty weeks. B Graph showing the CD45.2 levels in transplanted mice exhibiting normal progression of wild-type Jak2V617F and Dusp1−/− cells while Jak2V617F/Dusp1−/− cells are gradually depleted and completely removed by 30th week. Note, ruxolitinib treatment in contrast causes rapid progression of Jak2V617F cells which is likely due to higher IC50 values of Jak2V617F for ruxolitinib compared to normal cells. Shown are the WBC (C), RBC (D), HCT (E), and platelets (F) in transplanted mice with and without treatment. Mice transplanted with Jak2V617F/+/Dusp1−/− cells do not show any sign of MPN. As expected, mice transplanted with Jak2V617F/+ cells developed all signs of MPN, increased RBC, WBCs and HCT. Representative data are from two independent experiments (five mice per group) shown as the means ± SD. *p < 0.05, **p < 0.01 and ***p < 0.001.
Chemical inhibition of DUSP1 by BCI suppresses the MPN
To determine the potential of Dusp1 targeting towards clinical development, a small molecule DUSP1 inhibitor (BCI) [21] was evaluated alone and in combination with ruxolitinib in a competitive transplant model. One million primary bone marrow cells from the Jak2V617F/+/RosaCreER (CD45.2) mice or Jak2V617F/+/Dusp1−/−/RosaCreER (CD45.2) with one million normal bone marrow cells from the BoyJ mice (CD45.1) were transplanted in irradiated BoyJ mice (Fig. 4A). Mice recipients of Jak2V617F/+/RosaCreER BM cells treated with ruxolitinib (100 mg/kg daily) unexpectedly displayed increased Jak2V617F allele burden (CD45.2) with progressive MPN phenotype compared to the wild-type cells, CD45.1 (Fig. 3B–F). Thus, recapitulating the in vitro observation of ruxolitinib resistance in MPN cells (Fig. 1C). Even Jak2V617F cells lacking Dusp1 showed prolonged persistence with ruxolitinib treatment than the vehicle-treated control (Fig. 3B–F). Conversely, mice treated with BCI alone exhibited modest reduction in Jak2V617F allele burden while suppressing the WBC and HCT levels (Fig. 4B–E). Unfortunately, all treated mice displayed progressive disease. Even the combination treatment, BCI+Ruxolitinib, failed to exert clonal selectivity and all treated mice exhibited progressive disease (Fig. 4B–E). Given that BCI also inhibits DUSP6 besides inhibiting Dusp1. that causes phospho-Erk1/2 rebound, it is likely possible that a combination of BCI with MEK/ERK inhibitors may effectively deplete the JAK2 mutant cells. To test this, mice were treated with BCI+Trametinib combination showed more significant suppression of WBCs, RBCs, and HCT levels compared to BCI alone or BCI+ Ruxolitinib treatment. However, clonal selectivity and antileukemic effect of BCI+Trametinib treatment was not different than the BCI or BCI+ Ruxolitinib. Nonetheless, despite enhanced myelosuppressive response of BCI+Trametinib combination, all treated mice displayed progressive disease. Altogether, these data suggest that chemical inhibition of DUSP1 by BCI is not effective as noted with genetic deletion.
Fig. 4. Chemical inhibition of DUSP1 by BCI suppresses the MPN but lacks clonal selectivity.
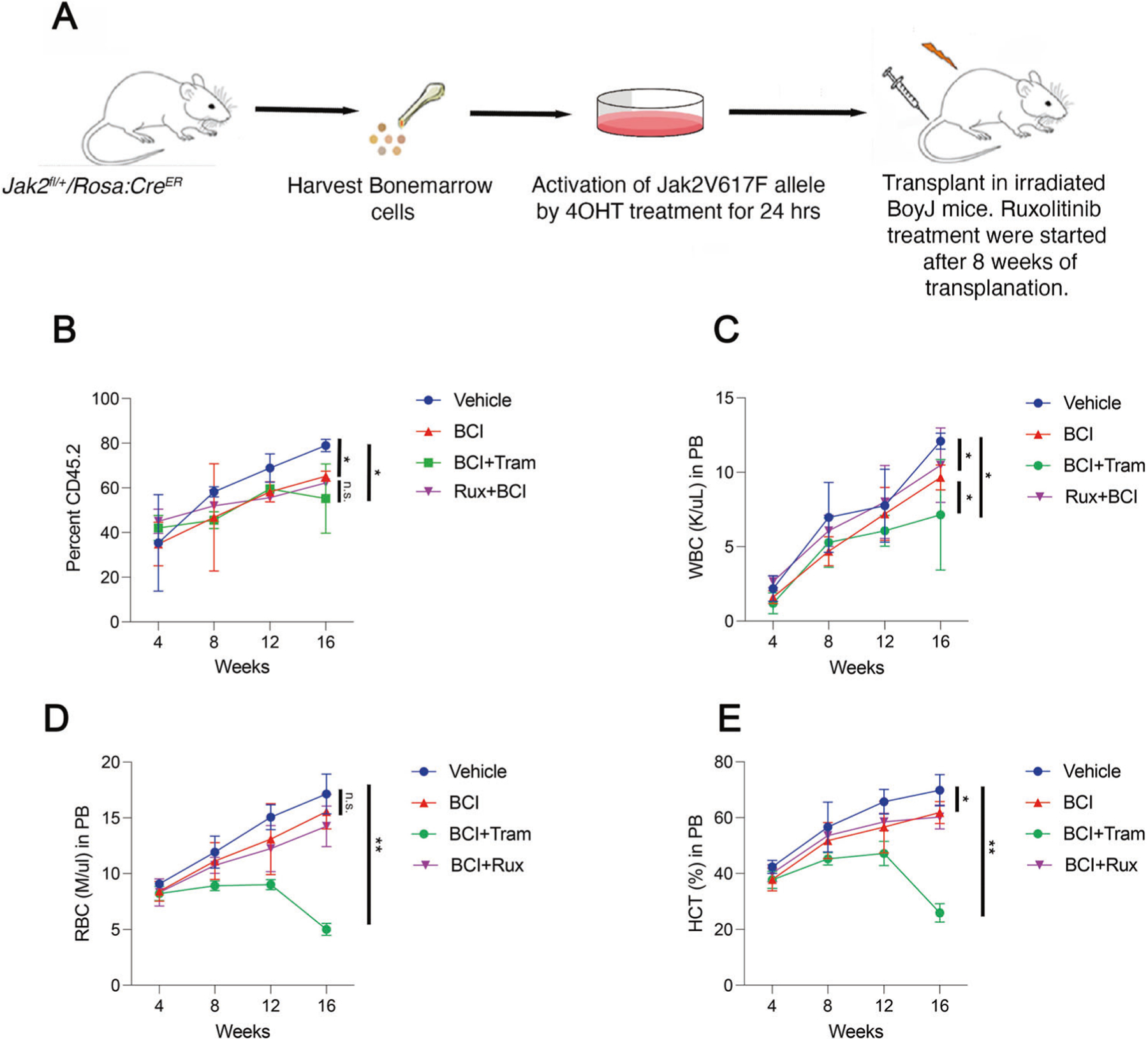
A An schema showing the evaluation of DUSP1 inhibitor (BCI) in Jak2V617F/+ induced MPN. B After 8 weeks of transplantation blood chimerism for CD45.2 were assessed, and mice were grouped for treatment. Graph showing the CD45.2 levels in drug treated and vehicle-treated mice. BCI treatment modestly suppressed the Jak2V617F/+ clone but all transplanted mice showing progressive disease. Even the combination of BCI with ruxolitinib or Trametinib failed to fully suppress the Jak2V617F/+ clone. C Graph showing WBC mice in treated and control mice. Note BCI treatment alone suppresses the WBCs but in combination with trametinib suppression is higher. Graphs are showing the RBC (D) and HCT (E) levels in treated and control mice. Treatment with BCI alone or with ruxolitinib is ineffective in suppressing the RBCs, however, in combination with trametinib it significantly suppressed the HCT and RBC levels. Representative data are from two independent experiments (three mice per group) shown as the means ± SD. *p < 0.05, **p < 0.01 and ***p < 0.001.
Enhanced pERK and BCL2 expression due to BCI treatment abrogate Jak2V617F clonal selectivity. Next, we sought to understand how Dusp1 deletion confers lethality to Jak2V617F expressing cells and why its chemical inhibition is ineffective. Because BCI inhibits both Dusp1 and Dusp6, we hypothesized that inhibition of Dusp6 abolished Dusp1 dependency in Jak2V617F cells. We reasoned that a comprehensive analysis of Dusp1 and Dusp6 substrates from the MAPK pathway in cells lacking Dusp1−/−, Dusp6−/− and Dusp1−/−/Dusp6−/− (genetic equivalence of BCI treatment) and cells treated with BCI will illuminate the underlying mechanisms driving synthetic lethality and why BCI treatment is ineffective in targeting Jak2V617F clones (Fig. 5A, C). Bone marrow derived Kit+ cells from the wild-type, Dusp1−/−, Dusp6−/− and Dusp1−/−/Dusp6−/− mice were transduced with either pMSCV-ires-GFP (pMIG) or pMSCV-Jak2V617F-ires-GFP retroviruses followed with cell sorting of GFP expressing cells. Total cell extracts from these GFP-positive cells were analyzed for MAPK signaling pathway components by western blotting. As reported earlier [21], deletion of Dusp1 resulted in higher phospho-p38 with modest increase in phospho-JNK while phospho-ERK levels were not changed compared to wild-type vector transduced control cells (Fig. 5A, B). Expression of Jak2V617F in wild-type cells displayed increased pERK levels and significantly suppressed the phopsho-p38 while pJnk levels were not significantly affected. In contrast, Jak2V617F expression in Dusp1 deficient cells exhibited greatly enhanced p-p38 with a modest increment in pJnk levels, while pErk levels remained the same (Fig. 5B). In contrast, the deficiency of Dusp6 in the context of Jak2V617F expression exhibited strong suppression of phopsho-p38 levels and modest reduction in pErk1/2 levels while pJnk levels remained the same compared to wild-type cells expressing pMIG or Jak2V617F (Fig. 5B). Interestingly, expression of Jak2V617F in double knock-out (Dusp1−/−/Dusp6−/−) cells displayed significantly enhanced pErk levels compared to wild-type and Dusp1−/− while pJnk levels were unaltered compared to Dusp1 deficient cells. Perhaps more importantly, phospho-p38 levels are significantly suppressed compared to Jak2V617F expressing Dusp1−/− cells. Together these data suggest that concomitant genetic inhibition of both Dusp1 and Dusp6 enhances Erk activation while suppressing the p38 activity in the context of Jak2V617F expression (Fig. 5B). In contrast, Dusp1−/−/Dusp6−/− expressing vector exhibited increased expression of both phospho-p38 and pErk1/2. Nonetheless, activation of Erk1/2 was lower in the normal cells compared to Dusp1−/−/Dusp6−/− expressing Jak2V617F (Fig. 5B). Given that both p38 and Erk regulate the intrinsic apoptotic activity by modulating the turnover of p53 and Bcl2 family members [25, 28, 31–34], their expression and phosphorylation were evaluated by immunoblotting. Expression of Jak2V617F induced the expression of Bcl-xl, Bax and Mcl1. We observed increased expression of proapoptotic protein Bax in Jak2V617F/Dusp1−/− cells while double knock-out cells exhibited lower Bax expression compared to Dusp1−/− alone. Importantly, we noted increased expression of Bcl2 and Bcl-xl in Jak2V617F/Dusp1−/−/Dusp6−/− cells and (Fig. 5B), which suggests simultaneous inhibition of both Dusp1 and Dusp6 would drive higher apoptotic threshold by inducing the expression of Bcl2 and Bcl-xl expression. While we noted induce expression of Mcl1 by Jak2V617F signaling, its expression was not affected by deletion of either Dusp1 or Dusp6. Strikingly, we observed a complete suppression of p53 phosphorylation at serine 15 (S15) in wild-type cells expressing Jak2V617F (Fig. 5B). Since p38 directly phosphorylates p53 at the S15, which prevents its interaction with MDM2 and degradation. Consequently, wild-type cells expressing Jak2V617F suppressed p38 activity by Dusp1 that resulted in enhanced p53 degradation due to reduced phosphorylation at serine 15 (Fig. 5B). In contrast, DUSP1 deficiency in the context of Jak2V617F expression displayed increased p38 activity and enhanced p53 phosphorylation that induced its expression, which led to selective demise of Jak2V617F clones (Fig. 5B).
Fig. 5. Activation of pERK1/2 due to off-target inhibition of DUSP6 by BCI abrogated its antileukemic response.

A A model depicting p38 and Jnk1/2 activation following Dusp1 deletion. B Immunoblots from primary BM-derived Kit+ cells showing enhanced activation of Erk1/2, suppression of 38 and downregulation of phospho-p53 at S15 in WT cells expressing Jak2V617F. Dusp1 deletion activates 38 that selectively induces the expression of total and phospho-p53 in Jak2V617F expressing cells compared to vector controls. C A model depicting chemical inhibition of DUSP1 and DUSP6 by BCI activated pErk1/2 and failed to selectively activate p38 in Jak2V617F expressing cells. D Immunoblots showing the BCI treatment alone, nonetheless, activated phospho-p38 but lacked selectivity as both vector and Jak2V617F expressing cells displayed equal phospho-p38 and p53 levels. Importantly, BCI treatment alone induced Bcl2 expression displayed significantly higher pErk1/2 levels in Jak2V617F expressing cells compare to vector control. Treatment with trametinib alone suppressed the pErk1/2 but in combination with BCI Jak2V617F expressing cells retained higher pErk1/2 possibly due to inhibition of Dusp6. E Immunoblots showing that treatment with BCI, Trametinib and in combination induce the expression of antiapoptotic proteins, Mcl1 and Bcl-xl. F–I MPN development in mice transplanted with BM-derived Kit+ cells from the wild-type and Dusp6−/− mice expressing Jak2V617F, MPLW515L, and CALRmut. F Graph showing progression of leukemic cells (GFP+ cells) in mice recipients of both wild-type and Dusp6−/− donors. Note, Dusp6 deficiency causes aggressive leukemia in mice transplanted with MPLW515L while mice recipients of Jak2V617F kit cells lacking Dusp6 do not show any difference compared to wild-type donor. G Graph showing WBC levels in Jak2V617F and MPLW515L transplanted mice. Note significantly higher WBC levels in MPLW515L transplanted mice lacking DUSP6. H Graph showing significantly higher RBC levels in both Jak2V617F and MPLW515L transplanted mice in the absence of DUSP6 compared to vector control. I Graph showing significantly increased HCT levels in both Jak2V617F and MPLW515L transplanted mice lacking DUSP6. J Graphs showing progressive disease transplanted with Jak2V617F lacking both Dusp1 and Dusp6. Representative data are from two independent experiments (three mice per group) shown as the means ± SD. *p < 0.05, **p < 0.01 and ***p < 0.001.
To examine why chemical inhibition of DUSP1 by BCI was ineffective in targeting the Jak2V617F expressing cells, MAPK signaling and levels of p53 and Bcl2 were evaluated in cells treated with BCI alone and with MEK/ERK inhibitors (Fig. 5C). BCI treatment alone exhibited increased phospho-p38 in both vector and Jak2V617F expressing cells compared to vehicle control (Fig. 5D). As expected, it also significantly increased pErk levels in Jak2V617F expressing cells likely due to the inhibition of Dusp6 (Fig. 5D). Surprisingly, BCI treatment alone significantly induced Bcl2, Bcl-xl, and Mcl1 levels (Fig. 5D, E). These data imply that sustained Erk activity in Jak2V617F expressing cells due to Dusp1 and Dusp6 inhibition induces antiapoptotic Bcl2 proteins and disrupted the selective activation of p38 mediated p53 stabilization. This resulted in a loss of therapeutic window and clonal selectivity, as an equal amount of p53 protein was observed in both vector and JakV617F-expressing cells (Fig. 5D). We sought to test whether inhibition of Mek/Erk with trametinib would restore the stabilization of p53. Cells treated with trametinib (Mek inhibitor) alone, nonetheless, exhibited modest increase in phospho-p38 and p53 levels in Jak2V617F expressing cells compared to vector control, but also displayed modest induction in Bcl2 and Mcl1 levels (Fig. 5D, E). Paradoxically, treatment with Trametinib+BCI combination resulted in pErk rebound (Fig. 5D). Consequently, it failed to achieve selective stabilization of p53 in Jak2V617F expressing cells. Similarly, Erk inhibitor in combination with BCI was ineffective in suppressing the pErk and selective stabilization of p53 in Jak2V617F expressing cells (data not shown). Altogether these data imply that Dusp6 functions as tumor suppressor in MPN. However, a recent study using BCI, which inhibits both Dusp1 and Dusp6 concluded that Dusp6 drives leukemogenesis and resistance to Jak2 inhibition in MPN [35]. To rule out this discrepancy, MPN development were assessed using bone marrow cells from Dusp6 deficient mice. Bone marrow derived c-Kit+ cells from the wild-type and Dusp6 deficient mice were transduced with retroviruses pMSCV-Ires-GFP, Jak2V617F-Ires-GFP and MPLW515L-ires-GFP. Eighty to one-hundred thousand FACS-sorted GFP expressing cells were transplanted in the recipient mice. As noted earlier, mice transplanted with wild-type c-Kit+ cells expressing Jak2V617F and MPLW515L developed MPN (Fig. 5F). Notably, mice recipients of Jak2V617F and MPLW515L cells lacking Dusp6 exhibited more aggressive MPN phenotype, significantly increased WBC, RBC and HCT (Fig. 5G–I). These data provide a direct and clear evidence that DUSP6, unlike DUSP1, functions as a tumor suppressor in MPN.
To ensure that the inhibition of DUSP6 abrogated BCI treatment, mice were transplanted with Jak2V617F expressing c-Kit+ cells lacking both Dusp1 and Dusp6. As expected, mice transplanted with Dusp1−/−/Dusp6−/− cells expressing Jak2V617F exhibited progressive disease (Fig. 5J). Thus, confirming that the inhibition of DUSP6 abrogates DUSP1 dependency in Jak2V617F induced MPN. Together, these data prove that the off-target inhibition of DUSP6 by BCI causes pErk rebound that resulted in higher expression of Bcl2 and Bcl-xl expression and non-selective p53 stabilization.
Ectopic expression of Dusp6 and BCI treatment selectively eradicated the Jak2V617F cells
To further confirm that the inhibition of DUSP6 abolishes Jak2V617F clonal selectivity, we transplanted Jak2V617F/+ RosaCreER c-Kit+ cells expressing pMSCV-Ires-cherry (vector) or pMSCV-Dusp6-cherry in C57Bl/6 recipient mice (Fig. 6A). Because Dusp6 overexpression will inhibit pERK levels, as expected, mice recipients of Jak2V617F/+ cells overexpressing Dusp6 displayed slower disease progression with a reduced leukemic burden (Fig. 6B). Strikingly, BCI treatment of mice transplanted with Dusp6 overexpressing Jak2V617F/+ cells not only normalized the WBC, RBC, HCT, and platelets levels but also selectively eradicated the Jak2V617F/+ cells (Fig. 6C-F). To confirm that the JAK2 mutant cells are fully eradicated, secondary transplantation was performed. Secondary bone marrow recipients of BCI-treated Jak2V617F/+ +Dusp6 overexpressing cells did not show any presence of cherry-positive cells determined by FACS while mice recipients of vehicle-treated mice Jak2V617F/++Dusp6 overexpression, exhibited robust engraftment (Fig. 6G). Thus, confirming that Dusp6 overexpression and BCI treatment selectively eradicated the Jak2V617F/+ cells. Altogether these data provide evidence that Dusp6 functions as a tumor suppressor in the context of Jak2V617F/+, and its off-target inhibition by BCI abolishes DUSP1 dependency in Jak2V617F/+ driven MPN.
Fig. 6. Dusp6 overexpression and BCI treatment eradicated the Jak2V617F clone.
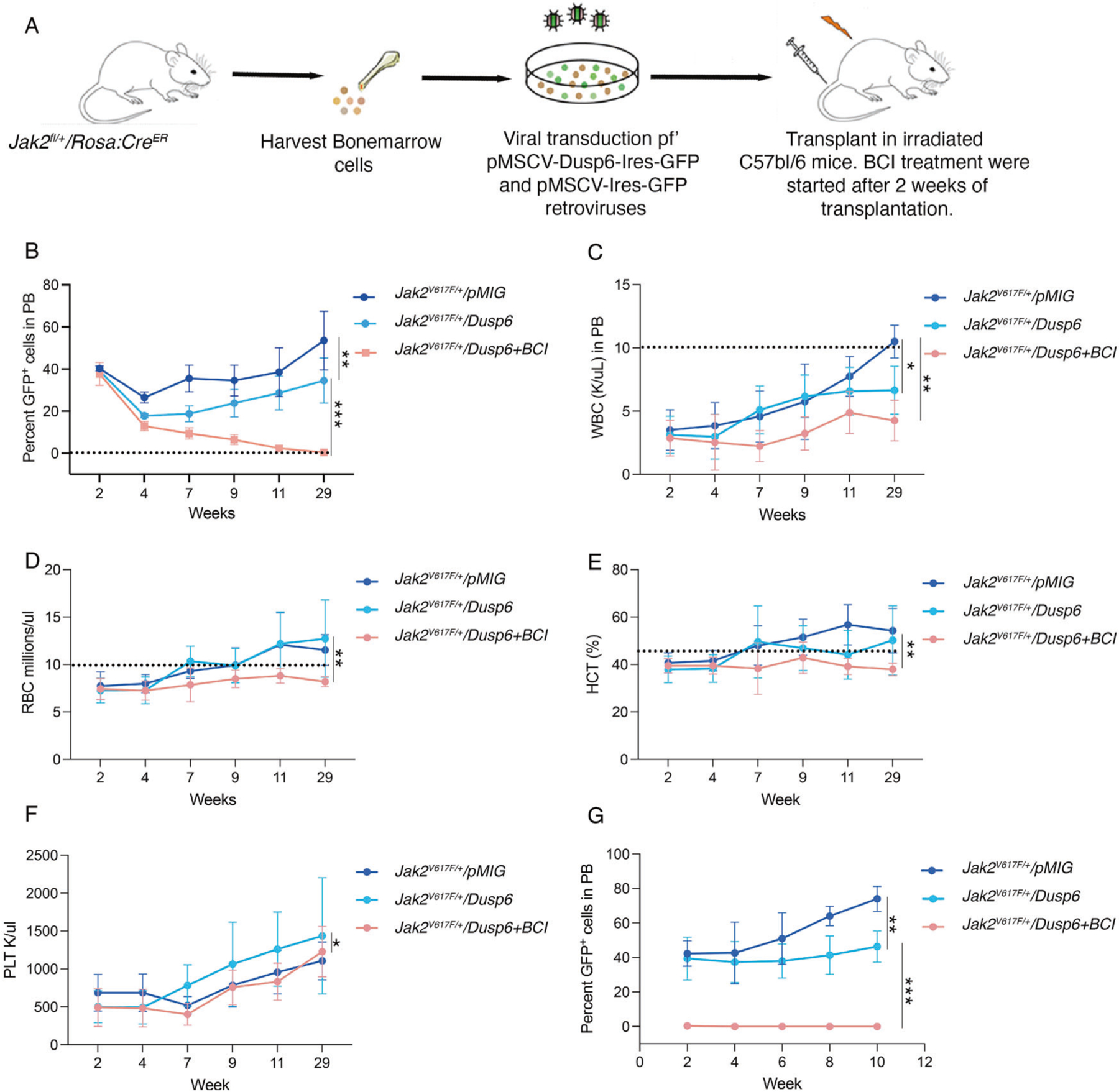
A An schema showing the experimental design for evaluating the role of Dusp6 in Jak2V617F transgenic mouse model. BM-derived Kit+ cells from Jak2V617F/+ mice were transduced with pMIC or pMSCV-Dusp6-Ires-cherry. One hundred thousand cherry-positive cells were transplanted in each lethally irradiated C57Bl/6 recipient mice. Engraftment and levels of Jak2V617F-pMIG or Jak2V617F+Dusp6 cells were determined after two weeks of transplantation by examining the cherry-positive cells. BCI treatment was started after two weeks of transplantation. B Graph showing the cherry-positive cells in transplanted mice exhibiting normal disease progression in mice transplanted with Jak2V617F-pMIG cells while it is significantly suppressed in mice recipients of Jak2V617F+Dusp6 cells. Strikingly, BCI treatment of Jak2V617F+Dusp6 eradicated the MPN cells by 14th week. And cured the mice as treatment discontinuation after week 14th did not result in disease relapse. Shown are the WBC (C), RBC (D), HCT (E), and platelets (F) in transplanted mice with and without BCI treatment. Note, overexpression of Dusp6 alone suppresses the WBC levels but not the RBCs, HCT and PLTs while BCI treatment in the context of Dusp6 overexpression exhibited normal blood counts (dotted lines are showing the normal range). G Peripheral blood chimerism of cherry-positive cells in secondary recipients of primary transplants shown in (B). Note slow progression of Dusp6 overexpressing Jak2V617F cells while BCI treatment cured the mice. Representative data are from two independent experiments (five mice per group) shown as the means ± SD. *p < 0.05, **p < 0.01 and ***p < 0.001.
DISCUSSION
Unlike the clinical efficacy of ABL inhibitors in CML, treatment outcomes to JAK2 inhibitors are limited to reducing the spleen size and alleviating the constitutional symptoms [36, 37]. Importantly, Jak inhibitors failed to induce cellular or molecular remission as they lacked clonal selectivity [38]. Besides, resistance, disease relapse, and treatment-related toxicities resulting in treatment discontinuation within months are common [39]. The persistence of Jak2V617F clones is mainly responsible for treatment failure. Therefore, therapeutic approaches are needed to eradicate the JAK2 mutant clones for durable or curative response.
Efficacy of TKI treatment is mediated by oncogene addiction [13, 40, 41]. However, genetic, cellular, and environmental contexts alter the oncogene dependence that results in treatment failure. For instance, cancer stem cells from both leukemia and solid tumors are intrinsically resistant to TKI treatment where growth-factor signaling abrogates oncogene dependence that causes intrinsic TKI resistance [16–18, 21]. MPNs are considered as oncoinflammatory disorders where malignant clones are the main source and target of cytokine storm acting both locally and systemically [42]. Significantly elevated levels of almost fifty different cytokines have been reported in MPN patients by different groups [42, 43]. Among these are hematopoietic cytokines, such as IL3, TPO, GM-CSF, HGF, and TGFβ that are implicated in abrogating TKI response were noted [21]. As a proof, we provide evidence that the addition of IL3 is sufficient to abrogate TKI response in BaF3 cells. Similarly, hematopoietic cytokines in combination significantly increased the IC50 of ruxolitinib in human and primary mouse MPN cells (Fig. 1C), which also implied that continued ruxolitinib treatment not only cause persistence rather it will exert Jak2V617F clonal dominance. As envisioned from the in vitro data, ruxolitinib treatment exhibited clonal dominance of Jak2V617F mutant clones in competitive transplants (Fig. 3). Comparative whole genome gene expression analysis of Jak2V617F and MplW515L in primary cells revealed that both oncogenes, besides commonly regulating a significant number of genes, also modulate the expression of a distinct set of genes. As reported earlier [23], we observed increased expression of key signaling intermediates driving JAK-STAT pathway by both oncogenes. In addition, we noted increased expression of, Csf1r, Csf3r and HRas implicated in driving JAK2V617F-negative MPN which is shown to be addicted to enhanced MAPK signaling [44, 45]. Collectively, these data support that oncogenic JAK2V617F and MPLW515L signaling beside activating JAK-STAT pathway also modulate MAPK signaling. Consequently, MAPK pathway inhibitors targeting BRAF, MEK and ERK alone or in combination with ruxolitinib have shown better therapeutic response than ruxolitinib alone [9, 11]. However, both MEK and ERK inhibitors in combination with ruxolitinib, nonetheless, exerted improved efficacy in ameliorating the disease phenotype but lacked clonal selectivity [9, 11, 45].
Pharmacological targeting of MEK/ERK and JAK2 either alone or in combination exert cytostatic response. Therefore, treatment outcomes to cytostatic drugs are short-lived as most patients develop resistance within short duration. The reasons for cytostatic response exerted by MER/ERK or JAK2 inhibitors is not fully understood. Enhanced MAPK-ERK activity is one of the central features in many cancers implicated in driving proliferation and survival by modulating the apoptotic threshold mainly by stabilizing and/or activating the antiapoptotic proteins, such as BCL2 family members and repressing the proapoptotic proteins, such as BIM [46–48]. Paradoxically, sustained ERK activation also induces a robust apoptotic response, which will be counter-selective for cellular transformation [25, 27, 49, 50]. To counter this, most cancers induce MAPK negative regulators to fine-tune the MAPK signaling output to suppress ERK-induced apoptotic signaling while selectively promoting survival and proliferation [51, 52]. We observed a significant induction of MAPK signaling genes in both JAK2V617F and MPLW515L cells where JAK2V617F signaling exhibited a more robust induction of MAPK network genes compared to MplW515L (Supplementary Fig. 2A, 18 genes are not induced in MplW515L cells from the MAPK network; Supplementary Fig. 3A, 7 genes are under-expressed in MplW515L cells from the ERK1/2 network). Consequently, both JAK2V617F and MPLW515L cells exhibited induced expression of several MAPK negative regulators, including DUSP family members (Supplementary Fig. 1H, I). Interestingly, a recent study implicated the role of DUSP1 in protecting the Jak2V617F MPN cells from cellular stress caused by the inflammatory cytokines [53]. Importantly, we observed induced expression MAPK negative regulators (Sfrp1, Sfrp5, Mapk8ip1 and Nppa) in Jak2V617F cells while their expressions are significantly suppressed in MPLW515L cells which may be a reason for conferring the dependence of JAK2V617F to DUSP1. SFRP proteins negatively regulate Wnt/β–catenine signaling by preventing the Wnt binding to its receptors, Fzds. There is extensive crosstalk between MAPK and WNT/β–catenine signaling [54]. Depending on the cellular context, a positive interaction between MAPK and WΝΤ/β–catenine drives cellular transformation, while a negative interaction promotes anti-tumor response [54]. For instance, overexpression of SFRP1 in colon cancer cells significantly decreased the protein levels of WNT, β-catenin and apoptosis-related proteins, including MMP2, MMP9, Twist, CDK1, TGF, and BCL2 [55]. Likewise, reduced expression of scaffold protein MAPL8ip1 would result in reduced JNK activation that will alter apoptotic threshold due to reduced AP1 activation [21]. Cumulatively induced expression of these negative regulators in Jak2V617F cells makes it more vulnerable to MAPK negative feedback perturbations. Consequently, the deletion of Dusp1 resulted in the selective demise of Jak2V617F but not the MplW515L cells. Future studies will uncover the molecular basis of altered dependence on MAPK negative feedback in JAK2V617F and MPLW515L. Perhaps suppressing β-catenin by overexpressing the Sfrp1 in combination with DUSP1 depletion may help in eradicating the MPLW515L cells.
Chemical inhibition of DUSP1 by BCI failed to recapitulate DUSP1 deletion-induced synthetic lethality to JAK2V617F. Because BCI inhibits both DUSP1 and DUSP6 [21], we hypothesized that simultaneous inhibition of DUSP6 induces ERK1/2 activity, neutralizing the effect of DUSP1 inhibition. As expected, BCI treatment or genetic deletion of Dusp1−/−/Dusp6−/− in the context of Jak2V617F expression significantly increased ERK1/2 activity (Fig. 5). In contrast, cells lacking Dusp1 only exhibited strong p38 activity compared to Jak2V617F expressing wild-type and Dusp6 deficient cells. Even cells lacking both Dusp1 and Dusp6 exhibited suppressed p38 activity in the context of Jak2V617F expression. This suggests a complex regulatory network between Dusp1 and Dusp6 modulating the ERK1/2 and p38 activity in normal and oncogenic contexts. Mechanistically, inhibition of DUSP1 activated p38 that phosphorylates p53 at S15, which blocks its MDM2 mediated degradation. Consequently, higher p53 expression selectively induced apoptosis in JAK2V617F expressing cells as vector expressing Dusp1 deficient cells did not show induced p53 expression. To ascertain the role of p38 in p53 stabilization, p38 inhibition and overexpression studies were performed (Supplementary Fig. 5A). As expected, p38 inhibition increased the CFU numbers in wild-type cells while expression of JAK2V617F significantly increased the CFU number and did not show any response to p38 inhibition as p38 is already inhibited into these cells. In contrast, inhibition of p38 in Jak2 expressing cells lacking Dusp1 exhibited increased CFU while its inhibition in control cells had no effect. Interestingly, ectopic expression of MAPK14 (p38-α) suppressed the CFU numbers in Jak2V617F expressing wild-type cells while its overexpression did not show any change in CFU numbers in control and Dusp1 deficient JAK2V617F expressing cells (Supplementary Fig. 5B). These results provide direct evidence that downregulation of p53 under JAK2V617F signaling is caused by Dusp1 mediated p-p38 blockade.
The enhanced pERK1/2 activity due to BCI treatment suggested that perhaps a combination of BCI with MEK or ERK inhibitor may likely restore clonal selectivity. Unfortunately, treatment with BCI or trametinib alone significantly induced the expression of BCL2 and lost selective stabilization of p53 likely due to pERK rebound in both normal and Jak2V617F expressing cells (Fig. 5D). This prompted us to test whether a combination of BCI with MDM2 inhibitor (idasanutlin) and BCL2 inhibitor (venetoclax) might be effective in depleting the Jak2V617F clones. In vitro evaluations showed significant suppression of Jak2V617F cells by BCI+Idasanitulin+venetoclax combination (Supplementary Fig. 6A). Mice treated with idasanutlin+venetocalx or BCI+idasanutlin+venetoclax showed significant suppression compared to vehicle-treated mice but all treated mice displayed progressive disease (Supplementary Fig. 6B). Unfortunately, venetoclax+ idasanutlin treatment exerted significant toxicity (lethargy and scruffy coat) that resulted in treatment discontinuation and prevented us from further evaluation. Our data suggest that the inefficacy of BCI treatment was due to DUSP6 inhibition. Dusp6 has been shown to have tumor suppressor as well as oncogenic role in different tumors [56–58]. A recent report during the preparation of this manuscript implicated DUSP6 in leukemic transformation of MF to secondary AML (sAML) and conferring resistance to JAK2 inhibitors [59]. They performed both shRNA-mediated knockdown and chemical inhibition for validation studies. Both shRNA-mediated knockdown and a higher concentration of BCI are prone to have off-target inhibitory effect. Using DUSP6 knock-out mice, we clearly demonstrate that the loss of DUSP6 had no effect in MPNs induced by JAK2V617F or MPLW515L rather its absence causes aggressive disease progression compared to wild-type BM donors, which supports its role as a tumor suppressor. Besides, using mice lacking both Dusp1 and Dusp6 mimicking the BCI treatment, we provide evidence that inhibition of DUSP6 either chemically or genetically abolishes DUSP1 dependency in Jak2V617F induced MPN. Moreover, ectopic overexpression of Dusp6 in Jak2V617F cells restored BCI sensitivity. Mice transplanted with Jak2V617F/+ cells overexpressing Dusp6 exhibited disease suppression likely due to inhibition of pERK1/2. Strikingly, BCI treatment of mice transplanted with Dusp6 overexpressing Jak2V617F cells cured the mice of disease. These data provide evidence that the inefficacy of BCI was due to pERK rebound because of Dusp6 inhibition. Importantly, our study demonstrates that the dynamic balance between Erk and p38 activation determines whether the cell survives or undergoes apoptosis. Mapk-negative regulator, DUSP1, regulates this balance to support the JAK2V617F-driven neoplasm.
In conclusion, we show that JAK2V617F signaling induces DUSP1 to fine-tune MAPK signaling output that suppresses p53 expression by enhancing its degradation. Consequently, DUSP1 confers dependence to JAK2V617F driven malignancies. Deletion of Dusp1 selectively induces p38 activity that promotes p53 phosphorylation and stabilization, which results in the selective eradication of Jak2V617F cells. In contrast, ERK1/2 negative regulator, DUSP6, functions as a tumor suppressor as its inhibition abrogated DUSP1 dependence, likely due to negative crosstalk between ERK and p38 [60]. Altogether these data provide evidence that the inhibitors selectively targeting DUSP1 would exert a durable or curative response to JAK2V617F driven MPN. Finally, our study exposes a new Achille’s heel to malignancies fueled by enhanced MAPK signaling and support for selective targeting of MAPK negative regulators for effective treatment outcomes.
Supplementary Material
ACKNOWLEDGEMENTS
Authors are thankful to Dr. Benjamin Ebert and Ann Mullaly for providing the Jak2V617F /+ mice and pMSCV-CALRwt and pMSCV-CALRmut retroviral constructs. This study was supported by grants to Mohammad Azam (M Azam) from the National Cancer Institutes at NIH (RO1CA211594) and (RO1CA250516) and to RL by MSKCC Support Grant/Core Grant P30 CA008748. MA is a recipient of the bridge award from the American Society of Hematology (ASH).
FUNDING
This study was supported by grants to MA from the National Cancer Institutes at NIH (RO1CA211594) and (RO1CA250516) and to RL.L. by MSKCC Support Grant/Core Grant P30 CA008748.
Footnotes
COMPETING INTERESTS
MA is a recipient of the bridge award from the American society of hematology (ASH). RLL is on the supervisory board of Qiagen and is a scientific advisor to Imago, Mission Bio, Bakx, Zentalis, Ajax, Auron, Prelude, C4 Therapeutics and Isoplexis. He has received research support from Abbvie, Constellation, Ajax, Zentalis and Prelude. He has received research support from and consulted for Celgene and Roche and has consulted for Syndax, Incyte, Janssen, Astellas, Morphosys and Novartis.
ADDITIONAL INFORMATION
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41375-023-01959-0.
DATA AVAILABILITY
RNA-seq data are publicly available at GSE229318. For materials and other resources, please contact the corresponding author; Mohammad.Azam@cchmc.org.
REFERENCES
- 1.Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, Huntly BJ, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7:387–97. [DOI] [PubMed] [Google Scholar]
- 2.James C, Ugo V, Le Couedic JP, Staerk J, Delhommeau F, Lacout C, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434:1144–8. [DOI] [PubMed] [Google Scholar]
- 3.Pikman Y, Lee BH, Mercher T, McDowell E, Ebert BL, Gozo M, et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 2006;3:e270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Klampfl T, Gisslinger H, Harutyunyan AS, Nivarthi H, Rumi E, Milosevic JD, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013;369:2379–90. [DOI] [PubMed] [Google Scholar]
- 5.Fisher DAC, Fowles JS, Zhou A, Oh ST. Inflammatory pathophysiology as a contributor to myeloproliferative neoplasms. Front Immunol. 2021;12:683401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wolf A, Eulenfeld R, Gäbler K, Rolvering C, Haan S, Behrmann I, et al. JAK2-V617F-induced MAPK activity is regulated by PI3K and acts synergistically with PI3K on the proliferation of JAK2-V617F-positive cells. JAKSTAT. 2013;2:e24574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bartalucci N, Guglielmelli P, Vannucchi AM. Rationale for targeting the PI3K/Akt/mTOR pathway in myeloproliferative neoplasms. Clin Lymphoma Myeloma Leuk. 2013;13:S307–9. [DOI] [PubMed] [Google Scholar]
- 8.Gerds AT, Bartalucci N, Assad A, Yacoub A. Targeting the PI3K pathway in myeloproliferative neoplasms. Expert Rev Anticancer Ther. 2022;22:835–43. [DOI] [PubMed] [Google Scholar]
- 9.Stivala S, Codilupi T, Brkic S, Baerenwaldt A, Ghosh N, Hao-Shen H, et al. Targeting compensatory MEK/ERK activation increases JAK inhibitor efficacy in myeloproliferative neoplasms. J Clin Investig. 2019;129:1596–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jayavelu AK, Schnoder TM, Perner F, Herzog C, Meiler A, Krishnamoorthy G, et al. Splicing factor YBX1 mediates persistence of JAK2-mutated neoplasms. Nature. 2020;588:157–63. [DOI] [PubMed] [Google Scholar]
- 11.Brkic S, Stivala S, Santopolo A, Szybinski J, Jungius S, Passweg JR, et al. Dual targeting of JAK2 and ERK interferes with the myeloproliferative neoplasm clone and enhances therapeutic efficacy. Leukemia. 2021;35:2875–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sawyers CL. Shifting paradigms: the seeds of oncogene addiction. Nat Med. 2009;15:1158–61. [DOI] [PubMed] [Google Scholar]
- 13.Weinstein IB. Cancer. Addiction to oncogenes–the Achilles heal of cancer. Science. 2002;297:63–4. [DOI] [PubMed] [Google Scholar]
- 14.Pagliarini R, Shao W, Sellers WR. Oncogene addiction: pathways of therapeutic response, resistance, and road maps toward a cure. EMBO Rep. 2015;16:280–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.O’Hare T, Zabriskie MS, Eiring AM, Deininger MW. Pushing the limits of targeted therapy in chronic myeloid leukaemia. Nat Rev Cancer. 2012;12:513–26. [DOI] [PubMed] [Google Scholar]
- 16.Corbin AS, Agarwal A, Loriaux M, Cortes J, Deininger MW, Druker BJ. Human chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of BCR-ABL activity. J Clin Investig. 2011;121:396–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wilson TR, Fridlyand J, Yan Y, Penuel E, Burton L, Chan E, et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature. 2012;487:505–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Straussman R, Morikawa T, Shee K, Barzily-Rokni M, Qian ZR, Du J, et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature. 2012;487:500–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Azhar M, Kincaid Z, Kesarwani M, Menke J, Schwieterman J, Ansari S, et al. Rational polypharmacological targeting of FLT3, JAK2, ABL, and ERK1 suppresses the adaptive resistance to FLT3 inhibitors in AML. Blood Adv. 2023;7:1460–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Azhar M, Kincaid Z, Kesarwani M, Ahmed A, Wunderlich M, Latif T, et al. Momelotinib is a highly potent inhibitor of FLT3-mutant AML. Blood Adv. 2022;6:1186–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kesarwani M, Kincaid Z, Gomaa A, Huber E, Rohrabaugh S, Siddiqui Z, et al. Targeting c-FOS and DUSP1 abrogates intrinsic resistance to tyrosine-kinase inhibitor therapy in BCR-ABL-induced leukemia. Nat Med. 2017;23:472–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fisher DAC, Miner CA, Engle EK, Hu H, Collins TB, Zhou A, et al. Cytokine production in myelofibrosis exhibits differential responsiveness to JAK-STAT, MAP kinase, and NFkappaB signaling. Leukemia. 2019;33:1978–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rampal R, Al-Shahrour F, Abdel-Wahab O, Patel JP, Brunel JP, Mermel CH, et al. Integrated genomic analysis illustrates the central role of JAK-STAT pathway activation in myeloproliferative neoplasm pathogenesis. Blood. 2014;123:e123–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Murphy LO, Blenis J. MAPK signal specificity: the right place at the right time. Trends Biochem Sci. 2006;31:268–75. [DOI] [PubMed] [Google Scholar]
- 25.Cagnol S, Chambard JC. ERK and cell death: mechanisms of ERK-induced cell death–apoptosis, autophagy and senescence. FEBS J. 2010;277:2–21. [DOI] [PubMed] [Google Scholar]
- 26.Marshall CJ. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell. 1995;80:179–85. [DOI] [PubMed] [Google Scholar]
- 27.Subramaniam S, Unsicker K. ERK and cell death: ERK1/2 in neuronal death. FEBS J. 2010;277:22–9. [DOI] [PubMed] [Google Scholar]
- 28.Martin P, Pognonec P. ERK and cell death: cadmium toxicity, sustained ERK activation and cell death. FEBS J. 2010;277:39–46. [DOI] [PubMed] [Google Scholar]
- 29.Zaleskas VM, Krause DS, Lazarides K, Patel N, Hu Y, Li S, et al. Molecular pathogenesis and therapy of polycythemia induced in mice by JAK2 V617F. PLoS ONE. 2006;1:e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mullally A, Lane SW, Ball B, Megerdichian C, Okabe R, Al-Shahrour F, et al. Physiological Jak2V617F expression causes a lethal myeloproliferative neoplasm with differential effects on hematopoietic stem and progenitor cells. Cancer Cell. 2010;17:584–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guo YJ, Pan WW, Liu SB, Shen ZF, Xu Y, Hu LL. ERK/MAPK signalling pathway and tumorigenesis. Exp Ther Med. 2020;19:1997–2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.De Chiara G, Marcocci ME, Torcia M, Lucibello M, Rosini P, Bonini P, et al. Bcl-2 Phosphorylation by p38 MAPK: identification of target sites and biologic consequences. J Biol Chem. 2006;281:21353–61. [DOI] [PubMed] [Google Scholar]
- 33.Whitaker RH, Cook JG. Stress Relief Techniques: p38 MAPK determines the balance of cell cycle and apoptosis pathways. Biomolecules. 2021;11:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bulavin DV, Saito S, Hollander MC, Sakaguchi K, Anderson CW, Appella E, et al. Phosphorylation of human p53 by p38 kinase coordinates N-terminal phosphorylation and apoptosis in response to UV radiation. EMBO J. 1999;18:6845–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kong T, Laranjeira ABA, Yang K, Fisher DAC, Yu L, Poittevin De La Fregonniere L, et al. DUSP6 mediates resistance to JAK2 inhibition and drives leukemic progression. Nat Cancer 2023;4:108–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Verstovsek S, Kantarjian H, Mesa RA, Pardanani AD, Cortes-Franco J, Thomas DA, et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N Engl J Med. 2010;363:1117–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pardanani A, Tefferi A, Jamieson C, Gabrail NY, Lebedinsky C, Gao G, et al. A phase 2 randomized dose-ranging study of the JAK2-selective inhibitor fedratinib (SAR302503) in patients with myelofibrosis. Blood Cancer J. 2015;5:e335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Talpaz M, Kiladjian JJ. Fedratinib, a newly approved treatment for patients with myeloproliferative neoplasm-associated myelofibrosis. Leukemia. 2021;35:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kirito K Recent progress of JAK inhibitors for hematological disorders. Immunol Med. 2022;45:1–12. [DOI] [PubMed] [Google Scholar]
- 40.Sharma SV, Settleman J. Oncogene addiction: setting the stage for molecularly targeted cancer therapy. Genes Dev. 2007;21:3214–31. [DOI] [PubMed] [Google Scholar]
- 41.Weinstein IB, Joe A. Oncogene addiction. Cancer Res. 2008;68:3077–80. discussion 80 [DOI] [PubMed] [Google Scholar]
- 42.Masselli E, Pozzi G, Gobbi G, Merighi S, Gessi S, Vitale M, et al. Cytokine profiling in myeloproliferative neoplasms: overview on phenotype correlation, outcome prediction, and role of genetic variants. Cells. 2020;9:1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang Y, Zuo X. Cytokines frequently implicated in myeloproliferative neoplasms. Cytokine X 2019;1:100005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maxson JE, Gotlib J, Pollyea DA, Fleischman AG, Agarwal A, Eide CA, et al. Oncogenic CSF3R mutations in chronic neutrophilic leukemia and atypical CML. N Engl J Med. 2013;368:1781–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rohrabaugh S, Kesarwani M, Kincaid Z, Huber E, Leddonne J, Siddiqui Z, et al. Enhanced MAPK signaling is essential for CSF3R-induced leukemia. Leukemia 2017;31:1770–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Balmanno K, Cook SJ. Tumour cell survival signalling by the ERK1/2 pathway. Cell Death Differ. 2009;16:368–77. [DOI] [PubMed] [Google Scholar]
- 47.Yue J, Lopez JM. Understanding MAPK signaling pathways in apoptosis. Int J Mol Sci. 2020;21:1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Clybouw C, Merino D, Nebl T, Masson F, Robati M, O’Reilly L, et al. Alternative splicing of Bim and Erk-mediated Bim(EL) phosphorylation are dispensable for hematopoietic homeostasis in vivo. Cell Death Differ. 2012;19:1060–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sugiura R, Satoh R, Takasaki T. ERK: a double-edged sword in cancer. ERK-dependent apoptosis as a potential therapeutic strategy for cancer. Cells. 2021;10:1–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Teixeiro E, Daniels MA. ERK and cell death: ERK location and T cell selection. FEBS J. 2010;277:30–8. [DOI] [PubMed] [Google Scholar]
- 51.Lake D, Correa SA, Muller J. Negative feedback regulation of the ERK1/2 MAPK pathway. Cell Mol Life Sci. 2016;73:4397–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Seternes OM, Kidger AM, Keyse SM. Dual-specificity MAP kinase phosphatases in health and disease. Biochim Biophys Acta Mol Cell Res. 2019;1866:124–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stetka J, Vyhlidalova P, Lanikova L, Koralkova P, Gursky J, Hlusi A, et al. Addiction to DUSP1 protects JAK2V617F-driven polycythemia vera progenitors against inflammatory stress and DNA damage, allowing chronic proliferation. Oncogene. 2019;38:5627–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Guardavaccaro D, Clevers H. Wnt/beta-catenin and MAPK signaling: allies and enemies in different battlefields. Sci Signal. 2012;5:pe15. [DOI] [PubMed] [Google Scholar]
- 55.Wang Z, Li R, He Y, Huang S. Effects of secreted frizzled-related protein 1 on proliferation, migration, invasion, and apoptosis of colorectal cancer cells. Cancer Cell Int. 2018;18:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ahmad MK, Abdollah NA, Shafie NH, Yusof NM, Razak SRA. Dual-specificity phosphatase 6 (DUSP6): a review of its molecular characteristics and clinical relevance in cancer. Cancer Biol Med. 2018;15:14–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Keyse SM. Dual-specificity MAP kinase phosphatases (MKPs) and cancer. Cancer Metastasis Rev. 2008;27:253–61. [DOI] [PubMed] [Google Scholar]
- 58.Okudela K, Yazawa T, Woo T, Sakaeda M, Ishii J, Mitsui H, et al. Down-regulation of DUSP6 expression in lung cancer: its mechanism and potential role in carcinogenesis. Am J Pathol. 2009;175:867–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kong T, Laranjeira ABA, Yang K, Fisher DAC, Yu L, Poittevin De La Fregonniere L, et al. DUSP6 mediates resistance to JAK2 inhibition and drives leukemic progression. Nat Cancer. 2022;4:108–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fey D, Croucher DR, Kolch W, Kholodenko BN. Crosstalk and signaling switches in mitogen-activated protein kinase cascades. Front Physiol. 2012;3:355. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq data are publicly available at GSE229318. For materials and other resources, please contact the corresponding author; Mohammad.Azam@cchmc.org.