

Abstract
Purpose:
activating missense mutations of KRAS are the most frequent oncogenic driver events in lung adenocarcinoma (LUAD). However, KRAS isoforms are highly heterogeneous, and data on the potential isoform-dependent therapeutic vulnerabilities are still lacking.
Experimental design:
we developed an isogenic cell-based platform to compare the oncogenic properties and specific therapeutic actionability of KRAS mutant isoforms. In parallel, we analyzed clinicopathologic and genomic data from 3560 patients with non-small cell lung cancer (NSCLC) to survey allele-specific features associated with oncogenic KRAS mutations.
Results:
In isogenic cell lines expressing different mutant KRAS isoforms, we identified isoform-specific biochemical, biological and oncogenic properties both in vitro and in vivo. These exclusive features correlated with different therapeutic responses to MEK inhibitors, with KRAS G12C and Q61H mutants being more sensitive compared to other isoforms. In vivo, combined KRAS G12C and MEK inhibition was more effective than either drug alone. Among patients with NSCLCs which underwent comprehensive tumor genomic profiling, STK11 and ATM mutations were significantly enriched among tumors harboring KRAS G12C, G12A, and G12V mutations. KEAP1 mutation was significantly enriched among KRAS G12C and KRAS G13X LUADs. KRAS G13X mutated tumors had the highest frequency of concurrent STK11 and KEAP1 mutations. Transcriptomic profiling revealed unique patterns of gene expression in each KRAS isoform, compared to KRAS wild-type tumors.
Conclusions:
This study demonstrates that KRAS isoforms are highly heterogeneous in terms of concurrent genomic alterations and gene expression profiles, and that stratification based on KRAS alleles should be considered in the design of future clinical trials.
Translational relevance
KRAS is the most commonly mutated driver oncogene in solid tumors, including non-small cell lung cancer (NSCLC), and KRAS mutations have traditionally been associated with poor prognosis and treatment resistance. Comprehensive genomic, biologic and clinicopathologic analysis of KRAS mutant variants in pre-clinical models and samples from patients with non-squamous NSCLC revealed isoform-dependent therapeutic vulnerabilities of these tumors. Our results suggest that the use of specific genetically defined in vitro tools together with the development of large patient-derived datasets facilitates the identification of potentially actionable vulnerabilities for KRAS mutant NSCLC.
Introduction
KRAS is one of the most commonly mutated oncogenes in human cancer, with selectively high frequency in tumors of the pancreas, colon, and lung. In non-small cell lung cancer (NSCLC), KRAS mutations are detected in about 30% of patients and are associated with poor prognosis (1,2). Several strategies have been investigated over the last decades to target KRAS mutation in lung cancer, including the inhibition of downstream effectors such as MEK (3,4). However, none of these strategies was effective in clinical trials, and the optimal first-line therapy for advanced KRAS-mutant lung adenocarcinoma (LUAD) still consists of PD-(L)1 blockade alone or in combination with platinum-based chemotherapy (5). More recently, alternative strategies targeting KRAS-mutant lung cancer have been investigated in preclinical models, and are in various stages of clinical development (1,6,7).
To date, the greatest success in targeting KRAS mutations is represented by the development of isoform-specific direct KRAS inhibitors. Recent data from early phase clinical trials of KRAS G12C direct inhibitors sotorasib and adagrasib have shown encouraging activity in KRAS G12C-mutant NSCLCs, with responses occurring in ~30–40% of patients (8–10). Based on the results of the CodeBreaK 100 study and KRYSTAL-1, sotorasib and adagrasib were granted accelerated FDA approval and breakthrough therapy designation (BTD), respectively, for patients with NSCLC who have received on prior systemic therapy. Nevertheless, an urgent need remains for better stratification and effective therapeutic strategies to improve outcomes for KRAS-mutant cancer patients.
Although RAS mutations were discovered over 30 years ago (11), the detailed understanding of the biological properties of oncogenic KRAS is still far to be complete. New aspects of KRAS biology have been recently described, including the requirement of dimerization for oncogenic signaling (12), lipid-sorting specificity into the membrane (13), tissue-specific co-mutation networks (14).
One intriguing aspect of KRAS biology is the potential impact of distinct KRAS activating mutations on downstream signaling and drug sensitivity (15,16). Until very recently, RAS proteins with alterations in codon 12, 13, or 61 have been considered oncogenic equivalents; however, emerging data suggest functional differences for each RAS mutation in colon cancer (17), melanoma (18) and NSCLC, where different mutant KRAS proteins are known to be heterogeneous in terms of clinical outcome (19–20). Recently studies in isogenic cell lines demonstrated that each individual mutation generates a distinct oncogenic and network response (14,15,21–23). In terms of translational impact, the study of differences among specific KRAS mutants by means of more informative experimental approaches will be fundamental to discover new therapeutic strategies for individual KRAS mutations.
Here, we developed an isogenic system where a series of mutant KRAS isoforms commonly found in lung cancer were directly compared to highlight distinct GTP-loading levels, differential drug sensitivity to MEK inhibitors and unique growth properties in vivo. To gather evidence on whether these differences also translated in different clinical outcomes we investigated the impact of the different KRAS isoforms on survival in a large cohort of patients with non-squamous NSCLC. Together, these results suggest that genomic co-alterations and residue-specific properties of KRAS mutations have important biologic and clinical implications.
Methods
Generation of KRaslox KRASMUT cells
KRaslox KRASMUT cells were generated as previously described (12). Briefly, KRASMUT retroviral constructs were created by point mutagenesis from pBABE HA-tagged KRASWT plasmid (a gift from Channing Der, Addgene plasmid #75282). Retroviruses were generated by co-transfection of pBABE plasmids together with pAmpho plasmid into 293T cells using FuGENE® HD Transfection Reagent (Promega). The retroviruses were transduced into HRas−/−; NRas−/−; KRaslox/lox MEFs (24) followed by puromycin selection (1 μg/ml). Selected cells were then cultured in the presence of 4-hydroxytamoxifen (4OHT) (Sigma, 600 nM) for at least 10 days in order to achieve complete deletion of endogenous KRas alleles. Cells were not cultured longer than 3 months after thawing from frozen stocks and were routinely checked for Mycoplasma as determined by the Mycoplasma Plus PCR Primer Set (Agilent).
Growth assays in vitro
Cells (1×103) were seeded in 96-well plates in 150 μl complete medium. The following day, cells were PBS-washed and 150 μl of medium with different concentrations of FBS was added to the plates. Cells were then incubated in the IncuCyte Zoom for real-time imaging, with three fields imaged per well under 10x magnification every two hours for a total of 60 to 96 hours. Data were analyzed using the IncuCyte Confluence version 1.5 software, which quantified cell surface area coverage as confluence values. IncuCyte experiments were performed in triplicate. A single representative growth curve is shown for each condition. For hypoxic conditions, cells (1×103) were seeded in 96-well plates in 150 μl complete medium. The following day, cells were PBS-washed and 150 μl of medium with different concentrations of FBS was added to the plates that were incubated in 1% O2 incubator. Cell viability was assessed by CellTiter-Glo® 2.0 (Promega) at the indicated time points.
Analysis of MEK/ERK inhibitor sensitivities
Data from a large-scale pharmacogenomics study, the Cancer Genome Project (CGP, dataset version 2020), were accessible from http://www.cancerrxgene.org. 310 lung, pancreas and colon cancer cell lines and their drug sensitivity values to MEK/ERK inhibitors in the CGP dataset were used for analysis. In these data, the natural logarithm of the IC50 value represents the drug sensitivity value. Cell lines were grouped according to KRAS mutational subtypes and pairwise comparisons were performed using the Mann-Whitney U test.
Drug-response assays
Cells (1×103) were seeded in 96-well plates. The following day, cells were treated with drugs using a ten-point dose titration scheme. After 72 hours, cell viability was assessed using the colorimetric MTS assay. Absolute inhibitory concentration (IC) values were calculated using four-parameter logistic curve fitting. All experimental points were a result of three to six replicates, and all experiments were repeated at least three times. The data was graphically displayed using GraphPad Prism 5 for Windows (GraphPad Software). Each point (mean ± standard deviation) represents growth of treated cells compared to untreated cells. The curves were fitted using a non-linear regression model with a sigmoidal dose response. For synergy distribution assays, cells were seeded at 1500 cells per well in a 384-well plate (Corning). The following day, cells were treated with selumetinib and SHP099, varying concentration from 0.25 μM to 15 μM and from 0.5 μM to 15 μM, respectively. After 72 hours, cell viability was measured by CellTiter-Glo 2D (Promega). Four replicates of each mutant were analyzed implementing LOEWE model (Combenefit software); experiments were repeated three times.
Ras-GTP Pull-down
Cell were grown in 0.1% FBS for 24 hours, stimulated with EFG (Thermo Fisher Cat#PHG0311) 50ng/ml for 5 minutes and Ras-GTP levels were assessed by Active Ras Detection Kit (Cell Signaling, Cat#8821) using Raf-RBD fused to GST to bind active (GTP-bound) Ras. Protein lysates (500 μg) were incubated with 30 μl glutathione resin and GST protein binding domains for one hour at 4ºC to capture active small GTPases according to the manufacturer’s protocol. After washing, the bound GTPase was recovered by eluting the GST-fusion protein from the glutathione resin. The purified GTPase was detected by Western blot using mouse monoclonal anti-KRAS (F234) (Santa Cruz Biotech, Cat#sc-30).
3D organoid culture and growth assay
After trypsinization, cells from the plastic culture were resuspended in ice-cold Matrigel (Corning) and incubated on pre-warmed 6 cm plate to solidify. Organoids were cultured in Renaissance Essential Tumor Medium (RETM; Cellaria) with B-27 supplement (Thermo Fisher Scientific), and passaged over three times before performing any experiments. For the growth assay, organoids in Matrigel were dissociated into single cells using TrypLE Express (Invitrogen) at 37°C. A thousand of cells were plated into each well of 384-well ultra-low attachment microplates (Corning) with the media and 10% Matrigel. On the next day, cells were treated with dose titration of trametinib, or selumetinib for three days (from 1 nM to 10 µM). Each dose had more than three replicates. The viability assay was tested using CellTiter-Glo 3D (Promega) according to the manufacturer’s protocol. The statistical significance was accessed using ANOVA and Turkey’s post-test for multiple comparison.
In vivo assays
Crl:NU-Foxn1nu mice (females, 8-week-old) were purchased from Charles River. For lung colonization assays, KRaslox KRASMUT cells (1×106) were injected tail vein as single-cell suspension in 200μl of sterile PBS. Mice were sacrificed synchronously at a single time point of one month and lungs collected in formalin. For drug treatment assays, KRaslox KRASMUT cells (2×106) were injected subcutaneously in a 1:1 mix of serum-free DMEM and Matrigel (phenol red–free; BD Biosciences) in both flanks of recipient mice. Once a palpable tumor formed, were randomly assigned to either selumetinib, sotorasib or vehicle treatment and measurements were taken daily using calipers. Selumetinib and sotorasib, resuspended in 2% Hydroxypropyl Methyl Cellulose (HPMC), 1% Tween 80 were administered daily by oral gavage at a dose of 50 mg/kg. For IVIS longitudinal monitoring, KRaslox KRASMUT cells were transduced with pFUGW-Pol2-ffLuc2-eGFP (Addgene plasmid #71394) and FACS-sorted for GFP expression. KRaslox KRASMUT-luciferase cells (2×106) were injected in the tail vein of 8-weeks old NCr nude mice (Taconic Biosciences, NY) as single-cell suspension in 200μl of sterile PBS and were monitored using an IVIS imaging system (Perkin Elmer, MA). All care and treatment of experimental animals were in strict accordance with Good Animal Practice as defined by the US Office of Laboratory Animal Welfare and approved by the Dana-Farber Cancer Institute Institutional Animal Care and Use Committee and by the Italian Health Minister (authorization n° 1227/2020-PR).
Study Population
Clinicopathologic and genomic data from patients with NSCLC whose tumors underwent targeted next generation sequencing at the Dana-Farber Cancer Institute who had provided written informed consent to institutional review board-approved protocols DF/HCC#02–180, #11–104, #13–364, and/or #17–000 were included in this study. This study was conducted in agreement to the Declaration of Helsinki.
Tumor mutational burden assessment and PD-L1 assessment
Tumor mutational burden (TMB), defined as the number of somatic, coding, base substitution and indel mutations per megabase (Mb) of genome examined was calculated from the DFCI OncoPanel next generation sequencing (NGS) platform. PD-L1 expression on tissue was assessed by immunohistochemistry using an anti-PD-L1 rabbit monoclonal antibody (clone E1L3N, Cell Signaling Technology).
Clinical outcomes
Overall survival (OS) was defined as the time from the date of initial diagnosis to death. Patients who were still alive at the data cut-off were censored at the date of last contact. Overall survival was compared between KRAS mutated and KRAS wild type cases, as well as across KRAS isoforms.
Co-occurrence and mutual exclusivity analysis
Fisher’s exact test p-values and conditional odds ratios were used to assess co-occurrence and mutual exclusivity for genes with at least 2% frequency in the relevant groups. Positive odds ratios represented tendency to co-occurrence and negative odds ratios represented tendency to mutually exclusivity. Multiple comparison correction was performed using the Benjamini-Hochberg procedure using the qvalue package in R.
Gene expression analysis from the TCGA
Gene expression data were downloaded from the Firehose website (TCGA Firehose Legacy version) while somatic mutation data were downloaded from cBioPortal website (cbioportal.org). The RSEM V2 values were used to represent gene expression and genes with counts less than 10 were filtered out. Gene expression profiles were analyzed according to KRAS mutation status. Median expression within each group was used to estimate expression fold-change (FC) to minimize the possible impact of outlier samples. Gene differential expression analyses across KRAS allele subgroups were conducted using R package DESeq2. P-values were corrected for multiple hypothesis testing via false discovery rate (FDR) adjustment. Fold-change threshold of an absolute value greater than 1.5 and FDR adjusted P-value threshold less than 0.1 were utilized to identify differentially expressed genes. Pathway enrichment analyses were conducted separately for up- and down-regulated genes using R package Reactome.
Statistical analysis
We summarized continuous and categorical variables using percentages and medians. To test for examine differences between continuous variables we used the Wilcoxon-Rank Sum test and Kruskal-Wallis, when appropriate. The Fisher’s exact test was used to compare associations between categorical variables. Estimate event-time distributions were examined using the Kaplan-Meier methodology, and the log-rank tests was used to test for differences in event-time distributions. Hazard ratios were estimated using the Cox regression models, as previously described (25). To investigate MEK/ERK inhibitor sensitivities for different KRAS mutant isoforms across cancers, we grouped cancer cell lines based on their KRAS mutation status into the different groups. Mann-Whitney U test was used to compare drug sensitivities between two groups. All P values were two-sided with confidence intervals set at the 95% level. P <0.05 were defined as significant. Multiple comparison correction was performed using the Benjamini-Hochberg procedure using the qvalue package in R. All statistical analyses were performed using R version 3.6.3.
Data availability statement
The Institutional clinicopathologic and genomic data that support the finding of our study are available upon reasonable request from the corresponding author. RNAseq data used in this study are publicly available from the TCGA (accession number: phs000178). Genomic data from the AACR project GENIE (Genomics Evidence Neoplasia Information Exchange) are publicly available (phs001337).
Results
Generation of KRasloxKRASMUT cell lines panel
Among KRAS-mutant lung cancer, codon 12 mutations predominate, accounting for nearly 90% of all KRAS mutations, followed by mutations in codons 13 and 61 (14). One question that arises from somatic genetic analysis is whether different KRAS mutations determine the clinical aspects of a given cancer. To investigate the role of individual KRAS isoforms on the biology of mutant KRAS avoiding context-dependent differences associated with different genetic backgrounds, we developed an isogenic system generated from HRas–/–; NRas–/–; KRaslox/lox mouse embryonic fibroblasts (MEFs) (24). In these cells, KRaslox/lox alleles excision is controlled by a resident 4-hydroxytamoxifen (4OHT)-dependent CREERT2 recombinase. We transduced HRas–/–; NRas–/–; KRaslox/lox MEFs with retroviruses encoding for different human HA-tagged KRAS mutants, including the most frequent mutant isoforms detected in human lung adenocarcinoma (G12C, G12D, G12V, G12A, G13D, Q61H) (12).
In all cell lines, the presence of the respective KRAS mutation was confirmed by Sanger sequencing (data not shown). Cells were treated with 4OHT to abolish the expression of endogenous wild-type KRas, thus allowing the characterization of specific oncogenic KRAS mutations in isogenic cell lines without the interference of H- or NRas isoforms or the endogenous wild-type KRas (herein referred to KRaslox KRASMUT system) (Figure 1A). Interestingly, all KRAS mutants showed increased expression upon deletion of the endogenous wild-type KRas allele. Therefore, we utilized only 4OHT-treated cells in order to eliminate any bias imposed by the wild-type allele. In full-serum condition, phosphorylation of ERK, MEK, SRC and AKT was slightly increased in all KRAS mutants compared to wild-type control cells (Figure 1B). In contrast, STAT3 and EGFR showed marked differences between mutants in terms of both phosphorylation and expression levels (Figure 1B). Remarkably, KRASGTP levels upon stimulation with EGF showed profound variability among mutants, with KRAS G12D and G12V being already saturated in untreated condition in contrast with the other mutants which are still responsive to mitogenic stimulation (Figure 1C).
Figure 1:
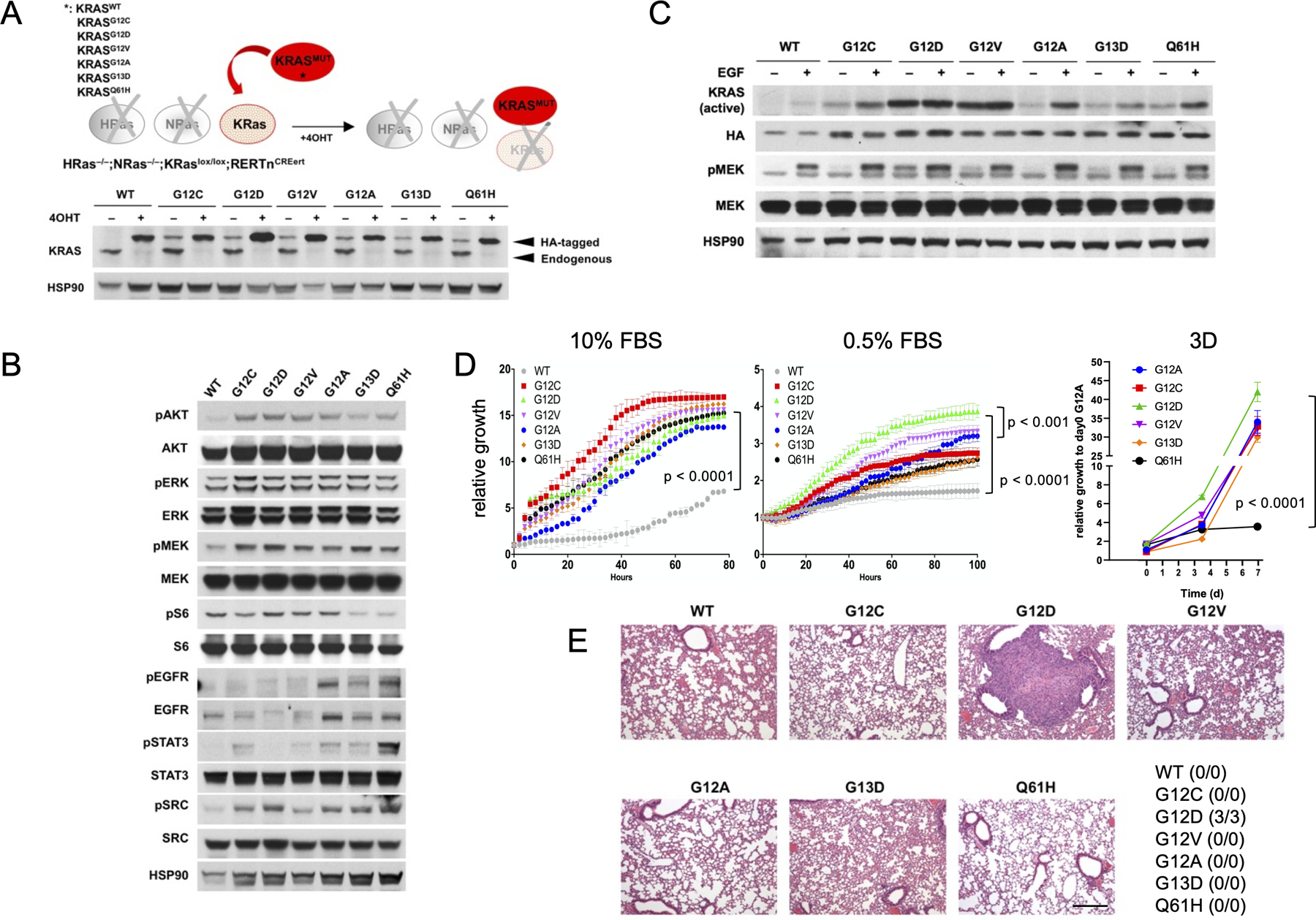
(A) schematic representation of the KRaslox KRASMUT system (top panel). HRas–/–; NRas–/–; KRaslox/lox mouse embryonic fibroblasts were infected with 5-MOI of retroviruses encoding for different human HA-tagged KRAS mutants, selected with puromycin (1μg/ml), treated with 4OHT (600nM) for at least 10 days and probed by Western Blot with the indicated antibodies (bottom panel). (B) KRaslox KRASMUT cells were treated with 4OHT for 10 days and probed by Western Blot with the indicated antibodies. Results are representative of one of three similar experiments. (C) KRas-GTP levels and activation of downstream pMEK signaling in KRaslox KRASMUT cells in 0.1% FBS medium upon stimulation with EGF (50 ng/mL). Results are representative of one of three similar experiments. (D) Growth rates of KRaslox KRASMUT cells in 10% or 0.5% fetal bovine serum (FBS) medium in 2D conditions as assessed by IncuCyte measurements (left panels, p < 0.0001 by unpaired Student’s t test). Results are representative of one of three similar experiments. Growth of KRaslox KRASMUT organoids was monitored by CTG assay. ANOVA analysis followed by Tukey’s multiple comparisons post-test was used for statistical analysis (p < 0.0001). (E) KRaslox KRASMUT cells (1×106) were injected into the tail vein of nude mice (n=3 per isoform). Animals were sacrificed after one month and lung colonization checked by serial sections stained with hematoxylin and eosin. The number of animals with evidence of lung colonization is indicated in the table. Scale bar: 100μM.
Growth properties of different KRAS mutants in vitro and in vivo
Next, we aimed at characterizing the proliferation kinetics of KRaslox KRASMUT cells both in vitro and in vivo. We tested growth properties of different KRAS mutants by Incucyte in full-serum and in starving conditions. All mutants displayed a strong proliferative advantage over wild-type KRAS controls in full-serum, nevertheless no significant differences were observed among mutants (Figure 1D). Under starving conditions however, a clear stratification was evident, with G12D, and G12V to a lesser extent, showing the best fitness over the other KRAS mutants (Figure 1D). Of note, G12D and G12V are the mutant isoforms displaying higher GTP-bound fraction in limited serum conditions (Figure 1C). Similar results were obtained in hypoxic conditions, with G12D mutant cells showing improved fitness compared to the other KRAS mutants upon concomitant starvation (Figure S1A). In 3D conditions, all KRAS mutant isoforms showed comparable growth kinetics with the exception of Q61H which was significantly less proliferative (Figure 1D). The advantage of G12D versus the other mutants was maintained also in tumor growth assays in vivo. We injected KRaslox KRASMUT cells expressing different mutants into the tail vein of nude mice and euthanized the animals after one month in order to check for colonization property into the lungs. At histological evaluation, G12D mutant cells displayed tumor nodules formation into the lungs, whereas all the other mutants did not (Figure 1E). Using the same mutant cells expressing luciferase gene, we noninvasively monitored tumor growth in the mice. As consistent to Figure 1E and in vitro results, G12D and G12V mutants were established in the mouse lung much quicker than other mutants (Figure S1B-C). These results confirmed an in vivo advantage for KRASG12D mutants (26), although further assessment at longer time points should be performed in order to detect delayed tumor onset in mutants other than G12D.
Sensitivity to MEK inhibitors and G12C inhibitors in vitro and in vivo
Due to the prominent role of the MAPK pathway in KRAS signaling, MEK inhibitors were thought to be able to achieve disease control in KRAS mutant tumors. However, phase III clinical trials demonstrated no overall benefit compared to chemotherapy (4). Retrospective analysis in different sub-populations of KRAS codon mutations showed that patients with KRAS G12C or G12V receiving selumetinib+docetaxel had improved overall survival compared with other KRAS mutations, suggesting that the type of KRAS mutations may impact on MEK inhibitors sensitivity in LUAD (20). We next surveyed the CGP pharmacogenomic dataset (27) to identify correlations between sensitivity to MEK inhibitors and KRAS mutant isoforms in a broad panel of lung, pancreas and colon cancer cell lines. We could not observe any obvious trend for increased sensitivity for specific isoforms other than G12R, likely due to the limited representation of small number of cell lines (Figure S2). Considering that cell lines heterogeneity poses challenging limits to the characterization of KRAS isoform as single variable to predict MEK inhibitors sensitivity, we tested a panel of MAPK inhibitors in our isogenic KRaslox KRASMUT system. GSK1120212 (trametinib) showed similar antiproliferative effects in all KRAS mutants, whereas selumetinib and the other MAPK inhibitors exhibited up to 10-fold IC50 variability in different KRAS isoforms, being G12C and Q61H among the most sensitive (Figure 2A and Figure S3) (28,29). The same trend was observed in 3D culture (Figure 2B). Western blot assessment of the MAPK pathway confirmed dose-dependent reduction of pERK in selumetinib-treated KRaslox KRASMUT cells, with the exception of G12A and G13D mutants (Figure 2C). To validate these findings into an in vivo setting, we implanted KRaslox KRASMUT cells into nude mice and followed tumor growth in presence or absence of selumetinib treatment (50mg/kg daily). In agreement with our findings in 2D and 3D conditions, we could stratify the outcome as tumor regression (G12C and Q61H), stable disease (G12D, G12V and G13D) and no response (G12A) based on tumor volume fold change on treatment (Figure 2D).
Figure 2:
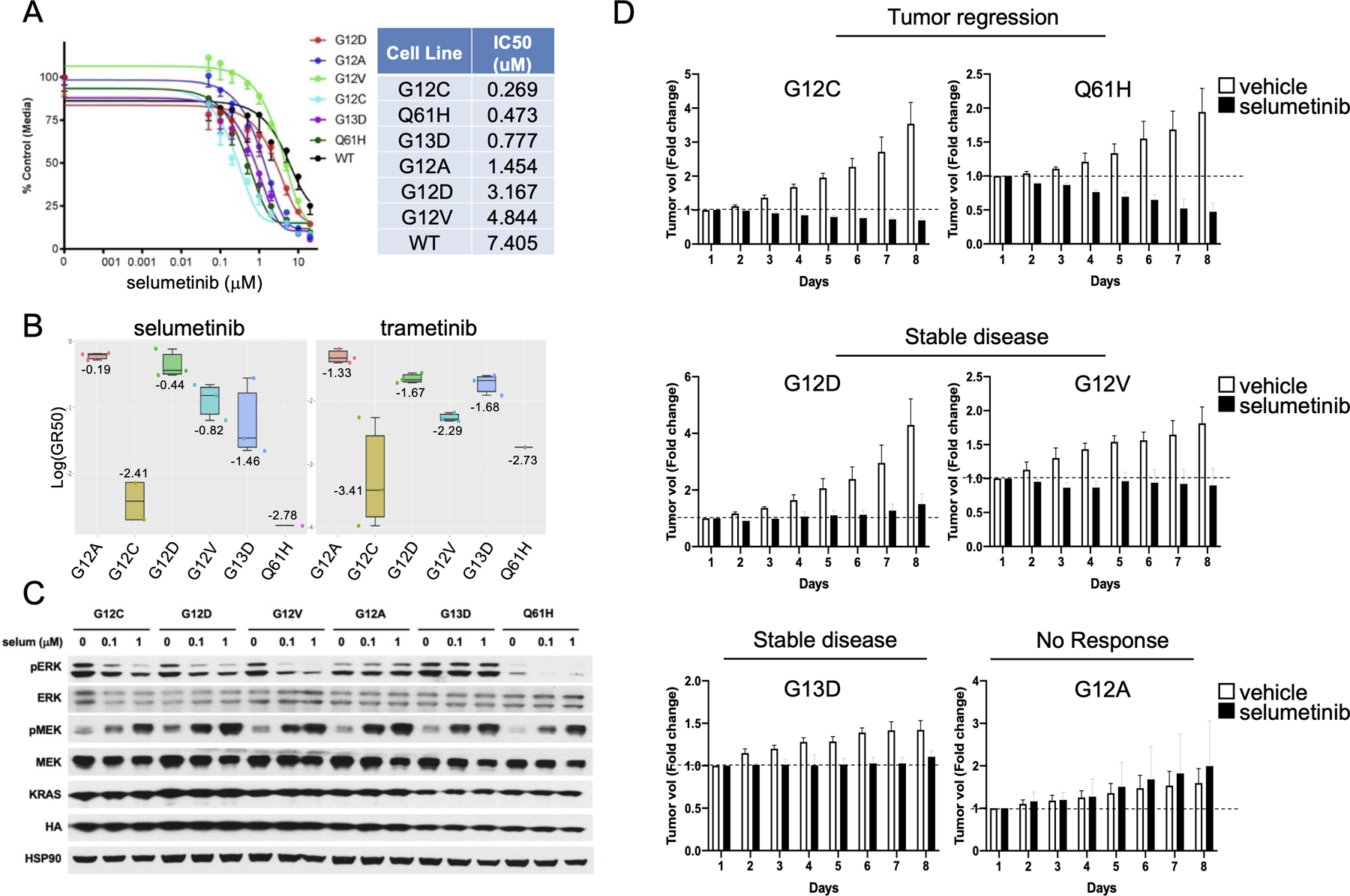
(A) Comparison of IC50 values to MEK inhibitor selumetinib between KRaslox KRASMUT cells expressing different KRAS isoforms. Error bars represent mean ± SD. Results are representative of one of three similar experiments. The table indicates cell lines ranked by IC50 values (lowest to highest). (B) Log GR50 values for selumetinib and trametinib are shown in the box plot. The numbers and vertical lines go through each box indicate the median. (C) KRaslox KRASMUT cells were seeded and treated with the indicated concentrations of selumetinib for 12 hours and then probed by Western Blot with the indicated antibodies. Results are representative of one of three similar experiments. (D) KRaslox KRASMUT cells (2×106) were injected subcutaneously with Matrigel (1:1) into nude mice. Mice with palpable tumors were randomized into two cohorts (n=6) and treated with vehicle or selumetinib (50mg/kg) daily by oral gavage for one week. Tumor size was measured every day with a caliper. Mean fold change in tumor volume relative to initial tumor volume is shown. Error bars represent mean ± SD.
Considering that pre-clinical in vivo studies demonstrated that in KRAS mutant LUAD, combined SHP2/MEK inhibition is therapeutically effective (30,31), we tested the same combination in KRaslox KRASMUT cells. Interestingly, we observed that combined SHP2/MEK blockade was more effective in G12C, G12D and G12V mutants compared to the other mutants, in agreement with previous findings (31,32) (Figure S4).
Given the very recent FDA approval of the selective G12C inhibitor sotorasib, (CodeBreaK 100, NCT03600883), we aimed at assessing the efficacy of G12C inhibition in our isoform-specific KRASG12C system, both in vitro and in vivo. As expected, drug-response curves with sotorasib demonstrated a clear selectivity for G12C mutant cells compared to the other KRAS mutant isoforms (Figure 3A). To determine how G12C inhibition compared with MEK inhibition in this model, we next implanted KRaslox KRASG12C cells into nude mice and randomized these mice to receive either vehicle, selumetinib (50mg/kg daily), sotorasib (50mg/kg daily), or selumetinib + sotorasib. Interestingly, selumetinib exerted better therapeutic response in comparison to sotorasib, possibly likely due to the delay in implementing feed-back mechanisms for MEK inhibitors as compared to selective G12C inhibitors as single agent. Importantly, combined treatment with selumetinib + sotorasib resulted in complete growth abrogation and disease control in mice treated with this combination (Figure 3B-E). This preliminary finding further supports the hypothesis that combined KRAS G12C and MEK inhibition may improve clinical outcomes in patients with KRAS mutant NSCLC over G12C inhibition alone. A phase Ib clinical trial of sotorasib in combination with other anticancer agents, including MEK inhibitors is currently ongoing (NCT04185883).
Figure 3:
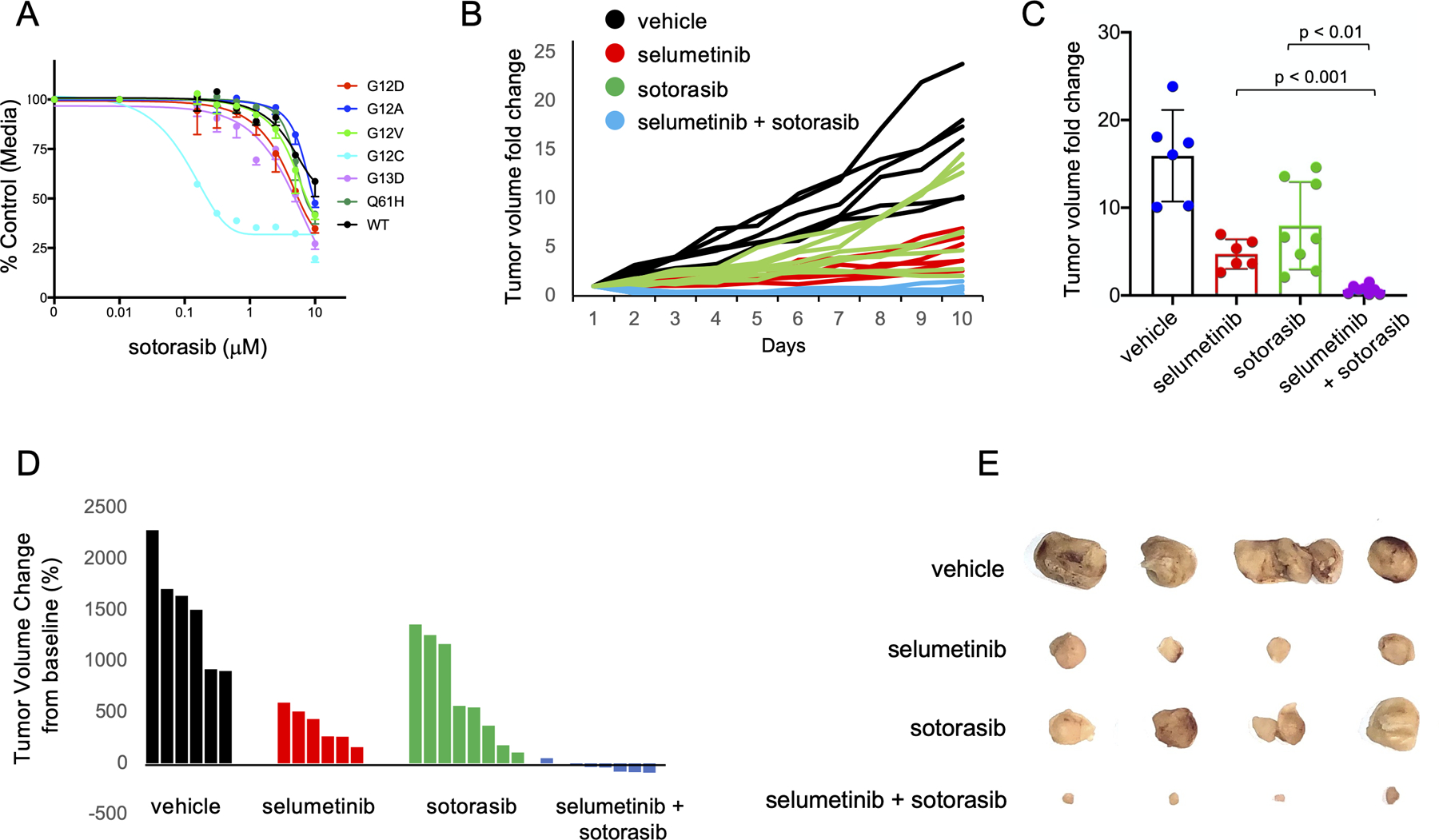
(A) Comparison of IC50 values to the selective G12C inhibitor sotorasib between KRaslox KRASMUT cells expressing different KRAS isoforms. Error bars represent mean ± SD. Results are representative of one of three similar experiments. (B) KRaslox KRASG12C cells (2×106) were injected subcutaneously with Matrigel (1:1) into nude mice. Mice with palpable tumors were randomized into four cohorts and treated with vehicle, selumetinib (50mg/kg), sotorasib (50mg/kg) or combo treatment daily by oral gavage. Tumor size was measured every day with a caliper. Tumor volume change relative to initial tumor volume is shown for individual tumors. (C) Bar graph showing average tumor volume fold change to baseline at end point. Error bars represent mean ± SD. (D) Waterfall plot showing individual tumor volume change from baseline (%) at end point. (E) Macroscopic photographs after 10 days of treatment and tumor volume tracking of KRaslox KRASG12C cells.
Clinicopathological and genomic correlation of KRAS variants in patients with non-squamous NSCLC
In order to translate our findings based on genetically defined cell lines to a clinically relevant context, a total of 3560 patients with NSCLCs which underwent successful comprehensive tumor genomic profiling at the Dana-Farber Cancer Institute were identified. Of these, 3126 (87.8%) were non-squamous NSCLC and were included in the final analysis (Figure S5A). The median age of the 3126 patients with non-squamous NSCLC was 66 (range: 18–99), 61.2% were female, 18.6% were current smokers, 58.4% were former smokers, and 18.6% were never smokers. KRAS mutation (KRASMUT) occurred in 1131 cases (36.2%), while the remaining 1995 cases (63.8%) had a KRAS wild type (KRASWT) genotype (Table S1). The most common KRAS mutations identified occurred at codon 12 (990/1131, 87.5%), of which the KRAS G12C was the most frequent (476/990, 47.2%) (Figure S5B).
We first investigated differences in clinicopathologic and genomic factors according to KRAS mutation status. Compared to KRASWT cases, patients with KRASMUT tumors were more likely to be older (P<0.001), females (P<0.0001), current/former smokers (P<0.0001), and to have adenocarcinoma histology (P<0.01), consistently with previous reports (33,34). Baseline clinicopathologic features of patients with KRASWT versus KRASMUT are shown in Table S2. We next focused on co-mutations in preselected genes of interest which define major subsets of NSCLC, including TP53, STK11, and KEAP1. Of note, while mutations in TP53 were more likely to occur in KRASWT tumors (P<0.0001), STK11 and KEAP1 were significantly enriched in KRAS mutated tumors (P<0.0001) (Figure S5C). We also examined whether PD-L1 expression and TMB distributions differed between KRASMUT and KRASWT Nsq-NSCLCs. We noted that both median PD-L1 expression (10 vs 5%, P<0.001) and median TMB (9.8 vs 8.4 mut/Mb, P<0.0001) were significantly higher in KRASMUT versus KRASWT tumors (Figure S5D).
Next, we examined the impact of KRAS mutation on survival among patients with Nsq-NSCLC. While we found no significant impact of KRAS mutation on OS in all comers (HR: 1.07, P=0.21), as well as among patients with stage I-III NSq-NSCLC (HR: 1.08, P=0.39), we observed a significant deleterious impact of KRAS mutation among patients with stage IV tumors (16.2 versus 25.5 months, HR:1.37, P<0.0001) (Figure 4A-C).
Figure 4:
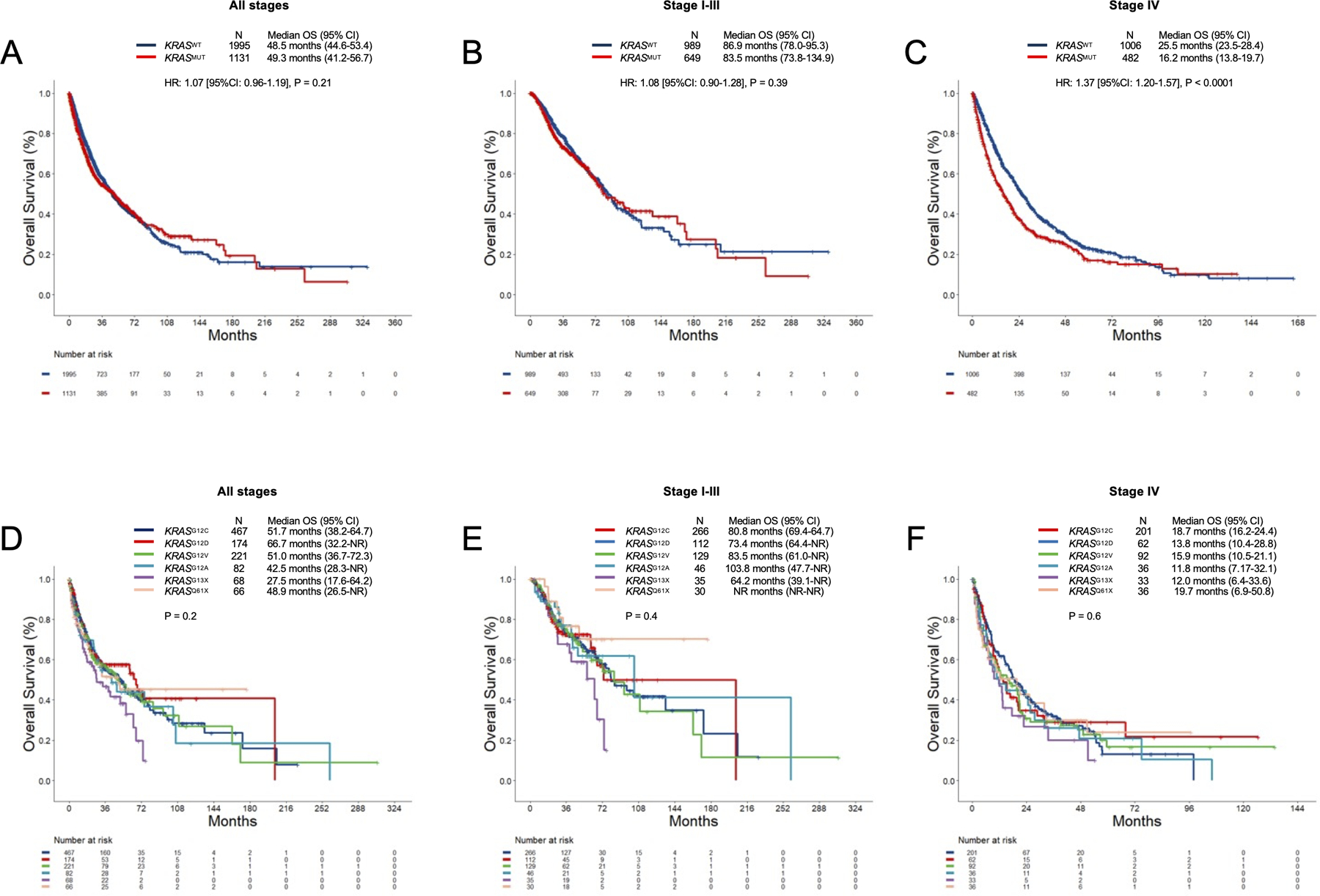
Overall survival in (A) all comers with non-squamous NSCLC, and (B) stage I-III, and (C) stage IV disease according to KRAS mutation status. Overall survival in (D) all comers with non-squamous NSCLC, and (E) stage I-III, and (F) stage IV disease according to KRAS alleles.
Although we identified a detrimental impact of KRAS mutation among patients with advanced NSq-NSCLC, this difference likely reflects the use of highly effective targeted therapies among patients with KRAS wild type tumors harboring EGFR mutations and ALK fusions.
Because we have shown that different KRAS mutation have different in vitro and in vivo growth properties, and differential responses to MEK inhibitors, we lastly investigated whether different KRAS mutation impacted survival in patients with Nsq-NSCLC. The clinicopathologic characteristics of patients according to KRAS alleles are shown in the Table S3. Consistently with previous reports (35) we found a significantly higher proportion of never smokers among patients with KRAS G12D mutation (21.8%) as opposed to the other KRAS variants (<10%). While there was no difference in terms of enrichment in TP53 mutation across the KRAS variants, the rates of concurrent STK11 and KEAP1 mutations were highest among KRAS codon 13 mutations (Figure S5C). Pairwise comparisons between KRAS alleles in terms of TP53, STK11, and KEAP1 co-mutation patterns are shown in Figure S6A. When we examined PD-L1 expression and TMB distributions in this across the different KRAS alleles, we found that the median TMB differed significantly across the seven groups, with KRAS G12D mutated tumors having the lowest number of non-synonymous mutations/megabase (Figure S5D). By contrast, no difference in PD-L1 expression was observed among these groups (Figure S5D). Pairwise comparisons between KRAS alleles in terms of PD-L1 and TMB distributions are shown in Figure S6B.
We lastly analyzed the impact of KRAS alleles on survival in patients with non-squamous NSCLC. We found no significant difference across KRAS mutation subtype in all comers, as well as among those with stage I-III or stage IV disease (Figure 4D-F).
KRAS variants have different co-mutation patterns and gene-expression profiles in patients with non-small cell lung cancer
Although we did not find differences in survival in patients with NSCLC with different KRAS alleles, we identified several differences in clinicopathologic and genomic factors among NSCLC harboring different KRAS alleles in the DFCI cohort. Therefore, we hypothesized that one of the reasons by which our findings in isogenic models did not translate into different clinical outcomes, could be the biological heterogeneity of KRAS-mutant LUADs. To further dissect this heterogeneity in patients with KRAS mutant LUADs, we interrogated a large publicly available cohort of 12.931 LUADs (GENIE v.9.0) which underwent tumor genomic profiling to comprehensively determine differences in co-mutation patterns across KRAS variants. After QC removing duplicate samples and gene annotation, 10,663 unique samples were included in the final analysis. KRAS mutation was detected in 3,843 of cases (36%). In this cohort, the most common genomic alterations included TP53 (33%), STK11 (16%), ATM (11%), KEAP1 (11%), and RBM10 (7%) (Figure 5A). When compared to KRAS wild-type cases, however, KRAS mutant LUADs were found to be significantly enriched in STK11, KEAP1, ATM, RBM10 mutations, while TP53, EGFR, BRAF, ERBB2 were less likely to co-occur with any KRAS mutation (Figure S7A-B).
Figure 5:
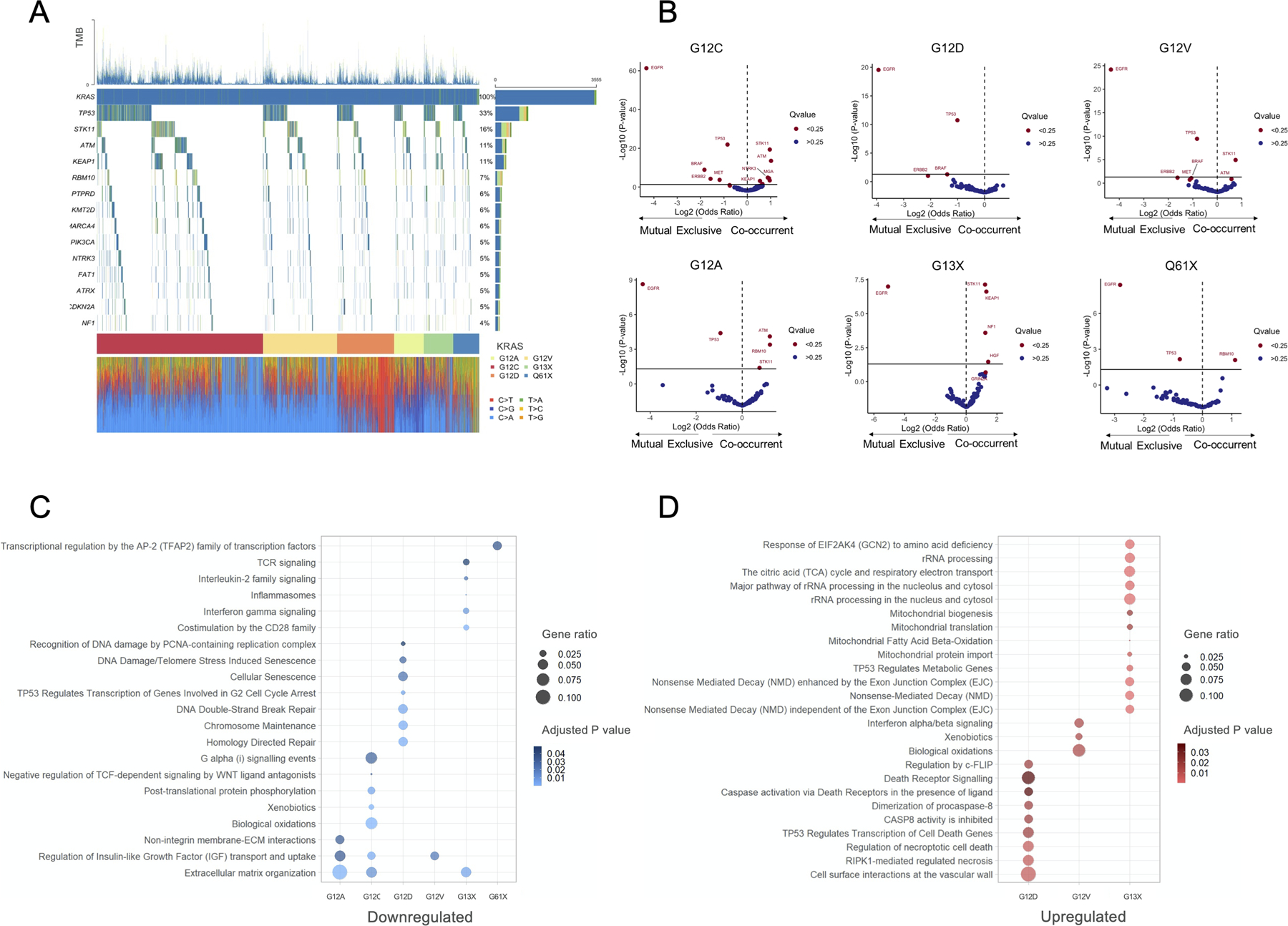
(A) Oncoprint plot of KRAS mutant LUADs in the GENIE v9.0 dataset. (B) Volcano plots showing co-mutation patterns in KRAS G12C, G12D, G12V, G12A, G13X, and G61X compared to KRAS wild type LUADs. Gene ontology analysis showing (C) down- and (D) up-regulated pathways in KRAS G12C, G12D, G12V, G12A, G13X, and G61X compared to KRAS wild type LUADs.
We next focused on the individual KRAS variants and compared the co-mutation patterns of each KRAS variant with KRAS wild-type LUADs, as reference. We identified that all KRAS variant tended to be mutually exclusive with EGFR mutation, and were more likely to lack concurrent TP53 mutations (Figure 5B). Of note, among LUADs with codon 12 mutations, STK11 and ATM mutations was significantly enriched among tumors harboring KRAS G12C, G12A, and G12V mutations, while such enrichment was not detected in LUADs with KRAS G12D (Figure 5B). Additionally, we also fund that KEAP1 mutation was significantly enriched among KRAS G12C and KRAS G13X LUADs. Importantly, the highest frequency of concurrent STK11 and KEAP1 mutation was observed in tumors with KRAS codon 13 mutations (Figure S7C). Pairwise comparisons in concurrent and mutually exclusive mutations between each KRAS variant and the remaining KRAS alleles are reported in Figure S8A-B.
Having shown that the KRAS mutant LUADs are highly heterogenous in term of concurrent genomic alterations, we lastly examined whether tumors with different KRAS mutations had also different gene expression profiles. To address this question, we performed a gene ontology analysis of differentially expressed genes in each KRAS variant versus KRAS wild-type tumors in the TCGA LUAD cohort. We found that several pathways were uniquely up- and downregulated in specific KRAS isoform (Figure 5C-D). Interestingly, KRAS G12D mutant LUADs showed a significant downregulation of pathways involved in chromosome maintenance, DNA double break repair, and cellular senescence (Figure 5C), while KRAS G13X LUADs had a significant downregulation of pathways involved in innate and adaptative immunity, including PD-1, CD28, INFγ, and IL-2 signaling, which is consistent with the high rate of concurrent STK11 and/or KEAP1 (36–37) mutations we identified in this group of LUADs. A full list of differentially regulated pathways in all KRAS isoforms is reported in Table S4.
Discussion
Activating mutations in the KRAS oncogene are known to trigger and sustain the development of tumors with different histology, being particularly frequent in lung, colon and pancreatic cancer (1). The comparison between predicted and observed frequency of different mutant KRAS isoforms suggests the existence of tissue-specific mechanisms of biologic selection (14), however the connection between KRAS-mutant isoforms and therapeutic vulnerabilities has not been fully addressed yet. Very recently, the development of two specific inhibitors of the KRAS-G12C allele, sotorasib and adagrasib, which were granted accelerated FDA approval and breakthrough therapy designation (BTD), respectively, in patients with previously treated advanced NSCLC (10,38–41) highlighted the need to identify new isoform-specific therapeutic strategies. Here we demonstrate that KRAS isoforms are characterized by different oncogenic properties and therapeutic sensitivities in isogenic models, and that KRAS-mutant LUADs are highly heterogenous in terms of clinicopathologic, genomic, and transcriptomic features.
Studies have been inconsistent in reporting whether KRAS isoforms differ in terms of oncogenic properties, preferred signaling pathways, genomics and clinical outcomes in patients with LUAD (1). Previously, NSCLC patient samples and cell lines harboring KRAS G12A have been shown to primarily rely on phosphatidylinositol 3-kinase (PI3K) and mitogen-activated protein/extracellular signal-regulated kinase kinase (MEK) signaling, while those with mutant KRAS G12C or mutant KRAS G12V are more likely to activate Ral signaling and decrease growth factor-dependent Akt activation (19). Similarly, in KRAS overexpressing clones from human cell lines harboring the three most common KRAS mutations in NSCLC (G12C, G12V, and G12D), the expression of a specific KRAS isoforms induced a different sensitivity pattern to cytotoxic agents, such as cisplatin, pemetrexed and taxanes (42). Nonetheless, subsequent retrospective analysis yielded highly variable results in terms of clinical outcomes to either chemotherapy or PD-(L)1 inhibition in patients with advanced NSCLC, as well as among those with resected tumors (19,42–43). However, data on KRAS co-mutation pattern have been underreported in these studies, therefore dissecting the biological differences among KRAS isoforms and their impact on clinical outcomes in patients with LUAD remains challenging.
To study the oncogenic properties of KRAS isoforms in a genetic-controlled system with the same genomic background, we generated isogenic models to dissect potential isoform-dependent therapeutic vulnerabilities. It has already been demonstrated that specific KRAS isoforms exhibit diverse biochemical and biological features in vitro and in vivo (19,21,25,44–45). However, the therapeutic impact of different KRAS alleles on treatment outcome has been only limitedly investigated so far (29,46,47). In our KRaslox KRASMUT platform expressing the most frequent KRAS mutant alleles (G12C, G12D, G12V, G12A, G13D and Q61H), we observed that G12D mutant cells show a significant growth advantage over the other mutants both in vitro and in vivo and are characterized, together with G12V, by the highest levels of GTP-bound fraction even in absence of upstream EGF stimulation.
To determine whether these differences translated in differential clinical outcomes we analyzed a large cohort of patients with LUAD. Although we did not identify significant differences in survival in patients with either early stage or advanced LUADs according to the different KRAS variants, we found that each KRAS isoform was associated with distinct clinicopathologic, genomic, and transcriptomic features, which may have impacted our ability to identify differences in outcomes. These data are in agreement with current knowledge confirming no significant differences in survival in LUAD patients according to specific KRAS mutant alleles. Still, KRAS isoform specificity could be used as a stratification criterion for better tailored therapies based on exclusive vulnerabilities. For instance, KRAS G12D mutated LUADs were more likely to develop in patients with no history of tobacco use. Consistently, tumors with KRAS G12D mutation had also the lowest TMB among KRAS isoforms, were less likely to harbor concurrent mutations in STK11 or KEAP1, and had a marked downregulation of pathways involved in cellular senescence and chromosome maintenance. In contrast, the highest rate of concurrent mutations in STK11 and KEAP1 was identified among KRAS G13X mutated tumors. Concurrent mutations in STK11 and KEAP1 define unique subsets of KRAS mutant LUADs, with distinct immunophenotype and transcriptomic profile, and are generally characterized by significantly worse survival with both chemotherapy and PD-(L)1 blockade (48,49). Consistently, we identified that G13X LUADs had a significant downregulation of pathways involved in innate and adaptative immunity, such as PD-1 signaling, TCR signaling, INFγ signaling, among others. It should be highlighted that gene expression profiling was performed NSCLC samples from the TCGA, rather than in our isogenic models. Therefore, these results should be interpreted in the context of the different genomic background which characterize each KRAS allele. Overall, our results support the hypothesis that in addition to the allele specific and intrinsic differences that we identified in our isogenic, each KRAS allele has a different spectrum of co-mutations and transcriptomic profiles in patient samples, which may not be dependent uniquely from KRAS alleles, but still have great biological relevance. Together, these results may have direct and immediate implications for the design and interpretation of panKRAS or multi/KRAS targeted therapies, as each individual allele has unique oncogenic properties and features.
To further dissect this question, we also investigated differential response to targeted therapy in KRaslox KRASMUT cells. As a proof of concept we chose MEK inhibitors, which showed promising results in Phase II clinical trial but failed to provide any significant benefit in addition to chemotherapy in KRAS-mutant LUAD in Phase III trial (4). Interestingly, retrospective analysis of Phase II trial showed a trend for KRAS G12C and G12V patients towards greater improvement in overall survival, progression free survival and overall response rate compared with other KRAS mutations (20). Indeed, we observed a consistently increased sensitivity of G12C and Q61H mutants with a panel of MEK inhibitors in vitro, which was maintained in a pre-clinical setting in vivo where G12C and Q61H implants showed greater tumor volume reduction compared to other mutants. This effect may partly rely on increased affinity to CRAF for some isoforms (29,50). Also, we validated the convenience of our pre-clinical tool in surveying combination therapies which can be easily tested in high-throughput screening, such as G12C/MEK inhibitors or SHP2/MEK inhibitors (30,31).
Our study has limitations inherent to the artificial generation of our isogenic model, and the retrospective assessment of clinical outcomes. However, we believe that our model does isolate the biological contribution of each isoform and allow the exploration of drug sensitivity in absence of potential confounding factors. This is of particular importance, given that our results indicate that each KRAS isoform is associated with unique co-mutation patterns and transcriptomic profiles which may affect drug sensitivity independently from KRAS intrinsic activity. Our results suggest that the use of specific genetically defined in vitro tools together with the development of large patient-derived datasets will facilitate the identification of potentially actionable vulnerabilities for KRAS mutant NSCLC.
In summary, this study indicates that KRAS isoforms are biologically and clinically heterogenous, and that such heterogeneity should be considered to optimize drug development strategies for KRAS mutant NSCLC.
Supplementary Material
Acknowledgments
This work was funded by the Giovanni Armenise–Harvard Foundation, the Lung Cancer Research Foundation (LCRF), the International Lung Cancer Foundation (ILCF), the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant agreement No. [101001288]) and AIRC under IG 2021 - ID. 25737 project (to C.A.). B.R.`s work was supported by the Conquer Cancer Foundation of ASCO YIA, and the IASLC Fellowship Award. C.A. is supported by the Zanon di Valgiurata family through Justus s.s.
Footnotes
Conflicts of interest
P.A. Janne reports grants from AstraZeneca, Boehringer Ingelheim, Daiichi Sankyo, Eli-Lilly, Takeda Oncology, Astellas, PUMA, and Revolution Medicines; personal fees from AstraZeneca, Boehringer Ingelheim Pfizer, Roche/Genentech, Eli-Lilly, Chugai, Ignyta, Loxo Oncology, from SFJ Pharmaceuticals, Voronoi, Daiichi Sankyo, Biocartis, Novartis, Sanofi Oncology, Takeda Oncology, Mirati Therapeutics, Trasncenta, Silicon Therapeutics, Syndax, Bayer, Esai, Allorion Therapeutics, Accutar Biotech and AbbVie outside the submitted work; and also receives postmarketing royalties from a DFCI-owned patent on EGFR Mutations issued and licensed to Lab Corp. Dr. Awad reports grants from The Mark Foundation for Cancer Research, during the conduct of the study; grants and personal fees from Genentech, grants and personal fees from Bristol-Myers Squibb, personal fees from Merck, grants and personal fees from AstraZeneca, grants from Lilly, personal fees from Maverick, personal fees from Blueprint Medicine, personal fees from Syndax, personal fees from Ariad, personal fees from Nektar, personal fees from Gritstone, personal fees from ArcherDX, personal fees from Mirati, personal fees from NextCure, personal fees from Novartis, personal fees from EMD Serono, personal fees from Panvaxal/NovaRx. J.K. has received consultant fees from and served on advisory boards for Boehringer-Ingelheim. A.J.R. has received personal fees from Boehringer-Ingelheim, Medtronic, Roche, Ariad and AstraZeneca. C.A. received research fees from Revolution Medicines, Verastem and Boehringer-Ingelheim.
The other authors declare no competing interests.
References
- 1.Salgia R, Pharaon R, Mambetsariev I, Nam A, Sattler M. The improbable targeted therapy: KRAS as an emerging target in non-small cell lung cancer (NSCLC). Cell Reports Med. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Judd J, Abdel Karim N, Khan H, Naqash AR, Baca Y, Xiu J, et al. Characterization of KRAS Mutation Subtypes in Non–small Cell Lung Cancer. Mol Cancer Ther. 2021; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Köhler J, Catalano M, Ambrogio C. Back to the bench? MEK and ERK inhibitors for the treatment of KRAS mutant lung adenocarcinoma. Curr Med Chem. 2017;24:1–17. [DOI] [PubMed] [Google Scholar]
- 4.Jänne P, van den Heuvel M, Barlesi F, Cobo M, Mazieres J, Crinò L, et al. Selumetinib Plus Docetaxel Compared With Docetaxel Alone and Progression-Free Survival in Patients With KRAS-Mutant Advanced Non-Small Cell Lung Cancer: The SELECT-1 Randomized Clinical Trial. JAMA. 2017;317:1844–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Planchard D, Popat S, Kerr K, Novello S, Smit EF, Faivre-Finn C, et al. Metastatic non-small cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2018; [DOI] [PubMed] [Google Scholar]
- 6.Ambrogio C, López-Gómez G, Falcone M, Vidal A, Nadal E, Crosetto N, et al. Combined inhibition of DDR1 and Notch signaling is a therapeutic strategy for KRAS-driven lung adenocarcinoma. Nat Med. 2016;22:270–7. [DOI] [PubMed] [Google Scholar]
- 7.Manchado E, Weissmueller S, Morris JP, Chen C-C, Wullenkord R, Lujambio A, et al. A combinatorial strategy for treating KRAS-mutant lung cancer. Nature. 2016;534:647–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Awad MM, Liu S, Rybkin II, Arbour KC, Dilly J, Zhu VW, et al. Acquired Resistance to KRAS G12C Inhibition in Cancer. N Engl J Med. 2021;384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Skoulidis F, Li BT, Dy GK, Price TJ, Falchook GS, Wolf J, et al. Sotorasib for Lung Cancers with KRAS p.G12C Mutation. N Engl J Med. 2021;384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Riely GJ, Ou S- HI, Rybkin I, Spira A, Papadopoulos K, Sabari JK, et al. 99O_PR KRYSTAL-1: Activity and preliminary pharmacodynamic (PD) analysis of adagrasib (MRTX849) in patients (Pts) with advanced non–small cell lung cancer (NSCLC) harboring KRASG12C mutation. J Thorac Oncol. 2021;16. [Google Scholar]
- 11.Santos E, Martin-Zanca D, Reddy EP, Pierotti M a, Della Porta G, Barbacid M. Malignant activation of a K-ras oncogene in lung carcinoma but not in normal tissue of the same patient. Science. 1984;223:661–4. [DOI] [PubMed] [Google Scholar]
- 12.Ambrogio C, Köhler J, Zhou Z, Wang H, Paranal R, Li J, et al. KRAS Dimerization Impacts MEK Inhibitor Sensitivity and Oncogenic Activity of Mutant KRAS. Cell. 2018;S0092–8674. [DOI] [PubMed] [Google Scholar]
- 13.Zhou Y, Prakash P, Liang H, Cho KJ, Gorfe AA, Hancock JF. Lipid-Sorting Specificity Encoded in K-Ras Membrane Anchor Regulates Signal Output. Cell. 2017;168:239–251.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cook J, Melloni G, Gulhan D, Park P, Haigis KM. The origins and genetic interactions of KRAS mutations are allele- and tissue-specific. Nat Commun. 2021; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hood FE, Klinger B, Newlaczyl AU, Sieber A, Dorel M, Oliver SP, et al. Isoform-specific Ras signaling is growth factor dependent. Mol Biol Cell. 2019; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tang R, Shuldiner EG, Kelly M, Murray CW, Hebert JD, Andrejka L, et al. Multiplexed identification of RAS paralog imbalance as a driver of lung cancer growth. bioRxiv. 2021; [Google Scholar]
- 17.Tejpar S, Celik I, Schlichting M, Sartorius U, Bokemeyer C, Van Cutsem E. Association of KRAS G13D tumor mutations with outcome in patients with metastatic colorectal cancer treated with first-line chemotherapy with or without cetuximab. J Clin Oncol. 2012; [DOI] [PubMed] [Google Scholar]
- 18.Burd CE, Liu W, Huynh MV., Waqas MA, Gillahan JE, Clark KS, et al. Mutation-specific RAS oncogenicity explains NRAS codon 61 selection in melanoma. Cancer Discov. 2014; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ihle NT, Byers LA, Kim ES, Saintigny P, Lee JJ, Blumenschein GR, et al. Effect of KRAS oncogene substitutions on protein behavior: Implications for signaling and clinical outcome. J Natl Cancer Inst. 2012;104:228–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jänne P a, Smith I, McWalter G, Mann H, Dougherty B, Walker J, et al. Impact of KRAS codon subtypes from a randomised phase II trial of selumetinib plus docetaxel in KRAS mutant advanced non-small-cell lung cancer. Br J Cancer [Internet]. 2015;113:199–203. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4506393&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hammond DE, Mageean CJ, Rusilowicz EV., Wickenden JA, Clague MJ, Prior IA. Differential reprogramming of isogenic colorectal cancer cells by distinct activating KRAS mutations. J Proteome Res. 2015; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.. Stolze B, Reinhart S, Bulllinger L, Fröhling S, Scholl C. Comparative analysis of KRAS codon 12, 13, 18, 61, and 117 mutations using human MCF10A isogenic cell lines. Sci Rep [Internet]. 2015;5:8535. Available from: http://www.nature.com/srep/2015/150223/srep08535/full/srep08535.html [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brubaker DK, Paulo JA, Sheth S, Poulin EJ, Popow O, Joughin BA, et al. Proteogenomic Network Analysis of Context-Specific KRAS Signaling in Mouse-to-Human Cross-Species Translation. Cell Syst. 2019;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.. Drosten M, Dhawahir A, Sum EYMM, Urosevic J, Lechuga CG, Esteban LM, et al. Genetic analysis of Ras signalling pathways in cell proliferation, migration and survival. EMBO J [Internet]. 2010;29:1091–104. Available from: 10.1038/emboj.2010.7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ricciuti B, Jones G, Severgnini M, Alessi JV, Recondo G, Lawrence M, Forshew T, Lydon C, Nishino M, Cheng M, Awad M. Early plasma circulating tumor DNA (ctDNA) changes predict response to first-line pembrolizumab-based therapy in non-small cell lung cancer (NSCLC). J Immunother Cancer. 2021;9(3):e001504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Winters I, Chiou S, Paulk N, McFarland C, Lalgudi P, Ma R, et al. Multiplexed in vivo homology-directed repair and tumor barcoding enables parallel quantification of Kras variant oncogenicity. Nat Commun. 2017;1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garnett MJ, Edelman EJ, Heidorn SJ, Greenman CD, Dastur A, Lau KW, et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature [Internet]. 2012;483:570–5. Available from: http://europepmc.org/articles/PMC3349233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li S, Liu S, Deng J, Akbay E, Hai J, Ambrogio C, et al. Assessing Therapeutic Efficacy of MEK Inhibition in a KRAS G12C-Driven Mouse Model of Lung Cancer. Clin Cancer Res. 2018; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou Z-W, Ambrogio C, Bera AK, Li Q, Li X-X, Li L, et al. KRASQ61H preferentially signals through MAPK in a RAF dimer-dependent manner in non-small cell lung cancer. Cancer Res. 2020;80. [DOI] [PubMed] [Google Scholar]
- 30.Mainardi S, Mulero-Sánchez A, Prahallad A, Germano G, Bosma A, Krimpenfort P, et al. SHP2 is required for growth of KRAS-mutant non-small-cell lung cancer in vivo letter. Nat Med. 2018; [DOI] [PubMed] [Google Scholar]
- 31.Ruess DA, Heynen GJ, Ciecielski KJ, Ai J, Berninger A, Kabacaoglu D, et al. Mutant KRAS-driven cancers depend on PTPN11/SHP2 phosphatase. Nat Med [Internet]. Springer US; 2018;24:954–60. Available from: 10.1038/s41591-018-0024-8 [DOI] [PubMed] [Google Scholar]
- 32.Gebregiworgis T, Kano Y, St-Germain J, Radulovich N, Udaskin M, Mentes A, et al. The Q61H mutation decouples KRAS from upstream regulation and renders cancer cells resistant to SHP2 inhibitors. Nat Commun. 2021; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.El Osta B, Behera M, Kim S, Berry LD, Sica G, Pillai RN, et al. Characteristics and Outcomes of Patients With Metastatic KRAS-Mutant Lung Adenocarcinomas: The Lung Cancer Mutation Consortium Experience. J Thorac Oncol. 2019;14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dogan S, Shen R, Ang DC, Johnson ML, D’Angelo SP, Paik PK, et al. Molecular epidemiology of EGFR and KRAS mutations in 3,026 lung adenocarcinomas: Higher susceptibility of women to smoking-related KRAS-mutant cancers. Clin Cancer Res. 2012;18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Riely GJ, Kris MG, Rosenbaum D, Marks J, Li A, Chitale DA, et al. Frequency and distinctive spectrum of KRAS mutations in never smokers with lung adenocarcinoma. Clin Cancer Res. 2008;14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Skoulidis F, Goldberg ME, Greenawalt DM, Hellmann MD, Awad MM, Gainor JF, et al. STK11/LKB1 mutations and PD-1 inhibitor resistance in KRAS-mutant lung adenocarcinoma. Cancer Discov. 2018;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen X, Su C, Ren S, Zhou C, Jiang T. Pan-cancer analysis of KEAP1 mutations as biomarkers for immunotherapy outcomes. Ann Transl Med. 2020;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li S, Balmain A, Counter CM. A model for RAS mutation patterns in cancers: finding the sweet spot. Nat. Rev. Cancer. 2018. [DOI] [PubMed] [Google Scholar]
- 39.Canon J, Rex K, Saiki A, Mohr C, Cooke K, Bagal D, et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature. 2019; [DOI] [PubMed] [Google Scholar]
- 40.Hallin J, Engstrom L, Hargis L, Calinisan A, Aranda R, Briere D, et al. The KRAS G12C Inhibitor, MRTX849, Provides Insight Toward Therapeutic Susceptibility of KRAS Mutant Cancers in Mouse Models and Patients. Cancer Discov. 2019; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hong DS, Fakih MG, Strickler JH, Desai J, Durm GA, Shapiro GI, et al. KRAS-G12C inhibition with sotorasib in advanced solid tumors. N Engl J Med. 2020; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Garassino M, Marabese M, Rusconi P, Rulli E, Martelli O, Farina G, et al. Different types of K-Ras mutations could affect drug sensitivity and tumour behaviour in non-small-cell lung cancer. Ann Oncol. 2011; [DOI] [PubMed] [Google Scholar]
- 43.Shepherd FA, Domerg C, Hainaut P, Jänne P, Pignon J, Graziano S, et al. Pooled analysis of the prognostic and predictive effects of KRAS mutation status and KRAS mutation subtype in early-stage resected non-small-cell lung cancer in four trials of adjuvant chemotherapy. J Clin Oncol. 2013;31:2173–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hunter JC, Manandhar A, Carrasco MA, Gurbani D, Gondi S, Westover KD. Biochemical and Structural Analysis of Common Cancer-Associated KRAS Mutations. Mol Cancer Res [Internet]. 2015;13:1325–35. Available from: 10.1158/1541-7786.MCR-15-0203 [DOI] [PubMed] [Google Scholar]
- 45.Zafra MP, Parsons MJ, Kim J, Alonso-Curbelo D, Goswami S, Schatoff EM, et al. An in vivo kras allelic series reveals distinct phenotypes of common oncogenic variants. Cancer Discov. 2020;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.De Roock W, Jonker DJ, Di Nicolantonio F, Sartore-Bianchi A, Tu D, Siena S, et al. Association of KRAS p.G13D mutation with outcome in patients with chemotherapy-refractory metastatic colorectal cancer treated with cetuximab. JAMA - J Am Med Assoc. 2010;304. [DOI] [PubMed] [Google Scholar]
- 47.Bournet B, Muscari F, Buscail C, Assenat E, Barthet M, Hammel P, et al. KRAS G12D Mutation Subtype Is A Prognostic Factor for Advanced Pancreatic Adenocarcinoma. Clin Transl Gastroenterol. 2016;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Skoulidis F, Byers LA, Diao L, Papadimitrakopoulou VA, Tong P, Izzo J, et al. Co-occurring genomic alterations define major subsets of KRAS-mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discov. 2015;5:861–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ricciuti B, Arbour K, Lin J, Vajdi A, Vokes N, Hong L, et al. Diminished Efficacy of Programmed Death-(Ligand)1 Inhibition in STK11- and KEAP1-Mutant Lung Adenocarcinoma Is Affected by KRAS Mutation Status. J Thorac Oncol. 2021; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mysore VP, Zhou ZW, Ambrogio C, Li L, Kapp JN, Lu C, et al. A structural model of a Ras–Raf signalosome. Nat Struct Mol Biol. 2021;28. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The Institutional clinicopathologic and genomic data that support the finding of our study are available upon reasonable request from the corresponding author. RNAseq data used in this study are publicly available from the TCGA (accession number: phs000178). Genomic data from the AACR project GENIE (Genomics Evidence Neoplasia Information Exchange) are publicly available (phs001337).