

Abstract
Background and Aims:
Relative roles of HSCs and portal fibroblasts in alcoholic hepatitis (AH) are unknown. We aimed to identify subpopulations of collagen type 1 alpha 1 (Col1a1)–expressing cells in a mouse AH model by single-cell RNA sequencing (scRNA-seq) and filtering the cells with the HSC (lecithin retinol acyltransferase [Lrat]) and portal fibroblast (Thy1 cell surface antigen [Thy1] and fibulin 2 [Fbln2]) markers and vitamin A (VitA) storage.
Approach and Results:
Col1a1–green fluorescent protein (GFP) mice underwent AH, CCl4, and bile duct ligation (BDL) procedures to have comparable F1-F2 liver fibrosis. Col1a1-expressing cells were sorted via FACS by VitA autofluorescence and GFP for single-cell RNA sequencing. In AH, approximately 80% of Lrat+Thy1−Fbln2− activated HSCs were VitA-depleted (vs. ~13% in BDL and CCl4). Supervised clustering identified a subset co-expressing Lrat and Fbln2 (Lrat+Fbln2+), which expanded 44-fold, 17-fold, and 1.3-fold in AH, BDL, and CCl4. Lrat+Fbln2+ cells had 3–15-times inductions of profibrotic, myofibroblastic, and immunoregulatory genes versus Lrat+Fbln2− cells, but 2–4-times repressed HSC-selective genes. AH activated HSCs had up-regulated inflammatory (chemokine [C-X-C motif] ligand 2 [Cxcl2], chemokine [C-C motif] ligand 2), antimicrobial (Il-33, Zc3h12a), and antigen presentation (H2-Q6, H2-T23) genes versus BDL and CCl4. Computational deconvolution of AH versus normal human bulk-liver RNA-sequencing data supported an expansion of LRAT+FBLN2+ cells in AH; AH patient liver immunohistochemistry showed FBLN2 staining along fibrotic septa enriched with LRAT+ cells; and in situ hybridization confirmed co-expression of FBLN2 with CXCL2 and/or human leukocyte antigen E in patient AH. Finally, HSC tracing in Lrat-Cre;Rosa26mTmG mice detected GFP+FBLN2+ cells in AH.
Conclusion:
A highly profibrotic, inflammatory, and immunoregulatory Lrat+Fbln2+ subpopulation emerges from HSCs in AH and may contribute to the inflammatory and immunoreactive nature of AH.
INTRODUCTION
Spatially defined liver mesenchymal cells with the common Mesp1+ mesoderm origin exist in the liver, including perisinusoidal HSCs, perivascular fibroblasts, and subcapsular fibroblasts.[1,2] They all respond to injury to initiate wound healing and liver fibrosis in the manner that is topographically governed by the site of injury by undergoing phenotypic changes commonly described as myofibroblastic activation.[3–9] To ascertain the relative roles and fate of these mesenchymal cells in liver fibrosis, the cell type–specific markers have been used. For instance, lecithin retinol acyltransferase (Lrat) is one of the accepted markers for quiescent and activated HSCs (aHSCs), while Thy1 cell surface antigen (Thy1) and fibulin-2 (Fbln2) are used to distinguish portal fibroblasts (PFs) from HSCs.[6]
Single-cell RNA sequencing (scRNA-seq) has identified heterogeneous liver mesenchymal cell subpopulations with distinct transcriptomic features allowing their classifications based on spatial/zonal and functional characteristics.[10–14] Several clusters of aHSCs and S100 calcium binding protein A6 (S100a6)–expressing myofibroblasts (MFBs)[10] were identified by scRNA-seq of CCl4 rodent models, consisting of fibrogenic, inflammatory, immunoregulatory, or regenerative clusters.[10,12] Identification of zonal HSC subpopulations revealed the major contribution by pericentral HSCs to CCl4-induced liver fibrosis in mice.[11] In healthy human liver, scRNA-seq identified perivascular HSCs with selective expression of glypican 3, dermatopontin, fibulin 1, and osteoglycin versus perisinusoidal HSCs expressing dopamin beta-hydroxylase, collectin-10 (COLEC10), and angiopoeitin like 6 (ANGPTL6).[15] aHSCs from a mouse NASH model were enriched with the expression of secreted molecules termed “stellakines,” which may mediate signaling network through the receptors for IL-11 and vasoactive molecules.[13] Using a NASH mouse model of foz/foz mice on Western diet, four aHSC clusters were identified such as classic MFBs, proliferating aHSCs, intermediate aHSCs, and immune and inflammatory aHSCs, as shown in other models but also identifying NASH-unique aHSCs expressing clusters of differentiation 36, lymphocyte antigen 6 and c-type lectin, which persisted even in recovery from NASH.[14]
In implementing the present study on an alcohol-associated hepatitis (AH) mouse model, we had two main rationales. The first and most obvious was the absence of prior scRNA-seq studies to characterize aHSCs and MFBs in an AH model. The second reason was our revelation that, uniquely in AH mice, there is a remarkable reduction in vitamin A–containing (VitA+) aHSC population with a reciprocal increase in VitA− aHSCs as detected by FACS analysis of Collagen α1(I) promoter (Col1a1)–green fluorescent protein (GFP) mice subjected to the AH regimen (Figure 1D), raising a natural question as to what subpopulations of aHSCs or activated PFs (aPFs) constitute this expanded pool of VitA− Col1a1-expressing cells in the model. To best approach these questions, we have collected VitA+GFP+ and VitA−GFP+ “activated” liver mesenchymal cells from the AH Col1a1-GFP mice via FACS gated by VitA autofluorescence and GFP and analyzed these two fractions by scRNA-seq.
FIGURE 1.
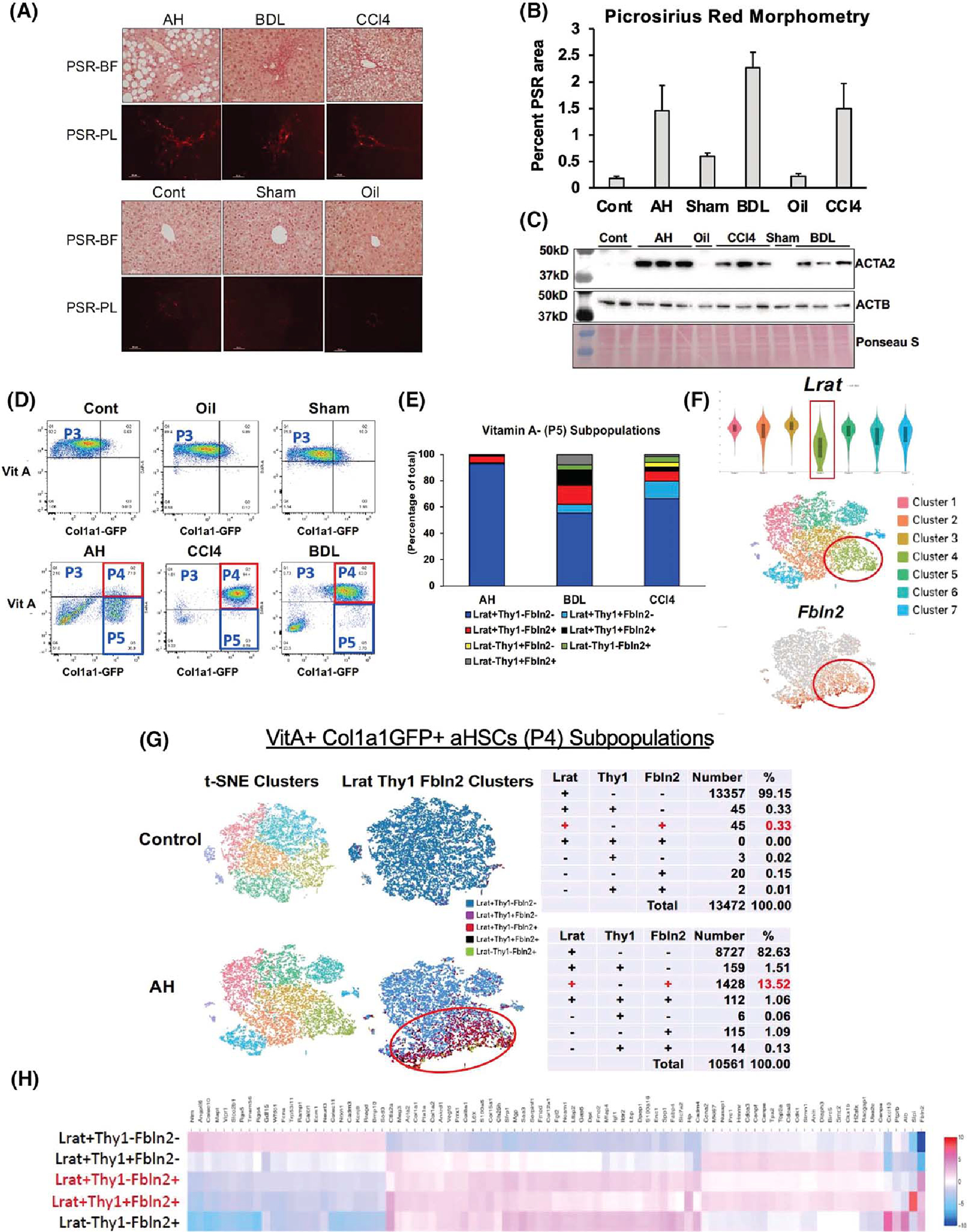
(A) Picrosirius red (PSR) staining of the alcoholic hepatitis (AH), bile duct ligation (BDL), and mouse CCl4 livers (upper panel) and respective control (Cont) livers (lower panel) imaged under bright field (PSR-BF) or polarized light (PSR-PL). Scale: ×20. (B) Morphometric analysis data of PSR-PL images of ten 400-μm2 random foci in perivenular or periportal zone per mouse using the image J software (n = 3 for each mouse group). (C) Immunoblot analysis of α-smooth muscle Actin (ACTA2), the accepted readout of liver fibrosis, with β-actin (ACTB) as a loading control. (D) FACS separation of liver mesenchymal cells gated by vitamin A (VitA) autofluorescence (y-axis) and collagen type 1 alpha 1 (Col1a1)–green fluorescent protein (GFP) (x-axis). (E) Percent distribution of various cell subpopulations in VitA− (P5) mesenchymal cells from AH, BDL, and CCl4 models based on lecithin retinol acyltransferase (Lrat), Thy1 cell surface antigen (Thy1), and fibulin-2 (Fbln2) expression. (F) Lrat expression of single-cell RNA-sequencing (scRNA-seq) clusters of the AH VitA+GFP+ P4 cells shown by violin plot (top), t-distributed stochastic neighbor embedding (t-SNE) plot of the clusters (middle), and distribution of Fbln2-expressing cells (bottom). (G) t-SNE plots of the control and AH mouse P4 activated HSCs (aHSCs; left), supervised clustering based on Lrat, Thy1, and Fbln2 expression (middle), and summary tables for the breakdown of subpopulations based on the expression of the three genes (right). (H) Heatmap of differentially expressed genes (DEGs) comparing the five subpopulations of P4 aHSCs based on Lrat, Thy1, and Fbln2 expression.
To specifically address our primary question on the relative contributions of aHSCs versus aPFs, we have performed a supervised clustering based on the expression of the HSC marker Lrat and the PF marker Thy1 and Fbln2. This approach has led to identification of a Lrat+Fbln2+ subpopulation that emerged and expanded more conspicuously in both VitA+GFP+ and VitA− GFP+ cells of the AH model as compared with CCl4 or bile duct ligation (BDL). The Lrat+Fbln2+ subset is most profibrotic, myofibroblastic and inflammatory, and its transcriptomic landscape and trajectory analysis suggested they are “transitional” cells between Lrat+Fbln2− aHSCs and Lrat−Fbln2+ MFBs/aPFs. Indeed, a lineage tracing analysis using Lrat− Cre;Rosa26mTmG mice on the AH regimen detected Lrat+ cell–derived FBLN2+ cells in the fibrotic septum, supporting the HSC origin of this unique subpopulation.
METHODS
Animals
Male Col1a1-GFP mice[16] on C57BL/6 background were used at 8 weeks of age for the following models produced by the Animal Core of the Southern California Research Center for ALPD and Cirrhosis. Mice were subjected to the 7-week AH regimen of hybrid feeding of intragastric ethanol diet infusion and ad libitum consumption of Western diet plus weekly alcohol binge,[17] 12 days of cholestasis caused by aseptic ligation of the common bile duct (BDL),[18] or five biweekly injections of CCl4 (1.5 μl/g diluted 2-fold with mineral oil). For genetic tracing of Lrat+ HSCs, male 8-week-old Lrat-Cre mice were obtained from Dr. Robert Schwabe of Columbia University and crossed with Rosa26mTmG (B6.129 (Cg)-Gt(ROSA)26Sor tm4(ACTB-tdTomato, -EGFP) Luo/J) mice (Jackson Laboratory) to generate the compound mice, which were subjected to the AH regimen. All mice were maintained in accordance with the National Institutes of Health guidelines for the use and care of live animals within the Association for Assessment and Accreditation of Laboratory Animal Care–accredited Department of Animal Resources of the University of Southern California. Animal protocols used for the present study were approved by the Institutional Animal Care and Use Committee (protocol #20068 and #20508).
Liver mesenchymal cell isolation
Mouse livers were sequentially perfused in situ via a catheter placed into superior vena cava with Ca2+-free Eagle's minimum essential medium with ethylene glycol tetraacetic acid, DMEM/F-12 containing 0.7% (wt/vol) pronase and DMEM/F-12 with 0.044% (wt/vol) collagenase (type IV). Non-parenchymal liver cells were collected by removal of hepatocytes by 1-min 50 g centrifugation followed by 8-min 800 g centrifugation and laid on top of discontinuous gradients (1.080, 1.054, 1.043, and 1.034) of OptiPrep for ultracentrifugation at 21,400 rpm for 20 min.[18] The cells from the top four gradient interfaces were collected and subjected to FACS performed by Flow Cytometry and Immune Monitoring Core of the USC Norris Comprehensive Cancer Center. The cells were sorted on ARIA IIu (Becton Dickinson) with solid state laser excited at 488 nm and measured with 510/20BP emission mirrors for GFP and excited at 355 nm and measured with 450/50BP mirrors for VitA.
RESULTS
VitA-depleted aHSCs in AH
For scRNA-seq analysis of HSC subpopulations in AH in comparison to liver fibrosis induced by BDL or CCl4, we performed preliminary studies to establish induction of comparable F1-F2 liver fibrosis in the three models as detected by picrosirius red staining (Figure 1A) and its morphometric data (Figure 1B). Immunoblotting of liver proteins for α-smooth muscle actin (ACTA2) also confirmed clear up-regulation of this bona-fide fibrosis marker in the three models as compared with the respective controls (Figure 1C). Liver mesenchymal cells from Col1a1-GFP mice subjected to the three fibrosis regimens and appropriate control treatments were separated by FACS via gating based on VitA autofluorescence and Col1a1-driven GFP to collect VitA+GFP− quiescent HSCs (P3), VitA+GFP+ aHSCs (P4), and VitA−GFP+ cells (P5), most likely comprising VitA-depleted aHSCs and aPFs (Figure 1D). As expected, the P3 population diminished while the P4 cells increased in all three models as compared with the respective controls, reflecting activation of HSCs. However, in AH, a marked increase in the P5 VitA−GFP+ population was noted, accounting for 78% of all the P3-P5 cells, as compared with < 13% in BDL or CCl4. Ninety three percent of these VitA− GFP+ cells in AH expressed Lrat but not Thy1 or Fbln2 by scRNA-seq analysis (Figure 1E), confirming that they are VitA-depleted aHSCs. In BDL and CCl4, VitA− Lrat+Thy1−Fbln2− cells accounted for 55%–65% of the P5 population, but their numbers were 3–4-fold lower than in AH due to much fewer VitA−GFP+ cells. We also noticed a VitA−GFP− population particularly in AH and BDL (Figure 1D). These cells may include other cell types such as macrophages, endothelial cells, even contaminating hepatocytes besides low Col1a1-expressing mesenchymal cells. As these cells are GFP− indicative of having minimal profibrotic potential, we did not include this population in our analysis.
Lrat+Thy1−Fbln2+ aHSCs
A recent scRNA-seq study revealed pericentral (PC) versus periportal (PP) zonal HSC subpopulations with unique expression of the signature genes and a major contribution of PC HSCs to CCl4 fibrosis.[11] Using three of the reported top signature genes for PC (R-spondin 3, spondin 2, and a disintegrin and metalloproteinase with thrompospondin motifs 12) and PP (insulin like growth factor binding protein 3, nerve growth factor receptor [Ngfr], and transgelin), we first assessed the distribution of the respective zonal HSCs in P3 VitA+GFP− quiescent HSCs (qHSCs). This analysis indeed revealed that the PC or PP HSC gene set was co-expressed, and the PC versus PP cells appeared segregated over three separate t-distributed stochastic neighbor embedding clusters (Figure S1A), supporting the existence of these zonal subpopulations. However, in P4 VitA+GFP+ aHSCs from AH, the PC or PP HSC gene set was not co-expressed in most clusters (Figure S1B) and was expressed in a few cells (Figure S1C, left and middle). In fact, many cells expressed both PC and PP HSC genes in clusters 3, 4, and 7 (Figure S1C, right), suggesting that there is no dominant contribution of either zonal HSCs to aHSCs in AH. Instead, our expression analysis for the HSC and PF marker genes in the P4 cells revealed attenuated expression of Lrat in cluster 4, where many cells also expressed the PF marker Fbln2 (Figure 1F), suggesting that a portion of aHSCs in AH may co-express the HSC and PF marker genes. To test this notion, we filtered the scRNA-seq data based on the expression of Lrat, Thy1, and Fbln2 to determine the distribution of VitA+ aHSCs, which expressed one or any of the combination of the three genes. This analysis showed that in a control mouse liver, 99.15% of VitA+GFP+ cells were Lrat+Thy1−Fbln2− aHSCs, and only 0.33% co-expressed Lrat and Thy1 or Fbln2 (Figure 1G, top). However, in AH, the Lrat+Thy1−Fbln2+ population expanded 41-fold to 13.5%, and Lrat+ cells co-expressing both Thy1 and Fbln2 also emerged (1.06%) (Figure 1G, bottom). We confirmed these changes in the second AH mouse (Figure S1D). To assess the transcriptomic landscape of these aHSC subpopulations in AH, we generated a heatmap of differentially expressed genes (DEGs) (Table S1), comparing the Lrat+Thy1−Fbln2− “pure” aHSC subpopulation, the Lrat+ subpopulations with co-expression of Thy1, Fbln2 or both, and Lrat−Thy1−Fbln2+ cells, which most likely are aPFs (Figure 1H). This heatmap depicted a transcriptomic shift from aHSCs to aPFs with Lrat+Thy1−Fbln2+ or Lrat+Thy1+Fbln2+ cells being in the middle of the shift, suggesting that these subpopulations co-expressing Lrat and Fbln2 have “transitional” transcriptional activities between aHSCs and aPFs. To this end, we have performed RNA velocity analysis to assess the transcriptional activity state based on spliced versus unspliced RNA counts, which revealed dynamic transcriptional activity of Lrat+Fbln2+ cells compared with Lrat+Fbln2− aHSCs (Figure S1E). We performed the identical scRNA-seq and supervised clustering to determine whether the Lrat+Fbln2+ subpopulation also exists in P4 cells in BDL or CCl4 liver fibrosis. As in the pair-fed control for AH, Lrat+Thy1−Fbln2+ cells were very rare (0.4%–0.6%) in Sham and Oil control mouse livers and increased to 5.83% and 9.23% of VitA+ aHSCs (P4) in BDL and CCl4, respectively (Figure S1F), revealing that this unique subpopulation also expands in these models. However, in CCl4, Lrat+Thy1−Fbln2+ cells did not cluster but were diffusely distributed.
Profibrotic Lrat+Thy1−Fbln2+ aHSCs
A more focused analysis of the Lrat+Fbln2+ subpopulation revealed that Lrat+Thy1−Fbln2+ or Lrat+Thy1+Fbln2+ cells had several-fold higher expressions of Col1a1, Col1a2, Col12a1, Col15a1, tissue inhibitor of metalloproteinase 1 (Timp1), lysyl oxidase (Lox), Lox1, Acta2, Tgfb2, Tgfbr1, and matrix metalloproteinase 2 (Mmp2) compared with Lrat+Thy1−Fbln2− “pure” aHSCs (Figure 2A). These genes were not significantly elevated in Lrat+Thy1+Fbln2− cells, suggesting that acquiring the expression of Fbln2 but not Thy1 makes Lrat+ aHSCs significantly more profibrotic. Conversely, the genes known to be HSC-specific were largely repressed in Lrat+Thy1−Fbln2+ or Lrat+Thy1+Fbln2+ cells (Figure 2B). In particular, Angptl6, Colec10, ficolin a (Fcna), and Ngfr showed pronounced repressions while desmin and cytoblobin b were not affected. Similar to the profibrotic genes, the MFB genes such as S100a6, N-methylpurine DNA glycosylase, growth arrest-specific 6, Colec12, but not vimentin were all induced 4–7 fold in the Lrat+Thy1− Fbln2+ and Lrat+Thy1+Fbln2+ subpopulations (Figure 2C). Collectively, these results suggested that the VitA+ Lrat+Fbln2+ aHSCs in AH have significantly heightened fibrotic potential and HSC cell–to–MFB cell fate shift. To place these results in a broader perspective, we analyzed an aggregate file containing scRNA-seq data from P3 (VitA+ qHSCs), P4 (VitA+ aHSCs), and P5 (VitA− aHSCs) fractions from an AH mouse and a pair-fed control mouse (Figure 2D). This analysis revealed that P4 Lrat+Fbln2+ cells with or without Thy1 expression had 10–70-times up-regulations of the profibrotic genes as compared with qHSCs, to the levels equal or even higher than Lrat−Thy1−Fbln2+ aPFs (Figure 2E). As the number of Lrat−Thy1−Fbln2+ aPF subpopulation was less than a tenth of Lrat+Thy1− Fbln2+ subpopulation in P4 (Figure 1G) and P5 (Figure 2G), Lrat+Fbln2+ aHSCs appear quantitatively most profibrotic in AH. We also assessed the HSC genes across P3–P5 and showed conspicuous repressions in the P4 and P5 Lrat+Fbln2+ subpopulations as predicted, but not in P4 Lrat+Thy1− Fbln2− “pure” aHSCs (Figure 2F), suggesting that these “more selective” HSC genes are sensitive markers of the HSC to MFB shift but are unaffected by activation of HSCs without Fbln2 expression.
FIGURE 2.
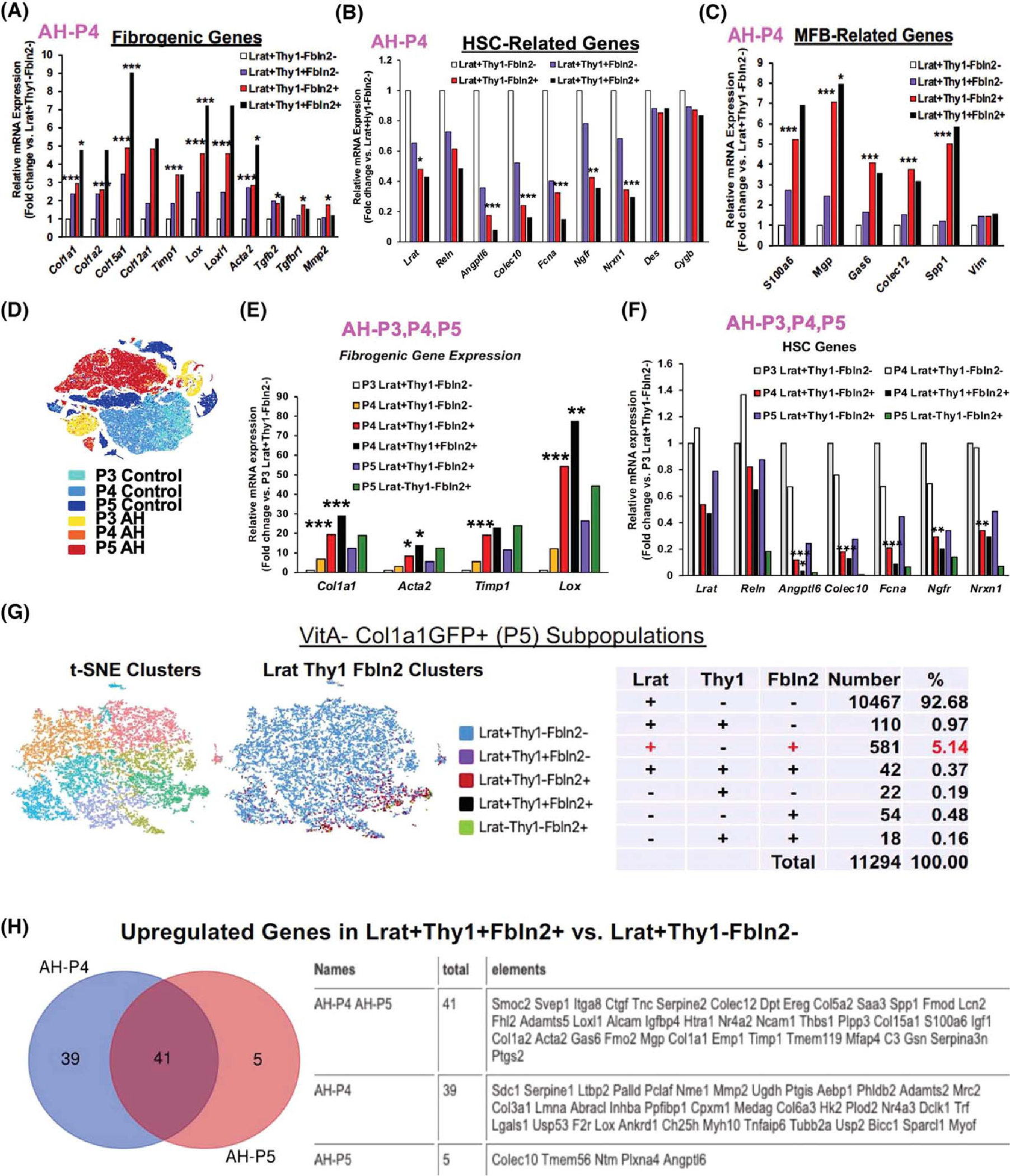
(A) Expression of profibrotic genes by AH-P4 aHSC subpopulations (*p < 0.05, ***p < 0.005). (B) Expression of HSC-related genes by AH-P4 aHSC subpopulations (*p < 0.05, **p < 0.01, ***p < 0.005 vs. Lrat+Thy1−Fbln2− cells). (C) Expression of myofibroblast (MFB)–related genes by AH-P4 aHSC subpopulations (*p < 0.05, ***p < 0.005 vs. Lrat+Thy1−Fbln2− cells). (D) t-SNE clusters of P3 (VitA+GFP−), P4 (VitA+GFP+), and P5 (VitA−GFP+) cells from both AH and control mice. (E) Expression of profibrotic genes by P4 and P5 subpopulations as compared with P3 Lrat+Thy1−Fbln2− quiescent HSCs (*p < 0.05, **p < 0.01, ***p < 0.005 vs. P3 Lrat+Thy1−Fbln2− cells). (F) Expression of HSC-related genes by P4 and P5 subpopulations as compared with P3 Lrat+Thy1−Fbln2− quiescent cells (*p < 0.05, **p < 0.01, ***p < 0.005 vs. P3 Lrat+Thy1−Fbln2− cells). (G) scRNA-seq t-SNE clusters (left), clusters based on Lrat, Thy1, and Fbln2 expression (middle), and a summary breakdown table for the subpopulations in VitA−GFP+ P5 cells (right) from the AH mouse. (H) Venn diagram for up-regulated genes in Lrat+-Thy1−Fbln2+ versus Lrat+Thy1−Fbln2− cells in AH P4 and P5 cells (left), showing 41 commonly up-regulated genes listed in the table (right). Ereg, epiregulin; Fmod, fibromodulin; Lcn2, lipocalin2; Mrc2, mannose receptor C type 2; Nme1, NME/NM23 nucleoside diphosphate kinase 1; Nrxn1, neurexin 1; Palld, paladin; Pclaf, PCNA clamp associated factor; Reln, reelin; Scd1, stearoyl-coenzyme A desaturease 1; Smoc2, SPARC related modular calcium binding 2; Thbs, thrombospondin; Tnc, tenascin C; Ugdh, UDP-glucose 6-dehydrogenase.
We also extended the same gene-expression analysis to Lrat+Thy1−Fbln2+ cells in BDL and CCl4 models and observed similar patterns of changes in profibrotic, MFB, and HSC genes (Figure S2A–C, Table S2), suggesting that the Lrat+Thy1−Fbln2+ subpopulation is generally highly profibrotic and myofibroblastic, regardless of the etiology.
Commonly regulated genes in Lrat+Thy1−Fbln2+ cells
To gain more insight into the phenotype and genesis of the Lrat+Thy1−Fbln2+ subpopulation, we compared the DEGs in Lrat+Thy1−Fbln2+ versus Lrat+Thy1−Fbln2− cells in P4 and P5 (Tables S1 and S3). There were 80 and 46 significantly up-regulated genes in Lrat+Thy1−Fbln2+ cells in P4 and P5, respectively, and 41 up-regulated genes were shared by both (Figure 2H). These 41 common DEGs included many known profibrotic and MFB genes as expected, but also genes that have not been explored in the context of liver fibrosis or HSC fate regulation such as sushi, von Willebrand factor type A, EGF and pentraxin domain containing 1 and nuclear receptor subfamily 4 group A member 2. As for down-regulated DEGs in Lrat+Thy1−Fbln2+ cells, there were 75 down-regulated genes in P4 but only four genes (Colec10, transmembrane protein 56 [Tmem56], neurotrimin [Ntm], and Angptl6) in P5, which were also shared by the P4 cells. Colec10 and Angptl6 are among the “more selective” HSC genes that were commonly suppressed in Lrat+Thy1−Fbln2+ cells (Figure 2B). These two genes and Tmem56 were shown to be expressed in a subset of aHSCs with differentiation trajectory by a recent scRNA-seq analysis of CCl4 mouse liver fibrosis.[12] Collectively, these findings suggested that the repression of these HSC differentiation genes might have contributed to the HSC-to-MFB shift in Lrat+Thy1−Fbln2+ cells.
We further assessed DEGs shared by P4 Lrat+-Thy1−Fbln2+ cells between AH and BDL. We did not include CCl4 data for this comparison, as they included only few significant DEGs as predicted by diffuse clustering and inconsistent regulations as discussed previously. This analysis identified 26 commonly up-regulated and 18 down-regulated genes between AH and BDL (Figure S2D). Many of these 26 commonly up-regulated DEGs were profibrotic and MFB genes but also included adipocyte enhancer binding protein 1 (Aebp1), which inhibits adipocyte differentiation through mitogen-activated protein kinase activation[19] and amplifies proinflammatory activation of macrophages by enhancing NF-κΒ through inhibition of IkappaB kinase.[20] As HSC activation is characterized by diminished adipogenesis regulation,[21] Aebp1 up-regulation observed in Lrat+Thy1−Fbln2+ may contribute to the HSC-to-MFB shift through its anti-adipogenesis actions. ABRA C-terminal like (Abracl) renders mitogenic and migratory signaling in cancer,[22] and transmembrane protein 119 (Tmem119) induces cancer cell proliferation through the PDGFRB-PI3K-AKT pathway[23] or Wnt-β-catenin signaling.[24] These cell-fate regulators up-regulated in Lrat+Thy1−Fbln2+ cells may also contribute to the profibrotic and MFB phenotype. Commonly repressed genes again included the four “more selective” HSC genes, Colec10, Tmem56, Ntm and Angptl6, further supporting that the aPF/MFB shift of Lrat+Thy1−Fbln2+cells may be caused by down-regulation of these HSC genes.
Impact of VitA loss on Lrat+Thy1−Fbln2− aHSCs
Our combinatorial FACS and scRNA-seq analysis revealed the presence of VitA− Lrat+Thy1−Fbln2− aHSCs in all three models but quantitatively more in AH as compared with BDL or CCl4 (Figure 1E). This raised an important question as to how this loss of vitamin A affected the transcriptomic landscape of aHSCs in the three models. For this, we compared the expression of the profibrotic and MFB genes in P5 versus P4 in each model (Tables S4–S6). To our surprise, the profibrotic genes Col1a1, Col1a2, Timp1, and Acta2, but not Tgfb1 and Mmp2, were repressed in P5 VitA− Lrat+Thy1−Fbln2− cells versus P4 VitA+ Lrat+Thy1−Fbln2− cells in AH, whereas in BDL and CCl4 they tended to be indifferent or induced (Figure 3A). Similarly, the MFB genes were repressed in AH-P5 cells but up-regulated in BDL-P5 and CCl4-P5 (Figure 3B). The HSC marker genes showed no significant differences in all three models. These results suggested that the uniquely expanded VitA− Lrat+Thy1−Fbln2− subpopulation in AH has a different transcriptomic profile versus VitA+ Lrat+Thy1−Fbln2− cells. To address this AH uniqueness, we directly assessed DEGs of VitA− (P5) versus VitA+ (P4) Lrat+Thy1−Fbln2− cells among the three models and subjected DEGs to the Go Biological Processes pathway analysis. This confirmed significantly down-regulated extracellular matrix (ECM) pathways in AH VitA− (P5) Lrat+Thy1−Fbln2− cells versus the same subpopulation in BDL or CCl4 (Figure 3C, black arrows). In contrast, leukocyte migration, antimicrobial cell killing, and antigen presentation pathways were up-regulated (Figure 3C, red arrows), as confirmed by up-regulations of inflammatory and immunoregulatory genes as denoted in red in a partial heatmap of DEGs in AH versus BDL or CCl4 (Figure 3D). Similar results were obtained from VitA+ (P4) Lrat+Thy1−Fbln2− cells in AH versus BDL and CCl4 (Figure 3E,F), suggesting that aHSCs in AH are relatively less profibrotic but uniquely more inflammatory and immunoregulatory compared with BDL or CCl4, regardless of VitA storage status. This observation prompted us to compare the expression of inflammatory and immunoregulatory genes in the Lrat+Thy1−Fbln2+ subpopulation in the three models. This analysis also confirmed less profibrotic but more inflammatory/immunoregulatory features in AH versus BDL or CCl4, regardless of VitA status (Figure S3A,B). Furthermore, the pathways responsive to pathogen-associated molecular patterns were up-regulated in VitA+ Lrat+Thy1−Fbln2+ cells, and those in antiviral and antimicrobial defense were activated in VitA− Lrat+Thy1−Fbln2+ cells in AH (Figure S3A,B, red arrows). These features, detected across the different aHSC subpopulations in AH, mirror the unique pathologic phenotype of AH mice such as increased hepatic bacterial load due to compromised gut barrier and neutrophilic inflammation,[17,25] suggesting the involvement of aHSCs in their adaptive response to the phenotype. To further characterize the signaling pathway associated with the inflammatory and immunoregulatory nature of Lrat+ cells in AH, we performed Gene Ontology analysis and showed that the NF-κB pathway was significantly activated (Figure S3C, red arrows) in both VitA+ and VitA− Lrat+cells in AH versus qHSCs.
FIGURE 3.
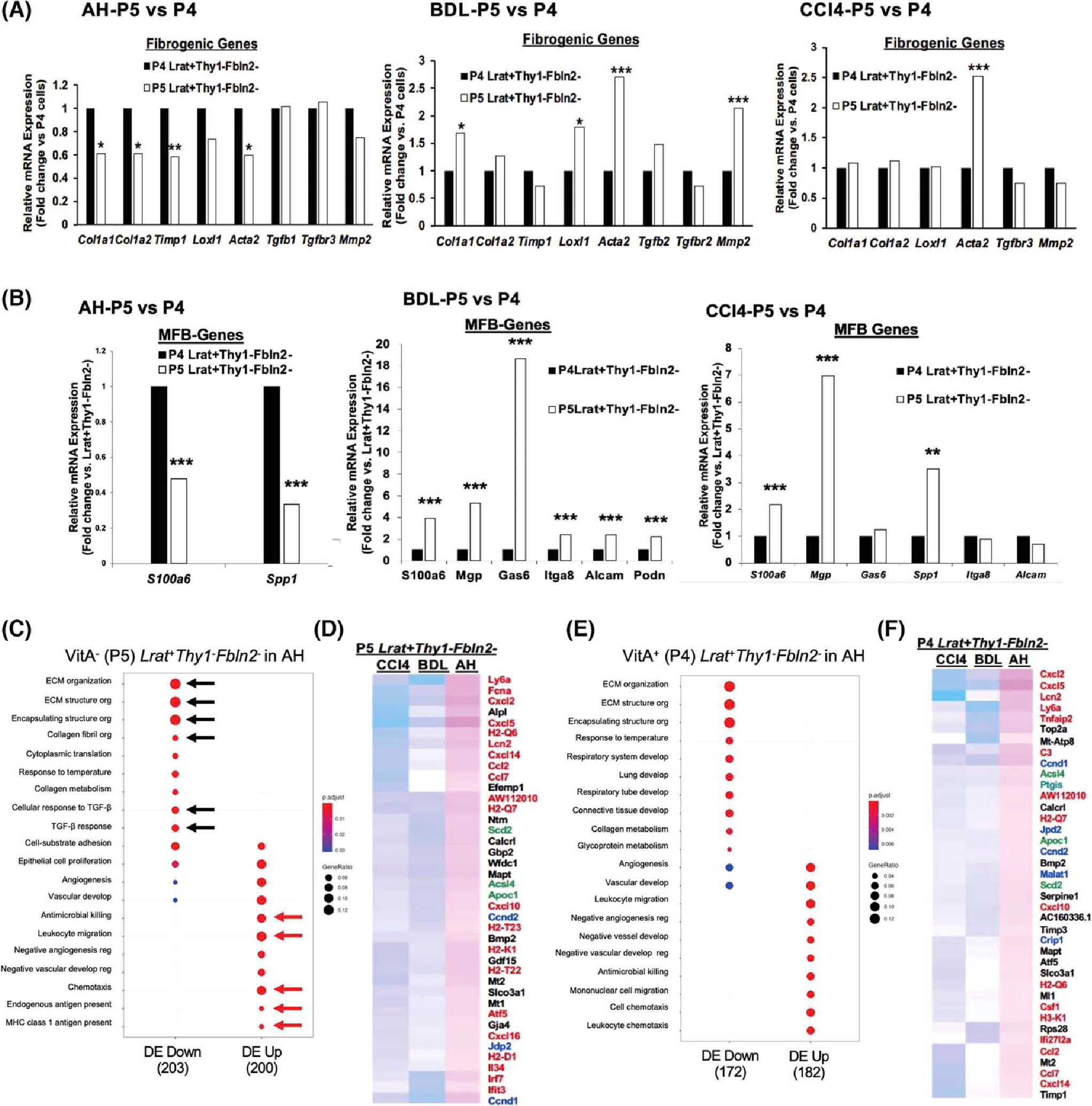
(A) Comparison of profibrotic gene expression between VitA+ (P4) and VitA− (P5). Lrat+Thy1−Fbln2− aHSCs in AH (left), BDL (middle), and CCl4 (right) (*p < 0.05, **p < 0.01, ***p < 0.005 vs. P4 aHSCs). (B) Comparison of MFB gene expression between VitA+ (P4) and VitA− (P5) Lrat+Thy1−Fbln2− aHSCs in AH (left), BDL (middle), and CCl4 (right) (**p < 0.01, ***p < 0.005 vs. P4 aHSCs). (C) Pathway analysis of DEGs in VitA− Lrat+Thy1−Fbln2− aHSCs in AH versus the same subset in BDL and CCl4 by the Go Biological Processes, identifying significantly down-regulated extracellular matrix (ECM) pathways (black arrows) and up-regulated antimicrobial, inflammatory, and antigen presentation pathways (red arrows) in AH versus BDL and CCl4. The gene ratio was calculated by dividing the number of DEGs that are significantly enriched in a given pathway by the total number of DEGs and depicted by the size of each circle. (D) Partial heatmap showing significantly up-regulated DEGs in VitA− (P5) Lrat+Thy1−Fbln2− aHSCs in AH versus BDL and CCl4. Inflammatory and immunoregulatory genes are denoted by red, cell proliferation genes by blue, and lipid metabolic genes by green. (E) The same Go Biological Processes pathway analysis for VitA+ (P4) Lrat+Thy1−Fbln2− aHSCs in AH versus BDL and CCl4 as shown for (C). (F) Partial heatmap showing significantly up-regulated DEGs in VitA+ (P4) Lrat+Thy1−Fbln2− aHSCs in AH versus BDL and CCl4 as shown for (D). Alcam, activated leukocyte cell adhesion molecule; Itga8, integrin subunit alpha 8; Loxl1, lysyl oxidase like 1; MHC, major histocompatibility complex; Mmp2, matrix metalloproteinase 2; Podn, podoplanin; Spp1, osteopontin; Timp1, tissue inhibitor of metalloproteinase 1.
We also noted that genes involved in cellular proliferation (cyclin D1 and cyclin D2) were also expressed higher in all Lrat+Thy1−Fbln2− and Lrat+Thy1−Fbln2+ populations in AH versus BDL and CCl4 (denoted in blue in Figure 3D, F). We searched for a proliferative cluster across three models by filtering for co-expression of Ki67 (Mki67) and proliferating cell nuclear antigen (Pcna) (Figure S3D, Table S7). Indeed, Lrat+Ki67+Pcna+ cells were observed most in the AH model, some in CCl4, but much fewer in BDL, confirming that AH liver uniquely harbors a proliferative aHSC cluster. Another category of uniquely up-regulated genes in AH was lipid metabolism, including acyl CoA synthetase long-chain isoform 4 (Acsl4), apolipoprotein C1 (Apoc1), and the isoform of stearoyl co-A desaturase (Scd2) (denoted in green in Figure 3D,F). Our previous study identified Scd2, as expressed in HSCs, is essential for activation of HSCs and liver fibrosis.[18] Acsl4 preferentially catalyzes the formation of arachidonoyl-CoA from arachidonic acid and is implicated in inflammation. Apoc1 regulates fatty acid flux into adipocytes and hepatocytes.[26] Prostaglandin-endoperoxide synthase 2 was also up-regulated in VitA+ and VitA− Lrat+Fbln2+ cells in AH versus BDL or CCl4 (Figure 3D), further supporting heightened arachidonic acid metabolism in these cells. Collectively, these results suggest that both aHSCs in AH are uniquely more inflammatory, immunoregulatory, and proliferative, and have more active eicosanoid metabolism.
Because we identified Angptnl6, Colec10, and Fcna as the most sensitive HSC differentiation markers (Figure 2B,F), we used this gene set to filter the aggregate file containing P3, P4, and P5 scRNA-seq data from both AH and control (Cont) mice to determine how qHSC clusters are distributed (Figure S3E). Indeed, the Angptl6higColec10hiFcnahi qHSCs were clustered mostly within Cont-P3 and Cont-P4 but much fewer in AH-P3, P4, and P5.
LRAT+FBLN2+ cells in patient AH
To test the translational relevance of Lrat+Fbln2+ aHSCs in AH mice, we first performed computational deconvolution using the PERT model.[27] Gene-expression profiles identified in murine AH HSC subsets by scRNA-seq data were used as the reference to estimate their relative constituent proportions in the bulk human AH versus normal-liver RNA-seq data from two separate cohorts.[28,29] Only the Lrat+Fbln2+ cells from the P4 and P5 cells were found to have higher proportions in AH livers relative to normal controls, suggesting that these cell populations indeed expanded in AH (depicted in orange, Figure 4A,B). We then performed sparse statistical regression to build an AH classifier based on the PERT proportions of the various HSC subsets. The Massey cohort[28] was used as a training cohort, while the Affo cohort[29] was used to validate the resulting HSC subset signature. The AH classifier was highly accurate in discriminating AH samples from normal controls in both the training and testing cohorts (area under the receiver operating characteristic curve [AUROC] = 95.8% and 97.1%, respectively; Figure S4A,B). We found that LRAT+FBLN2+ cells were associated with positive weights, suggesting that a high proportion of these cells is a characteristic feature of AH versus normal controls. As our scRNA-seq analysis of murine AH HSCs suggested the inflammatory nature, we determined whether inflammatory pathway activity was enriched in human AH samples by single-sample gene-set enrichment scores estimated for each sample in the human cohorts and trained a classifier to distinguish AH samples from normal controls. We identified a signature of six inflammatory gene sets in which five were positively weighted and highly predictive of AH samples in both the training and testing cohorts, indicating that inflammatory pathways, including the NF-κB pathway, are activated in human AH samples (AUROC = 100% and 94.3%, respectively). We then tested these inflammatory pathways in aHSC subpopulations of the murine AH model at the single-cell level and revealed that they were enriched in Lrat+ and especially in P4 and P5 Lrat+Fbln2+ cells (Figure S4C), suggesting that the activated inflammatory pathways in patient AH are also recapitulated in Lrat+Fbln2+ cells in murine AH.
FIGURE 4.
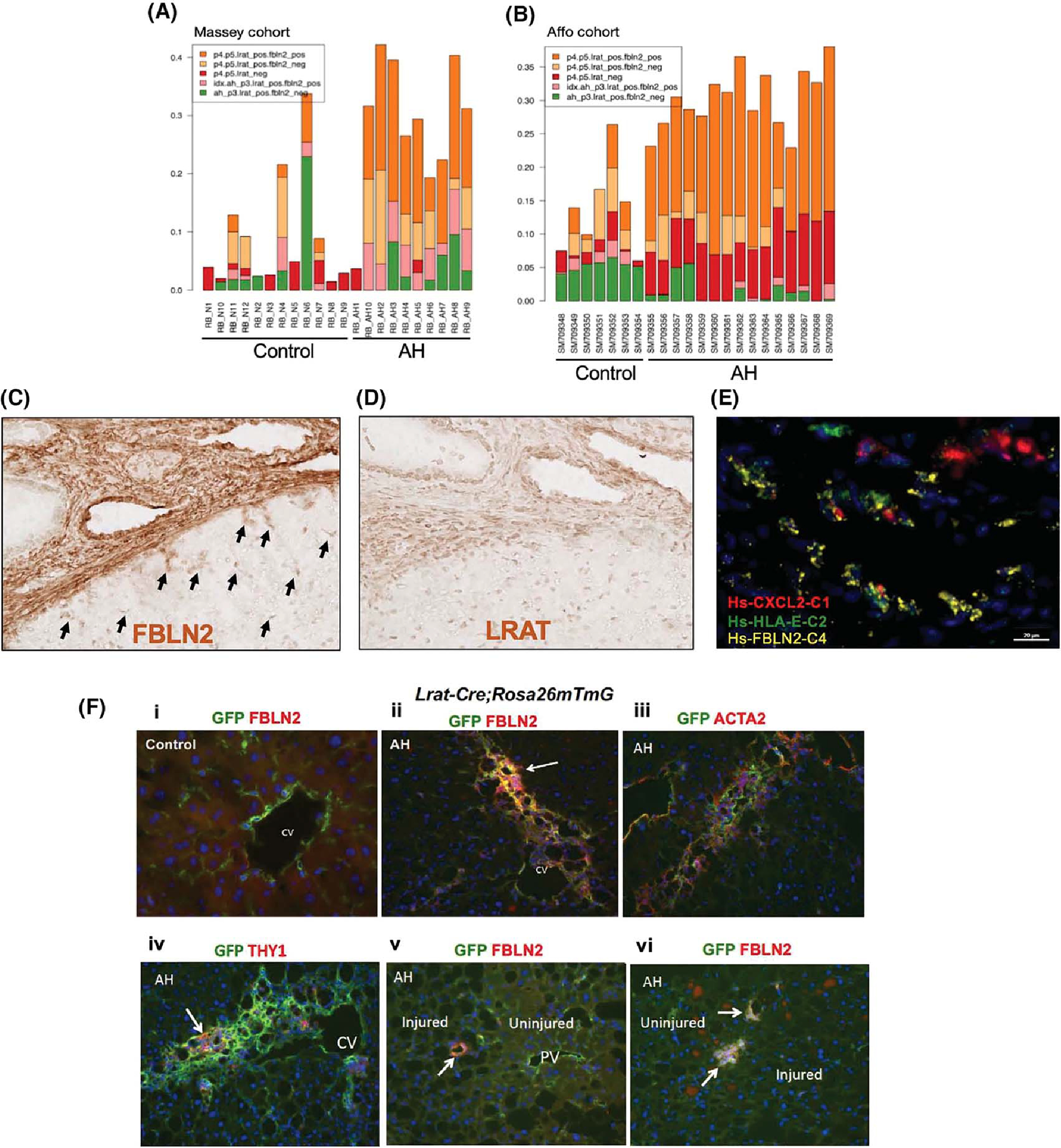
(A,B) The relative proportions of various HSC subpopulations inferred by their specific gene-expression programs represented in human normal and AH samples from the Massey cohort (A) or Affo cohort (B) as estimated by a computational deconvolution analysis of bulk liver RNA-seq data using the PERT algorithm. (C,D) Horseradish peroxidase (HRP) immunohistochemical (IHC) images (×20) of FBLN2 (C) and LRAT (D) staining of AH patient liver sections. Black arrows point to FBLN2+ star-shaped aHSCs. (E) Multiplex in situ hybridization (ISH) image of an AH patient liver section depicting FBLN2+ cells co-expressing chemokine (C-X-C motif) ligand 2 (CXCL2) and/or human leukocyte antigen E (HLA-E) (×62.5). (F) Double immunofluorescence (IF) staining microscopic images (×20) of Lrat-driven expression of GFP with FBLN2 (i, ii, v, vi), ACTA2 (iii), or THY1 (iv) in Lrat-Cre;Rosa26mTmG mouse subjected to the control (i) or AH (ii–vi) regimen. CV, central vein; PV, portal vein.
Patient AH immunohistochemistry and in situ hybridization
To directly test the presence of LRAT+FBLN2+ cells in AH patient, we performed immunostaining on AH patient liver sections. Because we could not identify good primary antibodies raised in different species, we could not perform dual staining. However, FBLN2 staining revealed strong staining along the edge of a fibrotic septum, but more importantly in many star-shaped HSC-like cells near the septum (Figure 4C, arrows). These cells in and near the septum were LRAT+ cells, most likely aHSCs (Figure 4D), suggesting the existence of LRAT+FBLN2+ aHSCs in patients with AH. We also performed in situ hybridization (ISH) to assess the expression of FBLN2 plus chemokine (C-X-C motif) ligand 2 (CXCL2), the inflammatory gene up-regulated in murine AH aHSC, and human leukocyte antigen E (HLA-E), the major histocompatibility complex (MHC) class I molecule and a functional homolog of murine H2-T23, which was also up-regulated in murine AH aHSC (Figure 3D).
Multiplex RNAscope ISH assay of AH patient liver cryosection revealed co-expression of FBLN2 with CXCL2 and/or HLA-E along the cirrhotic septum (Figure 4E), suggesting that FBLN2-expressing aHSCs also express the proinflammatory and immunoregulatory mediators in AH patient liver.
Lrat+ HSCs give rise to Lrat+Thy1−Fbln2+ cells in AH
Our scRNA-seq and its trajectory analysis data suggested that the Lrat+Thy1−Fbln2+ subpopulation might have emerged from Lrat+Thy1−Fbln2− “pure” aHSCs. To test this notion in AH, we used Lrat-Cre;Rosa26mTmG mice, which allowed GFP labeling of Lrat+ HSCs to trace their fate in the AH model. Our dual immunofluorescence staining for GFP and FBLN2 of a control mouse liver showed HSCs labeled with GFP but not FBLN2 staining in the perivenular zone (Figure 4F,i). In AH liver, cells co-expressing GFP and FBLN2 were detected in a fibrotic septum extending from the central vein (Figure 4F,ii), and these cells are mostly stained for ACTA2 (Figure 4F,iii), suggesting that Lrat+ HSCs give rise to FBLN2-expressing aHSCs to contribute to liver fibrosis in AH. Cells positively stained for GFP and THY1 were also found in the septum but with much less frequency (Figure 4F,iv). We also identified small clusters of GFP+FBLN2+ cells along the border of injured and adjacent uninjured regions of hepatic acini with minimal liver fibrosis (Figure 4F,v,vi), suggesting that LRAT+FBLN2+ cells may participate in the frontline of early wound healing.
DISCUSSION
The major role that aHSCs play in the genesis of MFBs in liver fibrosis has been supported by the fate-tracing study using Lrat-Cre;Rosa26 reporter mice, revealing a dominant contribution of HSCs to the generation of MFBs in various liver fibrosis models.[4] Our FACS data also showed that > 85% of Col1a1-GFP+ cells from BDL and CCl4 mice were VitA+, confirming the predominant role of aHSCs in these models. However, in the AH model, VitA−GFP+ cells markedly expanded at the expense of VitA+GFP+ cells and accounted for nearly 80% of Col1a1-GFP+ cells, suggesting possible contributions of VitA− mesenchymal cells in AH. To this end, we have taken a focused approach of supervised clustering of VitA+ and VitA− Col1a1-GFP+ cells based on the expression of the HSC marker Lrat and the PF markers Thy1 and Fbln2. This analysis led to two findings in AH. First, > 90% of VitA−GFP+ cells in AH were Lrat+Thy1−Fbln2− cells, identifying them as VitA-depleted aHSCs. Combined with the fact that about 80% of Col1a1-GFP+ cells were VitA−, VitA− Lrat+Thy1−Fbln2− aHSCs are the largest profibrotic subpopulation in AH. We also detected VitA−Lrat+Thy1−Fbln2− aHSCs in BDL and CCl4 mice, accounting for 55%–65% of VitA−GFP+ cells. However, VitA−GFP+ cells in these models were < 15% of all Col1a1-GFP+ cells, and for that reason the relative contribution of VitA− Lrat+Thy1−Fbln2− aHSCs may be less in these models. In contrast to the prevailing notion of VitA loss as a feature of HSC activation, VitA-depleted aHSCs were shown to be less profibrotic and myofibroblastic compared with VitA+ aHSCs in AH but not in BDL or CCl4. This phenotype may be caused by high blood ethanol concentration achievable in the intragastric feeding model of AH, as feeding alcohol-containing liquid diet—which achieves mild alcohol intake and minimal fibrosis—did not result in an increase in VitA−GFP+ aHSCs.[30]
Second, we detected the previously unreported aHSC subpopulation that co-expressed Lrat and Fbln2 in both VitA+ and VitA− Col1a1–expressing cells in AH and to a lesser extent in BDL and CCl4 mice. Although this subpopulation represents only 11%–13.5% of VitA+ and 5%–7% of VitA− Col1a1-expressing cells, due to its 4–12-fold-higher expression of the profibrotic and MFB genes, the potential contribution of this subset to fibrogenesis in AH may be significant. These Lrat+-Thy1− Fbln2+ cells with reduced expression of the HSC-specific genes and conversely up-regulated MFB are thought to be a transitional cell type from pure Lrat+Thy1−Fbln2− aHSCs toward Lrat−Thy1− Fbln2+ aPFs (Figure 1H and Figure S1E), the notion confirmed by the Lrat+ cell fate–tracing result (Figure 4F). Conceptually, this finding supports the plasticity of HSCs, which has been proposed for morphologic and phenotypic changes but more recently for metabolic reprogramming.[31] Our finding of Lrat+ aHSCs co-expressing Fbln2 also raises a caution for the use of Fbln2 as the bona fide marker of PFs as previously proposed. The same may be said for Thy1 as also emerged (~5%) in BDL and CCl4 fibrosis (Figure S1F).
Translationally, our computational deconvolution analysis of bulk-liver RNA-seq data inferred the expansion of the LRAT+FBLN2+aHSC subpopulation in AH patient livers, which was supported by the immunohistochemistry results. Furthermore, ISH of AH patient liver confirmed FBLN2+cells co-expressing the inflammatory mediator CXCL2 or/and the MHC type I antigen presentation molecule HLA-E. VitA+ and VitA− LRAT+FBLN2+ aHSCs in AH may be considered as a therapeutic target to suppress progression of liver fibrosis, as they appear to be a highly profibrotic, myofibroblastic, and proinflammatory subpopulation. However, targeting this subpopulation may prove to be detrimental if the inflammatory, antimicrobial, immunoregulatory functions these cells may offer are the manifestation of the liver's protective response in AH. Further preclinical studies on this specific question are warranted.
Supplementary Material
FUNDING INFORMATION
Supported by the National Institutes of Health (U01A018663, P50AA011999, R24 AA012885, R24AA02517, and P30CA014089); Department of Veterans Affairs (IK6BX004205 [BLR&D Research Career Scientist Award] and I01 BX001991 [VA Merit Review]); Lee Summer Research Fellowship of the Southern California Research Center for ALPD and Cirrhosis (to RF and SB); and American Liver Foundation Student Scholar Award (RF).
Abbreviations:
- Acta2
alpha smooth muscle actin
- AH
alcoholic hepatitis
- aHSC
activated HSC
- ANGPTL6
angiopoeitin like 6
- aPF
activated portal fibroblast
- BDL
bile duct ligation
- Col1a1
collagen type 1 alpha 1
- COLEC10
collectin-10
- CXCL2
chemokine (C-X-C motif) ligand 2
- DEG
differentially expressed gene
- Fbln2
fibulin 2
- Fcna
ficolin a
- GFP
green fluorescent protein
- HLA-E
human leukocyte antigen E
- ISH
in situ hybridization
- Lox
lysyl oxidase
- Lrat
lecithin retinol acyltransferase
- MFB
myofibroblast
- MHC
major histocompatibility complex
- PC
pericentral
- PP
periportal
- PF
portal fibroblast
- qHSC
quiescent HSC
- scRNA-seq
single-cell RNA sequencing
- Thy1
Thy1 cell surface antigen
- Timp1
tissue inhibitor of metalloproteinase 1
- Tmem56
transmembrane protein 56
- VitA
vitamin A
Footnotes
CONFLICTS OF INTEREST
Nothing to report.
Supplemental Digital Content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal’s website, www.hepjournal.com.
REFERENCES
- 1.Asahina K, Tsai SY, Li P, Ishii M, Maxson RE Jr, Sucov HM, et al. Mesenchymal origin of hepatic stellate cells, submesothelial cells, and perivascular mesenchymal cells during mouse liver development. Hepatology. 2009;49:998–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Asahina K, Zhou B, Pu WT, Tsukamoto H. Septum transversum-derived mesothelium gives rise to hepatic stellate cells and perivascular mesenchymal cells in developing mouse liver. Hepatology. 2011;53:983–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Friedman SL, Roll FJ, Boyles J, Bissell DM. Hepatic lipocytes: the principal collagen-producing cells of normal rat liver. Proc Natl Acad Sci U S A. 1985;82:8681–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mederacke I, Hsu CC, Troeger JS, Huebener P, Mu X, Dapito DH, et al. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat Commun. 2013;4:2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perepelyuk M, Terajima M, Wang AY, Georges PC, Janmey PA, Yamauchi M, et al. Hepatic stellate cells and portal fibroblasts are the major cellular sources of collagens and lysyl oxidases in normal liver and early after injury. Am J Physiol-Gastrointest Liver Physiol. 2013;304:G605–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Iwaisako K, Jiang C, Zhang M, Cong M, Moore-Morris TJ, Park TJ, et al. Origin of myofibroblasts in the fibrotic liver in mice. Proc Natl Acad Sci U S A. 2014;111:E3297–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nishio T, Hu R, Koyama Y, Liang S, Rosenthal SB, Yamamoto G, et al. Activated hepatic stellate cells and portal fibroblasts contribute to cholestatic liver fibrosis in MDR2 knockout mice. J Hepatol. 2019;71:573–85. [DOI] [PubMed] [Google Scholar]
- 8.Bhunchet E, Wake K. Role of mesenchymal cell populations in porcine serum-induced rat liver fibrosis. Hepatology. 1992;16:1452–73. [DOI] [PubMed] [Google Scholar]
- 9.Balog S, Li Y, Ogawa T, Miki T, Saito T, French SW, et al. Development of capsular fibrosis beneath the liver surface in humans and mice. Hepatology. 2020;71:291–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krenkel O, Hundertmark J, Ritz TP, Weiskirchen R, Tacke F. Single cell RNA sequencing identifies subsets of hepatic stellate cells and myofibroblasts in liver fibrosis. Cell. 2019;8:503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dobie R, Wilson-Kanamori JR, Henderson BEP, Smith JR, Matchett KP, Portman JR, et al. Single-cell transcriptomics uncovers zonation of function in the mesenchyme during liver fibrosis. Cell Rep. 2019;29:1832–47.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang W, Conway SJ, Liu Y, Snider P, Chen H, Gao H, et al. Heterogeneity of hepatic stellate cells in fibrogenesis of the liver: insights from single-cell transcriptomic analysis in liver injury. Cell. 2021;10:2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xiong X, Kuang H, Ansari S, Liu T, Gong J, Wang S, et al. Landscape of intercellular crosstalk in healthy and NASH liver revealed by single-cell Secretome gene analysis. Mol Cell. 2019;75:644–60.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rosenthal SB, Liu X, Ganguly S, Dhar D, Pasillas MP, Ricciardelli E, et al. Heterogeneity of HSCs in a mouse model of NASH. Hepatology. 2021;74:667–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Payen VL, Lavergne A, Sarika NA, Colonval M, Karim L, Deckers M, et al. Single-cell RNA sequencing of human liver reveals hepatic stellate cell heterogeneity. JHEP Rep. 2021;3:100278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yata Y, Scanga A, Gillan A, Yang L, Reif S, Breindl M, et al. DNase I-hypersensitive sites enhance alpha1(I) collagen gene expression in hepatic stellate cells. Hepatology. 2003;37:267–76. [DOI] [PubMed] [Google Scholar]
- 17.Lazaro R, Wu R, Lee S, Zhu N-L, Chen C-L, French SW, et al. Osteopontin deficiency does not prevent but promotes alcoholic neutrophilic hepatitis in mice. Hepatology. 2015;61:129–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lai KKY, Kweon S-M, Chi F, Hwang E, Kabe Y, Higashiyama R, et al. Stearoyl-CoA desaturase promotes liver fibrosis and tumor development in mice via a Wnt positive-signaling loop by stabilization of low-density lipoprotein-receptor-related proteins 5 and 6. Gastroenterology. 2017;152:1477–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim SW, Muise AM, Lyons PJ, Ro HS. Regulation of adipogenesis by a transcriptional repressor that modulates MAPK activation. J Biol Chem. 2001;276:10199–206. [DOI] [PubMed] [Google Scholar]
- 20.Majdalawieh A, Zhang L, Ro H-S. Adipocyte enhancer-binding protein-1 promotes macrophage inflammatory responsiveness by up-regulating NF-kappaB via IkappaBalpha negative regulation. Mol Biol Cell. 2007;18:930–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miyahara T, Schrum L, Rippe R, Xiong S, Yee HF, Motomura K, et al. Peroxisome proliferator-activated receptors and hepatic stellate cell activation. J Biol Chem. 2000;275:35715–22. [DOI] [PubMed] [Google Scholar]
- 22.Wang D, Liu H, Ren C, Wang L. High expression of ABRACL is associated with tumorigenesis and affects clinical outcome in gastric cancer. Genet Test Mol Biomark. 2019;23:91–7. [DOI] [PubMed] [Google Scholar]
- 23.Sun T, Bi F, Liu Z, Yang Q. TMEM119 facilitates ovarian cancer cell proliferation, invasion, and migration via the PDGFRB/PI3K/AKT signaling pathway. J Transl Med. 2021;19:111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang B, Wang F, Zheng G. Transmembrane protein TMEM119 facilitates the stemness of breast cancer cells by activating Wnt/β-catenin pathway. Bioengineered. 2021;12:4856–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Khanova E, Wu R, Wang W, Yan R, Chen Y, French SW, et al. Pyroptosis by caspase11/4-gasdermin-D pathway in alcoholic hepatitis in mice and patients. Hepatology. 2018;67:1737–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Muurling M, van den Hoek AM, Mensink RP, Pijl H, Romijn JA, Havekes LM, et al. Overexpression of APOC1 in obob mice leads to hepatic steatosis and severe hepatic insulin resistance. J Lipid Res. 2004;45:9–16. [DOI] [PubMed] [Google Scholar]
- 27.Qiao W, Quon G, Csaszar E, Yu M, Morris Q, Zandstra PW. PERT: a method for expression deconvolution of human blood samples from varied microenvironmental and developmental conditions. PLoS Comput Biol. 2012;8:e1002838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Massey V, Parrish A, Argemi J, Moreno M, Mello A, García-Rocha M, et al. Integrated multiomics reveals glucose use reprogramming and identifies a novel hexokinase in alcoholic hepatitis. Gastroenterology. 2021;160:1725–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Affò S, Dominguez M, Lozano JJ, Sancho-Bru P, Rodrigo-Torres D, Morales-Ibanez O, et al. Transcriptome analysis identifies TNF superfamily receptors as potential therapeutic targets in alcoholic hepatitis. Gut. 2013;62:452–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Page A, Paoli PP, Hill SJ, Howarth R, Wu R, Kweon S-M, et al. Alcohol directly stimulates epigenetic modifications in hepatic stellate cells. J Hepatol. 2015;62:388–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Trivedi P, Wang S, Friedman SL. The power of plasticity-metabolic regulation of hepatic stellate cells. Cell Metab. 2021;33:242–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.