

Abstract

Dearomatizations provide powerful synthetic routes to rapidly assemble substituted carbocycles and heterocycles found in a plethora of bioactive molecules. Harnessing the advantages of dearomatization typically requires vigorous reagents because of the difficulty in disrupting the stable aromatic core. A relatively mild dearomatization strategy is described that employs lithiated nitriles or isocyanides in a simple SNAr-type addition to form σ-complexes that are trapped by alkylation. The dearomatizations are diastereoselective and efficient and rapidly install two new carbon–carbon bonds, one of which is a quaternary center, as well as nitrile, isocyanide, and cyclohexadiene functionalities.
Keywords: dearomatization, interrupted SNAr, σ-complexes, quaternary center, lithiated nitriles
Aromatics rank among the most important organic molecules because of their high-volume deployment in industrial processes, their pervasive role in biological machinery, and their prevalence within active pharmaceutical ingredients.1 The centrality of aromatics has spurred numerous strategies to transform readily available precursors into high value products. Several methods effectively dearomatize fused aromatics,2 heteroaromatics,3,4 and phenols,3,5 whereas simple derivatives of benzene are more challenging because of the increased stability of the π-system.6 Despite the strongly reducing conditions,7 the classical dissolving metal reduction, and advantageous variants,8 remain the gold standard to access complex cyclohexadienes (Scheme 1A, 1 → 2 → 3).
Scheme 1. Strategies for Dearomatization.

Historically, nucleophilic aromatic substitutions have played a significant role in synthesis and in advancing the understanding of key σ-complexes 5 (Scheme 1B, 4 → 5 → 6).9 Originally posited as intermediates, σ-complexes 5 are now thought to more often occur as transition structures10 which is guiding the development of new dearomatization manifolds.11 Described here is a mild dearomatization strategy initiated by a nucleophilic dearomatization in which solvent effects are used to favor formation and trapping of intermediate σ-complexes 8 to assemble complex cyclohexadienes 9 (Scheme 1C, 7 → 8 → 9). The method efficiently dearomatizes substituted benzenes, naphthalenes, anthracenes, and pyridine to rapidly assemble complex cyclohexadiene scaffolds that are valuable precursors for synthesis in general12 and bioactive targets in particular.13
Discussion
Nucleophilic addition to an aromatic followed by alkylation of the resulting anionic σ-complex requires overcoming several challenges (Scheme 2). Typically, SNAr processes require electron deficient aromatics 10 substituted with strongly electron withdrawing groups (EWG) tolerant of strong nucleophiles. In practice, the optimal electron withdrawing groups polarize the aromatic and stabilize the intermediate σ-complex via resonance because inductive electron withdrawal is usually insufficient.11
Scheme 2. Generalized Interrupted SNAr Mechanism.

Among the best activating groups for SNAr reactions are nitro-substituted aromatics (10, EWG = NO2).2 In fact, nucleophilic additions to nitroaromatics occur more rapidly at positions substituted by hydrogen than at positions occupied by halogens (10 + R2 M → 11); subsequent ejection of the nucleophile from the σ-complex 11 to reform 10 is also fast which allows attack on carbons substituted with leaving groups leading to SNAr.14 Interrupting the dearomatization–rearomatization process requires that either the equilibrium between 10 and 11 (Scheme 2, path a) lies predominantly on the side of the σ-complex 11, or that the rate at which 11 forms 13 is dramatically faster than the rate of electrophilic trapping by the nucleophile R2 M to afford 12 (Scheme 2, path b).15
The feasibility of selectively trapping the intermediate σ-complex 11 was pursued with a lithiated nitrile and a substituted benzonitrile (Table 1). Lithiated nitriles are potent nucleophiles with a high charge density localized on the nucleophilic carbon which is ideal for disrupting the aromaticity.16 Inexpensive 2,6-difluorobenzonitrile 10a was selected as the aromatic because of the excellent ability of the nitrile and fluorine groups to stabilize the buildup of negative electron density in the σ-complex.17p-CF3–C6H4CH2Br was selected as the electrophile because of the presumed requirement for a reactive electrophile to trap the modestly nucleophilic, delocalized, cyclohexadienyl anion (cf. 11 EWG = CN).,18
Table 1. Interrupted SNAr Optimization Studya.

| entry | solvent | ratio 12a:13a | yield 13a |
|---|---|---|---|
| 1 | hexanes | 1.0:0 | 0% |
| 2 | toluene | 1.0:0 | 0% |
| 3 | Et2O | 1.0:0 | 0% |
| 4 | THF | 2.2:1.0 | 48% |
| 5 | THF + 18-C-6 | 1.7:1.0 | 50% |
| 6 | DME | 1.2:1.0 | 64% |
| 7 | diglyme | 1.1:1.0 | 91% |
Conditions: Deprotonation of cyclopentanecarboniirile (2.0 equiv) with LDA (2.1 equiv) at −78 °C followed by addition of 10a (1.0 equiv). After 1 h at-78 °C, added electrophile (2.1 equiv).
Exploratory forays revealed a significant solvent effect (Table 1). In hexanes, toluene, and ether (Table 1, entries 1–3, respectively), the only product was 12a derived from the deprotonation and benzylation of 14a. In THF, however, the 1,4-cyclohexadiene 13a was clearly detected with the identity being unequivocally determined by X-ray crystallography (Scheme 3). Although the yield was modest, the crude reaction mixture was clean, with unreacted 2,6-difluorobenzonitrile and nitrile 12a making up most of the remaining mass balance.
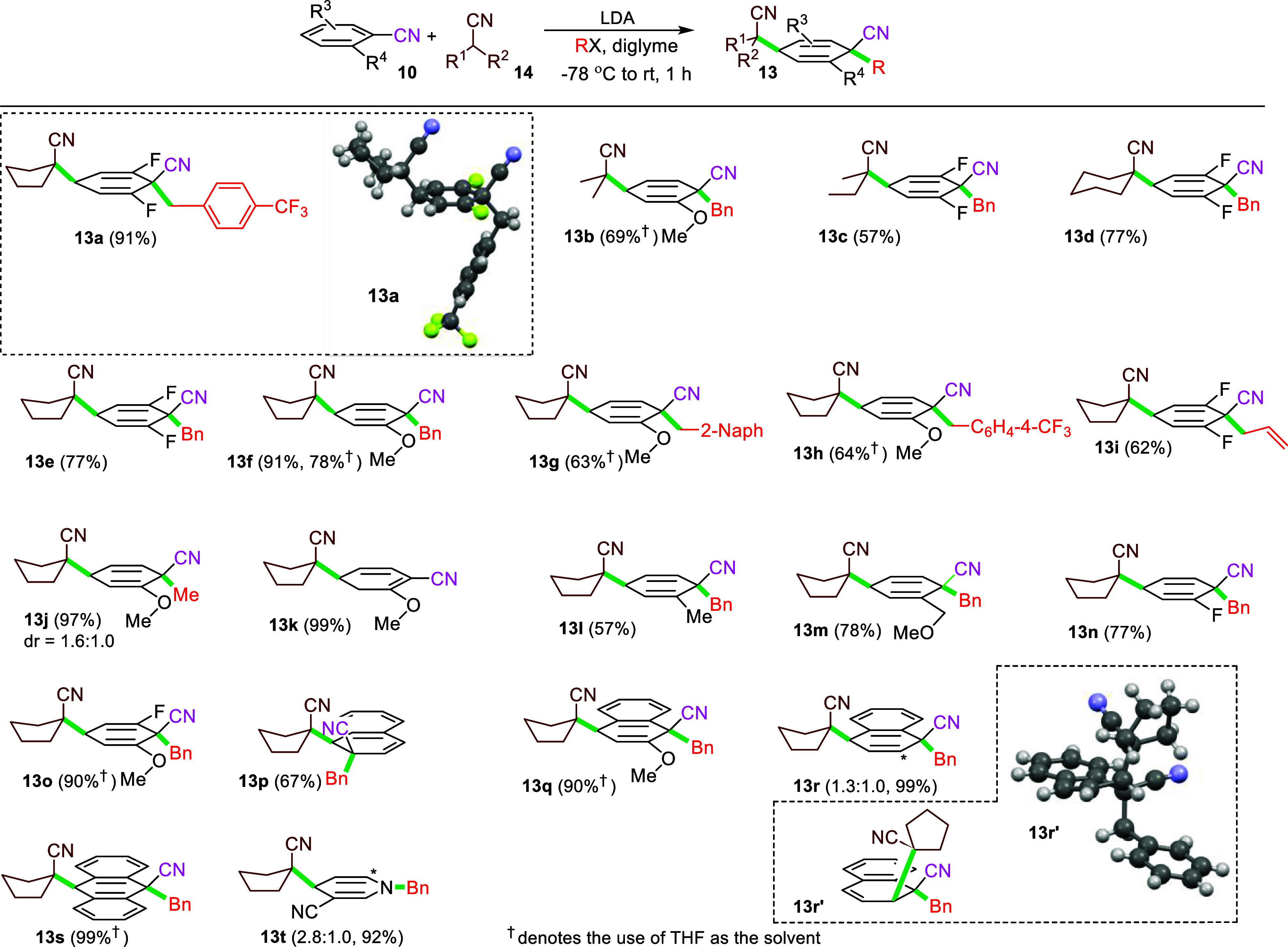
Scheme 3. Scope of the Interrupted SNAr-Alkylation of Substituted Benzonitriles with Nitrile Nucleophiles.
Efforts to promote full conversion focused on the temperature and time at which the electrophile, p-CF3–C6H4CH2Br, was added because of a strong dependence in a related dearomatization.19 Reducing the reaction temperature from −78 to −100 °C completely shut down the dearomatization, whereas temperatures higher than −78 °C produced less cyclohexadiene 13a and more of the benzylated nitrile 12a. Changing the time at which p-CF3–C6H4CH2Br was added from ten to less than 1 min led only to formation of the benzylated nitrile 12a, whereas extending the time between the addition of 2,6-difluorobenzonitrile and p-CF3–C6H4CH2Br substantially improved the conversion of 10a to 13a. The optimal time for aging the dearomatization in THF with 10a and 14a was 1 h, but the reaction did not go to complete conversion.
The aging was interpreted as the time required to establish the equilibrium in favor of the cyclohexadienyl anion (cf. Scheme 2, 10 → 11). An additional equivalent of cyclopentanecarbonitrile (14a) was added to coax the equilibrium toward the cyclohexadienyl anion with the combination of 2.1 equiv of 14a, 2.2 equiv of LDA, and a premixing time of 1 h affording a 48% yield of 13a (Table 1, entry 4). Interpreting the beneficial effect of THF as creating a better nucleophile through lithium solvation led to the addition of 18-crown-6. Although the influence of 18-C-6 was minimal (Table 1, compare entries 4 and 5), changing the lithium solvation by switching from THF to DME to diglyme progressively improved the yield (Table 1, 48, 64, 91%, entries 7, 8, 9, respectively).20
Having identified optimized conditions, the reaction scope was probed with a series of commercially available benzonitriles and lithiated nitriles (Scheme 3). Lithiated nitriles derived by deprotonating acyclic (isobutyronitrile, 2-methylbutyronitrile) and cyclic 5- and six-membered nitriles followed by addition of 2-methoxybenzonitrile or 2,6-difluorobenzonitrile and trapping with BnBr efficiently afforded the corresponding cyclohexadienes 13b–13f, respectively. Scaling the synthesis of 13e to a one mmol scale proceeded in 69% yield, an efficiency similar to that of a small-scale reaction (77%). Electrophilic trapping of the σ-complex formed from the addition of lithiated cyclopentanecarbonitrile to 2-methoxybenzonitrile or 2,6-difluorobenzonitrile proved equally efficient with benzyl bromide (13e and 13f), substituted benzyl bromides (13g and 13h), allyl bromide (13i), and methyl iodide (13j).21 Protonation with aqueous NH4Cl occurred exclusively at the γ-position to afford 13k, analogous to related protonations of cyclohexadienecarbonitrile anions.22 Only in the case of trapping with MeI were diastereomers observed resulting from attack on both faces of the cyclohexadienyl anion; in all other cases, a single diastereomer was obtained. Presumably, the small steric demand of MeI is insufficient to exert a large preference for addition to only one face of the σ-complex.
A range of substituted aromatic nitriles participate in the dearomatization.23 In addition to 2-methoxy- and 2,6-difluorobenzonitriles (13b–13k), lithiated cyclopentane-carbonitrile efficiently dearomatized 2-methyl-, 2-methoxymethyl- and 2-fluorobenzonitrile to afford the cyclohexadienes 13l, 13m, and 13n, respectively. The analogous dearomatization with 2-fluoro-6-methoxybenzonitrile very efficiently generated 13o.
The dearomatization of 2-methylbenzonitrile demonstrates that neither electron deficient substituents nor chelating ortho substituents are required; attempts to add lithiated cyclopentanecarbonitrile to benzonitrile (10, R3 = R4 = H) afforded only minimal amounts of the corresponding cyclohexadiene (vide infra). Lithiated cyclopentanecarbonitrile efficiently added to several fused aromatics: 2-cyanonaphthalene and 1-cyano-2-methoxynaphthalene regioselectively afforded 13p and 13q, respectively, whereas the analogous addition to 1-cyanonaphthalene afforded a 1.3:1 ratio of regioisomers 13r and 13r′. For 9-cyanoanthracene, dearomatization occurred on the central ring to afford 13s. The addition of lithiated cyclopentanecarbonitrile to 3-cyanopyridine afforded a mixture of regioisomers with divinylpyridine 13t predominating.
The success of lithiated nitriles in the interrupted SNAr-alkylation stimulated a search for other efficacious nucleophiles. While neither lithiated sulfides, sulfones, and sulfoxides nor the dianion derived from ethyl acetoacetate24 participated in the dearomatization, lithiated nucleophiles derived by adding BuLi to the Asmic-like25 isocyanide 15 efficiently afforded the corresponding isocyano-substituted cyclohexadienes 16a–16c (Scheme 4A).26 The lithiated benzylic isocyanide derived by deprotonating 17 dearomatized 9-cyanoanthracene to afford 16d; attempts to perform the analogous addition to 2,6-difluorobenzonitrile (10a) was not successful presumably because the delocalized, benzylic anion is insufficiently nucleophilic.29
Scheme 4. Interrupted SNAr-Alkylation Scope with Isocyanide-Containing Nucleophiles.

Dearomatization Mechanism
Mechanistic advances in nucleophilic aromatic substitution mechanisms have led to a revision of the classical SNAr process.14 Previously, the nucleophilic attack on an aromatic was thought to be particularly difficult, though now the nucleophilic addition is known to be facile with a faster attack at ortho and para positions occupied by hydrogen than at positions bearing leaving groups (cf. Scheme 5).27,28
Scheme 5. Mechanism of the Benzonitrile Dearomatization.

Lithiated tertiary nitriles15 and isocyanides29 are exceptional nucleophiles because the electron density is localized on the nucleophilic carbon.30 The nucleophilicity varies with solvation which dramatically accelerates the nucleophilic attack,31 particularly when dissociated ion pairs are formed that localize electron density without dissipating charge into the solvent.32 An attractive explanation for the efficacy of lithiated nitrile dearomatizations in diglyme is that diglyme encapsulates lithium cations in a cage-like structure 22(33) creating a more nucleophilic nitrile anion than the lithium dimer favored in THF.15 Support for the facile formation of the σ-complex 19 was obtained from a 19F NMR experiment with lithiated cyclopentanecarbonitrile and 2,6-difluorobenzonitrile (10a) in which the initial signal for 10a at −106.0 ppm was largely replaced by a signal at −105.4 ppm (6.4:1 ratio of the signals at −105.4 and −106.0, respectively).34
Conceptually, the conversion of the benzonitrile 10 to the cyclohexadiene 21 might arise by faster electrophilic trapping by 19 compared to the rate of trapping by the lithiated nitrile to give 20 (Scheme 5). The very efficient formation of 13k (99% yield) obtained by protonating the putative σ-complex implies that the equilibrium lies strongly toward the side of the σ-complex because protonation is likely to be much faster than an equilibration between 18 and 19 (Scheme 5).
Computational analyses employing the local attachment energy were particularly beneficial in identifying the carbons most likely to be attacked in SNAr processes;35 the calculations point to a mechanism in which the formation of σ-complexes at aromatic sites substituted by hydrogen is faster than attack at a position substituted by a halogen. Calculations at the M06-2X/jun-cc-pVTZ level with an implicit SMD-PCM solvation correction for the attack of cyclopentanecarbonitrile anion on the para position of 2,6-difluorobenzonitrile show an exergonic attack passing through a transition structure TS1 with a free energy of 5.5 kcal/mol at −78 °C in THF solution (Figure 1, top left). Incorporating a lithium cation in the analogous calculation raises the energy of the transition structure considerably to 12.1 kcal/mol [TS1(Li+)], consistent with the putative lithium solvation for the experiments in diglyme (Figure 1, bottom left). The lithium cation is not destabilizing TS1 or the anionic intermediate per se but the barrier is lower because lithium binds more strongly to deprotonated cyclopentanecarbonitrile than to the TS; this is the result of the negative charge being more localized in the anion than in TS1.
Figure 1.

Stationary points for the dearomatization of 2,6-difluorobenzonitrile by deprotonated cyclopentylcarbonitrile followed by nucleophilic substitution with p-CF3benzyl bromide in THF at −78 °C. Free energies have been computed at the M06-2X/jun-cc-pVTZ level with the implicit SMD-PCM solvation correction. The lower potential surface is for complexes with Li+, where DG(Li+)Cmpl is the free energy of complexation between deprotonated cyclopentylcarbonitrile and Li+ in THF at −78 °C. The latter energy is very difficult to estimate accurately by computational means, because of a high dependence on the solvation energy of the bare Li+ ion, which varies strongly with the radius assigned to Li+.
Thus, the rate enhancing effect of diglyme derives from the favorable complexation to the lithium cation which activates the deprotonated nitrile toward attack on the π-cloud; the subsequent attack of the σ-complex onto p-CF3–C6H4CH2Br is calculated to be the most favorable through an electrophilic trajectory TS2 to the face opposite the cyclopentylcarbonitrile substituent, i.e. an SN2 type mechanism with the σ-complex as the nucleophile (Figure 1, top right).36 Noteworthy is that the second step was identified to be rate-determining with a computed activation free energy of 18.4 kcal/mol at −78 °C relative to the σ-complex and free p-CF3–C6H4CH2Br.
Calculations using the local electron attachment energy33 to predict the regioselective attack on the aryl nitriles are largely consistent with the observed addition–alkylations. For nitriles 10a and 10s, attack at the position para to the nitrile was predicted, whereas for 10f and 10l, attack at the positions ortho and para to the nitrile was calculated to be favorable (Figure 2); attack on the parent benzonitrile (10z) and 10q were predicted to occur at the ortho position, though experimentally 10z afforded minimal dearomatization–alkylation under all the conditions examined. Potentially, the attack preference as predicted by the electron attachment energy is accurate, but if alkylation of the nitrile-stabilized cyclohexadienyl anion is rate-determining, then the regioselectivity is independent of the initial ortho or para attack on the aromatic (cf. Scheme 5). Consequently, the reaction pathways for forming cyclohexadiene from 10l by initial attack at both the ortho and para positions were computed to provide a point of comparison with 10a.
Figure 2.

Arylnitriles evaluated with the local electron attachment energy (ES(r)) to determine the addition regioselectivity. The asterisk marks the preferred position(s) for nucleophilic attack according to ES(r). The figure on the right shows ES(r) on the molecular surface of 10a. The lowest ES(r) (red) is found at the para-position, which is also the position of the initial attack during the dearomatization. More information about the predictions of the ES(r) calculations are found in Table S1.
Table 2 reports the relative free energies at −78 °C of the stationary points along the potential energy surfaces of 10l together with the corresponding energies for 10a. The activation free energy of the initial attack of 10l is of similar magnitude for the ortho and para pathways, but the latter is slightly favored, i.e., by 0.8 kcal/mol. In contrast, the σ-complex resulting from attack at the para position is stabilized by 2.2 kcal/mol relative to the ortho σ-complex. The stabilization is the result of a more favorable solvation of the para σ-complex; the solvation energy of the para σ-complex is lower than that of the ortho σ-complex by 4.8 kcal/mol. The difference in solvation energy is reduced to 2.7 kcal/mol for the rate determining TS2, a difference that can only partially explain the lower free energy (−4.7 kcal/mol) of TS2 for the para pathway compared to the ortho pathway. The origin of the additional stabilization of TS2 in the para pathway by 2 kcal/mol is difficult to deduce but is speculated to stem from the increased steric compression for conversion of the ortho sp2 hybridized carbon into an sp3 center in the resulting σ-complex.
Table 2. Computed Relative Free Energies (in kcal/mol) of the Stationary Points for the Dearomatization of 10 Followed by Nucleophilic Substitution with p-CF3benzylbromide to Form 13 in THF at −78 οC.
| entry | EWG | position | TS1 | int | TS2 | product |
|---|---|---|---|---|---|---|
| 10a | 2,6-diF | para | 5.5 | –13.7 | 4.7 | –29.0 |
| 10l | 2-Me | ortho | 12.0 (12.3)a | –0.7 | 15.1 | –23.4 |
| 10l | 2-Me | para | 11.2 | –2.9 | 10.4 | –27.9 |
The energy in parentheses refers to a TS conformer that will proceed directly to the lowest energy intermediate. The lowest energy TS proceeds to a conformer of higher energy, and the barrier of the conformational transition between the intermediate structures have not been computed.
The regioselectivity of the dearomatizations was probed with a series of monofluorobenzonitriles. Addition of lithiated cyclopentanecarbonitrile to o-fluorobenzonitrile and trapping with BnBr afforded 13n (Scheme 3), whereas p-fluorobenzonitrile 10b afforded 23, the product of anionic SNAr (Scheme 6A); no dearomatization was observed with m-fluorobenzonitrile. Collectively, activation with an ortho or para fluorine substituent appears to be required, consistent with an SNAr process, whereas the absence of a reaction with m-fluorobenzonitrile implies that an SET mechanism is unlikely. Probing the potential for SET-generated radical intermediates by performing the addition of lithiated cyclopentanecarbonitrile with 2,6-difluorobenzonitrile in the presence of TEMPO, and separately with diphenylethylene, decreased the yield from 91 to 48% (see 13a in Table 1) but did not completely suppress the reaction.37
Scheme 6. Mechanistic Experiments.

A related reaction with lithiated cyclopentanecarbonitrile and p-methoxybenzonitrile (10c) gave only nitrile 12b arising from alkylation and no dearomatization, consistent with a more difficult attack at the para position (Scheme 6B). Consequently, benzylation of the lithiated nitrile becomes faster, leading to only formation of 12b.
Conclusions
Lithiated nitriles add to variously substituted benzonitriles, triggering an unusual interrupted dearomatization–alkylation to afford complex cyclohexadienes. Use of diglyme as the solvent is particularly effective in facilitating the dearomatization which correlates with encapsulation of the lithium cation, thereby creating a potent nucleophile that readily disrupts the aromaticity. Calculations and mechanistic experiments indicate that the lithiated nitrile addition establishes an equilibrium between lithiated nitrile and a σ-complex. Subsequent rate-determining trapping of the σ-complex is efficient and highly diastereoselective with activated electrophiles, such as benzyl bromide and allyl bromide. The facile dearomatization of readily accessible arylnitriles makes the process potentially valuable for rapidly accessing complex cyclohexadienes.
Methods
General Dearomatization Procedure
Neat benzonitrile (1.0 equiv) was rapidly added to a −78 °C, THF solution (0.1 M) of the lithiated nitrile [prepared by deprotonating the nitrile (2.0 equiv) with LDA (2.1 equiv)] or the lithiated isocyanide [prepared by adding BuLi (1.1 equiv) to a −78 °C, THF solution (0.1 M) of the isocyanide]. After 1 h at −78 °C for the lithiated nitrile or 60 s for the lithiated isocyanide, a neat electrophile (2.1 equiv) was rapidly added. The cooling bath was removed and after 1 h, saturated, aqueous NH4Cl was added, the phases were separated, and the aqueous phase was extracted with CH2Cl2 (3 × 25 mL). The combined organic extract was dried (Na2SO4) and concentrated. The crude cyclohexadiene was dry loaded onto Celite and purified by automated flash column chromatography on silica gel using a Reveleris X2 purification system with 0–10% EtOAc/hexanes as the mobile phase.
Acknowledgments
Financial support from NSF (#1953128) is gratefully acknowledged. HRMS analyses conducted by Drexel University staff Timothy P. Wade, Andrew Greene, and Hannah Palmer, and Dr. Chuck Ross at the University of Pennsylvania are gratefully acknowledged. C.A. acknowledges NSF (#CHE-2215854) for support of crystallographic instrumentation and T.B. acknowledges support from the Swedish Research Council (VR), grant number 2021-05881.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacsau.3c00813.
Author Contributions
‡ B.A. and J.-P.R.M. contributed equally. T.B. for computational analysis. The manuscript was written through contributions of all authors who have given approval to the final version of the manuscript. CRediT: Bilal Altundas formal analysis, investigation, writing-review & editing; John-Paul R. Marrazzo investigation, methodology, writing-review & editing; Tore Brinck conceptualization, software, validation, writing-review & editing; Christopher Absil data curation, formal analysis, resources, visualization; Fraser F. Fleming conceptualization, data curation, formal analysis, funding acquisition, investigation, methodology, project administration, resources, writing-original draft, writing-review & editing.
The authors declare no competing financial interest.
Supplementary Material
References
- Industrial Arene Chemistry: Markets, Technologies, Sustainable Processes and Cases Studies of Aromatic Commodities; Mortier J., Ed.; Wiley VCH: Weinheim, Germany, 2023. [Google Scholar]
- López Ortiz F.; Iglesias M. J.; Fernández I.; Andújar Sánchez C. M.; Ruiz Gómez G. Nucleophilic Dearomatizing (DNAr) Reactions of Aromatic C,H-Systems. A Mature Paradigm in Organic Synthesis. Chem. Rev. 2007, 107, 1580–1691. 10.1021/cr030207l. [DOI] [PubMed] [Google Scholar]
- Roche S. P.; Porco J. A. Jr. Dearomatization Strategies in the Synthesis of Complex Natural Products. Angew. Chem., Int. Ed. 2011, 50, 4068–4093. 10.1002/anie.201006017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma U. K.; van der Ranjan P.; Van der Eycken E. V.; You S.-L. Sequential and direct multicomponent reaction (MCR)-based dearomatization strategies. Chem. Soc. Rev. 2020, 49, 8721–8748. 10.1039/d0cs00128g. [DOI] [PubMed] [Google Scholar]
- Wengryniuk S. E.; Xiao X. Recent Advances in the Selective Oxidative Dearomatization of Phenols to o-Quinones and o-Quinols with Hypervalent Iodine Reagents. Synlett 2021, 32, 752–762. 10.1055/s-0037-1610760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemière G.; Clayden J. Dearomatization Reactions Using Organolithiums. Sci. Synth. 2011, 4, 139–190. [Google Scholar]
- Zimmerman H. E. A Mechanistic Analysis of the Birch Reduction. Acc. Chem. Res. 2012, 45, 164–170. 10.1021/ar2000698. [DOI] [PubMed] [Google Scholar]
- a Cheng Y. Z.; Feng Z.; Zhang X.; You S.-L. Visible-light induced dearomatization reactions. Chem. Soc. Rev. 2022, 51, 2145–2170. 10.1039/c9cs00311h. [DOI] [PubMed] [Google Scholar]; b Burrows J.; Kamo S.; Koide K. Scalable Birch reduction with lithium and ethylenediamine in tetrahydrofuran. Science 2021, 374, 741–746. 10.1126/science.abk3099. [DOI] [PubMed] [Google Scholar]; c Lv S.; Zhang G.; Chen J.; Gao W. Electrochemical Dearomatization: Evolution from Chemicals to Traceless Electrons. Adv. Synth. Catal. 2020, 362, 462–477. 10.1002/adsc.201900750. [DOI] [Google Scholar]
- Terrier F.Modern Nucleophilic Aromatic Substitution; Wiley VCH: Weinheim, 2013. [Google Scholar]
- Lennox A. J. J. Meisenheimer Complexes in SNAr Reactions: Intermediates or Transition States?. Angew. Chem., Int. Ed. 2018, 57, 14686–14688. 10.1002/anie.201809606. [DOI] [PubMed] [Google Scholar]
- Kwan E. E.; Zeng Y.; Besser H. A.; Jacobsen E. N. Concerted nucleophilic aromatic substitutions. Nat. Chem. 2018, 10, 917–923. 10.1038/s41557-018-0079-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Okumura M.; Nakamata Huynh S. M.; Pospech J.; Sarlah D. Arenophile-Mediated Dearomative Reduction. Angew. Chem., Int. Ed. 2016, 55, 15910–15914. 10.1002/anie.201609686. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Butters M.; Beetstra D. J.; Elliott M. C.; Hill-Cousins J.; Kariuki B. M. Directed epoxidation of cyclohexa-1,4-dienes–stereoselective formation of up to six contiguous stereogenic centres. Org. Biomol. Chem. 2008, 6, 4426–4434. 10.1039/b814131m. [DOI] [PubMed] [Google Scholar]; c Ikeuchi K.; Ido S.; Yoshimura S.; Asakawa T.; Inai M.; Hamashima Y.; Kan T. Catalytic Desymmetrization of Cyclohexadienes by Asymmetric Bromolactonization. Org. Lett. 2012, 14, 6016–6019. 10.1021/ol302908a. [DOI] [PubMed] [Google Scholar]
- a Elliott M. C.; El Sayed N. N. E.; Paine J. S. Concise synthesis of the tricyclic core of lycoposerramine S. Org. Biomol. Chem. 2008, 6, 2611–2618. 10.1039/b804664f. [DOI] [PubMed] [Google Scholar]; b Ivkovic A.; Matovic R.; Saicic R. N. Ring-Closing Metathesis/Fragmentation Route to Geometrically Defined Medium-Ring Cycloalkenes: Total Synthesis of (±)-Periplanone C. Org. Lett. 2004, 6, 1221–1224. 10.1021/ol049875z. [DOI] [PubMed] [Google Scholar]; c Callier-Dublanchet A.-C.; Cassayre J.; Gagosz F.; Quiclet-Sire B.; Sharp L. A.; Zard S. Z. Amidyls in radical cascades. The total synthesis of (±)-aspidospermidine and (±)-13-deoxyserratine. Tetrahedron 2008, 64, 4803–4816. 10.1016/j.tet.2008.02.107. [DOI] [Google Scholar]; d Trullinger T. K.; Qi J.; Roush W. R. Studies on the Synthesis of Quartromicins A3 and D3: Synthesis of the Vertical and Horizontal Bis- Spirotetronate Fragments. J. Org. Chem. 2006, 71, 6915–6922. 10.1021/jo061059c. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Schultz A. G.; Macielag M.; Sundararaman P.; Taveras A. G.; Welch M. An enantioselective method for reductive alkylation of aromatic carboxylic acid derivatives. Examination of the factors that provide stereoselectivity. J. Am. Chem. Soc. 1988, 110, 7828–7841. 10.1021/ja00231a038. [DOI] [Google Scholar]
- Makosza M. Nucleophilic substitution of hydrogen in electron-deficient arenes, a general process of great practical value. Chem. Soc. Rev. 2010, 39, 2855–2868. 10.1039/b822559c. [DOI] [PubMed] [Google Scholar]
- Makosza M.; Staliński K. Oxidative Nucleophilic Substitution of Hydrogen in Nitroarenes. Chem.—Eur. J. 1997, 3, 2025–2031. 10.1002/chem.19970031217. [DOI] [Google Scholar]
- Yang X.; Fleming F. F. C- and N-Metalated Nitriles: The Relationship Between Structure and Selectivity. Acc. Chem. Res. 2017, 50, 2556–2568. 10.1021/acs.accounts.7b00329. [DOI] [PubMed] [Google Scholar]
- Richard J. P.; Williams G.; Gao J. Experimental and Computational Determination of the Effect of the Cyano Group on Carbon Acidity in Water. J. Am. Chem. Soc. 1999, 121, 715–726. 10.1021/ja982692l. [DOI] [Google Scholar]
- Schultz A. G.; Macielag M.; Sundararaman P.; Taveras A. G.; Welch M. An enantioselective method for reductive alkylation of aromatic carboxylic acid derivatives. Examination of the factors that provide stereoselectivity. J. Am. Chem. Soc. 1988, 110, 7828–7841. 10.1021/ja00231a038. [DOI] [Google Scholar]
- Altundas B.; Alwedi E.; Song Z.; Gogoi A. R.; Dykstra R.; Gutierrez O.; Fleming F. F. Dearomatization of Aromatic Asmic Isocyanides to Complex Cyclohexadienes. Nat. Commun. 2022, 13, 6444. 10.1038/s41467-022-33807-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subsequent experiments demonstrated an optimal aging time of ten minutes in diglyme before the addition of the electrophile (cf. One h for THF). Determined by monitoring the formation of the dienyl anion by 19F NMR.
- Attempted trapping with diphenyldisulfide, methyl cyanoformate, benzophenone, and benzaldehyde only afforded products from alkylation with the lithiated nitrile.
- Schultz A. G.; Macielag M. Birch Reduction and Reductive Alkylation of Benzonitriles and Benzamides. J. Org. Chem. 1986, 51, 4983–4987. 10.1021/jo00375a043. [DOI] [Google Scholar]
- Attempted addition of lithiated cyclopentanecarbonitrile to 1-(butylsulfonyl)-2-fluorobenzene and 1-fluoro-4-nitrobenzene were not successful.
- Weiler L. Alkylation of the dianion of β-keto esters. J. Am. Chem. Soc. 1970, 92, 6702–6704. 10.1021/ja00725a088. [DOI] [Google Scholar]
- Fleming F. F.; Altundas B.. Asmic, Anisylsulfanylmethylisocyanide; eEROS, 2020.
- The reactions were found to be significantly more efficient in THF than diglyme.
- Błaziak K.; Danikiewicz W.; Mąkosza M. How Does Nucleophilic Aromatic Substitution Really Proceed in Nitroarenes? Computational Prediction and Experimental Verification. J. Am. Chem. Soc. 2016, 138, 7276–7281. 10.1021/jacs.5b13365. [DOI] [PubMed] [Google Scholar]
- Makosza M. Nucleophilic substitution in nitroarenes: a general corrected mechanism. ChemTexts 2019, 5, 10. 10.1007/s40828-019-0084-5. [DOI] [Google Scholar]
- Altundas B.; Marrazzo J.-P. R.; Fleming F. F. Metalated Isocyanides: Formation, Structure, and Reactivity. Org. Biomol. Chem. 2020, 18, 6467–6482. 10.1039/D0OB01340D. [DOI] [PubMed] [Google Scholar]
- Makosza M.; Winiarski J. Reaction of organic anions. 96. Vicarious substitution of hydrogen in aromatic nitro compounds with acetonitrile derivatives. J. Org. Chem. 1980, 45, 1534–1535. 10.1021/jo01296a045. [DOI] [Google Scholar]
- Rohrbach S.; Smith A. J.; Pang J. H.; Poole D. L.; Tuttle T.; Chiba S.; Murphy J. A. Concerted Nucleophilic Aromatic Substitution Reactions. Angew. Chem., Int. Ed. 2019, 58, 16368–16388. 10.1002/anie.201902216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pliego Jr J. R. Jr.; Pilo-Veloso D. Effects of ion-pairing and hydration on the SNAr reaction of the F–with p-chlorobenzonitrile in aprotic solvents. Phys. Chem. Chem. Phys. 2008, 10, 1118–1124. 10.1039/b716159j. [DOI] [PubMed] [Google Scholar]
- a Rogachev A. Y.; Alkan M.; Li J.; Liu S.; Spisak S. N.; Filatov A. S.; Petrukhina M. A. Mono-reduced Corannulene: To Couple and Not to Couple in One Crystal. Chem.—Eur. J. 2019, 25, 14140–14147. 10.1002/chem.201902992. [DOI] [PubMed] [Google Scholar]; b Michel R.; Herbst-Irmer R.; Stalke D. Revealing Coordination Patterns in C5-Cyclic Lithium Organics. Organometallics 2011, 30, 4379–4386. 10.1021/om200471e. [DOI] [Google Scholar]; c Iravani E.; Neumüller B. Reaktionen von Nitrilen mit tBuAsLi2. Z. Anorg. Allg. Chem. 2004, 630, 1811–1815. 10.1002/zaac.200400116. [DOI] [Google Scholar]
- Analogous experiments in THF and DME afforded 2.7:1 and 2.1:1 ratios, respectively, consistent with lower overall yields (cf Table 1, entry 7 with entries 4 and 6, respectively).
- Stenlid J. H.; Brinck T. Nucleophilic Aromatic Substitution Reactions Described by the Local Electron Attachment Energy. J. Org. Chem. 2017, 82, 3072–3083. 10.1021/acs.joc.7b00059. [DOI] [PubMed] [Google Scholar]
- The trajectory shown with the benzyl bromide underneath the σ-complex was found to be more favorable than an analogous exocyclic trajectory with the benzyl bromide rotated by 180°.
- An analogous reaction in THF in which lithiated cyclopentanecarbonitrile was reacted with 2-methoxybenzonitrile and TEMPO followed by the addition of BnBr reduced the yield from 78% to 25%.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.