

Abstract
Lung cancer’s intractability is enhanced by its frequent resistance to (chemo)therapy and often high relapse rates that make it the leading cause of cancer death worldwide. Improvement of therapy efficacy is a crucial issue that might lead to a significant advance in the treatment of lung cancer. Oncolytic viruses are desirable combination partners in the developing field of cancer immunotherapy due to their direct cytotoxic effects and ability to elicit an immune response. Systemic oncolytic virus administration through intravenous injection should ideally lead to the highest efficacy in oncolytic activity. However, this is often hampered by the prevalence of host-specific, anti-viral immune responses. One way to achieve more efficient systemic oncolytic virus delivery is through better protection against neutralization by several components of the host immune system. Carrier cells, which can even have innate tumor tropism, have shown their appropriateness as effective vehicles for systemic oncolytic virus infection through circumventing restrictive features of the immune system and can warrant oncolytic virus delivery to tumors. In this overview, we summarize promising results from studies in which carrier cells have shown their usefulness for improved systemic oncolytic virus delivery and better oncolytic virus therapy against lung cancer.
Keywords: lung cancer, oncolytic viral therapy, antibody neutralization, cell-mediated carrier, target delivery
Graphical abstract
Meuwissen and colleagues present a review of how carrier cells can protect and optimize the systemic delivery of oncolytic viruses (OVs) during therapeutic applications. OVs have great potential for immunotherapy use due to their preferential cytotoxic effects on cancer cells and are important new tools to fight lung cancer.
Introduction
Designing therapeutic agents with a high therapeutic index (i.e., efficacy against malignant cells) and minimal or no toxicity to normal cells is the main current objective for the development of novel therapies against cancer. Future therapeutic approaches must be more focused and targeted, enabling the delivery of efficient medication dosages to every tumor cell.1 Despite undeniable advancements in cancer treatment, certain cancers resist effective treatment directly or through therapy-induced remission. Although drastically better results may not be possible just now, incremental improvements are predicted with innovative or enhanced therapies using immunotherapy, cell-based medicines, oncolytic virotherapy, and hybrid techniques thereof.2 Lung tumors are among the most recalcitrant solid cancers and, although the last decade has seen a steady development of immunotherapeutic approaches against lung cancer such as immune checkpoint inhibitors (ICIs) becoming first-line standard therapies alone or in combination with long-established chemo- and radiotherapies,3 clinical course and prognosis of especially advanced lung cancer remains rather poor (Table 1). The need for new or better therapies for lung cancer is urgent and one way to alleviate this necessity could be the application of oncolytic virotherapy.
Table 1.
Treatment modality for different stages of lung cancer
| Modality | Process | Stage of disease | Reference |
|---|---|---|---|
| Non-small cell lung cancer | |||
| Surgery | Patients with entirely resectable tumors and in a good position for resection should consider surgery. When the cancer is small enough to ensure that resection will be possible, there has been little or no dissemination to nearby lymph nodes, and the patient and other tumor variables are favorable, surgical resection is advised. Thoracotomy surgery is the most popular type of surgery. The less-invasive surgery, a limited anterior thoracotomy, requires a small opening through the front of the chest. On the other hand, lung cancer patients frequently undergo a lobectomy. Segmentectomy or wedge resection may be an option for individuals with exceedingly small, early-stage lung tumors or those who cannot tolerate having a lobe removed because of compromised lung function. | Surgery is the preferred and most effective treatment for stage I and II diseases. Up to 60% of patients with stage I illness survive for 5 years. Although surgery is still the preferred treatment option, 5-year survival rates for stage II tumors are typically 30%. Resection may be viable for some stage III tumors; this should be determined individually. Regrettably, many of these patients experience recurrence despite resection. Due to its variability, it is challenging to specify specific therapy choices for cancer in stage III. Since the survival benefits of surgery have not been established, it is not recommended for individuals with stage IV illness. | Herbst et al.4,5,6; Alexander et al.5; Duma et al.4,5,6 |
| Radiotherapy | Radiation therapy (RT) is used to treat cancer to raise the likelihood of curing the disease, improve local tumor control, and provide palliative care (e.g., improve symptoms and quality of life). Brachytherapy and external beam radiation therapy are RT’s two basic delivery systems. The therapeutic ratio is determined by comparing the maximum damage that the surrounding healthy tissue can withstand to the total damage that radiation is intended to cause to the malignant cells. | At any stage, RT can be used as the first-line, curative, adjuvant, or palliative treatment for lung cancer. Even though they have stage I and stage II disease, patients judged inoperable typically receive RT (e.g., due to age). The current "gold standard" therapy for patients with stage III illness consists of RT and chemotherapy. Combining chemotherapy and radiotherapy can be considered a curative therapy for stage IIIB illness. |
Herbst et al.4,5,6; Alexander et al.5; Duma et al.4,5,6 |
| Chemotherapy |
|
According to studies, postoperative adjuvant therapy improved 5-year survival rates for patients with stage IB to stage III illness. At the same time, chemotherapy appeared to harm patients with stage IA disease. Adjuvant cisplatin-based chemotherapy is the gold standard of care for some stage I and the majority of stage II and III patients with completely resected NSCLC. Since surgery is typically not an option for patients in the stage IIIB, chemo-radiation is the only treatment method used. Despite meager survival rates, chemotherapy is the primary treatment for people with stage IV illness. | Herbst et al.4,5,6; Alexander et al.4,5 |
| Small cell lung cancer | |||
| Chemotherapy | Chemotherapy agent mixtures are typically used to treat SCLC. One drug, topotecan, a topoisomerase I inhibitor, has been FDA approved for use in SCLC as a second-line treatment. In addition, the National Comprehensive Cancer Network has approved immunotherapy drugs such as nivolumab and nivolumab plus ipilimumab as a course of treatment for SCLC patients who have progressed after receiving one or more prior regimens or who have relapsed within 6 months of receiving initial therapy. | Both mild and severe SCLC are mostly treated with chemotherapy. | van Meerbeeck et al.7; Zugazagoitia and Paz-Ares7,8,9Yang et al.7,8,9 |
| Radiotherapy | Radiation treatment and chemotherapy boost median survival to about 1.5 years for limited-stage diseases. Patients with early-stage illness are also advised to have radiation therapy to the brain. This is administered when there is no indication that cancer has progressed to the brain and chest therapy is the only treatment. | Mortality was reduced when radiation was used with chemotherapy for both extensive-stage and limited-stage illnesses. | Zugazagoitia et al.8,9,10; Yang et al.8,9,10; Wang et al.8,9,10 |
| Surgery | Most cases of SCLC cannot be treated with surgery. | Surgery, in addition to chemotherapy, may be an option for patients with stage I SCLC and no nodal involvement. | Zugazagoitia et al.8,9,10; Yang et al.8,9,10; Wang et al.8,9,10 |
Intriguingly, the first clinical observations of occasional viral infections that lead to partial and sometimes complete tumor regression in cancer patients date back more than a hundred years.11 These initial observations were then pursued by more specified clinical and preclinical experiments with various well-defined viral strains.12 Although results clearly showed the potential of virotherapy, since a distinct class of now-called oncolytic viruses (OVs) did infect and lyse cancer cells much more efficiently than healthy tissues.13 But there the problem arose. Most early oncolytic virotherapy made use of wild-type virus strains with high titers that could cause viremia resulting in severe infections and after affecting the vital organs, sometimes ended in lethality through organ failure or sepsis.13,14 However, in those early days, not only lethal virulence posed a major problem but especially the poor understanding of viral tropism for tumor cells did seriously hamper the clinical efficacy of oncolytic virotherapy.15 Only after the onset of knowledge on how to genetically modify viruses to make them conditionally replicate in specific host tumor cells and hence control their toxicity, could these newly engineered OV strains start to fulfill the original expectations of oncolytic virotherapy. Significant clinical results could then be obtained with various attenuated OV strains16 optimized for specific tumor types. However, the ultimate challenge for OV therapy is not only the eradication of locally injected tumor lesions but also all metastases, especially in advanced cancers. Yet eradication of metastases necessitates systemic OV infections, although clinical applications thereby have not been very successful so far.17 Even though OVs work well after direct injection into tumors, their systemic application shows a far lower anti-tumor efficacy. The latter effect is mainly due to OV’s susceptibility to factors of the innate and adaptive immune systems such as complement proteins, antibodies, and the reticuloendothelial system that surveys the blood circulation for oncolytic virions. More specifically, complement proteins have been found to compromise oncolytic functions of OVs.18 Nonetheless, some of the complement protein effects can be antagonized by intrinsic factors encoded by specific OVs, such as glycoprotein C of herpes simplex virus (HSV) and complement control protein VCP encoded by vaccinia virus (VV).19,20 In addition, it became clear that systemic anticancer treatment efficacies of OV platforms derived from measles virus,21 vesicular stomatitis virus (VSV),22 reovirus,23 HSV,24 adenovirus,25 and parvovirus26 are significantly hampered or eliminated by pre-existing or therapy-induced neutralizing antibodies. Table 2 presents an overview of several viruses that have been used in (pre)clinical applications against lung cancer, indicating potential (dis)advantages for the therapeutic use of each OV type.
Table 2.
Preclinical studies on OV therapy mechanisms in lung cancer
| Oncolytic virus | Virus constructs | Applications and eventual combined (chemo)therapies | Carrier cell type | Results | Reference |
|---|---|---|---|---|---|
| Vaccinia virus | vvDD-IL2-RG, vvDD | Intratumoral | IL-2 linked with glycosylphosphatidylinositol anchor (IL2-RG) expression induces an increased immune response and almost total tumor clearing in the subcutaneous transplanted syngeneic LLC model. Increased CD4+/CD8+ and TNF-α in TME. | Liu et al.27 | |
| VV.mIFN-β | Systemic and intratumoral | Drastic 40% tumor reduction in both NSCLC syngeneic models using TC-1 and LKRM2 cells as subcutaneous transplant or orthotopic. Albeit virus replication was substantially low in LKRM2 compared with TC-1, both models showed high cytokine induction due to ectopic IFN-β expression. | Wang et al.28 | ||
| vvDD | Systemic, intravenously with anti-PD1 and -TIM3 treatment | Urethane-induced endogenous as well as syngeneic, subcutaneous transplanted NSCLC models were used. vvDD infection synergized with blockade of both PD-1 and TIM-3 through the efficient direct killing of lung cancer tumor cells and recruiting and activating T cells for indirect tumor killing, vvDD was shown to induce higher expression of both PD-1 and TIM-3 in refractory lung cancer. Therefore, the triple combination therapy is more effective for refractory lung cancer. | Yang et al.29 | ||
| vvDD-IL23, derived from VV-WR strain modified for expressing recombinant IL23 | Intratumoral | LLC (Lewis lung cancer) NSCLC cell line was used as a subcutaneous transplant in the syngeneic model. Intratumoral infection with IL-23-armed vaccinia virus can induce potent antitumor effects through increased tumor cell death. Oncolysis combined with expression of membrane-bound IL-23 induces elevated expression of chemokines and other antitumor factors that cause increased antitumor immunity. | Chen et al.30 | ||
| TG4010, a modified vaccinia strain Ankara (MVA), expressing human mucin1 (MUC1) and IL-2 | Systemic, intravenously with anti-PD1 treatment | Intravenous injection with CT26 (expressing human MUC1) colon cancer cells induced extensive tumor growth in the lung. TG4010 application combined with anti-PD1 caused a better antitumor immune response and tumor regression compared with a single TG4010 treatment. | Remy-Ziller et al.31 | ||
| Oncopox-trail, derived from VV-WR strain, expressing recombinant TRAIL | Both intravenous and intratumoral injections | Tumor regression of A549 xenotransplant and LLC syngeneic models. TRAIL-induced increase in apoptosis and necrosis. | Hu et al.32 | ||
| GLV-1h68, replication-competent recombinant vaccinia virus derived from the Lister strain | Intravenous injections and systemic cyclophosphamide (CPA) | Systemic application of GLV-1h68 and CPA have synergistic effects against human lung adenocarcinoma PC14PE6 cell line in subcutaneous xenograft models. | Hofmann et al.33 | ||
| WR A34R(IHD-J) TK-Luc+ recombinant virus derived from WR strain. This VV recombinant produces high levels of extracellular enveloped virus (EEV) | Intravenous | EEV is covered by host-cell-derived lipid bilayer with anti-complement proteins that protect against immune clearance. | WR A34R(IHD-J) TK-Luc+ injected in two syngeneic models. Subcutaneous transplanted lung cancer cell line CMT64 and lung metastases producing mammary tumor cell line JC. In both models, systemic infection with WR A34R(IHD-J) TK-Luc+ induced a significantly higher tumor clearance level as compared with the WR strain. | Kirn et al.34 | |
| vvDD | Intravenous | Cytokine-induced killer (CIK) cells expressing NKG2D receptor. | CIKs loaded with vvDD were injected into syngeneic lung metastases producing mammary tumor (cell line JC) model causing almost complete clearance of primary tumor as well as lung metastases. | Thorne et al.35 | |
| VVΔTKΔN1L Recombinant virus with TK and N1L deletion in VVL15 strain |
Intratumoral | LLC NSCLC cell line was used as a subcutaneous transplant in the syngeneic model. Intratumoral infection with VVΔTKΔN1L marked tumor regression with increased intratumoral CD4+ and CD8+T cells and neutrophil accumulation. TME changed with increased systemic NK cells and augmented IL-α, IL1-β, and GCSF. | Ahmed et al.36 | ||
| Recombinant GLV-1h107, GLV-1h108 GLV-1h109 contains the GLAF-1 gene under the control of the VV SE, SEL, and SL promoters, respectively. All were inserted at the J2R locus in the parental virus GLV-1h68. | Intravenous | Intravenous injection of GLV-1h107, GLV-1h108, and GLV-1h109, in subcutaneous xenotransplanted A549 cell line tumor, caused tumor regression. GLAF-1 is a scAb against VEGF. Upon i.v. infection GLAF-1 is found inside the tumor and TME, causing a decrease in blood vessel formation occurs. | Frentzen et al.37 | ||
| EphA2-TEA-VV, a T cell engager armed oncolytic VV (TEA-VV) encoding secretory bispecific. T cell engagers (TEs) that bind both to human CD3 and a tumor cell surface antigen EphA2. Derived from original vvDD, WR strain. | Intravenous | After lung tumors were formed in systemic (i.v. injected A549 cell line) xenotransplanted NSCLC model, EphA2-TEA-VV was i.v. injected with or without unstimulated PBMCs. Strong tumor clearance and activation of T cells together with creased IFN-γ and IL-2 secretion. | Yu et al.38 | ||
| Myxoma virus | Oncolytic myxoma virus (MYXV) | Intraperitoneal and intratumoral application of MYXV, single or combined with a low dose of cisplatin | Intratumoral delivery of MYXV to the syngeneic immunocompetent murine SCLC model induces extensive tumor necrosis with marked host immune cell infiltration. Intratumoral injection of human SCLC PDX models in NSG mice background showed severe impairment of tumor growth | Kellish et al.39 | |
| Recombinant MYXV expressing IL-15 | Intravenous | Bone marrow-derived MSCs | i.v. injected B16-F10 melanoma cell line forming tumor foci in the lung of syngeneic C57BL6 mice, followed by i.v. injection with MYXV-IL-15 loaded MSCs. Formation of pulmonary melanoma foci was largely prevented, resulting in longer survival. Treated lungs showed high infiltration with NK and CD8+ T cells. | Jazowiecka-Rakus et al.40 | |
| Recombinant MYXV expressing TNF-α | Intravenous, combined with ICI anti- PD1/PD-L1 and CTLA-4 | Autologous peripheral blood mononuclear cells (PBMCs) | i.v. injected K7M2-Luc lung metastatic osteosarcoma cell line in syngeneic BALB/cJ mice, followed by i.v. injection with MYXV-TNF-loaded PBMCs. MYXV-loaded PBMCs caused efficient lung lesion regression and longer survival. Combined ICIs anti-PD1/PD-L1 and anti-CTLA-4 synergized well with MYXV-TNF. | Christie et al.41 | |
| Recombinant MYXV-expressing tumor necrosis factor family member TNFSF14 (LIGHT) | Retro-orbital intravenous, combined with ICI anti-PD1 | PBMCs | i.v. injected K7M2-Luc lung metastatic osteosarcoma cell line in syngeneic BALB/cJ mice, followed by systemic application of MYXV-LIGHT-loaded PBMCs. Treatment led to overall longer survival and tumor regression. Very efficient LIGHT expression and onset of innate and adaptive immune responses. | Christie et al.42 | |
| Reovirus | Reovirus type 3 Dearing strain (T3D) | Intravenous | H1299 human NSCLC cell line was used for the subcutaneous xenotransplanted lung cancer model. Reovirus i.v. injected caused tumor regression and induced a decrease in HIF-1α expression thereby lowering VEGFA levels in the tumor. | Hotani et al.43 | |
| Reovirus T3D | Intravenous | Adipose-derived MSCs | TC1 NSCLC cell line was used for the subcutaneous syngeneic lung cancer model. MSCs loaded with reovirus were i.v. injected causing tumor regression and growth arrest, followed by an increase of IFN-γ secretion. | Seyed-Khorrami et al.44 | |
| Measles virus | Attenuated measles virus (MV) HU-191 strain | Intratumoral | Intratumoral injection with MV Hu-191 in a syngeneic model with subcutaneously transplanted A549 and LLC tumor cell lines caused clear tumor cell death and regression with increased T cell infiltration of TME. | Zhao et al.45 | |
| Attenuated Schwartz vaccinal strain of measles virus expressing recombinant GFP (MV-eGFP) | Intratumoral | MV-eGFP infection of subcutaneous xenotransplanted human NSCLC cell lines ADK3, ADK117, ADK153, and A549 caused high levels of tumor cell death and tumor regression. MV oncolysis is associated with in vivo activation of caspase-3. | Boisgerault et al.46 | ||
| Influenza virus | Low pathogenic oncolytic influenza virus IAV | Intranasal injections | IAV infection of somatic NSCLC in Raf-BxB mice leads to reversal of immunosuppressed tumor-associated lung macrophage function to an M1-like pro-inflammatory active phenotype. | Masemann et al.47 | |
| Herpes simplex virus | AP27i145 HSV-1, recombinant HSV-1 expressing complement miRNA145 sequences in 3′ UTR of ICP27 | In vitro only | The combination of radiotherapy and AP27i145 infection was significantly more potent in killing NSCLC cell lines (A549, H1975, H460, and H838) than each therapy alone. | Li et al.48 | |
| R-LM249, recombinant HSV-1 retargeted to exclusively infect HER2 expressing cells | Intravenously | Fetal membrane (FM)-derived MSCs | Single i.v. injection with R-LM249-loaded FM-MSCs efficiently prevented metastatic tumor formation in the lungs of subcutaneous xenotransplanted human ovarian cancer cell line SK-OV-3. | Leoni et al.49 | |
| Cf33-GFP | Chimeric poxvirus generated through homologous recombination among nine strains/species of poxviruses, expressing GFP | Intratumoral injections | Tumor regression in both xenotransplant and syngeneic NSCLC models. Infiltration of tumors by CD8+ T cells. | Chaurasiya et al.50 | |
| Coxsackievirus | Coxsackievirus B3 (CVB3) strain | Intratumoral, injections | CVB3 was injected in a subcutaneous xenotransplant NSCLC (A549 or EBC-1 cells) model. Tumor regression and clearance with NK and granulocyte inside tumor and TME. Tumor cells express calreticulin and secreted ATP as well as HMGB1. | Miyamoto et al.51 | |
| CVB3 strain | Intratumoral injections, with wild-type or UV-inactivated CVB3 | Single-dose CVB3 injection in subcutaneous xenotransplanted, KRASmut human NSCLC cell line (A549, H2030, and H23) tumors. A specific increase in tumor cell death and regression occurred, suggesting that CVB3 is a potent OV for preferential KRAS-mutant lung adenocarcinoma. | Deng et al.52 | ||
| microRNA-modified CVB3 (miR-CVB3) strain containing multiple miR-145/miR-143 sequences | Intraperitoneal | Infection of miR-CVB3 in both NSCLC, KRASmut (cell lines A549, H2030, and H23) and SCLC, Trp53mut/RBmut (cell lines H524 and H526) causes significant tumor regression in both lung tumor types, expanding CVB3 tropism to SCLC, independent from KRAS status. | Liu et al.53 | ||
| Bovine herpes virus | Bovine herpes virus 1 (BoHV-1) strain | Intratumoral with trichostatin A treatment | Infection of BoHV-1 in subcutaneous xenotransplanted A549 cell line tumor caused tumor regression. HDAC levels are repressed, and BoHV-1 shows synergy with HDAC inhibitor trichostatin A. | Qiu et al.54 | |
| Newcastle disease virus | Wild-type NDV strain (NDV/Altai/pigeon/770/201) | Intratumoral | NDV infection of A549 human lung tumor cells in subcutaneous xenotransplant model. Increased necrotic effect on tumor cells but not on non-tumor cells and PBMCs. | Yurchenko et al.55 | |
| Wild-type FMW strain (NDV/FMW) | Intratumoral | FMW infection in subcutaneous xenotransplanted tumor cell lines A549 and H460 induced tumor regression and tumor cell death mainly via autophagy. | Ye et al.56 | ||
| Wild-type FMW strain (NDV/FMW) | NDV-FMW triggers caspase-dependent apoptosis in lung cancer spheroids and promotes autophagic degradation in lung cancer spheroids by inhibiting the AKT/mTOR pathway. NDV-FMW injected in subcutaneous xenotransplanted H460 spheroid cell clusters induced tumor regression. | Hu et al.57 | |||
| Attenuated NDV-HUJ strain derived from the original NDV B1 strain | Intravenous | NDV-HUJ was injected in a syngeneic model with metastasizing lung adenocarcinoma cell line 3LL-D122 as a subcutaneous and orthotopic lung transplant. Virus-selective oncolysis is dependent on apoptosis and is associated with higher levels of viral transcription, translation, and progeny virus formation. | Yaacov et al.58 | ||
| Recombined NDV strain (NDV-D90) | Intratumoral | NDV-D90 maintains tumor-selective replication properties and induces tumor cell apoptosis. A549 human lung tumor cells in subcutaneous xenotransplant model showing impairment of tumor growth. | Chai et al.59 | ||
| Adenovirus | H101, generated by both deleting E1B and E3 in adenovirus type 5 (Ad5) | Intratumoral | Human lung adenocarcinoma cell line XWLC-05 subcutaneous xenotransplant model. High levels of cytotoxicity, efficient cell lysis, and G2/M arrest cause significant tumor regression. | Lei et al.60 | |
| Ad-apoptin, recombinant Ad5-expressing apoptin | Intratumoral | Ad-apoptin injected in subcutaneous xenotransplant lung tumor (A549 cells) model showing impairment of tumor growth, and increased apoptosis. Ad-apoptin targets AMPK and inhibits glycolysis, migration, and invasion of lung cancer cells through the AMPK/mTOR signaling pathway. | Song et al.61 | ||
| Ad.hTERT-E1A-TK, recombinant Ad5 expressing HSV-TK and hTERT driving | Intratumoral | Ad.hTERT-E1A-TK infection combined with administration of prodrug gancyclovir (GCV) resulted in more potent cytotoxicity on A549 cells and synergistically suppressed human lung cancer A549 tumor growth in the subcutaneous xenotransplant model. | Zhang et al.62 | ||
| Complete E1B-deleted conditionally replicating Ad (CRAd) Adhz60 | Intratumoral and intrape, with temozolomide (TMZ) | H441 human lung tumor cell line in subcutaneous xenotransplant model. Adhz60 acted synergistically with TMZ in suppressing tumor growth. | Gomez-Gutierrez et al.63 | ||
| OBP-301 (telomelysin) is an attenuated Ad5 with a hTERT promoter driving both E1A and E1B to regulate viral replication. OBP-301 sensitizes human cancer cells to ionizing radiation by inhibiting DNA repair | Intratumoral, with gemcitabine | OBP-301 and gemcitabine have synergistic effects causing increased regression of tumor lesions in subcutaneous xenografts of human lung cancer cell lines H460, H322, and H358. | Liu et al.64 | ||
| Ad5/3-Δ24aCTLA4 Expressing Ig2 type anti-CTLA4 mAb in E1A-deleted Ad5/3 chimeric virus |
Intratumor | Ad5/3-Δ24aCTLA4, injected in a subcutaneous xenotransplant lung tumor (A549 cells) model. Severe tumor regression and inhibition of Tregs and increased CD8+/Treg ratios; increased T cell activity. | Dias et al.65 | ||
| Capsid-modified, Ad5-derived virus | Intravenous | Mesenchymal stem cells (MSCs) | MSCs loaded with modified Ad5 homed primarily to lungs in orthotopic xenotransplanted NSCLC cell line model. Significantly increased tumor regression. Very efficient systemic delivery of Ad5 in various organs besides the liver. | Hakkarainen et al.66 | |
| Ad-IAI.3b, derived from Ad5 | Intravenous | A549 cells were first infected with Ad-IAI.3b and then irradiated | Irradiated and with Ad-IAI.3b-loaded A549 tumor cells into orthotopic xenotransplanted lung squamous cell carcinoma cell line (KLN205) model. Accumulation of Ad-IAI.3b in the lung with strong tumor regression. Increased tumor-infiltrating lymphocytes (TILs) with high Th1-related cytokine expression. | Saito et al.67 | |
| Ad-uPAR-MMP-9, expressing antisense RNAs against uPAR and MMP-9 | Intratumoral and intravenous | Ad-uPAR-MMP-9 was injected into a subcutaneous xenotransplant lung tumor (H1299). Severe tumor regression and impaired angiogenesis. Ad-uPAR-MMP-9 was i.v. injected into a subcutaneous xenotransplant metastasizing lung tumor (A549). A strong decrease in angiogenesis but also metastasizing capacity is shown by the low number and size of lung metastases. | Rao et al.68 | ||
| AdE3-SCCA1 derived from Ad E3. Here the squamous cell carcinoma (SCC) specific promoter SCCA1 drives E1A expression | Intratumoral | A549 cells infected with AdE3-SCCA1 | A549 tumor cells loaded with AdE3-SCCA1 were injected into a syngeneic subcutaneous SCC (SCC7 cells) model. Mice were preimmunized against AdE3 and only the loaded A549 cells induced complete tumor regression. Co-loading A549 with AdE3-SCCA1 and Ad-mGM-CSF augmented the anti-tumor effect. | Hamada et al.69 | |
| ICO15K-FBiTE, expressing FBiTE, a bispecific T cell engager against FAP. Fibroblast activation protein-α (FAP) is highly overexpressed in cancer-associated fibroblasts (CAFs) |
Intravenous combined with preactivated T cells from human PBMCs | ICO15K-FBiTE, injected in subcutaneous xenotransplant lung tumor (A549 cells) model. Clearance of tumor lesion and activation and proliferation of T cells; resulting in CAF targeting. Increased tumor T cell retention and accumulation in tumor and TME. | Sostoa et al.70 Freedman et al.71 |
||
| Recombinant adenovirus KGHV500, expressing anti-p21Ras scFv | Intravenous | CIK cells | CIKs loaded with KGHV500 were intravenously injected into an A549 tumor xenotransplant model leading to significantly increased tumor regression. Note that A549 has high expression of KRASV12 and KGHV500 and anti-p21Ras scFv were observed in tumor tissue but were nearly undetectable in normal tissues. | Lin et al.72 | |
| Recombinant adenovirus ZD55 harboring tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL), manganese-containing superoxide dismutase (MnSOD), and TRAIL-isoleucine-aspartate-threonine-glutamate (IETD)-MnSOD |
Intravenous | CIK cells | CIKs loaded with ZD55 were intravenously injected into an A549 tumor xenotransplant model inducing a significantly higher level of tumor cell death and lower tumor volumes as compared with the non-transgene expressing control ZD55. Tumor tropism of ZD55-loaded CIKs was very high. | Jiang et al.73 | |
| ICOVIR-15 with E2F responsive palindromes in E1A promoter, as CRAd derived from AdΔ24-RGD; replication-incompetent Ad-iC9, expressing inducible caspase-9 | Intravenous | Bone marrow-derived MSCs loaded with combined ICOVIR-15 and Ad-iC9 | MSC loaded with ICOVIR-15/Ad-iC9 were systemically applied in a subcutaneous A549 tumor xenotransplant model After 48–72 h, MSCS could be detected in local tumor tissue resulting in consistent tumor clearance and longer survival of treated mice. | Hoyos et al.74 | |
| Single ICOVIR-15 | Intravenous with allogeneic PBMCs | Human menstrual blood-derived MSCs loaded with ICOVİR-15 | MSCs loaded with ICOVIR-15, combined with allogeneic PBMCs, systemically infected in a subcutaneous A549 cell xenotransplant model. Efficient clearance of tumor mass and antitumor efficacy partially mediated by monocytes and NK cells. | Moreno et al.75 | |
| ICOVIR-5, strain analog to ICOVIR-15 | Intraperitoneal | Murine bone marrow-derived MSCs | MSCs loaded with ICOVIR-5 (Celyvir therapy), i.p. infected in syngeneic murine lung adenocarcinoma (CMT64 cells) model. Significant tumor clearance occurred, loaded MSCs homing into the CMT64 tumor followed by an invasion of the tumor bed by a high number of CD8+ and CD4+ cells. | Rincón et al.76 | |
| ICOVIR-5, strain analog to ICOVIR-15 | Intravenous | Murine bone marrow-derived MSCs, either syngeneic or allogeneic | MSCs loaded with ICOVIR-5 (Celyvir therapy), systemically infected in syngeneic murine lung adenocarcinoma (CMT64 cells) model. Tumor clearance after Celyvir treatment. Both syngeneic and allogeneic Celyvir induce systemic activation of the immune system, similar antitumor effect, and a higher intratumoral infiltration of leukocytes, as shown by high infiltration of CD45+ cells in the tumor core. | Morales-Molina et al.77 | |
| ICOVIR15-cBiTE, expressing an epidermal growth factor receptor (EGFR)-targeting bispecific T cell engager (cBiTE) | Intraperitoneal with allogeneic PBMCs | Human menstrual blood-derived MSCs | MSCs loaded with ICOVIR15-cBiTE, systemically infected in subcutaneous (A549) xenotransplanted human adenocarcinoma model. Enhanced tumor clearance compared with unarmed IVOVIR-15. | Barlabé et al.78 | |
| Vesicular stomatitis virus | Recombinant oncolytic vesicular stomatitis virus (VSV) pseudo-typed with LCMV-GP expressing tumor-associated antigens, termed VSV-GP-TAA | Intravenous with KISIMA-TAA as a self-adjuvant cancer vaccine in combination with anti-Pd1 antibody | Priming vaccination with KISIMA-TAA followed by VSV-GP-TAA resulted in strong tumor regression and even increased with anti-PD1, in the syngeneic NSCLC model using TC-1 cell line as subcutaneous transplant. | Das et al.79 | |
| Attenuated, recombinant Av3 strain, expressing GFP | Intravenous | Murine CT26 colon carcinoma and L1210 leukemia cell lines. Human A549 lung adenocarcinoma cell line | Tumor cell lines loaded with Av3 were injected in orthotopic transplanted (CT26 lung tumor cell line) syngeneic models. Both murine tumor cell lines delivered high doses of VSV, even when recipient tumor-bearing mice were pre-immunized against VSV. The xenogeneic human A549 cell line showed similar carrier efficiency. Lung tumor carrier cells end up preferentially in the lung while leukemic carrier cells disperse systemically. Cell-mediated delivery of VSV can be achieved using allogeneic or xenogeneic carrier tumor cell lines, proving that carrier cells can evade immune responses against OVs. | Power et al.22 | |
| Voyager-V1 (VV1) VSV strain expressing IFN-β and sodium iodide symporter (NIS) |
Intravenous, combined with anti- CTLA4 and PD1 Abs | VV1, combined with anti-CTLA4 and anti-PD-1, were intravenously injected in syngeneic CMT64 adenocarcinoma cell line models and induced durable tumor regression, high level of TILs and efficient CD8 TL response against CMT64 neo epitopes. | Ram et al.80 | ||
| VSV-IFN-β, recombinant VSV expressing interferon-β | Intravenous | Blood outgrowth endothelial cells (BOECs) | Syngeneic lung metastatic LM2 mammary tumor model was intravenously injected with VSV-IFN-β loaded BOECs causing a major regression of lung metastases. Similar i.v. application in an A549 xenotransplantation model led to severely increased antitumor activity and survival. | Patel et al.81 |
On the other hand, improvements in molecular engineering have made it possible to alter the viral genome to increase its anticancer activity by inserting novel transgenes and attenuating their virulence by removing viral genes associated with pathogenesis.82 Since the FDA recently approved a modified HSV strain, talimogene laherpaprevec (T-vec), for the treatment of melanoma, oncolytic virotherapy has become more accepted.83 Patients with intradermal metastatic melanoma are administered this OV via intratumoral injections that initiate anti-tumor immune responses leading to a sustainable clinical efficacy.84,85 However, the overall response to intratumoral injections of primarily visceral illnesses, such as late-stage metastasizing non-small cell lung cancer (NSCLC), is quite low. Visceral malignancies can be targeted through systemic OV application, but efficient tumor cell infection might still be impaired due to the lowering of the viral load by complement and antiviral antibodies.86 The possibility of more significant systemic toxicity may be another drawback to intravenous OV application.87 Several different methods have been tried to reduce these risks, such as immunological suppression to inhibit the neutralizing antibody response or alterations to the virus to prevent detection using concealed common antigens. Specifically, for VV, several methods have been described to suppress antibody-mediated viral clearance. In one study, the extracellular enveloped virus (EEV) form of VV was covered by a lipid layer with anti-complement proteins after which systemic use of these modified EEVs generated a far lower viral clearance.88 In a more recent study from Nakatake et al.,89 partial deletion of vaccinia surface glycoprotein B5R generated an EEV form with a higher antibody-dependent neutralization resistance than the wild-type EEV. The latter mechanism can be used by VV via its EEV form, consisting of a normal virion covered with a host cell-derived outer membrane that enables its spread via circulation while evading host immune mechanisms.90 Even so, immune suppression might turn out to be a two-edged sword since it might as well negatively affect the OV-induced antitumor immune response.
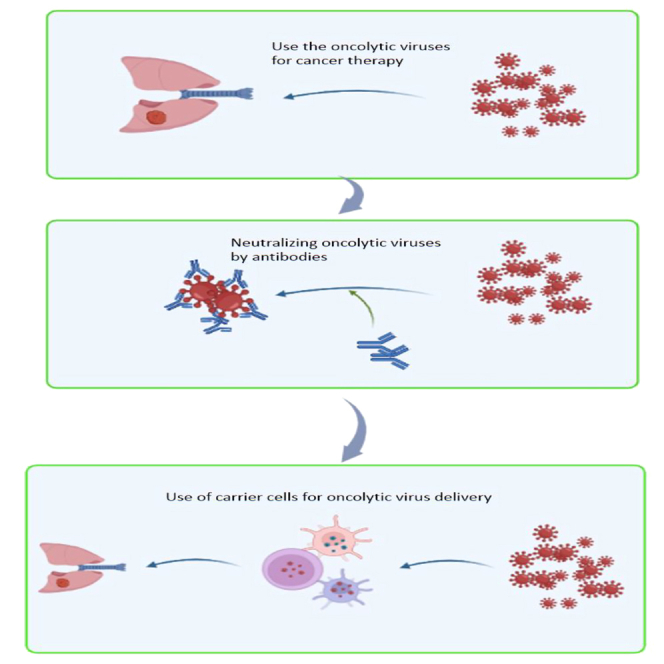
Apart from all these methods, there is a need for alternative techniques to facilitate shielded OV delivery91,92 since systemic infection with naked virions has so far been shown to insufficiently escape the above-described humoral immunity factors. One proposed delivery method to solve OV’s issues with both blood clearance and tumor penetration is based on the use of a patient’s own cells as a "Trojan horse"-like delivery vehicle for OVs. Here, the patient’s own “carrier” cells are first infected ex vivo, followed by systemic injection of these OV-carrier cells, which then are ideally transported into the tumor beds causing efficient tumor infection and lysis. Considering that cells are the viruses’ natural hosts, seeing them as stealth OV carriers is indeed very tempting. Even more so, the immune system typically ignores OV carriers until antigen production begins in the latter stages of infection. However, note that not every cell type can harbor or replicate a specific virus as efficiently as to serve as an ideal carrier. Research using intravenous administration of naked reovirus to patients revealed that all viral particles found in the blood were cell associated, indicating that some viruses may naturally convey themselves via cell carriers (Table 3). So, cells can act as OV carriers.93 One study intriguingly demonstrated that human monocytes loaded with preformed reovirus-antibody complexes (neutralizing the reovirus) could ultimately deliver replication-competent reovirus to melanoma cells in vitro.94 Here we present an overview of OV therapy used against lung cancer and how to improve its efficacy using carrier cells for the systemic delivery of OVs. We discuss the use, advances, and future promises that such improved techniques hold for therapy against lung cancer, especially when combined with established (immune) therapeutic approaches.
Table 3.
Clinical applications of systemic OV therapy for lung cancer
| Oncolytic virus | Virus construct | Combination therapy | Phase of development | Method and results | Clinical trials identifier and references |
|---|---|---|---|---|---|
| Adenovirus | Ad-HSVtk | Stereotactic body radiotherapy and pembrolizumab | II window of opportunity, | Metastatic non-small cell lung cancer (NSCLC). In situ, intratumoral, OV therapy consists of adenovirus-mediated expression of herpes simplex virus thymidine kinase (ADV/HSVtk) plus valacyclovir therapy. Initial 28.5% complete or partial response. The study is ongoing. | NCT03004183 |
| Ad-HSVtk | Stereotactic body radiotherapy and nivolumab | II window of opportunity terminated | Intratumoral injection with Ad-HSVtk in metastatic squamous and non-squamous NSCLC. No results, the study is terminated. | NCT02831933 | |
| Adapted Ad-HSVtk (CAN-2409) | Pembrolizumab | II | Intratumoral injection of CAN-2409 in patients with stage III/IV, refractory NSCLC. The study is ongoing. | NCT04495153 | |
| ColoAd1, chimeric adenovirus strain (enadenotucirev) | I | i.v. infusion with enadenotucirev to assess delivery in patients with NSCLC. A study completed: enadenotucirev was delivered in most tumor samples following i.v. infusion, with almost no activity in normal tissue. Virus delivery coincided with high local CD8+ cell infiltration in 80% of tested tumor samples, suggesting a potential enadenotucirev-driven immune response. Enadenotucirev delivery was well tolerated, with no serious adverse events. |
NCT02053220 Garcia-Carbonero et al.95 |
||
| Ad-MAGEA3, adenovirus vaccine expressing MAGE-A3 | Pembrolizumab | I/II | IO-resistant stage IV NSCLC. Study is ongoing. | NCT02879760 | |
| MEM-288, adenovirus expressing recombinant CD40L and human IFN-β | I | Intratumoral injection in stage III/IV NSCLC patients. Initial determination of MTD and recommended phase II dose for the planned combination of MEM-288 with an immune checkpoint inhibitor. The study is ongoing. | NCT05076760 | ||
| Combination of oncolytic Onc.Ad5Δ24 and helper -dependent HDΔ28E4 PD-L1, expressing PD-L1 antibody | Onc.Ad5Δ24 and HDΔ28E4 PD-L1 (CAd-VECPDL1) in combination with HER2-specific CAR T cell therapy | I | Patients with HER2-positive (including NSCLC) solid tumors. Intratumoral injection with CAdVEC followed by HER2-CAR T cell application. Determining MTD and influence of tumor microenvironment changes on CAR T therapy efficacy. The study is ongoing. |
NCT03740256 Tanoue et al.96 |
|
| Recombinant adenovirus expressing human IFN-λ, Onc.Ad.L-IFN | Onc.Ad.L-IFN (YSCH-01) single use | I | Intratumoral injection with YSCH-01 in solid tumors (including NSCLC) determining MTD and safety. Study ongoing. | NCT05180851 | |
| ICOVIR-5 (CELYVIR) | MSCs loaded with ICOVIR-5 | II | Trial to determine the toxicity and clinical outcome of infusion of autologous MSCs infected with the oncolytic adenovirus ICOVIR5 (CELYVIR) systemically applied in children with refractory or recurrent metastatic solid tumors. Results not posted. Ended prematurely. |
EudraCT no. 2008-000364-16 | |
| CELYVIR | MSCs loaded with ICOVIR-5 | I/II | Evaluation of the safety and clinical response of weekly (n = 6) infusions of CELYVIR in children and adults with metastatic and refractory solid tumors. Well-tolerated treatment, with only mild toxicity, with the potential to achieve clinical responses in patients with advanced tumors. The study is completed. |
NCT01844661 Ruano et al.97 |
|
| CELYVIR | MSCs loaded with ICOVIR-5 | I/II | Studies the feasibility of the combination of AloCELYVIR with chemotherapy and radiotherapy for the treatment of children and adolescents with relapsed or refractory extracranial solid tumors. The study is ongoing, and results are not posted. | EudraCT no. 2019-001154-26 | |
| Measles virus | MV-NIS | Atezolizumab | I | Intratumoral injection with measles virus, expressing sodium iodide symporter (MV-NIS) for recurrent and metastatic NSCLC. Study is ongoing. | NCT02919449 |
| Maraba virus | MG1 strain expressing tumor-associated antigen MAGE-A3 (MG1-MAGEA3) | MG1-MAGEA3 with vaccine Ad-MAGEA3 | I/II | Initial vaccination with Ad-MAGEA3 is followed by intravenous injections with MG1-MAGEA3 in refractory NSCLC after complete platinum-based chemotherapy and PD-1 or PD-L1 antibody-targeted therapy. Study is ongoing. | NCT02879760 |
| Coxsackie virus | Coxsackie A21 strain (CVA21) | CVA21 with pembrolizumab | I | Intravenous injection in refractory NSCLC patients. Study is ongoing. | NCT02043665 |
| Vaccinia virus | Recombinant vaccinia virus VV-GL-ONC1, derived from Lister strain. Expressing luciferase and β-galactosidase | GLV-1h68 or GL-ONC1 alone | I | Intra-pleural administration in NSCLC patients with malignant pleural effusion. The study is ongoing. | NCT01766739 |
| GL-ONC1 | GL-ONC1 alone | I | Systemic application for NSCLC patients. Dose safety profile and viral delivery were monitored. Study is ongoing. | NCT00794131 | |
| TG4010 (Mva-Muc1-Il2), a modified vaccinia strain Ankara (Mva), expressing human mucin1 (MUC1) and IL-2 | TG4010 alone or after combined vinorelbine/cisplatin | II | i.v. infusion with TG4010 in stage IIIB or IV NSCLC patients. A total of 65 patients were treated: 35.1% of TG4010 (combined) showed PR and 14.1% of TG4010 (alone) had CR. The OS for TG4010 (combined) was 12.7 months and for TG4010 (alone) 14.9 months. The combination of TG4010 with chemotherapy was well-tolerated and gave promising results. | Ramlau et al.98 | |
| TG4010 | TG4010 in combination with first-line chemotherapy | IIB/III | Intravenous infusion with TG4010 in stage IV NSCLC patients. A total of 222 patients were treated: median PFS in TG4010 patients was 5.9 months and in the placebo group 5.1 months. No adverse treatment effects were noted. Overall TG4010 combined with chemotherapy improves PFS vs. single chemotherapy treatment. The study is completed. |
NCT01383148 Quoix et al.99 |
|
| BT-001, modified vaccinia strain expressing anti- CTLA4 and human GM-CSF | BT-001 and pembroluzimab | I/II | Intratumoral injection BT-001 in patients with metastatic and advanced NSCLC. The study is ongoing. | NCT04725331 | |
| JX-594, modified vaccinia virus expressing human GM-CSF | I | Intravenous infusion of JX-594 in advanced NSCLC patients. The study is ongoing. MTD was determined but virus delivery efficacy was moderate. Good safety/toxicity profile. |
NCT00625456 Breitbach et al.100 |
||
| Vesicular stomatitis virus | Voyager-V1 (VV1) VSV strain expressing IFN-β and NIS |
VV1 and pembroluzimab | II | Intravenous injection with VV1 in refractory NSCLC or pulmonary neuroendocrine cancer (NEC, including SCLC) patients after initial treatment with pembroluzimab. | NCT03647163 |
| Reovirus | reolysin: human reovirus type 3- Dearing strain | reolysin combined with docetaxel and pemetrexed | II | Systemic intravenous injection of reolysin in patients with recurrent or metastatic NSCLC; 166 patients were enrolled (14 to the safety run-in). The study is completed: reolysin did not improve the progress-free survival (PFS vs. single agent chemotherapy (median PFS 3.0 months, 95% confidence interval [CI], 2.6–4.1) vs. 2.8 months (95% CI, 2.5–4.0), hazard ratio (HR) 0.90 (95% CI, 0.65–1.25), p = 0.53). Neither KRAS nor EGFR mutation was associated with improved PFS, but STK11 mutations did appear to have an association with improved PFS (HR 0.29 [0.12–0.67); as did PIK3CA mutation (HR 0.45 [0.22–0.93]). The combination was tolerable, although associated with increased rates of neutropenic fever. |
NCT01708993 Bradbury et al.101 |
| reolysin | reolysin combined with paclitaxel and carboplatin | II | Systemic intravenous injection of reolysin in patients with recurrent or metastatic NSCLC. Out of 37 patients enrolled, 20 patients had detected K-Ras mutations, 3 patients had EGFR mutations, 10 patients had EGFR amplifications alone, and 4 patients had BRAF V600E mutations. The study is completed: median PFS was 4 months (95% CI, 2.9–6.1), median OS was 13.1 months (95% CI, 9.2–21.6), and 1-year OS rate was 57% (95% CI, 39%–72%). |
NCT00861627 Gong et al.102 Villalona-Carrero et al.103 |
|
| reolysin | reolysin combined with paclitaxel and carboplatin | II | Systemic intravenous injection of reolysin in patients with recurrent or metastatic SCC. Out of 25 patients who received more than 1 cycle of therapy, the best overall response was PR in 12 patients (48%) and SD in 10 patients (40%) for a CBR of 88%. Of 21 patients with >6 months follow-up 7 had PFS ≥6 months (33.3%). |
NCT00998192 Gong et al.102 |
|
| Senecavirus | Seneca Valley virus (NTX-010) | NTX-010 after 4 cycles of platinum-based chemotherapy | II | Systemic i.v. injection with NTX-010 in patients with extensive stage SCLC after completion of first-line chemotherapy. A total of 50 patients were treated. Study completed: median PFS was 1.7 months for both the NTX-010 group and the placebo group; therefore, OV as a single agent could not generate obvious clinical responses in patients with advanced SCLC. |
NCT01017601 Schenk et al.104 |
| Non-specified | RT-01 | RT-01 combined with durvalumab | I | Intravenous injection of RT-01 followed by durvalumab treatment of patients with extensive-stage SCLC. Determining efficacy MTD and safety. Study is ongoing. | NCT05205421 |
| Herpes simplex virus | T3011, a genetically modified oncolytic herpes simplex virus (HSV-1) strain | T3011 and pembroluzimab | I/IIA | Intratumoral injection of T3011 followed by pembrolizumab. Total 64 patients with advanced solid tumors, including NSCLC. Safety and MTD measurements. The study is ongoing. | NCT04370587 |
| R130 genetically modified HSV-1 strain | I | Intratumoral or intraperitoneal injection in patients with advanced, refractory solid tumors (including NSCLC). Initial MTD and safety/toxicity evaluation. Study is ongoing. |
NCT05886075 NCT05860374 |
Lung cancer
According to GLOBOCAN 2020, lung cancer has worldwide an expected 2.2 million new cases (11.4%) and is the major cause of cancer mortality, with an estimated 1.8 million deaths (18%).105 There are two main types of lung cancer: small cell lung cancer (SCLC) and NSCLC, whereby NSCLC makes up over 85% of cases overall, while SCLC makes up just about 15%. Based on histological characteristics, NSCLC is further categorized into lung adenocarcinomas (40%), squamous cell carcinoma (25%–30%), giant cell carcinoma (10%–15%), and some not otherwise specified (15%–20%)106,107,108 (Figure 1). Lung cancer is typically asymptomatic in its early stages, and this is one of the main reasons why most lung cancer patients are diagnosed relatively late with stage III or stage IV. Moreover, lung cancer patients are often diagnosed between the ages of 60 and 80, with the most frequent occurrence between 65 and 75. Of these individuals, 50%–70% have locally progressed or fully metastasized cancer. The International Association for the Study of Lung Cancer claims that the 5-year survival rates in NSCLC stages IIIA, IIIB, IIIC, and IV, were 36%, 26%, 13%, and 6%, respectively. The older population is ineligible for intensive treatment because of the age-related functional deterioration of several organs, which poses a problem in treating lung cancer.109,110 Lung cancer therapy relies on the tumor stage at the time of diagnosis (Table 1, types of treatment for various stages). Surgical resection is the recommended course of therapy for individuals with NSCLC in stages I and II. For stage II malignancies, platinum-based adjuvant treatment is advised after complete resection. Patients whose tumors cannot be surgically removed are administered radiation and chemotherapy with a curative goal (stage IIIB). Palliative treatments and radio-chemotherapy are provided to patients who are in advanced stages (stage IV).108,111,112,113 Patients with SCLC have a significantly lower life expectancy compared with those with NSCLC.114 Typically, SCLC has an early onset of metastasis whereby early, limited lesions already have similar histopathological features as extensive lesions. Therefore, only lesion size and presence of metastasis determine if SCLC is of limited or extended stage (Nicholson, 2021). For both SCLC stages, the standard of care is radiation with platinum-based chemotherapy. Although SCLC initially responds well to radio-chemotherapy, almost all tumors eventually relapse leading to high mortality rates. Recently, some advances in anti-SCLC treatment have been achieved using immune checkpoint blockers in combination with chemotherapy.115 Overall, the lethality of SCLC remains much higher than for NSCLC patients. According to reports, patients with late-stage lung cancer have a shorter life expectancy than those diagnosed with early stages. Therefore, it is essential to develop new methods for early lung cancer diagnosis as well as novel efficient therapies to treat lung cancers in their advanced stages.
Figure 1.

Pie chart illustrating the overall prevalence of common lung cancer (sub)types
About 80% of SCLC patients are ever-smokers vs. only 16% never-smokers. This differs significantly for NSCLC where about 40% are never-smokers compared with 60% ever-smokers. Especially lung adenocarcinoma (90% vs. 35%) was higher among never-smokers than among ever-smokers, suggesting fundamental differences in lung cancer ontology between these two different groups of patients.
OVs for cancer therapy
Oncolytic virotherapy is a method of treating cancer by utilizing an attenuated virus with increased tropism for tumor cells. In general, viruses are harmful germs that infect cells, engage their DNA, RNA, and protein-synthesis machinery to reproduce, and then lyse their host cell to distribute their offspring, spreading the infection across a tissue (Figure 2). OVs, however, must have certain characteristics, such as not being pathogenic, capable of targeting and eradicating cancer cells specifically, and having the ability to be genetically engineered to release tumor-killing proteins.116 Table S1 shows an overview of specific features from OVs that were most frequently used against lung cancer in various experimental settings.
Figure 2.

Strategies for improving oncolytic virus efficacy
Oncolytic viruses (OVs) are designed for distinct expansion within the tumor niche. At least seven important modes of action can be elicited in tumor cells after infection with optimized, genetically engineered OVs. The introduction of three types of transgenic payloads in OV genomes results in the expression of angiogenesis inhibitors, immunostimulatory factors, and pro-drug converting enzymes. Direct genetic manipulation of the OV genome might alter its intrinsic capacity to transduce host cells and affect the viral replicative life cycle through alternations in transcription, translation, and induction of pro-apoptosis activities. The latter intrinsic OV genome mutation can also affect the maintenance and viral life cycle of OVs in various carrier cell types.
The cellular tropism of each virus dictates which tissues are most frequently infected. Different OVs with various tropism and tumor selectivity may be influenced by the level of receptor-mediated cell entrance, intracellular antiviral responses, or restriction factors that control the sensitivity of the infected cell to viral gene expression and replication.117 Nevertheless, the ability to infect, multiply, and lyse cells is a trait of naturally occurring lytic viruses.
This means that many OVs have their specific cell receptors, and upon cell entry can interact with host cell factors to facilitate viral genome replication, after which many viruses' replication cycles take advantage of altered biological pathways in cancer cells.118
With advances in biomolecular techniques and our improved understanding of cancer biology and virology, it became possible to create viruses with higher tumor selectivity and enhanced oncolytic activity.
Thus intracellular aberrations that arise in gene expression or signaling pathways (such as RB/E2F/p16, p53, IFN, PKR, EGFR, Ras, Wnt, anti-apoptosis, or hypoxia) in cancer cells or tumor microenvironment have been studied and dissected with a variety of OVs such as adenovirus, HSV, poxvirus, VSV, measles virus, Newcastle disease virus (NDV), influenza virus, and reovirus.117 Each of the different virus types has its own specific set of genes responsible for viral replication and in some cases immune evasion of the infected host cell. This knowledge facilitated the genetic engineering of diverse OVs to target cancer cells and spare normal cells. This especially holds for the three OV types most used against lung cancer, namely HSV, adenovirus, and pox virus. For example, in adenovirus, transcription of both viral immediate-early E1A and E1B genes can be controlled by tumor-specific promoters, excluding adenovirus replication in normal cells, such as deletion of the Delta-24 (D24) sequence in the constant region 2 (CR2) of the E1A coding region caused an even more attenuated virus. Here, the D24 nucleotide sequence of CR2 encodes the RB-binding domain of E1A, which in infected normal cells enables the release of E2F and subsequent viral replication. Thereby, the Delta-24 deletion in E1A renders the adenovirus deficient in replication in normal cells that are out of the cell cycle in a post-mitotic status. Importantly, the 24-base pair deletion does not compromise Ad replication in cancer cells without functional RB expression. Resulting conditionally replicating adenovirus (CRAd) strains could then be further optimized by inserting a short peptide sequence with an Arg-Gly-Asp RGD motif into the HI loop of the adenovirus knob protein, which significantly raises its affinity with its receptor on the cell surface and causes a more efficient internalization in the host cell.119 For HSV, conditionally replicating HSV-1 strains were specifically designed to target cancer cells.120,121 Here, three viral genes γ34.5, UL39, and α47 were deleted that encode infected-cell protein ICP34.5, ICP6, and ICP47, respectively. Normally these three genes render wild-type HSV the ability to evade the host antiviral response and continue its replication cycle.
Normally infection with wild-type HSV causes the activation of protein kinase R (PKR), which then phosphorylates eukaryotic initiation factor 2α (eIF-2α) rendering it inactive and thereby leading to the inhibition of protein synthesis. Expression of ICP34.5 circumvents this by dephosphorylating eIF-2α and thus allowing viral protein synthesis in the infected cells.
Since PKR activity is a downstream inhibiting target of RAS and is also otherwise often impaired in most cancers, deleting ICP34.5 will severely attenuate HSV in normal cells only. Deletion of UL39 has a different effect since it encodes ICP6, a large subunit in viral ribonuclease reductase, which converts ribonucleotides into deoxyribonucleotides that are utilized in viral genome synthesis. Lacking ICP6 restricts viral replication to dividing cells, as mature, post-mitotic cells lack ribonucleotide reductase expression and enough deoxyribonucleotides.121 On the other hand, HSV immediately early protein ICP47 interacts with the transporter associated with antigen presentation (TAP), blocking the antigenic peptide transport into the endoplasmic reticulum and subsequent loading onto major histocompatibility complex class I (MHC class I) molecules and presentation on the cell surface. ICP47 therefore impairs MHC class I-dependent CD8+ T cell response against HSV-infected cells,122 improving HSV capacity to evade viral-induced immune response. Deletion of ICP47 attenuates HSV virulence but this can be beneficial for the removal of infected tumor cells due to the more efficient presentation of tumor-associated antigens (TAAs) as we discuss below. Comparable mechanisms to modify virulence are applied in VV, belonging to the family of poxviruses. VV encodes a double-stranded RNA binding protein (E3L) that prevents activation of protein kinase PKR,123 and an eIF-2a homolog (K3L) that blocks phosphorylation of eIF2a.124 Complementary to these mechanisms, VV can weaken the binding of complement cascade through the VV expression of the viral complement control protein (VCP) encoded by C3L125 and membrane-bound glycoprotein B5R,126 which is essential for the formation of an EEV form of VV. Moreover, the B5R extracellular domain shares a high similarity with VCP and together they might be involved in protection against host immune responses.
However, an alternative way for attenuating vaccinia virulence was introduced through the deletion of the VV thymidine kinase (TK) gene. TK is involved in the synthesis of deoxyribonucleotides to facilitate DNA replication in cells with suboptimal nucleotide precursor pools. While the TK gene is necessary for replication in normal cells, where intracellular nucleotide concentration tends to be low, it is not necessary for cancer cells, which have relatively high concentrations of intracellular nucleotides.127 Thus, replication of the TK-deleted virus is dependent on the growth status of the host cells. Another way of generating tumor specificity of VV through genetic engineering was found through the deletion of the vaccinia growth factor (VGF) gene. VGF is expressed early during the VV infection cycle and is secreted from infected cells. It then binds growth factor receptors on surrounding resting cells and stimulates cell proliferation, thus preparing them for subsequent VV infection. This function is important for VV replication in normal tissues, but dispensable in tumors because tumor cells are naturally proliferating. Therefore, VGF deletion creates VV that preferentially replicates in tumors. Deletion of both the TK and VGF genes (double deleted VV or vvDD) was shown to significantly decrease pathogenicity compared with wild-type virus.128 Interestingly and contrary to most other OVs (Table 2), wild-type unmodified reovirus did already show an enhanced viral replication preference in cancer cells as compared with normal cells. This enhanced reovirus tropism is most likely linked to activated EGFR-RAS pathway activity connected with PKR inactivation.129
On the other hand, both preclinical investigations and clinical trials have demonstrated that anti-tumor therapy employing naturally oncolytic NDV is safe and efficacious.130,131,132 In a preclinical experiment using athymic mice implanted with lung cancer, Chai et al.59 showed that a reverse genetics system based on the oncolytic NDV D90 strain (rNDV-GFP, recombinant NDV carrying an enhanced green fluorescent protein gene), as well as the parental D90 virus, significantly suppressed body weight loss and tumor growth (Table 2).
Essential elements of OV therapy, such as systemic dissemination and intratumoral replication, are easily monitored in vivo by adding molecular reporters into viral genomes, which has dramatically advanced our understanding of the intricate dynamics of this strategy.133,134 These encouraging developments have raised the possibility that OVs may be administered intravenously to patients with advanced and otherwise incurable illnesses to "seek and kill" metastatic deposits. Optimal therapeutic efficacy of OVs as a systemic administration reagent is, however, constrained because, in most situations, the evoked anti-viral immune response restricts the lethal effects of OVs, and the effectiveness remains low.135,136,137 Four hypotheses might explain this lack of efficacy: (1) patients carry antiviral antibodies. Pre-existing antibodies quickly remove OVs after systemic injection, which reduces OV therapy efficacy138,139. (2) Macrophages in the liver and spleen eliminate OVs. (3) Physical barriers present a substantial hindrance to viral transmission because, in solid tumors, OVs must penetrate the endothelium layer to reach target cells. (4) OVs are quickly eliminated by the host’s immune system as a result of interactions between OVs and antigen-presenting cells, strong antiviral immunity, pre-existing circulating antibodies, and blood factors such as platelets140 and complement proteins.141,142 All these various combined factors make it very difficult to predict if enough OV particles can reach the tumor location after systemic infection. One contemporary solution to evade some of the delivery problems for OVs is the use of cellular carriers (CCs) as we discuss further below.
Mechanisms of OV action
Even though the mechanisms of OV function are still not fully understood, it appears that OV administration can safely cause regression in several human cancers through both: (1) direct oncolysis in which both infected tumor and, only by some specific OV types, simultaneous infected tumor-associated stroma cells143 undergo local cell death, and (2) promotion of the systemic immunological activity to the tumor’s virally induced cell death144,145 (Figure 3).
Figure 3.

Mechanisms of OV therapy
(A) OVs either naturally or after genetic manipulation selectively multiply in cancerous cells. Due to viral clearance, normal cells are unaffected. Lysis of tumor cells is caused by viral replication and the activation of cell death mechanisms. By releasing viral offspring, oncolysis makes it possible for fresh tumor cells to become infected. (B) Immuno-stimulating effects. Oncolysis, which is brought on by virus replication, results in the production of pathogen- and damage-associated molecular pattern molecules (PAMPs and DAMPs, respectively), as well as antigens associated with tumors (TAAs) and viruses. These antigens are subsequently absorbed by antigen-presenting cells (APCs) such as dendritic cells, which then induce the formation of tumor- and virus-specific T cells. At the same time, viral infection and replication trigger an inflammatory response, which results in the production of chemokines and thereby attracts T cells. The latter action facilitates tumor- and virus-specific T lymphocytes to infiltrate into the tumor and perform their immune function. (C) OVs as a platform for transgene delivery. Adenovirus and vaccinia virus are two examples of OVs that may be altered to carry transgenes (armed OVs), after which transgene products can be specifically delivered to the tumor microenvironment and further stimulate an anticancer immune response.
Intrinsic mechanisms
Multiple methods, including pyroptosis, apoptosis, necrosis, and autophagic cell death, are used by OVs to kill cancer cells after infection. Except for apoptosis, the remaining modes of cell death mentioned above are very immunogenic and trigger both non-specific and specific immune responses. When viral-infected cancer cells die, TAAs together with danger signals, such as danger-associated molecular pattern (DAMP) and pathogen-associated molecular pattern (PAMP) molecules, are released.146 Antitumor effects through stimulating an adaptive immune response can be seen in distant tumor sites that were not locally treated with OV, as a result of cytotoxic CD8+ T cell activation.147
A range of cytokines, including interleukins, interferons, and tumor necrosis factor-α (TNF-α), are released by dying cells into the immediate environment, further enhancing cell-mediated immunity. PAMPs, TAAs, DAMPs, and cytokines work synergistically to promote antigen-presenting cell (APC) maturation, which in turn primes CD4+ and CD8+ T lymphocytes for the adaptive host immune response through cross-presentation.148 The innate immune system is also implicated in the antitumor reaction following OV therapy, as shown by the ability of type I IFNs and DAMPs to directly promote natural killer (NK) cell activity against cancer cells.149 In addition, certain OVs also target the tumor vasculature, which results in the loss of the tumor’s blood supply and the death of uninfected tumor cells.150
Enhancing OV anti-tumoral response
Just as viral gene deletion has been applied to increase the selective infection of tumor cells by OVs, the insertion of therapeutic genes through genetic engineering has been utilized to increase OVs' anti-tumoral responses. In this section, we emphasize the arming of OVs with immune-stimulating molecules, such as cytokines, molecules that improve immune system cross-priming, and T cell co-stimulatory molecules, as a critical method to improve anti-tumoral responses after OV infection.145 Numerous studies with cytokine-expressing OVs produced promising outcomes.151,152 Several OVs expressing cytokine granulocyte macrophage colony-stimulating factor (GM-CSF), which stimulates APC maturation and increases cytotoxic T lymphocyte responses against malignancies, are presently applied in clinical studies (Table 3). The most researched GM-CSF expressing OV is T-VEC. Sample examination from melanoma lesions treated with intratumoral T-VEC injection revealed that these treatments resulted in both local and systemic T lymphocyte responses.153 OVs expressing heat shock proteins (HSPs) are an alternative strategy to boost anti-tumoral immune responses. OV-infected tumor cells undergoing oncolysis produce HSPs, which trigger the generation of chemokines and activate dendritic cells (DCs) via the TLR4 pathway. The innate and adaptive immune systems are cross-primed by HSPs; hence OVs were developed to overexpress HSPs, particularly HSP70. Adenoviral vectors that express HSP70 have shown anticancer activity in human solid tumors from a phase I study and in patients with xenograft models of hepatocellular carcinoma.154 Another strategy to boost cytotoxic T lymphocyte activation against tumor cells is to target T lymphocyte activation by designing OVs to produce T lymphocyte co-stimulatory molecules. Costimulatory molecules, including CD40 and CD80, are present in professional APCs. Arming OVs by expressing CD40 ligand (CD40L) evokes an innate response and enhances DC maturation and activation, which in turn activates T cells. The latter effect occurs when a T-helper response is elicited and results in cytotoxic lymphocyte (CTL) activation when CD40L, a transmembrane protein produced on CD4+ T cells, interacts with its receptor on an APC.155 However, the functional efficacy of OV infection can be severely hampered by the presence of stroma surrounding solid tumor lesions. The stroma is a multi-componential tissue, which consists of tumor-associated macrophages (TAMs), the (compact) extracellular matrix, tumor vasculature, and, to a large degree, cancer-associated fibroblasts (CAFs), which all together as non-epithelial, tumor-associated stromal cells (TACs) create a proficient tumor environment. TACs are of decisive importance for tumor progression, metastasis, and therapy resistance, by forming a barrier against infiltrating immune cells and anti-tumor drugs. Some native OVs are known to target stromal components such as CAFs or vascular endothelial cells. Only VSV has been shown to have a natural tropism for CAFs. VSV could infect CAFs that were associated with pancreatic tumor cells in patient-derived xenograft models.156 Interestingly, VSV replication in both pancreatic tumor cells and stroma is enhanced by reciprocal signaling between tumor cells and CAFs. Tumor cells secrete TGF-β1, which promotes VSV infection in CAFs, and CAFs secrete FGF2, which reduces innate anti-viral retinoic acid-inducible gene I (RIG-I) expression in pancreatic tumor cells. In turn, the reduced expression of RIG-I makes these cells more permissive to viral infection.156
Although only VV and VSV have an intrinsic capacity to target tumor-associated endothelial cells and thereby disrupt vascularization, other OVs need to adapt their viral genomes. In the latter case, OVs are “armed” and capable of transcriptional or transductional endothelial targeting by way of downregulating angiogenic factors or expressing antiangiogenic molecules157 (Table 2).68 A study from Arulanandam et al.158 showed that tumor-dependent levels of vascular endothelial growth factor (VEGF) repressed type I interferon-mediated antiviral signaling in endothelial cells, after which endothelial cells were sensitized for VV infection. These results showed that intrinsic high levels of VEGF inside the tumor vasculature could increase OV tropism for endothelial cells158 and exemplify how vascular-targeted OVs can improve cancer treatment efficacy.
The last two decades have shown a steady advance in the targeted use of other and above-mentioned engineered OVs against a broad range of solid tumors in an extensive number of (pre)clinical trials that confirmed OVs’ potential as immunotherapeutic tools too.
These promising developments coincide with the remarkable breakthrough of immunotherapy using ICIs in the treatment of lung cancer. ICI therapies ultimately aim to vaccinate against lung cancer through the induction of a lasting adaptive immune response against lung tumor cells. Various vaccination technologies are currently being developed and optimized as promising new strategies for lung cancer therapy. Oncolytic virotherapy could certainly take its place among these new types of immunotherapeutics since, because of their selective lysis of tumor cells, OVs can induce a lasting adaptive immune response. In Tables 2 and 3 we present an overview of most (pre)clinical studies with various OVs against lung cancer. Although many OVs show clear anti-tumor efficacy in nearly all preclinical studies, progress in clinical phase I/II studies is nevertheless slow and takes a long time to evaluate but promising results are obtained (Table 3).
Moreover, systematic reviews and meta-analyses evaluated the efficacy and safety of OV in lung cancer and showed that the objective response rate was significantly higher in patients receiving oncolytic adenovirus H101 monotherapy or combination with chemotherapy than in patients only receiving chemotherapy.159 There still is a long way to go before the use of optimized OVs will be sophisticated enough to become a standard part of a likely combined (immuno)therapy against lung cancer. For this, the pitfall of systemic OV application must be overcome.
Cellular carriers for OVs
As stated above, an entirely different way of augmenting systemic OV therapy effectiveness can be achieved by using CCs for OVs. Ideally, CCs protect their OV load against innate and adaptive immune responses and thereby lead to a more efficient OV delivery in any tumor bed. Not only OV but also CC characteristics determine delivery efficacy and tropism and, even after optimizing these different factors, the cellular OV delivery system’s ultimate success or failure depends on the precise coordination of three crucial steps in space and time, as shown in Figure 4. The CCs are first loaded ex vivo; secondly, cells are delivered to the tumor location through the circulatory system; and, finally, viral release inside the tumor bed is required. Once this process is started, it must be synchronized with the OV life cycle inside the specific carrier cell type employed for delivery. Therefore, to guarantee that OV-loaded CCs reach their destination at the appropriate moment, a thorough understanding of the process dynamics is necessary.134 This strategy resembles how some viruses have evolved to propagate throughout their host or obtain access to other tissues. For instance, the human immunodeficiency virus has been shown to bind to circulating DCs and macrophages, which subsequently move spontaneously to lymph nodes and spread the virus to its intended target cell type, i.e., CD4+ T cells.160 Preclinical research has shown that most cell types investigated for use as oncolytic viral delivery systems belong to one of the three groups listed below: progenitor cells, immune cells, and transformed cells. For instance, several cell types, including mesenchymal stem cells (MSCs), cancer cells, DCs, and blood outgrowth endothelial cells (BOECs), have been employed to treat solid tumors161,162 and in particular for lung cancer as shown in Tables 2 and 4.
Figure 4.

Carrier cells deliver their replicating OV load via a three-stage kinetic model
(A) Typical kinetics of OV delivery using permissive cancer cells (CCs). OV-infected cellular carriers undergo an eclipse phase after the virus is ex vivo added at time zero (t = 0), which comes before the release phase in which viral protein production, exponential amplification, and the release of offspring virions take place. (B) Three stages of CC/OV delivery that are delivered sequentially and are mapped to the duration of the viral development cycle depicted in (A) at the optimal times. Slower replicating OVs in specific CCs can elongate the eclipse phase significantly but might also produce a lower number of virions. The eclipse and release periods are therefore critically dependent on the OV life cycle in its CC and unique for every specific CC/OV combination. The complete eclipse period must be long enough to facilitate a proper stealth delivery. The release phase on the other hand should be fast enough to prevent a long time expression of viral proteins by the CCs to successfully evade adaptive barriers such as antiviral antibody-dependent cellular clearance.
Each of these cell types has specific benefits and possible drawbacks. In principle, the perfect cell carrier should not only shield its viral payload from neutralization and target it directly to the tumor site, reducing harm to normal tissues, but the cell carrier should also possess anticancer efficacy. Additional properties, such as a favorable safety profile, ease of isolation, and/or manufacturing, are essential features to consider when deciding which carrier cell type is most suitable for clinical application. An optimal CC therefore requires three features: (1) CCs should be easily infected by virus, (2) CCs carry the virus specifically to the tumor bed while hiding it from immune recognition, and (3) CCs should release progeny virus to act upon distant tumor sites. All these features must be considered for determining the CC systems best suited for each different tumor indication, especially considering the need for systemic OV applications against lung cancer metastases.
MSCs
MSCs are immature multi-potent stem cells that can self-renew and differentiate into various cell types such as fat cells that give rise to marrow adipose tissue (adipocytes), muscle cells (myocytes), bone cells (osteoblasts), and cartilage cells (chondrocytes). Apart from placental umbilical cord tissue, MSCs can be derived from distinct adult tissues such as bone marrow, peripheral blood, and adipose tissue. MSCs infected with OVs can enhance the transport of the therapeutic payload to cancer sites because of their innate tumor tropism.163 These cells are potential delivery vehicles to even difficult-to-reach metastatic neoplastic foci because they in principle protect OVs from antiviral host immune response (Figure 5). Numerous studies demonstrated that injected MSCs can migrate in a targeted manner (homing) to specific tissues163 (Tables 2 and 4), including damaged and tumor regions. A signaling cascade similar to that found in wound healing and intrinsic characteristics of the tumor sites, such as the degree of vascularization, the level of oxygenation, the degree of inflammation, etc., causes MSCs to migrate toward the tumor bed.164
Figure 5.

Advantage of systemic delivery of an MSC-shielded OV
Systemically injected unprotected OVs, such as the virus, cause an antiviral response through innate (NK cells, cytokines, mononuclear phagocyte system (MPS), complement activation) and eventual adaptive (antibodies, T cell mediated) immunity, which clears the virus and prevents any oncolytic effects. On the other hand, efficient transport to the tumor bed and oncolytic activity are made possible by viruses being protected by an appropriate protective carrier, such as mesenchymal stem cells (MSCs). The therapeutic system, or "Trojan horse," consists of MSCs infected with OVs, which improves oncolysis and raises the acquired anti-tumor immune response, enhancing the total anticancer impact. This figure is adapted from Hadryś et al.2
Studies showed that administering MSCs intravenously causes significant initial trapping in the lungs,165,166 most likely due to the large size of MSCs and their accumulation in the capillary beds of the lung. Following a single intravenous injection of infected MSCs, Leoni et al.167 found that the combination of MSCs from various sources infected with a HER2-retargeted oncolytic HSV and tested in murine models of metastatic cancers had the highest concentration of carrier cells and viral genomes in the lungs (Table 2). For at least 2 days, viral genomes remained throughout the body. The HSV-MSC application considerably slowed the formation of lung metastases from subcutaneously (s.c.) transplanted ovarian cancer in athymic nude mice and decreased the burden of brain metastases from breast cancer s.c. transplants in NOD SCID gamma (NSG) mice by more than half. In addition, studies on the antitumor efficacy of syngeneic or allogeneic murine MSCs infected with the oncolytic adenovirus ICOVIR5 (i.e., Celyvir system) have shown that both types of Celyvir increase the infiltration of CD45+ cells and leukocytes in the center of murine lung adenocarcinoma tumors77 (Table 2). In addition, more than a decade ago, MSCs loaded with oncolytic adenoviruses were shown to increase the bioavailability of systemically injected oncolytic adenoviruses in orthotopic murine models of breast and lung cancer.66 The use of MSCs in combination with different adenoviral designs has been widely investigated in (non)systemic therapy. Rincón et al.76 used the syngeneic murine CMT64 lung cancer cell line to produce a human adenovirus semi-permissive tumor model. They showed the ability of adenovirus-loaded murine MSCs (mCelyvir) homing to the transplanted CMT64 lung tumors. It was shown that intratumoral injections of ICOVIR5, the actual adenoviral construct, and mCelyvir together caused a higher tumor clearance than they did separately (Table 2). Another notable benefit of this combination therapy was the enhanced recruitment and boost of tumor-infiltrating CD8+ and CD4+ T cells. Therefore, MSCs can effectively transport genetically engineered OVs to solid tumors such as lung cancer,165 and overall (pre)clinical results suggest that systemic OV therapies using MSC carrier systems have a perspective in lung cancer treatment (Tables 2 and 3).
Seyed-Khorrami et al.44 used MSCs loaded with oncolytic reovirus in the syngeneic mouse TC-1 lung cancer model and tested their migratory potency in vivo after systemic infection and assessed the efficacy and quality of lung cancer treatment. The study’s findings showed that the effect of reovirus infection on adipose-derived MSCs does not seriously impair their ability to migrate into the tumor bed and that MSCs can act effectively as a vehicle for oncolytic viral delivery into the tumor site. Another encouraging development was shown in a study by Wang et al.168 Here, advanced genetically engineered enucleated MSCs kept their loading capacity for cytoplasmic-replicating OVs in combination with refined homing mechanisms for tumor tissues. Furthermore, enucleated MSCs were prevented from proliferating and stably engrafting in host tissues, thereby diminishing unwanted side effects after systemic use of MSCs.
Immune cells
A major problem for the effective use of immune cells as carrier cells lies in the rather poor replication capacity of OVs within these cells.169 While immune cells were once believed to be nothing more than primary, albeit not efficient, viral transporters to tumor locations, more recent research has concentrated on developing a synergy between the virus and immune cells to increase the anticancer effects of both entities. The prominent role of the host immune system in controlling and eradicating tumors is well known. So it is tempting to use immune cells in such a way that their natural features can be combined with a role as OV carrier cells170 and stimulate an even more robust antitumor response.171
Cytokine-induced killer (CIK) cells have been extensively used to gain a better understanding of this synergy. CIK cells locate their targets through the use of their own NKG2D receptor and its ligands, such as the stress response ligands MICA and MICB. Multiple pressures imposed by the immune environment during its fight against tumor formation cause the elevated expression of these ligands in human cancers172 (Table 2). Remarkably, viral infection is a stressor as well and thereby enhances NKG2D ligand expression. CIK cells behave like NK cells and try to eliminate tumor cells when they encounter them. Adoptive transfer of OV-infected CIK cells into a tumor-bearing host led to approximately 20% of CIK cells lysing at the tumor location and the remaining 80% attacking the tumor.172
In addition, immuno-stimulatory mediators released by activated CIK cells assist in generating a memory response and trigger the host immune response.172 Jiang et al.73 (Table 2) infected CIKs with oncolytic adenovirus ZD55-bearing TNF-related apoptosis-inducing ligand (TRAIL), manganese-containing superoxide dismutase (MnSOD), and TRAIL-isoleucine-aspartate-threonine-glutamate-MnSOD. They showed that co-cultivation of lung cancer cell lines with CIK cells expressing oncolytic adenoviruses dramatically decreased the colony formation, proliferation, and invasion of these tumor cells. The co-cultivated lung cancer cells also produced more TNF-α, IFN-γ, and lactate dehydrogenase than normal cells. Moreover, systemic injection of CIK cells harboring oncolytic adenoviruses in a lung tumor xenograft model led to a significant decrease in tumor load and lower levels of Ki67-expressing tumor cells.
Other studies investigated the use of DCs as delivery vectors for OV. Although DCs can preferentially move to tumor areas, they have a relatively limited inherent capacity to destroy tumor cells.173,174 However, as with other OVs, the fast generation of neutralizing antibodies poses a significant challenge to effective systemic administration. Therefore, the capacity of T cells, DCs, and peripheral blood mononuclear cells (PBMCs) as some types of CCs was evaluated, in how far they could shield reovirus and myxoma virus from systemic neutralization41,42,94 (Table 2).
In addition, studies with reovirus94,175 investigated how far these CCs might influence the balance of the ensuing antiviral vs. antitumor immune response. Reovirus was injected intravenously into C57Bl/6 mice harboring melanoma metastases, either in purified form or loaded onto DCs or T cells. In both cases for naive mice, infected by either purified OV or OV-carrier cells, OVs were detected in the tumor-draining lymph nodes a few days after treatment. However, clearance of lung metastases was only attained when the virus was delivered on mature DCs or T cells; purified reovirus or loaded immature DCs only caused partial early tumor clearance.94
BOECs
BOECs facilitate their use as CCs because they can be easily acquired from a peripheral blood draw, rapidly produced in cell culture to large numbers, and consistently cryopreserved without changing their genome or phenotype,176 which makes them very well suited for clinical applications. In lung cancer and melanoma models, BOECs have previously shown their value as gene therapy vectors for systemic delivery to tumors.177 VSV is an OV that triggers a potent immunological response, which makes it a good candidate for testing systemic OV therapy in the context of naturally occurring immunity. Patel et al.81 (Table 2) used ex-vivo-infected BOECs to deliver VSV in preclinical models of NSCLC, in which BOECs were acquired from either C57/Bl6 mice or human donors. VSV was engineered to contain either exogenous GFP or IFN-β, as VSV-GFP or IFN-β, respectively. This was followed by infecting both human and murine BOECs with either VSV-GFP or VSV-IFN-β. In vitro, both human and murine VSV-loaded BOECs killed NSCLC cells, whereas VSV-IFN-β was protected against antibody neutralization. The in vivo experiments with immune-competent mice harboring syngeneic LM2 lung metastases81 (Table 2) clearly showed murine BOECs localizing in the lungs and VSV-infected murine BOECs decreased the tumor burden in this model. The VSV-IFN-β-infected human BOECs showed improved antitumor activity and survival in an A549 cell line xenograft model as well since innate immunity is still largely intact in the athymic nude mouse background.
OV-loaded tumor cells
Transformed cells have been tested as carriers for OV delivery to tumors for more than two decades. Tumor cells have certain advantages since they are easier to grow and infect with OVs compared with normal cells, and their metastatic capacity renders them able to home to specific organs where they can infiltrate metastatic tumor beds. This would be ideal for the systemic delivery of large amounts of viruses to metastatic tumors. Yet this is not the case for OV-loaded transformed cells that form solid tumors such as A549 lung adenocarcinoma, MCF-7 breast carcinoma, HeLa cervical carcinoma, and others. Despite their predicted efficacy, the abilities of these tumor cells to home to specific body and tumor locations after intravenous injection are rather limited due to their tendency to mostly end up in the small capillary beds of the lung178 as a result of their large size. Nevertheless, their limited capacity to reach metastatic tumors in these experimental settings did not diminish their use as OV carriers to target primary lung tumors.22 The use of leukemia cells as carriers had a somewhat better effect, although it was not clear whether the delivery into distal tumors was a result of random accumulation of the infected leukemia cells rather than specific homing of these carrier cells to the local tumor beds.22 Still, transformed tumor cells do have the capacity to metastasize and preferentially home into specific organs for that matter. An example are myeloma cells expressing CXC chemokine receptor 4 (CXCR4) and thereby facilitating their homing into bone marrow, as has been shown in a study by Liu et al.179 with myeloma cells infected with the measles virus. But then again, each tumor cell type would have its characteristic tissue tropism, making it difficult to use as a more general carrier cell. A personalized approach would, however, be more tempting using a patient’s autologous tumor cells as carriers. This would then incur the additional effect from these infected, and after OV delivery lysed, tumor (carrier) cells as vaccines during their systemic application.180 How far these procedures could be applied in clinical therapy depends obviously on the proper safety warrants and attenuation of any tumor cell line before use.
Optimization of cell-mediated oncolytic viral therapy
It became evident that systemic application of cell carriers loaded with OVs holds a promise for significantly improved OV therapy against primary lung cancer as shown in numerous (pre)clinical settings (Tables 2 and 3). Primary lung tumor lesions are well reached when OV-loaded carrier cells accumulate inside the lung. However, this is mostly due to lung blood vessels acting as a physical barrier to especially OV-loaded MSCs albeit to a lesser extent for other carrier cell types. Late-stage NSCLC and almost always SCLC are nevertheless overwhelmingly linked with aggressive metastasis formation, which results in limited treatment options and therefore poor prognosis. So, indeed, metastases are the main cause of high overall lethality in lung cancer. OV therapy can only reach sufficient clinical efficacy if the problem of targeting most if not all metastases has been addressed and that is why there is an urgent need to optimize systemic (intravenous) OV application against lung cancer. The latter implies a further necessity to improve the use of cell-mediated OV therapy for optimal clinical applications against lung cancer.
This means that the above-described direct genetically engineered OVs with more selective tropisms and/or therapeutic gene expression are crucial but insufficient to decidedly improve the much-needed systemic OV therapy against metastasizing lung cancer. One evident way of circumventing liver and lung barriers would be the use of an intra-arterial (IA) application route for OV-loaded carrier cells. IA administration has been shown to significantly expand the availability and bio-distribution of MSCs.163 In a recent phase I study (NCT03896568), Chen et al.181 used Ad Delta 24RGD loaded on allogeneic bone marrow-derived human mesenchymal stem cells via IA in patients with recurrent glioblastoma or astrocytoma. The first results showed that OV-MSC perfusion into brain tissue worked far superior compared with the i.v. applications.181 This method of IA administration is potentially useful against brain metastases that often occur during extensive-stage SCLC. Similarly, intrapleural OV administration is much more efficient than its systemic application in causing major regression of malignant pleural disease.182 Given that some 50% of advanced lung cancers develop metastases that manifest themselves as malignant pleural effusions, intrapleural administration of OV-loaded carrier cells could be a very effective therapeutic application, which needs to be further explored. Another important aspect that should be emphasized in the improvement of cell-mediated OV therapy, is the preferential use of allogenic or even better autologous carrier cells that can be derived from the patient without extensive ex vivo procedures. Together all these measures of specific systemic application routes and choice of carrier cell types should be complemented by an improved collaboration between OV and carrier cells, which can be achieved by advanced genetic engineering of either or both partners. For instance, carrier cells can gain increased tropism for tumors through transgenic expression of appropriate chemo- or cytokines. As we briefly discussed above, CXCR4 is a significant candidate and CXCR4-expressing MSCs manifest a more efficient homing toward tumors in their inflamed microenvironment.183 However, increased tropism toward tumor or specific organ tissues can lead to some unwanted viral infection and replication inside healthy tissue too, which then necessitates an adaptation of OVs so that their capacity to infect normal tissue cells is vehemently blocked.
Notably, the myxoma virus bypasses these side effects due to its remarkably safety profile and cancer tropism. Myxoma belongs to the pox virus family and only infects rabbits but no humans or other non-leporid animals, while at the same time selectively infects and lyses cancer cells from humans and various other species.184
Apart from MSCs, other carrier systems based on immune cells have the advantage of not only increasing OV bioavailability but also combining efficient tumor tropism with adoptive cell transfer. This would then consequently link carrier-immune cell effector functions with OV-induced oncolysis. In Table 2, we described two recent examples of this approach in preclinical lung tumor models. In the first approach, Sostoa et al.70 used modified adenovirus ICO15K-FBiTE, expressing FBiTE, a bispecific T cell engager against fibroblast activation protein-α (FAP), which is highly overexpressed in CAFs. In a model using transplanted human lung tumor cell line A549, ICO15K-FBiTE together with pre-activated T cells were i.v. injected and caused strong regression of tumor lesions and simultaneous clearance of surrounding CAFs. CAFs can impose a major hindrance on the infiltration of OVs in the tumor microenvironment and this elegant combination of OV-directed oncolysis with FBiTE-mediated cytotoxicity against FAP-expressing CAFs might overcome this hindrance on OV function.
In a second example, Barlabé et al.78 used modified adenovirus ICOVIR15-cBiTE, expressing an epidermal growth factor receptor (EGFR)-targeting bispecific T cell engager (cBiTE) loaded on human MSCs and systemically administered it together with allogenic PBMCs into an A549 transplanted human lung tumor model. Due to the high EGFR expression in A549 lung tumor cells, ICOVIR15-cBiTE induced a much higher tumor clearance than the unarmed ICOVIR15. Further preclinical experiments in which chimeric antigen receptor-engineered T cells161,185 or tumor-infiltrating lymphocytes were used as carrier cells186 showed that this combinational therapy improved tumor clearance as well as adaptive anti-tumor immunity. These studies demonstrated the potential of immune cell carriers of OVs and should be followed up with further (pre)clinical tests in suitable lung tumor settings.
While some research groups recognized the potential of chiefly T lymphocytes and secondarily myeloid cells, proof-of-concept studies were not pursued by therapeutic refinement and a progression toward clinical trials. We propose that, to bring forward a real integration between carrier cells and oncolytic virotherapy, the researchers’ attention should be focused on autologous cells that can be easily recovered from the patient without lengthy ex vivo culture or differentiation steps. Furthermore, genetic engineering of OVs could be further exploited to enhance the “collaboration” between viruses and carrier cells, for example, to boost carrier cell migration into the tumor bed or to achieve prolonged release of OVs from carriers without cytopathic effects. Even if carrier cells have a very high tropism for tumors, they will likely also accumulate in non-tumoral tissue, which limits the viral dose delivered to the tumor and puts the healthy tissue at risk of viral infection and replication. Therefore, if OV-loaded carrier cells have a tropism toward a specific organ (for example, the lung or the liver) it may be necessary to modify the virus to make the normal cells from that organ specifically resistant to infection.
We believe that these approaches are highly promising and that we will be seeing more examples of immune cell carriers for OV therapies in clinical studies soon.
Given the variety of biological tools at our disposal and our expanding knowledge of tumor biology and cell migration, it is conceivable that, by biologically modifying the carrier cells and/or OVs, a significantly improved boost in cell trafficking and systemic OV delivery can be attained.
Even with tumor-targeted cell carriers, Reale et al.169 noted that, at best, about 10% of all supplied cell carriers reach the target region. Although this is a significant improvement over the expected intravenous delivery of "naked" VSV, where just 0.001% of virions reach the tumor, there is still potential for improvement. New strategies are already presented such as one method in which molecules are expressed or conjugated that attach to exposed tumor antigens or tumor vasculature. The latter effect occurs after a local change in inflammatory condition inside the tumor vasculature causes an increased expression of several adhesion molecules, such as integrins and selectins. This most likely has an important impact on OV infections since tumor vasculature is a common characteristic of most cancer types and several OVs are already known to infect the tumor vasculature, reducing blood supply to the tumor because of vascular collapse. Thus, tumor vasculature is an appealing element of tumor biology to target OV infection.100,187 Indeed, several studies using VV JX-954 that specifically targets RAS/MAPK pathway188 have been published in which OV infections were successfully directed into the tumor vasculature causing regression of NSCLC lesions,100,158 and that is why cell carriers could benefit from similar targeting techniques. An example of such a technique is the use of P-selectin as a suitable conjugate. P-selectin functions as a cell adhesion molecule on the surfaces of activated endothelial cells, which line the inner surface of inflamed blood vessels and activated platelets in the tumor microenvironment. The capacity of MSCs to bind to P-selectin in vitro is enhanced by polymer coating189 with sialyl-Lewis X and chemical conjugation190 of the MSC cell surface. Similarly, enzymatic conversion of carbohydrate groups on the MSC and T cell subset-expressed glycoprotein CD44 to the sialyl-Lewis X epitope allowed CD44 to bind to E-selectin. Interestingly, systemic application of engineered MSCs expressing P-selectin glycoprotein ligand-1 (PSGL-1) and Sialyl-Lewis X (SLeX)191,192 led to a specifically increased delivery of MSCs into inflamed tissues. Although the latter experiments were not performed on tumor-bearing mice, similar increased MSC delivery should likely occur in highly inflamed lung tumor lesions too. Besides this, these engineered MSCs showed a comparable high in vivo turnover with normal control MSCs and had markedly higher tropisms for inflamed tissues only. Therefore, ectopic expression of PSGL-1 and SLeX in engineered MSCs does not induce an increased loss of MSCs during passage through the lung capillary vessels but merely causes a more efficient MSC delivery in inflamed tissues. Further research is necessary to substantiate if CD44 and other alternated conjugates can increase the preferential attachment of circulating carrier cells to the tumor vasculature.
Another appealing strategy that may be used to treat various tumor types is magnetic targeting since it does not rely on the carrier cell or the expression of tumor-specific conjugates. Magnetic targeting has been demonstrated to be feasible in guiding a wide variety of cell types to multiple tissues for diverse purposes,193 even though it is more frequently used to target smaller agents to tumors, such as drugs194 or liposomes.195 Modifying the tumor microenvironment is another possible strategy to increase the trafficking of carrier cells to tumors. Using drugs or radiation, it is possible to cause the tumor to become inflamed. This can raise the expression of adhesion molecules, stimulate leakiness within the tumor’s blood vessels, and cause the release of cytokines, which may facilitate the homing or extravasation of carrier cells from blood vessels into the tumor.
In addition to boosting the trafficking of cells to tumors, cell carriers, and their viral payloads, can be altered to enhance many elements of cell-mediated OV delivery, such as loading capacity, virus generation, and delivery to tumor cells. After all, the preferred OV might not replicate or infect a cell carrier that reaches tumors optimally. Here, OVs can be modified or pseudo-typed if needed in such a way that they attach to a different entrance receptor expressed by the cell carrier. Many drugs improving viral replication can be utilized to increase virus production by the carrier cell. Of course, it must be certain that the applied drugs do not change the carrier cell’s trafficking patterns during the OV-CC infection process. Besides this, ectopic expressed immunological modulatory peptides might be designed into cell carriers to either inhibit the immune response, prepare the tumor for viral infection, or activate the immune system for the second wave of attack on cancer. Next-generation cell carriers have the potential to develop into sophisticated, mobile biological factories that can launch a coordinated, multifaceted attack on tumors as our knowledge of cell trafficking, tumor biology, and virology advances.
Conclusion and perspectives
There is an urgent need for improved diagnosis and treatment of lung cancer. Since it is one of the major causes of cancer-related deaths globally, swift progress in developing new and better therapies is of crucial importance. OVs have great potential to reverse immunological tumor tolerance and activate anti-tumor immunity. They can be used as a promising new immunotherapy approach alone or in conjunction with other treatment modalities.196
In this review, we focus on how OVs with improved use of carrier cells can refine (immuno)therapy for lung cancer. However, the number and type of clinical studies that were specifically performed with systemically administered OV-loaded carrier cells against lung cancer are still limited (Table 3) and mainly used MSCs as carrier cells. Even so, experimental settings of preclinical tests showed a high variation in both OV and carrier cell types, although a major weakness in these studies is the limited resemblance that most preclinical models have with the complex biology of heterogeneous lung cancer. Clonal lung cancer cell lines such as A549 are indeed a poor substitute for advanced lung cancer, whereas replacing them with advanced somatic mouse models for lung cancer in proper syngeneic backgrounds can significantly increase the physiological relevance of the preclinical models. Besides, the use of patient-derived xenotransplant (PDX) models ensures a better mimicking of human lung cancer, although a drawback hereby is the need for an immune-compromised host that impairs our potential for measuring a qualified host immune response against applied OVs. However, some of the latter problems can by now be bypassed through advances in genetically engineered mouse models that generate humanized mice with a (partially) reconstituted human adaptive and/or innate immunity.197,198 Although still far from perfect, humanized mice have the advantage of allowing OV-loaded carrier cells to be used in variable ways against orthotopically transplanted human lung cancer PDXs and measure specific human adaptive or innate immune responses after OV infection. More recent developments even presented humanized mice in which intrinsic murine HLA genes were replaced by human ones so that these mice were recipients of PDX tumors as well as autologous PBMCs from the same patient.199 In summary, we can say that indeed preclinical lung cancer models can be improved for better translatable results with already available tools. We should also be realistic in accepting that murine and human immunity does have some intrinsic differences, which apply to advanced syngeneic lung cancer models. The humanized lung cancer models, although under continuous development and improvement, still have their limitations too. Nonetheless, a proper refinement of the type and use of lung cancer models can lead to an important improvement in the quality and efficacy of preclinical studies on OV applications with or without carrier cells.
We also should make better use of our growing insight into lung cancer biology in which the concept of tumor plasticity is not only a major determining factor for intrinsic lung cancer heterogeneity and changing tumor microenvironment (TME) interactions but also influences treatment response of most if not all lung cancer types.200 This tumor plasticity is common for lung cancer and is influenced if not governed by distinct molecular mechanisms such as epithelial-mesenchymal transition, which leads to a dynamic shift in the differentiated state of lung cancer cells. This can have major implications for both OV and carrier cell tropism alike and should therefore be considered during the design of each specific OV therapy for lung cancer. OVs that infect through interaction with specific receptors on their host cells are especially affected by tumor plasticity. For instance, the SVV virus infects SCLC cell lines very efficiently due to its direct interaction with anthrax toxin receptor 1201 which is only expressed in a subclass of NEUROD1-expressing SCLC.202 Tumor plasticity influences the presence and number of these NEUROD1 subclass tumor cells during SCLC progression and that is the likely reason that phase II trials with SVV on SCLC patients were not successful104 (Table 3). Contrary to this, myxoma virus tropism for all SCLC subtypes is not hindered by the need for a specific cellular receptor and preclinical results of its use against SCLC PDX as well as syngeneic murine SCLC models showed significant tumor clearance. So far OV immunotherapy against lung cancer has been mainly tested in early-stage clinical trials with modest efficacy. However, it becomes clear that OVs are a very promising platform for combination therapy in the treatment of lung cancer. As we discussed, the steadily improved clinical use of ICIs against various human lung cancer types presents itself as an excellent choice for combination therapy with OVs.
Apart from the latter therapeutic synergy, we should, however, bear in mind how canonical lung cancer therapies such as radio- but especially chemotherapy are steadily improving using newly developed nanocarriers. Both the specificity and efficacy of these therapies advanced significantly, including those applied for lung cancer.203,204 Chemotherapy can have an even more profound influence on changing the immunosuppressive nature of the tumor microenvironment after disruption of the complete tumor bed through more efficient killing of cancer cells. Further to this, a synergy of ICI and chemotherapy has already shown its improved efficacy in clinical applications.205 So chemotherapy certainly remains an important partner in combination with OV therapy too, although more clinical data are needed to substantiate this.206 Another important aspect of combinational ICI with chemotherapy or OV therapies in clinical trials so far is the occurrence of patients undergoing complete tumor regression (so-called "elite responders"). Although these elite responders are often only a small fraction of the total amount of patients in each clinical trial, they are nevertheless very valuable and can offer us a way to get important new insights into how immunotherapeutic responses can be improved.
Since ICIs promote anti-tumor adaptive responses by removing blocks on immune activation, which is known to break tolerance to self-antigens and induce autoantibody formation,207,208 a subset of these autoantibodies may thus mediate anti-tumor responses and enhance ICI efficacy. Changes in B cells and tertiary lymphoid structures have been shown to contribute to ICI efficacy too,209,210 whereby specific ICI-induced humoral (auto)antibody responses in elite responders change the overall cellular adaptive immune response against a solid cancer. Therefore, interrogation of humoral responses in cancers from elite responders is an attractive strategy for the discovery of novel targets and therapeutic antibodies for the treatment of cancer to exploit synergism between ICI, chemotherapy and OV therapy.
Other encouraging prospects arise for early-stage, non-metastasized lung cancer through local application of OVs in either the pleural cavity or supported by advanced bronchoscopic techniques in the case of parenchymal lung tumor lesions. Nevertheless, when lung cancer presents itself in its most deadly metastasizing form, OV therapy can only secure some efficacy after systemic application. And, even so, only induction of a powerful anti-tumor response through OV’s immunotherapeutic effect can guarantee lasting remission of most if not all metastatic lesions. Here, cell carriers play an important role in enabling systemic delivery of OVs. As we discussed, essential features of carrier cells are their specific tropism for tumor cells, capacity to internalize viruses or attach them to their cell membrane, facilitate proper viral replication, and at the same time remain viable long enough to enable a systemic spread. These features pose a serious challenge in the advancement of carrier cell use in OV therapy, but genetic engineering of different carrier cell types together with selective use of adapted OVs can alleviate some of the encountered difficulties in (pre)clinical use. For lung cancer, like most solid cancers, this means optimization of intrinsic features of CCs such as increased tropism for pro-inflammatory tissues in the case of MSCs. Increased and broadening tropism for various cell types will, however, bear invariable risk of accumulation in non-tumor-bearing tissue so extra care should be given to the use of attenuated OVs with strictly limited pathogenicity to normal tissues. Also for lung cancer (Table 2), the use of immune cells for OV carriers remains tempting; e.g., CAR T cells can be loaded with OVs for successfully combining both therapies.185 CAR T cells have a direct strong activity to kill cancer cells expressing TAAs on the cell surface but are susceptible to the TME with its immune suppressive modulators among others, such as immunosuppressive cytokines, immune checkpoint inhibitory receptors and ligands (e.g., PD-1, PD-L1, CTLA-4), and M2 phenotype TAMs. Moreover, pre-existing TME can severely impair the level and function of tumor-infiltrating lymphocytes such as CAR T cells. As we already discussed above, OVs remodel the TME through upregulating immune checkpoint costimulatory receptors and ligands (e.g., 4-1BB, 4-1BBL, OX40, OX40L), proinflammatory cytokines (e.g., IL-12, IFN, IL-6, TNF), M1 phenotype TAMs, mature DCs, NK cells, etc. OV-mediated oncolysis then further promotes immune activation by TAAs spreading, which results in the proliferation of CTLs targeting other TAAs presented by MHC in addition to CAR T cells. On the whole, combining CAR T cell therapy and oncolytic virotherapy in a TME should therefore benefit from an immediate function of CAR T cells followed by OV-stimulated immune activation that causes a more effective lysis of heterogeneous cancer cell populations, which could mitigate tumor relapse encountered by CAR T cell therapy due to antigen escape.211,212,213 However, one has to reckon with the most common toxicities of CAR T cell therapy, which consist of release syndrome (CRS) often concurrent with or shortly followed by immune cell-associated neurotoxicity syndrome (ICANS). Also whether a combination with OV therapy mitigates CRS and ICANS is not clear, also due to the fact that so far this combinational therapy is only being investigated in one clinical trial, in which HER2-CAR virus-specific T cells and a binary oncolytic adenovirus are being used to treat patients with advanced HER2-positive solid tumors (NCT03740256, Table 3). More research is needed to optimize combinations of CAR T cells and OVs, including their dosage, delivery, and schedule, to maximize their efficacy while minimizing toxicity.
On the other hand, oncolytic VSV, NDV, and VV induce vasculature disruption to enhance tumor destruction,100,214 but this may limit the ability of intravenously delivered CAR T cells to reach the targeted tumor cells. This latter defect might at least be partially reduced by concomitant systemic application of OVs loaded on CAR T carrier cells.185,215 These studies show that CAR T cells loaded with albeit low OV doses do not interfere with the function or expression of receptors in both murine and human T cells. CAR T cells readily deposit OVs in a broad range of tissues and tumor targets and thereby improve the applicability of CAR T and OV combinational therapies.
Many different types of OVs with tumor-selective replication and cytolysis have been tested against lung cancer (Tables 2 and 3), but optimal systemic application during clinical use was hampered by the host immune system.
To warrant a more efficient delivery of significant doses of OVs to lung cancer, diverse types of carriers have been tested that include a variety of cell lines and immune and progenitor cells (Table 2) for lung cancer examples. Arguably, MSCs possibly have the best potential as cell carriers for systemic OV delivery. In vitro and in vivo experiments with MSC-based carrier cells have demonstrated their capacity for viral infection and multiplication, immune evasion, and tumor tropism production.216 Further to this, MSC-derived carriers have shown great promise for cancer therapy based on the results obtained in a variety of preclinical tumor models (Table 2, ICOVIR-5/CELYVIR). The eventual translation of these preclinical results into clinical usage will be revealed by the outcomes of trials such as an ongoing phase I/II investigation (NCT02068794), in which MSCs infected with oncolytic measles virus are being administered217 or MSCs loaded with ICOVIR-5 is being investigated in phase I/II trials (Table 3, NCT01844661 and EudraCT no. 2008-000364-16) against refractory and metastatic solid tumors in children and adults. Also, immune cells such as CIKs have been successfully tested against lung cancer in a recent preclinical study.72 Here, CIKs served as CIK cells as delivery vehicles for recombinant adenovirus KGHV500 that expresses anti-p21Ras scFv. According to their findings, systemic infection with combined CIK-KGHV500 considerably slowed the growth of KRas mutated lung cancer xenografts when compared with mice given KGHV500 therapy alone. KGHV500 and anti-p21Ras scFv were also detected in tumor tissue but were hardly noticeable in normal tissues.
These results and those from many other studies continue to emphasize the importance of carrier cells for the improved use of OV therapy against not only lung cancer but most other solid cancers too.
However, as natural carrier cells are not optimized for delivering their (viral) cargo to a particular location, other techniques, such as modifying their tumor-specific ligand on the membrane surface,169,218 have been utilized to improve their performance. Furthermore, genetic engineering of OVs will lead to new OV strains with increased oncolytic capacity and decreased pathogenicity.219,220
Despite the potential of this approach, there are clear restrictions on the use of cell carriers in therapy. The main issues are the necessity of ex vivo cell infection and the cost, scalability, manufacture, and regulatory requirements associated with combining two extremely complex biologic therapeutics into one product.17 This is nevertheless not much more restrictive when compared with other cancer immunotherapies such as CAR T cell therapies. Of key importance will be the improved efficacy of carrier cell-based OV delivery and, in doing so, induce potent immunotherapy against various cancers. By extending our knowledge about the molecular mechanism of solid tumors such as lung cancer and also improved tools for genome editing such as CRISPR-Cas9,221 we will be able to produce new generations of viruses with improved innate oncolytic capacity as well as better ability to infect broad-range carrier cell types. In parallel to this, cell-mediated carrier techniques are being improved using carrier cell-surface-modifying techniques.169,218 Ultimately, all these efforts should be combined to create optimal oncolytic viral therapies that can be safely used in the clinic for eradicating lung solid tumors.
Acknowledgments
We thank Ayşe Caner and Venus Zafari for their careful reading of the manuscript and their constructive remarks.
Author contributions
G.E.N., M.F., and E.N.P. collected the literature and wrote the manuscript. R.M. and A.B. designed, edited, and prepared the manuscript for submission. All authors read and approved the final manuscript.
Declaration of interests
The authors declare no conflict of interest. This work was supported in part by TÜSEB grant no. 2022-B-01-16483 (to R.M.), for the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript or in the decision to publish the results.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omton.2024.200788.
Contributor Information
Azam Bolhassani, Email: a_bolhasani@pasteur.ac.ir.
Ralph Meuwissen, Email: ralph.meuwissen@ege.edu.tr.
Supplemental information
References
- 1.Dorer D.E., Nettelbeck D.M. Targeting cancer by transcriptional control in cancer gene therapy and viral oncolysis. Adv. Drug Deliv. Rev. 2009;61:554–571. doi: 10.1016/j.addr.2009.03.013. [DOI] [PubMed] [Google Scholar]
- 2.Hadryś A., Sochanik A., McFadden G., Jazowiecka-Rakus J. Mesenchymal stem cells as carriers for systemic delivery of oncolytic viruses. Eur. J. Pharmacol. 2020;874 doi: 10.1016/j.ejphar.2020.172991. [DOI] [PubMed] [Google Scholar]
- 3.Mountzios G., Remon J., Hendriks L.E.L., García-Campelo R., Rolfo C., Van Schil P., Forde P.M., Besse B., Subbiah V., Reck M., et al. Immune-checkpoint inhibition for resectable non-small-cell lung cancer — opportunities and challenges. Nat. Rev. Clin. Oncol. 2023;20:664–677. doi: 10.1038/s41571-023-00794-7. [DOI] [PubMed] [Google Scholar]
- 4.Herbst R.S., Morgensztern D., Boshoff C. The biology and management of non-small cell lung cancer. Nature. 2018;553:446–454. doi: 10.1038/nature25183. [DOI] [PubMed] [Google Scholar]
- 5.Alexander M., Kim S.Y., Cheng H. Update 2020: Management of Non-Small Cell Lung Cancer. Lung. 2020;198:897–907. doi: 10.1007/s00408-020-00407-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Duma N., Santana-Davila R., Molina J.R. Non-Small Cell Lung Cancer: Epidemiology, Screening, Diagnosis, and Treatment. Mayo Clin. Proc. 2019;94:1623–1640. doi: 10.1016/j.mayocp.2019.01.013. [DOI] [PubMed] [Google Scholar]
- 7.van Meerbeeck J.P., Fennell D.A., De Ruysscher D.K.M. Small-cell lung cancer. Lancet. 2011;378:1741–1755. doi: 10.1016/S0140-6736(11)60165-7. [DOI] [PubMed] [Google Scholar]
- 8.Zugazagoitia J., Paz-Ares L. Extensive-Stage Small-Cell Lung Cancer: First-Line and Second-Line Treatment Options. J. Clin. Oncol. 2022;40:671–680. doi: 10.1200/JCO.21.01881. [DOI] [PubMed] [Google Scholar]
- 9.Yang S., Zhang Z., Wang Q. Emerging therapies for small cell lung cancer. J. Hematol. Oncol. 2019;12:47. doi: 10.1186/s13045-019-0736-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Y., Zou S., Zhao Z., Liu P., Ke C., Xu S. New insights into small-cell lung cancer development and therapy. Cell Biol. Int. 2020;44:1564–1576. doi: 10.1002/cbin.11359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dock G. The influence of complicating diseases upon leukemia. Am. J. Med. Sci. 1904;127:563–592. [Google Scholar]
- 12.Moore A.E. The destructive effect of the virus of Russian Far East encephalitis on the transplantable mouse sarcoma 180. Cancer. 1949;2:525–534. doi: 10.1002/1097-0142(194905)2:3<525::aid-cncr2820020317>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 13.Moore A.E. Effects of viruses on tumors. Annu. Rev. Microbiol. 1954;8:393–410. doi: 10.1146/annurev.mi.08.100154.002141. [DOI] [PubMed] [Google Scholar]
- 14.Southam C.M. History and prospects of immunotherapy of cancer: an introduction. Ann. N. Y. Acad. Sci. 1976;277:1–6. doi: 10.1111/j.1749-6632.1976.tb41687.x. [DOI] [PubMed] [Google Scholar]
- 15.Kelly E., Russell S.J. History of oncolytic viruses: genesis to genetic engineering. Mol. Ther. 2007;15:651–659. doi: 10.1038/sj.mt.6300108. [DOI] [PubMed] [Google Scholar]
- 16.Russell S.J., Peng K.W. Viruses as anticancer drugs. Trends Pharmacol. Sci. 2007;28:326–333. doi: 10.1016/j.tips.2007.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hill C., Carlisle R. Achieving systemic delivery of oncolytic viruses. Expert Opin. Drug Deliv. 2019;16:607–620. doi: 10.1080/17425247.2019.1617269. [DOI] [PubMed] [Google Scholar]
- 18.Wakimoto H., Ikeda K., Abe T., Ichikawa T., Hochberg F.H., Ezekowitz R.A.B., Pasternack M.S., Chiocca E.A. The complement response against an oncolytic virus is species-specific in its activation pathways. Mol. Ther. 2002;5:275–282. doi: 10.1006/mthe.2002.0547. [DOI] [PubMed] [Google Scholar]
- 19.Girgis N.M., Dehaven B.C., Xiao Y., Alexander E., Viner K.M., Isaacs S.N. The Vaccinia virus complement control protein modulates adaptive immune responses during infection. J. Virol. 2011;85:2547–2556. doi: 10.1128/JVI.01474-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hook L.M., Lubinski J.M., Jiang M., Pangburn M.K., Friedman H.M. Herpes simplex virus type 1 and 2 glycoprotein C prevents complement-mediated neutralization induced by natural immunoglobulin M antibody. J. Virol. 2006;80:4038–4046. doi: 10.1128/JVI.80.8.4038-4046.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Iankov I.D., Blechacz B., Liu C., Schmeckpeper J.D., Tarara J.E., Federspiel M.J., Caplice N., Russell S.J. Infected cell carriers: a new strategy for systemic delivery of oncolytic measles viruses in cancer virotherapy. Mol. Ther. 2007;15:114–122. doi: 10.1038/sj.mt.6300020. [DOI] [PubMed] [Google Scholar]
- 22.Power A.T., Wang J., Falls T.J., Paterson J.M., Parato K.A., Lichty B.D., Stojdl D.F., Forsyth P.A.J., Atkins H., Bell J.C. Carrier cell-based delivery of an oncolytic virus circumvents antiviral immunity. Mol. Ther. 2007;15:123–130. doi: 10.1038/sj.mt.6300039. [DOI] [PubMed] [Google Scholar]
- 23.Hirasawa K., Nishikawa S.G., Norman K.L., Coffey M.C., Thompson B.G., Yoon C.S., Waisman D.M., Lee P.W.K. Systemic reovirus therapy of metastatic cancer in immune-competent mice. Cancer Res. 2003;63:348–353. [PubMed] [Google Scholar]
- 24.Ikeda K., Ichikawa T., Wakimoto H., Silver J.S., Deisboeck T.S., Finkelstein D., Harsh G.R., 4th, Louis D.N., Bartus R.T., Hochberg F.H., Chiocca E.A. Oncolytic virus therapy of multiple tumors in the brain requires suppression of innate and elicited antiviral responses. Nat. Med. 1999;5:881–887. doi: 10.1038/11320. [DOI] [PubMed] [Google Scholar]
- 25.Tsai V., Johnson D.E., Rahman A., Wen S.F., LaFace D., Philopena J., Nery J., Zepeda M., Maneval D.C., Demers G.W., Ralston R. Impact of human neutralizing antibodies on antitumor efficacy of an oncolytic adenovirus in a murine model. Clin. Cancer Res. 2004;10:7199–7206. doi: 10.1158/1078-0432.CCR-04-0765. [DOI] [PubMed] [Google Scholar]
- 26.Lang S.I., Giese N.A., Rommelaere J., Dinsart C., Cornelis J.J. Humoral immune responses against minute virus of mice vectors. J. Gene Med. 2006;8:1141–1150. doi: 10.1002/jgm.940. [DOI] [PubMed] [Google Scholar]
- 27.Liu Z., Ge Y., Wang H., Ma C., Feist M., Ju S., Guo Z.S., Bartlett D.L. Modifying the cancer-immune set point using vaccinia virus expressing re-designed interleukin-2. Nat. Commun. 2018;9:4682. doi: 10.1038/s41467-018-06954-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang L.-C.S., Lynn R.C., Cheng G., Alexander E., Kapoor V., Moon E.K., Sun J., Fridlender Z.G., Isaacs S.N., Thorne S.H., Albelda S.M. Treating Tumors With a Vaccinia Virus Expressing IFNβ Illustrates the Complex Relationships Between Oncolytic Ability and Immunogenicity. Mol. Ther. 2012;20:736–748. doi: 10.1038/mt.2011.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang M., Giehl E., Feng C., Feist M., Chen H., Dai E., Liu Z., Ma C., Ravindranathan R., Bartlett D.L., et al. IL-36γ-armed oncolytic virus exerts superior efficacy through induction of potent adaptive antitumor immunity. Cancer Immunol. Immunother. 2021;70:2467–2481. doi: 10.1007/s00262-021-02860-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen L., Chen H., Ye J., Ge Y., Wang H., Dai E., Ren J., Liu W., Ma C., Ju S., et al. Intratumoral expression of interleukin 23 variants using oncolytic vaccinia virus elicit potent antitumor effects on multiple tumor models via tumor microenvironment modulation. Theranostics. 2021;11:6668–6681. doi: 10.7150/thno.56494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Remy-Ziller C., Thioudellet C., Hortelano J., Gantzer M., Nourtier V., Claudepierre M.C., Sansas B., Préville X., Bendjama K., Quemeneur E., Rittner K. Sequential administration of MVA-based vaccines and PD-1/PD-L1-blocking antibodies confers measurable benefits on tumor growth and survival: Preclinical studies with MVA-βGal and MVA-MUC1 (TG4010) in a murine tumor model. Hum. Vaccin. Immunother. 2018;14:140–145. doi: 10.1080/21645515.2017.1373921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hu J., Wang H., Gu J., Liu X., Zhou X. Trail armed oncolytic poxvirus suppresses lung cancer cell by inducing apoptosis. Acta Biochim. Biophys. Sin. 2018;50:1018–1027. doi: 10.1093/abbs/gmy096. [DOI] [PubMed] [Google Scholar]
- 33.Hofmann E., Weibel S., Szalay A.A. Combination treatment with oncolytic Vaccinia virus and cyclophosphamide results in synergistic antitumor effects in human lung adenocarcinoma bearing mice. J. Transl. Med. 2014;12:197. doi: 10.1186/1479-5876-12-197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kirn D.H., Wang Y., Liang W., Contag C.H., Thorne S.H. Enhancing poxvirus oncolytic effects through increased spread and immune evasion. Cancer Res. 2008;68:2071–2075. doi: 10.1158/0008-5472.CAN-07-6515. [DOI] [PubMed] [Google Scholar]
- 35.Thorne S.H., Negrin R.S., Contag C.H. Synergistic antitumor effects of immune cell-viral biotherapy. Science. 2006;311:1780–1784. doi: 10.1126/science.1121411. [DOI] [PubMed] [Google Scholar]
- 36.Ahmed J., Chard L.S., Yuan M., Wang J., Howells A., Li Y., Li H., Zhang Z., Lu S., Gao D., et al. A new oncolytic V accinia virus augments antitumor immune responses to prevent tumor recurrence and metastasis after surgery. J. Immunother. Cancer. 2020;8 doi: 10.1136/jitc-2019-000415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Frentzen A., Yu Y.A., Chen N., Zhang Q., Weibel S., Raab V., Szalay A.A. Anti-VEGF single-chain antibody GLAF-1 encoded by oncolytic vaccinia virus significantly enhances antitumor therapy. Proc. Natl. Acad. Sci. USA. 2009;106:12915–12920. doi: 10.1073/pnas.0900660106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yu F., Wang X., Guo Z.S., Bartlett D.L., Gottschalk S.M., Song X.T. T-cell engager-armed oncolytic vaccinia virus significantly enhances antitumor therapy. Mol. Ther. 2014;22:102–111. doi: 10.1038/mt.2013.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kellish P., Shabashvili D., Rahman M.M., Nawab A., Guijarro M.V., Zhang M., Cao C., Moussatche N., Boyle T., Antonia S., et al. Oncolytic virotherapy for small-cell lung cancer induces immune infiltration and prolongs survival. J. Clin. Invest. 2019;129:2279–2292. doi: 10.1172/JCI121323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jazowiecka-Rakus J., Sochanik A., Rusin A., Hadryś A., Fidyk W., Villa N., Rahman M.M., Chmielik E., Franco L.S., McFadden G. Myxoma Virus-Loaded Mesenchymal Stem Cells in Experimental Oncolytic Therapy of Murine Pulmonary Melanoma. Mol. Ther. Oncolytics. 2020;18:335–350. doi: 10.1016/j.omto.2020.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Christie J.D., Appel N., Canter H., Achi J.G., Elliott N.M., de Matos A.L., Franco L., Kilbourne J., Lowe K., Rahman M.M., et al. Systemic delivery of TNF-armed myxoma virus plus immune checkpoint inhibitor eliminates lung metastatic mouse osteosarcoma. Mol. Ther. Oncolytics. 2021;22:539–554. doi: 10.1016/j.omto.2021.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Christie J.D., Appel N., Zhang L., Lowe K., Kilbourne J., Daggett-Vondras J., Elliott N., Lucas A.R., Blattman J.N., Rahman M.M., McFadden G. Systemic Delivery of mLIGHT-Armed Myxoma Virus Is Therapeutic for Later-Stage Syngeneic Murine Lung Metastatic Osteosarcoma. Cancers. 2022;14:337. doi: 10.3390/cancers14020337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hotani T., Mizuguchi H., Sakurai F. Systemically Administered Reovirus-Induced Downregulation of Hypoxia Inducible Factor-1α in Subcutaneous Tumors. Mol. Ther. Oncolytics. 2019;12:162–172. doi: 10.1016/j.omto.2018.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Seyed-Khorrami S.M., Soleimanjahi H., Soudi S., Habibian A. MSCs loaded with oncolytic reovirus: migration and in vivo virus delivery potential for evaluating anti-cancer effect in tumor-bearing C57BL/6 mice. Cancer Cell Int. 2021;21:244. doi: 10.1186/s12935-021-01848-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhao D., Chen P., Yang H., Wu Y., Zeng X., Zhao Y., Wen Y., Zhao X., Liu X., Wei Y., Li Y. Live attenuated measles virus vaccine induces apoptosis and promotes tumor regression in lung cancer. Oncol. Rep. 2013;29:199–204. doi: 10.3892/or.2012.2109. [DOI] [PubMed] [Google Scholar]
- 46.Boisgerault N., Guillerme J.B., Pouliquen D., Mesel-Lemoine M., Achard C., Combredet C., Fonteneau J.F., Tangy F., Grégoire M. Natural oncolytic activity of live-attenuated measles virus against human lung and colorectal adenocarcinomas. BioMed Res. Int. 2013;2013 doi: 10.1155/2013/387362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Masemann D., Köther K., Kuhlencord M., Varga G., Roth J., Lichty B.D., Rapp U.R., Wixler V., Ludwig S. Oncolytic influenza virus infection restores immunocompetence of lung tumor-associated alveolar macrophages. OncoImmunology. 2018;7 doi: 10.1080/2162402X.2017.1423171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li J.-M., Kao K.-C., Li L.-F., Yang T.-M., Wu C.-P., Horng Y.-M., Jia W.W.G., Yang C.-T. MicroRNA-145 regulates oncolytic herpes simplex virus-1 for selective killing of human non-small cell lung cancer cells. Virol. J. 2013;10:241. doi: 10.1186/1743-422X-10-241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Leoni V., Gatta V., Palladini A., Nicoletti G., Ranieri D., Dall’Ora M., Grosso V., Rossi M., Alviano F., Bonsi L., et al. Systemic delivery of HER2-retargeted oncolytic-HSV by mesenchymal stromal cells protects from lung and brain metastases. Oncotarget. 2015;6:34774–34787. doi: 10.18632/oncotarget.5793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chaurasiya S., Chen N.G., Lu J., Martin N., Shen Y., Kim S.-I., Warner S.G., Woo Y., Fong Y. A chimeric poxvirus with J2R (thymidine kinase) deletion shows safety and anti-tumor activity in lung cancer models. Cancer Gene Ther. 2020;27:125–135. doi: 10.1038/s41417-019-0114-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Miyamoto S., Inoue H., Nakamura T., Yamada M., Sakamoto C., Urata Y., Okazaki T., Marumoto T., Takahashi A., Takayama K., et al. Coxsackievirus B3 is an oncolytic virus with immunostimulatory properties that is active against lung adenocarcinoma. Cancer Res. 2012;72:2609–2621. doi: 10.1158/0008-5472.CAN-11-3185. [DOI] [PubMed] [Google Scholar]
- 52.Deng H., Liu H., de Silva T., Xue Y., Mohamud Y., Ng C.S., Qu J., Zhang J., Jia W.W.G., Lockwood W.W., Luo H. Coxsackievirus Type B3 Is a Potent Oncolytic Virus against KRAS-Mutant Lung Adenocarcinoma. Mol. Ther. Oncolytics. 2019;14:266–278. doi: 10.1016/j.omto.2019.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu H., Xue Y.C., Deng H., Mohamud Y., Ng C.S., Chu A., Lim C.J., Lockwood W.W., Jia W.W.G., Luo H. MicroRNA Modification of Coxsackievirus B3 Decreases Its Toxicity, while Retaining Oncolytic Potency against Lung Cancer. Mol. Ther. Oncolytics. 2020;16:207–218. doi: 10.1016/j.omto.2020.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Qiu W., Ding X., Li S., He Y., Zhu L. Oncolytic Bovine Herpesvirus 1 Inhibits Human Lung Adenocarcinoma A549 Cell Proliferation and Tumor Growth by Inducing DNA Damage. Int. J. Mol. Sci. 2021;22 doi: 10.3390/ijms22168582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yurchenko K.S., Zhou P., Kovner A.V., Zavjalov E.L., Shestopalova L.V., Shestopalov A.M. Oncolytic effect of wild-type Newcastle disease virus isolates in cancer cell lines in vitro and in vivo on xenograft model. PLoS One. 2018;13 doi: 10.1371/journal.pone.0195425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ye T., Jiang K., Wei L., Barr M.P., Xu Q., Zhang G., Ding C., Meng S., Piao H. Oncolytic Newcastle disease virus induces autophagy-dependent immunogenic cell death in lung cancer cells. Am. J. Cancer Res. 2018;8:1514–1527. [PMC free article] [PubMed] [Google Scholar]
- 57.Hu L., Sun S., Wang T., Li Y., Jiang K., Lin G., Ma Y., Barr M.P., Song F., Zhang G., Meng S. Oncolytic newcastle disease virus triggers cell death of lung cancer spheroids and is enhanced by pharmacological inhibition of autophagy. Am. J. Cancer Res. 2015;5:3612–3623. [PMC free article] [PubMed] [Google Scholar]
- 58.Yaacov B., Eliahoo E., Lazar I., Ben-Shlomo M., Greenbaum I., Panet A., Zakay-Rones Z. Selective oncolytic effect of an attenuated Newcastle disease virus (NDV-HUJ) in lung tumors. Cancer Gene Ther. 2008;15:795–807. doi: 10.1038/cgt.2008.31. [DOI] [PubMed] [Google Scholar]
- 59.Chai Z., Zhang P., Fu F., Zhang X., Liu Y., Hu L., Li X. Oncolytic therapy of a recombinant Newcastle disease virus D90 strain for lung cancer. Virol. J. 2014;11:84. doi: 10.1186/1743-422X-11-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lei J., Li Q.H., Yang J.L., Liu F., Wang L., Xu W.M., Zhao W.X. The antitumor effects of oncolytic adenovirus H101 against lung cancer. Int. J. Oncol. 2015;47:555–562. doi: 10.3892/ijo.2015.3045. [DOI] [PubMed] [Google Scholar]
- 61.Song G., Fang J., Shang C., Li Y., Zhu Y., Xiu Z., Sun L., Jin N., Li X. Ad-apoptin inhibits glycolysis, migration and invasion in lung cancer cells targeting AMPK/mTOR signaling pathway. Exp. Cell Res. 2021;409 doi: 10.1016/j.yexcr.2021.112926. [DOI] [PubMed] [Google Scholar]
- 62.Zhang J.-F., Wei F., Wang H.-P., Li H.-M., Qiu W., Ren P.-K., Chen X.-F., Huang Q. Potent anti-tumor activity of telomerase-dependent and HSV-TK armed oncolytic adenovirus for non-small cell lung cancer in vitro and in vivo. J. Exp. Clin. Cancer Res. 2010;29:52. doi: 10.1186/1756-9966-29-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gomez-Gutierrez J.G., Nitz J., Sharma R., Wechman S.L., Riedinger E., Martinez-Jaramillo E., Sam Zhou H., McMasters K.M. Combined therapy of oncolytic adenovirus and temozolomide enhances lung cancer virotherapy in vitro and in vivo. Virology. 2016;487:249–259. doi: 10.1016/j.virol.2015.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liu D., Kojima T., Ouchi M., Kuroda S., Watanabe Y., Hashimoto Y., Onimatsu H., Urata Y., Fujiwara T. Preclinical evaluation of synergistic effect of telomerase-specific oncolytic virotherapy and gemcitabine for human lung cancer. Mol. Cancer Ther. 2009;8:980–987. doi: 10.1158/1535-7163.MCT-08-0901. [DOI] [PubMed] [Google Scholar]
- 65.Dias J.D., Hemminki O., Diaconu I., Hirvinen M., Bonetti A., Guse K., Escutenaire S., Kanerva A., Pesonen S., Löskog A., et al. Targeted cancer immunotherapy with oncolytic adenovirus coding for a fully human monoclonal antibody specific for CTLA-4. Gene Ther. 2012;19:988–998. doi: 10.1038/gt.2011.176. [DOI] [PubMed] [Google Scholar]
- 66.Hakkarainen T., Särkioja M., Lehenkari P., Miettinen S., Ylikomi T., Suuronen R., Desmond R.A., Kanerva A., Hemminki A. Human mesenchymal stem cells lack tumor tropism but enhance the antitumor activity of oncolytic adenoviruses in orthotopic lung and breast tumors. Hum. Gene Ther. 2007;18:627–641. doi: 10.1089/hum.2007.034. [DOI] [PubMed] [Google Scholar]
- 67.Saito A., Morishita N., Mitsuoka C., Kitajima S., Hamada K., Lee K.M., Kawabata M., Fujisawa M., Shirakawa T. Intravenous injection of irradiated tumor cell vaccine carrying oncolytic adenovirus suppressed the growth of multiple lung tumors in a mouse squamous cell carcinoma model. J. Gene Med. 2011;13:353–361. doi: 10.1002/jgm.1578. [DOI] [PubMed] [Google Scholar]
- 68.Rao J.S., Gondi C., Chetty C., Chittivelu S., Joseph P.A., Lakka S.S. Inhibition of invasion, angiogenesis, tumor growth, and metastasis by adenovirus-mediated transfer of antisense uPAR and MMP-9 in non-small cell lung cancer cells. Mol. Cancer Ther. 2005;4:1399–1408. doi: 10.1158/1535-7163.MCT-05-0082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hamada K., Zhang T., Desaki J., Nakashiro K.i., Itoh H., Tani K., Koyama Y., Hamakawa H. Carrier cell-mediated cell lysis of squamous cell carcinoma cells by squamous cell carcinoma antigen 1 promoter-driven oncolytic adenovirus. J. Gene Med. 2010;12:545–554. doi: 10.1002/jgm.1467. [DOI] [PubMed] [Google Scholar]
- 70.Sostoa J., Fajardo C.A., Moreno R., Ramos M.D., Farrera-Sal M., Alemany R. Targeting the tumor stroma with an oncolytic adenovirus secreting a fibroblast activation protein-targeted bispecific T-cell engager. J. Immunother. Cancer. 2019;7:19. doi: 10.1186/s40425-019-0505-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Freedman J.D., Duffy M.R., Lei-Rossmann J., Muntzer A., Scott E.M., Hagel J., Campo L., Bryant R.J., Verrill C., Lambert A., et al. An Oncolytic Virus Expressing a T-cell Engager Simultaneously Targets Cancer and Immunosuppressive Stromal Cells. Cancer Res. 2018;78:6852–6865. doi: 10.1158/0008-5472.CAN-18-1750. [DOI] [PubMed] [Google Scholar]
- 72.Lin X.R., Zhou X.L., Feng Q., Pan X.Y., Song S.L., Fang H., Lei J., Yang J.L. CIK cell-based delivery of recombinant adenovirus KGHV500 carrying the anti-p21Ras scFv gene enhances the anti-tumor effect and safety in lung cancer. J. Cancer Res. Clin. Oncol. 2019;145:1123–1132. doi: 10.1007/s00432-019-02857-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jiang R., Zhang Z., Liao X., Huang L., Liao Y., Deng W. Combination of oncolytic adenovirus ZD55 harboring TRAIL-IETD-MnSOD and cytokine-induced killer cells against lung cancer. Ann. Transl. Med. 2021;9:1527. doi: 10.21037/atm-21-4479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hoyos V., Del Bufalo F., Yagyu S., Ando M., Dotti G., Suzuki M., Bouchier-Hayes L., Alemany R., Brenner M.K. Mesenchymal Stromal Cells for Linked Delivery of Oncolytic and Apoptotic Adenoviruses to Non-small-cell Lung Cancers. Mol. Ther. 2015;23:1497–1506. doi: 10.1038/mt.2015.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Moreno R., Fajardo C.A., Farrera-Sal M., Perisé-Barrios A.J., Morales-Molina A., Al-Zaher A.A., García-Castro J., Alemany R. Enhanced Antitumor Efficacy of Oncolytic Adenovirus-loaded Menstrual Blood-derived Mesenchymal Stem Cells in Combination with Peripheral Blood Mononuclear Cells. Mol. Cancer Ther. 2019;18:127–138. doi: 10.1158/1535-7163.MCT-18-0431. [DOI] [PubMed] [Google Scholar]
- 76.Rincón E., Cejalvo T., Kanojia D., Alfranca A., Rodríguez-Milla M.Á., Gil Hoyos R.A., Han Y., Zhang L., Alemany R., Lesniak M.S., García-Castro J. Mesenchymal stem cell carriers enhance antitumor efficacy of oncolytic adenoviruses in an immunocompetent mouse model. Oncotarget. 2017;8:45415–45431. doi: 10.18632/oncotarget.17557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Morales-Molina Á., Gambera S., Cejalvo T., Moreno R., Rodríguez-Milla M.Á., Perisé-Barrios A.J., García-Castro J. Antitumor virotherapy using syngeneic or allogeneic mesenchymal stem cell carriers induces systemic immune response and intratumoral leukocyte infiltration in mice. Cancer Immunol. Immunother. 2018;67:1589–1602. doi: 10.1007/s00262-018-2220-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Barlabé P., Sostoa J.d., Fajardo C.A., Alemany R., Moreno R. Enhanced antitumor efficacy of an oncolytic adenovirus armed with an EGFR-targeted BiTE using menstrual blood-derived mesenchymal stem cells as carriers. Cancer Gene Ther. 2020;27:383–388. doi: 10.1038/s41417-019-0110-1. [DOI] [PubMed] [Google Scholar]
- 79.Das K., Belnoue E., Rossi M., Hofer T., Danklmaier S., Nolden T., Schreiber L.-M., Angerer K., Kimpel J., Hoegler S., et al. A modular self-adjuvanting cancer vaccine combined with an oncolytic vaccine induces potent antitumor immunity. Nat. Commun. 2021;12:5195. doi: 10.1038/s41467-021-25506-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ram P., Tung N., Sharma H., Fulton B., Peng K.-W., Russell S., Mohrs M., Thurston G., Kyratsous C., Baum A., et al. 851 Voyager V1 (VV1) oncolytic virus combined with immune checkpoint therapy boosts CTL responses to multiple tumor antigens and correspondingly deepens tumor responses in murine models of melanoma, lung and colon cancer. Journal for ImmunoTherapy of Cancer. 2022;10:A890. [Google Scholar]
- 81.Patel M.R., Jacobson B.A., Ji Y., Hebbel R.P., Kratzke R.A. Blood Outgrowth Endothelial Cells as a Cellular Carrier for Oncolytic Vesicular Stomatitis Virus Expressing Interferon-β in Preclinical Models of Non-Small Cell Lung Cancer. Transl. Oncol. 2020;13 doi: 10.1016/j.tranon.2020.100782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Patel M.R., Kratzke R.A. Oncolytic virus therapy for cancer: the first wave of translational clinical trials. Transl. Res. 2013;161:355–364. doi: 10.1016/j.trsl.2012.12.010. [DOI] [PubMed] [Google Scholar]
- 83.Andtbacka R.H.I., Kaufman H.L., Collichio F., Amatruda T., Senzer N., Chesney J., Delman K.A., Spitler L.E., Puzanov I., Agarwala S.S., et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. J. Clin. Oncol. 2015;33:2780–2788. doi: 10.1200/JCO.2014.58.3377. [DOI] [PubMed] [Google Scholar]
- 84.Bommareddy P.K., Patel A., Hossain S., Kaufman H.L. Talimogene Laherparepvec (T-VEC) and Other Oncolytic Viruses for the Treatment of Melanoma. Am. J. Clin. Dermatol. 2017;18:1–15. doi: 10.1007/s40257-016-0238-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Shmuylovich L., McEvoy A.M., Fields R.C., Hernandez-Aya L., Ansstas G., Chen D.Y. Durable melanoma control following disseminated talimogene laherparepvec herpetic infection. JAAD Case Rep. 2022;29:131–133. doi: 10.1016/j.jdcr.2022.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tesfay M.Z., Kirk A.C., Hadac E.M., Griesmann G.E., Federspiel M.J., Barber G.N., Henry S.M., Peng K.W., Russell S.J. PEGylation of vesicular stomatitis virus extends virus persistence in blood circulation of passively immunized mice. J. Virol. 2013;87:3752–3759. doi: 10.1128/JVI.02832-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cook M., Chauhan A. Clinical Application of Oncolytic Viruses: A Systematic Review. Int. J. Mol. Sci. 2020;21:7505. doi: 10.3390/ijms21207505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kirn D.H., Thorne S.H. Targeted and armed oncolytic poxviruses: a novel multi-mechanistic therapeutic class for cancer. Nat. Rev. Cancer. 2009;9:64–71. doi: 10.1038/nrc2545. [DOI] [PubMed] [Google Scholar]
- 89.Nakatake M., Kurosaki H., Kuwano N., Horita K., Ito M., Kono H., Okamura T., Hasegawa K., Yasutomi Y., Nakamura T. Partial Deletion of Glycoprotein B5R Enhances Vaccinia Virus Neutralization Escape while Preserving Oncolytic Function. Mol. Ther. Oncolytics. 2019;14:159–171. doi: 10.1016/j.omto.2019.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kirn D.H., Wang Y., Liang W., Contag C.H., Thorne S.H. Enhancing Poxvirus Oncolytic Effects through Increased Spread and Immune Evasion. Cancer Res. 2008;68:2071–2075. doi: 10.1158/0008-5472.CAN-07-6515. [DOI] [PubMed] [Google Scholar]
- 91.Peng K.W., Myers R., Greenslade A., Mader E., Greiner S., Federspiel M.J., Dispenzieri A., Russell S.J. Using clinically approved cyclophosphamide regimens to control the humoral immune response to oncolytic viruses. Gene Ther. 2013;20:255–261. doi: 10.1038/gt.2012.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Almstätter I., Mykhaylyk O., Settles M., Altomonte J., Aichler M., Walch A., Rummeny E.J., Ebert O., Plank C., Braren R. Characterization of magnetic viral complexes for targeted delivery in oncology. Theranostics. 2015;5:667–685. doi: 10.7150/thno.10438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.García-Castro J., Martínez-Palacio J., Lillo R., García-Sánchez F., Alemany R., Madero L., Bueren J.A., Ramírez M. Tumor cells as cellular vehicles to deliver gene therapies to metastatic tumors. Cancer Gene Ther. 2005;12:341–349. doi: 10.1038/sj.cgt.7700801. [DOI] [PubMed] [Google Scholar]
- 94.Ilett E.J., Prestwich R.J., Kottke T., Errington F., Thompson J.M., Harrington K.J., Pandha H.S., Coffey M., Selby P.J., Vile R.G., Melcher A.A. Dendritic cells and T cells deliver oncolytic reovirus for tumour killing despite pre-existing anti-viral immunity. Gene Ther. 2009;16:689–699. doi: 10.1038/gt.2009.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Garcia-Carbonero R., Salazar R., Duran I., Osman-Garcia I., Paz-Ares L., Bozada J.M., Boni V., Blanc C., Seymour L., Beadle J., et al. Phase 1 study of intravenous administration of the chimeric adenovirus enadenotucirev in patients undergoing primary tumor resection. J. Immunother. Cancer. 2017;5:71. doi: 10.1186/s40425-017-0277-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tanoue K., Rosewell Shaw A., Watanabe N., Porter C., Rana B., Gottschalk S., Brenner M., Suzuki M. Armed Oncolytic Adenovirus-Expressing PD-L1 Mini-Body Enhances Antitumor Effects of Chimeric Antigen Receptor T Cells in Solid Tumors. Cancer Res. 2017;77:2040–2051. doi: 10.1158/0008-5472.CAN-16-1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ruano D., López-Martín J.A., Moreno L., Lassaletta Á., Bautista F., Andión M., Hernández C., González-Murillo Á., Melen G., Alemany R., et al. First-in-Human, First-in-Child Trial of Autologous MSCs Carrying the Oncolytic Virus Icovir-5 in Patients with Advanced Tumors. Mol. Ther. 2020;28:1033–1042. doi: 10.1016/j.ymthe.2020.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ramlau R., Quoix E., Rolski J., Pless M., Lena H., Lévy E., Krzakowski M., Hess D., Tartour E., Chenard M.-P., et al. A Phase II Study of Tg4010 (Mva-Muc1-Il2) in Association with Chemotherapy in Patients with Stage III/IV Non-small Cell Lung Cancer. J. Thorac. Oncol. 2008;3:735–744. doi: 10.1097/JTO.0b013e31817c6b4f. [DOI] [PubMed] [Google Scholar]
- 99.Quoix E., Lena H., Losonczy G., Forget F., Chouaid C., Papai Z., Gervais R., Ottensmeier C., Szczesna A., Kazarnowicz A., et al. TG4010 immunotherapy and first-line chemotherapy for advanced non-small-cell lung cancer (TIME): results from the phase 2b part of a randomised, double-blind, placebo-controlled, phase 2b/3 trial. Lancet Oncol. 2016;17:212–223. doi: 10.1016/S1470-2045(15)00483-0. [DOI] [PubMed] [Google Scholar]
- 100.Breitbach C.J., Arulanandam R., De Silva N., Thorne S.H., Patt R., Daneshmand M., Moon A., Ilkow C., Burke J., Hwang T.H., et al. Oncolytic vaccinia virus disrupts tumor-associated vasculature in humans. Cancer Res. 2013;73:1265–1275. doi: 10.1158/0008-5472.CAN-12-2687. [DOI] [PubMed] [Google Scholar]
- 101.Bradbury P.A., Morris D.G., Nicholas G., Tu D., Tehfe M., Goffin J.R., Shepherd F.A., Gregg R.W., Rothenstein J., Lee C., et al. Canadian Cancer Trials Group (CCTG) IND211: A randomized trial of pelareorep (Reolysin) in patients with previously treated advanced or metastatic non-small cell lung cancer receiving standard salvage therapy. Lung Cancer. 2018;120:142–148. doi: 10.1016/j.lungcan.2018.03.005. [DOI] [PubMed] [Google Scholar]
- 102.Gong J., Sachdev E., Mita A.C., Mita M.M. Clinical development of reovirus for cancer therapy: An oncolytic virus with immune-mediated antitumor activity. World J. Methodol. 2016;6:25–42. doi: 10.5662/wjm.v6.i1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Villalona-Calero M.A., Lam E., Otterson G.A., Zhao W., Timmons M., Subramaniam D., Hade E.M., Gill G.M., Coffey M., Selvaggi G., et al. Oncolytic reovirus in combination with chemotherapy in metastatic or recurrent non–small cell lung cancer patients with KRAS-activated tumors. Cancer. 2016;122:875–883. doi: 10.1002/cncr.29856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Schenk E.L., Mandrekar S.J., Dy G.K., Aubry M.C., Tan A.D., Dakhil S.R., Sachs B.A., Nieva J.J., Bertino E., Lee Hann C., et al. A Randomized Double-Blind Phase II Study of the Seneca Valley Virus (NTX-010) versus Placebo for Patients with Extensive-Stage SCLC (ES SCLC) Who Were Stable or Responding after at Least Four Cycles of Platinum-Based Chemotherapy: North Central Cancer Treatment Group (Alliance) N0923 Study. J. Thorac. Oncol. 2020;15:110–119. doi: 10.1016/j.jtho.2019.09.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sung H., Ferlay J., Siegel R.L., Laversanne M., Soerjomataram I., Jemal A., Bray F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA. Cancer J. Clin. 2021;71:209–249. doi: 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- 106.Yuan M., Huang L.L., Chen J.H., Wu J., Xu Q. The emerging treatment landscape of targeted therapy in non-small-cell lung cancer. Signal Transduct. Target. Ther. 2019;4:61. doi: 10.1038/s41392-019-0099-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wang C., Long Y., Li W., Dai W., Xie S., Liu Y., Zhang Y., Liu M., Tian Y., Li Q., Duan Y. Exploratory study on classification of lung cancer subtypes through a combined K-nearest neighbor classifier in breathomics. Sci. Rep. 2020;10:5880. doi: 10.1038/s41598-020-62803-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Cortés-Jofré M., Uranga R., Torres Pombert A., Arango Prado M.D.C., Caballero Aguirrechu I., Pacheco C., Ortiz Reyes R.M., Chuecas F., Mas Bermejo P.I. Therapeutic vaccines for advanced non-small cell lung cancer. Cochrane Database Syst. Rev. 2019:CD013377. doi: 10.1002/14651858.CD013377.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Losanno T., Gridelli C. Recent advances in targeted advanced lung cancer therapy in the elderly. Expert Rev. Anticancer Ther. 2017;17:787–797. doi: 10.1080/14737140.2017.1348232. [DOI] [PubMed] [Google Scholar]
- 110.Cáceres-Lavernia H.H., Nenínger-Vinageras E., Varona-Rodríguez L.M., Olivares-Romero Y.A., Sánchez-Rojas I., Mazorra-Herrera Z., Basanta-Bergolla D., Duvergel-Calderín D., Torres-Cuevas B.L., Castillo-Carrillo C. Racotumomab in Non-Small Cell Lung Cancer as Maintenance and Second-Line Treatment. MEDICC Rev. 2021;23:21–28. doi: 10.37757/MR2021.V23.N3.5. [DOI] [PubMed] [Google Scholar]
- 111.Stevens D., Ingels J., Van Lint S., Vandekerckhove B., Vermaelen K. Dendritic Cell-Based Immunotherapy in Lung Cancer. Front. Immunol. 2020;11 doi: 10.3389/fimmu.2020.620374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Osmani L., Askin F., Gabrielson E., Li Q.K. Current WHO guidelines and the critical role of immunohistochemical markers in the subclassification of non-small cell lung carcinoma (NSCLC): Moving from targeted therapy to immunotherapy. Semin. Cancer Biol. 2018;52:103–109. doi: 10.1016/j.semcancer.2017.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hirsch F.R., Scagliotti G.V., Mulshine J.L., Kwon R., Curran W.J., Jr., Wu Y.L., Paz-Ares L. Lung cancer: current therapies and new targeted treatments. Lancet. 2017;389:299–311. doi: 10.1016/S0140-6736(16)30958-8. [DOI] [PubMed] [Google Scholar]
- 114.Raso M.G., Bota-Rabassedas N., Wistuba I.I. Pathology and Classification of SCLC. Cancers. 2021;13:820. doi: 10.3390/cancers13040820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Korde R., Veluswamy R., Allaire J.C., Barnes G. Small cell lung cancer patients treated with immune checkpoint inhibitor: a systematic literature review of treatment efficacy, safety and quality of life. Curr. Med. Res. Opin. 2022;38:1361–1368. doi: 10.1080/03007995.2022.2078101. [DOI] [PubMed] [Google Scholar]
- 116.Maroun J., Muñoz-Alía M., Ammayappan A., Schulze A., Peng K.W., Russell S. Designing and building oncolytic viruses. Future Virol. 2017;12:193–213. doi: 10.2217/fvl-2016-0129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Seymour L.W., Fisher K.D. Oncolytic viruses: finally delivering. Br. J. Cancer. 2016;114:357–361. doi: 10.1038/bjc.2015.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kaufman H.L., Maciorowski D. Advancing oncolytic virus therapy by understanding the biology. Nat. Rev. Clin. Oncol. 2021 doi: 10.1038/s41571-021-00490-4. [DOI] [PubMed] [Google Scholar]
- 119.Yang Y., Xu H., Shen J., Yang Y., Wu S., Xiao J., Xu Y., Liu X.Y., Chu L. RGD-modifided oncolytic adenovirus exhibited potent cytotoxic effect on CAR-negative bladder cancer-initiating cells. Cell Death Dis. 2015;6 doi: 10.1038/cddis.2015.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Martuza R.L. Conditionally replicating herpes vectors for cancer therapy. J. Clin. Invest. 2000;105:841–846. doi: 10.1172/JCI9744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Jahan N., Ghouse S.M., Martuza R.L., Rabkin S.D. Situ Cancer Vaccination and Immunovirotherapy Using Oncolytic HSV. Viruses. 2021;13 doi: 10.3390/v13091740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Goldsmith K., Chen W., Johnson D.C., Hendricks R.L. Infected cell protein (ICP)47 enhances herpes simplex virus neurovirulence by blocking the CD8+ T cell response. J. Exp. Med. 1998;187:341–348. doi: 10.1084/jem.187.3.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Chang H.W., Jacobs B.L. Identification of a conserved motif that is necessary for binding of the vaccinia virus E3L gene products to double-stranded RNA. Virology. 1993;194:537–547. doi: 10.1006/viro.1993.1292. [DOI] [PubMed] [Google Scholar]
- 124.Carroll K., Elroy-Stein O., Moss B., Jagus R. Recombinant vaccinia virus K3L gene product prevents activation of double-stranded RNA-dependent, initiation factor 2 alpha-specific protein kinase. J. Biol. Chem. 1993;268:12837–12842. [PubMed] [Google Scholar]
- 125.Kotwal G.J., Isaacs S.N., McKenzie R., Frank M.M., Moss B. Inhibition of the complement cascade by the major secretory protein of vaccinia virus. Science. 1990;250:827–830. doi: 10.1126/science.2237434. [DOI] [PubMed] [Google Scholar]
- 126.Herrera E., Lorenzo M.M., Blasco R., Isaacs S.N. Functional analysis of vaccinia virus B5R protein: essential role in virus envelopment is independent of a large portion of the extracellular domain. J. Virol. 1998;72:294–302. doi: 10.1128/jvi.72.1.294-302.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Buller R.M., Smith G.L., Cremer K., Notkins A.L., Moss B. Decreased virulence of recombinant vaccinia virus expression vectors is associated with a thymidine kinase-negative phenotype. Nature. 1985;317:813–815. doi: 10.1038/317813a0. [DOI] [PubMed] [Google Scholar]
- 128.McCart J.A., Ward J.M., Lee J., Hu Y., Alexander H.R., Libutti S.K., Moss B., Bartlett D.L. Systemic cancer therapy with a tumor-selective vaccinia virus mutant lacking thymidine kinase and vaccinia growth factor genes. Cancer Res. 2001;61:8751–8757. [PubMed] [Google Scholar]
- 129.Norman K.L., Hirasawa K., Yang A.D., Shields M.A., Lee P.W.K. Reovirus oncolysis: the Ras/RalGEF/p38 pathway dictates host cell permissiveness to reovirus infection. Proc. Natl. Acad. Sci. USA. 2004;101:11099–11104. doi: 10.1073/pnas.0404310101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Lorence R.M., Roberts M.S., O'Neil J.D., Groene W.S., Miller J.A., Mueller S.N., Bamat M.K. Phase 1 clinical experience using intravenous administration of PV701, an oncolytic Newcastle disease virus. Curr. Cancer Drug Targets. 2007;7:157–167. doi: 10.2174/156800907780058853. [DOI] [PubMed] [Google Scholar]
- 131.Hotte S.J., Lorence R.M., Hirte H.W., Polawski S.R., Bamat M.K., O'Neil J.D., Roberts M.S., Groene W.S., Major P.P. An optimized clinical regimen for the oncolytic virus PV701. Clin. Cancer Res. 2007;13:977–985. doi: 10.1158/1078-0432.CCR-06-1817. [DOI] [PubMed] [Google Scholar]
- 132.Csatary L.K., Gosztonyi G., Szeberenyi J., Fabian Z., Liszka V., Bodey B., Csatary C.M. MTH-68/H oncolytic viral treatment in human high-grade gliomas. J. Neuro Oncol. 2004;67:83–93. doi: 10.1023/b:neon.0000021735.85511.05. [DOI] [PubMed] [Google Scholar]
- 133.Yu Y.A., Shabahang S., Timiryasova T.M., Zhang Q., Beltz R., Gentschev I., Goebel W., Szalay A.A. Visualization of tumors and metastases in live animals with bacteria and vaccinia virus encoding light-emitting proteins. Nat. Biotechnol. 2004;22:313–320. doi: 10.1038/nbt937. [DOI] [PubMed] [Google Scholar]
- 134.Power A.T., Bell J.C. Taming the Trojan horse: optimizing dynamic carrier cell/oncolytic virus systems for cancer biotherapy. Gene Ther. 2008;15:772–779. doi: 10.1038/gt.2008.40. [DOI] [PubMed] [Google Scholar]
- 135.Howard F., Muthana M. Designer nanocarriers for navigating the systemic delivery of oncolytic viruses. Nanomedicine. 2020;15:93–110. doi: 10.2217/nnm-2019-0323. [DOI] [PubMed] [Google Scholar]
- 136.Rosewell Shaw A., Suzuki M. Oncolytic Viruses Partner With T-Cell Therapy for Solid Tumor Treatment. Front. Immunol. 2018;9:2103. doi: 10.3389/fimmu.2018.02103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Phan M., Watson M.F., Alain T., Diallo J.S. Oncolytic Viruses on Drugs: Achieving Higher Therapeutic Efficacy. ACS Infect. Dis. 2018;4:1448–1467. doi: 10.1021/acsinfecdis.8b00144. [DOI] [PubMed] [Google Scholar]
- 138.Hwang C.C., Igase M., Sakurai M., Haraguchi T., Tani K., Itamoto K., Shimokawa T., Nakaichi M., Nemoto Y., Noguchi S., et al. Oncolytic reovirus therapy: Pilot study in dogs with spontaneously occurring tumours. Vet. Comp. Oncol. 2018;16:229–238. doi: 10.1111/vco.12361. [DOI] [PubMed] [Google Scholar]
- 139.Mok D.Z.L., Chan K.R. The Effects of Pre-Existing Antibodies on Live-Attenuated Viral Vaccines. Viruses. 2020;12 doi: 10.3390/v12050520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Seyoum M., Enawgaw B., Melku M. Human blood platelets and viruses: defense mechanism and role in the removal of viral pathogens. Thromb. J. 2018;16:16. doi: 10.1186/s12959-018-0170-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Harrington K., Freeman D.J., Kelly B., Harper J., Soria J.-C. Optimizing oncolytic virotherapy in cancer treatment. Nat. Rev. Drug Discov. 2019;18:689–706. doi: 10.1038/s41573-019-0029-0. [DOI] [PubMed] [Google Scholar]
- 142.Lemos de Matos A., Franco L.S., McFadden G. Oncolytic Viruses and the Immune System: The Dynamic Duo. Molecular therapy. Mol. Ther. Methods Clin. Dev. 2020;17:349–358. doi: 10.1016/j.omtm.2020.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Everts A., Bergeman M., McFadden G., Kemp V. Simultaneous Tumor and Stroma Targeting by Oncolytic Viruses. Biomedicines. 2020;8 doi: 10.3390/biomedicines8110474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Choi A.H., O'Leary M.P., Fong Y., Chen N.G. From Benchtop to Bedside: A Review of Oncolytic Virotherapy. Biomedicines. 2016;4 doi: 10.3390/biomedicines4030018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Rahman M.M., McFadden G. Oncolytic Viruses: Newest Frontier for Cancer Immunotherapy. Cancers. 2021;13 doi: 10.3390/cancers13215452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Cary Z.D., Willingham M.C., Lyles D.S. Oncolytic vesicular stomatitis virus induces apoptosis in U87 glioblastoma cells by a type II death receptor mechanism and induces cell death and tumor clearance in vivo. J. Virol. 2011;85:5708–5717. doi: 10.1128/JVI.02393-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Ghiringhelli F., Apetoh L., Tesniere A., Aymeric L., Ma Y., Ortiz C., Vermaelen K., Panaretakis T., Mignot G., Ullrich E., et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat. Med. 2009;15:1170–1178. doi: 10.1038/nm.2028. [DOI] [PubMed] [Google Scholar]
- 148.Engeland C.E., Bell J.C. Introduction to Oncolytic Virotherapy. Methods Mol. Biol. 2020;2058:1–6. doi: 10.1007/978-1-4939-9794-7_1. [DOI] [PubMed] [Google Scholar]
- 149.Jiang H., Fueyo J. Healing after death: antitumor immunity induced by oncolytic adenoviral therapy. OncoImmunology. 2014;3 doi: 10.4161/21624011.2014.947872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Breitbach C.J., Paterson J.M., Lemay C.G., Falls T.J., McGuire A., Parato K.A., Stojdl D.F., Daneshmand M., Speth K., Kirn D., et al. Targeted inflammation during oncolytic virus therapy severely compromises tumor blood flow. Mol. Ther. 2007;15:1686–1693. doi: 10.1038/sj.mt.6300215. [DOI] [PubMed] [Google Scholar]
- 151.Pearl T.M., Markert J.M., Cassady K.A., Ghonime M.G. Oncolytic Virus-Based Cytokine Expression to Improve Immune Activity in Brain and Solid Tumors. Mol. Ther. Oncolytics. 2019;13:14–21. doi: 10.1016/j.omto.2019.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Nguyen H.-M., Guz-Montgomery K., Saha D. Oncolytic Virus Encoding a Master Pro-Inflammatory Cytokine Interleukin 12 in Cancer Immunotherapy. Cells. 2020;9:400. doi: 10.3390/cells9020400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Kaufman H.L., Kim D.W., DeRaffele G., Mitcham J., Coffin R.S., Kim-Schulze S. Local and distant immunity induced by intralesional vaccination with an oncolytic herpes virus encoding GM-CSF in patients with stage IIIc and IV melanoma. Ann. Surg Oncol. 2010;17:718–730. doi: 10.1245/s10434-009-0809-6. [DOI] [PubMed] [Google Scholar]
- 154.Hu H., Qiu Y., Guo M., Huang Y., Fang L., Peng Z., Ji W., Xu Y., Shen S., Yan Y., et al. Targeted Hsp70 expression combined with CIK-activated immune reconstruction synergistically exerts antitumor efficacy in patient-derived hepatocellular carcinoma xenograft mouse models. Oncotarget. 2015;6:1079–1089. doi: 10.18632/oncotarget.2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Wang R., Chen J., Wang W., Zhao Z., Wang H., Liu S., Li F., Wan Y., Yin J., Wang R., et al. CD40L-armed oncolytic herpes simplex virus suppresses pancreatic ductal adenocarcinoma by facilitating the tumor microenvironment favorable to cytotoxic T cell response in the syngeneic mouse model. J. Immunother. Cancer. 2022;10 doi: 10.1136/jitc-2021-003809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Ilkow C.S., Marguerie M., Batenchuk C., Mayer J., Ben Neriah D., Cousineau S., Falls T., Jennings V.A., Boileau M., Bellamy D., et al. Reciprocal cellular cross-talk within the tumor microenvironment promotes oncolytic virus activity. Nat. Med. 2015;21:530–536. doi: 10.1038/nm.3848. [DOI] [PubMed] [Google Scholar]
- 157.Toro Bejarano M., Merchan J.R. Targeting tumor vasculature through oncolytic virotherapy: recent advances. Oncolytic Virother. 2015;4:169–181. doi: 10.2147/OV.S66045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Arulanandam R., Batenchuk C., Angarita F.A., Ottolino-Perry K., Cousineau S., Mottashed A., Burgess E., Falls T.J., De Silva N., Tsang J., et al. VEGF-Mediated Induction of PRD1-BF1/Blimp1 Expression Sensitizes Tumor Vasculature to Oncolytic Virus Infection. Cancer Cell. 2015;28:210–224. doi: 10.1016/j.ccell.2015.06.009. [DOI] [PubMed] [Google Scholar]
- 159.Gao P., Ding G., Wang L. The efficacy and safety of oncolytic viruses in the treatment of intermediate to advanced solid tumors: a systematic review and meta-analysis. Transl. Cancer Res. 2021;10:4290–4302. doi: 10.21037/tcr-21-905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Martin N., Sattentau Q. Cell-to-cell HIV-1 spread and its implications for immune evasion. Curr. Opin. HIV AIDS. 2009;4:143–149. doi: 10.1097/COH.0b013e328322f94a. [DOI] [PubMed] [Google Scholar]
- 161.Melzer M.K., Zeitlinger L., Mall S., Steiger K., Schmid R.M., Ebert O., Krackhardt A., Altomonte J. Enhanced Safety and Efficacy of Oncolytic VSV Therapy by Combination with T Cell Receptor Transgenic T Cells as Carriers. Mol. Ther. Oncolytics. 2019;12:26–40. doi: 10.1016/j.omto.2018.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Na Y., Nam J.P., Hong J., Oh E., Shin H.C., Kim H.S., Kim S.W., Yun C.O. Systemic administration of human mesenchymal stromal cells infected with polymer-coated oncolytic adenovirus induces efficient pancreatic tumor homing and infiltration. J. Control. Release. 2019;305:75–88. doi: 10.1016/j.jconrel.2019.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Yoon A.R., Rivera-Cruz C., Gimble J.M., Yun C.O., Figueiredo M.L. Immunotherapy by mesenchymal stromal cell delivery of oncolytic viruses for treating metastatic tumors. Mol. Ther. Oncolytics. 2022;25:78–97. doi: 10.1016/j.omto.2022.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Spaeth E., Klopp A., Dembinski J., Andreeff M., Marini F. Inflammation and tumor microenvironments: defining the migratory itinerary of mesenchymal stem cells. Gene Ther. 2008;15:730–738. doi: 10.1038/gt.2008.39. [DOI] [PubMed] [Google Scholar]
- 165.Jazowiecka-Rakus J., Sochanik A., Rusin A., Hadryś A., Fidyk W., Villa N., Rahman M.M., Chmielik E., Franco L.S., McFadden G. Myxoma virus-loaded mesenchymal stem cells in experimental oncolytic therapy of murine pulmonary melanoma. Mol. Ther. Oncolytics. 2020;18:335–350. doi: 10.1016/j.omto.2020.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Fischer U.M., Harting M.T., Jimenez F., Monzon-Posadas W.O., Xue H., Savitz S.I., Laine G.A., Cox C.S., Jr. Pulmonary passage is a major obstacle for intravenous stem cell delivery: the pulmonary first-pass effect. Stem Cells Dev. 2009;18:683–692. doi: 10.1089/scd.2008.0253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Leoni V., Gatta V., Palladini A., Nicoletti G., Ranieri D., Dall'Ora M., Grosso V., Rossi M., Alviano F., Bonsi L., et al. Systemic delivery of HER2-retargeted oncolytic-HSV by mesenchymal stromal cells protects from lung and brain metastases. Oncotarget. 2015;6:34774–34787. doi: 10.18632/oncotarget.5793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Wang H., Alarcón C.N., Liu B., Watson F., Searles S., Lee C.K., Keys J., Pi W., Allen D., Lammerding J., et al. Genetically engineered and enucleated human mesenchymal stromal cells for the targeted delivery of therapeutics to diseased tissue. Nat. Biomed. Eng. 2022;6:882–897. doi: 10.1038/s41551-021-00815-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Reale A., Calistri A., Altomonte J. Giving Oncolytic Viruses a Free Ride: Carrier Cells for Oncolytic Virotherapy. Pharmaceutics. 2021;13 doi: 10.3390/pharmaceutics13122192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Reale A., Krutzke L., Cadamuro M., Vitiello A., von Einem J., Kochanek S., Palù G., Parolin C., Calistri A. Human Monocytes Are Suitable Carriers for the Delivery of Oncolytic Herpes Simplex Virus Type 1 In Vitro and in a Chicken Embryo Chorioallantoic Membrane Model of Cancer. Int. J. Mol. Sci. 2023;24:9255. doi: 10.3390/ijms24119255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Eisenstein S., Chen S.H., Pan P.Y. Immune cells: more than simple carriers for systemic delivery of oncolytic viruses. Oncolytic Virother. 2014;3:83–91. doi: 10.2147/OV.S47143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Thorne S.H., Contag C.H. Integrating the biological characteristics of oncolytic viruses and immune cells can optimize therapeutic benefits of cell-based delivery. Gene Ther. 2008;15:753–758. doi: 10.1038/gt.2008.42. [DOI] [PubMed] [Google Scholar]
- 173.Kim Y., Clements D.R., Sterea A.M., Jang H.W., Gujar S.A., Lee P.W.K. Dendritic Cells in Oncolytic Virus-Based Anti-Cancer Therapy. Viruses. 2015;7:6506–6525. doi: 10.3390/v7122953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Ghasemi M., Abbasi L., Ghanbari Naeini L., Kokabian P., Nameh Goshay Fard N., Givtaj N. Dendritic cells and natural killer cells: The road to a successful oncolytic virotherapy. Front. Immunol. 2022;13 doi: 10.3389/fimmu.2022.950079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Ilett E.J., Bárcena M., Errington-Mais F., Griffin S., Harrington K.J., Pandha H.S., Coffey M., Selby P.J., Limpens R.W.A.L., Mommaas M., et al. Internalization of oncolytic reovirus by human dendritic cell carriers protects the virus from neutralization. Clin. Cancer Res. 2011;17:2767–2776. doi: 10.1158/1078-0432.CCR-10-3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Lin Y., Weisdorf D.J., Solovey A., Hebbel R.P. Origins of circulating endothelial cells and endothelial outgrowth from blood. J. Clin. Invest. 2000;105:71–77. doi: 10.1172/JCI8071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Dudek A.Z., Bodempudi V., Welsh B.W., Jasinski P., Griffin R.J., Milbauer L., Hebbel R.P. Systemic inhibition of tumour angiogenesis by endothelial cell-based gene therapy. Br. J. Cancer. 2007;97:513–522. doi: 10.1038/sj.bjc.6603883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Power A.T., Bell J.C. Cell-based delivery of oncolytic viruses: a new strategic alliance for a biological strike against cancer. Mol. Ther. 2007;15:660–665. doi: 10.1038/sj.mt.6300098. [DOI] [PubMed] [Google Scholar]
- 179.Liu C., Russell S.J., Peng K.W. Systemic therapy of disseminated myeloma in passively immunized mice using measles virus-infected cell carriers. Mol. Ther. 2010;18:1155–1164. doi: 10.1038/mt.2010.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Lemay C.G., Rintoul J.L., Kus A., Paterson J.M., Garcia V., Falls T.J., Ferreira L., Bridle B.W., Conrad D.P., Tang V.A., et al. Harnessing oncolytic virus-mediated antitumor immunity in an infected cell vaccine. Mol. Ther. 2012;20:1791–1799. doi: 10.1038/mt.2012.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Chen S.R., Chen M.M., Ene C., Lang F.F., Kan P. Perfusion-guided endovascular super-selective intra-arterial infusion for treatment of malignant brain tumors. J. Neurointerv. Surg. 2022;14:533–538. doi: 10.1136/neurintsurg-2021-018190. [DOI] [PubMed] [Google Scholar]
- 182.Ekeke C.N., Russell K.L., Murthy P., Guo Z.S., Soloff A.C., Weber D., Pan W., Lotze M.T., Dhupar R. Intrapleural interleukin-2-expressing oncolytic virotherapy enhances acute antitumor effects and T-cell receptor diversity in malignant pleural disease. J. Thorac. Cardiovasc. Surg. 2022;163:e313–e328. doi: 10.1016/j.jtcvs.2020.11.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Morein D., Erlichman N., Ben-Baruch A. Beyond Cell Motility: The Expanding Roles of Chemokines and Their Receptors in Malignancy. Front. Immunol. 2020;11 doi: 10.3389/fimmu.2020.00952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Rahman M.M., McFadden G. Oncolytic Virotherapy with Myxoma Virus. J. Clin. Med. 2020;9 doi: 10.3390/jcm9010171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.VanSeggelen H., Tantalo D.G., Afsahi A., Hammill J.A., Bramson J.L. Chimeric antigen receptor-engineered T cells as oncolytic virus carriers. Mol. Ther. Oncolytics. 2015;2 doi: 10.1038/mto.2015.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Santos J., Heiniö C., Quixabeira D., Zafar S., Clubb J., Pakola S., Cervera-Carrascon V., Havunen R., Kanerva A., Hemminki A. Systemic Delivery of Oncolytic Adenovirus to Tumors Using Tumor-Infiltrating Lymphocytes as Carriers. Cells. 2021;10:978. doi: 10.3390/cells10050978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Benencia F., Courreges M.C., Conejo-García J.R., Buckanovich R.J., Zhang L., Carroll R.H., Morgan M.A., Coukos G. Oncolytic HSV exerts direct antiangiogenic activity in ovarian carcinoma. Hum. Gene Ther. 2005;16:765–778. doi: 10.1089/hum.2005.16.765. [DOI] [PubMed] [Google Scholar]
- 188.Breitbach C.J., Burke J., Jonker D., Stephenson J., Haas A.R., Chow L.Q.M., Nieva J., Hwang T.H., Moon A., Patt R., et al. Intravenous delivery of a multi-mechanistic cancer-targeted oncolytic poxvirus in humans. Nature. 2011;477:99–102. doi: 10.1038/nature10358. [DOI] [PubMed] [Google Scholar]
- 189.Sarkar D., Spencer J.A., Phillips J.A., Zhao W., Schafer S., Spelke D.P., Mortensen L.J., Ruiz J.P., Vemula P.K., Sridharan R., et al. Engineered cell homing. Blood. 2011;118:e184–e191. doi: 10.1182/blood-2010-10-311464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Sarkar D., Vemula P.K., Teo G.S.L., Spelke D., Karnik R., Wee L.Y., Karp J.M. Chemical engineering of mesenchymal stem cells to induce a cell rolling response. Bioconjug. Chem. 2008;19:2105–2109. doi: 10.1021/bc800345q. [DOI] [PubMed] [Google Scholar]
- 191.Levy O., Zhao W., Mortensen L.J., Leblanc S., Tsang K., Fu M., Phillips J.A., Sagar V., Anandakumaran P., Ngai J., et al. mRNA-engineered mesenchymal stem cells for targeted delivery of interleukin-10 to sites of inflammation. Blood. 2013;122:e23–e32. doi: 10.1182/blood-2013-04-495119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Liao W., Pham V., Liu L., Riazifar M., Pone E.J., Zhang S.X., Ma F., Lu M., Walsh C.M., Zhao W. Mesenchymal stem cells engineered to express selectin ligands and IL-10 exert enhanced therapeutic efficacy in murine experimental autoimmune encephalomyelitis. Biomaterials. 2016;77:87–97. doi: 10.1016/j.biomaterials.2015.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Muthana M., Scott S.D., Farrow N., Morrow F., Murdoch C., Grubb S., Brown N., Dobson J., Lewis C.E. A novel magnetic approach to enhance the efficacy of cell-based gene therapies. Gene Ther. 2008;15:902–910. doi: 10.1038/gt.2008.57. [DOI] [PubMed] [Google Scholar]
- 194.Wahajuddin A.S., Arora S. Superparamagnetic iron oxide nanoparticles: magnetic nanoplatforms as drug carriers. Int. J. Nanomedicine. 2012;7:3445–3471. doi: 10.2147/IJN.S30320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Fattahi H., Laurent S., Liu F., Arsalani N., Vander Elst L., Muller R.N. Magnetoliposomes as multimodal contrast agents for molecular imaging and cancer nanotheragnostics. Nanomedicine (Lond) 2011;6:529–544. doi: 10.2217/nnm.11.14. [DOI] [PubMed] [Google Scholar]
- 196.Hennessy M.L., Bommareddy P.K., Boland G., Kaufman H.L. Oncolytic Immunotherapy. Surg. Oncol. Clin. N. Am. 2019;28:419–430. doi: 10.1016/j.soc.2019.02.007. [DOI] [PubMed] [Google Scholar]
- 197.Pyo K.H., Kim J.H., Lee J.M., Kim S.E., Cho J.S., Lim S.M., Cho B.C. Promising preclinical platform for evaluation of immuno-oncology drugs using Hu-PBL-NSG lung cancer models. Lung Cancer. 2019;127:112–121. doi: 10.1016/j.lungcan.2018.11.035. [DOI] [PubMed] [Google Scholar]
- 198.Guil-Luna S., Sedlik C., Piaggio E. Humanized Mouse Models to Evaluate Cancer Immunotherapeutics. Annu. Rev. Cancer Biol. 2021;5:119–136. [Google Scholar]
- 199.Zeng Y., Liu B., Rubio M.-T., Wang X., Ojcius D.M., Tang R., Durrbach A., Ru Z., Zhou Y., Lone Y.-C. Creation of an immunodeficient HLA-transgenic mouse (HUMAMICE) and functional validation of human immunity after transfer of HLA-matched human cells. PLoS One. 2017;12 doi: 10.1371/journal.pone.0173754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Shi Z.-D., Pang K., Wu Z.-X., Dong Y., Hao L., Qin J.-X., Wang W., Chen Z.-S., Han C.-H. Tumor cell plasticity in targeted therapy-induced resistance: mechanisms and new strategies. Signal Transduct. Target. Ther. 2023;8:113. doi: 10.1038/s41392-023-01383-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Miles L.A., Burga L.N., Gardner E.E., Bostina M., Poirier J.T., Rudin C.M. Anthrax toxin receptor 1 is the cellular receptor for Seneca Valley virus. J. Clin. Invest. 2017;127:2957–2967. doi: 10.1172/JCI93472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Caeser R., Egger J.V., Chavan S., Socci N.D., Jones C.B., Kombak F.E., Asher M., Roehrl M.H., Shah N.S., Allaj V., et al. Genomic and transcriptomic analysis of a library of small cell lung cancer patient-derived xenografts. Nat. Commun. 2022;13:2144. doi: 10.1038/s41467-022-29794-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Yu X.-Y., Jin X., Shou Z.-X. Surface-engineered smart nanocarrier-based inhalation formulations for targeted lung cancer chemotherapy: a review of current practices. Drug Deliv. 2021;28:1995–2010. doi: 10.1080/10717544.2021.1981492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Sun L., Liu H., Ye Y., Lei Y., Islam R., Tan S., Tong R., Miao Y.-B., Cai L. Smart nanoparticles for cancer therapy. Signal Transduct. Target. Ther. 2023;8:418. doi: 10.1038/s41392-023-01642-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Gandhi L., Rodríguez-Abreu D., Gadgeel S., Esteban E., Felip E., De Angelis F., Domine M., Clingan P., Hochmair M.J., Powell S.F., et al. Pembrolizumab plus Chemotherapy in Metastatic Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2018;378:2078–2092. doi: 10.1056/NEJMoa1801005. [DOI] [PubMed] [Google Scholar]
- 206.Liu X., Zhang J., Feng K., Wang S., Chen L., Niu S., Lu Q., Fang Y. Efficacy and safety of oncolytic virus combined with chemotherapy or immune checkpoint inhibitors in solid tumor patients: A meta-analysis. Front. Pharmacol. 2022;13 doi: 10.3389/fphar.2022.1023533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Schoenfeld J., Jinushi M., Nakazaki Y., Wiener D., Park J., Soiffer R., Neuberg D., Mihm M., Hodi F.S., Dranoff G. Active immunotherapy induces antibody responses that target tumor angiogenesis. Cancer Res. 2010;70:10150–10160. doi: 10.1158/0008-5472.CAN-10-1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Wu X., Giobbie-Hurder A., Connolly E.M., Li J., Liao X., Severgnini M., Zhou J., Rodig S., Hodi F.S. Anti-CTLA-4 based therapy elicits humoral immunity to galectin-3 in patients with metastatic melanoma. OncoImmunology. 2018;7 doi: 10.1080/2162402X.2018.1440930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Helmink B.A., Reddy S.M., Gao J., Zhang S., Basar R., Thakur R., Yizhak K., Sade-Feldman M., Blando J., Han G., et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature. 2020;577:549–555. doi: 10.1038/s41586-019-1922-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Cabrita R., Lauss M., Sanna A., Donia M., Skaarup Larsen M., Mitra S., Johansson I., Phung B., Harbst K., Vallon-Christersson J., et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature. 2020;577:561–565. doi: 10.1038/s41586-019-1914-8. [DOI] [PubMed] [Google Scholar]
- 211.He J., Munir F., Ragoonanan D., Zaky W., Khazal S.J., Tewari P., Fueyo J., Gomez-Manzano C., Jiang H. Combining CAR T Cell Therapy and Oncolytic Virotherapy for Pediatric Solid Tumors: A Promising Option. Immuno. 2023;3:37–56. [Google Scholar]
- 212.Park A.K., Fong Y., Kim S.-I., Yang J., Murad J.P., Lu J., Jeang B., Chang W.-C., Chen N.G., Thomas S.H., et al. Effective combination immunotherapy using oncolytic viruses to deliver CAR targets to solid tumors. Sci. Transl. Med. 2020;12:eaaz1863. doi: 10.1126/scitranslmed.aaz1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Porter C.E., Rosewell Shaw A., Jung Y., Yip T., Castro P.D., Sandulache V.C., Sikora A., Gottschalk S., Ittman M.M., Brenner M.K., Suzuki M. Oncolytic Adenovirus Armed with BiTE, Cytokine, and Checkpoint Inhibitor Enables CAR T Cells to Control the Growth of Heterogeneous Tumors. Mol. Ther. 2020;28:1251–1262. doi: 10.1016/j.ymthe.2020.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Breitbach C.J., De Silva N.S., Falls T.J., Aladl U., Evgin L., Paterson J., Sun Y.Y., Roy D.G., Rintoul J.L., Daneshmand M., et al. Targeting Tumor Vasculature With an Oncolytic Virus. Mol. Ther. 2011;19:886–894. doi: 10.1038/mt.2011.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Evgin L., Kottke T., Tonne J., Thompson J., Huff A.L., van Vloten J., Moore M., Michael J., Driscoll C., Pulido J., et al. Oncolytic virus-mediated expansion of dual-specific CAR T cells improves efficacy against solid tumors in mice. Sci. Transl. Med. 2022;14 doi: 10.1126/scitranslmed.abn2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Shi Y., Hu G., Su J., Li W., Chen Q., Shou P., Xu C., Chen X., Huang Y., Zhu Z., et al. Mesenchymal stem cells: a new strategy for immunosuppression and tissue repair. Cell Res. 2010;20:510–518. doi: 10.1038/cr.2010.44. [DOI] [PubMed] [Google Scholar]
- 217.Mader E.K., Maeyama Y., Lin Y., Butler G.W., Russell H.M., Galanis E., Russell S.J., Dietz A.B., Peng K.W. Mesenchymal stem cell carriers protect oncolytic measles viruses from antibody neutralization in an orthotopic ovarian cancer therapy model. Clin. Cancer Res. 2009;15:7246–7255. doi: 10.1158/1078-0432.CCR-09-1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Kim J., Hall R.R., Lesniak M.S., Ahmed A.U. Stem Cell-Based Cell Carrier for Targeted Oncolytic Virotherapy: Translational Opportunity and Open Questions. Viruses. 2015;7:6200–6217. doi: 10.3390/v7122921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Cristi F., Gutiérrez T., Hitt M.M., Shmulevitz M. Genetic Modifications That Expand Oncolytic Virus Potency. Front. Mol. Biosci. 2022;9 doi: 10.3389/fmolb.2022.831091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Tian Y., Xie D., Yang L. Engineering strategies to enhance oncolytic viruses in cancer immunotherapy. Signal Transduct. Target. Ther. 2022;7:117. doi: 10.1038/s41392-022-00951-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Yuan M., Webb E., Lemoine N.R., Wang Y. CRISPR-Cas9 as a Powerful Tool for Efficient Creation of Oncolytic Viruses. Viruses. 2016;8:72. doi: 10.3390/v8030072. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.