

Abstract
Expression quantitative trait loci (eQTLs) are used to inform the mechanisms of transcriptional regulation in eukaryotic cells. However, the specificity of genome-wide eQTL identification is limited by stringent control for false discoveries. Here, we described a method based on the non-homogeneous Poisson process to identify 125 489 regions with highly frequent, multiple eQTL associations, or ‘eQTL-hotspots’, from the public database of 59 human tissues or cell types. We stratified the eQTL-hotspots into two classes with their distinct sequence and epigenomic characteristics. Based on these classifications, we developed a machine-learning model, E-SpotFinder, for augmented discovery of tissue- or cell-type-specific eQTL-hotspots. We applied this model to 36 tissues or cell types. Using augmented eQTL-hotspots, we recovered 655 402 eSNPs and reconstructed a comprehensive regulatory network of 2 725 380 cis-interactions among eQTL-hotspots. We further identified 52 012 modules representing transcriptional programs with unique functional backgrounds. In summary, our study provided a framework of epigenome-augmented eQTL analysis and thereby constructed comprehensive genome-wide networks of cis-regulations across diverse human tissues or cell types.
Keywords: eQTL-hotspots, non-homogeneous Poisson process, epigenome-augmented eQTL mapping, transcriptional programs, cis-regulatory network
INTRODUCTION
Expression quantitative trait loci (eQTLs) provide many important clues to the regulatory programs of gene expression [1, 2], and facilitate the characterization of cis-elements and trans-acting factors [3–6]. Moreover, eQTLs serve as important instrumental variables for the trait-associated loci and help us better understand the genetic background of complex human diseases [7–11]. An eQTL is usually a haplotype-block containing a series of single-nucleotide polymorphisms (eSNPs) in linkage disequilibrium (LD), the genotype of which is associated with the transcript abundance of genes. Accordingly, the gene affected by an ‘eSNP’ are known as ‘eGene’ [2].
By far, more than 4.2 million eQTL associations (2 006 095 eSNPs and 21 253 eGenes, P < 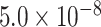 ) have been identified in different tissues or cell types, most of which are located in non-coding regions of the genome [12, 13]. Many more potential associations with statistical significance are still pending multiple-testing correction [12, 13]. Only a very small portion of eQTLs have been empirically verified for their function in transcriptional regulation [3]. In most cases, causal variants interrupt the binding of either proteins or non-coding RNAs to cis-regulatory elements, thus altering the transcriptional program [6, 14, 15]. Characterizing functional eQTLs has proven to be highly important for understanding the transcriptional regulation underlying the complex etiology of inheritable diseases [8–11, 16–18].
) have been identified in different tissues or cell types, most of which are located in non-coding regions of the genome [12, 13]. Many more potential associations with statistical significance are still pending multiple-testing correction [12, 13]. Only a very small portion of eQTLs have been empirically verified for their function in transcriptional regulation [3]. In most cases, causal variants interrupt the binding of either proteins or non-coding RNAs to cis-regulatory elements, thus altering the transcriptional program [6, 14, 15]. Characterizing functional eQTLs has proven to be highly important for understanding the transcriptional regulation underlying the complex etiology of inheritable diseases [8–11, 16–18].
Although eQTLs inform transcriptional regulation directly, the known eQTLs are still not enough to fully explain the dynamics of transcriptional regulation [19]. First, the specificity of genome-wide eQTL analysis is often limited due to the lack of statistical power and the stringent control of false positives [12]. Most of the eQTLs are identified by the statistical significance of associations in a cohort, of which the sample size is far smaller than that of the SNPs tested. Consequently, after correction for multiple-testing errors, the results are sparsely located in the genome, representing few individual cis-regulatory events [1, 20–23]. Previous studies have described approaches for eQTL analysis by either incorporating additional allelic data or using LD to correct the multiple-testing errors. Nevertheless, these methods can only partially improve the specificity of the test [24]. Besides, eQTLs are highly specific to tissues or cell types. Tissue heterogeneity and sampling biases often confound eQTL analysis, thus hindering the discovery of new transcriptional programs [13]. In addition, performing eQTL analysis for each of the known cell types is technically challenging and costly [25].
As an alternative approach, most recent studies have used epigenomic features such as histone modification marks to identify regulatory elements, which are less burdened by multiple-testing errors [26, 27]. However, chromatin immunoprecipitation sequencing (ChIP-Seq) can only be applied to a limited number of cells or tissues and hence cannot reveal the populational variation in the cistrome [26, 28]. In addition, the cis-regulatory elements are represented by peaks of ChIP-Seq, which are also subject to all experimenter biases and false discoveries [26, 27, 29, 30].
Here, we described a framework of augmented eQTL discovery by integrating epigenomic data to enhance the specificity of pan-tissue eQTL mapping and thereby generated a comprehensive map of the cis-regulatory programs in 36 human tissues or cell types. We first defined pan-tissue eQTL-hotspots from limited, well-controlled, published eQTLs. Then, we retrieved the consensus signatures of genomic and epigenomic characteristics for the eQTL-hotspots, which were used to train a machine-learning model to predict tissue or cell-type-specific hotspots. We validated the predicted hotspots for eQTL association strengths and known transcription regulation activities. Finally, we constructed comprehensive maps of the transcriptional programs of 36 tissue or cell types.
RESULTS
Deriving eQTL-hotspots
Despite being identified from specific tissues or cell types, most eQTLs act similarly by disrupting regulatory programs involving specific cis-elements and trans-factors. Here, we collected uniformly processed 127 574 148 cis-eQTL associations (6 936 091 eSNPs, 33 338 eGenes, P < 0.05) from 59 tissues or cell types (51 tissues and eight cell types) from eQTL Catalogue [13] (Table S1, Supplemental Methods). To control for false positives, we selected 2 006 095 eSNPs with test P < 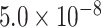 . We considered the occurrence of eSNPs following the non-homogeneous Poisson process (NHPP) [31] and determined the rate parameter,
. We considered the occurrence of eSNPs following the non-homogeneous Poisson process (NHPP) [31] and determined the rate parameter, 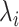 , for each moving window
, for each moving window 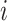 . To account for the background rate of genetic variations in the genome, we defined each window by a genomic region encompassing 18 SNPs (Supplemental Methods).
. To account for the background rate of genetic variations in the genome, we defined each window by a genomic region encompassing 18 SNPs (Supplemental Methods).
Based on the distribution of 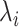 , we identified two distinct classes of genomic regions: the eQTL-hotspots were defined by
, we identified two distinct classes of genomic regions: the eQTL-hotspots were defined by 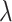 ≥ 0.176 (Supplemental Methods) and consisted of 125 489 regions (54–149 590 bp; Figure S1; Table S2), covering 20.2% of the genome but 79.4% of known eSNPs (n = 1 592 343); the rest of the genome were defined as non-hotspot regions (
≥ 0.176 (Supplemental Methods) and consisted of 125 489 regions (54–149 590 bp; Figure S1; Table S2), covering 20.2% of the genome but 79.4% of known eSNPs (n = 1 592 343); the rest of the genome were defined as non-hotspot regions (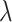 < 0.176). The extremely inactive regions with
< 0.176). The extremely inactive regions with 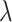 = 0 were defined as cold regions (Figure 1A; Supplemental Methods). Each eQTL-hotspot contained 4–629 eSNPs with P <
= 0 were defined as cold regions (Figure 1A; Supplemental Methods). Each eQTL-hotspot contained 4–629 eSNPs with P < 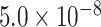 , which were associated with 2–52 eGenes.
, which were associated with 2–52 eGenes.
Figure 1.

Derivation and quality assessment of eQTL-hotspots. (A) Schematic representation of the process for deriving eQTL-hotspots in the human genome. (B) The eQTL-hotspots showed significant colocalization with epigenomic marks of transcriptional activation but repellence to marks of transcriptional suppression. The tendency was evidenced by the fractions of eQTL-hotspots and non-hotspots intersecting each epigenomic mark. P-values were calculated using the permutation test. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; ns, not significant.
The epigenomic characteristics of the eQTL-hotspots
We retrieved nine known epigenomic marks for the eQTL-hotspots from matched tissues or cell types, including H3K27ac, H3K4me1, H3K4me3, H3K9ac, H3K36me3, H3K27me3 and H3K9me3 [32]; chromatin accessibility (CA); and transcription factor binding site (TFBS) [26] (Figure 1B; Table S1; Supplemental Methods). For comparison, we used randomly sampled regions of the same lengths from the non-hotspot regions as the control (Supplemental Methods). As expected, the eQTL-hotspots showed strong tendencies of colocalizing with epigenomic marks associated with transcriptional activation, such as H3K27ac (P < 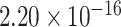 ) and H3K4me1 (P <
) and H3K4me1 (P < 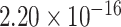 ), which are indicative of enhancers and promoters [32]. Furthermore, eQTL-hotspots displayed repellency to epigenomic marks associated with transcriptional repression, such as H3K27me3 (P = 0.076) and H3K9me3 (P <
), which are indicative of enhancers and promoters [32]. Furthermore, eQTL-hotspots displayed repellency to epigenomic marks associated with transcriptional repression, such as H3K27me3 (P = 0.076) and H3K9me3 (P < 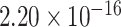 ), which are commonly indicative of inactive genomic regions [32] (Figure 1B).
), which are commonly indicative of inactive genomic regions [32] (Figure 1B).
eQTL-hotspots consisted of two distinct classes of cis-elements
As we showed that the eQTL-hotspots colocalize with poised cis-elements, we investigated whether these eQTL-hotspots could be further stratified into subgroups with distinct genomic characteristics and surrogates for regulatory activities that involve the recognition and binding of specific DNA motifs by transcription factors [2]. We used 396 k-mers (k = 6, Table S3) [33] to represent the genomic features of the eQTL-hotspots and performed kernel-PCA [34], followed by Leiden clustering [35] (Methods). As a result, the eQTL-hotspots were clustered into two categories, namely, hotspot-C1 (n = 105 820) and hotspot-C2 (n = 19 669) (Figure 2A; Table S2). To obtain a non-hotspot control set, we randomly sampled fragments from the cold regions with the same length distribution, C0.
Figure 2.

Characterizing two distinct classes of eQTL-hotspots. (A) UMAP projection of the genomic features based on k-mers (k = 6) showing the two classes of eQTL-hotspots. (B) The consensus landscape of relevant epigenomic features of hotspot-C1, hotspot-C2 and non-hotspot (C0). The x-axis represents the genomic region of 2.5 kb on either side of the center of the segments of specific class, and the y-axis represents the signal values for an epigenomic mark. (C–E) The distributions of the GC content (C), log-transformed number of eSNP–eGene pairs per kilobase (D) and log-transformed signal of in situ HiC on H1-hESC per kilobase (E) in hotspot-C1, hotspot-C2 and non-hotspot (C0). P-values were calculated using the one-sided Wilcoxon rank-sum test. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; ns, not significant. (F, G) Heatmaps demonstrate the fold of enrichment/depletion for chromatin states (F) and cCREs (G) across hotspot-C1, hotspot-C2 and non-hotspot C0.
To further characterize the two subtypes of eQTL-hotspots, we retrieved the consensus landscape [36] of nine epigenomic features for hotspot-C1, hotspot-C2 and non-hotspot C0 (Figure 2B; Supplemental Methods). Notably, within 2.5 kb from the centers, hotspot-C1 and C2 each demonstrated distinct epigenomic landscapes and differed from those of the non-hotspot C0. Hotspot-C2 showed the highest activity of the active marks (H3K27ac, H3K4me1, H3K4me3, H3K9ac and H3K36me3; CA; and TFBS) and relatively low activity of the repressive marks (H3K27me3 and H3K9me3). In addition, the signals of certain epigenomic features tended to peak at the center of hotspot-C2 and decline with distance. Hotspot-C1 showed a similar but moderate increase in active marks and a strong decrease in repressive marks. However, the landscapes of all features in hotspot-C1 are relatively flat and resemble those in the non-hotspot C0. In addition, we observed significant increase in GC content from C0 to hotspot-C1 and then C2 (Figure 2C). Coupled with the changes above, we also note the increased proportion of known eSNP–eGene pairs, and chromatin interactions (in situ Hi-C and Micro-C [37]) in hotspot-C1 to C2 (Figure 2D and E; Figure S2, one-sided Wilcoxon rank-sum test).
We annotated the two classes of eQTL-hotspots for enrichment of inferred chromatin states and cis-elements from previous studies (Figure 2F and G; Supplemental Methods) [27, 28, 32]. As a result, hotspot-C2 sites were significantly enriched for active TSS and enhancers as well as many bivalent chromatin states such as Bivalent Enhancer (12_EnhBiv) and Flanking Bivalent TSS/Enh (11_BivFlnk). Then, hotspot-C1 exhibited a significant depletion in bivalent chromatin states and moderate enrichment for active chromatin states. Our findings suggest that hotspot-C2 are involved mainly in transcription starting and enhancer activities, especially in response to stimulation, whereas hotspot-C1 involved inactive promoters. We also annotated the eQTL-hotspots for ENCODE Registry of candidate cis-Regulatory Elements (cCREs) [27, 28]. As a result, hotspot-C2 were significantly enriched in promoter-like signature (prom, 9.47-fold) and proximal enhancer-like signature (enhP, 7.49-fold), whereas hotspot-C1 were enriched in prom (1.35-fold) and enhP (1.38-fold) with lower magnitudes. Notably, hotspot-C1 are depleted in DNase-H3K4me3 (K4m3), which is concordant with hotspot-C1 being the less active promoter (Figure 2F and G). In summary, eQTL-hotspots consist of two distinct classes with recognizable sequence characteristics that are coupled with highly unique epigenomic landscapes, chromatin states and potential regulatory roles.
Development of E-SpotFinder, a machine-learning model for predicting tissue- or cell-type-specific eQTL-hotspots
With the full characterization of hotspot-C1 and C2, we developed a classifier capable of identifying eQTL-hotspots specific to certain tissues or cell types (Figure 3A). Our training dataset consisted of 19 669 sites from hotspot-C2, 105 580 sites from hotspot-C1 (positive samples) and 125 489 sites from non-hotspot (C0, negative samples), where each site is represented by 406 genomic and epigenomic features (Table S4). Subsequently, we trained a set of machine-learning models to classify hotspot-C1, hotspot-C2 and non-hotspot (C0); from these models, we selected a gradient-boosted tree–based classification algorithm, XGBoost [38], which achieved the best performance in 10-fold cross-validation and named it E-SpotFinder (Figure 3B). The areas under the ROC curves (AUCs) of the validation were 0.81, 0.99 and 0.78 for hotspot-C1, C2 and non-hotspot (C0), respectively (Figure 3C). Besides, for all the metrics, we used to evaluate the model; the best predictive performance was reached only when all three types of input features were used, namely, the GC content, the DNA sequence (k-mers) and the epigenomic marks (Figure 3D; Table S4). We employed SHAP (Shapley Additive exPlanations) analysis [39] to calculate the global impact of all 406 features on prediction results. As a result, the top 20 most important features were nine epigenomic marks, 10 k-mers and GC content, of which GC content was the most important feature, followed by H3K36me3 (Figure 3E).
Figure 3.

The performance evaluation of E-SpotFinder and the importance of input features. (A) Schematic depiction outlining the development of E-SpotFinder. (B) The prediction performance of four methods, XGBoost (XGBClassifier), random forest, decision tree, and ridge regression for eQTL-hotspots-C1, hotspotC2 and non-hotspot (C0) based on 10-fold cross-validation. (C) The prediction performance of ESpotFinder for eQTL-hotspot-C1, hotspot-C2, and non-hotspot (C0) as shown by ROC curves. (D) The prediction performance of E-SpotFinder is compared among models based on each of three types of input features including GC content, k-mers, epigenomic marks and the combination of all. (E) The 20 most important input features for E-SpotFinder as determined by mean absolute SHAP values.
Next, we used E-SpotFinder to predict eQTL-hotspots from whole genomes in 36 tissues or cell types (31 tissues and five cell types). This analysis yielded an average of 57 370 (15 403–109 653) hotspot-C1 and 5992 (1086–12 507) hotspot-C2 in each tissue or cell type (Figure S3; Table S5). Notably, non-hotspots covered most of the genome (79.0–97.1%), while the predicted hotspot-C1 and C2 covered 2.7–19.1% and 0.19–2.2%, respectively, of the genome.
To further verify the predicted eQTL-hotspots, we retrieved the consensus epigenomic landscape for three classes of predicted regions in each tissue or cell type. To this end, we focused on the three tissues (blood, iPSC and suprapubic skin) with complete feature sets, and we observed the epigenomic landscapes for hotspot-C1 and C2 highly consistent with those in the pan-tissue analysis (Figure 4A; Figure S4A and B). Among all 36 tissue or cell types, we observed a significant increase in GC content from the predicted non-hotspot C0 to hotspots-C1 and then C2 (Figure 4B; Figure S5). We also noticed a substantial increase in chromatin interaction [27] activity in the predicted hotspot-C1 and C2 in all of 12 tissues or cell types with published Hi-C data (Figure 4C; Figure S6).
Figure 4.

Characterizing predicted tissue- or cell-type-specific eQTL-hotspots. (A) The consensus landscape of relevant epigenomic features of predicted eQTL-hotspot-C1, hotspot-C2 and non-hotspot (C0) in blood. The x-axis represents the genomic region of 2.5 kb on either side of the center of the segments of the specific class, and the y-axis represents the signal values for an epigenomic mark. (B) The distributions of GC content in predicted eQTL-hotspots in blood and (C) the distributions of Hi-C signal in adrenal gland in hotspot-C1, hotspot-C2 and non-hotspot (C0). P-values were calculated using the one-sided Wilcoxon rank-sum test. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; ns, not significant. (D, E) Heatmaps demonstrate the fold of enrichment/depletion for chromatin states (D) and cCREs (E) in hotspot-C1 and C2 across 36 tissues or cell types.
We annotated the predicted eQTL-hotspots for chromatin states and cCREs in each of the 36 tissues or cell types. Notably, we found that the predicted hotspot-C2 and C1 exhibited highly consistent and conserved patterns of enrichment for specific chromatin states and cCREs, as we previously observed in pan-tissue analyses (Figure 4D and E; Figure S7).
Taken together, using the epigenomic and genomic characteristics of the pan-tissue eQTL-hotspots, we developed a machine-learning model that can identify genomic regions with highly consistent epigenomic features in specific tissues or cell types, thereby inferring potential eQTL activities.
Augmented eQTL-discovery by E-SpotFinder
Typical eQTL mapping is based on the significance of genome-wide associations and is thus burdened by multiple-testing errors [1, 12, 13, 20–23]. In contrast, with E-SpotFinder, we can segregate the genome into regions with distinct regulatory potentials. We first used eQTLs with allelic fold-change (aFC) [40], fine-mapping eQTLs [12] and GWAS SNPs [41] to benchmark the eQTL-hotspot-C1 and C2 inferred by E-SpotFinder in 36 tissues or cell types. As a result, all three sets of benchmarks were significantly enriched in hotspot-C1 and C2 (Figure 5A–C). As for the folds of enrichment, hotspot-C2 (eQTLs with aFC: 2.10–3.30-fold, fine-mapping eQTLs: 1.49–2.41-fold, GWAS SNPs: 2.08–2.38-fold) were strongly enriched for all benchmark sets, which is similar to that of enhP (eQTLs with aFC: 2.33–3.38-fold, fine-mapping eQTLs: 1.69–1.92-fold, GWAS SNPs: 2.08–2.23-fold) but lower than that of prom. The enrichment is lower in hotspot-C1 (eQTLs with aFC: 1.40–1.90-fold, fine-mapping eQTLs: 1.37–2.08-fold, GWAS SNPs: 1.28–1.65-fold) but still significantly higher than that in CTCF, K4me3 and enhD (except for ‘GWAS SNPs’). These data suggest that the predicted eQTL-hotspots show a high level of eQTL activity most resembling those of annotated enhancers.
Figure 5.

Augmented eQTL identification by refined thresholding for P-values in tissue- or cell-type-specific hotspots. The fold of enrichment for three benchmark sets in seven types of genomic regions defined by cCREs (CTCF: CTCF-only, K4m3: DNase-H3K4me3, enhD: distal enhancer-like signature, enhP: proximal enhancer-like signature, prom: promoter-like signature) and eQTL-hotspots (C1 and C2) were compared across 36 tissues or cell types. (A) eQTLs with allelic fold change (aFC), (B) fine-mapping eQTLs and (C) GWAS SNPs. (D) A Q-Q plot illustrates the deviation of the distributions of the original association test P-values for eSNPs in hotspot-C1 and C2 against C0 (expected) in blood. The x-axis represents the −log10P of non-hotspot (C0) as ‘expected’, and the y-axis represents the observed −log10P. The dashed lines correspond to −log10FDR and with a slope equal to 1, the FDR was set to 0.001 (Methods). The y-coordinate of the intersection point of the quantiles of the observed P-values indicates the refined thresholds for significant eSNP associations. (E) The fold of enrichment for fine-mapping eQTL associations in the eSNP–eGene pairs based on four different calling schemes. (F) The fold of enrichment for GWAS SNPs in eSNP sets based on four different calling schemes. Each colored dot represents a tissue or cell type; P-values were calculated using the one-sided Wilcoxon rank-sum test and were adjusted using ‘bonferroni’. ∙, adjusted P < 0.1; *, adjusted P < 0.05; **, adjusted P < 0.01; ***, adjusted P < 0.001; ****, adjusted P < 0.0001; ns, not significant. Hotspot: eSNPs defined within each type of hotspot by refined thresholds; Same ranking, WG: eSNPs defined by the same number of top-ranking SNPs in the whole genome as that defined by hotspots; Empirical threshold, WG: eSNPs defined by the whole genome, empirical thresholds of 5.0 × 10–8; FDR < 0.001, WG: eSNPs defined by the whole genome thresholds corresponding to the same level of FDR (0.001).
Next, we tried to recover more eQTLs from the predicted hotspots. We found that the distributions of the test P-values for eSNP–eGene pairs within hotspot-C1 and C2 deviated from those of non-hotspots, which allowed for a less stringent correction of P-values at the FDR of 0.001 [42] (Figure 5D; Figure S8; Table S6). By applying refined thresholds of P-values for hotspot-C1 and C2, 164–97 347 eSNPs were recovered in each tissue or cell type, those SNPs were located in 103–32 266 predicted eQTL-hotspots (Table S6). We used a set of fine-mapping eQTLs [12] to evaluate various eSNP calling schemes, including ours and those based on genome-wide ranking and thresholding of the P-values (Supplemental Methods). As a result, we found that eSNP–eGene pairs based on hotspots exhibited greater enrichment (6.01–35.22-fold) for fine-mapping eQTLs across all 36 tissues or cell types than did any of the genome-wide methods (Figure 5E, one-sided Wilcoxon rank-sum test). Similarly, eSNPs based on hotspots also exhibited the highest enrichment for GWAS risk loci [41] across 36 tissues or cell types compared to the genome-wide mapping methods, exhibiting an average of 2.43-fold (1.95–3.20-fold) of enrichment (Figure 5F, one-sided Wilcoxon rank-sum test). To summarize, we described an augmented eQTL analysis based on hotspots predicted by E-SpotFinder.
Reconstruction of the cis-regulatory networks based on augmented eQTLs
By obtaining a total of 1 347 945 recovered eSNP–eGene pairs in addition to the existing data, we now have a range of eGenes (1–33) associated with the eSNPs in each eQTL-hotspot (Table S6). We consider the majority of these eQTL-hotspots to be potential cis-regulatory elements. Therefore, hotspots targeting the same eGene are likely involved in the same regulatory program. In this study, we constructed interaction networks among eQTL-hotspots based on the commonality of eGenes. Two hotspots were connected if their corresponding eGene sets exhibited a similarity corresponding to a Jaccard index (JI) > 0.2 (Figure 6A). The threshold of the JI was determined based on alignment with the Hi-C signal across 12 tissues or cell types (Figure S9A and B, one-sided Wilcoxon rank-sum test). The threshold of JI also showed significant concordance with known promoter–promoter (P-P), enhancer–promoter (E-P), and enhancer–enhancer (E-E) [43] interactions across 36 tissues or cell types (Figure S9C–E, one-sided Wilcoxon rank-sum test). The resulting interaction network of eQTL-hotspots consisted of 116 238 nodes and 2 725 380 edges corresponding to cis-associations across the 36 tissues or cell types.
Figure 6.

Reconstruction of the cis-regulatory networks using inferred eQTL-hotpots. (A) Two eQTL-hotspots are considered to be interacted if the corresponding eGene sets show a JI > 0.2. (B) The fractions of three types of interactions (P-P, E-P and E-E) are presented at the edges corresponding to C1-C1, C1-C2 and C2-C2 across 36 tissues or cell types. Each colored dot represents one tissue or cell type. See Figure 5 for the legend of tissue colors. P-values were calculated using the one-sided Wilcoxon rank-sum test, and were adjusted using ‘bonferroni’. ∙, adjusted P < 0.1; *, adjusted P < 0.05; **, adjusted P < 0.01; ***, adjusted P < 0.001; ****, adjusted P < 0.0001; ns, not significant. (C) Heatmaps demonstrate the similarity of cis-regulatory networks among 36 tissues or cell types. Tissues are ordered by agglomerative hierarchical clustering. (D) The distribution of the total number of TPMs in different tissues or cells. (E, F) The significant positive correlation between transcription factor binding activities within TPM hotspots and the scaled expression of the corresponding eGenes as demonstrated by IRF4 in blood (E) and MYC in suprapubic skin (F). Each dot represents one TPM. The x-axis represents the average binding activity of the transcription factor in TPM, while the y-axis represents the average of Z-score scaled eGene expression in TPM. The eGenes of known targets of IRF4 and MYC are labeled.
We also noticed that within the network, edges between hotspot-C2 were strongly enriched for P-P interactions (8.02–41.67%); and E-P interactions were primarily enriched in C2-C2 edges (0–4.36%) and C1-C2 edges (0–1.83%) (Figure 6B, one-sided Wilcoxon rank-sum test). These findings are consistent with the potential chromatin states of hospot-C1 and C2. Furthermore, the interaction network can also reveal unknown cis-regulatory events.
In addition, the edges of the network represent interactions within specific tissues or cell types. The pairwise similarity of 36 subgraphs consisting only of tissue- or cell-type-specific edges, strongly correlates with the tissue origins, which is consistent with the findings in the GTEx study [12, 23] (Figure 6C).
We focused on a set of network modules that were extracted using network clustering (Leiden) [35]. Based on our previous findings, these modules represent a series of cis-regulatory interactions that act on the same set of eGenes and hence are surrogates for specific transcription programs (TPs). We identified a total of 52 012 such modules from 36 tissues or cell types (Figure 6D; Supplemental Methods), which we named TP-modules, or TPMs. Among the TPMs, 8938 (17.18%) exclusively consisted of 30 401 eQTL-hotspots of recovered eSNPs (recovered TPM, Figure 6D; Table S7). We identified 11–232 highly interconnected TPMs from each tissue or cell type (highly connected TPM, Table S7). Notably, 17.88% of the TPMs are tissue-specific, with their edges present in not more than 10% (n = 4) [44] of the tissues or cell types (tissue-specific TPM, Table S7). However, only 0.05% of these TPMs are tissue-shared, with their edges present in more than 90% (n = 32) [44] of the tissues or cell types (tissue-shared TPM, Table S7).
We further annotated the TPMs to identify potential trans-acting factors, including transcription factors [26, 45], and non-coding RNAs [46]. Notably, in TPMs where certain transcription factors were overrepresented, we observed significant correlations between the binding activities of the transcription factors and eGene expression levels (Figure 6E and F; Figure S10A–C). For example, in blood, IRF4 is represented in seven TPMs, of which the scaled expression levels of the eGenes are significantly correlated (Pearson’s R = 0.84, P = 0.037) with IRF4 binding activities in the hotspots. Among these eGenes, ACY3 (TPM 1033) was previously annotated as a target gene of IRF4 [47] (Figure 6E). Similarly, MYC is represented in 11 TPMs in suprapubic skin. MYC binding activity was correlated with the expression of the eGenes in these TPMs (Pearson’s R = 0.66, P = 0.026), including some notable targets of MYC (CD151 in TPM 1470 and AEN in TPM 1501) [47] (Figure 6F).
In summary, we developed a framework of epigenome-augmented eQTL analysis and prioritized genomic regions with regulatory potential. Based on this framework, we constructed a genome-wide cis-regulatory network and suggested new transcription programs.
DISCUSSION
Genome-wide mapping of eQTLs has yielded a plethora of genetic variants that are involved in transcription regulation and contributed to a better understanding of the etiology of complex diseases [8–11, 16–18]. However, eQTL studies always face the dilemma of the control of Type I (false discovery) and Type II errors (statistical power). Most of the current studies tend to use stringent adjustment of test P-values to ensure that the resulting loci are empirically verifiable regulatory elements [3, 6, 10, 12]. Nevertheless, for many more associations that did not meet the significance criteria, questions remain about whether these associations represent true biology or random effects.
Recent studies, such as QTLtools [48], FastQTL [20] and EPISPOT [49], have used computational models to enhance the sensitivity and specificity in QTL discovery. Many of these models leverage epigenomic features to prioritize variants with promising functional impacts [49]. Nevertheless, annotations based on epigenomic features are subject to substantial non-specific variations such as sampling, environmental and sequencing biases, let alone cell types and tissues [50–52]. Other studies, have focused on individual loci associated with multiple responses, or ‘hotspots’ [49, 53, 54]. Inspired by previous studies, our approach aimed to identify genomic regions with highly frequent, multiple eQTL associations, namely, ‘eQTL-hotspot’. Our study utilized well-curated eQTL databases from various tissues or cell types. We consider that the occurrence of significant genetic associations across tissues or cell types follows the NHPP, which is governed by locus-specific parameters, 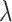 . Our data suggest that in multiple normal tissues or cell types, there are thousands of such hotspots of highly active eQTL associations. To account for genetic conservation, we estimated
. Our data suggest that in multiple normal tissues or cell types, there are thousands of such hotspots of highly active eQTL associations. To account for genetic conservation, we estimated 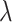 based on DNA segments with an equal number of SNPs.
based on DNA segments with an equal number of SNPs.
Most eQTL associations are cell-specific, although there are exceptions [12]. Based on the limited functional evidence, the causal variants of eQTLs interrupt with a cis-element and thus alter the transcription program of the target genes [3, 55]. Nevertheless, the interruption of a transcription program may occur in cis, in trans or even between cells; hence, the biological background of eQTLs in tissue is usually complicated by many conditions [3, 56]. Our analysis is based on the NHPP, which considers the eSNPs reported in each independent study as a random point process [31]. By integrating eQTL data from different independent studies at a pan-tissue level, and thresholding for significance, our analysis was less affected by tissue-specific conditions and biases. Our findings revealed that multiple variants located in a confined locus (eQTL-hotspot), typically an LD block, demonstrate highly frequent eQTL activities in multiple tissues or cell types, which imply conserved transcription programs. Indeed, the eQTL-hotspots were characterized by a consensus landscape of relevant epigenomic marks, which informed the identification of genomic regions with similar regulatory potentials in specific tissues or cell types.
In most regulons, trans-acting proteins, such as transcription factors, recognize and bind to cis-elements with conserved sequence motifs and histone marks, thus initiating transcription [57, 58]. Therefore, in the process of transcriptional programming, proteins, cis-elements and transcriptional activity are highly specific. In this study, eQTL-hotspots were considered as potential cis-elements, and the variability of the sequence context provided critical information for the underlying transcriptional programming. We reported two distinct classes of eQTL-hotspots characterized by distinct genomic and epigenomic features. In the characterization, eQTL-hotspot-C2 showed colocalized with chromatin states and cCREs related to active TSS and enhancers, while hotspot-C1 is less active and correspond to inactive promoters.
Identification of cis-elements is key to understanding gene expression regulation [59]. However, individual eQTL data are too sparse to reveal the landscape of cis-elements [13], whereas epigenomic marks lack specificity [26]. Our study combined multiple sparse eQTL data to define eQTL-hotspots and thereby retrieved highly specific genomic and epigenomic signatures for the prediction of potential cis-elements at the whole genome level. This study provides an alternative approach to accurately depict the regulatory landscape based on existing data. Furthermore, it ought to be mentioned that the hotspots identified by pan-tissue eQTL activity do not necessarily represent those tissue- or cell-type-specific cis-element but offer distinct genomic and epigenomic signatures that help to identify more specific eQTLs.
The present study offered two major advances in the field. First, by integrating pan-tissue eQTLs and epigenomic data, we retrieved a set of eQTL-hotspots with distinct genomic and epigenomic signatures. The results provided insight into the epigenomic landscape of eQTLs and were highly consistent with the up-to-date knowledge of cis-elements involved in gene expression. Then, by augmented eQTL-analysis and the corresponding interaction map, we provided a comprehensive view of the cis-regulatory map of gene expression in normal tissues or cell types, which serves as a foundational knowledge base for advancing future studies on transcriptional regulation.
Nevertheless, the current study is based on post hoc analysis of existing eQTLs and epigenomic data and hence is subject to all biases in the original studies, such as the sensitivity of ChIP-seq data [29, 30] and the sampling biases in the original eQTL studies [12, 13]. Moreover, trans-eQTLs, which represent a major class of genetic regulation of gene expression [60, 61], were not included in the current study because of poor representation in the database. Other QTLs related to transcription activities, such as methylation QTLs [62] and splicing QTLs [13], were also excluded. However, the current method can also be applied to these QTLs when larger databases are available in the future. Finally, the current NHPP model was based on test significance only. Prompted by previous studies, our future model can incorporate more statistics, such as effect size, response types, allele frequency and LD among SNPs.
In summary, we described an analytical framework of augmented eQTL mapping and thereby performed genome-wide identification of cis-elements in different tissues or cell types, and reconstrued a comprehensive interaction map of the poised cis-elements, which contributed to a better understanding of the dynamics of transcriptional regulation.
METHODS
The classification of eQTL-hotspots
To classify eQTL-hotspots, we first identified hotspot-specific k-mers through a sequence comparison between the hotspots and their surrounding regions. Using bedtools shuffle, we generated random genomic locations in hotspots’ surrounding regions that resemble actual hotspots of the same size. Subsequently, we generated sequences for both the hotspots and random genomic locations based on the hg38 using bedtools getfasta [63]. We generated six bases k-mers count matrix Seekr [33] and performed a two-sided Wilcoxon rank-sum test to identify the k-mers that counts exhibited significant differences (FDR-adjusted P < 0.05) between the hotspots and random genomic locations. We then conducted permutation tests to calculate expected values for k-mers with fold changes ≥1.5 or ≤0.9 (Figure S11). Expected values in the random genomic locations were determined based on 1000 random datasets. We also used the permutation test to calculate empirical P-values.
Next, we conducted hotspot-specific k-mers dimensionality reduction using kernel principal component analysis (kpca) with rbfdot kernel [34]. Then we selected the top three principal components (PCs) with the largest variation as features (Figure S12). We selected 50 neighbors for each hotspot using the R hnsw_knn package [64], and constructed a graph using the R igraph package [35].
Finally, we effectively classified the hotspots in the graph using the Leiden algorithm implemented through the R cluster_leiden package with a resolution of 0.00002 [35].
Refining new thresholds of significant eQTLs in hotspots
We generated Q-Q plots for each tissue or cell type using test P-values for eSNP–eGene pairs that were randomly sampled from hotspot-C1, C2 and pan-tissue non-hotspot (C0). We used test P-values from hotspot-C1 and C2 as observed values and the test P-values from non-hotspot (C0) as expected values. To establish the significance thresholds for eSNP–eGene pairs in hotspot-C1 and C2, we identified them as the y-coordinates of the intersection points between the curves and a line characterized by an intercept of −log10FDR and a slope of 1, where the FDR was set at 0.001 [42]. The coordinates of the intersections were determined by fitting polynomial regression to the hotspot-C1 and C2 curves, respectively.
Key Points
We used the non-homogeneous Poisson process to analyze a consortium of eQTL datasets and thus identified 125 489 eQTL-hotspots with highly frequent, pan-tissue eSNP activities.
We stratified the eQTL-hotspots into two classes based on the genomic features and further characterized these eQTL-hotspots for regulatory potential by distinct epigenomic signatures and the selective enrichment of annotated cis-elements.
We developed ‘E-SpotFinder’, a machine learning model trained by the consensus genomic and epigenomic features of eQTL-hotspots and capable of inferring genomic regions with high regulatory potential in specific tissues or cell types.
We established an augmented eQTL mapping based on ‘E-SpotFinder’ predicted segments of the genome and with refined P-value adjustment for eQTL, thus recovering 655 402 eSNPs, which strongly enriched for gene-expression regulation activity.
We generated a comprehensive cis-regulatory map spanning 36 unique human tissues or cell types and identified modules of transcriptional programming associated with specific transcription factors that influence the expression of genes.
Supplementary Material
Author Biographies
Huanhuan Liu is a PhD candidate at the Department of Hematology, The First Affiliated Hospital of Xiamen University and Institute of Hematology, School of Medicine, Xiamen University.
Qinwei Chen is a PhD at the Department of Hematology, The First Affiliated Hospital of Xiamen University and Institute of Hematology, School of Medicine, Xiamen University.
Jintao Guo is a Postdoctoral Fellow at the National Institute for Data Science in Health and Medicine, School of Medicine, Xiamen University.
Ying Zhou is a Research Scientist at the National Institute for Data Science in Health and Medicine, School of Medicine, Xiamen University.
Zhiyu You is a PhD candidate at the National Institute for Data Science in Health and Medicine, School of Medicine, Xiamen University.
Jun Ren is a PhD candidate at the School of Informatics, Xiamen University.
Yuanyuan Zeng is a PhD candidate at the National Institute for Data Science in Health and Medicine, School of Medicine, Xiamen University.
Jing Yang is a PhD candidate at the National Institute for Data Science in Health and Medicine, School of Medicine, Xiamen University.
Jialiang Huang is a Professor at the School of Life Sciences, Xiamen University.
Qiyuan Li is a Professor at the Department of Hematology, The First Affiliated Hospital of Xiamen University and Institute of Hematology, School of Medicine, Xiamen University.
Contributor Information
Huanhuan Liu, Department of Hematology, The First Affiliated Hospital of Xiamen University and Institute of Hematology, School of Medicine, Xiamen University, Xiamen, 361102, China; National Institute for Data Science in Health and Medicine, School of Medicine, Xiamen University, Xiamen, 361102, China; Department of Pediatrics, Women and Children's Hospital, School of Medicine, 361003, Xiamen University, Xiamen, China.
Qinwei Chen, Department of Hematology, The First Affiliated Hospital of Xiamen University and Institute of Hematology, School of Medicine, Xiamen University, Xiamen, 361102, China; National Institute for Data Science in Health and Medicine, School of Medicine, Xiamen University, Xiamen, 361102, China.
Jintao Guo, Department of Hematology, The First Affiliated Hospital of Xiamen University and Institute of Hematology, School of Medicine, Xiamen University, Xiamen, 361102, China; National Institute for Data Science in Health and Medicine, School of Medicine, Xiamen University, Xiamen, 361102, China; Department of Pediatrics, Women and Children's Hospital, School of Medicine, 361003, Xiamen University, Xiamen, China.
Ying Zhou, Department of Hematology, The First Affiliated Hospital of Xiamen University and Institute of Hematology, School of Medicine, Xiamen University, Xiamen, 361102, China; National Institute for Data Science in Health and Medicine, School of Medicine, Xiamen University, Xiamen, 361102, China; Department of Pediatrics, Women and Children's Hospital, School of Medicine, 361003, Xiamen University, Xiamen, China.
Zhiyu You, Department of Hematology, The First Affiliated Hospital of Xiamen University and Institute of Hematology, School of Medicine, Xiamen University, Xiamen, 361102, China; National Institute for Data Science in Health and Medicine, School of Medicine, Xiamen University, Xiamen, 361102, China; Department of Pediatrics, Women and Children's Hospital, School of Medicine, 361003, Xiamen University, Xiamen, China.
Jun Ren, Department of Hematology, The First Affiliated Hospital of Xiamen University and Institute of Hematology, School of Medicine, Xiamen University, Xiamen, 361102, China; School of Informatics, Xiamen University, Xiamen, 361102, China.
Yuanyuan Zeng, Department of Hematology, The First Affiliated Hospital of Xiamen University and Institute of Hematology, School of Medicine, Xiamen University, Xiamen, 361102, China; National Institute for Data Science in Health and Medicine, School of Medicine, Xiamen University, Xiamen, 361102, China; Department of Pediatrics, Women and Children's Hospital, School of Medicine, 361003, Xiamen University, Xiamen, China.
Jing Yang, Department of Hematology, The First Affiliated Hospital of Xiamen University and Institute of Hematology, School of Medicine, Xiamen University, Xiamen, 361102, China; National Institute for Data Science in Health and Medicine, School of Medicine, Xiamen University, Xiamen, 361102, China; Department of Pediatrics, Women and Children's Hospital, School of Medicine, 361003, Xiamen University, Xiamen, China.
Jialiang Huang, State Key Laboratory of Cellular Stress Biology, School of Life Sciences, Faculty of Medicine and Life Sciences, Xiamen University, Xiamen, Fujian 361102, China.
Qiyuan Li, Department of Hematology, The First Affiliated Hospital of Xiamen University and Institute of Hematology, School of Medicine, Xiamen University, Xiamen, 361102, China; National Institute for Data Science in Health and Medicine, School of Medicine, Xiamen University, Xiamen, 361102, China; Department of Pediatrics, Women and Children's Hospital, School of Medicine, 361003, Xiamen University, Xiamen, China.
FUNDING
This work was supported by National Natural Science Foundation of China (82272944 to Q.L. and 82203420 to J.G.).
DATA AVAILABILITY
All datasets analyzed in this study were published previously [12, 13, 26, 27, 32, 41, 43]. The code of E-SpotFinder was available via GitHub https://github.com/Lhhuan/E-SpotFinder. Supplementary Tables are available online at http://bib.oxfordjournals.org/.
AUTHOR CONTRIBUTIONS
Q.L. conceived the study. Q.L. and H.L. designed algorithms. H.L. performed all computational experiments. Q.L. and H.L. contributed to the manuscript writing. Q.L. was responsible for the decision to submit the manuscript. J.H., Y.Z. and Q.C. contributed scientific expertise. J.H., J.G. and Q.C. contributed to the review of the manuscript. All the authors read and approved the manuscript.
References
- 1. Gamazon ER, Segrè AV, Bunt M, et al. Using an atlas of gene regulation across 44 human tissues to inform complex disease- and trait-associated variation. Nat Genet 2018;50(7):956–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Flynn ED, Tsu AL, Kasela S, et al. Transcription factor regulation of eQTL activity across individuals and tissues. PLoS Genet 2022;18(1):e1009719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jung I, Schmitt A, Diao Y, et al. A compendium of promoter-centered long-range chromatin interactions in the human genome. Nat Genet 2019;51(10):1442–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hong D, Lin H, Liu L, et al. Complexity of enhancer networks predicts cell identity and disease genes revealed by single-cell multi-omics analysis. Brief Bioinform 2023;24(1):1–13. [DOI] [PubMed] [Google Scholar]
- 5. Brown CD, Mangravite LM, Engelhardt BE. Integrative modeling of eQTLs and cis-regulatory elements suggests mechanisms underlying cell type specificity of eQTLs. PLoS Genet 2013;9(8):e1003649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chandra V, Bhattacharyya S, Schmiedel BJ, et al. Promoter-interacting expression quantitative trait loci are enriched for functional genetic variants. Nat Genet 2021;53(1):110–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Taylor K, Davey Smith G, Relton CL, et al. Prioritizing putative influential genes in cardiovascular disease susceptibility by applying tissue-specific Mendelian randomization. Genome Med 2019;11(1):6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yang H, Liu D, Zhao C, et al. Mendelian randomization integrating GWAS and eQTL data revealed genes pleiotropically associated with major depressive disorder. Transl Psychiatry 2021;11(1):225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bryois J, Calini D, Macnair W, et al. Cell-type-specific cis-eQTLs in eight human brain cell types identify novel risk genes for psychiatric and neurological disorders. Nat Neurosci 2022;25(8):1104–12. [DOI] [PubMed] [Google Scholar]
- 10. Zhu Z, Zhang F, Hu H, et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat Genet 2016;48(5):481–7. [DOI] [PubMed] [Google Scholar]
- 11. Hormozdiari F, van de Bunt M, Segrè AV, et al. Colocalization of GWAS and eQTL signals detects target genes. Am J Hum Genet 2016;99(6):1245–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Consortium GT. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 2020;369(6509):1318–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kerimov N, Hayhurst JD, Peikova K, et al. A compendium of uniformly processed human gene expression and splicing quantitative trait loci. Nat Genet 2021;53(9):1290–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li Q, Seo JH, Stranger B, et al. Integrative eQTL-based analyses reveal the biology of breast cancer risk loci. Cell 2013;152(3):633–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li W, Xu C, Guo J, et al. Cis- and trans-acting expression quantitative trait loci of long non-coding RNA in 2,549 cancers with potential clinical and therapeutic implications. Front Oncol 2020;10:602104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sheng Q, Samuels DC, Yu H, et al. Cancer-specific expression quantitative loci are affected by expression dysregulation. Brief Bioinform 2020;21(1):338–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Geeleher P, Nath A, Wang F, et al. Cancer expression quantitative trait loci (eQTLs) can be determined from heterogeneous tumor gene expression data by modeling variation in tumor purity. Genome Biol 2018;19(1):130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gillies CE, Putler R, Menon R, et al. An eQTL landscape of kidney tissue in human nephrotic syndrome. Am J Hum Genet 2018;103(2):232–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lawrenson K, Li Q, Kar S, et al. Cis-eQTL analysis and functional validation of candidate susceptibility genes for high-grade serous ovarian cancer. Nat Commun 2015;6:8234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ongen H, Buil A, Brown AA, et al. Fast and efficient QTL mapper for thousands of molecular phenotypes. Bioinformatics 2016;32(10):1479–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gong J, Mei S, Liu C, et al. PancanQTL: systematic identification of cis-eQTLs and trans-eQTLs in 33 cancer types. Nucleic Acids Res 2018;46(D1):D971–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chen C, Liu Y, Luo M, et al. PancanQTLv2.0: a comprehensive resource for expression quantitative trait loci across human cancers. Nucleic Acids Res 2023;52:D1400–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Consortium GT, et al. Genetic effects on gene expression across human tissues. Nature 2017;550(7675):204–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Abell NS, DeGorter MK, Gloudemans MJ, et al. Multiple causal variants underlie genetic associations in humans. Science 2022;375(6586):1247–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bossini-Castillo L, Glinos DA, Kunowska N, et al. Immune disease variants modulate gene expression in regulatory CD4(+) T cells. Cell Genom 2022;2(4):100117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zheng R, Wan C, Mei S, et al. Cistrome data browser: expanded datasets and new tools for gene regulatory analysis. Nucleic Acids Res 2019;47(D1):D729–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. ENCODE Project Consortium, Moore JE, Purcaro MJ, et al. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 2020;583(7818):699–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Consortium EP. An integrated encyclopedia of DNA elements in the human genome. Nature 2012;489(7414):57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nakato R, Shirahige K. Sensitive and robust assessment of ChIP-seq read distribution using a strand-shift profile. Bioinformatics 2018;34(14):2356–63. [DOI] [PubMed] [Google Scholar]
- 30. Park PJ. ChIP-seq: advantages and challenges of a maturing technology. Nat Rev Genet 2009;10(10):669–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cebrian AC, Abaurrea J, Asin J. NHPoisson: an R package for fitting and validating nonhomogeneous Poisson processes. J Stat Softw 2015;64(6):1–25. [Google Scholar]
- 32. Roadmap Epigenomics C, Kundaje A, Meuleman W, et al. Integrative analysis of 111 reference human epigenomes. Nature 2015;518(7539):317–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kirk JM, Kim SO, Inoue K, et al. Functional classification of long non-coding RNAs by k-mer content. Nat Genet 2018;50(10):1474–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Scholkopf B, Smola A, Muller KR. Nonlinear component analysis as a kernel eigenvalue problem. Neural Comput 1998;10(5):1299–319. [Google Scholar]
- 35. Csardi G, Nepusz T. The igraph software package for complex network research. InterJournal, Complex Systems 2006;1695. [Google Scholar]
- 36. Ramírez F, Ryan DP, Grüning B, et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 2016;44(W1):W160–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Reiff SB, Schroeder AJ, Kırlı K, et al. The 4D Nucleome Data Portal as a resource for searching and visualizing curated nucleomics data. Nat Commun 2022;13(1):2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chen T, Guestrin C. XGBoost: a scalable tree boosting system. In: Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining. San Francisco, California, USA: Association for Computing Machinery, 2016, 785–94. [Google Scholar]
- 39. Lundberg SM, Erion G, Chen H, et al. From local explanations to global understanding with explainable AI for trees. Nat Mach Intell 2020;2(1):56–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mohammadi P, Castel SE, Brown AA, Lappalainen T. Quantifying the regulatory effect size of cis-acting genetic variation using allelic fold change. Genome Res 2017;27(11):1872–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Welter D, MacArthur J, Morales J, et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res 2014;42(Database issue):D1001–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Galwey NW. A Q-Q plot aids interpretation of the false discovery rate. Biom J 2023;65(1):e2100309. [DOI] [PubMed] [Google Scholar]
- 43. Li X, Shi L, Wang Y, et al. OncoBase: a platform for decoding regulatory somatic mutations in human cancers. Nucleic Acids Res 2019;47(D1):D1044–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fagny M, Paulson JN, Kuijjer ML, et al. Exploring regulation in tissues with eQTL networks. Proc Natl Acad Sci U S A 2017;114(37):E7841–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Castro-Mondragon JA, Riudavets-Puig R, Rauluseviciute I, et al. JASPAR 2022: the 9th release of the open-access database of transcription factor binding profiles. Nucleic Acids Res 2022;50(D1):D165–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Consortium RN. RNAcentral 2021: secondary structure integration, improved sequence search and new member databases. Nucleic Acids Res 2021;49(D1):D212–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rouillard AD, Gundersen GW, Fernandez NF, et al. The harmonizome: a collection of processed datasets gathered to serve and mine knowledge about genes and proteins. Database (Oxford) 2016;2016:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Delaneau O, Ongen H, Brown AA, et al. A complete tool set for molecular QTL discovery and analysis. Nat Commun 2017;8:15452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ruffieux H, Fairfax BP, Nassiri I, et al. EPISPOT: an epigenome-driven approach for detecting and interpreting hotspots in molecular QTL studies. Am J Hum Genet 2021;108(6):983–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rivera CM, Ren B. Mapping human epigenomes. Cell 2013;155(1):39–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Vandereyken K, Sifrim A, Thienpont B, Voet T. Methods and applications for single-cell and spatial multi-omics. Nat Rev Genet 2023;24(8):494–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rahmani E, Schweiger R, Rhead B, et al. Cell-type-specific resolution epigenetics without the need for cell sorting or single-cell biology. Nat Commun 2019;10(1):3417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Barmukh R, Roorkiwal M, Dixit GP, et al. Characterization of ‘QTL-hotspot’ introgression lines reveals physiological mechanisms and candidate genes associated with drought adaptation in chickpea. J Exp Bot 2022;73(22):7255–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wu P-Y, Yang MH, Kao CH. A statistical framework for QTL hotspot detection. G3 Genes|Genomes|Genetics 2021;11(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Battle A, Montgomery SB. Determining causality and consequence of expression quantitative trait loci. Hum Genet 2014;133(6):727–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Alasoo K, Rodrigues J, Mukhopadhyay S, et al. Shared genetic effects on chromatin and gene expression indicate a role for enhancer priming in immune response. Nat Genet 2018;50(3):424–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yang MG, Ling E, Cowley CJ, et al. Characterization of sequence determinants of enhancer function using natural genetic variation. Elife 2022;11:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Inukai S, Kock KH, Bulyk ML. Transcription factor-DNA binding: beyond binding site motifs. Curr Opin Genet Dev 2017;43:110–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kim S, Wysocka J. Deciphering the multi-scale, quantitative cis-regulatory code. Mol Cell 2023;83(3):373–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Yao C, Joehanes R, Johnson AD, et al. Dynamic role of trans regulation of gene expression in relation to complex traits. Am J Hum Genet 2017;100(4):571–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Brynedal B, Choi JM, Raj T, et al. Large-scale trans-eQTLs affect hundreds of transcripts and mediate patterns of transcriptional co-regulation. Am J Hum Genet 2017;100(4):581–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zheng Z, Huang D, Wang J, et al. QTLbase: an integrative resource for quantitative trait loci across multiple human molecular phenotypes. Nucleic Acids Res 2020;48(D1):D983–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 2010;26(6):841–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Malkov YA, Yashunin DA. Efficient and robust approximate nearest neighbor search using hierarchical navigable small world graphs. IEEE Trans Pattern Anal Mach Intell 2020;42(4):824–36. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All datasets analyzed in this study were published previously [12, 13, 26, 27, 32, 41, 43]. The code of E-SpotFinder was available via GitHub https://github.com/Lhhuan/E-SpotFinder. Supplementary Tables are available online at http://bib.oxfordjournals.org/.