

Abstract

All solid-state batteries (SSBs) are considered the most promising path to enabling higher energy-density portable energy, while concurrently improving safety as compared to current liquid electrolyte solutions. However, the desire for high energy necessitates the choice of high-voltage cathodes, such as nickel-rich layered oxides, where degradation phenomena related to oxygen loss and structural densification at the cathode surface are known to significantly compromise the cycle and thermal stability. In this work, we show, for the first time, that even in an SSB, and when protected by an intact amorphous coating, the LiNi0.5Mn0.3Co0.2O2 (NMC532) surface transforms from a layered structure into a rocksalt-like structure after electrochemical cycling. The transformation of the surface structure of the Li3B11O18 (LBO)-coated NMC532 cathode in a thiophosphate-based solid-state cell is characterized by high-resolution complementary electron microscopy techniques and electron energy loss spectroscopy. Ab initio molecular dynamics corroborate facile transport of O2– in the LBO coating and in other typical coating materials. This work identifies that oxygen loss remains a formidable challenge and barrier to long-cycle life high-energy storage, even in SSBs with durable, amorphous cathode coatings, and directs attention to considering oxygen permeability as an important new design criteria for coating materials.
Introduction
The rising demand for long-range, safe, electrified transportation has highlighted the need for further advancements in rechargeable Li-ion batteries.1−4 Currently, a promising technology to meet these goals is solid-state batteries (SSBs), which provide a safer solution to utilizing higher energy-density electrode materials. Such materials include a Li metal anode and a high-energy density cathode, often found in the family of Ni-rich layered cathodes LiNixMnyCo1–x–yO2 (NMC, x ≥ 0.5), owing to their high specific capacities and power densities. However, despite the many merits of Ni-rich layered cathodes, numerous studies have shown that when employed in liquid-electrolyte cells, at a high state of charge (SoC), electrochemical oxidation of oxygen and oxygen loss occur in the surface regions of the cathode, leading to destabilization of the transition metal cations at the surface.5−8 This destabilization, in turn, leads to surface densification from the initial layered structure (R3̅m) into an undesirable rocksalt-like (Fm3̅m) or spinel-like (Fd3̅m) structure.7−10 The resulting structural degradation and interfacial mismatch with the underlying bulk material degrade lithium transport, causing impedance build-up, bulk fatigue,1 and poor high-voltage cycling performance.
Indeed, the formation of a structural reconstruction layer (a reduced surface transition-metal layer) has been reported even after simply exposing NMC materials to LiPF6-based organic electrolytic solutions.11 The most prevalent strategy to protect the cathode surface in traditional liquid electrolyte cells is to coat the cathode with a protective “buffer” layer.12−14 This layer typically comprises a solid ceramic15−20 and to date, various surface coatings, including metal oxides (e.g., Al2O3, ZnO, Ta2O5),16,17,21,22 metal fluorides (e.g., AlF3),19,20 and metal phosphates (e.g., LaPO4)17 have been shown to enhance the cyclability of NMC materials. However, recently, Zheng et al. reported that although the application of an AlF3 coating on Li1.2Ni0.15Co0.10Mn0.55O2 was found to protect the cathode surface from severe etching/corrosion and alleviated or delayed the surface reconstruction of the cathode compared with that without the coating, phase transformation from the layered to spinel-like structure still occurred after extended cycling.19 Similarly, Croy et al.23 reported that the use of an atomic layer-deposited Al2O3 coating as a physical barrier on a Ni-rich cathode was insufficient to overcome its surface instabilities. It is fair to conclude that cathode oxygen loss still occurs in liquid electrolyte cells and that the released oxygen may enhance electrolyte degradation, leading to the formation of high-impedance surface layers. Surprisingly, researchers have recently shown that similar surface structure reconstruction and transition metal reduction can be probed on a bare Ni-rich layered cathode when employed in a thiophosphate-based solid-electrolyte environment.24 Ultimately, the final question is whether this surface structure rearrangement can be prohibited with the introduction of a stable, state-of-the-art surface coating25 in SSBs.
In this work, we used high-resolution scanning transmission electron microscopy (STEM), transmission electron microscopy (TEM), and electron energy loss spectroscopy (EELS) complemented with ab initio molecular dynamics (AIMD) to investigate the surface structure evolution of a coated Ni-rich layered cathode in a thiophosphate-based SSB environment. An amorphous Li3B11O18 (LBO) coating was selected as an example because, in previous work, it was observed and computed to be chemically/electrochemically stable even after extended cycling in an SSB.26 In this work, we show that even when protected by an intact amorphous surface coating, the surface region of LiNi0.5Mn0.3Co0.2O2 (NMC532) transforms from a layered structure into a rocksalt-like structure after electrochemical cycling in a SSB. More importantly, we demonstrate the essential role of O2– transport in the ceramic coating. To date, surface coatings have been selected based on their facile Li-ion transport, low electron transport, chemical compatibility with both the SE and cathode, and electrochemical stability; however, oxygen transport in the coating material has never been considered as a criterion in their selection. We emphasize that if the surface coating exhibits a facile O2– transport, the cathode can still lose oxygen, densify, and increase its impedance, even when coated by an amorphous surface coating. Using AIMD simulations, we systematically evaluated the O2– diffusion rate in amorphous LBO as well as in other typical coating materials reported in the literature. The O2– flux estimated using the Onsager transport models reveals facile transport of O2– through amorphous LBO, rationalizing our observation that a rocksalt structure still forms on the surface of the NMC532 cathode in a solid-state cell. In summary, we provide the first evidence of the formation of a surface-reduced layer in NMC532 in the presence of an intact, amorphous oxide coating after cycling in a thiophosphate-based SSB. Our work highlights the importance of designing coating materials with a low O2– diffusivity to mitigate cathode degradation, even in SSBs.
Methods
Material Synthesis and Coating Method
The Li3B11O18 (LBO)-coated LiNi0.5Mn0.3Co0.2O2 (NMC532) samples were prepared by Samsung Research Japan using the sol–gel method reported in previous work.26 For the LBO-coated NMC532 sample (0.5 mol % LBO coated NMC), the LBO coating solution was first prepared by dissolving a stoichiometric amount of Li acetate and triisopropyl borate in super dehydrated ethanol at 60 °C. Next, the NMC532 powder was dispersed in the as-prepared coating solution with stirring. Then, the solvent from the flask was removed using a rotary evaporator in a hot water bath (60 °C) with ultrasound sonication followed by a heat treatment at 350 °C in air. The 75Li2S–25P2S5 (LPS) was synthesized by ball-milling stoichiometric amounts of Li2S (99.98% Sigma-Aldrich) and P2S5 (99% Sigma-Aldrich) in a 50 mL ZrO2 jar for 200 min using a SPEX 8000 M mixer mill. The LPS with a small particle size used in the composite cathode was prepared by wet ball milling the LPS solid electrolyte (SE) with heptane and dibutyl ether using a Retsch PM200 ball mill for 40 h. All of the LPS synthesis steps were conducted in an Ar atmosphere.
Cell Fabrication and Electrochemical Characterization
SSBs were fabricated in an Ar-filled glovebox with H2O < 2 ppm and O2 < 0.1 ppm. The LPS, uncoated or LBO-coated NMC532, and graphite were used as the SE, cathode, and anode active materials, respectively. The cathode and anode composites were prepared by hand mixing 60% active material, 35% LPS, and 5% CNF. The full cells were assembled using an in-house-designed pressure cell (13 mm inner diameter). First, 100 mg of LPS powder was added to the cell and cold pressed under ∼100 MPa pressure. Next, 10 mg of the cathode composite powders and anode composite powders were carefully spread evenly on the top and bottom sides of the LPS pellet, respectively. A pressure of ∼350 MPa was applied for 5 min to compact the cell and ensure good interfacial contact between the different components.27−29
All the electrochemical tests were conducted at room temperature under an Ar atmosphere, and a pressure of 5 MPa was applied to the cells during cycling. The full SSBs were cycled at a constant current (0.1 mA cm–2 for charge and 0.05 mA cm–2 for discharge) between 2.5 and 4.3 V versus Li/Li+.
Electron Microscopy Experiments
The TEM sample preparation was conducted in an Ar-filled glovebox. The LBO-coated cathodes were extracted from the disassembled cells after cycling. The cycled and uncycled composite cathode powder samples were diluted in hexane and sonicated to obtain good particle dispersion. The TEM samples were prepared by drop casting the solution onto a copper mesh TEM grid with an ultrathin carbon and lacey carbon support. The TEM grids were transferred from the glovebox into the microscope under an inert Ar atmosphere using a Gatan 648 double-tilt vacuum-transfer holder. The high-resolution STEM and EELS analyses were performed using the TEAM I microscope (a modified FEI Titan 80–300 microscope with a double-aberration-corrected (scanning) transmission electron microscope) with an accelerating voltage of 300 kV.
For the EELS analysis, a power law background subtraction was performed for the Mn L3/L2 edge. Then, the multiple scattering was then removed by Fourier-ratio deconvolution using the low-loss spectrum obtained from the same scanning region using the dual EELS mode. To quantify the L3/L2 intensity ratio, a step function threshold with the ratio of step heights of 2:1 was first applied in the background subtraction to account for the multiplicity of the 3p1/2 and 3p3/2 initial states.30 After the step function subtraction, the L3/L2 intensity ratios were determined by area integration beneath the L3 and L2 peaks.
Density Functional Theory Methods
All the density functional theory (DFT) electronic structure calculations were performed using the Vienna Ab initio Simulation Package31 with projector-augmented wave potentials.32 The Perdew–Burke–Ernzerhof generalized gradient approximation functional was adopted for the exchange–correlation functional.33 For the AIMD simulations, we used Γ-point-only Brillouin zone integration at a plane-wave cutoff energy of 400 eV and a time step of 2 fs.
To generate the amorphous structures, we employed a “liquid-quench” process in which heating, equilibration, and quenching were performed using an AIMD workflow, which can be found as part of the open-source mpmorph package at http://github.com/materialsproject/mpmorph. We used the Packmol package34 to generate the initial amorphous structures. To generate the “liquid” phase of the amorphous structures, the structures were “heated” at 3000 K, and a sequence of 4 ps AIMD simulations in the NVT ensemble was performed until the external pressure and energy converged. Then, the equilibrated “liquid” amorphous structures were simulated for an additional 10 ps, from which three independent configurations were selected, i.e., 3.3 ps apart from each other, and quenched to 0 K to obtain three, representative amorphous structures to exemplify varying atomic environments. The radial distribution functions of B–O, Al–O, and Nb–O pairs in Li3B11O18, LiAlO2, and LiNbO3, respectively, at 0 K are plotted in Figure S1, which illustrates the distinct ordering behavior between amorphous and crystalline structures. For each configuration, we equilibrated the structures at T = 1800, 2000, 2200, 2400, 2600, and 2800 K, respectively, following the same procedure of obtaining the “liquid” amorphous structures mentioned above and then simulated an 80 ps diffusion trajectory at each corresponding temperature. Therefore, from the three representative amorphous structures, there are three independent diffusion trajectories at each corresponding temperature. Further details about the AIMD and DFT workflows can be found in ref.13
From the obtained
diffusion trajectories, we calculated the self-diffusion
coefficients (D) of Li+ and O2– ions using the Einstein relation: 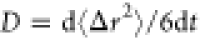 , where t, r, and
, where t, r, and  are the time, ion position, and mean square
displacement (MSD) that is averaged over all relevant ions (Li+ or O2– ions), respectively. Figure S2 illustrates
the MSD of the Li+ and O2– ions in amorphous
Li3B11O18, LiAlO2, and
LiNbO3. From this data, we obtained the D values at T = 1800, 2000, 2200, 2400, 2600, and
2800 K. At each temperature, there are three D values
calculated from three independent AIMD diffusion trajectories obtained
earlier for each representative amorphous structure. Figure S3 illustrates the density of the amorphous Li3B11O18 structures versus the oxygen
diffusivity (DO). As expected, higher
temperatures lead to a lower density due to an expanded volume, which
in turn leads to an increase in oxygen diffusivity. The room-temperature Drt were extrapolated from the D values at high temperatures using the Arrhenius relation, D = D0 exp(−Ea/kBT), where kB is the Boltzmann constant, D0 is the pre-exponential factor, and Ea is the activation energy of ion diffusion. D0 and Ea were determined by
fitting log D vs 1/T as the Arrhenius
relation. Compared with the diffusion trajectories at higher temperatures
(e.g., 2800 K), the trajectories at lower temperatures (e.g., 1800
K) typically exhibit fewer ion hops, thus yielding fitted D values with higher statistical uncertainty. Therefore,
we considered the statistical uncertainty of the D value at each temperature when fitting the Arrhenius relation by
assigning the standard deviation of log D as the
uncertainty for each averaged D. In this study, the
Arrhenius relation was fitted by the curve_fit function
in the SciPy package.35 It should be noted
that pure B2O3 glass belongs to a strong glassformer,
and its fragility increases with Li2O content.36−38 Therefore, even though the temperature-dependent D in Li3B11O18 follows an Arrhenius-like
relation at high temperatures, such as near the glass-transition temperature,
it may not follow the same Arrhenius relation at low temperatures,
such as room temperature. As a result, our estimated Li+ and O2– diffusivities in Li3B11O18 could be overestimated. In addition, we fitted the
temperature-dependent diffusivities with a non-Arrhenius Vogel–Fulcher–Tammann
(VFT) equation39 (see Figure S4),
are the time, ion position, and mean square
displacement (MSD) that is averaged over all relevant ions (Li+ or O2– ions), respectively. Figure S2 illustrates
the MSD of the Li+ and O2– ions in amorphous
Li3B11O18, LiAlO2, and
LiNbO3. From this data, we obtained the D values at T = 1800, 2000, 2200, 2400, 2600, and
2800 K. At each temperature, there are three D values
calculated from three independent AIMD diffusion trajectories obtained
earlier for each representative amorphous structure. Figure S3 illustrates the density of the amorphous Li3B11O18 structures versus the oxygen
diffusivity (DO). As expected, higher
temperatures lead to a lower density due to an expanded volume, which
in turn leads to an increase in oxygen diffusivity. The room-temperature Drt were extrapolated from the D values at high temperatures using the Arrhenius relation, D = D0 exp(−Ea/kBT), where kB is the Boltzmann constant, D0 is the pre-exponential factor, and Ea is the activation energy of ion diffusion. D0 and Ea were determined by
fitting log D vs 1/T as the Arrhenius
relation. Compared with the diffusion trajectories at higher temperatures
(e.g., 2800 K), the trajectories at lower temperatures (e.g., 1800
K) typically exhibit fewer ion hops, thus yielding fitted D values with higher statistical uncertainty. Therefore,
we considered the statistical uncertainty of the D value at each temperature when fitting the Arrhenius relation by
assigning the standard deviation of log D as the
uncertainty for each averaged D. In this study, the
Arrhenius relation was fitted by the curve_fit function
in the SciPy package.35 It should be noted
that pure B2O3 glass belongs to a strong glassformer,
and its fragility increases with Li2O content.36−38 Therefore, even though the temperature-dependent D in Li3B11O18 follows an Arrhenius-like
relation at high temperatures, such as near the glass-transition temperature,
it may not follow the same Arrhenius relation at low temperatures,
such as room temperature. As a result, our estimated Li+ and O2– diffusivities in Li3B11O18 could be overestimated. In addition, we fitted the
temperature-dependent diffusivities with a non-Arrhenius Vogel–Fulcher–Tammann
(VFT) equation39 (see Figure S4),  , where T0 is
a temperature that is ∼50 K below the glass-transition temperature.
However, because of the time limitations of AIMD simulations in calculating
the diffusivities of Li3B11O18 at
low temperatures, i.e., 400–800 K, we are not able to obtain
the fitted VFT equations with sufficient accuracy.
, where T0 is
a temperature that is ∼50 K below the glass-transition temperature.
However, because of the time limitations of AIMD simulations in calculating
the diffusivities of Li3B11O18 at
low temperatures, i.e., 400–800 K, we are not able to obtain
the fitted VFT equations with sufficient accuracy.
To estimate the ionic flux under the driving force of the chemical potential gradient across the coating layer, we applied the Onsager transport equation:
| 1 |
where Ji, Lii, and ∇μi are the flux, Onsager transport coefficient, and chemical potential gradient of species i, respectively. It should be noted that in this study, we ignore the contributions from cross-correlations between two distinct species, such as LOLi, and between distinct sites, such as LOOdistinct, to JO. Assuming steady-state conditions, we can reasonably approximate the chemical potential gradient ∇μi to be a constant throughout the thickness of the coating, which renders the above equation as
| 2 |
where μic and μie are the chemical potentials at the cathode and electrolyte sides, respectively. Ignoring the Liidistinct term, Lii can be directly related to the self-diffusion coefficient Di:
| 3 |
where ci is the concentration of species i in the coating. The room temperature Liirt is extrapolated from Lii values at high temperatures by fitting eq 3.
In this work, we estimate the time t required for O2– to diffuse through the LBO coating in order to reach the observed surface rocksalt phase. We assume the surface rocksalt layer of the LBO-coated NMC532 mainly consists of NiO phase, which results from the surface NiO2 to lose 50% of its oxygen. Let cOmax denote the upper bound value of the O2– concentration in NiO2, and t can be calculated as
| 4 |
where A and V are the surface area of the LBO-coated NMC532 cathode particle and the shell volume of the surface rocksalt phase,
respectively, and can be expressed as  , A = 4π(r + lc)2, where r is the radius of the cathode particle, ls is the thickness of the rocksalt phase, and lc is the coating thickness. Combining eqs 2 and 4, we obtain
, A = 4π(r + lc)2, where r is the radius of the cathode particle, ls is the thickness of the rocksalt phase, and lc is the coating thickness. Combining eqs 2 and 4, we obtain
| 5 |
As r ≫ ls, lc, the first term in eq 5 can be considered to be a constant for a given r. Therefore, the time t required to transport the same amount of O2– through the LBO coating is mainly governed by the values of lc and ∇μO. Hence, we consider a range of conditions to estimate t by varying the lc and ∇μO.
Results and Discussion
Appearance of a Surface-Reduced Layer in LBO-Coated NMC upon Cycling
We investigated LBO-coated LiNi0.5Mn0.3Co0.2O2 (LBO-NMC532) from uncycled and cycled cathode composites (Figures S5–S10). The cathode composites were prepared by hand mixing 60% LBO-NMC, 35% 75Li2S–25P2S5 (LPS), and 5% CNF, with the cycled material undergoing 10 charge–discharge cycles from 2.5 to 4.3 V versus Li. The composite cathode material was then diluted in hexane and sonicated to obtain a good particle dispersion before drop casting on a TEM grid.
High-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) images of the uncycled LBO-NMC along the [1–10] zone axis are presented in Figure 1a,c. The bright and dark regions in the HAADF-STEM images correspond to atomic columns of transition-metal cations and Li ions, respectively. Despite coming into contact with the amorphous LPS solid electrolyte (Figure S11), the HAADF-STEM image and corresponding fast-Fourier transform (FFT) image (Figure 1b) indicate that the uncycled nanoscale LBO-coated NMC maintains a well-defined R3̅m layered structure with separated transition metal and lithium sites in both the edge and bulk of the NMC particle (Figure S9).
Figure 1.

Structural characterization of the uncycled Li3B11O18-coated LiNi0.5Mn0.3Co0.2O2 cathode composite. a, HAADF-STEM image of pristine LBO-NMC cathode composite. b, FFT image obtained from the selected surface region, showing the layered rhombohedral R3̅m structure along the [1–10] zone axis. c, Enlarged HAADF-STEM image of the surface region, confirming the well-defined layered structure prior to cycling. The inset image shows the ball models of NMC532 viewing along [1–10]. The red, blue, and yellow balls represent mixed transition metal (Ni, Co, Mn), O and Li atoms. The d, e, EELS line scan spectra of (d) the O–K edge and (e) the Mn, Co, and Ni L-edge collected on pristine LBO-NMC cathode composite along the scanning pathway shown in the HAADF-STEM image in (f).
Depth-profiled EELS line scan spectra collected on the surface of LBO-NMC from the cathode composite were used to probe the surface and bulk electronic structure of the uncycled LBO-NMC. The oxygen K-edge spectra shown in Figure 1d show a characteristic40,41 pre-edge feature onset at ∼525 eV alongside the main edge at 532 eV, which corresponds to the promotion of O 1s electrons to hybridized TM3d-O 2p and TM4sp-O 2p orbitals.42 The location and shape of these K-edge features are consistent from the surface to the bulk of the NMC, as are the L edges of the transition metals (Mn, Co, and Ni). These features suggest that the average oxidation states of the transition metal cations remain unchanged in the uncycled LBO-NMC after exposure to the solid-electrolyte thiophosphate environment in the composite cathode.
A similar analysis of cycled LBO-NMC reveals the presence of a thick rocksalt-like surface layer in the NMC (Figure 2a), despite the nanoscale LBO amorphous coating (Figures 2b, S10, and S12). High-resolution HAADF-STEM imaging (Figure 2a–c) shows the atomic cation arrangement on the surface of the cycled LBO-NMC along the [100] zone axis of the bulk layered rhombohedral R3̅m structure. Even with the protection of the LBO coating, cation mixing within a few atomic layers (∼2 nm) is readily observed in the surface region of the cycled LBO-NMC (Figures S8 and S10). FFT analysis of the surface of the LBO-NMC after electrochemical cycling confirms that the surface region consists primarily of an Fm3̅m rock-salt-structured phase (Figure 2d). In addition, the STEM-EELS characterization of the cycled cathode composite confirms a fairly uniform distribution of oxygen in the SE after 10 cycles (Figure S13). Surface reconstruction of the cathode and a change in the oxidation state of the transition metal are observed after cycling (Figures 2a–h, S8, and S10), which supports the coating’s O2– permeability. The presence of oxygen is likely to oxidize Li3PS4, leading to the formation of a small amount of P–Ox species.43 Further evidence for the formation of a surface-reduced layer on the coated NMC is provided by EELS analysis of the cycled LBO-NMC (Figure 2e–h). As shown in the O K-edge spectra (Figure 2e), the pre-edge of the O K-edge in the surface region shows a significantly reduced intensity compared with that for the uncycled LBO-NMC or the inner region of the cycled NMC particle. Because the pre-edge intensity of the O K-edge strongly correlates with the density of available empty states in the hybridized metal 3d-O 2p orbitals in layered LiTMO2,42 the area under the oxygen pre-edge is generally found to show a linear correlation with the oxidation state of the TM (Mn, Co, Ni).44,45 Thus, the significantly decreased pre-edge intensity on the surface of the cycled LBO-NMC suggests reduced oxidation states of the transition metals in the surface region of the LBO-NMC. The shift toward higher energy of the peak positions in the TM L edge peaks (particularly the Mn and Co L3 peaks) at the surface (Figure 2f) also indicates reduced oxidation states near the surface of LBO-NMC after cycling. Quantitative analysis of the Mn L3/L2 edge intensity ratios in the cycled LBO-NMC shows that in the particle’s bulk, the valence of Mn is 4+, but is reduced to ∼2.2+ in the surface reconstruction region. The formation of a Fm3̅m rocksalt structure, the changes in the oxygen pre-edge intensities, and the shift of TM L-edge peaks in the HAADF-STEM and STEM-EELS profiles are all consistent with the presence of a densified surface area comprised of reduced cations on the LBO-coated layered NMC cathode after cycling.6−8,11
Figure 2.

Structural characterization of cycled Li3B11O18-coated LiNi0.5Mn0.3Co0.2O2. (a) HAADF-STEM image of LBO-NMC after cycling from 2.5 to 4.3 V versus Li for 10 cycles; (b) the same region displayed with false color for better visualization of the lithium borate thin-film coating. (c) Enlarged HAADF-STEM image of the surface region, showing the surface reconstruction layer. (d) FFT image obtained from the surface region, showing the Fm3̅m rocksalt structure along the [110] zone axis. (e, f) EELS line scan spectra of (e) O–K edge and (f) Mn, Co, and Ni L-edge collected on cycled LBO-NMC along the scanning pathway shown in (h). (g) L3/L2 intensity ratios of Mn L edges deduced from the EELS spectra in (f). h, HAADF-STEM image, showing the EELS line scan pathway.
Confirmation of Ample Oxygen Mobility in Amorphous Coatings
To validate our hypothesis that oxygen diffusion plays a role in surface densification, we used ab initio methods to evaluate whether sufficient oxygen mobility is present in the coating materials to allow oxygen to escape from the layered cathode surface. We investigated the ionic diffusivity, flux, and transport time through the coating material using AIMD simulations and Onsager transport models. Because of the amorphous nature of the LBO and the typical coatings, we specifically targeted amorphous coatings in our calculations. The equilibrated amorphous structures were generated using a “liquid-quench” process, which was simulated using a series of AIMD calculations, as described in the Methods. From the AIMD-calculated diffusion trajectories, we extracted the self-diffusion coefficients and estimated the O2– diffusion rate.
The room temperature self-diffusion coefficients of Li+ (DLi) and O2– (DO) in LBO are estimated by extrapolating the D values from high temperatures using the Arrhenius relation (Figure S14). For comparison, we also compute DLi and DO of two commonly used amorphous cathode coatings, LiAlO2 and LiNbO3, and include three previously calculated13 amorphous Al2O3, ZnO, and Li2O, see Figure 3. Figure S14 presents Arrhenius plots of the Li+ and O2– self-diffusivity D in Li3B11O18, LiAlO2, and LiNbO3 as a function of temperature T. We note that Al2O3, LiAlO2, LiNbO3, and ZnO have all been reported to form an amorphous cathode coating and improve the cell cycling stability.46−48 The calculated values for DLi and DO in LBO are 1 × 10–14 and 7 × 10–19 cm2/s, respectively, both of which are lower than the respective values in LiAlO2 and LiNbO3. Tables 1 and 2 list the calculated activation energies Ea, D, and the Onsager transport coefficient Lii of Li+ and O2– diffusion for the amorphous structures. More details on the calculation of Lii can be found in the Methods. In Figure S15, we also plotted the calculated DLi and DO at 300 K in various LixByOz compositions among which LBO exhibits the smallest DO.
Figure 3.

Self-diffusion coefficients in selected amorphous materials. Calculated room-temperature self-diffusion coefficients of Li+ and O2– in Li3B11O18, Al2O3, LiAlO2, ZnO, LiNbO3, and Li2O. The inserted figure shows the oxygen loss from the surface of the Li3B11O18-coated NMC532. r is the radius of an NMC532 particle, lc is the coating thickness, and ls is the thickness of the surface rocksalt phase. The error bars represent the standard deviation of extrapolated self-diffusion coefficients at 300 K. Amorphous 0.23 Li2O·0.77 Al2O3 and 0.11 Li2O·0.89 ZnO were used to simulate Li+ and O2–diffusion in Al2O3 and ZnO, respectively.
Table 1. Calculated Activation Energy Ea, Extrapolated Room-Temperature Diffusivity D, and Onsager Transport Coefficient LLiLi for Li+ Diffusiona.
| compounds | ELia (eV) | DLi(cm2/s) | error bound DLi | LLiLi (eV–1 · cm–1 · s–1) | error bound LLiLi |
|---|---|---|---|---|---|
| Li3B11O18 | 0.68 ± 0.03 | 1 × 10–14 | 9 × 10–15, 2 × 10–14 | 6 × 109 | 4 × 109, 9 × 109 |
| LiAlO2 | 0.54 ± 0.02 | 2 × 10–12 | 1 × 10–12, 2 × 10–12 | 1 × 1012 | 9 × 1011, 2 × 1012 |
| LiNbO3 | 0.47 ± 0.03 | 2 × 10–11 | 1 × 10–11, 4 × 10–11 | 1 × 1013 | 8 × 1012, 2 × 1013 |
| Al2O3 | 0.74 ± 0.08 | 1 × 10–15 | 3 × 10–16, 4 × 10–15 | 5 × 108 | 1 × 108, 2 × 109 |
| ZnO | 0.58 ± 0.07 | 6 × 10–13 | 2 × 10–13, 2 × 10–12 | 2 × 1011 | 5 × 1010, 7 × 1011 |
| Li2O | 0.26 ± 0.01 | 3 × 10–8 | 2 × 10–8, 3 × 10–8 | 7 × 1016 | 6 × 1016, 9 × 1016 |
The error bars correspond to the standard deviation of activation energy from the Arrhenius relation. The error bounds correspond to the standard deviation of extrapolated diffusivities and transport coefficients at 300 K. Amorphous 0.23 Li2O·0.77 Al2O3 and 0.11 Li2O·0.89 ZnO were used to simulate Li+ diffusion in Al2O3 and ZnO, respectively
Table 2. Calculated Activation Energy Ea, Extrapolated Room-Temperature Diffusivity Drt, and Onsager Transport Coefficient LOO for O2– Diffusiona.
| compounds | EOa (eV) | DO (cm2/s) | error bound DO | LOO (eV–1 · cm–1 · s–1) | error bound LOO |
|---|---|---|---|---|---|
| Li3B11O18 | 0.92 ± 0.05 | 7 × 10–19 | 3 × 10–19, 2 × 10–18 | 1 × 106 | 5 × 105,4 × 106 |
| LiAlO2 | 0.86 ± 0.05 | 5 × 10–18 | 2 × 10–18, 1 × 10–17 | 9 × 106 | 3 × 106,2 × 107 |
| LiNbO3 | 0.61 ± 0.02 | 5 × 10–14 | 4 × 10–14, 7 × 10–14 | 9 × 1010 | 7 × 1010,1 × 1011 |
| Al2O3 | 1.04 ± 0.07 | 9 × 10–21 | 3 × 10–21, 3 × 10–20 | 2 × 104 | 5 × 103,6 × 104 |
| ZnO | 0.67 ± 0.09 | 9 × 10–15 | 2 × 10–15, 4 × 10–14 | 1 × 1010 | 3 × 109,6 × 1010 |
| Li2O | 0.37 ± 0.02 | 5 × 10–11 | 3 × 10–11, 8 × 10–11 | 7 × 1013 | 5 × 1013,1 × 1014 |
The error bars correspond to the standard deviation of the activation energy from the Arrhenius relation. The error bounds correspond to the standard deviation of extrapolated diffusivities and transport coefficients at 300 K. Amorphous 0.23 Li2O·0.77 Al2O3 and 0.11 Li2O·0.89 ZnO were used to simulate O2– diffusion in Al2O3 and ZnO, respectively.
We evaluate the effectiveness of the LBO coating in suppressing oxygen loss-induced surface reconstructions of the layered cathode by estimating the O2– flux under steady-state conditions. To estimate the time t required for O2– to diffuse through the LBO coating, we consider a range of conditions by varying the coating thickness (lc) and oxygen chemical potential gradient (∇μO). The oxygen chemical potential on the cathode side (μOc) can be estimated from the cathode densification reaction consistent with the phase diagram. For example, at 4.3 V, layered NiO2 would densify to rocksalt NiO, with oxygen being released at μOc = −4.95 eV. The estimate for the oxygen chemical potential on the electrolyte side (μOe) depends on how the oxygen reacts on the electrolyte side of the coating. We evaluate two limiting conditions: (1) a lower bound for the chemical potential obtained from reaction of oxygen with the electrolyte to form a new oxidized compound (at μOe = −8.39 eV, Li3PS4 reacts with oxygen and forms Li3PO4); (2) an upper bound assuming oxygen loses electrons to the carbon network and is released as O2 (at μOe=–5.24 eV at room temperature and PO2e = 0.21 atm). Therefore, the range of μOe is assumed to be −8.39 ≤ μOe ≤ −5.24 eV. The O2– concentration in NiO2, denoted as cOmax, is ≈4.2 × 1022 cm–3. Thus, densification to NiO requires about 2.1 × 1022 cm–3 of O2– to diffuse through the coating. Based on the experimental observations, we set the radius of the NMC532 cathode particle to r = 5 μm and the thickness of the surface rocksalt phase to be ls = 2 nm. Finally, we estimated the time t required for the surface NiO2 to lose 50% of its oxygen. Table 3 shows that the released oxygen can diffuse through the LBO coating in 1.4–165 min, depending on lc and ∇μO. We emphasize that this time is considerably shorter than the time an electrode spends at high voltage, where its driving force for oxygen release is the highest.
Table 3. Estimated Time t for O2– to Diffuse through the LBO Coating for Various lc Values and ∇μia.
| r (μm) | ls (nm) | μOc (eV) | μOe (eV) | lc (nm) | t (min) | error bound t |
|---|---|---|---|---|---|---|
| 5 | 2 | –4.95 | –8.39 | 1 | 1.4 | 0.5, 3.8 |
| 10 | 14 | 5, 38 | ||||
| –5.24 | 1 | 16.6 | 6.5, 45 | |||
| 10 | 165 | 64,452 |
lc is the LBO coating thickness. μOc and μOe are the oxygen chemical potentials on the cathode and electrolyte side, respectively. We assume an NMC532 cathode particle radius (r) of 5 μm that forms a surface layer of densified NiO rocksalt phase with thickness (ls) of 2 nm.
Implications for Cathode Coating Design
In this article, we provide the first evidence of the formation of a surface-reduced layer in NMC532 in the presence of an amorphous LBO coating after cycling in a thiophosphate-based SSB. The fact that a reduced cation-disordered region forms at the surface of LBO-NMC532 indicates that the strategy of using a chemically/electrochemically stable amorphous surface coating with low electronic conductivity remains insufficient to inhibit the surface densification of the cathode particles, even in a SSB. We propose that the origin of the surface transformation of NMC532 in the presence of an amorphous coating originates in oxygen transport through the coating. Using AIMD and the Onsager transport model, we demonstrate that under a reasonable gradient range of oxygen chemical potential and coating thickness, transport of the O2– through the LBO coating can indeed occur on the time scale of electrochemical cycling. Such oxygen loss triggers the formation of a densified rocksalt phase at the surface of NMC532, even in the presence of an intact and amorphous surface coating. Besides increasing the impedance of the cathode, oxygen transport through the coating may damage the solid electrolyte by creating phases with lower conductivity or by mechanical decohesion related to the reaction-induced volume change.
As revealed by the modeling, the effectiveness of a surface coating in retaining oxygen in the cathode is determined by both kinetic and thermodynamic factors, primarily the oxygen diffusion rate in the coating materials and the oxygen chemical potential gradient across the coating layer. The high oxygen chemical potential of a charged cathode drives the O2– to diffuse from the cathode surface to the SE. On the cathode surface side, O2– can either lose electrons to the carbon network and be released as O2 or react with the SE. In an SSB with a thiophosphate SE, the presence of oxygen is likely to oxidize Li3PS4.49,50 Although previous research has shown that for an uncoated Ni-rich layered cathode, the surface oxygen loss and surface structure rearrangement is strongly correlated to the environmental conditions,51 our calculations confirm that it is possible to create a surface reduced layer even when direct contact between the cathode and the SE is prevented by an amorphous coating. It is worth noting that although the amorphous LBO coating significantly improves the cycling performance of full SSBs compared with an uncoated NMC cathode (Figures S16 and S17), a capacity loss of ∼20% is still observed after 100 cycles. The reduced Li kinetics due to cathode surface densification and the oxidation of the surrounding thiophosphate SE all play roles in this performance degradation.
Although our key findings in this study are based on a thiophosphate system, we argue that oxygen diffusion in the coating material is broadly relevant in solid-electrolyte and liquid-electrolyte systems. For a solid electrolyte that does not react with oxygen, such as Li7La3Zr2O12, μOe can be estimated from the oxygen partial pressure (PO2e) of air at room temperature and therefore corresponds to the calculated upper bound of μOe = −5.24 eV. In this case, we find that the driving force for oxygen diffusion through the coating becomes smaller than in the case of a reactive SE such as Li3PS4. However, in this scenario, we still estimate that only 17–165 min is needed at a high state of charge for enough oxygen to diffuse through a 1–10 nm LBO coating to create a 2 nm fully densified layer. This diffusion time is comparable to the thiophosphate case and, in the context of SSB operation, is expected to occur within a few cycles.
To demonstrate the broader consequences of our findings, we extend our modeling to other reported coating materials. Following the same procedure and assumption outlined above, we determine O2– transport through a 1–10 nm Al2O3 coating. We find that the O2– diffusivity (DO) and Onsager transport coefficient (LOO) in Al2O3 are approximately 2 orders of magnitude less than those in an LBO coating (see Table 2). As a result, the estimated time t required to transport the same amount of O2– through an Al2O3 coating varies between 2 and 230 h, depending on lc and ∇μO (see Table S1). Experimentally, Croy et al.23 reported that Al2O3 surface coating is not sufficient to stabilize an NMC cathode surface, while David et al.16 showed an Al2O3 ALD coating can effectively prevent surface reconstruction of an NMC cathode. Our calculations demonstrate that an NMC cathode coated with a thin Al2O3 coating layer, such as 1 nm, is still prone to surface oxygen loss, especially when cycled at a low C-rate. On the other hand, a thicker Al2O3 coating layer can effectively mitigate O2– transport, which results in better cathode surface protection. In addition, our calculations show that the Li+ and O2– diffusivities in commonly used LiAlO2 and LiNbO3 coatings are higher than those of LBO, which suggests a faster oxygen loss should occur from LiAlO2- or LiNbO3-coated NMC. Other inorganic Li+-containing compounds, such as Li2CO3, have been suggested as viable components in a protective surface coating layer due to their electronic insulating properties and overall acceptable ionic conductivity. A previous theoretical study has reported that the Li+ diffusivity in Li2CO3 is ∼10–7 cm2/s.52 Given the correlated diffusion between Li+ and O2–, we approximate that Li2CO3 exhibits a similar O2– diffusivity with Li2O, ∼10–10 cm2/s. It should be noted that our diffusion analysis neglects the reaction kinetics of surface structural transitions and back diffusion of transition metals whose effects contribute to the surface reconstruction into the densified layer.9 The bulk region of the cathode particle remains as a layered phase due to the slow kinetics associated with the layered-to-spinel transition.9
Using ab initio calculations and the Onsager transport theory, we propose that oxygen transport in coating materials plays an essential role in the surface reconstruction and oxygen loss of the layered cathode in various coating and electrolyte systems. In addition to the criteria of providing facile Li+ transport and preventing chemical reactions between the cathode and electrolyte, we highlight the importance of designing coating materials with low O2– diffusivity to block oxygen diffusion and mitigate lattice densification at the cathode surface. However, based on the coating materials investigated in this study, we note that there can be a trade-off between the O2– diffusivity and Li+ diffusivity, as shown in Figure 3. Generally, the diffusion of Li+ and O2– are correlated.14 This is because Li+ is bonded to its neighboring O2– ions, and its diffusion through the amorphous coating is governed by discrete hops between two adjacent sites, which are initiated by the Li–O bond breaking/formation process. Therefore, more sluggish O2– diffusion generally accompanies a slower Li+ diffusion. An ideal amorphous coating should maintain a low O2– diffusivity and a high Li+ diffusivity to achieve oxygen-retaining and surface-protective functions while avoiding significant losses in rate capacity. To guide the selection of coatings with adequate Li+ diffusion as well as reasonably low O2– diffusion, we refer to ref14 which presents an extensive high-throughput computational study of 20 common coating materials and their self-diffusion coefficients.14 In ref,14 design guidelines for Li+ and O2– diffusivities are provided; specifically recommending that Li+ diffusivity should be higher than 7 × 10–16 cm2/s and O2– diffusivity should be lower than 10–17 cm2/s. In particular, it is reported that BOxy–, SiOxy–, POxy–, and SbOxy– exhibit improved oxygen retention,14 which focuses the attention on cathode coating materials that contain one or more of these anion groups.
Conclusions
In this work, we use STEM and EELS to conclusively show that even in an SSB, and when protected by an intact, amorphous coating, the surface of a high-energy density oxide cathode still transforms from a layered structure into a rock-salt-like structure after electrochemical cycling. We propose that the reason a stable surface coating cannot inhibit the cathode surface degradation lies in oxygen transport in the surface coating. Using AIMD calculations, we systematically evaluated the O2– diffusion rate in amorphous LBO as well as in typical cathode coating materials reported in the literature. Our results demonstrate the facile O2– transport through the LBO coating and explain similar surface densification observed in liquid-cell systems on coated layered oxide cathodes in the literature. This work identifies oxygen loss as a significant barrier to long-cycle life high-energy storage, even in SSBs with coated cathodes, and highlights the need to design durable, amorphous cathode coatings with optimized lithium/oxygen diffusivity.
Acknowledgments
We acknowledge support from the Samsung Advanced Institute of Technology. The STEM and EELS experiments were performed at the Molecular Foundry, LBNL. Work at the Molecular Foundry was supported by the Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. The authors also gratefully acknowledge Dr. Taku Watanabe, Dr. Yuichi Aihara, Dr. Tomoyuki Tsujimura, and the Samsung R&D Institute of Japan for providing the LBO-coated cathode materials.
Data Availability Statement
All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supporting Information.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.chemmater.3c02351.
Radial distribution functions, MSD of Li+ and O2– ions, density versus oxygen diffusivity, fitting diffusivities using VFT and Arrhenius equations, TEM images of the uncycled and cycled cathode, structural characterization of the uncycled and cycled cathode, X-ray diffraction pattern of LPS, thickness distribution of the LBO coating layer, composition characterization of the cycled cathode, Arrhenius plots of Li+ and O2– diffusivity, calculated room-temperature diffusivities of Li+ and O2–, cycling performance comparison, electrochemical cycling data, and estimated time for O2– to diffuse through the Al2O3 coating (PDF)
Accession Codes
The codes used in this study are available either publicly (mpmorph; http://github.com/materialsproject/mpmorph) or through subscription (Vienna Ab-initio Simulation Package; https://www.vasp.at). For a detailed description of input parameters used for each code, refer to the “Density functional theory methods” section in the Methods.
Author Contributions
⊥ J.C., X.P., and Y.-Q.Z.: co-authorship.
Author Contributions
G.C., K.A.P., and M.S. planned the project and supervised all aspects of the research. Y.-Q.Z., X.P., and J.C. contributed equally to this work. Y.-Q.Z., X.P., T.O., A.H., and A.D. designed the experiment and collected and analyzed the high-resolution STEM, TEM, EELS, and X-ray diffraction data. J.C. performed AIMD calculations. Y.T. fabricated the cells and performed the electrochemical cycling test.
The authors declare no competing financial interest.
Supplementary Material
References
- Xu C.; et al. Bulk fatigue induced by surface reconstruction in layered Ni-rich cathodes for Li-ion batteries. Nat. Mater. 2021, 20, 84–92. 10.1038/s41563-020-0767-8. [DOI] [PubMed] [Google Scholar]; (2020).
- Tian Y.; et al. Promises and Challenges of Next-Generation ‘beyond Li-ion’ Batteries for Electric Vehicles and Grid Decarbonization. Chem. Rev. 2021, 121, 1623–1669. 10.1021/acs.chemrev.0c00767. [DOI] [PubMed] [Google Scholar]
- Li W.; Zhu J.; Xia Y.; Gorji M. B.; Wierzbicki T. Data-Driven Safety Envelope of Lithium-Ion Batteries for Electric Vehicles. Joule 2019, 3, 2703–2715. 10.1016/j.joule.2019.07.026. [DOI] [Google Scholar]
- Cheng J.; Ouyang B.; Persson K. A. Mitigating the High-Charge Detrimental Phase Transformation in LiNiO2 Using Doping Engineering. ACS Energy Lett. 2023, 8, 2401–2407. 10.1021/acsenergylett.3c00169. [DOI] [Google Scholar]
- Cheng J.; et al. Enhancing surface oxygen retention through theory-guided doping selection in Li1–xNiO2 for next-generation lithium-ion batteries. J. Mater. Chem. A Mater. 2020, 8, 23293–23303. 10.1039/D0TA07706B. [DOI] [Google Scholar]
- Armstrong A. R.; et al. Demonstrating Oxygen Loss and Associated Structural Reorganization in the Lithium Battery Cathode Li [ Ni Li Mn ] O. J. Am. Chem. Soc. 2006, 128, 8694–8698. 10.1021/ja062027+. [DOI] [PubMed] [Google Scholar]
- Tran N.; et al. Mechanisms associated with the ‘plateau’ observed at high voltage for the overlithiated Li1.12(Ni0.425Mn 0.425Co0.15)0.88O2 system. Chem. Mater. 2008, 20, 4815–4825. 10.1021/cm070435m. [DOI] [Google Scholar]
- Boulineau A.; Simonin L.; Colin J. F.; Bourbon C.; Patoux S. First evidence of manganese-nickel segregation and densification upon cycling in Li-rich layered oxides for lithium batteries. Nano Lett. 2013, 13, 3857–3863. 10.1021/nl4019275. [DOI] [PubMed] [Google Scholar]
- Xiao P.; Shi T.; Huang W.; Ceder G. Understanding surface densified phases in Ni-rich layered compounds. ACS Energy Lett. 2019, 4, 811–818. 10.1021/acsenergylett.9b00122. [DOI] [Google Scholar]
- Yu Z.; et al. Synthesis and Mechanism of High Structural Stability of Nickel-Rich Cathode Materials by Adjusting Li-Excess. ACS Appl. Mater. Interfaces 2020, 12, 40393–40403. 10.1021/acsami.0c12541. [DOI] [PubMed] [Google Scholar]
- Lin F.; et al. Surface reconstruction and chemical evolution of stoichiometric layered cathode materials for lithium-ion batteries. Nat. Commun. 2014, 5 (1), 3259. 10.1038/ncomms4529. [DOI] [PubMed] [Google Scholar]
- Xiao Y.; Miara L. J.; Wang Y.; Ceder G. Computational Screening of Cathode Coatings for Solid-State Batteries. Joule 2019, 3, 1252–1275. 10.1016/j.joule.2019.02.006. [DOI] [Google Scholar]
- Cheng J.; Sivonxay E.; Persson K. A. Evaluation of Amorphous Oxide Coatings for High-Voltage Li-Ion Battery Applications Using a First-Principles Framework. ACS Appl. Mater. Interfaces 2020, 12, 35748–35756. 10.1021/acsami.0c10000. [DOI] [PubMed] [Google Scholar]
- Cheng J.; Fong K. D.; Persson K. A. Materials design principles of amorphous cathode coatings for lithium-ion battery applications. J. Mater. Chem. A Mater. 2022, 10, 22245–22256. 10.1039/D2TA06051E. [DOI] [Google Scholar]
- (2018).Hu E.; et al. Evolution of redox couples in Li- and Mn-rich cathode materials and mitigation of voltage fade by reducing oxygen release. Nature Energy 2018, 3 (8), 690–698. 10.1038/s41560-018-0207-z. [DOI] [Google Scholar]
- David L.; et al. Unveiling the Role of Al2O3 in Preventing Surface Reconstruction during High-Voltage Cycling of Lithium-Ion Batteries. ACS Appl. Energy Mater. 2019, 2, 1308–1313. 10.1021/acsaem.8b01877. [DOI] [Google Scholar]
- Xiong D. J.; Hynes T.; Ellis L. D.; Dahn J. R. Effects of Surface Coating on Gas Evolution and Impedance Growth at Li[Ni x Mn y Co 1-x-y ]O 2 Positive Electrodes in Li-Ion Cells. J. Electrochem. Soc. 2017, 164, A3174–A3181. 10.1149/2.0991713jes. [DOI] [Google Scholar]
- Wise A. M.; et al. Effect of Al2O3 Coating on Stabilizing LiNi0.4Mn0.4Co0.2O2 Cathodes. Chem. Mater. 2015, 27, 6146–6154. 10.1021/acs.chemmater.5b02952. [DOI] [Google Scholar]
- Zheng J.; et al. Functioning mechanism of AlF3 coating on the Li- and Mn-rich cathode materials. Chem. Mater. 2014, 26, 6320–6327. 10.1021/cm502071h. [DOI] [Google Scholar]
- Zheng J. M.; et al. The Effects of AlF3 Coating on the Performance of Li [ Li0.2Mn0.54Ni0.13Co0.13 ] O2 Positive Electrode Material for Lithium-Ion Battery. J. Electrochem. Soc. 2008, 155, A775. 10.1149/1.2966694. [DOI] [Google Scholar]
- Kong J. Z.; et al. Ultrathin ZnO coating for improved electrochemical performance of LiNi0.5Co0.2Mn0.3O2 cathode material. J. Power Sources 2014, 266, 433–439. 10.1016/j.jpowsour.2014.05.027. [DOI] [Google Scholar]
- Zhang X.; et al. Improving the Structure Stability of LiNi0.8Co0.15Al0.05O2by Double Modification of Tantalum Surface Coating and Doping. ACS Appl. Energy Mater. 2021, 4, 8641–8652. 10.1021/acsaem.1c01811. [DOI] [Google Scholar]
- Croy J. R.; et al. Insights on the Stabilization of Nickel-Rich Cathode Surfaces: Evidence of Inherent Instabilities in the Presence of Conformal Coatings. Chem. Mater. 2019, 31, 3891–3899. 10.1021/acs.chemmater.8b04332. [DOI] [Google Scholar]
- Li X.; et al. Unravelling the Chemistry and Microstructure Evolution of a Cathodic Interface in Sulfide-Based All-Solid-State Li-Ion Batteries. ACS Energy Lett. 2019, 4, 2480–2488. 10.1021/acsenergylett.9b01676. [DOI] [Google Scholar]
- Li J.; et al. The Impact of Electrolyte Additives and Upper Cut-off Voltage on the Formation of a Rocksalt Surface Layer in LiNi 0.8 Mn 0.1 Co 0.1 O 2 Electrodes. J. Electrochem. Soc. 2017, 164, A655–A665. 10.1149/2.0651704jes. [DOI] [Google Scholar]
- Zhang Y. Q.; et al. Direct Visualization of the Interfacial Degradation of Cathode Coatings in Solid State Batteries: A Combined Experimental and Computational Study. Adv. Energy Mater. 2020, 10, 1903778 10.1002/aenm.201903778. [DOI] [Google Scholar]
- Doux J. M.; et al. Pressure effects on sulfide electrolytes for all solid-state batteries. J. Mater. Chem. A Mater. 2020, 8, 5049–5055. 10.1039/C9TA12889A. [DOI] [Google Scholar]
- Zhang J.; et al. All-solid-state batteries with slurry coated LiNi0.8Co0.1Mn0.1O2 composite cathode and Li6PS5Cl electrolyte: Effect of binder content. J. Power Sources 2018, 391, 73–79. 10.1016/j.jpowsour.2018.04.069. [DOI] [Google Scholar]
- Yubuchi S.; et al. Preparation of high lithium-ion conducting Li6PS5Cl solid electrolyte from ethanol solution for all-solid-state lithium batteries. J. Power Sources 2015, 293, 941–945. 10.1016/j.jpowsour.2015.05.093. [DOI] [Google Scholar]
- Pearson D. H.; Fultz B.; Ahn C. C. Measurements of 3d state occupancy in transition metals using electron energy loss spectrometry. Appl. Phys. Lett. 1988, 53, 1405–1407. 10.1063/1.100457. [DOI] [Google Scholar]
- Kresse G.; Furthmüller J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. 10.1103/PhysRevB.54.11169. [DOI] [PubMed] [Google Scholar]
- Kresse G.; Joubert D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758. 10.1103/PhysRevB.59.1758. [DOI] [Google Scholar]
- Perdew J. P.; Burke K.; Ernzerhof M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865. 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- Martinez L.; Andrade R.; Birgin E. G.; Martínez J. M. PACKMOL: A package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 2009, 30, 2157–2164. 10.1002/jcc.21224. [DOI] [PubMed] [Google Scholar]
- (2020).Virtanen P.; et al. SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat. Methods 2020, 17 (3), 261–272. 10.1038/s41592-019-0686-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima S.; Novikov V. N.; Kodama M. Fast relaxation, boson peak, and anharmonicity in Li2O–B2O3 glasses. J. Chem. Phys. 2000, 113, 6344–6350. 10.1063/1.1309530. [DOI] [Google Scholar]
- Matsuda Y.; Fukawa Y.; Kawashima M.; Kojima S. Fragility Variation of Lithium Borate Glasses Studied by Temperature-Modulated DSC. AIP Conf. Proc. 2008, 982, 207–210. 10.1063/1.2897785. [DOI] [Google Scholar]
- Chryssikos G. D.; et al. Lithium borate glasses: a quantitative study of strength and fragility. J. Non Cryst. Solids 1994, 172–174, 378–383. 10.1016/0022-3093(94)90460-X. [DOI] [Google Scholar]
- (2020).Zhao Q.; Stalin S.; Zhao C. Z.; Archer L. A. Designing solid-state electrolytes for safe, energy-dense batteries. Nature Reviews Materials 2020, 5 (3), 229–252. 10.1038/s41578-019-0165-5. [DOI] [Google Scholar]
- Carroll K. J.; et al. Probing the electrode/electrolyte interface in the lithium excess layered oxide Li1.2Ni0.2Mn0.6O2. Phys. Chem. Chem. Phys. 2013, 15, 11128–11138. 10.1039/c3cp51927a. [DOI] [PubMed] [Google Scholar]
- Hwang S.; et al. Determination of the mechanism and extent of surface degradation in Ni-based cathode materials after repeated electrochemical cycling. APL Mater. 2016, 4, 096105 10.1063/1.4963723. [DOI] [Google Scholar]
- Yoon W. S.; et al. Investigation of the charge compensation mechanism on the electrochemically Li-ion deintercalated Li1-xCo1/3Ni1/3Mn 1/3O2 electrode system by combination of soft and hard X-ray absorption spectroscopy. J. Am. Chem. Soc. 2005, 127, 17479–17487. 10.1021/ja0530568. [DOI] [PubMed] [Google Scholar]
- Koerver R.; et al. Capacity Fade in Solid-State Batteries: Interphase Formation and Chemomechanical Processes in Nickel-Rich Layered Oxide Cathodes and Lithium Thiophosphate Solid Electrolytes. Chem. Mater. 2017, 29, 5574–5582. 10.1021/acs.chemmater.7b00931. [DOI] [Google Scholar]
- House R. A.; et al. Lithium manganese oxyfluoride as a new cathode material exhibiting oxygen redox. Energy Environ. Sci. 2018, 11, 926–932. 10.1039/C7EE03195E. [DOI] [Google Scholar]
- House R. A.; et al. What Triggers Oxygen Loss in Oxygen Redox Cathode Materials?. Chem. Mater. 2019, 31, 3293–3300. 10.1021/acs.chemmater.9b00227. [DOI] [Google Scholar]
- Zhang X.; et al. Structural and Electrochemical Study of Al2O3 and TiO2 Coated Li1.2Ni0.13Mn0.54Co0.13O2 Cathode Material Using ALD. Adv. Energy Mater. 2013, 3, 1299–1307. 10.1002/aenm.201300269. [DOI] [Google Scholar]
- Park J. S.; et al. Ultrathin lithium-ion conducting coatings for increased interfacial stability in high voltage lithium-ion batteries. Chem. Mater. 2014, 26, 3128–3134. 10.1021/cm500512n. [DOI] [Google Scholar]
- Zhao J.; Wang Y. Ultrathin surface coatings for improved electrochemical performance of lithium ion battery electrodes at elevated temperature. J. Phys. Chem. C 2012, 116, 11867–11876. 10.1021/jp3010629. [DOI] [Google Scholar]
- Bartsch T.; et al. Gas Evolution in All-Solid-State Battery Cells. ACS Energy Lett. 2018, 3, 2539–2543. 10.1021/acsenergylett.8b01457. [DOI] [Google Scholar]
- Walther F.; et al. Influence of Carbon Additives on the Decomposition Pathways in Cathodes of Lithium Thiophosphate-Based All-Solid-State Batteries. Chem. Mater. 2020, 32, 6123–6136. 10.1021/acs.chemmater.0c01825. [DOI] [Google Scholar]
- Karki K.; et al. Tuning the Activity of Oxygen in LiNi0.8Co0.15Al0.05O2 Battery Electrodes. ACS Appl. Mater. Interfaces 2016, 8, 27762–27771. 10.1021/acsami.6b09585. [DOI] [PubMed] [Google Scholar]
- Shi S.; Qi Y.; Li H.; Hector L. G. Defect thermodynamics and diffusion mechanisms in Li2CO 3 and implications for the solid electrolyte interphase in Li-ion batteries. J. Phys. Chem. C 2013, 117, 8579–8593. 10.1021/jp310591u. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supporting Information.