

Abstract
Innate immune pathways early after pulmonary transplantation have been shown to cause primary graft dysfunction (PGD) and also predispose to late graft failure. Recent studies in animal models have elucidated critical mechanisms governing such innate immune responses. Here, we discuss pathways of inflammatory cell death, triggers for sterile and infectious inflammation, and signaling cascades that mediate lung injury early after transplantation. These studies highlight potential avenues for lung-specific therapies early following lung transplantation to dampen innate immune responses and improve outcomes. @WashUSurgeryIn this article, Shepherd, Gauthier and Kreisel discuss recent studies that highlight potential avenues for lung-specific therapies early following transplantation to dampen innate immune responses and improve outcomes.
Keywords: lung transplantation, cell death, damage-associated molecular patterns, innate immunity, graft rejection
For many patients with end stage lung disease, pulmonary transplantation plays a vital role as the only viable treatment option once all other therapeutic alternatives have been exhausted. However, the majority of lung allografts fail in the first decade, largely because of chronic rejection. Thus, the survival benefit after lung transplantation remains markedly limited compared to other solid organ transplants.1 Innate immune responses immediately following reperfusion play a critical role in the development of primary graft dysfunction (PGD), which affects up to 80% of lung recipients and remains the leading cause of death in the first month after pulmonary transplantation.2–4 In contrast to adaptive immune responses where the rearrangement of gene segments yields a highly diverse array of specific B and T cell receptors, the innate immune system is comprised of cells with surface receptors encoded by genes in the germline DNA which remain constant throughout the organism’s lifespan. Innate immune responses provide an early, robust trigger that can also augment adaptive alloimmunity, ultimately promoting the development of chronic lung allograft dysfunction (CLAD), the leading cause of overall mortality after lung transplantation.4–7 Identification of the key immune mediators in these processes remains highly relevant to clinical practice and an important consideration for ongoing research and the development of novel targeted therapies.
This review focuses on the pathways involved in the innate immune response following lung transplantation. Specifically, we discuss the primary mechanisms responsible for an early compromise of the integrity of cells within the graft, resulting in the release of endogenous ligands and activation of signaling pathways that trigger graft injury and failure.
Cell death
After lung transplantation, both apoptotic and non-apoptotic cell death occurs. Several forms of non-apoptotic cell death have been identified as key mechanisms of cellular injury in the setting of pulmonary ischemia-reperfusion injury (IRI) (Figure 1). Collectively referred to as forms of ‘inflammatory cell death’, the loss of plasma membrane integrity and cell dissolution along with the release of pro-inflammatory intracellular contents is fundamentally different from the organized formation of apoptotic bodies seen in apoptosis.
Figure 1.
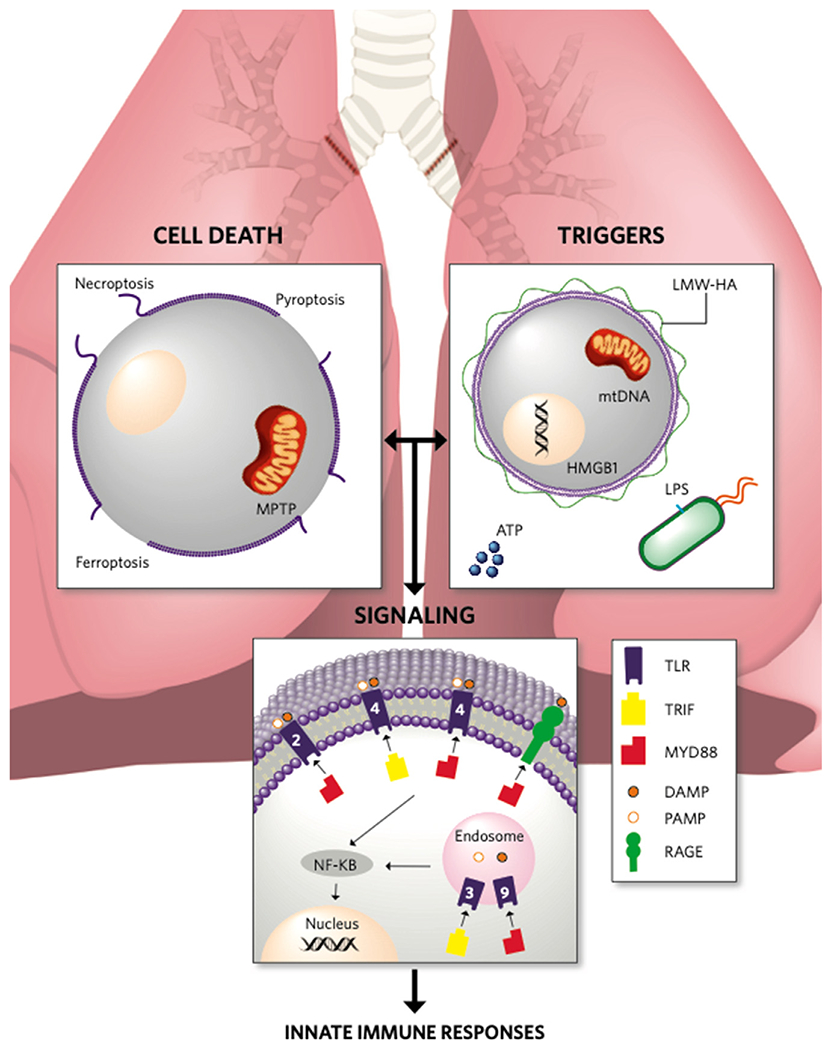
Activation of innate immune responses following lung transplantation. Inflammatory cell death leads to the release of endogenous ligands that propagate inflammatory responses through activation of innate immune signaling pathways. Illustration credit: Erin Neill. ATP, Adenosine triphosphate; DAMP, Damage-associated molecular pattern; HMGB1, High-mobility group box 1; LMW-HA, Low-molecular-weight hyaluronic acid; LPS Lipopolysaccharide; MPTP, Mitochondrial permeability transition pore; mtDNA, Mitochondrial DNA; MyD88, Myeloid differentiation factor 88; NF-κB, Nuclear factor kappa-light-chain enhancer of activated B cells; PAMP, Pathogen-associated molecular pattern; RAGE, Receptor for advanced glycation end products; TLR, Toll-like receptor; TRIF, Toll/Interleukin-1 receptor domain-containing adaptor-inducing interferon-beta.
Apoptotic cell death in human lung tissues following transplantation is virtually undetectable during cold ischemic preservation and increases significantly after reperfusion, though its correlation with clinical outcomes has not been clearly demonstrated.8 In a rat single-lung transplant model of IRI, rates of apoptotic cell death did not significantly correlate with increasing cold ischemic time or post-transplant graft function.9 While apoptosis alone has not been shown to induce clinically significant allograft injury, there may be a contributory role in IRI-induced injury and the development of PGD, as shown in mouse lung transplant models.10
Necroptosis is a regulated form of necrosis which involves the activation of two receptor-interacting protein kinases (RIPK1 and RIPK3) to form a ‘necrosome’ which phosphorylates the mixed lineage kinase domain-like (MLKL) pseudokinase. Activated MLKL inserts into bilipid membranes of cells and results in the expulsion of intracellular contents. In a human lung epithelial cell culture model of IRI, prolonged cold ischemia resulted in receptor-independent necroptosis triggered by increased intracellular calcium and activation of calpain, with subsequent phosphorylation of STAT3 (signal transducer and activator of transcription 3) resulting in increased RIPK3 expression.11 Cell cultures treated with a calpain inhibitor (N-acetyl-Leu-Leu-norleucinal) or a RIPK1 inhibitor (necrostatin-1) showed reduced cell death.11 Necroptosis has been shown to contribute to the development of multi-hit PGD induced by cold ischemia, donor brain death, and hemorrhagic shock in a syngeneic mouse lung transplant model, with reduced necroptosis activation after treatment of the recipient with necrostatin-1 prior to transplantation.10 In a rat transplant model of IRI, necrostatin-1 administration was associated with improved graft function, with reduction of necroptosis via inhibition of phosphorylation of RIPK1 and expression of RIPK3.12 However, this effect was only observed with necrostatin-1 administration to both the recipient and donor due to the continuous activation of the RIPK1 pathway during both ischemia and reperfusion.12 Thus, targeting necroptosis signaling pathways may be a promising strategy to reduce PGD after pulmonary transplantation.
Mitochondrial permeability transition pore (MPTP) insertion into the inner mitochondrial membrane is another form of programmed cell death which may lead to cellular attrition following lung transplantation. Cellular hypoxia during tissue ischemia leads to increased intracellular calcium which overwhelms the capacity of mitochondrial efflux channels, prompting translocation of cytosolic p53 to the mitochondrial membrane with formation of a permeability pore.13 This allows for efflux of protons and loss of the pH gradient required for oxidative phosphorylation, resulting in necrotic cell death13. In mouse cardiac microvascular endothelial cells, cold ischemic storage followed by warm reperfusion triggers MPTP opening and results in increased immunogenicity to allogeneic CD8+ T lymphocytes.14 In cultured human lung epithelial cells subjected to cold ischemia and reperfusion, a similar mechanism of necrosis occurred via translocation of protein kinase C-δ (PKCδ) into the mitochondrial membrane.15 Treatment with a PKCδ inhibitor (δV1-1) reduced necrotic cell death by inhibiting PKCδ and p53 translocation to mitochondria.15 Furthermore, in a rabbit model of pulmonary IRI, attenuated lung injury was observed following treatment with Cyclosporine A, which inhibited MPTP opening via cyclophilin D inhibition.16
Pyroptosis, a form of inflammatory cell death observed in innate immune cells (e.g. monocytes, macrophages), involves the formation of an ‘inflammasome’ which promotes cleavage of Caspase-1 and gasdermin D and promotes secretion of pro-inflammatory cytokines such as interleukin-1β (IL-1β). This ultimately results in the formation of large pores in the plasma membrane and cell death. In rat and mouse models of acute lung injury induced by cardiopulmonary bypass or lipopolysaccharide (LPS), pyroptosis in alveolar macrophages is mitigated by inhibition of caspase-1.17,18 In a rat model of ex-vivo lung perfusion, capsase-1-mediated pyroptotic cell death of donor leukocytes resulted in substantially impaired pulmonary function, and treatment with a caspase-1 inhibitor decreased inflammatory responses and resulted in improved graft function.19 High-mobility group box 1 (HMGB1) is a nuclear protein that is released after inflammasome activation and may contribute to alveolar pyroptosis after IRI.20 Interestingly, preconditioning with recombinant HMGB1 appeared to ameliorate IRI by suppressing alveolar macrophage pyroptosis in a mouse model of hilar clamping, possibly due to down-regulation of endogenous HMGB1 release and up-regulation of protective anti-oxidant enzyme activity, including superoxide dismutase, glutathione peroxidase, and catalase.20
We have recently reported that ferroptosis, a form of inflammatory cell death characterized by iron-dependent phospholipid peroxidation, plays an important role in regulating leukocyte trafficking early after heart transplantation.21 Ferroptotic cell death has also been implicated in mediating lung IRI and therefore may play a role in triggering inflammatory responses following lung transplantation. To this end, a recent study showed that mouse lungs subjected to IRI exhibited increased tissue iron content and accumulation of lipid peroxidation products, and pharmacological inhibition of ferroptosis resulted in amelioration of lung injury.22 Similarly, ferroptosis was found to contribute to acute lung injury following intestinal IRI.23
While the study of regulated cell death has been ongoing for over 100 years, the identification of non-apoptotic cell death pathways have only emerged within the past few decades. As recently as 2018, two novel forms of cellular death (alkaliptosis and oxeiptosis) were identified. Alkaliptosis, a pH-dependent form of regulated cell death, and oxeiptosis, a form of cell death induced by reactive oxygen species, have been identified as potential targets in cancer therapy and viral infections, but their pathological significance in human disease remains largely unknown.24,25 Thus, it is possible that some forms of cell death that impact outcomes after transplantation have not yet been identified, and more research may delineate such pathways. Additionally, the mechanism of cell death which becomes activated is not only trigger-dependent, but also context-dependent. Thus, while an effector molecule may trigger a destructive inflammatory cascade under certain influences, it may play a protective, housekeeping role when other factors are present.
Triggers
The release of endogenous ligands from dying cells elicits an inflammatory immune response commonly referred to as necroinflammation26 (Figure 1). Mitochondrial DNA (mtDNA) is one such ligand which has been recognized as an important trigger of innate immune activation after lung transplantation. In mouse and human pulmonary transplant recipients, our group has demonstrated that increased levels of circulating mtDNA are associated with the development of severe PGD.27 We observed that damaged mitochondria are released into the graft airways during lung transplantation-mediated IRI, and engulfment of these damaged mitochondria by graft-infiltrating neutrophils results in the production of reactive oxygen species, which propagates graft injury.27 Similarly, acute lung injury can trigger the release of HMGB1, a nuclear protein which interacts with pattern recognition receptors and upregulates pro-inflammatory cytokines.28,29 In human donor lungs that are subjected to ex vivo perfusion, increased HMGB1 levels in the perfusate were associated with the development of severe PGD.29 Elevated HMGB1 levels were also associated with lungs rejected for transplantation due to poor physiologic function, supporting the potential use of HMGB1 as a biomarker for organ evaluation and selection prior to transplantation. Notably, neutralizing HMGB1 has been shown to abrogate lung IRI in mouse models of pulmonary hilar clamping and may thus be a potential target to prevent or ameliorate PGD after lung transplantation.30
Extracellular adenosine triphosphate (ATP) can elicit inflammatory responses by engaging purinergic receptors. Our group showed that intravenous administration of soluble recombinant apyrase, which catalyzes the hydrolysis of purigenic nucleotides, resulted in reduced extracellular ATP levels and attenuated IRI after lung transplantation in rats thereby providing evidence that extracellular purigenic nucleotides contribute to the pathogenesis of PGD.31 Extracellular purigenic nucleotides may also enhance alloimmune responses, as illustrated by a mouse allogenic lung transplant study which demonstrated that targeting the P2X purinoreceptor 7 (P2XR7) results in blunted activation of T cells and attenuated acute rejection.32 Hyaluronan is an extracellular matrix glycosaminoglycan that forms a high-molecular-weight polymer under homeostatic conditions and has been shown to attenuate inflammation and protect from acute lung injury.33,34 Following tissue damage, pro-inflammatory low-molecular-weight hyaluronic acid (LMW-HA) fragments are generated by enzymatic degradation.33,34 In a murine orthotopic lung transplant model, we have shown that hyaluronan fragments of low, but not high, molecular weight trigger acute allograft rejection in recipients with established tolerance.35 Thus, reducing sterile inflammation following lung transplantation by inhibition of pro-inflammatory triggers, perhaps during ex-vivo lung perfusion or early after implantation, may prove to be a promising avenue to reduce early graft injury and enhance long term graft survival.
In addition to damage-associated molecular patterns (DAMPs), pathogen-associated molecular patterns (PAMPs) can activate innate immune responses that can have deleterious effects on transplanted lungs. For example, low levels of bacterial endotoxin within donor lungs can trigger neutrophil recruitment and the development of PGD through signaling pathways mediated by tissue-resident alveolar macrophages.36 We have also recently shown that the presence of a synthetic bacterial lipopeptide in donor airways at the time of transplantation enhances alloimmunity resulting in acute rejection, a process mediated through activation of recipient monocytes37. These observations raise concern for the use of donor lungs that are known to harbor pneumonia.
Bacterial PAMPs can also activate innate immune responses to abolish established tolerance in transplanted lungs. In a mouse model of lung transplantation, we previously reported that a respiratory infection with Pseudomonas aeruginosa triggers neutrophilic graft infiltration which abrogates previously established tolerance through activation of alloreactive T cells within the pulmonary graft.38 In human lung transplant recipients, the isolation of Pseudomonas aeruginosa from respiratory samples has been identified as an independent risk factor for the development of donor-specific antibodies.39 Collectively, these studies provide strong evidence that innate immune responses, triggered through either DAMPs or PAMPs, can potentiate adaptive alloimmune responses that can result in graft rejection.
Signaling pathways
The innate immune system utilizes a variety of pattern recognition receptors (PRRs) which can be expressed on the cell surface, in intracellular compartments, or secreted (Figure 1). One class of PRRs, known as toll-like receptors (TLRs), is responsible for recognition of both PAMPs and DAMPs and influence the expression of a variety of host defense genes.40 Once TLRs are engaged by a ligand this results in the recruitment of adaptor proteins that trigger downstream signaling pathways to promote inflammation. The best characterized TLR adaptor molecules are the myeloid differentiation factor 88 (MyD88) and the Toll/IL-1R domain-containing adaptor-inducing interferon-beta (TRIF). MyD88 is directly involved in all TLR signaling with the exception of TLR3, which utilizes only TRIF. TLR signaling pathways result in the activation of nuclear factor kappa-light-chain enhancer of activated B cells (nuclear factor-κB), which translocates to the nucleus to drive the transcription of genes that encode inflammatory cytokines and cellular adhesion molecules. Unique among TLRs is TLR4, which can signal through either MyD88 or TRIF. TLR4 signaling through TRIF, a pathway that induces type I interferon gene expression, has been shown to contribute to inflammatory responses during IRI after heart transplantation.21,41
Several studies utilizing hilar clamping models in mice and rats have indicated that TLR4 expression contributes to lung IRI.42–45 TLR4 was first described as an LPS receptor in mice but has since been shown to recognize other PAMPs from gram-positive bacteria, gram-negative bacteria and viruses, as well as multiple DAMPs, such as heat shock proteins and HMGB1.46 As such, it appears that the role for TLR4 signaling is context-dependent and varies based on the organ system and relative abundance of these ligands in the microenvironment. In the lungs, Zanotti has shown that MyD88-independent signaling via TLR4 on pulmonary vascular endothelial cells promotes edema due to alterations in the cytoskeleton.43 Importantly, Altemeier demonstrated that MyD88-deficient mice were more protected from lung IRI than TLR4-deficient mice, suggesting that TLR4-independent but MyD88-dependent signaling contributes to PGD, implicating the role of other TLRs in pulmonary innate immune responses.44 Furthermore, through the use of bone marrow chimeric mice the authors demonstrated that MyD88 signaling in hematopoietic cells mediated the injury response.44
Neutrophilic graft infiltration plays a critical role in mediating PGD after lung transplantation as well.47 Our group has recently reported that innate immune signaling in various donor and recipient-derived cell populations regulates the trafficking of neutrophils to lung grafts immediately after reperfusion. MyD88/TRIF-dependent production of the chemokine (C-X-C motif) ligand 2 (CXCL2) by donor intravascular non-classical monocytes is critical for the recruitment of neutrophils from the periphery to reperfused pulmonary grafts.48 Extravasation of neutrophils into the graft tissue depends on MyD88-dependent production of IL-1β by spleen-derived recipient classical monocytes.49 In this context, it is interesting that MyD88 expression may regulate the differentiation of graft-infiltrating monocytes, which may have an impact on alloimmunity.50 Signaling through TLR4 or DNAX activation protein of 12 kDa (DAP12), a cell membrane-associated protein that can associate with several receptors including various triggering receptors expressed on myeloid cells, in donor macrophages can also result in the production of neutrophil chemokines and contribute to PGD.51 Similar to TLRs, transmembrane proteins called receptors for advanced glycation end products (RAGE) recruit MyD88 and activate NF-κB to upregulate transcription of inflammatory genes. In lungs, RAGE is abundantly expressed on type I epithelial cells and soluble RAGE detected in plasma has been shown to be a marker of acute lung injury and PGD after lung transplantation.52–54
Neutrophil extracellular traps (NETs), chromatin-enzyme complexes released into the extracellular space by neutrophils, have been observed after lung transplantation and are associated with PGD in both humans and mice.55,56 A recent study suggested that NET formation can be triggered through mtDNA and depends on TLR9 signaling.57 Although the breakdown of NETs through administration of DNAse attenuates IRI, we have recently shown that the resultant NET fragments can augment alloimmune responses through MyD88-dependent maturation of antigen presenting cells.56 Combined with our previous observations that neutrophils can prime alloreactive T cells within lung grafts through activation of antigen presenting cells or by providing costimulation-in-trans, these studies indicate that neutrophilic graft infiltration not only mediates PGD, but may also predispose to rejection.38, 58 Several studies have explored the potential significance of innate immune signaling pathways after lung transplantation in humans. For example, a TLR-based transcript analysis of donor tissue can aid in predicting the risk for developing PGD.59 A separate study found that, recipient, but not donor, TLR4 polymorphisms that are associated with hypo-responsiveness are tied to reductions in acute rejection rates following lung transplantation, suggesting that innate immunity following lung transplantation in humans may lead to changes in adaptive immunity.60 This finding is consistent with our report of TLR2/4- and MyD88-deficient tolerant recipients that demonstrated protection from allograft rejection after treatment with LMW-HA.35
Human translation
Preclinical models are essential to address knowledge gaps in our understanding of the complex immune pathways which contribute to lung allograft failure. A variety of models have been developed, such as in vitro culture systems, hilar clamp models, and animal models of tracheal and lung transplantation. The introduction of the vascularized, aerated mouse orthotopic lung transplant model in 2007 has yielded valuable insights into the immune pathways activated after lung transplantation, which correlate to processes observed in humans.61,62 For example, an association between PGD and increased serum biomarkers, such as circulating mtDNA, was identified in mouse lung transplant recipients and demonstrated in human lung transplant patients as well.27 It is important to bear in mind the limitations inherent to animal models, as many of the pathways and responses to therapy observed in animal models unfortunately fail to translate into clinical practice. For example, species-specific differences exist with regard to the expression of major histocompatibility complex class II antigens on stromal cells that may impact immune responses to allografts.63 Therefore, it is important to have an understanding of the limitations of experimental preclinical models, and to utilize well-designed, complementary approaches to accurately address questions that are relevant to human pathology.
Future directions
The valuable insights gained over recent years regarding innate immune responses that are activated after lung transplantation have identified potential therapeutic targets and provided guidance for future studies. It is likely that we will discover novel pathways which, if targeted pharmacologically, may ameliorate deleterious immunologic responses. For example, a recent study suggested that circulating exosomes that develop in human lung transplant patients who sustain certain viral infections can activate cGAS-STING and RIG-1 pathways.64 Further understanding on the interspecies differences in immunologic responses after transplantation may lead to advancements in xenotransplantation or initiate a paradigm shift in preclinical models, such as utilization of biomimetic microsystems with human pulmonary tissue.65 Also, insights into innate immunity following lung transplantation may aid with the evaluation, selection and potentially treatment of donor organs as well.
Conclusion
There is an expanding body of literature that demonstrates the vital role of the innate immune system in mounting inflammatory responses after lung transplantation, leading to acute graft injury and also enhanced alloimmunity. These immunologic signals may be triggered by both infectious and non-infectious ligands. Triggers of innate immune pathways activate inflammatory signaling cascades which can regulate the migratory behavior of leukocytes to the graft and their activation. This is highly relevant as lung grafts are constantly exposed to the external environment and also provide a suitable site for the generation of immune responses. Further research is needed to elucidate the signaling pathways involved in cell death, ischemia-reperfusion injury and respiratory infections following lung transplantation so that novel therapies may be developed to abrogate graft injury and improve outcomes.
Acknowledgement
DK is supported by National Institutes of Health grants 1P01AI116501, R01HL094601, R01HL151078, Veterans Administration Merit Review grant 1I01BX002730, The Cystic Fibrosis Foundation and The Foundation for Barnes-Jewish Hospital.
Abbreviations:
- ATP
Adenosine triphosphate
- CLAD
Chronic lung allograft dysfunction
- CXCL2
Chemokine (C-X-C motif) ligand 2
- DAMP
Damage-associated molecular pattern
- DAP12
DNAX activation protein of 12 kDa
- HMGB1
High-mobility group box 1
- IL-1β
Interleukin-1β
- IRI
Ischemia-reperfusion injury
- LMW-HA
Low-molecular-weight hyaluronic acid
- LPS
Lipopolysaccharide
- MLKL
Mixed lineage kinase domain-like
- MPTP
Mitochondrial permeability transition pore
- mtDNA
Mitochondrial DNA
- MyD88
Myeloid differentiation factor 88
- NET
Neutrophil extracellular trap
- NF-κB
Nuclear factor kappa-light-chain enhancer of activated B cells
- P2XR7
P2X purinoreceptor 7
- PAMP
Pathogen-associated molecular pattern
- PGD
Primary graft dysfunction
- PKCδ
Protein kinase C-δ
- PRR
Pattern recognition receptor
- RAGE
Receptor for advanced glycation end products
- RIPK1
Receptor-interacting protein kinase 1
- RIPK3
Receptor-interacting protein kinase 3
- STAT3
Signal transducer and activator of transcription 3
- TLR
Toll-like receptor
- TRIF
Toll/Interleukin-1 receptor domain-containing adaptor-inducing interferon-beta
Footnotes
Conflict of interest
DK has a pending patent entitled “Compositions and methods for detecting CCR2 receptors” (application number 15/611,577). The other authors have declared no conflict.
@WashUSurgeryIn this article, Shepherd, Gauthier and Kreisel discuss recent studies that highlight potential avenues for lung-specific therapies early following transplantation to dampen innate immune responses and improve outcomes.
References
- 1.Rana A, Gruessner A, Agopian VG, et al. Survival benefit of solid-organ transplant in the United States. JAMA Surg 2015;150:252–9. [DOI] [PubMed] [Google Scholar]
- 2.Gelman AE, Fisher AJ, Huang HJ, et al. Report of the ISHLT working group on primary lung graft dysfunction part III: mechanisms: a 2016 consensus group statement of the International Society for Heart and Lung Transplantation. J Heart Lung Transplant 2017;36:1114–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Christie JD, Kotloff RM, Ahya VN, et al. The effect of primary graft dysfunction on survival after lung transplantation. Am J Resp Crit Care Med 2005;171:1312–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chambers DC, Cherikh WS, Harhay MO, et al. The international thoracic organ transplant registry of the international society for heart and lung transplantation: thirty-sixth adult lung and heart-lung transplantation report-2019; focus theme: donor and recipient size match. J Heart Lung Transplant 2019;38:1042–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kulkarni HS, Cherikh WS, Chambers DC, et al. bronchiolitis obliterans syndrome-free survival after lung transplantation: an international society for heart and lung transplantation thoracic transplant registry analysis. J Heart Lung Transpl 2018;38:5–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bharat A, Narayanan K, Street T, et al. Early posttransplant inflammation promotes the development of alloimmunity and chronic human lung allograftrejection. Transplantation 2007;83:150–8. [DOI] [PubMed] [Google Scholar]
- 7.Bharat A, Kuo E, Steward N, et al. Immunological link between primary graft dysfunction and chronic lung allograft rejection. Ann Thorac Surg 2008;86:189–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fischer S, Cassivi SD, Xavier AM, et al. Cell death in human lung transplantation: apoptosis induction in human lungs during ischemia and after transplantation. Ann Surg 2000;231:424–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fischer S, MacLean AA, Liu M, et al. Dynamic changes in apoptotic and necrotic cell death correlate with severity of ischemia-reperfusion injury in lung transplantation. Am J Respir Crit Care Med 2000;162:1932–9. [DOI] [PubMed] [Google Scholar]
- 10.Wang X, O’Brien ME, Yu J, et al. Prolonged cold ischemia induces necroptotic cell death in ischemia-reperfusion injury and contributes to primary graft dysfunction after lung transplantation. Am J Resp Cell Mol Biol 2019;61:244–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim H, Zamel R, Bai XH, et al. Ischemia-reperfusion induces death receptor-independent necroptosis via calpain- STAT3 activation in a lung transplant setting. Am J Physiol Lung Cell Mol Physiol 2018;315:L595–608. [DOI] [PubMed] [Google Scholar]
- 12.Kanou T, Ohsumi A, Kim H, et al. Inhibition of regulated necrosis attenuates receptor-interacting protein kinase 1-mediated ischemia-reperfusion injury after lung transplantation. J Heart Lung Transplant 2018;37:1261–70. [DOI] [PubMed] [Google Scholar]
- 13.Vaseva AV, Marchenko ND, Ji K, et al. p53 opens the mitochondrial permeability pore to trigger necrosis. Cell 2012;149:1536–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tran DT, Esckilsen S, Mulligan J, Mehrotra S, Atkinson C, Nadig SN. Impact of mitochondrial permeability on endothelial cell immunogenicity in transplantation. Transplantation 2018;102:935–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim H, Zhao J, Zhang Q, et al. δV1–1 reduces pulmonary ischemia reperfusion-induced lung injury by inhibiting necrosis and mitochondrial localization of PKCδ and p53. Am J Transplant 2015;16:83–98. [DOI] [PubMed] [Google Scholar]
- 16.Li J, Yan Z, Fang W. A mechanism study underlying the protective effects of cyclosporine-a on lung ischemia-reperfusion injury. Pharm 2017;100:83–90. [DOI] [PubMed] [Google Scholar]
- 17.Hou L, Yang Z, Wang Z, et al. NLRP3/ASC-mediated alveolar macrophage pyroptosis enhances HMGB1 secretion in acute lung injury induced by cardiopulmonary bypass. Lab Invest 2018;98:1052–64. [DOI] [PubMed] [Google Scholar]
- 18.Wu DD, Pan PH, Liu B, et al. Inhibition of alveolar macrophage pyroptosis reduces lipopolysaccharide-induced acute lung injury in mice. Chin Med J 2015;128:2638–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Noda K, Tane S, Haam S, et al. Targeting circulating leukocytes and pyroptosis during ex vivo lung perfusion improves lung preservation. Transplantation 2017;101:2841–9. [DOI] [PubMed] [Google Scholar]
- 20.Fei L, Jingyuan X, Fangte L, et al. Preconditioning with rHMGB1 ameliorates lung ischemia–reperfusion injury by inhibiting alveolar macrophage pyroptosis via the Keap1/Nrf2/HO-1 signaling pathway. J Transl Med 2020;18:301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li W, Feng G, Gauthier JM, et al. Ferroptotic cell death and TLR4/Trif signaling initiate neutrophil recruitment after heart transplantation. J Clin Invest 2019;129:2293–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu Y, Li X, Cheng Y, Yang M, Wang R. Inhibition of ACSL4 attenuates ferroptotic damage after pulmonary ischemia-reperfusion. FASEB J 2020;00:1–14. [DOI] [PubMed] [Google Scholar]
- 23.Dong H, Qiang Z, Chai D, et al. Nrf2 inhibits ferroptosis and protects against acute lung injury due to intestinal ischemia reperfusion via regulating SLC7A11 and HO-1. Aging 2020;12:12943–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Holze C, Michaudel C, Mackowiak C, et al. Oxeiptosis, a ROS-induced caspase-independent apoptosis-like cell-death pathway. Nat Immunol 2018;19:130–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Song X, Zhu S, Xie Y, et al. JTC801 Induces pH-dependent death specifically in cancer cells and slows growth of tumors in mice. Gastroenterology 2018;154:1480–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Linkermann A, Hackl MJ, Kunzendorf U, Walczak H, Krautwald S, Jevnikar AM. Necroptosis in immunity and ischemia-reperfusion injury. Am J Transplant 2013;13:2797–804. [DOI] [PubMed] [Google Scholar]
- 27.Scozzi D, Ibrahim M, Liao F, et al. Mitochondrial damage–associated molecular patterns released by lung transplants are associated with primary graft dysfunction. Am J Transplant 2019;19:1464–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ueno H, Matsuda T, Hashimoto S, et al. Contributions of high mobility group box protein in experimental and clinical acute lung injury. Am J Respir Crit Care Med 2004;170:1310–6. [DOI] [PubMed] [Google Scholar]
- 29.Hashimoto K, Cypel M, Juvet S, et al. Higher M30 and high mobility group box 1 protein levels in ex vivo lung perfusate are associated with primary graft dysfunction after human lung transplantation. J Heart Lung Transplant 2018;37:240–9. [DOI] [PubMed] [Google Scholar]
- 30.Sharma AK, LaPar DJ, Stone ML, et al. Receptor for Advanced Glycation End Products (RAGE) on iNKT cells mediates lung ischemia-reperfusion injury. Am J Transplant 2013;13:2255–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sugimoto S, Lin X, Lai J, et al. Apyrase treatment prevents ischemia–reperfusion injury in rat lung isografts. J Thorac Cardiovasc Surg 2009;138:752–9. [DOI] [PubMed] [Google Scholar]
- 32.Liu K, Vergani A, Zhao P, et al. Inhibition of the purinergic pathway prolongs mouse lung allograft survival. Am J Respir Cell Mol Biol 2014;51:300–10. [DOI] [PubMed] [Google Scholar]
- 33.Jiang D, Liang J, Fan J, et al. Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nat Med 2005;11:1173–9. [DOI] [PubMed] [Google Scholar]
- 34.Jiang D, Liang J, Noble PW. Regulation of noninfectious lung injury, inflammation, and repair by the extracellular matrix glycosaminoglycan hyaluronan. Anatomical Rec 2010;293:982–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Todd JL, Wang X, Sugimoto S, et al. Hyaluronan contributes to bronchiolitis obliterans syndrome and stimulates lung allograft rejection through activation of innate immunity. Am J Resp Crit Care Med 2014;189:556–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Akbarpour M, Lecuona E, Chiu SF, et al. Residual endotoxin induces primary graft dysfunction through ischemia/reperfusion-primed alveolar macrophages. J Clin Invest 2020;130:4456–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tanaka S, Gauthier JM, Terada Y, et al. Bacterial products in donor airways prevent the induction of lung transplant tolerance. Am J Transplant 2021;21:353–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yamamoto S, Nava RG, Zhu J, et al. Cutting edge: Pseudomonas aeruginosa abolishes established lung transplant tolerance by stimulating B7 expression on neutrophils. J Immunol 2012;189:4221–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kulkarni HS, Tsui K, Sunder S, et al. Pseudomonas aeruginosa and acute rejection independently increase the risk of donor-specific antibodies after lung transplantation. Am J Transplant 2020;20:1028–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Arancibia SA, Beltrán CJ, Aguirre IM, et al. Toll-like receptors are key participants in innate immune responses. Biol Res 2007;40:97–112. [DOI] [PubMed] [Google Scholar]
- 41.Kaczorowski DJ, Nakao A, Vallabhaneni R, et al. mechanisms of Toll-like receptor 4 (TLR4)-mediated inflammation after cold ischemia/reperfusion in the heart. Transplantation 2009;87:1455–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shimamoto A, Pohlman TH, Shomura S, et al. Toll-like receptor 4 mediates lung ischemia-reperfusion injury. Ann Thorac Surg 2006;82:2017–23. [DOI] [PubMed] [Google Scholar]
- 43.Zanotti G, Casiraghi M, Abano JB, et al. Novel critical role of Tolllike receptor 4 in lung ischemia-reperfusion injury and edema. Am J Physiol Lung Cell Mol Physiol 2009;297:L52–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Altemeier WA, Liles WC, Villagra-Garcia A, et al. Ischemia-reperfusion lung injury is attenuated in MyD88-deficient mice. PLoS ONE 2013;8:e77123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Merry HE, Phelan P, Doak MR, Zhao M, Hwang B, Mulligan MS. Role of toll-like receptor-4 in lung ischemia-reperfusion injury. Ann Thorac Surg 2015;99:1193–9. [DOI] [PubMed] [Google Scholar]
- 46.Poltorak A, He X, Smirnova I, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 1998;282:2085–8. [DOI] [PubMed] [Google Scholar]
- 47.Kreisel D, Sugimoto S, Tietjens J, et al. Bcl3 prevents acute inflammatory lung injury in mice by restraining emergency granulopoiesis. J Clin Invest 2011;121:265–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zheng Z, Chiu S, Akbarpour M, et al. Donor pulmonary intravascular nonclassical monocytes recruit recipient neutrophils and mediate primary lung allograft dysfunction. Sci Transl Med 2017;9:eaal4508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hsiao HM, Fernandez R, Tanaka S, et al. Spleen-derived classical monocytes mediate lung ischemia-reperfusion injury through IL-1b. J Clin Invest 2018;128:2833–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sugimoto S, Lin X, Okazaki M, et al. Monocyte differentiation is controlled by MyD88 after mouse orthotopic lung transplantation. Transplant Proc 2009;41:388–90. [DOI] [PubMed] [Google Scholar]
- 51.Spahn JH, Li W, Bribriesco AC, et al. DAP12 expression in lung macrophages mediates ischemia/reperfusion injury by promoting neutrophil extravasation. J Immunol 2015;194:4039–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fehrenbach H, Kasper M, Tschernig T, Shearman MS, Schuh D, Müller M. Receptor for advanced glycation endproducts (RAGE) exhibits highly differential cellular and subcellular localisation in rat and human lung. Cell Mol Biol 1998;44:1147–57. [PubMed] [Google Scholar]
- 53.Uchida T, Shirasawa M, Ware LB, et al. Receptor for advanced glycation end-products is a marker of type I cell injury in acute lung injury. Am J Respir Crit Care Med 2006;173:1008–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Christie JD, Shah CV, Kawut SM, et al. Plasma levels of receptor for advanced glycation end products, blood transfusion, and risk of primary graft dysfunction. Am J Respir Crit Care Med 2009;180:1010–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sayah DM, Mallavia B, Liu F, et al. Neutrophil extracellular traps are pathogenic in primary graft dysfunction after lung transplantation. Am J Respir Crit Care Med 2015;191:455–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Scozzi D, Wang X, Liao F, et al. Neutrophil extracellular trap fragments stimulate innate immune responses that prevent lung transplant tolerance. Am J Transplant 2019;19:1011–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mallavia B, Liu F, Lefrançais E, et al. Mitochondrial DNA stimulates TLR9-dependent neutrophil extracellular trap formation in primary graft dysfunction. Am J Respir Cell Mol Biol 2020;62:364–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kreisel D, Sugimoto S, Zhu J, et al. Emergency granulopoiesis promotes neutrophil-dendritic cell encounters that prevent mouse lung allograft acceptance. Blood 2011;118:6172–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cantu E, Yan M, Suzuki Y, et al. Preprocurement In situ donor lung tissue gene expression classifies primary graft dysfunction risk. Am J Respir Crit Care Med 2020;202:1046–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Palmer SM, Burch LH, Davis RD, et al. The role of innate immunity in acute allograft rejection after lung transplantation. Am J Respir Crit Care Med 2003;168:628–32. [DOI] [PubMed] [Google Scholar]
- 61.Gelman AE, Li W, Richardson SB, et al. Cutting edge: Acute lung allograft rejection is independent of secondary lymphoid organs. J Immunol 2009;182:3969–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Okazaki M, Krupnick AS, Kornfeld CG, et al. A mouse model of orthotopic vascularized aerated lung transplantation. Am J Transplant 2007;7:1672–9. [DOI] [PubMed] [Google Scholar]
- 63.Houser SL, Benjamin LC, Wain JC, Madsen JC, Allan JS. Constitutive expression of major histocompatibility complex class II antigens in pulmonary epithelium and endothelium varies among different species. Transplantation 2004;77:605–7. [DOI] [PubMed] [Google Scholar]
- 64.Bansal S, Limaye A, Bharat A, et al. Circulating exosomes from human lung transplant recipients having respiratory viral infections contain nucleic acids and activate signaling pathways CGAS/STING and RIG-1. J Heart Lung Transplant 2020;39:S113–4. [Google Scholar]
- 65.Huh D, Matthews BD, Mammoto A, et al. Reconstituting organ-level lung functions on a chip. Science 2010;328:1662–8. [DOI] [PMC free article] [PubMed] [Google Scholar]