

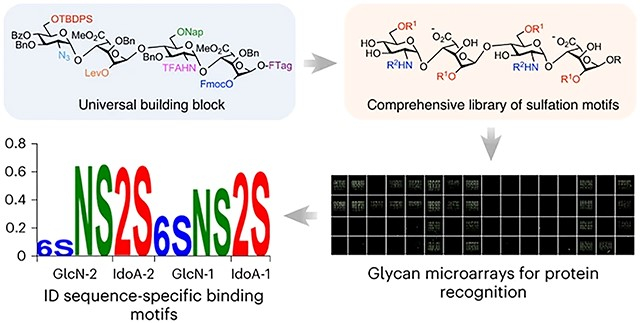
Abstract
Glycosaminoglycans (GAGs) are abundant, ubiquitous carbohydrates in biology, yet their structural complexity has limited an understanding of their biological roles and structure–function relationships. Synthetic access to large collections of well defined, structurally diverse GAG oligosaccharides would provide critical insights into this important class of biomolecules and represent a major advance in glycoscience. Here we report a new platform for synthesizing large heparan sulfate (HS) oligosaccharide libraries displaying comprehensive arrays of sulfation patterns. Library synthesis is made possible by improving the overall synthetic efficiency through universal building blocks derived from natural heparin and a traceless fluorous tagging method for rapid purification with minimal manual manipulation. Using this approach, we generated a complete library of 64 HS oligosaccharides displaying all possible 2-O-, 6-O- and N-sulfation sequences in the tetrasaccharide GlcN–IdoA–GlcN–IdoA. These diverse structures provide an unprecedented view into the sulfation code of GAGs and identify sequences for modulating the activities of important growth factors and chemokines.
Graphical Abstract
Heparan sulfate glycosaminoglycans (HS GAGs) are ubiquitous, complex polysaccharides that mediate diverse biological processes, ranging from cell proliferation and viral invasion to neural development and immune regulation1–4. Their linear polysaccharide chains are composed of repeating disaccharides that undergo sulfation at various positions, resulting in an enormous number of different sulfation sequences5. The sulfation motifs enable HS GAGs to interact with more than 2,000 proteins6 and thus are central to their biological activities. Moreover, alterations in HS sulfation patterns are associated with human diseases such as cancer, Alzheimer’s disease, osteoarthritis and cardiovascular diseases7–10. However, an in-depth understanding of their physiological and pathological roles has been hampered by limited access to defined HS structures. Large collections of HS oligosaccharides representing a diversity of sulfation motifs will be essential for elucidating the structure–function relationships of HS and expanding the development of GAG-based therapeutics.
As HS GAGs are among the most structurally complex carbohydrates in nature, their chemical synthesis remains an immense challenge11. For example, HS can display 16 distinct sulfation motifs within its disaccharide unit alone. Its synthesis requires regioselective differentiation of five hydroxyls and one amino group within each disaccharide, stereoselective glycosylation of low-reactivity glycosyl donors and acceptors, access to l-iduronic acid (IdoA) monosaccharides that are not commercially available and are tedious to prepare, and multi-step purifications of highly charged, polar intermediates. As such, the synthesis of HS oligosaccharides is notoriously difficult and labour-intensive, requiring specialized expertise in carbohydrate chemistry. For example, a single HS tetrasaccharide typically takes 35–50 chemical steps depending on the sulfation pattern11–14. Thus, accessing large libraries of sulfated HS oligosaccharides represents a formidable barrier. Indeed, comprehensive GAG libraries representing all possible sulfation patterns have not been achieved except in the case of HS disaccharides15,16. Despite key synthetic advances and elegant targeted syntheses12–30, the majority of HS sequence space remains both chemically and biologically unexplored.
In response to these challenges, efficient platforms have been developed to accelerate the synthesis of GAGs31. Chemical methods have focused primarily on automating glycan assembly and on less structurally complex GAGs such as chondroitin sulfate (CS), keratan sulfate (KS) and hyaluronan32–35 rather than on generating large libraries of sulfated GAGs. Moreover, the automated synthesis of HS oligosaccharides has not yet been reported, presumably due to challenges with the additional late-stage modification steps (for example, N-sulfation). Thus, the development of new synthetic platforms for rapid GAG production, particularly the synthesis of large HS oligosaccharide libraries, remains an important, unrealized goal. Such platforms are much needed to provide broad access to a wide range of glycan structures, similar to the now routine production of peptides and oligonucleotides.
In this Article we report a new method for the expedient solution-phase synthesis of HS oligosaccharides displaying comprehensive arrays of diverse sulfation motifs. We employ disaccharide synthons derived from natural heparin and design a universal tetrasaccharide building block to substantially reduce the total number of synthetic steps. Diversification of this building block provided a comprehensive library of 64 sulfated tetrasaccharides (1-64; Fig. 1a) representing all possible products from differential sulfation of six functional groups (the 2-O-, 6-O- and N-positions) within the tetrasaccharide. To simplify the late-stage modification steps required for HS library synthesis, we developed a traceless, fluorous tagging method to allow for rapid purification of the highly charged, sulfated intermediates by fluorous solid-phase extraction (FSPE). The comprehensive HS library enabled systematic investigations into the unique sulfation dependencies of HS-binding proteins and identified position-dependent, sequence-specific modifications critical for HS recognition by growth factors and chemokines important for morphogenesis, cell growth and inflammatory responses.
Fig. 1 |. Synthesis of a comprehensive HS tetrasaccharide library.
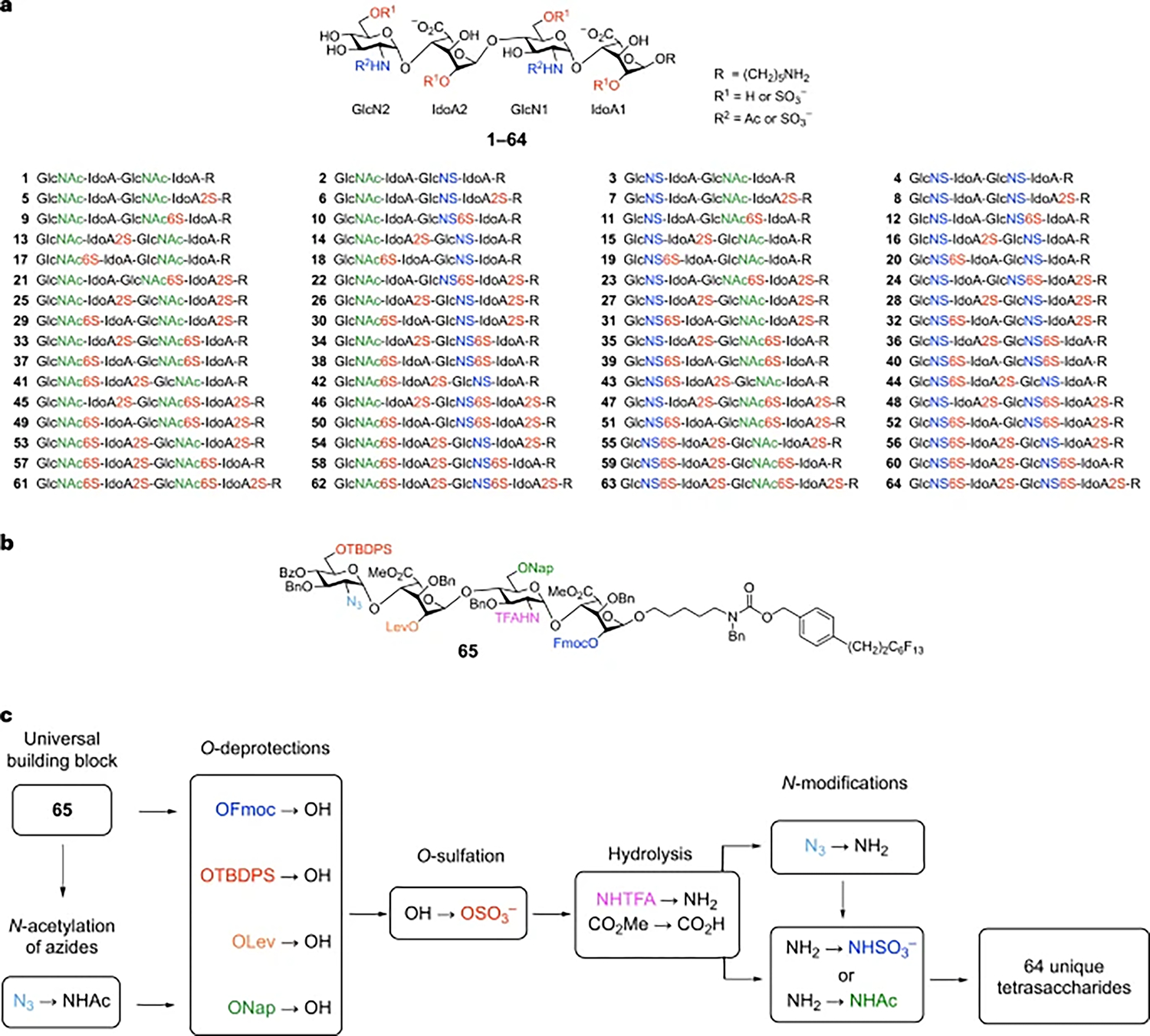
a, 64-compound library representing all possible N-, 2-O- and 6-O-sulfate modifications of the tetrasaccharide GlcN–IdoA–GlcN–IdoA. b, Universal building block for HS library synthesis. The six orthogonal functional groups are shown in colour. c, General strategy and late-stage modification steps. IdoA, l-iduronic acid; GlcN, d-glucosamine; GlcNAc, N-acetyl-d-glucosamine; Fmoc, 9-fluorenylmethoxycarbonyl; Lev, levulinoyl; TBDPS, tert-butyldiphenylsilyl; Nap, 6-O-(2-naphthyl)methyl; TFA, trifluoroacetyl; Bz, benzoyl; Bn, benzyl.
Results and discussion
General synthetic strategy of the library
In contrast to peptides and nucleic acids, universal building blocks for the synthesis of GAGs have not been developed. Moreover, the majority of the synthesis is dedicated to preparing suitably protected mono- and disaccharide precursors. For example, starting from commercially available monosaccharides, 18–30 chemical steps are typically required to produce each HS disaccharide used for glycosylation. Ideally, a small set of building blocks would be employed to generate a large number of different sulfation motifs. Towards this end, we designed tetrasaccharide 65 as a versatile building block (Fig. 1b). The IdoA-containing backbone glucosamine (GlcN)–IdoA–GlcN–IdoA was chosen because the conformational flexibility of IdoA is crucial for many HS–protein interactions and often induces a kink in the HS structure that is important for protein binding36,37. We selected a tetrasaccharide because four monosaccharide residues are typically the minimum length required to engage high-affinity binding sites in many proteins, including fibroblast growth factors (FGFs) and chemokines such as CCL538,39. Furthermore, a comprehensive library of sulfation sequences at the major positions (N-, 2-O- and 6-O-) would consist of 64 tetrasaccharides, an ambitious yet achievable goal for chemical synthesis and biological investigations.
To facilitate library synthesis, the 2-O, 6-O and N-positions in tetrasaccharide 65 were differentially protected with six orthogonal functionalities (Fig. 1b). The orthogonality of these functional groups allows for their selective removal to unmask each position for sulfation. Site-specific O-deprotection steps, followed by O-sulfation, would install sulfate groups at the desired 2-O- and 6-O-positions (Fig. 1c). Hydrolysis to saponify the methyl esters and selectively remove the N-trifluoroacetyl (N-TFA) groups, followed by N-sulfation or N-acetylation, would provide different amino group modifications in the tetrasaccharide. Thus, a single universal building block would enable access to all 64 sequences (1–64), the largest number of sulfation motifs generated from a single HS precursor so far.
A major challenge to HS library synthesis is the substantial number of late-stage modification steps (274 steps for 64 tetrasaccharides). Each step also requires the labour-intensive, multi-step purification of highly polar, charged intermediates using a combination of silica gel, size-exclusion and/or strong anion-exchange chromatography. We envisioned using fluorous chemistry to overcome these challenges. Although fluorous-assisted synthesis has been elegantly applied to many organic compounds including a limited number of glycans40–45, it has not been extensively exploited for HS synthesis. We reasoned that fluorous chemistry would greatly accelerate the synthesis of large GAG libraries by reducing the purification of highly charged intermediates to a single FSPE step. Moreover, fluorous-assisted synthesis would afford the efficiency, convenience and flexibility of solution-phase synthesis by permitting easy reaction monitoring using standard spectroscopic methods (for example, thin-layer chromatography (TLC), MS and NMR) and by circumventing challenges associated with solid-phase synthesis (for example, solvent constraints for resin swelling and difficult reaction monitoring). Finally, fluorous-assisted synthesis is amenable to automation45, which could facilitate the first automated synthesis of HS oligosaccharides larger than disaccharides. We thus appended a C6F13 fluorous tag to the reducing end of tetrasaccharide 65 (Fig. 1b) via a benzyloxycarbonyl (Cbz)-like aminopentyl linker, which, upon cleavage, would expose an amine handle for versatile conjugation of the tetrasaccharides to small molecules, polymers, proteins and microarray surfaces.
Synthesis of tetrasaccharide 65
Synthesis of 65 began with the controlled hydrolysis of heparin using aqueous triflic acid, followed by esterification, NH2-to-N3 conversion, and acetylation to give the peracetylated disaccharide 66 in 20% overall yield (Fig. 2)18. The use of disaccharides derived from natural heparin greatly simplified the synthesis by eliminating the need to synthesize IdoA monosaccharides and to perform the challenging 1,2-cis glycosylation reaction. Disaccharide 66 was elaborated to form 68 in a total of only nine steps and five purifications from heparin, about half the number of steps previously required11,28. Compound 68 is a versatile intermediate that was readily converted to glycosyl acceptor 72 and donor 74. Acceptor 72 was obtained in six steps from 68 (38% overall yield) by converting the N3 group to an N-TFA group to form thioglycoside 69, glycosylation to append the fluorous tag, exchange of the 2-OBz group of IdoA with an Fmoc group, and regioselective opening of the Naph acetal of 71. Donor 74 was generated in five steps from 68 (72% overall yield) by exchanging the 2-O-Bz group of IdoA with a Lev group, removal of the Naph acetal, and installation of tert-butyldiphenylsilyl (TBDPS) and benzoyl (Bz) groups at the resulting 6-OH and 4-OH of GlcN, respectively.
Fig. 2 |. Synthesis of universal building block 65.
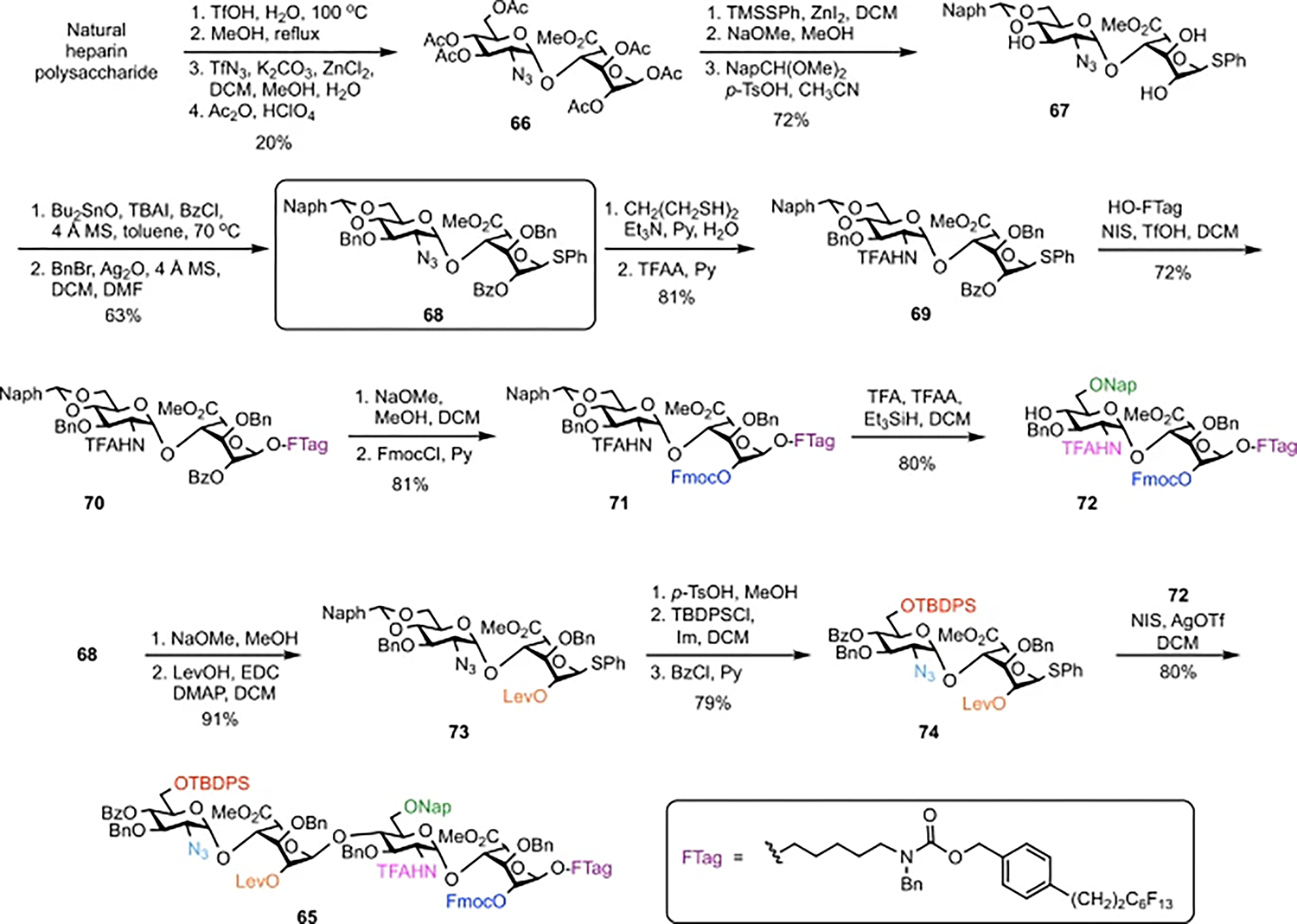
Preparation of orthogonally protected tetrasaccharide 65 starts with acidic hydrolysis of natural heparin to form a core disaccharide, which is converted to versatile intermediate 68 in nine steps. Installation of the fluorous tag (69 to 70) and subsequent protecting-group manipulations give disaccharide acceptor 72. TFAA, trifluoroacetic anhydride; Im, imidazole. Conversion of key intermediate 68 along another route yields disaccharide donor 74, which, upon glycosylation with 72, affords universal building block 65 in good yield.
Glycosylation of disaccharide donor 74 and acceptor 72 using N-iodosuccinimide (NIS) and silver trifluoromethanesulfonate (AgOTf) at room temperature delivered 65 with exclusively the desired α-stereochemistry46 (1JC–H = 173.7 Hz) in 80% yield. Importantly, this concise, scalable route furnished the universal building block on a multi-gram scale in only 21 steps, approximately half the number of steps previously reported for HS oligosaccharides of similar complexity11.
Divergent synthesis of the HS library
To access the 64-compound library, we envisaged a divergent strategy in which 65 was elaborated into two separate pools of 32 compounds via intermediates 75 and 76 (Fig. 3a and Supplementary Fig. 1). Intermediate 75, obtained via selective removal of the C2 Fmoc group in 65, served as a precursor for all structures with 2-O-sulfation at the reducing end IdoA-1 (library 2OS(1), 32 compounds). Intermediate 76, obtained via benzoylation of 75, served as a precursor for all structures containing an unsulfated 2-OH at IdoA-1 (Library 2OH(1), 32 compounds).
Fig. 3 |. Divergent synthesis of the HS tetrasaccharide library.
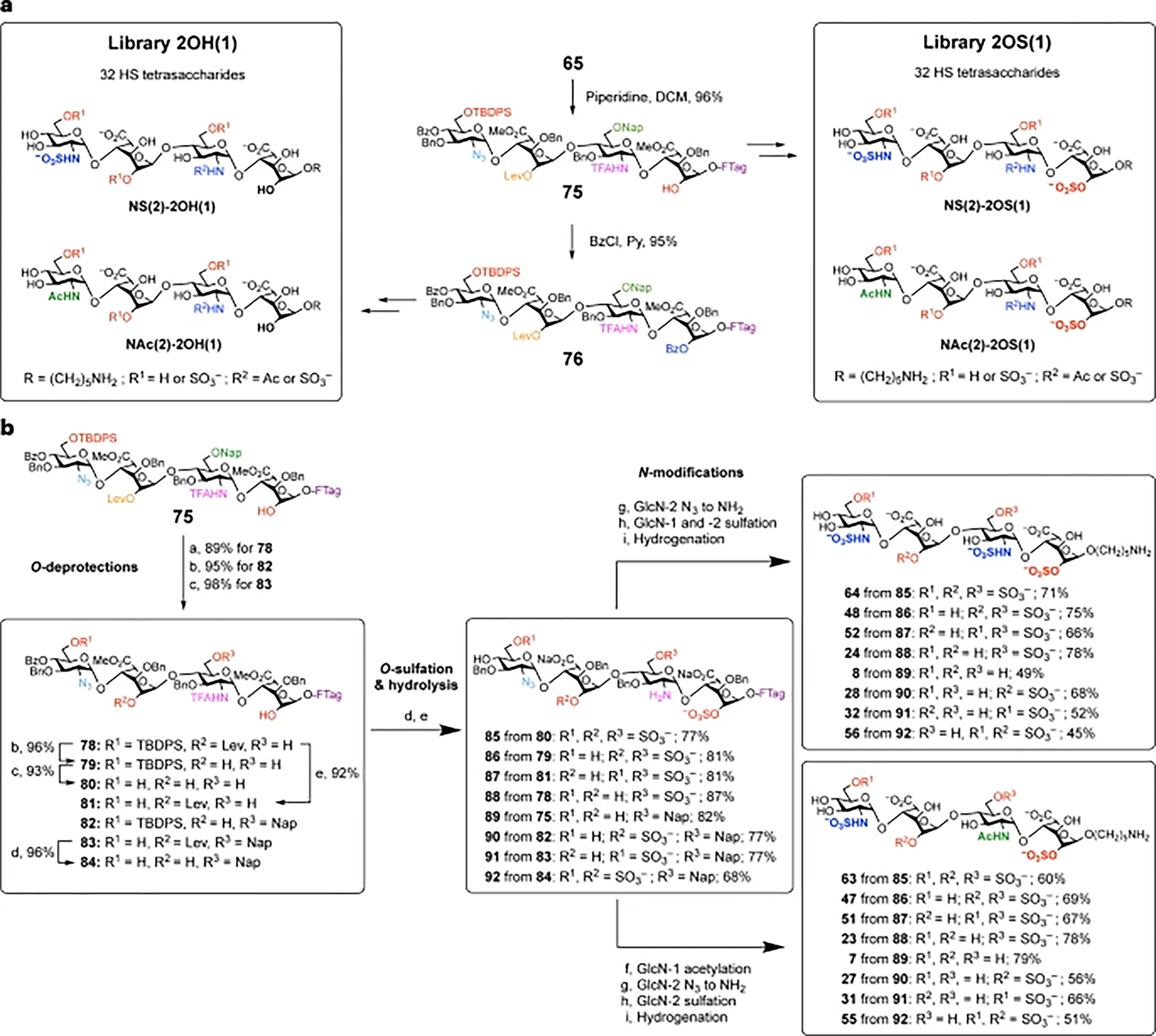
a, Strategy for the synthesis of libraries 2OH(1) and 2OS(1) from 65. b, Synthesis of the 16-compound NS(2)-2OS(1) sub-library. Reagents and conditions: aDDQ, DCE, MeOH, PBS, room temperature (r.t.); bhydrazine acetate, MeOH, DCM, r.t.; cHF·Py, py, 0 °C to r.t.; dSO3·Et3N, DMF, 50 °C; e1 M LiOH, MeOH, THF, 40 °C; fAc2O, Et3N, MeOH, r.t.; gPMe3, NaOH, THF, r.t.; hSO3·Py, NaOH, Et3N, TFE, MeOH, r.t.; iPd(OH)2, H2, t-BuOH, H2O, r.t.
For the 2OS(1) library, two sub-libraries with either N-sulfation (sub-library NS(2)-2OS(1), 16 compounds) or N-acetylation (sub-library NAc(2)-2OS(1), 16 compounds) at the nonreducing end GlcN-2 were obtained from tetrasaccharides 75 and 77, respectively (Fig. 3 and Supplementary Figs. 2 and 3). Treatment of compound 75 with thioacetic acid (AcSH) converted the azide to the corresponding acetamide directly, forming 77 in 83% yield. Chemoselective cleavage of the TBDPS, Lev and/or Nap groups of 75 and 77 was accomplished using hydrogen fluoride in pyridine (HF·Py) or tetra-n-butylammonium fluoride (TBAF), hydrazine acetate (NH2NH2·AcOH) or 2,3-dichloro-5,6 -dicyano-1,4-benzoquinone (DDQ), respectively. These various O-deprotections of 75 furnished compounds 78–84 with different combinations of hydroxyl groups unmasked for O-sulfation (Fig. 3b). After O-sulfation using SO3·Et3N in DMF, LiOH hydrolysis led to removal of the N-TFA, Bz groups and methyl esters to give compounds 85–92. Under these strongly basic conditions, the TBDPS group was also simultaneously cleaved, which saved one step (TBDPS deprotection) for compounds lacking 6-O-sulfation at GlcN-2 (for example, 75, 78, 79 and 82). Staudinger reduction of the azido group in compounds 85–92, followed by N-sulfation of both amines using SO3·Py in an aqueous trifluoroethanol (TFE)/MeOH co-solvent mixture, afforded the corresponding hydrogenation-ready tetrasaccharides. However, initial attempts at LiOH hydrolysis and N-sulfation using the fluorous-tagged tetrasaccharides required longer reaction times and more equivalents of SO3·Py to achieve reaction completion (Supplementary Figs. 4 and 5). After investigating both C6F13 and C8F17 tags, we found that the C6F13 tag was better suited for GAG synthesis due to its greater solubility in polar solvents. TFE further enhanced the solubility of the fluorous-tagged tetrasaccharides in the aqueous MeOH solvent used for N-sulfation, thereby avoiding aggregation and improving the overall yields.
Global hydrogenolysis cleaved all benzyl and Nap groups to generate the eight tetrasaccharides with di-N-sulfation in the NS(2)-2OS(1) sub-library (64, 48, 52, 24, 8, 28, 32 and 56; Fig. 3b). Alternatively, N-acetylation of intermediates 85–92, followed by azide reduction, N-sulfation and hydrogenolysis afforded the other eight mono-N-sulfated tetrasaccharides (63, 47, 51, 23, 7, 27, 31 and 55) in the sub-library. Similar procedures were performed using tetrasaccharide 77 to give the 16-compound NAc(2)-2OS(1) sub-library (Fig. 3a and Supplementary Fig. 3) and using tetrasaccharide 76 to generate the 32-compound 2OH(1) library (Supplementary Figs. 6 and 7).
Overall, 64 different HS tetrasaccharides were prepared on a 5–15-mg scale in 86–95% purity from 3.5 g of the universal building block 65 (Supplementary Table 1). This comprehensive library of HS oligosaccharides containing all possible 2-O-, 6-O- and N-sulfation sequences in the tetrasaccharide GlcN–IdoA–GlcN–IdoA represents the most diverse collection of HS tetrasaccharides synthesized so far. Most of the structures are irregular, ‘mixed’ sequences where each disaccharide unit displays a different sulfation motif. Such sequences cannot be obtained through chemoenzymatic synthesis. Our results highlight the power of chemical synthesis to generate both natural and non-natural HS structures with a wide range of regiodefined sulfation patterns.
Elucidating GAG structure–function relationships
The sulfation preferences of GAG-binding proteins are typically described in general terms (for example, 6-O-sulfation is required)10,47–49, with little information regarding the relative importance of specific modifications at each monosaccharide residue in the sequence. As our library covers the entire chemical space of N-acetyl, N-sulfate, 2-O-sulfate and 6-O-sulfate modifications within the GlcN–IdoA–GlcN–IdoA tetrasaccharide, we can systematically and comprehensively evaluate the impact of modifications at each position and obtain critical, sequence-specific information that was previously unattainable. To assess protein binding to the HS library, we constructed glycan microarrays in which the amine on the reducing end of tetrasaccharides 1–64 was covalently attached to N-hydroxysuccinimide (NHS)-functionalized glass slides. Droplets (<1 nl) of each compound were spotted in nonuplicate at nine different concentrations (0.8–200 μM) using robotic printing technology.
We first examined the binding of fibroblast growth factor 2 (FGF2), a well-studied HS-binding protein involved in a diverse range of developmental processes and human diseases such as asthma, cancer and cardiovascular diseases50. FGF2 binding was detected using an Alexa Fluor 647-labelled anti-His-tag antibody, and relative binding to the 64 tetrasaccharides was represented as both a heatmap (Fig. 4) and bar graph (Supplementary Fig. 8). Loss of a single 2-O-sulfate group at IdoA-1 or IdoA-2 led to a substantial reduction in FGF2 binding (for example, 64 (100%) versus 60 (39%) and 52 (45%), respectively; Figs. 4b and 5), whereas loss of both 2-O-sulfate groups abolished binding (40 (3%)). Similar trends were observed across multiple series of compounds (for example, 63 (70%) versus 51 (3%) and 39 (2%); 47 (57%) versus 35 (38%), 23 (2%) and 11 (0.7%)), indicating that the 2-O-sulfate groups at both IdoA positions are critical for FGF2 recognition. Although both N-sulfate groups were also key contributors (Fig. 5; 64 (100%) versus 61 (2%)), N-sulfation was generally more important at GlcN-2 than GlcN-1 (for example, 64 (100%) versus 62 (28%) and 63 (70%), respectively). In contrast, loss of either or both of the 6-O-sulfate groups led to a relatively small reduction in binding (Fig. 5; 64 (100%) versus 48 (77%), 56 (63%) and 28 (68%)), indicating that 6-O-sulfation is not essential for FGF2 recognition.
Fig. 4 |. FGF2 recognizes specific HS sulfation sequences.
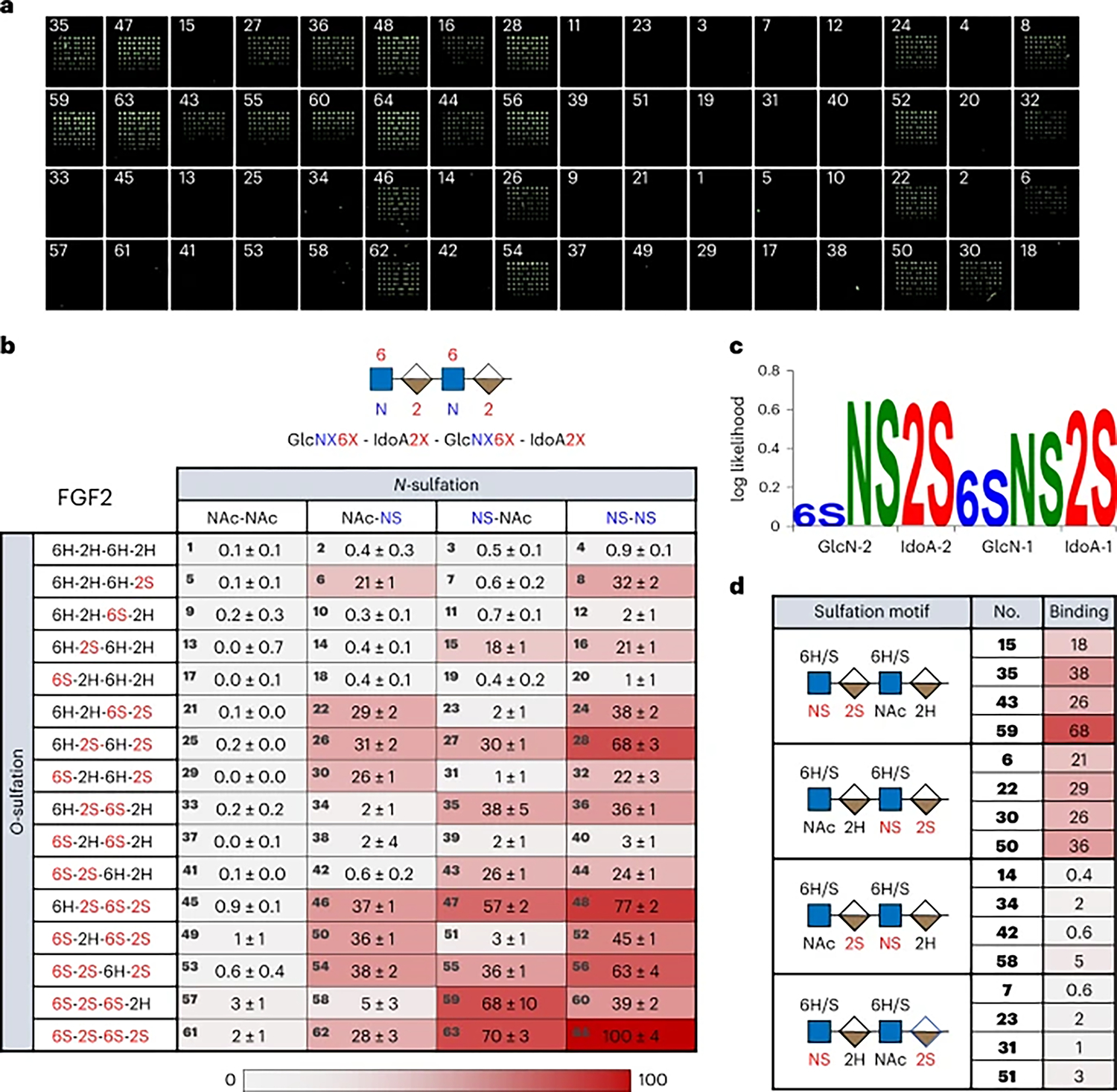
a, Representative fluorescence image of FGF2 binding to the HS tetrasaccharide microarray. Compound numbers are indicated on the array. b, Heatmap of the relative binding of FGF2 to each member of the 64-compound library. Binding signals were normalized with respect to the compound with greatest binding. The tetrasaccharide backbone structure is schematically represented with sites of modification labelled by ‘X.’ The error is the standard deviation to the first digit of uncertainty for the mean across nine replicates. c, The sulfation logo of FGF2. d, Selected tetrasaccharides highlight the regiospecific contributions of N-sulfation and 2-O-sulfation to FGF2 binding.
Fig. 5 |. Comparison of binding of selected glycans to FGF2.
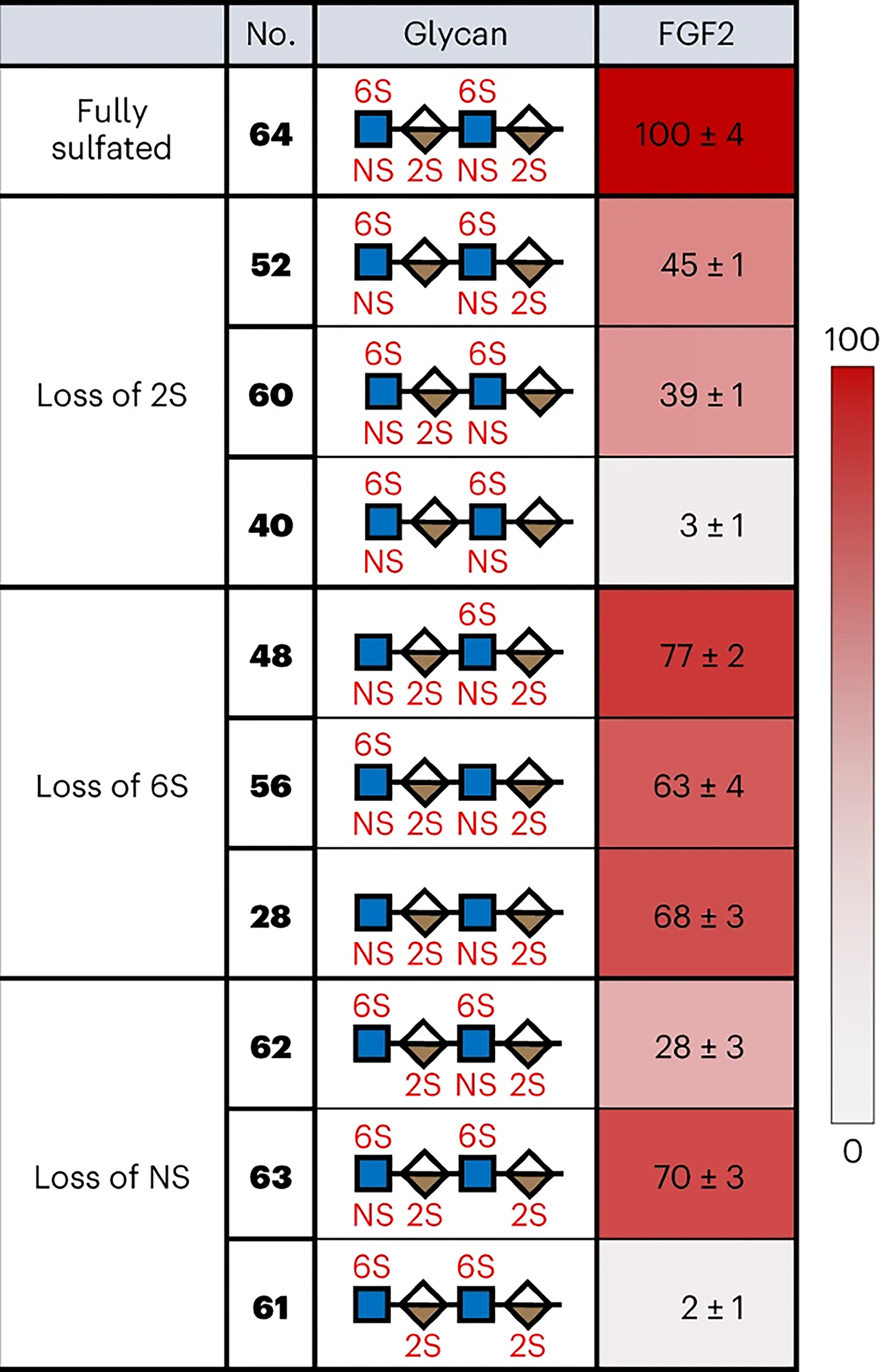
The shown glycans facilitate systematic analysis of the importance of individual sulfate modifications to FGF2 binding.
Our findings highlight an interesting parallel between the molecular recognition of HS GAGs and DNA. DNA-binding transcription factors, like HS-binding proteins, are capable of binding a subset of closely related sequences, and their binding specificities have been characterized using DNA sequence logos51. Drawing on these parallels, we developed a general method to quantify and visualize the preferred sulfation sequences of GAG-binding proteins. Briefly, we first determined the relative frequencies of each HS modification across the full set of sequences in the library and weighted them by the normalized binding values in the heatmaps (Supplementary Methods). The log ratio of the observed frequency to the expected frequency produced position weight matrices, which were then visualized as ‘sulfation logos’. These logos depict the enrichment (or log-likelihood) of sulfation (S), acetylation (Ac) or no modification (H) at each position on a logarithmic scale within the bound HS sequences, where a positive value indicates a higher probability of occurrence than by chance (Fig. 4c). Sulfation logos thus represent the range of HS modifications bound by a given protein and highlight the extent to which each modification is preferred at each position in the sequence. As GAG-binding proteins often tolerate multiple sequences yet prefer distinct motifs, these logos help to determine and depict the key elements of HS–protein recognition.
In the case of FGF2, the N-sulfate (NS) modification of GlcN-2 is the most overrepresented modification among the bound sequences. The computed sulfation logo reveals that N-sulfation at GlcN-1 is less important overall than at GlcN-2, consistent with the general trends observed. Similarly, 2-O-sulfation (2S) at both IdoA-1 and IdoA-2 is also highly overrepresented. In contrast, 6-O-sulfate (6S) modifications at GlcN-2 and to a lesser extent, GlcN-1, are only slightly more enriched than by chance, indicating that 6-O-sulfation is not critical to FGF2 binding. Together with the heatmap analysis, our results lend further support to a large body of structural and biochemical studies demonstrating the crucial requirements of HS 2-O- and N-sulfation, but not 6-O-sulfation, for FGF2 recognition47,48. Indeed, crystal structures of FGF2 complexed with heparin oligosaccharides show that the 6-O-sulfate groups of heparin point away from the binding site and do not make key contacts with FGF236,52.
Our results also expand an understanding of HS–FGF2 interactions. Interestingly, FGF2 binding was not highly correlated with the overall negative charge of the tetrasaccharides. Tetrasaccharides bearing the same number of sulfate groups displayed a wide range of binding efficiencies. For example, binding to the tetrasulfated compounds 28, 47, 54 and 61 ranged from 2% to 68%, depending on the precise location of the sulfate groups. The pentasulfated tetrasaccharides 60 and 63 also exhibited substantially different interaction with FGF2 (39% and 70%, respectively). These observations reinforce the notion that HS-binding proteins such as FGF2 bind in a sequence-specific manner and that HS–FGF2 interactions are driven by the specific pairing of electrostatic, hydrogen-bonding and van der Waals interactions, rather than cumulative, nonspecific charge effects.
We also obtained position-resolved, sequence-specific information regarding HS recognition by FGF2 that was previously unknown. Although the importance of the NS and 2S modifications had been shown, it was not known that FGF2 binding required a precise sequence of these modifications. We found that the NS and 2S modifications must be adjacent to one another within the same GlcN–IdoA disaccharide unit, and the 2-O-sulfated IdoA must be on the reducing end relative to the N-sulfated GlcN (Fig. 4d; GlcNS-2-IdoA2S-2, compounds 15, 35, 43, 59 or GlcNS-1-IdoA2S-1, compounds 6, 22, 30, 50). Separating the NS and 2S modifications (GlcNS-2-IdoA-2-GlcN-1-IdoA2S-1, compounds 7, 23, 31, 51) or changing their reducing end orientation to a nonreducing end orientation (IdoA2S-2-GlcNS-1, compounds 14, 34, 42, 58) abolished FGF2 binding. These unexpected observations suggest that FGF2 binds the HS tetrasaccharides in a preferred orientation, and the FGF2–HS interaction requires a specific sequence of nonreducing-to-reducing end modifications. Thus, our HS libraries enable a deeper understanding of HS–protein interactions, permitting the identification of sequence-specific modifications and specific combinations of modifications critical for interaction. Detailed sequence-specific information has not been previously attainable for HS GAGs, even though it has been vital to the understanding of other major biopolymers such as DNA, RNA and proteins.
Comparisons between FGFs
Previous studies have proposed that each FGF family member may recognize a distinct sulfation sequence due to differences in the spatial arrangement of basic residues in their HS-binding sites36,53. We thus chose to examine the sulfation preferences of another FGF, FGF4, which plays critical roles in biological functions such as angiogenesis and neurogenesis50. We found that the HS-binding heatmaps and sulfation logos for FGF4 and FGF2 are strikingly different (Figs. 4b,c and 6a,b). In contrast to FGF2, FGF4 showed the highest binding preference for tetrasaccharide 63, which lacks N-sulfation at GlcN-1. Interestingly, loss of the N-sulfate group and N-acetylation at GlcN-1 generally increased FGF4 binding (for example, 64 (81%) versus 63 (100%); 60 (38%) versus 59 (83%); 48 (49%) versus 47 (95%)), and the computed sulfation logo showed no overall preference for N-sulfation or N-acetylation at GlcN-1 in the bound sequences (Fig. 6b). On the other hand, loss of the N-sulfate group at GlcN-2 generally decreased FGF4 binding (for example, 64 (81%) versus 62 (27%); 60 (38%) versus 58 (10%); 48 (49%) versus 46 (32%)). Although previous studies have suggested that N-sulfation is important for HS-binding to FGF2 and FGF454, our results indicate that N-sulfation is not fully required for strong FGF4 binding and reveal the distinct contributions of each N-sulfate group in the tetrasaccharide sequence. We also identify other crucial differences in HS recognition between FGF4 and FGF2, including a stronger dependence on 6-O-sulfation for FGF4. Loss of either or both 6-O-sulfate groups substantially reduced FGF4 binding (for example, 63 (100%) versus 55 (43%) and 27 (35%); 64 (81%) versus 56 (56%), 48 (49%) and 28 (42%); 59 (83%) versus 35 (44%), 43 (16%) and 15 (16%)), with 6-O-sulfation generally being more important at GlcN-1 (Fig. 6b), particularly when GlcN-1 lacks N-sulfation (for example, 47 (95%) versus 55 (43%)). Similar to FGF2, loss of 2-O-sulfation at both IdoA-1 and IdoA-2 substantially reduced binding to FGF4 (39 (43%) and 40 (52%)). Overall, the sulfation preferences of FGF4 are remarkably distinct from those of FGF2, suggesting that these FGF family members likely recognize unique HS sequences in vivo. Interestingly, several tetrasaccharides were preferentially recognized by FGF4 but not FGF2 (for example, 51 (67% versus 3%) and 40 (52% versus 3%)), raising the possibility of selective manipulation of FGFs in vivo by endogenous HS expression patterns or exogenous HS mimetics.
Fig. 6 |. HS sulfation specificities of FGF4 and CXCL8.
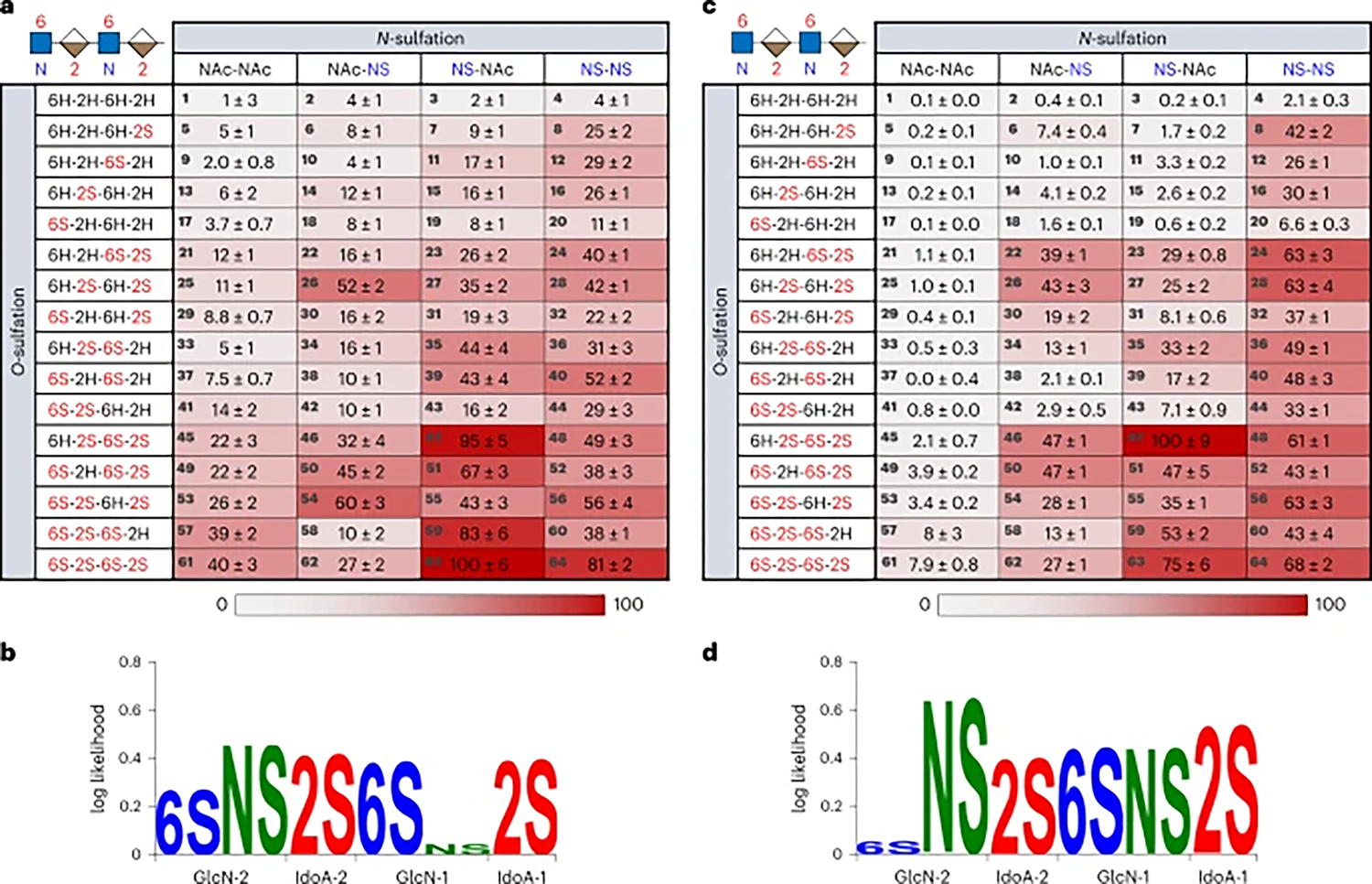
a, Heatmap of the relative binding of FGF4 to the 64-compound HS library. Binding signals were normalized with respect to the compound with greatest binding. Error is the standard deviation to the first digit of uncertainty for the mean across nine replicates. b, Sulfation logo for FGF4. c, Heatmap of the relative binding of CXCL8. Error is the standard deviation to the first digit of uncertainty for the mean across nine replicates. d, Sulfation logo for CXCL8.
HS–chemokine interactions
HS regulates numerous chemokines important for the immune response, such as CXCL8 (interleukin-8), which is involved in leukocyte migration during bacterial and viral infections, as well as chronic inflammation55,56. In stark contrast to FGF2 and FGF4, the top binding sequence for CXCL8 was compound 47, which lacks 6-O-sulfation at GlcN-2 and N-sulfation at GlcN-1 (Fig. 6c). Contrary to views that greater negative charge on HS leads to higher binding affinity, further increases in charge density were generally detrimental to CXCL8 binding (for example, 47 (100%) versus 63 (75%) and 48 (61%); 24 (63%) versus 52 (43%)), indicating that CXCL8–HS interactions are driven by specific interactions rather than overall negative charge. CXCL8 recognition depended strongly on 2-O-sulfation, as loss of either or both 2S groups substantially decreased binding (47 (100%) versus 23 (29%), 35 (33%) and 11 (3.3%); 63 (75%) versus 51 (47%), 59 (53%) and 39 (17%)). Although previous studies have reported that 6-O-sulfation and N-sulfation are favourable to CXCL8 binding57, our results demonstrate important position- and context-dependent effects for these modifications. For example, 6-O-sulfation at GlcN-1 enhanced CXCL8–HS interactions (27 (25%) versus 47 (100%)), whereas 6-O-sulfation at GlcN-2 was detrimental or neutral to binding (47 (100%) versus 63 (75%); 36 (49%) versus 60 (43%)). Accordingly, the sulfation logo for CXCL8 showed a striking lack of dependence on the 6S modification at GlcN-2 (Fig. 6d). We also observed more nuanced effects of 6-O-sulfation. For example, when GlcN-1 lacked N-sulfation, 6-O-sulfation at GlcN-1 became more important for binding and contributed to the optimal recognition sequence (47 (100%) versus 27 (25%)). On the other hand, when GlcN-1 was N-sulfated, 6-O-sulfation at GlcN-1 had little effect on binding (46 (47%) versus 26 (43%) and 48 (61%) versus 28 (63%)). Notably, N-sulfation was critical for binding, particularly at GlcN-2 (for example, NS–NS column versus NAc–NS column), whereas N-sulfation/N-acetylation at GlcN-1 reduced CXCL8 binding for some highly sulfated sequences (47 (100%) versus 48 (61%); 59 (53%) versus 60 (43%)). Overall, these findings highlight the ability of comprehensive HS oligosaccharide libraries to provide insights into the precise HS modifications required for optimal interaction with CXCL8, a protein that has been challenging to characterize and reported to rely on the domain structure of HS rather than specific modifications27,37,58.
Together, our studies reveal the different sulfate modification dependencies of CXCL8, FGF4 and FGF2 with residue-specific, sequence resolution that has been previously unattainable. We observed that each protein is characterized by a distinct set of HS-binding interactions represented as a heatmap ‘fingerprint’. Furthermore, binding enrichment analyses visualized as sulfation logos highlight the unique features exploited by each protein for molecular recognition of HS.
Conclusion
We have developed a new strategy to prepare large, structurally diverse libraries of HS oligosaccharides. Our approach exploits a universal HS building block derived from natural heparin to minimize the number of synthetic transformations and a fluorous tagging method to expedite the purification of charged HS intermediates. This streamlines the late-stage diversification steps and substantially reduces the time and labour required for GAG library synthesis. Overall, this platform greatly accelerates the production of large, comprehensive collections of HS oligosaccharides and represents an important step toward providing broad access to synthetic GAG oligosaccharides on demand.
We applied the approach to generate a complete library of HS oligosaccharides representing all possible modifications at the most commonly sulfated N-, 2-O- and 6-O-positions in the tetrasaccharide GlcN–IdoA–GlcN–IdoA. This library allowed for detailed, systematic investigations into the structural determinants important for molecular recognition by HS-binding proteins. We also developed powerful methods to analyse, visualize and compare the HS sequences bound by proteins, and dissected the importance of individual modifications at each position in the oligosaccharide sequence. Our sulfation sequence logos demonstrate that FGFs and chemokines exhibit unique binding preferences across a broad range of HS sequences. Notably, our synthetic approach and sulfation logos can be readily expanded in the future to incorporate other modifications such as epimerization (that is, GlcA or IdoA) or to represent other GAG classes.
Overall, the streamlined synthesis of comprehensive libraries of defined HS oligosaccharides provides a wide diversity of both natural and non-natural structures for elucidating structure–function relationships. When combined with powerful, high-throughput microarray technologies, these libraries offer an unparalleled view into the sulfation code of GAGs, demonstrating the unique preferences of proteins for specific subsets of closely related sequences and revealing interesting analogies to DNA-binding proteins. Notably, the incorporation of short GAG oligosaccharides into multivalent polymer scaffolds can serve as effective mimetics for GAG polysaccharides59–63. Thus, we anticipate that these libraries and the ability to decode GAG structure–function relationships will open new opportunities to target therapeutically important proteins.
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41557-023-01248-4.
Supplementary Material
Acknowledgements
Financial support was provided by the National Institutes of Health (NIH U01 GM116262 to L.C.H.-W. and X.H., R44 GM134738 to V. Pagadala, L.C.H.-W. and X.H., U01 GM116248 to N.L.B.P., and T32 GM109825 and T32 GM131994 to M.K.K.) and the National Science Foundation (DGE-1745301 to A.W.S.). We thank M. Shahgholi in the CCE Division Mass Spectrometry Facility and D. Vander Velde in the CCE Division NMR Facility at Caltech for technical support.
Footnotes
Competing interests
J.L. is a founder and chief scientific officer of Glycan Therapeutics. G.S. is an employee of Glycan Therapeutics and has stock options from the company. The remaining authors declare no competing interests.
Methods
All experimental methods and procedures are provided in the Supplementary Information.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this Article.
Code availability
All code associated with this report is publicly available at https://doi.org/10.5281/zenodo.7787616.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41557-023-01248-4.
Peer review information Nature Chemistry thanks Hongzhi Cao, Chi-Huey Wong and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Reprints and permissions information is available at www.nature.com/reprints.
Data availability
All data associated with this report are contained within the manuscript or the Supplementary Information. Source data are provided with this Paper.
References
- 1.Zhang P et al. Heparan sulfate organizes neuronal synapses through neurexin partnerships. Cell 174, 1450–1464 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Taylor KR & Gallo RL Glycosaminoglycans and their proteoglycans: host-associated molecular patterns for initiation and modulation of inflammation. FASEB J. 20, 9–22 (2006). [DOI] [PubMed] [Google Scholar]
- 3.Parish CR The role of heparan sulphate in inflammation. Nat. Rev. Immunol. 6, 633–643 (2006). [DOI] [PubMed] [Google Scholar]
- 4.Bulow HE & Hobert O The molecular diversity of glycosaminoglycans shapes animal development. Annu. Rev. Cell Dev. Biol. 22, 375–407 (2006). [DOI] [PubMed] [Google Scholar]
- 5.Kusche-Gullberg M & Kjellen L Sulfotransferases in glycosaminoglycan biosynthesis. Curr. Opin. Struct. Biol. 13, 605–611 (2003). [DOI] [PubMed] [Google Scholar]
- 6.Vallet SD, Berthollier C & Ricard-Blum S The glycosaminoglycan interactome 2.0. Am. J. Physiol. Cell Physiol. 322, C1271–C1278 (2022). [DOI] [PubMed] [Google Scholar]
- 7.Chanalaris A et al. Heparan sulfate proteoglycan synthesis is dysregulated in human osteoarthritic cartilage. Am. J. Pathol. 189, 632–647 (2019). [DOI] [PubMed] [Google Scholar]
- 8.Gordts P & Esko JD The heparan sulfate proteoglycan grip on hyperlipidemia and atherosclerosis. Matrix Biol. 71–72, 262–282 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Soares da Costa D, Reis RL & Pashkuleva I Sulfation of glycosaminoglycans and its implications in human health and disorders. Annu. Rev. Biomed. Eng. 19, 1–26 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Mah D et al. The sulfation code of tauopathies: heparan sulfate proteoglycans in the prion like spread of tau pathology. Front. Mol. Biosci. 8, 671458 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mende M et al. Chemical synthesis of glycosaminoglycans. Chem. Rev. 116, 8193–8255 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Zong C et al. Integrated approach to identify heparan sulfate ligand requirements of Robo1. J. Am. Chem. Soc. 138, 13059–13067 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zulueta MM et al. α-Glycosylation by d-glucosamine-derived donors: synthesis of heparosan and heparin analogues that interact with mycobacterial heparin-binding hemagglutinin. J. Am. Chem. Soc. 134, 8988–8995 (2012). [DOI] [PubMed] [Google Scholar]
- 14.Noti C, de Paz JL, Polito L & Seeberger PH Preparation and use of microarrays containing synthetic heparin oligosaccharides for the rapid analysis of heparin-protein interactions. Chem. Eur. J. 12, 8664–8686 (2006). [DOI] [PubMed] [Google Scholar]
- 15.Ramadan S et al. Automated solid phase assisted synthesis of a heparan sulfate disaccharide library. Org. Chem. Front. 9, 2910–2920 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hu YP et al. Divergent synthesis of 48 heparan sulfate-based disaccharides and probing the specific sugar-fibroblast growth factor-1 interaction. J. Am. Chem. Soc. 134, 20722–20727 (2012). [DOI] [PubMed] [Google Scholar]
- 17.Jain P et al. Discovery of rare sulfated N-unsubstituted glucosamine based heparan sulfate analogs selectively activating chemokines. Chem. Sci. 12, 3674–3681 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pawar NJ et al. Expedient synthesis of core disaccharide building blocks from natural polysaccharides for heparan sulfate oligosaccharide assembly. Angew. Chem. Int. Ed. 58, 18577–18583 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu B et al. Facile chemoenzymatic synthesis of biotinylated heparosan hexasaccharide. Org. Biomol. Chem. 13, 5098–5101 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hansen SU et al. Tetrasaccharide iteration synthesis of a heparin-like dodecasaccharide and radiolabelling for in vivo tissue distribution studies. Nat. Commun. 4, 2016 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen Y et al. Tailored design and synthesis of heparan sulfate oligosaccharide analogues using sequential one-pot multienzyme systems. Angew. Chem. Int. Ed. 52, 11852–11856 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu Y et al. Chemoenzymatic synthesis of homogeneous ultralow molecular weight heparins. Science 334, 498–501 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hu YP et al. Synthesis of 3-O-sulfonated heparan sulfate octasaccharides that inhibit the herpes simplex virus type 1 host–cell interaction. Nat. Chem. 3, 557–563 (2011). [DOI] [PubMed] [Google Scholar]
- 24.Wang Z et al. Preactivation-based, one-pot combinatorial synthesis of heparin-like hexasaccharides for the analysis of heparin-protein interactions. Chem. Eur. J. 16, 8365–8375 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen C & Yu B Efficient synthesis of Idraparinux, the anticoagulant pentasaccharide. Bioorg. Med. Chem. Lett. 19, 3875–3879 (2009). [DOI] [PubMed] [Google Scholar]
- 26.Dey S & Wong CH Programmable one-pot synthesis of heparin pentasaccharides enabling access to regiodefined sulfate derivatives. Chem. Sci. 9, 6685–6691 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zong C et al. Heparan sulfate microarray reveals that heparan sulfate-protein binding exhibits different ligand requirements. J. Am. Chem. Soc. 139, 9534–9543 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hansen SU et al. Synthesis of l-iduronic acid derivatives via [3.2.1] and [2.2.2] l-iduronic lactones from bulk glucose-derived cyanohydrin hydrolysis: a reversible conformationally switched superdisarmed/rearmed lactone route to heparin disaccharides. J. Org. Chem. 80, 3777–3789 (2015). [DOI] [PubMed] [Google Scholar]
- 29.Codee JD et al. A modular strategy toward the synthesis of heparin-like oligosaccharides using monomeric building blocks in a sequential glycosylation strategy. J. Am. Chem. Soc. 127, 3767–3773 (2005). [DOI] [PubMed] [Google Scholar]
- 30.Munoz-Garcia JC et al. Effect of the substituents of the neighboring ring in the conformational equilibrium of iduronate in heparin-like trisaccharides. Chem. Eur. J. 18, 16319–16331 (2012). [DOI] [PubMed] [Google Scholar]
- 31.Panza M, Pistorio SG, Stine KJ & Demchenko AV Automated chemical oligosaccharide synthesis: novel approach to traditional challenges. Chem. Rev. 118, 8105–8150 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hahm HS et al. Automated glycan assembly of oligo-N-acetyllactosamine and keratan sulfate probes to study virus-glycan interactions. Chem 2, 114–124 (2017). [Google Scholar]
- 33.Eller S, Collot M, Yin J, Hahm HS & Seeberger PH Automated solid-phase synthesis of chondroitin sulfate glycosaminoglycans. Angew. Chem. Int. Ed. 52, 5858–5861 (2013). [DOI] [PubMed] [Google Scholar]
- 34.Budhadev D et al. Using automated glycan assembly (AGA) for the practical synthesis of heparan sulfate oligosaccharide precursors. Org. Biomol. Chem. 17, 1817–1821 (2019). [DOI] [PubMed] [Google Scholar]
- 35.Walvoort MT et al. Automated solid-phase synthesis of hyaluronan oligosaccharides. Org. Lett. 14, 3776–3779 (2012). [DOI] [PubMed] [Google Scholar]
- 36.Raman R, Venkataraman G, Ernst S, Sasisekharan V & Sasisekharan R Structural specificity of heparin binding in the fibroblast growth factor family of proteins. Proc. Natl Acad. Sci. USA 100, 2357–2362 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu D & Esko JD Demystifying heparan sulfate-protein interactions. Annu. Rev. Biochem. 83, 129–157 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guglier S et al. Minimum FGF2 binding structural requirements of heparin and heparan sulfate oligosaccharides as determined by NMR spectroscopy. Biochem. 47, 13862–13869 (2008). [DOI] [PubMed] [Google Scholar]
- 39.Shaw JP et al. The X-ray structure of RANTES: heparin-derived disaccharides allows the rational design of chemokine inhibitors. Structure 12, 2081–2093 (2004). [DOI] [PubMed] [Google Scholar]
- 40.Studer A et al. Fluorous synthesis: a fluorous-phase strategy for improving separation efficiency in organic synthesis. Science 275, 823–826 (1997). [DOI] [PubMed] [Google Scholar]
- 41.Kern MK & Pohl NLB Automated solution-phase synthesis of S-glycosides for the production of oligomannopyranoside derivatives. Org. Lett. 22, 4156–4159 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bhaduri S & Pohl NL Fluorous-tag assisted syntheses of sulfated keratan sulfate oligosaccharide fragments. Org. Lett. 18, 1414–1417 (2016). [DOI] [PubMed] [Google Scholar]
- 43.Cai C et al. Fluorous-assisted chemoenzymatic synthesis of heparan sulfate oligosaccharides. Org. Lett. 16, 2240–2243 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zong C, Venot A, Dhamale O & Boons GJ Fluorous supported modular synthesis of heparan sulfate oligosaccharides. Org. Lett. 15, 342–345 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jaipuri FA & Pohl NL Toward solution-phase automated iterative synthesis: fluorous-tag assisted solution-phase synthesis of linear and branched mannose oligomers. Org. Biomol. Chem. 6, 2686–2691 (2008). [DOI] [PubMed] [Google Scholar]
- 46.Bock K & Pedersen C A study of 13CH coupling constants in hexopyranoses. J. Chem. Soc. Perkin Trans. 2, 293–297 (1974). [Google Scholar]
- 47.Ishihara M, Kariya Y, Kikuchi H, Minamisawa T & Yoshida K Importance of 2-O-sulfate groups of uronate residues in heparin for activation of FGF-1 and FGF-2. J. Biochem. 121, 345–349 (1997). [DOI] [PubMed] [Google Scholar]
- 48.Guimond S, Maccarana M, Olwin BB, Lindahl U & Rapraeger AC Activating and inhibitory heparin sequences for FGF-2 (basic FGF). Distinct requirements for FGF-1, FGF-2 and FGF-4. J. Biol. Chem. 268, 23906–23914 (1993). [PubMed] [Google Scholar]
- 49.Ashikari-Hada S et al. Characterization of growth factor-binding structures in heparin/heparan sulfate using an octasaccharide library. J. Biol. Chem. 279, 12346–12354 (2004). [DOI] [PubMed] [Google Scholar]
- 50.Beenken A & Mohammadi M The FGF family: biology, pathophysiology and therapy. Nat. Rev. Drug. Discov. 8, 235–253 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schneider TD & Stephens RM Sequence logos: a new way to display consensus sequences. Nucleic Acids Res. 18, 6097–6100 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Faham S, Hileman RE, Fromm JR, Linhardt RJ & Rees DC Heparin structure and interactions with basic fibroblast growth factor. Science 271, 1116–1120 (1996). [DOI] [PubMed] [Google Scholar]
- 53.Faham S, Linhardt RJ & Rees DC Diversity does make a difference: fibroblast growth factor-heparin interactions. Curr. Opin. Struct. Biol. 8, 578–586 (1998). [DOI] [PubMed] [Google Scholar]
- 54.Ishihara M Structural requirements in heparin for binding and activation of FGF-1 and FGF-4 are different from that for FGF-2. Glycobiology 4, 817–824 (1994). [DOI] [PubMed] [Google Scholar]
- 55.Vora SM, Lieberman J & Wu H Inflammasome activation at the crux of severe COVID-19. Nat. Rev. Immunol. 21, 694–703 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Moore BB & Kunkel SL Attracting attention: discovery of IL-8/CXCL8 and the birth of the chemokine field. J. Immunol. 202, 3–4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.de Paz JL et al. Profiling heparin-chemokine interactions using synthetic tools. ACS Chem. Biol. 2, 735–744 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Spillmann D, Witt D & Lindahl U Defining the interleukin-8-binding domain of heparan sulfate. J. Biol. Chem. 273, 15487–15493 (1998). [DOI] [PubMed] [Google Scholar]
- 59.Rawat M, Gama CI, Matson JB & Hsieh-Wilson LC Neuroactive chondroitin sulfate glycomimetics. J. Am. Chem. Soc. 130, 2959–2961 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sheng GJ, Oh YI, Chang SK & Hsieh-Wilson LC Tunable heparan sulfate mimetics for modulating chemokine activity. J. Am. Chem. Soc. 135, 10898–10901 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Oh YI, Sheng GJ, Chang SK & Hsieh-Wilson LC Tailored glycopolymers as anticoagulant heparin mimetics. Angew. Chem. Int. Ed. 52, 11796–11799 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Naticchia MR, Laubach LK, Honigfort DJ, Purcell SC & Godula K Spatially controlled glycocalyx engineering for growth factor patterning in embryoid bodies. Biomater. Sci. 9, 1652–1659 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Loka RS et al. Heparan sulfate mimicking glycopolymer prevents pancreatic β cell destruction and suppresses inflammatory cytokine expression in islets under the challenge of upregulated heparanase. ACS Chem. Biol. 17, 1387–1400 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this report are contained within the manuscript or the Supplementary Information. Source data are provided with this Paper.