

Abstract

Luminescence quenching by hole transport layers (HTLs) is one of the major issues in developing efficient perovskite light-emitting diodes (PeLEDs), which is particularly prominent in blue-emitting devices. While a variety of material systems have been used as interfacial layers, the origin of such quenching and the type of interactions between perovskites and HTLs are still ambiguous. Here, we present a systematic investigation of the luminescence quenching of CsPbBr3 by a commonly employed hole transport polymer, poly[(9,9-dioctylfluorenyl-2,7diyl)-co-(4,4′-(N-(4-sec-butylphenyl) diphenylamine)] (TFB), in LEDs. Strong and weak quantum-confined CsPbBr3 (nanoplatelets (NPLs)/nanocrystals (NCs)) are rationally selected to study the quenching mechanism by considering the differences in their morphology, energy level alignments, and quantum confinement. The steady-state and time-resolved Stern–Volmer plots unravel the dominance of dynamic and static quenching at lower and higher concentrations of TFB, respectively, with a maximum quenching efficiency of 98%. The quenching rate in NCs is faster than that in NPLs owing to their longer PL lifetimes and weak quantum confinement. The ultrafast transient absorption results support these dynamics and rule out the involvement of Forster or Dexter energy transfer. Finally, the 1D 1H and 2D nuclear overhauser effect spectroscopy nuclear magnetic resonance (NOESY NMR) study confirms the exchange of native ligands at the NCs surface with TFB, leading to dark CsPbBr3-TFB ensemble formation accountable for luminescence quenching. This highlights the critical role of the triarylamine functional group on TFB (also the backbone of many HTLs) in the quenching process. These results shed light on the underlying reasons for the luminescence quenching in PeLEDs and will help to rationally choose the interfacial layers for developing efficient LEDs.
Keywords: PL quenching, LEDs, CsPbBr3, nanoplatelets, perovskite, TFB, ligand exchange, interfacial interactions
Introduction
Colloidal lead halide perovskites (APbX3, where A = cesium, methylammonium, and formamidinium; X = Cl, Br, and I) are explored as red and green downconverters in liquid crystal displays (LCDs)1−3 and as emitters in electrically driven perovskite light-emitting diodes (PeLEDs).4−9 The rapid progression after the realization of the first room-temperature PeLED10 in 2014 paved the path for a new generation of low-cost and high-performance LEDs.9,11−16 Especially colloidal cesium lead bromide (CsPbBr3) nanocrystals (NCs), owing to unique and exceptional optoelectronic properties like high photoluminescence quantum yield (PLQY), narrow emission line widths (less than 20 nm), high carrier mobility, and long exciton diffusion lengths (>200 nm), are favorites for light-emitting applications.17−19 With a particle size slightly larger (10–20 nm) than the exciton Bohr radius (∼7 nm), these NCs exhibit weak quantum confinement. However, the defect-tolerant nature of CsPbBr3 NCs facilitates efficient radiative recombination and results in efficient and bright green (∼515 nm) emission.20 Owing to such unique features, these NCs are well-explored for bright green LEDs with external quantum efficiencies of over 20%.12 Recent advancements in their controlled synthesis allow one to tailor the band gap and the emission wavelength without halide mixing or compositional tunning.21−23 Such controlled one-dimensional size reduction forms 2D nanoplatelets (NPLs) with a thickness less than that of the Bohr diameter (∼7 nm) and favors quantum confinement.21,24,25 As a result, thickness-dependent bright blue emission (450–480 nm) can easily be obtained from CsPbBr3.22
Despite such advancements, the CsPbBr3-based devices are still far behind commercial applications and need more fundamental studies to tackle several key barriers. Although increased exciton binding energy (>300 meV)26 and recombination rate in CsPbBr3 NPLs are well suited for high-efficiency blue LEDs, increased surface traps and film inhomogeneity restrict their performance in the device. In contrast, weakly confined and green-emitting CsPbBr3 with small exciton binding energy close to the thermal energy suffer from the dissociation of excitons to the free charge careers and slow recombination rate under the LED operating conditions.27,28 However, these are not the only reasons limiting their advancement. To achieve high-efficiency and possible commercialization, other key aspects include, but are not limited to, interfacial interactions, efficiency roll-off, balanced and efficient charge injection, and high light outcoupling efficiency.29,30 Therefore, device engineering plays an important role in defining the performance of the PeLEDs.
Generally, in LEDs, the choice of transport layers and the type of device architecture play a critical role in achieving efficient charge carrier balance, energy level alignment, light outcoupling, and hence high efficiency.31 Regardless of the device structure, the inherent nature of the emissive layer in a device can limit the efficiency of LEDs. For example, in blue perovskites, despite their high PL quantum yields in solution, the efficiency is severely hindered when employed in a device. This is primarily due to (i) the challenges with carrier injection and carrier balance on the emissive layer owing to the wide band gap nature, (ii) physical and/or chemical interactions with underlining layers, and (iii) ion migration. In blue-emitting CsPbBr3 NPLs, although the ion migration is not a major issue, unlike Cl/Br perovskites, challenges (i) and (ii) still significantly impact the device performance. For instance, the physical interactions between CsPbBr3 and the transport layers may alter the crystal growth and uniform coverage. In contrast, the chemical interaction can affect the energy level alignment and charge carrier mobilities and create new defect states at the interfaces.32 While the crystallinity and uniform coverage can be achieved through thin-film processing and fabrication methods, the chemical interactions are hard to understand due to the complexity of their chemical composition. Typical CsPbBr3 NPLs consist of long-chain surface passivating ligands such as oleic acid and oleyl amines, A- and B-site ions, and halide ions. Any of these can form bonds with the transport layers and create nonradiative channels at the interface.31,33,34 Often, in PeLEDs, poly(3,4-ethylenedioxythiophene) polystyrenesulfonate (PEDOT:PSS), poly[(9,9-dioctylfluorenyl-2,7diyl)-co-(4,4′-(N-(4-sec-butylphenyl) diphenylamine)] (TFB), poly(N-vinylcarbazole) (PVK), poly(N,N′-bis-4-butylphenyl-N,N′-bisphenyl)benzidine (PolyTPD), 1,1-Bis[(di-4-tolylamino)phenyl]cyclohexane (TAPC), and poly(bis(4-phenyl)(2,4,6-trimethylphenyl)amine) (PTAA) are employed as hole transport layers (HTLs).6,9,35−38 Notably, many of these HTLs, such as TFB, PolyTPD, PTAA, and TAPC, possess triarylamine functionality and are known for interfacial luminescence quenching.6,34,39−41
Therefore, the interfaces play a crucial role in PL and EL quenching,34,42,43 and understanding these interfacial interactions to overcome the luminescence quenching is challenging but crucial to achieving high-efficiency PeLEDs.40,44 While several strategies exist to overcome such quenching, introducing a molecular interlayer at the perovskite/HTL interface is a simple yet effective way to mitigate the quenching. Wang et al. used a molecular modifier, 1,3,5-tris (bromomethyl) benzene (TBB), between polyTPD and the emitting layer to improve charge injection, charge balance, and efficiency, despite the reduction in the PL.41 Nafion perfluorinated ionomer (PFI) is widely known to reduce exciton quenching by preventing the charge transport from the emitting layer to TFB.7 Recently, a thin layer of phenethylammonium bromide has been explored as an interlayer over PTAA that helps to balance the charge carrier transport.9 Similarly, amphiphilic poly[(9,9-bis(3′-(N,N-dimethylamino)propyl)-2,7-fluorene)-alt-2,7-(9,9-dioctylfluorene)] (PFN) was also utilized as an interfacial layer to improve quality of the film and device performance.45 Apart from this, a few studies demonstrated the treatment/doping of HTLs to overcome the luminescence quenching.42,46 While numerous interfacial layers are available, their applicability cannot be extended across a broad spectrum of HTL variations. The effective prevention of quenching by certain molecules, as opposed to others, lacks a clear rationale. This discrepancy stems from a fundamental gap in our understanding of the interfaces. Consequently, it becomes imperative to illuminate the origins of interfacial quenching, unraveling the specific interactions between HTLs and CsPbBr3. Such insight is crucial for the judicious utilization of interfacial layers to enhance the efficiency of LEDs.
Here, we systematically elucidate the luminescence quenching of colloidal blue- and green-emitting CsPbBr3 (NPLs and NCs) by TFB using time-resolved absorption and emission spectroscopies and nuclear magnetic resonance (NMR) spectroscopy. TFB is an electron-blocking, hole transport-conjugated polymer and belongs to a family of widely used triarylamine-based HTLs with a high hole mobility of 0.01 cm2 V–1 s–1 at room temperature. Several reports demonstrated that TFB acts as a luminescence quencher due to interfacial interactions.47,48 To understand luminescence quenching, we treated colloidal CsPbBr3 NPLs and CsPbBr3 NCs with increasing concentrations of TFB while monitoring their UV–visible absorption, photoluminescence, and time-resolved photoluminescence at room temperature. Owing to the wide band gap of CsPbBr3 NPLs, their UV–visible absorption and emission features significantly overlap with the emission of the TFB. At the same time, the optical characteristics of the green-emitting NCs are significantly different than that of TFB. This allowed us to investigate the effect of quantum confinement, PL lifetime, and energy levels of NPLs and NCs on quenching kinetics by employing the classical Stern–Volmer model based on the change in PL intensity and PL decay dynamics with increasing TFB concentration. Excited-state decay dynamics obtained from ultrafast transient absorption support the systematic quenching with increasing TFB amount, ruling out the Foster/Dexter energy transfer processes. Further, in-depth 1D 1H and 2D NOESY NMR spectroscopic investigations provide evidence for the formation of the dark TFB-CsPbBr3 adduct through the exchange of native ligands, mainly responsible for the static quenching process. The unique combination of optical spectroscopy and NMR analysis presented here enables us to elucidate the origin and mechanism of luminescence quenching, which are crucial for designing high-efficiency CsPbBr3-based blue and green LEDs.
Results
Characteristics of CsPbBr3 and the Quencher (TFB)
The blue-emitting CsPbBr3 NPLs were synthesized at room temperature by the ligand-assisted precipitation (LARP) method49 with slight modifications, and the green-emitting CsPbBr3 NCs were synthesized by a hot-injection protocol.20 The detailed synthesis procedure is given in the Materials and Methods section. From now on, blue- and green-emitting CsPbBr3 will be represented as NPLs and NCs, respectively. One-dimensional size control yields NPLs that show strong excitonic absorption at 445 nm and blue emission at 454 nm with a Stokes shift of 9 nm and a full width at half-maximum (FWHM) of 20 nm (Figure 1a, blue-dotted and smooth lines). While NCs show an absorption peak at 502 nm and a PL peak at 516 nm with a Stokes shift of 14 nm and an FWHM of 22 nm. The time-resolved PL (TRPL) decay dynamics suggest average lifetimes of 7.2 ns for NPLs and 10.4 ns for NCs (Figure 1b).
Figure 1.

Characterization of CsPbBr3. (a) UV–visible absorption (dotted lines) and photoluminescence (smooth lines) spectra of CsPbBr3 NPLs and NCs. (b) Comparison between time-resolved photoluminescence of NPLs, NCs, and TFB. (c) TEM image of CsPbBr3 NPLs. (d) HRTEM image of CsPbBr3 NCs. (e) Chemical structure of TFB. (f) Energy level diagram of TFB, NPLs, and NCs.
From the TEM and HRTEM images shown in Figure 1c,1d and Figure S1a–e, the morphologies of NPLs and NCs can be easily differentiated. TEM images of NPLs show stacked particles with an average length of 5 (±0.1) nm and a width of 1.8 (±0.3) nm, while the average edge length for NCs is 12 (±3) nm (Figure S1c,f). Further, these colloidal NPLs and NCs are used for the luminescence quenching studies by treating with the known concentration of TFB. Before diving into the detailed quenching study, the optical properties of TFB are studied and compared with those of NPLs and NCs. TFB is a wide band gap organic HTL with a HOMO (highest occupied molecular orbital)-LUMO (lowest unoccupied molecular orbital) gap of ∼3 eV,6,50 whereas the band gap of NPLs is ∼2.8 eV and that of NCs is ∼2.5 eV. A chemical structure of TFB and an energy level diagram of TFB, NPLs, and NCs are shown in Figure 1e,1f.6,9,51 The absorption and emission spectra of the TFB, along with the optical features of NPLs and NCs, are shown in Figure S2 for comparison. Both absorption (excitonic peak at 445 nm) and emission (peak at 454 nm) spectra of NPLs significantly overlap with the broad TFB emission, which is centered at 440 nm (Figure S2a). In contrast, the TFB absorption (centered at 390 nm) shows negligible spectral overlap with the emission from NPLs and NCs (Figure S2a,b). Therefore, to verify the possibility of energy exchange between TFB and NPLs/NCs, we performed optical spectroscopy.
Photophysical Aspects of CsPbBr3 Luminescence Quenching
To probe the interactions, we begin treating colloidal dispersion of NPLs in toluene with TFB and monitor the changes in their UV–visible absorption and emission. With the increase in TFB addition, the absorbance of TFB (black upward arrow in Figure S3a) increased systematically, as expected. The excitonic absorption peak of the NPLs showed a slight red shift after the excessive addition of TFB (Figure S3b). Interestingly, the corresponding PL measurements show an unusual trend in the emission intensity of both TFB and NPLs, and these trends are observed to be dependent on the concentration of the TFB in the mixture. The addition of TFB results in two main changes to the PL spectra of both NPLs and TFB: (i) an increase in the emission intensity of both NPLs and TFB at very low concentrations ranging from 0.030 to 0.090 μM (shown in black-colored upward arrows in Figure 2a) and (ii) with further increase in TFB concentrations from 0.10 to 3.30 μM, a systematic decrease in the emission intensity (indicated by red downward arrows in Figure 2a,b) of both NPLs and TFB. In the first case, while an increase in the concentration of TFB leads to an increase in TFB emission intensity, the reason for the increase in NPLs emission with TFB addition is unclear. Note that there is no change in the absorption of NPLs. This increase in emission of NPLs could be due to (a) energy or electron transfer from TFB to NPLs or (b) surface passivation of NPLs by TFB. However, the possibility of energy transfer is ruled out as one can expect a decrease in the emission intensity of the donor and an increase in the emission intensity of the acceptor, but in this case, the emission intensity of both TFB and NPLs is increasing. A systematic decrease in emission intensity of both NPLs and TFB with increasing TFB concentration in case (ii) could be due to degradation or nonradiative energy loss. However, the unchanged absorbance of NPLs, even at higher concentrations of TFB, confirms the colloidal stability of NPLs, thus ruling out degradation. There may be a formation of a nonradiative species from the interaction of NPLs and TFB, responsible for the decrease in intensity in case (ii). As the TFB concentration increases, the chances of TFB encountering the NPL increase, potentially leading to the formation of the dark complex. Clearly, changes observed in PL alone cannot explain the mechanism of quenching. Therefore, we investigated this further using time-resolved PL measurements, transient absorption studies, and NMR analysis.
Figure 2.

Quenching of CsPbBr3 emission. (a, b) Photoluminescence quenching of CsPbBr3 NPLs upon increasing addition of TFB. (c, d) Time-resolved photoluminescence spectra monitored at 454 nm for CsPbBr3 NPLs with increasing concentrations of TFB. IRF stands for the instrumental response function. (e, f) Photoluminescence quenching of CsPbBr3 NCs upon increasing addition of TFB. (g, h) Time-resolved photoluminescence spectra monitored at 516 nm for CsPbBr3 NCs emission with increasing concentrations of TFB. The black and red arrows guide the eyes, highlighting an increase or decrease in the PL intensity or lifetime.
The time-resolved photoluminescence (TRPL) of all of the reaction mixtures is recorded and coupled with PL measurements. The unusual trend continued in the lower concentration range (0.030–0.155 μM); with an increase in TFB, the average lifetime values decreased systematically. NPLs without TFB showed an average PL lifetime of 7.2 ns, which decreased to as low as 4 ns when treated with 0.155 μM TFB (Figure 2c,d). Such faster decay, as compared to the untreated NPLs, indicates an emergence of nonradiative decay channels with increasing TFB concentration. Surprisingly, when the TFB concentrations increased >0.155 μM, the decay in NPLs became slower (∼5 ns) and remained constant despite the continuous decrease in the emission intensity.
The best-fit parameters and average lifetime values for NPLs are given in Table S1. These unusual trends in PL and TRPL could be due to (1) energy or electron transfer from NPLs to TFB (or vice versa), (2) the effect of quantum confinement, and (3) formation of transient NPL-TFB complex from the chemical interactions between NPLs and TFB under excitation. To unravel this, we examine all of the potential causes systematically.
There have been numerous reports on energy or electron transfer from perovskites to chromophores in the literature.52−54 Generally, if such transfer occurs, the emission intensity of one compound (donor) decreases, the other (acceptor) increases, and corresponding changes will be reflected in their TRPL profile. In the current case, since the emission of TFB strongly overlaps with the excitonic absorption peak of NPLs, there is a possibility of energy transfer from TFB to NPLs. When such transfer occurs, a decrease in the emission of TFB (donor) and an increase in the emission from NPLs (acceptor) are expected. However, from the above experiments, we observe a decrease in the emission of both NPLs and TFB, particularly in the higher TFB concentration range. Therefore, the possibility of energy or electron transfer from TFB to NPL can be ruled out in this concentration range. The transfer from NPL to TFB is also not possible, considering the minimal overlap in the emission of NPL and absorption of TFB. Recently, several reports elucidated the energy transfer from perovskites to triplet states of chromophores.52−54 To verify if such transfer is responsible for the luminescence quenching in NPLs, we employed weakly confined (average size = ∼12 ± 3 nm), narrow band gap, green-emitting CsPbBr3 NCs with TFB, whose VB is expected to be below triplet state (∼2.2 eV) of TFB.50 In addition, the green NCs exhibit no spectral overlap with the TFB. Similar experiments were performed by adding TFB into colloidal NCs that showed a slight red shift in the absorption peak (∼508 nm) that is more prominent at a higher TFB concentration (0.790 μM) (Figure S3c,d). Most importantly, the trends observed in PL and TRPL of NCs are similar to those of NPLs (Figure 2e–h and Table S2), irrespective of the particle size, dimensionality, band gap, and energy levels, indicating that the quenching mechanism is the same for both NPLs and NCs. However, to understand if the luminescence quenching is caused by the particle destruction upon TFB addition, we performed a TEM analysis of the pure and TFB-treated NPLs/NCs. TFB-treated (0.155 and 3.30 μM) NPLs and TFB-treated (0.155 and 0.790 μM) NCs showed similar morphology to untreated NPLs and NCs (Figure S4). Also, size distribution histograms for untreated and treated NPLs and NCs suggest negligible changes. This confirms that the luminescence quenching in NPLs and NCs is not driven by particle destruction/decomposition.
Overall, the above photophysical aspects clarify that no transfer of energy/electrons occurs from TFB to NPL/NCs and vice versa, as the PL intensity of both TFB and NPLs/NCs decreases when mixed. Owing to the wide band gap, TFB can act as a donor while NPLs/NCs act as acceptors due to their narrow band gap. However, the possibility of PL quenching by energy transfer (Forster/Dexter) is neglected because of (i) no enhancement in emission intensity of acceptor and a systematic decrease in the PL intensity of both TFB and NPLs/NCs, (ii) a systematic faster decay with increasing TFB concentration, and (iii) a significant gap between absorption peaks/onset of TFB and NPLs (0.4 eV) or NCs (0.7 eV). Therefore, a decrease in PL intensity and unchanged PL decay with increasing TFB concentration supports the static nonradiative complex formation between TFB and CsPbBr3.
Elucidation of the Quenching Mechanism by the Classical Stern–Volmer Model
Next, we turned to steady-state and time-resolved Stern–Volmer plots to identify different types of quenching processes involved in both NPLs and NCs.55−58 To begin with, the Stern–Volmer eq (eq 1) is employed to seek insights into the TFB-induced emission quenching of NPLs and NCs.
| 1 |
where I0 corresponds to integrated PL intensity in the absence of TFB, I is the integrated PL intensity of NPLs/NCs at varied concentrations of TFB, KSV is the Stern–Volmer constant, and Q represents the concentration of the TFB. The plot I0/I vs concentration of TFB (steady-state Stern–Volmer plot) in the low TFB concentration range of 0.030–0.155 μM resulted in a second-order polynomial (nonlinear) curve with an upward curvature (red-colored points in Figure 3a,3b). Such quadratic dependence of I0/I on the concentration of TFB in both cases of NPLs and NCs suggests that there is an existence of dual dynamic and static quenching.55−57,59
Figure 3.

The
Stern–Volmer Plots. (a, b) Stern–Volmer plots
for the low concentration region of TFB (c, d) Stern–Volmer
plots for the high concentration region of TFB obtained for CsPbBr3 NPLs and CsPbBr3 NCs. Black and red lines indicate
a linear fit and second-order polynomial fit for τ0/τ vs [TFB] and I0/I vs [TFB], respectively. (e, f) PL quenching efficiency calculated
using the equation 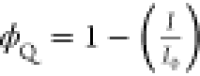 , where ϕQ is the quenching efficiency, I0 and I are the integrated PL intensity before and after the addition
of TFB for CsPbBr3 NPLs and NCs, respectively.
, where ϕQ is the quenching efficiency, I0 and I are the integrated PL intensity before and after the addition
of TFB for CsPbBr3 NPLs and NCs, respectively.
Since the PL quenching includes the contribution of both static complex and dynamic collision, the emission ratio of I0/I cannot differentiate between static and dynamic quenching. To separate the contributions of dynamic and static quenching, we used emission lifetimes and plotted the PL lifetime ratio (τ0/τ) vs the concentration of TFB in Figure 3a,3b (black-colored points). The emission lifetime, an inherent characteristic of the compound, is not generally affected by the formation of a dark static complex. Therefore, the changes observed in emission decay are mostly due to bimolecular collisional quenching. Corresponding collisional constant kq can be derived from the τ0/τ vs the concentration of TFB plots. It is noteworthy that, unlike the steady-state Stern–Volmer plots, the time-resolved Stern–Volmer plots52,56 show a linear relation with the concentration of the TFB when fitted with eq 2
| 2 |
where τ0 corresponds to the PL lifetime of NPLs/NCs in the absence of TFB while τ is the lifetime of NPLs/NCs emission in the presence of the TFB, KD is the dynamic quenching constant, and kq is the bimolecular quenching rate constant. Such a linear dependence or the decreasing trend in the lifetime values (Table S1) at low concentrations of the TFB (0.030–0.155 μM) in both NPLs and NCs is an indication of the presence of a dynamic quenching process in addition to static quenching.56,59 The values of KD obtained for NPLs and NCs as the fit parameters for τ0/τ vs [TFB] in Figure 3a,3b are used to extract the contribution of the static quenching process using a modified Stern–Volmer equation (eq 3).60
| 3 |
Here, KS is the
static quenching constant, and the values of Stern–Volmer quenching
constants, KS, KD, and kq are given in Table 1.
are given in Table 1.
Table 1. Best-Fitted Values of Dynamic (KD) and Static (KS) Quenching Constants, the Bimolecular Quenching Rate Constant (kq), and Maximum Quenching Efficiency (ϕQ) Calculated for the Highest Concentration of TFB for the CsPbBr3 NPLs and NCs.
| sample | KD × 106 [M–1] | kq × 1015 [M–1 S–1] | KS [M–1] | maximum ϕQ (%) |
|---|---|---|---|---|
| NPLs | 5.52 | 0.76 | 3.32 | 98 |
| NCs | 16.15 | 1.55 | 21 | 97 |
A common feature of a larger dynamic quenching constant (KD) than the static quenching constant (KS) confirms the dominance of dynamic quenching over static in both NPLs and NCs when treated with TFB in the concentration range of 0.030–0.155 μM. We note that the calculated kq values are in the order of 1015 [M–1 S–1] and are much higher than the typical diffusion-induced bimolecular quenching constant (kdiff = 1010 [M–1 S–1]).52,60 The kq values larger than this limit suggest the presence of surface binding interactions between NPLs/NCs and TFB and rule out purely diffusion-mediated PL quenching. PL quenching by charge transfer or energy transfer is another possibility that can occur at the same time scales, leading to dynamic quenching.59,60 Overall, in the low concentration range of 0.030–0.155 μM, the steady-state and time-resolved Stern–Volmer plots revealed that the quenching is dominated by dynamic interactions between NPLs/NCs and TFB that are leading to a nonradiative charge transfer/trapping mechanism.60
Interestingly, Stern–Volmer constants (Table 1) estimated for NPLs and NCs show some deviations in the quenching rates in the presence of a quencher (TFB). Note that NPLs and NCs exhibit different degrees of quantum confinement due to their electronic dimensionality and the carrier lifetime. The PL lifetime of NCs (τavg: 10.4 ns) is slightly longer compared to that of NPLs (τavg: 7.2 ns). These differences can have an impact on the luminescence quenching rates. Indeed, dynamic quenching observed in colloidal NCs (KD = 16.15 × 106 M–1) is more prominent than that observed in the NPLs (KD = 5.52 × 106 M–1). In addition, we observed two main distinct features for NCs: (i) a high degree of nonlinearity/quadratic nature in steady-state Stern–Volmer plot (Figure 3a) compared to NPLs and (ii) large Stern–Volmer quenching constants (KSV) as compared to NPLs (Table 1). These characteristics indicate that the PL quenching is faster in NCs and is attributed to the weak quantum confinement (slightly longer PL lifetime of the NCs) of the excited charge carriers in NCs (average particle size: 12 nm). Since NPLs are strong quantum-confined, the excitons generated after photoexcitation recombine radiatively at a faster rate while competing with the rates of collisional quenching, whereas in NCs, the generated excitons are weak quantum-confined and can live relatively longer, leading to a slower recombination rate. Therefore, upon photoexcitation, the excitons live rather longer to interact with the quencher, which occurs in nanoseconds to the tens of nanosecond time scales, leading to enhanced dynamic quenching in NCs.60,61 In addition, due to the weak quantum confinement, the charge carriers are more delocalized in NCs compared to NPLs that can easily reach the nonradiative traps on the surfaces generated by “dark” static complex, eventually leading to enhanced static quenching in NCs.60,61 Therefore, the faster PL quenching in the NCs-TFB ensemble (large Stern–Volmer quenching constants (KS, KD, and kq)) is a synergistic effect of the longer PL lifetime and weak quantum confinement.
Further, we investigated the quenching mechanism at higher concentrations by repeating the same analyses. At a higher concentration of the TFB (0.80–3.30 μM in NPLs and 0.170–0.790 μM in NCs), the plot of I0/I vs the concentration of TFB shows linear dependence in both NPLs and NCs (red-colored points in Figure 3c,d). Also, it is noted from the PL decay dynamics (Table S1) that the average PL lifetime remains constant, irrespective of the TFB concentration. The plots of PL lifetime ratio τ0/τ against TFB concentration are constant (black-colored points in Figure 3c,3d) and are independent of TFB concentration and type of the particles (NCs vs NPLs). Such a linear dependence of I0/I and independency of τ0/τ on increased TFB loading is concrete evidence of the static PL quenching process.56,57,59 The slope of the steady-state Stern–Volmer plot is equal to the equilibrium constant for the formation of the ground-state complex. The obtained value of the equilibrium constant in NPLs is 4.40 × 106 M–1, while in NCs, it is 81.2 × 106 M–1 at room temperature, implying that the static quenching in NCs is more prominent as compared to that of NPLs and agrees well with the earlier discussion on the quantum confinement. The calculated quenching efficiency52 with respect to the TFB concentration is presented in Figure 3e for NPLs and in Figure 3f for NCs. The highest quenching efficiency obtained in the case of NPLs is 98% at 3.30 × 106 M–1 of TFB concentration, which is 97% in NCs at 0.790 × 106 M–1 of TFB. This highlights a unique difference: the amount of TFB needed to quench NC emission is very low compared to that in NPLs. Despite low required TFB concentration and large Stern–Volmer constants in NCs, both NPLs and NCs exhibit almost complete emission quenching by forming the nonemissive ensemble with TFB. Therefore, static quenching observed at higher TFB concentrations is attributed to the CsPbBr3-TFB adduct formation via strong interactions/adsorption at the surface, enabling nonradiative decay channels. To summarize, the Stern–Volmer analysis of PL and TRPL data revealed the underlying mechanism of PL quenching by contributing to the dynamic and static quenching processes. However, the nature of dark states involved in the quenching process and the type of interactions between TFB and CsPbBr3 leading to the static complex are still unclear from this analysis. This information is crucial for rationally choosing the interfacial layers that can cease the TFB-CsPbBr3 interactions and prevent emission quenching by TFB in device applications.
Further evidence of the interactions between CsPbBr3 and TFB can be obtained through transient absorption (TA) spectroscopy. Figure 4a shows the transient absorption spectra at different pump–probe delays for colloidal CsPbBr3 NCs. The pump wavelength for this experiment is tuned to 420 nm. We observe a clear positive differential transmission (ΔT/T) signal at the optical edge of the system, which is assigned to bleaching of the ground state (GSB). Note that the carrier thermalization is complete within the resolution of the experiment. In Figure 4b, we show the ultrafast dynamics of this GSB feature at different excitation fluences obtained via integration of the signal over the entire spectral range for a better signal-to-noise ratio. We observe a clear intensity dependence in the dynamics with a faster component emerging at higher excitation densities. This component is associated with the nonradiative, Auger-like recombination process, typically driven by multiexciton species manifesting at high excitation densities. Such dynamics in the perovskite nanocrystals have been quantitatively rationalized by several previous works.26,62,63
Figure 4.

Transient absorption (TA) spectroscopy. (a) Transient absorption spectra of colloidal CsPbBr3 NCs. Spectrally integrated ground-state bleach dynamics at different pump excitation fluences in (b) CsPbBr3 and (c) a CsPbBr3:TFB mixture.
We now analyze the transient absorption signal of the CsPbBr3:TFB mixture. First, we note that no significant absorption is associated with TFB at the pump wavelength used in this experiment. Second, direct optical excitation of the TFB solution with a more blue-shifted pump resulted in an evident photoinduced absorption (PA) band in the visible spectral range with a peak around 620 nm, which is associated with the excited-state absorption of TFB.64 Given that there is direct excitation of TFB in the mixture, we can see that the transient absorption spectral line shape is similar to that of the pristine NCs (Figure S5). If there was any interaction between the nanocrystals and TFB and subsequent charge or energy transfer to TFB, we would have observed the PA band. On the contrary, we do not observe any such spectroscopic signature, at least within the sensitivity of our measurement. While this may appear to contradict the observations we have previously made, temporal dynamics offer a more supportive perspective.
We show the intensity-dependent GSB dynamics of the CsPbBr3:TFB mixture in Figure 4c. Note that the concentration of the CsPbBr3 NCs is kept the same as that of the pristine system to enable a robust comparison, and the concentration of TFB is kept high to ensure TFB-NCs interaction. For the same excitation fluence, we observe a substantial reduction in the intensity dependence of the dynamics with the dynamics, showing a hint of faster recombination only at the highest excitation fluence. Given that the nanocrystal concentration is comparable across both samples, we expect similar initial excitation density in both samples. In the presence of TFB molecules, however, the dynamics indicate much lower excitation density, low enough to exclude any Auger-like quenching in the subnanosecond time scales. This observation can be rationalized by invoking an additional ultrafast (<200 fs) quenching of the photoexcited population in the CsPbBr3:TFB mixture that results in a much lower excitation density within the nanocrystal ensemble available for recombination. Notably, this quenching occurs within the time resolution of the experiment. Given the lack of any spectroscopic evidence of an ultrafast transfer process to TFB, we hypothesize that the ultrafast quenching is associated with a transfer of the photoexcited population to an optically inaccessible composite (nanocrystal-TFB) state. Given the limited spectral bandwidth of the probe pulses used in this experiment, this observation should only be taken as speculation and not as proof of the existence of a composite state. More concrete evidence, however, emerges in the 2D-NMR studies presented in later sections of the manuscript.
Validation of Static Quenching by Nuclear Magnetic Resonance (NMR) Spectroscopy
We use nuclear magnetic resonance (NMR) techniques to gain more insight into the surface chemistry of colloidal NPLs and their interactions with TFB. We conducted one-dimensional (1D) and two-dimensional (2D) NMR experiments to understand the changes over the NPLs surface due to TFB addition. The synthesized NPLs contain oleic acid and oleyl amine as ligands (X-type ligands) on their surfaces. Therefore, before diving directly to investigate NPLs-TFB adduct, we started with 1H NMR experiments to identify corresponding oleic acid and oleyl amine resonances from NPLs by comparing them with pure oleic acid and oleyl amine. The 1D 1H spectrum of oleic acid and oleyl amine showed all expected resonances for the aliphatic protons in the upfield region (δ 0.8–3 ppm), olefinic protons at δ 5.4 ppm, and a broad singlet for acidic proton (−COOH) near δ 11.7 ppm. These resonances and corresponding protons in their chemical structure shown in Figures S6 and S7 are in good agreement with the previous reports.65−69 Further, to confirm their presence on the NPLs surface, 1H NMR of colloidal NPLs in toluene-d8 was recorded and compared with the proton NMR of the oleic acid and oleyl amine (Figure S8). The presence of all characteristic peaks, including broad doublet for olefinic protons near δ 5.4 ppm, corroborates the passivation by oleic acid and/or oleyl amine ligands.68,69
Since the photophysical studies suggest the NPLs-TFB complex formation with increasing TFB loading, one can expect the following changes on the NPLs surface: (i) binding of TFB on the surface and/or (ii) exchange of dynamic oleic acid and oleyl amine ligands from NPLs surface by TFB. In either case, the changes should be reflected in the proton resonances of ligands and TFB. However, these changes can only be traced if TFB resonances are distinguished from the ligand resonances. Figure S9 shows the 1D 1H NMR spectrum of TFB, where aliphatic and aromatic protons of TFB are identified and labeled in its chemical structure, as shown in the inset. Further, by comparing the 1H NMR spectrum of TFB with colloidal NPLs, we identified distinct resonances and monitored them by increasing the amount of the TFB, as shown in Figure S10 (cyan-colored bullets). The methyl resonance (−CH3) at 0.8 ppm, methylene (−CH2−) resonances near 1, 1.5, and 2.2 ppm in shielded regions, and aromatic 1H resonances between 7 ppm to 7.8 ppm are identified as signals of interest for the TFB and ignored the rest because they significantly overlap with the ligand resonances. 1H NMR of colloidal NPLs after incremental addition of the TFB in colloidal NPLs is shown in Figure S11a, and the enlarged view of the shielded region is presented in Figure 5a. Note that most TFB resonances in the shielded region (aliphatic protons) show no shift with an increase in the TFB amount, as denoted by the dotted black line in Figure 5a. Conversely, we observed a systematic upfield shift for the resonances corresponding to the aromatic protons of TFB. Especially, a broad singlet near 7.76 ppm and a multiplet between 7.5 and 7.6 ppm show a systematic upfield shift with an increase in TFB loading (Figure 5b,5c). These observations imply that the aliphatic group in TFB remains unaffected, but the protons of the triarylamine group in TFB experience a change in the electronic environment when CsPbBr3 NPLs are present. Therefore, it is most likely that TFB binds to the surface of NPLs through the triarylamine group, resulting in an upfield shift of aromatic proton resonances. However, no shift in the aliphatic protons indicates that they remain away from the NPL surface due to a lack of binding site. In addition to the TFB resonances, the aliphatic and olefinic protons of the oleic acid and oleyl amine ligands appeared, and their peak position remained the same despite the addition of TFB (Figure S11b). The ligands could be bound to the surface or detached from the surface to accommodate the TFB. Generally, resonances of bound ligands are characterized by line broadening that distinguishes them from the free ligands.65,68 However, various relaxation mechanisms can also affect the line width; therefore, predicting if the ligands are bound or unbound based on the line widths is difficult.69 This is a critical limitation of 1D 1H experiments, and in this case, it is restricting to establish a mechanism for PL quenching via NPLs-TFB adduct formation.
Figure 5.

1D 1H and 2D NOESY nuclear magnetic resonance spectroscopy. (a) Shielded region of 1H NMR spectra of TFB and colloidal CsPbBr3 NPLs in toluene-d8 with different concentrations of TFB. (b, c) Enlarged view of the deshielded region of TFB and colloidal CsPbBr3 NPLs in toluene-d8 with varying concentrations of TFB. The dotted lines are guides to the eyes to monitor the peak positions. (d) 2D NOESY spectrum of NPLs. (e) 2D NOESY spectrum of colloidal CsPbBr3 NPLs treated with 3.30 μM TFB. (f) Enlarged view of the shielded region in (e). The red-colored dots indicate the peaks corresponding to the TFB. Schematic (g) shows the NPLs surface capped with ligands. (h) Possible mechanism of ligand exchange on the NPL surface. (i) Dark CsPbBr3-TFB complex formation after ligand exchange. Inset of (i) is the chemical structure of TFB.
Nuclear overhauser effect spectroscopy (NOESY) based on interactions “through space” between dipolar coupled 1H nuclei is widely used to differentiate bound and free ligands.69,70 The dipolar spin relaxation pathways depend on the molecular mobility or tumbling in solvent expressed as the rotational correlation time (τc). Mainly, cross-relaxation pathways cause the transfer of spin polarization between two sets of protons (Hα and Hβ) embedded in the same molecule and help to distinguish the free ligands from the bound ones.71−74 Freely floating ligands behave like small molecules (τc ≪ 1/ω0, ω0 is the angular frequency of the spectrometer) and build positive NOEs or negative cross peaks between dipolar coupled protons with opposite sign/color to the diagonal peaks.69 At the same time, the ligands that are bound to the nanocrystals (restricted mobility or slow tumbling) behave like rigid/large molecules that develop large negative NOEs or positive cross peaks with a faster rate due to larger τc than 1/ω0.69,70 Such negative NOEs show the same sign/color as the diagonal peaks in the NOESY spectrum. Therefore, 2D NOESY experiments of pure ligands, TFB, and a TFB-NPLs mixture are carried out to illustrate the surface interactions and their impact on PL quenching. A 2D NOESY spectrum of the pure NPLs features strong negative NOE cross peaks for ligands (oleic acid and/or oleyl amine), as shown in Figure 5d. Notably, the negative NOE of the olefinic protons of ligands at 5.4 ppm dipolar coupled (through space) toward methylene and methyl protons is prominent (Figures 5d and S12), suggesting their surface-bound nature.69,72,74 This is attributed to the increased τc due to the tumbling of bound ligands with the characteristics of NPLs.69,74 A 2D NOESY spectrum of pure TFB in toluene-d8 (Figure S13) shows weak NOE for aromatic protons toward the neighboring aromatic and methylene protons due to its large molecule nature.
2D NOESY spectrum of the NPLs-TFB has a strikingly different appearance (Figure 5e), and comparison emphasizes two main changes: (i) the disappearance of strong negative NOEs for ligands and (ii) the appearance of additional negative NOEs for the TFB. Notably, the absence of negative NOE for alkene protons of the ligands (shown in the red-colored box in Figure 5e) suggests that they are no more bound to NPLs surface, but resonances in 1D 1H (Figure S11b) discussed above confirm their presence in colloidal solution as free ligands. It is noteworthy that the strong negative NOEs initially present in NPLs in the aliphatic region (Figures 5d and S12) are now absent but show increased zero-quantum coherence artifacts due to scalar coupled spins,72 further confirming their unbound nature (Figure 5e,f). Conversely, we notice additional positive cross peaks (negative NOEs) for aromatic and aliphatic protons of TFB (Figure 5e,5f), which indicates the binding of TFB with NPLs. For better understanding, we labeled TFB resonances that are distinct from ligands with red dots on the spectrum (see the spectra on the left and top axes in Figure 5e,5f). The aromatic protons of TFB at 7.8 ppm show an additional negative NOE toward the methylene protons of TFB near 1.2 and 2.1 ppm (denoted by the black-dotted lines in Figure 5e). Moreover, the methylene protons at 2.1 ppm show positive cross peaks with methylene protons of ligands at 1.2 ppm and overlapping end methyl protons of the ligands and TFB near 0.8 ppm (shown by the black-dotted lines in Figure 5f). Additional weak negative NOEs are also observed near 1.1 ppm between the methylene protons of the TFB.
Possible Mechanism of Luminescence Quenching through Ligand Exchange
All of the above observations hint toward the surface attachment of TFB and the detachment of ligands from the NPLs surface potentially by the ligand exchange process.72 Interestingly, a strong negative NOE for labile hydrogen (residual water) at 0.45 ppm (Figure 5f) features the cross peaks with methylene resonances of the TFB and ligands at 0.9 ppm, suggesting the proton exchange between the amine and residual water in the sample.74 Such an exchange could take place with either triarylamine-based TFB or oleylammonium bromide. Most importantly, the residual water resonance is not observed in 2D NOESY or 1D 1H NMR of pure ligands and TFB, and it appears only when the TFB is added to the colloidal NPLs solution (Figure S11a). Also, this water resonance shows a systematic upfield shift with TFB loading (Figure 3a, blue-dotted line). Therefore, it is apparent that the triarylamine group of TFB is exchanging a proton with oleylammonium bromide. The proton exchange between oleylammonium bromide and TFB can be depicted by the following reaction:
where R is the alkyl substitution in TFB, Ar is the benzene rings, and R′ is the long carbon chain (C18H35). Although the pKa of the triarylamine features a weak base character, the bulky alkyl groups attached to the aromatic rings, due to their positive inductive effect, promote protonation of the triarylamine group. Such protonation converts TFB to triarylammonium cation (RAr3NH+Br– in the above reaction), making it suitable for binding to the CsPbBr3 surface like typical X-type ligands. The bromine atoms over the CsPbBr3 surface can interact with the protonated TFB, leading to TFB:CsPbBr3 adduct formation. This is like the binding of oleylammonium ligands to the CsPbBr3 surface through a hydrogen bridge as illustrated by previous 2D-NMR studies.68,75 It is, therefore, appropriate to consider that protonated TFB can act as an X-type ligand76 similar to oleylammonium bromide, thus favoring the ligand exchange process as depicted schematically in Figure 5g–i. Therefore, NMR experiments provide concrete evidence for (i) the triarylamine functional group as an active site on the TFB responsible for interactions with the surface of NPLs, (ii) protonation of triarylamine functionality transforming to an X-type ligand, and (iii) binding of protonated TFB on NPLs surface through exchange of native ligands leading to a dark static complex. These all-important observations support our initial assessment based on Stern–Volmer analysis, confirming static PL quenching by the NPLs-TFB ensemble. Owing to the same chemical composition, ligands, PL quenching trend, and mechanism, we close with the same possible mechanism for luminescence quenching in NCs, albeit with different morphology.
Discussion
By compiling data from steady-state, time-resolved photoluminescence (PL), Stern–Volmer plots, ultrafast spectroscopy, and NMR spectroscopic analyses, we establish a concrete understanding of luminescence quenching in CsPbBr3. Our findings reveal a dynamic quenching process prevailing at lower TFB concentrations, while elevated TFB loading induces the formation of dark CsPbBr3-TFB adducts, resulting in static quenching of the CsPbBr3 emission. The quenching dynamics remain consistent across variations in size, shape, energy level alignments, and quantum confinement, particularly when exposed to TFB.
Comparative analysis underscores a swifter and more pronounced quenching in nanocrystals (NCs) compared to that in nanoplatelets (NPLs), attributed to slightly larger PL lifetimes and weak quantum confinement in NCs, leading to higher Stern–Volmer quenching constants (KS, KD, and kq). Ultrafast transient absorption spectroscopy dismisses the involvement of Forster and Dexter energy transfer in quenching processes, supporting our conclusion of static quenching at higher concentrations as per the Stern–Volmer model.
Further substantiating our findings, 1D 1H and 2D NOESY NMR experiments demonstrate ligand exchange on the CsPbBr3 surface, resulting in CsPbBr3-TFB adduct formation through robust interactions at the surface. In the case of colloidal NPLs treated with TFB, 2D NOESY confirms the exchange of hydrogen from residual water with TFB and oleylammonium bromide. This implies protonation of TFB, promoting ligand exchange and demonstrating its X-type ligand nature. These results offer compelling evidence of luminescence quenching via CsPbBr3-TFB adduct formation at high TFB loading, thereby presenting a comprehensive understanding of the underlying mechanism between CsPbBr3 and triarylamine-based hole transport materials. This knowledge holds significant promise for advancing the technology of PeLEDs.
Materials and Methods
Materials
Lead bromide (≥98%), cesium bromide (99.99%), cesium carbonate (Cs2CO3), oleic acid (90%), oleyl amine (≥98%), anhydrous N, N′-dimethylformamide (DMF, 99.8%), ethyl acetate, octane (≥99%), hexane (95%), anhydrous toluene (99.8%), and phenethylammonium bromide (PEABr, ≥98%) were procured from Sigma-Aldrich. Poly[(9,9-dioctylfluorenyl-2,7diyl)-co-(4,4′-(N-(4-sec-butylphenyl) diphenylamine)] (TFB, molecular weight >30,000) was purchased from Luminescence Technology Company, Taiwan (Lumtec). All of the chemicals were used as received without further purification.
Methods
Synthesis of CsPbBr3 Nanoplatelets
The NPLs were synthesized using the ligand-assisted reprecipitation (LARP) method at room temperature. The precursor solution was prepared by dissolving CsBr (0.5 mM) and PbBr2 (1.0 mM) in 20 mL of anhydrous DMF. In a separate vial, a solution of oleic acid (100 μL), oleyl amine (100 μL), and anhydrous toluene (10 mL) was prepared by vortexing for 10 s at room temperature. Subsequently, 1000 μL of the precursor solution was swiftly added into a vial containing toluene and ligands. Immediately, the color of the mixture turns yellowish green, indicating the formation of the product. The reaction was then quenched by the addition of ethyl acetate (10 mL), and the resulting product was centrifuged at 5000 rpm for 5 min to remove larger platelets. The supernatant was collected separately in a 50 mL centrifugation tube and treated with oleic acid, oleyl amine, and PEABr (10 μL each). For PEABr treatment, 200 mg of PEABr was dissolved in 1 mL of DMF. The mixture was then vortexed for 5 s followed by the addition of 15 mL of ethyl acetate. Immediately, the mixture was centrifuged at 7800 rpm for 15 min, and the precipitate was collected and dispersed in 1 mL of anhydrous hexane. Subsequently, oleic acid and oleyl amine (2 μL each) were further added, and nanoplatelets were again reprecipitated using 2 mL of ethyl acetate. Finally, the mixture was centrifuged at 7800 rpm for 15 min, and the precipitate was redispersed in anhydrous toluene/toluene-d8 (1 mL) and stored in the glovebox.
Synthesis of CsPbBr3 Nanocrystals
The synthesis of CsPbBr3 nanocrystals was carried out by a hot-injection method. Typically, for the Cs-oleate preparation, 0.814 g of Cs2CO3 was loaded into a three-neck round-bottom flask containing 30 mL of 1-octadecene and 2.5 mL of oleic acid. The mixture was degassed at 120 °C for 60 min and then heated at 150 °C, making sure complete dissolution of Cs2CO3. The Cs-oleate precursor solution is stored under nitrogen and used whenever necessary by preheating at 100 °C. For the synthesis of CsPbBr3 NCs, a three-neck round-bottom flask was loaded with 1-octadecene (10 mL), PbBr2 (138 mg), oleic acid (1 mL), and oleyl amine (1 mL) under nitrogen atmosphere. The mixture was then degassed at 120 °C for 60 min. After complete solubilization of PbBr2, the temperature was raised to 160 °C under a nitrogen atmosphere. Preheated Cs-oleate solution (∼100 °C) was then swiftly injected (0.8 mL) into the above mixture, and immediately within 5 s later, the reaction mixture was cooled by an ice–water bath to attain room temperature. The reaction mixture was collected in centrifuge tubes, and an equal amount of ethyl acetate was added. The mixture was then centrifuged at 7000 rpm for 7 min, and the precipitate was collected. The precipitate was dispersed in 2 mL of hexane and washed again with 3 mL of ethyl acetate. The resulting mixture was centrifuged again at 7000 rpm for 10 min, and the precipitate was collected and redispersed in toluene/toluene-d8 (1 mL) and stored in the glovebox for further use.
Luminescence Quenching Experiment
For the quenching experiment, the dispersion of NPLs or NCs in toluene was prepared by mixing 200 μL of as-synthesized NPLs/NCs in 2 mL of anhydrous toluene and labeled as a reference. In a separate vial, a stock solution of TFB (125 μM) was prepared by dissolving 50 mg of TFB in 5 mL of toluene. To probe the optical properties, the reference dispersion of NPLs/NCs was treated with 0.5 μL of TFB (0.0325 μM) from the stock solution. The treated solution was first used for UV–visible absorption measurements followed by PL and TRPL measurements. Further, the same solution was treated with increasing amounts of the TFB (1–70 μL), and after each addition, absorption, PL, and TRPL measurements were carried out.
Characterization
UV–Visible Absorption Spectroscopy
UV–visible absorption measurements were done using a Varion Cary 100 Bio UV–visible spectrophotometer.
Photoluminescence (PL) and Time-Resolved Photoluminescence (TRPL) Measurements
Photoluminescence and TRPL measurements were carried out using a Deltaflex modular fluorescence lifetime system (Horiba Scientific) equipped with a 356 nm LED source. The PL lifetime was monitored at fixed emission wavelengths for NPLs (454 nm) and NCs (516 nm).
1D 1H and 2D NOESY Nuclear Magnetic Resonance Spectroscopy Measurements
NMR experiments were carried out using a Bruker AVANCE III operating at a 1H frequency of 500 MHz, at 298 K. The 1H measurements were carried out by fixing the spectral width to 20 ppm. 2D NOESY experiments were conducted by using 2048 data points in the direct dimensions and 256 data points in the indirect dimensions with a mixing time of 500 ms. The 1H and NOESY spectra were processed by using Bruker TopSpin NMR data analysis software.
Transmission Electron Microscopy (TEM)
TEM and HRTEM imaging were carried out using an FEI Technai F-20 Transmission Electron Microscope (200 kV). Samples were prepared by drop-casting a colloidal solution on carbon-coated TEM grids. The images were analyzed by using ImageJ image processing software.
Ultrafast Transient Absorption Measurements
The experimental system is based on a 20 W femtosecond Yb:KGW amplifier generating 200 fs pulses at 1030 nm (Pharos from Light Conversion) and operating at 100 kHz. About 12 W of the output is used to pump a tunable optical parametric amplifier (Orpheus from Light Conversion) to generate pump pulses at the desired wavelengths. About 2 W of the output is used to pump a sapphire substrate to generate white light continuum pulses to be used as probe pulses. The pump pulses are modulated using a mechanical chopper (Thorlabs), temporally delayed using a mechanical translation stage (PI), and focused on the sample at a slight angle with respect to the probe pulses. The probe pulses are collected after the sample interaction and directed toward the Fourier transform spectroscopy system based on a translating-wedge-based identical pulse encoding system (GEMINI from Nireos srl).77 The transient absorption signal is then demodulated by using a photodetector (Femto.de) connected to a lock-in amplifier (Zurich Instruments). The GEMINI system provides a temporal interferogram of the TA spectrum, which is a Fourier transform to obtain the experimental spectrum. To obtain the spectrally integrated dynamics with a high signal-to-noise ratio, we used optical filters to spectrally filter the desired spectral ranges and detected the signal with the photodiode:lock-in system without GEMINI.
Acknowledgments
We acknowledge the support of the National Science Foundation (Grant No. OIA-1757220) for providing support. We thank DOE (Award No: DE-SC0024214) for partially supporting Anuraj in studying the optical properties of perovskites. We are thankful for the SRI seed grant supported by the Mississippi State University.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacsau.4c00083.
TEM/HRTEM images, UV–visible absorption, transient absorption plots, NMR spectra, and TRPL dynamics (PDF)
Author Contributions
CRediT: Anuraj S. Kshirsagar conceptualization, formal analysis, investigation, methodology, writing-original draft; Katherine A Koch data curation, writing-review & editing; Ajay Ram Srimath Kandada formal analysis, investigation, resources, supervision, writing-review & editing; Mahesh K Gangishetty conceptualization, data curation, funding acquisition, investigation, resources, supervision, writing-review & editing.
The authors declare no competing financial interest.
Supplementary Material
References
- Lu M.; Zhang Y.; Wang S.; Guo J.; Yu W. W.; Rogach A. L. Metal Halide Perovskite Light-Emitting Devices: Promising Technology for Next-Generation Displays. Adv. Funct. Mater. 2019, 29, 1902008 10.1002/adfm.201902008. [DOI] [Google Scholar]
- Yoon H. C.; Lee H.; Kang H.; Oh J. H.; Do Y. R. Highly efficient wide-color-gamut QD-emissive LCDs using red and green perovskite core/shell QDs. J. Mater. Chem. C 2018, 6, 13023–13033. 10.1039/C8TC04537B. [DOI] [Google Scholar]
- Wei Y.; Cheng Z.; Lin J. An overview on enhancing the stability of lead halide perovskite quantum dots and their applications in phosphor-converted LEDs. Chem. Soc. Rev. 2019, 48, 310–350. 10.1039/C8CS00740C. [DOI] [PubMed] [Google Scholar]
- Li C. H. A.; Zhou Z.; Vashishtha P.; Halpert J. E. The Future Is Blue (LEDs): Why Chemistry Is the Key to Perovskite Displays. Chem. Mater. 2019, 31, 6003–6032. 10.1021/acs.chemmater.9b01650. [DOI] [Google Scholar]
- Chiba T.; Hayashi Y.; Ebe H.; Hoshi K.; Sato J.; Sato S.; Pu Y.-J.; Ohisa S.; Kido J. Anion-exchange red perovskite quantum dots with ammonium iodine salts for highly efficient light-emitting devices. Nat. Photonics 2018, 12, 681–687. 10.1038/s41566-018-0260-y. [DOI] [Google Scholar]
- Gangishetty M. K.; Hou S.; Quan Q.; Congreve D. N. Reducing Architecture Limitations for Efficient Blue Perovskite Light-Emitting Diodes. Adv. Mater. 2018, 30, 1706226 10.1002/adma.201706226. [DOI] [PubMed] [Google Scholar]
- Hou S.; Gangishetty M. K.; Quan Q.; Congreve D. N. Efficient Blue and White Perovskite Light-Emitting Diodes via Manganese Doping. Joule 2018, 2, 2421–2433. 10.1016/j.joule.2018.08.005. [DOI] [PubMed] [Google Scholar]
- Mir W. J.; Alamoudi A.; Yin J.; Yorov K. E.; Maity P.; Naphade R.; Shao B.; Wang J.; Lintangpradipto M. N.; Nematulloev S.; Emwas A. H.; Genovese A.; Mohammed O. F.; Bakr O. M. Lecithin Capping Ligands Enable Ultrastable Perovskite-Phase CsPbI(3) Quantum Dots for Rec. 2020 Bright-Red Light-Emitting Diodes. J. Am. Chem. Soc. 2022, 144, 13302–13310. 10.1021/jacs.2c04637. [DOI] [PubMed] [Google Scholar]
- Wang H.; Ye F.; Sun J.; Wang Z.; Zhang C.; Qian J.; Zhang X.; Choy W. C. H.; Sun X. W.; Wang K.; Zhao W. Efficient CsPbBr3 Nanoplatelet-Based Blue Light-Emitting Diodes Enabled by Engineered Surface Ligands. ACS Energy Lett. 2022, 7, 1137–1145. 10.1021/acsenergylett.1c02642. [DOI] [Google Scholar]
- Tan Z.-K.; Moghaddam R. S.; Lai M. L.; Docampo P.; Higler R.; Deschler F.; Price M.; Sadhanala A.; Pazos L. M.; Credgington D.; Hanusch F.; Bein T.; Snaith H. J.; Friend R. H. Bright light-emitting diodes based on organometal halide perovskite. Nat. Nanotechnol. 2014, 9, 687–692. 10.1038/nnano.2014.149. [DOI] [PubMed] [Google Scholar]
- Zhao B.; Bai S.; Kim V.; Lamboll R.; Shivanna R.; Auras F.; Richter J. M.; Yang L.; Dai L.; Alsari M.; She X.-J.; Liang L.; Zhang J.; Lilliu S.; Gao P.; Snaith H. J.; Wang J.; Greenham N. C.; Friend R. H.; Di D. High-efficiency perovskite–polymer bulk heterostructure light-emitting diodes. Nat. Photonics 2018, 12, 783–789. 10.1038/s41566-018-0283-4. [DOI] [Google Scholar]
- Lin K.; Xing J.; Quan L. N.; de Arquer F. P. G.; Gong X.; Lu J.; Xie L.; Zhao W.; Zhang D.; Yan C.; Li W.; Liu X.; Lu Y.; Kirman J.; Sargent E. H.; Xiong Q.; Wei Z. Perovskite light-emitting diodes with external quantum efficiency exceeding 20%. Nature 2018, 562, 245–248. 10.1038/s41586-018-0575-3. [DOI] [PubMed] [Google Scholar]
- Shynkarenko Y.; Bodnarchuk M. I.; Bernasconi C.; Berezovska Y.; Verteletskyi V.; Ochsenbein S. T.; Kovalenko M. V. Direct Synthesis of Quaternary Alkylammonium-Capped Perovskite Nanocrystals for Efficient Blue and Green Light-Emitting Diodes. ACS Energy Lett. 2019, 4, 2703–2711. 10.1021/acsenergylett.9b01915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie M.; Guo J.; Zhang X.; Bi C.; Zhang L.; Chu Z.; Zheng W.; You J.; Tian J. High-Efficiency Pure-Red Perovskite Quantum-Dot Light-Emitting Diodes. Nano Lett. 2022, 22, 8266–8273. 10.1021/acs.nanolett.2c03062. [DOI] [PubMed] [Google Scholar]
- Sun C.; Jiang Y.; Cui M.; Qiao L.; Wei J.; Huang Y.; Zhang L.; He T.; Li S.; Hsu H.-Y.; Qin C.; Long R.; Yuan M. High-performance large-area quasi-2D perovskite light-emitting diodes. Nat. Commun. 2021, 12, 2207 10.1038/s41467-021-22529-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J.; Chu Z.; Yin Z.; Li J.; Yang Y.; Chen J.; Wu J.; You J.; Zhang X. Red Perovskite Light-Emitting Diodes with Efficiency Exceeding 25% Realized by Co-Spacer Cations. Adv. Mater. 2022, 34, 2204460 10.1002/adma.202204460. [DOI] [PubMed] [Google Scholar]
- Becker M. A.; Scarpelli L.; Nedelcu G.; Rainò G.; Masia F.; Borri P.; Stöferle T.; Kovalenko M. V.; Langbein W.; Mahrt R. F. Long Exciton Dephasing Time and Coherent Phonon Coupling in CsPbBr2Cl Perovskite Nanocrystals. Nano Lett. 2018, 18, 7546–7551. 10.1021/acs.nanolett.8b03027. [DOI] [PubMed] [Google Scholar]
- Quan L. N.; Rand B. P.; Friend R. H.; Mhaisalkar S. G.; Lee T. W.; Sargent E. H. Perovskites for Next-Generation Optical Sources. Chem. Rev. 2019, 119, 7444–7477. 10.1021/acs.chemrev.9b00107. [DOI] [PubMed] [Google Scholar]
- Sutherland B. R.; Sargent E. H. Perovskite photonic sources. Nat. Photonics 2016, 10, 295–302. 10.1038/nphoton.2016.62. [DOI] [Google Scholar]
- Protesescu L.; Yakunin S.; Bodnarchuk M. I.; Krieg F.; Caputo R.; Hendon C. H.; Yang R. X.; Walsh A.; Kovalenko M. V. Nanocrystals of cesium lead halide perovskites (CsPbX3, X = Cl, Br, and I): Novel optoelectronic materials showing bright emission with wide color gamut. Nano Lett. 2015, 15, 3692–3696. 10.1021/nl5048779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamsi J.; Dang Z.; Bianchini P.; Canale C.; Di Stasio F.; Brescia R.; Prato M.; Manna L. Colloidal Synthesis of Quantum Confined Single Crystal CsPbBr3 Nanosheets with Lateral Size Control up to the Micrometer Range. J. Am. Chem. Soc. 2016, 138, 7240–7243. 10.1021/jacs.6b03166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohn B. J.; Tong Y.; Gramlich M.; Lai M. L.; Döblinger M.; Wang K.; Hoye R. L. Z.; Müller-Buschbaum P.; Stranks S. D.; Urban A. S.; Polavarapu L.; Feldmann J. Boosting Tunable Blue Luminescence of Halide Perovskite Nanoplatelets through Postsynthetic Surface Trap Repair. Nano Lett. 2018, 18, 5231–5238. 10.1021/acs.nanolett.8b02190. [DOI] [PubMed] [Google Scholar]
- Shamsi J.; Kubicki D.; Anaya M.; Liu Y.; Ji K.; Frohna K.; Grey C. P.; Friend R. H.; Stranks S. D. Stable Hexylphosphonate-Capped Blue-Emitting Quantum-Confined CsPbBr3 Nanoplatelets. ACS Energy Lett. 2020, 5, 1900–1907. 10.1021/acsenergylett.0c00935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otero-Martínez C.; Ye J.; Sung J.; Pastoriza-Santos I.; Pérez-Juste J.; Xia Z.; Rao A.; Hoye R. L. Z.; Polavarapu L. Colloidal Metal-Halide Perovskite Nanoplatelets: Thickness-Controlled Synthesis, Properties, and Application in Light-Emitting Diodes. Adv. Mater. 2022, 34, 2107105 10.1002/adma.202107105. [DOI] [PubMed] [Google Scholar]
- Bekenstein Y.; Koscher B. A.; Eaton S. W.; Yang P.; Alivisatos A. P. Highly Luminescent Colloidal Nanoplates of Perovskite Cesium Lead Halide and Their Oriented Assemblies. J. Am. Chem. Soc. 2015, 137, 16008–16011. 10.1021/jacs.5b11199. [DOI] [PubMed] [Google Scholar]
- Vale B. R. C.; Socie E.; Burgos-Caminal A.; Bettini J.; Schiavon M. A.; Moser J.-E. Exciton, Biexciton, and Hot Exciton Dynamics in CsPbBr3 Colloidal Nanoplatelets. J. Phys. Chem. Lett. 2020, 11, 387–394. 10.1021/acs.jpclett.9b03282. [DOI] [PubMed] [Google Scholar]
- Xing G.; Wu B.; Wu X.; Li M.; Du B.; Wei Q.; Guo J.; Yeow E. K. L.; Sum T. C.; Huang W. Transcending the slow bimolecular recombination in lead-halide perovskites for electroluminescence. Nat. Commun. 2017, 8, 14558 10.1038/ncomms14558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao X.; Wang Y.; Wang L.; Wang X.; Bao Y. The Dissociation of Exciton During the Lasing of a Single CsPbBr3Microplate. J. Phys. Chem. Lett. 2022, 13, 10851–10857. 10.1021/acs.jpclett.2c03242. [DOI] [PubMed] [Google Scholar]
- Cho C.; Zhao B.; Tainter G. D.; Lee J.-Y.; Friend R. H.; Di D.; Deschler F.; Greenham N. C. The role of photon recycling in perovskite light-emitting diodes. Nat. Commun. 2020, 11, 611 10.1038/s41467-020-14401-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y.; Cheng L.-P.; Li Y.-Q.; Li W.; Chen J.-D.; Lee S.-T.; Tang J.-X. High-Efficiency Perovskite Light-Emitting Diodes with Synergetic Outcoupling Enhancement. Adv. Mater. 2019, 31, 1901517 10.1002/adma.201901517. [DOI] [PubMed] [Google Scholar]
- Liu X.-K.; Xu W.; Bai S.; Jin Y.; Wang J.; Friend R. H.; Gao F. Metal halide perovskites for light-emitting diodes. Nat. Mater. 2021, 20, 10–21. 10.1038/s41563-020-0784-7. [DOI] [PubMed] [Google Scholar]
- Zhang L.; Yang X.; Jiang Q.; Wang P.; Yin Z.; Zhang X.; Tan H.; Yang Y. M.; Wei M.; Sutherland B. R.; Sargent E. H.; You J. Ultra-bright and highly efficient inorganic based perovskite light-emitting diodes. Nat. Commun. 2017, 8, 15640 10.1038/ncomms15640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Z.; Miao Y.; Hu Z.; Xu W.; Kuang C.; Pan K.; Liu P.; Lai J.; Sun B.; Wang J.; Bai S.; Gao F. Unveiling the synergistic effect of precursor stoichiometry and interfacial reactions for perovskite light-emitting diodes. Nat. Commun. 2019, 10, 2818 10.1038/s41467-019-10612-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Lin H.; Huang H.; Reckmeier C.; Zhang Y.; Choy W. C. H.; Rogach A. L. Enhancing the Brightness of Cesium Lead Halide Perovskite Nanocrystal Based Green Light-Emitting Devices through the Interface Engineering with Perfluorinated Ionomer. Nano Lett. 2016, 16, 1415–1420. 10.1021/acs.nanolett.5b04959. [DOI] [PubMed] [Google Scholar]
- Wang H.; Zhang X.; Wu Q.; Cao F.; Yang D.; Shang Y.; Ning Z.; Zhang W.; Zheng W.; Yan Y.; Kershaw S. V.; Zhang L.; Rogach A. L.; Yang X. Trifluoroacetate induced small-grained CsPbBr(3) perovskite films result in efficient and stable light-emitting devices. Nat. Commun. 2019, 10, 665 10.1038/s41467-019-08425-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma D. K.; Hirata S.; Vacha M. Single-particle electroluminescence of CsPbBr(3) perovskite nanocrystals reveals particle-selective recombination and blinking as key efficiency factors. Nat. Commun. 2019, 10, 4499 10.1038/s41467-019-12512-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ban M.; Zou Y.; Rivett J. P. H.; Yang Y.; Thomas T. H.; Tan Y.; Song T.; Gao X.; Credgington D.; Deschler F.; Sirringhaus H.; Sun B. Solution-processed perovskite light emitting diodes with efficiency exceeding 15% through additive-controlled nanostructure tailoring. Nat. Commun. 2018, 9, 3892 10.1038/s41467-018-06425-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.; Han T. H.; Tan S.; Xue J.; Zhao Y.; Liu Y.; Wang H.; Hu W.; Bao C.; Mazzeo M.; Wang R.; Duan Y.; Yang Y. Efficient Flexible Inorganic Perovskite Light-Emitting Diodes Fabricated with CsPbBr(3) Emitters Prepared via Low-Temperature in Situ Dynamic Thermal Crystallization. Nano Lett. 2020, 20, 4673–4680. 10.1021/acs.nanolett.0c01550. [DOI] [PubMed] [Google Scholar]
- Hu L.; Ye Z.; Wu D.; Wang Z.; Wang W.; Wang K.; Cui X.; Wang N.; An H.; Li B.; Xiang B.; Qiu M. Marked Efficiency Improvement of FAPb(0.7)Sn(0.3)Br(3) Perovskite Light-Emitting Diodes by Optimization of the Light-Emitting Layer and Hole-Transport Layer. Nanomaterials 2022, 12, 154 10.3390/nano12091454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y.-H.; Cho H.; Heo J. H.; Kim T.-S.; Myoung N.; Lee C.-L.; Im S. H.; Lee T.-W. Multicolored Organic/Inorganic Hybrid Perovskite Light-Emitting Diodes. Adv. Mater. 2015, 27, 1248–1254. 10.1002/adma.201403751. [DOI] [PubMed] [Google Scholar]
- Wang H.; Gong X.; Zhao D.; Zhao Y.-B.; Wang S.; Zhang J.; Kong L.; Wei B.; Quintero-Bermudez R.; Voznyy O.; Shang Y.; Ning Z.; Yan Y.; Sargent E. H.; Yang X. A Multi-functional Molecular Modifier Enabling Efficient Large-Area Perovskite Light-Emitting Diodes. Joule 2020, 4, 1977–1987. 10.1016/j.joule.2020.07.002. [DOI] [Google Scholar]
- Peng X.-F.; Wu X.-Y.; Ji X.-X.; Ren J.; Wang Q.; Li G.-Q.; Yang X.-H. Modified Conducting Polymer Hole Injection Layer for High-Efficiency Perovskite Light-Emitting Devices: Enhanced Hole Injection and Reduced Luminescence Quenching. J. Phys. Chem. Lett. 2017, 8, 4691–4697. 10.1021/acs.jpclett.7b01992. [DOI] [PubMed] [Google Scholar]
- Groenendaal L.; Jonas F.; Freitag D.; Pielartzik H.; Reynolds J. R. Poly(3,4-ethylenedioxythiophene) and Its Derivatives: Past, Present, and Future. Adv. Mater. 2000, 12, 481–494. . [DOI] [Google Scholar]
- Zou W.; Li R.; Zhang S.; Liu Y.; Wang N.; Cao Y.; Miao Y.; Xu M.; Guo Q.; Di D.; Zhang L.; Yi C.; Gao F.; Friend R. H.; Wang J.; Huang W. Minimising efficiency roll-off in high-brightness perovskite light-emitting diodes. Nat. Commun. 2018, 9, 608 10.1038/s41467-018-03049-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Y.; Ban M.; Yang Y.; Bai S.; Wu C.; Han Y.; Wu T.; Tan Y.; Huang Q.; Gao X.; Song T.; Zhang Q.; Sun B. Boosting Perovskite Light-Emitting Diode Performance via Tailoring Interfacial Contact. ACS Appl. Mater. Interfaces 2018, 10, 24320–24326. 10.1021/acsami.8b07438. [DOI] [PubMed] [Google Scholar]
- Wang Z.; Li Z.; Zhou D.; Yu J. Low turn-on voltage perovskite light-emitting diodes with methanol treated PEDOT:PSS as hole transport layer. Appl. Phys. Lett. 2017, 111, 233304 10.1063/1.5010245. [DOI] [Google Scholar]
- Ngai K. H.; Wei Q.; Chen Z.; Guo X.; Qin M.; Xie F.; Chan C. C. S.; Xing G.; Lu X.; Chen J.; Wong K. S.; Xu J.; Long M. Enhanced Electrochemical Stability by Alkyldiammonium in Dion–Jacobson Perovskite toward Ultrastable Light-Emitting Diodes. Adv. Opt. Mater. 2021, 9, 2100243 10.1002/adom.202100243. [DOI] [Google Scholar]
- Bruno A.; Reynolds L. X.; Dyer-Smith C.; Nelson J.; Haque S. A. Determining the Exciton Diffusion Length in a Polyfluorene from Ultrafast Fluorescence Measurements of Polymer/Fullerene Blend Films. J. Phys. Chem. C 2013, 117, 19832–19838. 10.1021/jp404985q. [DOI] [Google Scholar]
- Dong Y.; Wang Y.-K.; Yuan F.; Johnston A.; Liu Y.; Ma D.; Choi M.-J.; Chen B.; Chekini M.; Baek S.-W.; Sagar L. K.; Fan J.; Hou Y.; Wu M.; Lee S.; Sun B.; Hoogland S.; Quintero-Bermudez R.; Ebe H.; Todorovic P.; Dinic F.; Li P.; Kung H. T.; Saidaminov M. I.; Kumacheva E.; Spiecker E.; Liao L.-S.; Voznyy O.; Lu Z.-H.; Sargent E. H. Bipolar-shell resurfacing for blue LEDs based on strongly confined perovskite quantum dots. Nat. Nanotechnol. 2020, 15, 668–674. 10.1038/s41565-020-0714-5. [DOI] [PubMed] [Google Scholar]
- Kang N. S.; Ju B.-K.; Kim J. W.; Kim J.-J.; Yu J.-W.; Chin B. D. Correlation of photoluminescent quantum efficiency and device characteristics for the soluble electrophosphorescent light emitter with interfacial layers. J. Appl. Phys. 2008, 104, 024511 10.1063/1.2959817. [DOI] [Google Scholar]
- Ravi V. K.; Markad G. B.; Nag A. Band Edge Energies and Excitonic Transition Probabilities of Colloidal CsPbX3 (X = Cl, Br, I) Perovskite Nanocrystals. ACS Energy Lett. 2016, 1, 665–671. 10.1021/acsenergylett.6b00337. [DOI] [Google Scholar]
- Cortés-Villena A.; Caminos D. A.; Galian R. E.; Pérez-Prieto J. Singlet Sensitization of a BODIPY Rotor Triggered by Marriage with Perovskite Nanocrystals. Adv. Opt. Mater. 2023, 11, 2300138 10.1002/adom.202300138. [DOI] [Google Scholar]
- DuBose J. T.; Kamat P. V. How Pendant Groups Dictate Energy and Electron Transfer in Perovskite-Rhodamine Light Harvesting Assemblies. J. Am. Chem. Soc. 2023, 145, 4601–4612. 10.1021/jacs.2c12248. [DOI] [PubMed] [Google Scholar]
- DuBose J. T.; Kamat P. V. Energy Versus Electron Transfer: Managing Excited-State Interactions in Perovskite Nanocrystal-Molecular Hybrids. Chem. Rev. 2022, 122, 12475–12494. 10.1021/acs.chemrev.2c00172. [DOI] [PubMed] [Google Scholar]
- Boaz H.; Rollefson G. K. The Quenching of Fluorescence. Deviations from the Stern-Volmer Law. J. Am. Chem. Soc. 1950, 72, 3435–3443. 10.1021/ja01164a032. [DOI] [Google Scholar]
- Fraiji L. K.; Fraiji K.; Werner T. C. Static and Dynamic Fluorescence Quenching Experiments for the Physical Chemistry Laboratory. J. Chem. Educ. 1992, 69, 424–428. 10.1021/ed069p424. [DOI] [Google Scholar]
- Tanwar A. S.; Parui R.; Garai R.; Chanu M. A.; Iyer P. K. Dual ″Static and Dynamic″ Fluorescence Quenching Mechanisms Based Detection of TNT via a Cationic Conjugated Polymer. ACS Meas. Sci. Au 2022, 2, 23–30. 10.1021/acsmeasuresciau.1c00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehlen M. H. The centenary of the Stern-Volmer equation of fluorescence quenching: From the single line plot to the SV quenching map. J. Photochem. Photobiol. C 2020, 42, 100338 10.1016/j.jphotochemrev.2019.100338. [DOI] [Google Scholar]
- Pal A.; Srivastava S.; Saini P.; Raina S.; Ingole P. P.; Gupta R.; Sapra S. Probing the Mechanism of Fluorescence Quenching of QDs by Co(III)-Complexes: Size of QD and Nature of the Complex Both Dictate Energy and Electron Transfer Processes. J. Phys. Chem. C 2015, 119, 22690–22699. 10.1021/acs.jpcc.5b06795. [DOI] [Google Scholar]
- Al-Maskari S.; Issac A.; Varanasi S. R.; Hildner R.; Sofin R. G. S.; Ibrahim A. R.; Abou-Zied O. K. Dye-induced photoluminescence quenching of quantum dots: role of excited state lifetime and confinement of charge carriers. Phys. Chem. Chem. Phys. 2023, 25, 14126–14137. 10.1039/D3CP00715D. [DOI] [PubMed] [Google Scholar]
- Issac A.; Al-Maskari S.; Sofin R. G. S.; Ibrahim A. R.; Abou-Zied O. K. Photoexcited Charge Trapping Induced Quenching of Radiative Recombination Pathways in CuInS2/ZnS-Dye Nanoassemblies. J. Lumin. 2021, 239, 118402 10.1016/j.jlumin.2021.118402. [DOI] [Google Scholar]
- Aneesh J.; Swarnkar A.; Kumar Ravi V.; Sharma R.; Nag A.; Adarsh K. V. Ultrafast Exciton Dynamics in Colloidal CsPbBr3 Perovskite Nanocrystals: Biexciton Effect and Auger Recombination. J. Phys. Chem. C 2017, 121, 4734–4739. 10.1021/acs.jpcc.7b00762. [DOI] [Google Scholar]
- Yarita N.; Tahara H.; Ihara T.; Kawawaki T.; Sato R.; Saruyama M.; Teranishi T.; Kanemitsu Y. Dynamics of Charged Excitons and Biexcitons in CsPbBr(3) Perovskite Nanocrystals Revealed by Femtosecond Transient-Absorption and Single-Dot Luminescence Spectroscopy. J. Phys. Chem. Lett. 2017, 8, 1413–1418. 10.1021/acs.jpclett.7b00326. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Xia Y.; Friend R. H.; Silva C. Sequential absorption processes in two-photon-excitation transient absorption spectroscopy in a semiconductor polymer. Phys. Rev. B 2006, 73, 245201 10.1103/PhysRevB.73.245201. [DOI] [Google Scholar]
- De Roo J.; Yazdani N.; Drijvers E.; Lauria A.; Maes J.; Owen J. S.; Van Driessche I.; Niederberger M.; Wood V.; Martins J. C.; Infante I.; Hens Z. Probing Solvent–Ligand Interactions in Colloidal Nanocrystals by the NMR Line Broadening. Chem. Mater. 2018, 30, 5485–5492. 10.1021/acs.chemmater.8b02523. [DOI] [Google Scholar]
- De Roo J.; Zhou Z.; Wang J.; Deblock L.; Crosby A. J.; Owen J. S.; Nonnenmann S. S. Synthesis of Phosphonic Acid Ligands for Nanocrystal Surface Functionalization and Solution Processed Memristors. Chem. Mater. 2018, 30, 8034–8039. 10.1021/acs.chemmater.8b03768. [DOI] [Google Scholar]
- Chen P. E.; Anderson N. C.; Norman Z. M.; Owen J. S. Tight Binding of Carboxylate, Phosphonate, and Carbamate Anions to Stoichiometric CdSe Nanocrystals. J. Am. Chem. Soc. 2017, 139, 3227–3236. 10.1021/jacs.6b13234. [DOI] [PubMed] [Google Scholar]
- De Roo J.; Ibanez M.; Geiregat P.; Nedelcu G.; Walravens W.; Maes J.; Martins J. C.; Van Driessche I.; Kovalenko M. V.; Hens Z. Highly Dynamic Ligand Binding and Light Absorption Coefficient of Cesium Lead Bromide Perovskite Nanocrystals. ACS Nano 2016, 10, 2071–2081. 10.1021/acsnano.5b06295. [DOI] [PubMed] [Google Scholar]
- Hens Z.; Martins J. C. A Solution NMR Toolbox for Characterizing the Surface Chemistry of Colloidal Nanocrystals. Chem. Mater. 2013, 25, 1211–1221. 10.1021/cm303361s. [DOI] [Google Scholar]
- Ravi V. K.; Santra P. K.; Joshi N.; Chugh J.; Singh S. K.; Rensmo H.; Ghosh P.; Nag A. Origin of the Substitution Mechanism for the Binding of Organic Ligands on the Surface of CsPbBr3 Perovskite Nanocubes. J. Phys. Chem. Lett. 2017, 8, 4988–4994. 10.1021/acs.jpclett.7b02192. [DOI] [PubMed] [Google Scholar]
- Jeener J.; Meier B. H.; Bachmann P.; Ernst R. R. Investigation of exchange processes by two-dimensional NMR spectroscopy. J. Chem. Phys. 1979, 71, 4546–4553. 10.1063/1.438208. [DOI] [Google Scholar]
- Moreels I.; Fritzinger B.; Martins J. C.; Hens Z. Surface Chemistry of Colloidal PbSe Nanocrystals. J. Am. Chem. Soc. 2008, 130, 15081–15086. 10.1021/ja803994m. [DOI] [PubMed] [Google Scholar]
- Macura S.; Huang Y.; Suter D.; Ernst R. R. Two-dimensional chemical exchange and cross-relaxation spectroscopy of coupled nuclear spins. J. Magn. Reson. 1981, 43, 259–281. 10.1016/0022-2364(81)90037-8. [DOI] [Google Scholar]
- Fritzinger B.; Moreels I.; Lommens P.; Koole R.; Hens Z.; Martins J. C. In Situ Observation of Rapid Ligand Exchange in Colloidal Nanocrystal Suspensions Using Transfer NOE Nuclear Magnetic Resonance Spectroscopy. J. Am. Chem. Soc. 2009, 131, 3024–3032. 10.1021/ja809436y. [DOI] [PubMed] [Google Scholar]
- Fiuza-Maneiro N.; Sun K.; López-Fernández I.; Gómez-Graña S.; Müller-Buschbaum P.; Polavarapu L. Ligand Chemistry of Inorganic Lead Halide Perovskite Nanocrystals. ACS Energy Lett. 2023, 8, 1152–1191. 10.1021/acsenergylett.2c02363. [DOI] [Google Scholar]
- Dey A.; Ye J.; De A.; Debroye E.; Ha S. K.; Bladt E.; Kshirsagar A. S.; Wang Z.; Yin J.; Wang Y.; Quan L. N.; Yan F.; Gao M.; Li X.; Shamsi J.; Debnath T.; Cao M.; Scheel M. A.; Kumar S.; Steele J. A.; Gerhard M.; Chouhan L.; Xu K.; Wu X.-g.; Li Y.; Zhang Y.; Dutta A.; Han C.; Vincon I.; Rogach A. L.; Nag A.; Samanta A.; Korgel B. A.; Shih C.-J.; Gamelin D. R.; Son D. H.; Zeng H.; Zhong H.; Sun H.; Demir H. V.; Scheblykin I. G.; Mora-Seró I.; Stolarczyk J. K.; Zhang J. Z.; Feldmann J.; Hofkens J.; Luther J. M.; Pérez-Prieto J.; Li L.; Manna L.; Bodnarchuk M. I.; Kovalenko M. V.; Roeffaers M. B. J.; Pradhan N.; Mohammed O. F.; Bakr O. M.; Yang P.; Müller-Buschbaum P.; Kamat P. V.; Bao Q.; Zhang Q.; Krahne R.; Galian R. E.; Stranks S. D.; Bals S.; Biju V.; Tisdale W. A.; Yan Y.; Hoye R. L. Z.; Polavarapu L. State of the Art and Prospects for Halide Perovskite Nanocrystals. ACS Nano 2021, 15, 10775–10981. 10.1021/acsnano.0c08903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oriana A.; Réhault J.; Preda F.; Polli D.; Cerullo G. Scanning Fourier transform spectrometer in the visible range based on birefringent wedges. J. Opt. Soc. Am. A 2016, 33, 1415–1420. 10.1364/JOSAA.33.001415. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.