

Summary
Multi-enhancer hubs are spatial clusters of enhancers which have been recently characterized across numerous developmental programs. Yet, the functional relevance of these three-dimensional structures is poorly understood. Here we show that the multiplicity of enhancers interacting with the Ets1 gene controls the expression level of Ets1 in response to cellular cues. We identified a highly connected multi-enhancer hub at the Ets1 locus, comprising a noncoding regulatory element that is a hotspot for sequence variation associated with allergic diseases. Deleting this hotspot revealed that the multi-enhancer connectivity is required for CD4+ T helper 1 (Th1) differentiation. Mice lacking this hotspot are thus protected from Th1-mediated colitis but demonstrate overt allergic responses. Mechanistically, the multi-enhancer hub controls the Ets1 dosage that is required for the Th1-specific genome topology. Together, we establish a paradigm for the functional and mechanistic relevance of multi-enhancer hubs controlling cellular competence to respond specifically to an inductive cue.
Introduction
The spatiotemporal control of gene-expression programs relies on nuclear organization of noncoding regulatory elements called enhancers. Recent advances in genomics and imaging technologies attest to the formation of spatial clusters of enhancers, called interchangeably as multi-enhancer hubs1–8, 3D cliques9,10, cis-regulatory domains11, interacting triplets12, connected gene communities13, or architectural stripes14,15. Despite numerous efforts profiling multi-enhancer interactions across diverse developmental programs, the functional relevance of these structures is poorly understood.
We reasoned that multi-enhancer hubs enriched with disease-associated sequence variation can put forward candidates for functional and mechanistic investigations. Using T cells as a model, we mathematically defined the higher-order structure of multi-enhancer interactions in mouse T cells10. Our unbiased strategy revealed that one of the most hyperconnected regions in T cells occurs at a locus harboring Ets1 and Fli1 genes. ETS1 and FLI1 contain a conserved winged helix-turn-helix DNA-binding domain16,17 and bind to a consensus DNA sequence with a core GGA(A/T) motif18. ETS1, which has diverse roles in multiple biological processes19, is most highly expressed in lymphoid organs20,21 and is required for the development of T cells, natural killer cells, and innate lymphoid cells19,20,22–27. FLI1 is not as highly expressed as ETS1 in lymphoid organs, yet its contribution to effector CD8+ T cell function has been described28. While numerous single-nucleotide polymorphisms (SNPs) associated with immune-mediated diseases are distributed across the human ETS1-FLI1 locus, a noncoding region within this locus forms a hotspot for SNPs associated with type 2 immune diseases including allergy, asthma, and atopic dermatitis29,30. The complete ablation of Ets1 has been linked to immune-mediated diseases22,23, yet mechanisms through which noncoding sequence variation at this locus contributes to allergic responses are unknown.
Here we report that the genetic deletion of a regulatory element in the Ets1 locus using the CRISPR–Cas9 system in mice does not affect T cell development but impairs CD4+ T helper 1 (Th1) cell differentiation, leading to protection against colitis and an overt allergic response. Our molecular and cellular analyses propose a mechanism through which the multiplicity of enhancer interactions at the Ets1 locus controls the sharp increase in the expression of Ets1 in response to changes in the cytokine environment which in turn is required for the recruitment of CTCF to specify three-dimensional (3D) nuclear organization of Th1 cells. Considering that Th1 differentiation is a critical mechanism by which type 2 immune responses are dampened31–33, our findings imply the molecular processes through which sequence variation within noncoding elements at the Ets1 locus predisposes individuals to allergic responses. These findings further establish a paradigm for understanding the importance of multi-enhancer hubs in dynamically regulating gene expression in response to changes in cellular environment.
Results
Multi-enhancer connectivity at T cell-lineage genes
We aimed to identify multi-enhancer hubs in T cells in an unbiased manner and leveraged human genetics to ascertain whether sequence variation within the top densely connected multi-enhancer hubs is linked to immune-mediated diseases. We reasoned that mapping multi-enhancer interactions in thymocytes, which represent T cells before any antigen exposure, can delineate critical regulatory units shared across T cell populations. Hence, we mapped enhancer interactions in double-positive (DP) thymocytes and used H3K27ac HiChIP10, which is a protein-centric assay for 3D mapping of enhancer interactions34,35. We algorithmically searched for groups of densely connected multi-enhancers, which have also been referred to as 3D cliques9,10 (Figure 1A). The degree of enhancer connectivity was asymmetrical, reminiscent of asymmetrical histone acetylation at super-enhancers36,37, where only fewer than 18% of regulatory elements (2,372) spatially converged into “hyperconnected” multi-enhancer hubs defined based on the slope of 1 in the plot ranking their connectivity (Figure 1A, Table S1). Super-enhancers were significantly enriched in hyperconnected hubs, suggesting extensive spatial connectivity among highly acetylated genomic elements (Figure 1B). Although not statistically significant, around 20% of hyperconnected hubs overlapped with annotated noncoding RNAs and approximately 70% of hyperconnected hubs were characterized as architectural stripes14,15 (Figure S1A). Overall, genes associated with T cell biology, including the “T cell receptor signaling pathway” and “adaptive immune system”, were highly enriched at hyperconnected hubs (Figure 1C), thus suggesting that our analytical approach can prioritize regions harboring genes critical for T cell function.
Figure 1: Exceptional enhancer connectivity at the Ets1-Fli1 locus.
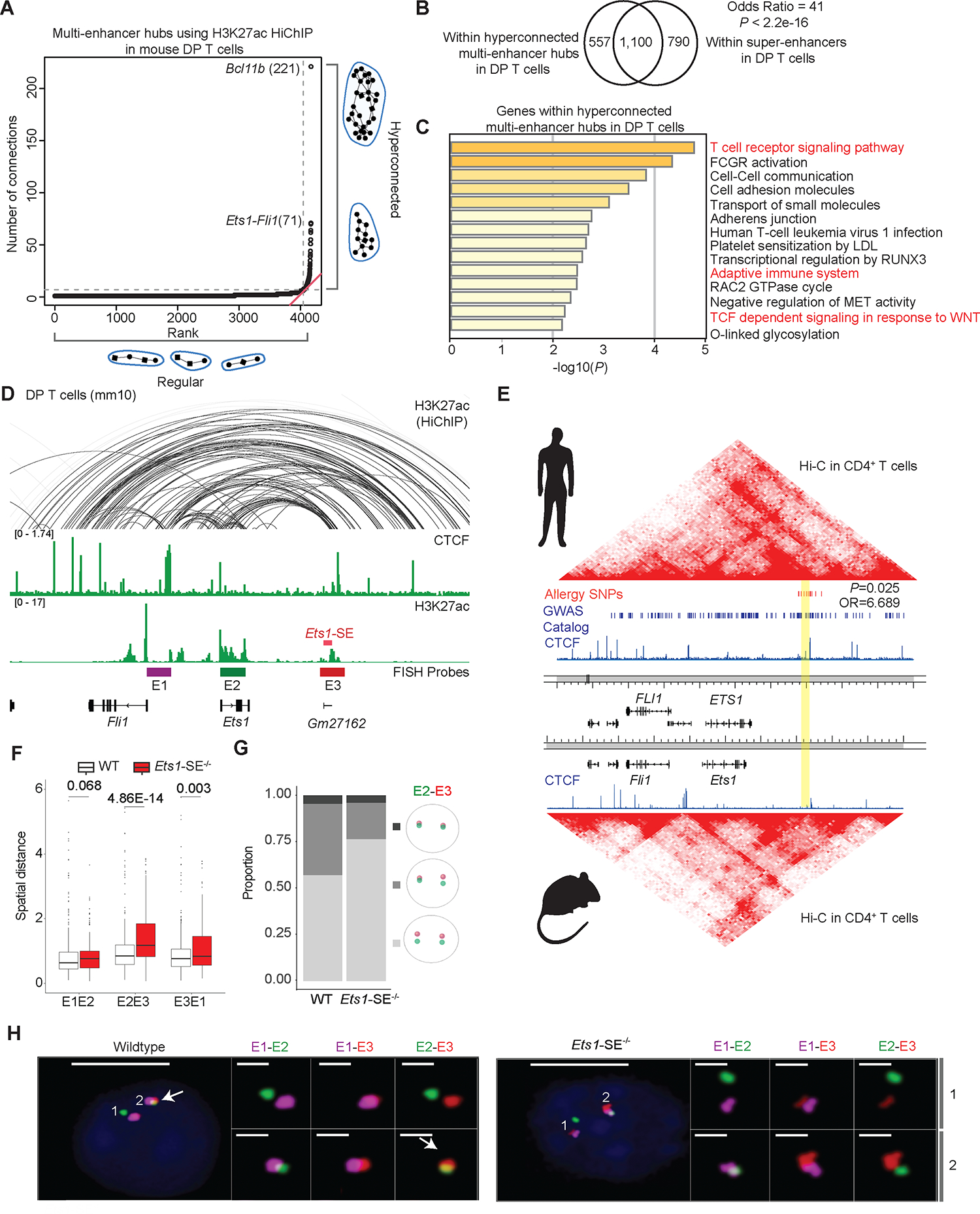
A, Plot depicts ranking versus number of connections in multi-enhancer hubs also referred to as 3D cliques9,10 detected in double-positive (DP) T cells using H3K27ac HiChIP measurements generated in our previous study10. Hyperconnected multi-enhancer hubs are defined as the ones above the elbow of the total connectivity ranking. Top two hyperconnected multi-enhancer hubs Bcl11b and Ets1-Fli1 are labeled. Number of interactions in each hub is provided in parenthesis.
B, Venn-diagram depicts the overlaps of genomic regions within hyperconnected multi-enhancer hubs detected using H3K27ac HiChIP when compared to super-enhancers defined by H3K27ac ChIP-seq in DP T cells. Super-enhancers were defined based on H3K27ac ChIP-seq in DP T cells as described before36. Odds ratio and Fisher’s exact test were used for statistical analysis.
C, Bar-plot demonstrates the significance of gene-ontology terms enriched in genes encompassing multi-enhancer hubs. Metascape56 was used for gene-ontology analysis. Terms relating to immune response pathways are marked in red.
D, The genome browser view demonstrates H3K27ac and CTCF ChIP-seq, as well as H3K27ac HiChIP 3D interactions at the Ets1-Fli1 locus in DP T cells, and the exact genomic location of E1-E3 probes used for “Oligopaint” DNA-FISH. The 25kbp super-enhancer which is the focus of this study is called Ets1-SE and is marked in red. FISH probes depicted in the browser view include E1 (enhancer upstream of Ets1 and proximal to Fli1 promoter, magenta), E2 (Ets1 promoter and gene-body, green) and E3 (Ets1-SE and a 25kbp enhancer downstream of Ets1-SE, red), representing the three independent 50kbp genomic regions for which Oligopaint probes were designed.
E, Heatmaps demonstrate contact-frequency maps measured by Hi-C in CD4+ T cells in humans and mice. ChIP-seq tracks demonstrate CTCF binding in human CD4+ T cells and mouse DP T cells. SNPs associated with asthma and allergic diseases highlighted in red were curated from the GWAS catalog and meta-analysis studies for allergy, asthma, and atopic dermatitis29,30. Blue bars demonstrate statistically significant GWAS SNPs associated with diverse traits. The orthologous human coordinate of Gm27162 is highlighted in yellow (chr11:128,303,536–128,330,986). P-value and odds ratio were estimated by Fisher’s exact test.
F, Box plots depict the pairwise spatial distance formed between E1, E2 and E3 probes (Mann-Whitney U test). Oligopaint 3D FISH40,57 in 428 and 460 thymocytes were imaged using widefield microscopy from one wildtype and one Ets1-SE−/− mouse, respectively. Spots for each probe in Oligopaint 3D FISH data were analyzed in a semi-manual manner (Methods).
G, Divided bar-plots demonstrating the proportion of mono vs. biallelic spatial contacts for E2 and E3 probes, in wildtype and Ets1-SE−/− cells. Only cells in which both alleles of both probes were detected, were used (112 wildtype cells and 53 Ets1-SE−/− cells). The distance cutoff used for E2 and E3 spatial contact was 0.7 μm. Pale gray represents no allele showing spatial contact, gray represents mono-allelic and dark gray represents bi-allelic contacts.
H, Representative images of the Oligopaint FISH probes hybridization in one wildtype and one Ets1-SE−/− thymocytes, with magnification of pairwise contacts for each allele. DAPI: blue, E1: magenta, E2: green, E3: red. Scale-bar in whole cell: 5μm and scale-bar in magnification of allele: 1μm. White arrow represents the spatial overlap between E2 and E3.
The top hyperconnected locus in thymocytes encompassed the transcription factor Bcl11b (Figure 1A). Bcl11b is expressed in T cell progenitors and controls a T cell lineage-specific program by suppressing alternative cell fates38. One of the major enhancer elements in this hyperconnected locus repositions from the peripheral lamina to the nuclear interior, a process which is required for T cell development and lymphomas39. The identification of Bcl11b as the most hyperconnected locus in T cells highlights the sensitivity of our analytical approach in identifying genes with key biological roles in this lineage. Hence, we aimed to investigate whether hyperconnectivity of enhancers at other genomic loci is essential for T cell functional competence.
Exceptional enhancer connectivity at a type 2 immune disease-associated risk locus
The second most hyperconnected multi-enhancer hub was detected at the locus encompassing two ETS-family transcription factors, Ets1 and Fli1, and demonstrated conserved chromatin folding patterns between human and mouse T cells (Figure 1A,D,E). Whereas numerous SNPs associated with diverse diseases were distributed within the ~700kbp region encompassing ETS1 and FLI1 in the human genome, variants associated with type 2 immune diseases, namely self-reported allergy29, asthma29, and atopic dermatitis30, were significantly enriched within the ~25kbp orthologous DNA segment that is ~250 kbp downstream of the ETS1 promoter (SNPs in red, segment marked in yellow, Figure 1E). This regulatory segment was a major node of enhancer connectivity, overlapped with a noncoding RNA annotated as Gm27162, and scored as a super-enhancer, and is hereafter referred to as “Ets1-SE” (red block, Figure 1D).
The unusual enrichment of type 2 immune disease-associated SNPs around the Ets1-SE element provided the rationale for us to test its functional relevance. Hence, we generated a new mouse strain in which the 25 kbp Ets1-SE is deleted on the C57BL/6J background (Figure 1D). Together, the high expression of Ets1 during T cell development along with the hyperconnectivity of the Ets1 locus in T cells was the motivation to study the effect of the Ets1-SE deletion on T cell development and function.
Ets1-SE deletion does not affect T cell development
Since ETS1 is required for the T cell lineage19 and our multi-enhancer hub profiling was performed in DP T cells, we first assessed the effect of the Ets1-SE deletion on the expression of Ets1 and Fli1 in DP T cells using bulk RNA sequencing. We found that Fli1 expression did not change but Ets1 expression was reduced by ~29% in Ets1-SE−/− DP T cells (Figure S1B). To visualize genome reorganization of the Ets1 locus in the absence of the Ets1-SE element in DP T cells, we used the “Oligopaint” DNA fluorescence in situ hybridization (FISH) approach40,41. We painted 3 anchors of the Ets1 multi-enhancer hub using probes from our earlier study10: a 50kbp region spanning the Fli1 promoter (E1), a 50kbp region spanning the Ets1 promoter and gene-body (E2), and a 50kbp region spanning the 25kbp Ets1-SE element and its downstream 25kbp region (E3) (Figure 1D). We found that the average spatial distance between the Ets1 promoter (E2) and proximal region to the Ets1-SE (E3) increased in Ets1-SE−/− compared with wildtype DP T cells in a mono-allelic manner (Figure 1 F–H). Moreover, multi-enhancer interactions (or 3D cliques) where all three regulatory elements converged into a hub in the same cell were detected in 2 times fewer alleles in Ets1-SE−/− compared with wildtype DP T cells, suggesting that the Ets1-SE deletion can rewire the Ets1 locus (Figure S1C). Representative cells from wildtype and Ets1-SE−/− mice further corroborated the mono-allelic spatial localization of the Ets1 promoter (E2) and (Ets1-SE) E3 (Figures 1H and S1D). Together, our imaging experiments in DP T cells suggested the reorganization of the Ets1 multi-enhancer hub in the absence of Ets1-SE.
Despite partial changes in the Ets1 level and genome reorganization in DP T cells, T cell development remained intact in Ets1-SE−/− mice as the proportion of cells in the different thymic T cell developmental stages as well as the numbers of natural regulatory T cells (Tregs) were not significantly different when compared to wildtype mice (Figures 2A and S1 E–G). At steady-state, the majority of CD4+ and CD8+ T cell populations in peripheral tissues remained comparable between wildtype and Ets1-SE−/− mice (Figure S2 H–J), apart from a moderate reduction in CD4+ T cell numbers in the lungs (Figure 2B). Thus, unlike the Bcl11b locus39, perturbation of the hyperconnected Ets1 locus was tolerated during T cell development.
Figure 2: Ets1-SE is dispensable for thymic T cell generation but is required for CD4+ Th1 differentiation.
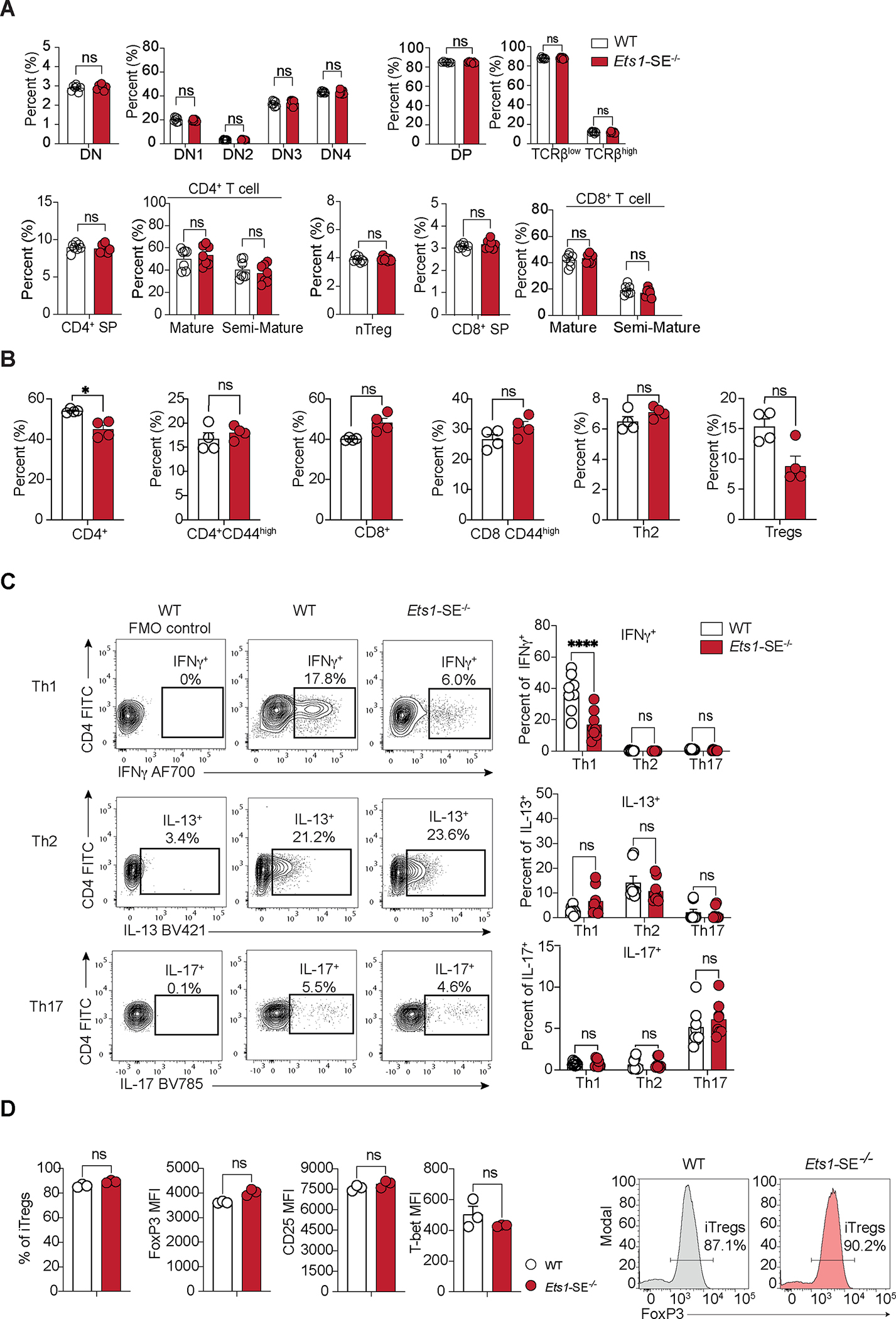
A, Plots demonstrate percentage of cells defined by flow cytometry analysis in the thymus from age matched wildtype and Ets1-SE−/− female mice. Data are representative of three independent experiments. Each dot represents an individual mouse (wildtype; n=7 - Ets1-SE−/−; n=7). Error bars = SEM; and P: ns = not significant, (DN, DP, nTregs, CD4+/CD8+ SP: Mann-Whitney U test; DN1-DN4, TCRβlow/high, Semi-Mature/Mature CD4+/CD8+: Two-way ANOVA with multiple comparisons and Bonferroni correction).
B, Plots demonstrate percentages of cells defined by flow cytometry analysis in the lung Frequencies at steady state of lungs parenchyma CD4+ T cells (TCRβ+, CD4+), activated CD4+ T cells (TCRβ+, CD4+, CD44+), CD8+ T cells (TCRβ+, CD8+), activated CD8+ T cells (TCRβ+, CD8+, CD44+), CD4+ Th2 cells (TCRβ+, CD4+, GATA-3+) from age-matched wildtype and Ets1-SE−/− male mice. Data are representative of two independent experiments. Each dot represents an individual mouse (wildtype; n=4 - Ets1-SE−/−; n=4). Error bars = SEM; and P: ns = not significant, * =P≤0.05 (Mann-Whitney U test).
C, (left) Representative flow cytometry contour plot of naive CD4+ T cells from wildtype or Ets1-SE−/− mice cultured under Th1, Th2 or Th17 polarizing conditions for 6 days. Unstained wildtype cells are shown for each polarizing condition as a negative control (wildtype FMO Control). (right) Frequencies of Th1 (IFNγ+), Th2 (IL-13+) or Th17 (IL-17+) cytokines producing CD4+ T cells cultured under Th1, Th2 or Th17 polarizing conditions for 6 days. All populations were pre-gated on SSC-A/FSC-A, Singlets and Viability− (Live), TCRβ+ and CD4+ cells. Two independent experiments were pooled and were repeated five times. Each dot represents an individual mouse (wildtype; n=8 - Ets1-SE−/−; n=8). Error bars = SEM; and P: ns = not significant, * = P ≤0.05, ** = P ≤0.01, *** = P ≤0.0005, **** = P ≤0.0001 (Two-way ANOVA with multiple comparisons and Bonferroni correction).
D, (left) Frequencies of induced Tregs (iTregs, FoxP3+ CD4+ cells) and mean fluorescence intensity (MFI) of FoxP3, CD25 and T-bet from naïve CD4+ T cells cultured under iTreg polarizing conditions for 3 days. (right) Representative histogram of the proportion of FoxP3+ CD4+ T cells 3 days after culturing in iTregs polarizing conditions. Data are representatives of two independent experiments. Each dot represents an individual mouse (wildtype; n=3 - Ets1-SE−/−; n=3). Error bars = SEM; and P: ns = not significant, * = P ≤0.05, ** = P ≤0.01, *** = P ≤0.0005, **** = P ≤0.0001 (Mann and Whitney).
Multi-enhancer interactions in the Ets1 locus control CD4+ T helper 1 differentiation
The concentration of genetic variants associated with dysregulated type 2 immune responses within Ets1-SE implies a link between this region and CD4+ T differentiation, which is a process mediated by changes in the extracellular cytokine milieu evoked by pathogenic stimuli42. We next explored whether perturbation in the Ets1 multi-enhancer hub affected the capacity of CD4+ T cells to differentiate into distinct T helper functional subsets in vitro31. Strikingly, naïve CD4+ T cells from Ets1-SE−/− mice had a significantly reduced capacity to differentiate into interferon-gamma (IFNγ) producing Th1 cells when compared to wildtype counterparts (Figure 2C). In contrast, Th2, Th17 and iTreg differentiation measured by IL-13, IL-17, and FoxP3 expression, respectively, remained comparable between wildtype and Ets1-SE−/− CD4+ T cells (Figure 2 C–D). Thus, our results suggest that the Ets1-SE element is required for CD4+ Th1 differentiation in vitro.
Ets1-SE element promotes the development of severe colitis in vivo
To study the in vivo relevance of Ets1-SE region, we tested the capacity of Ets1-SE−/− CD4+ T cells to induce colitis using a well-established adoptive transfer Th1-driven model43. To do so, we adoptively transferred CD4+CD45RBhigh T cells from wildtype or Ets1-SE−/− mice into Rag1−/− mice and monitored their body weights as well as colitis development for 6 weeks (Figure 3A). As expected43, Rag1−/− mice that received wildtype CD4+ T cells lost ~10% of their initial body weights by week 6 post-T cell transfer while Rag1−/− mice that received Ets1-SE−/− CD4+ T cells steadily maintained their body weights (Figure 3B). In concordance, Rag1−/− mice that received Ets1-SE−/− CD4+ T cells had significantly longer colon lengths and reduced severity in colonic histopathology when compared to Rag1−/− recipient of wildtype T cells, thus indicating that Th1-driven colon inflammation was drastically reduced in presence of Ets1-SE−/− CD4+ T cells (Figures 3C–E and S2A).
Figure 3: Ets1-SE deletion limits Th1-mediated inflammation in vivo.
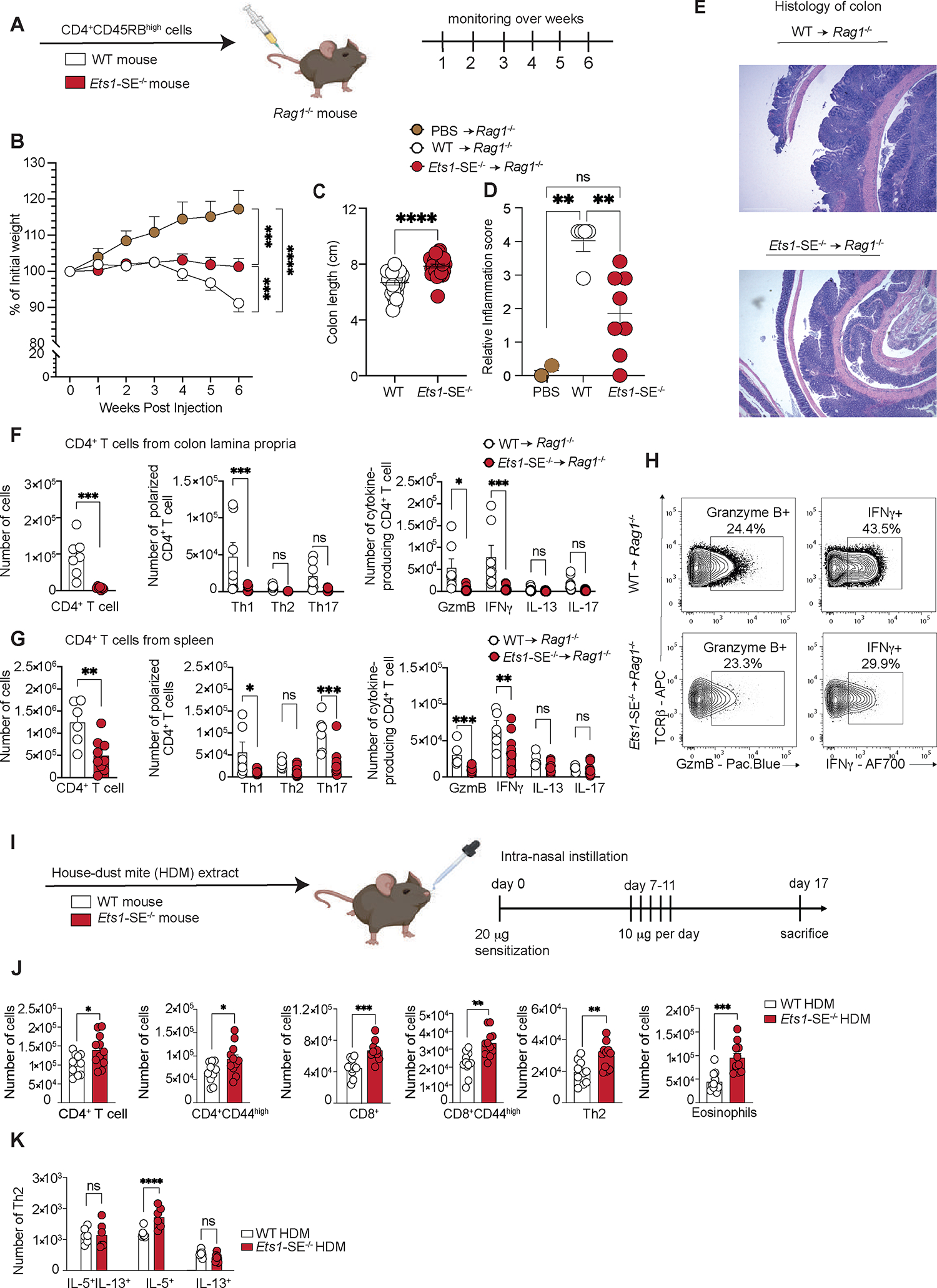
A, Schematic representation of the CD45RBHigh-induced colitis model. 1×106 FACS sorted TCRβ+, CD4+, CD45RBHigh naïve CD4+ T cells from wildtype or Ets1-SE−/− were transferred into Rag1−/− recipients to induce colitis.
B, Weight loss tracking of Rag1−/− mice as compared to PBS-injected animals (controls) during 6 weeks post transfer. Three independent experiments were pooled and repeated four times. Dots represent the mean of individual mouse (PBS -> Rag1−/−; n= 5; wildtype -> Rag1−/−; n=25 and Ets1-SE−/− -> Rag1−/−; n=27). Error bars = SEM; and P: ns = not significant, * = P≤0.05, ** = P≤0.01, *** = P≤0.0005, **** = P≤0.0001 (Two-way ANOVA; Mixed-effect REML model with Fisher’s LSD Test).
C, Quantification of colon length (cm) of Rag1−/− mice that received either 1×106 FACS sorted TCRβ+, CD4+, CD45RBHigh naïve wildtype or Ets1-SE−/− CD4+ T cells at week 6 post transfer. Three independent experiments were pooled and repeated four times. Dots represent the mean of individual mouse (wildtype -> Rag1−/−; n=25 and Ets1-SE−/− -> Rag1−/−; n=27). Error bars = SEM; and P: ns = not significant, * = P ≤0.05, ** = P ≤0.01, *** = P ≤0.0005, **** = P ≤0.0001 (Mann-Whitney U Test).
D, E Quantification of the histological score of paraffin-embedded colon rolls from Rag1−/− mice that received 1×106 FACS sorted TCRβ+, CD4+, CD45RBHigh naïve CD4+ T cells from either wildtype or Ets1-SE−/− mice at week 6 post transfer and as compared to PBS-injected mice (control). Histological sections were obtained from two independent experiments and were scored in a blinded manner. Each dot represents an individual mouse (PBS -> Rag1−/−; n= 2, wildtype -> Rag1−/−; n=5 and Ets1-SE−/− -> Rag1−/−; n=8). Error bars = SEM; and P: ns = not significant, * = P≤0.05, ** = P ≤0.01, *** = P ≤0.0005, **** = P ≤0.0001 (One-way ANOVA with multiple comparisons and Bonferroni correction). E, Histological section of colon rolls stained with H&E of Rag1−/− mice that received either 1×106 FACS sorted TCR+, CD4+, CD45RBHigh naïve wildtype or Ets1-SE−/− CD4+ T cells 6 weeks post transfer. Scale = 100μm; Magnification = 100X.
F, (left) Quantification of infiltrating colon lamina propria (cLP) colitogenic CD4+ T cells of Rag1−/− mice that received either wildtype or Ets1-SE−/− CD4+ T cells. (middle) Quantification of Th1 (T-bet+), Th2 (GATA-3+), Th17 (RORγ-t+) CD4+ T cells among infiltrating cLP CD4+ T cells of Rag1−/− mice that received either wildtype or Ets1-SE−/− CD4+ T cells. (right) Quantification of Th1 (IFNγ+), Th2 (IL-13+), Th17 (IL-17+) and Granzyme B (GzmB+) producing CD4+ T cells among infiltrating cLP CD4+ T cells of Rag1−/− mice that received either wildtype or Ets1-SE−/− CD4+ T cells. Two independent experiments were pooled. Dots represent an individual mouse (Ets1-SE+/+ -> Rag1−/−; n=7 and Ets1-SE−/− -> Rag1−/−; n=7). Error bars = SEM; and P: ns = not significant, * = P ≤0.05, ** = P ≤0.01, *** = P ≤0.0005, **** = P≤0.0001 (CD4+ T cells: Mann-Whitney U Test; Th1/Th2/Th17/Treg and cytokines production: Two-way ANOVA with multiple comparisons and Bonferroni correction).
G, (left) Quantification CD4+ T cells in the spleen of Rag1−/− mice that received wildtype or Ets1-SE−/− CD4+ T cells. (middle) Quantification in the spleen of Th1 (T-bet+), Th2 (GATA-3+), Th17 (RORγ-t+) CD4+ T cells of Rag1−/− mice that received either wildtype or Ets1-SE−/− CD4+ T cells. (right) Quantification of ex vivo stimulated splenic Th1 (IFNγ+), Th2 (IL-13+), Th17 (IL-17+) and Granzyme B (Gzm B+) producing CD4+ T cells of Rag1−/− mice that received either wildtype or Ets1-SE−/− CD4+ T cells. Data represent one experiment which was repeated three times. Dots represent an individual mouse (wildtype -> Rag1−/−; n=7 and Ets1-SE−/− -> Rag1−/−; n=7). Error bars = SEM; and p-values: ns = not significant, * = P≤0.05, ** = P ≤0.01, *** = P ≤0.0005, **** = P ≤0.0001 (CD4+ T cells: Mann-Whitney U Test; Th1/Th2/Th17/Treg and cytokines production: Two-way ANOVA with multiple comparisons and Bonferroni correction).
H, Representative flow cytometry contour plot of Granzyme B- and IFNγ-producing ex vivo stimulated cLP CD4+ T cells of Rag1−/− mice that received either wildtype or Ets1-SE−/− CD4+ T cells.
I, Schematic representation of the house dust mite (HDM) extract challenge. Arrows represent days by which intranasal HDM was administered. Mice were euthanized 16 days after the initial sensitization and immune cell infiltration was checked. Two independent experiments were used to measure immune cell infiltration in the lung parenchyma.
J, Quantification of lung parenchyma infiltrating CD4+ T cells (TCRβ+, CD4+), activated CD4+ T cells (TCRβ+, CD4+, CD44+), CD8+ T cells (TCRβ+, CD8+), activated CD4+ T cells (TCRβ+, CD8+, CD44+), CD4+ Th2 cells (TCRβ+, CD4+, GATA-3+) and eosinophils (MHC-II−, Siglec-F+) from wildtype or Ets1-SE−/− mice 16 days after HDM challenge. Two independent experiments were pooled. Each Dot represents an individual mouse (wildtype n=11 and Ets1-SE−/− n=11). Error bars = SEM; and ns = not significant, * = P ≤0.05, ** = P ≤0.01, *** = P ≤0.0005, **** = P ≤0.0001 (Mann-Whitney).
K, Quantification of Th2 cells numbers from wildtype or Ets1-SE−/− mice producing IL-5, IL-13 or IL-5/IL-13 four hours after PMA and Ionomycin stimulation. Data are representative of one independent experiment and repeated twice. Each dot represents an individual mouse (wildtype n=6 and Ets1-SE−/− n=6). Error bars = SEM; and P: ns = not significant, * = P ≤0.05, ** = P ≤0.01, *** = P ≤0.0005, **** = P ≤0.0001 (Two-way ANOVA with multiple comparison and Bonferroni correction).
To evaluate the functional profiles of CD4+ T cells in Rag1−/− recipient mice, we isolated CD4+ T cells from the colon lamina propria (cLP) and spleen of these animals at least 6 weeks after the CD4+CD45RBhigh T cell transfers and measured the frequency and numbers of Th1, Th2, Th17, and Treg populations. In concordance with a decrease in capacity of Ets1-SE−/− CD4+ T cells to differentiate into IFNγ-producing Th1 cells, Rag1−/− mice that received wildtype CD4+ T cells had significantly higher total numbers of CD4+ T cells in the cLP and spleen as compared to Rag1−/− mice that received Ets1-SE−/− CD4+ T cells (Figure 3F–G). Moreover, numbers of Granzyme B- and IFNγ-producing CD4+ T cells were lower in Ets1-SE−/− injected Rag1−/− animals as compared to wildtype (Figure 3F–H), while numbers of Th2 or Th17 remained comparable (Figure 3F). Of note, our analysis revealed that there were no significant differences in total FoxP3+ or T-bet+ FoxP3+ CD4+ T cells in the spleen or the cLP of Rag1−/− mice injected with wildtype or Ets1-SE−/− cells (Figure S2B), suggesting that the decreased frequencies of CD4+ Th1 cells as a result of Ets1-SE deficiency were not likely caused by significant alterations in the Treg population. Hence, Ets1-SE in CD4+ T cells is specifically required for Th1 differentiation in vivo in the context of a Th1-induced colitis model.
Ets1-SE deletion induced type 2 immune responses in vivo
IFNγ production by Th1 cells is a critical mechanism that dampens Th2 responses31–33. Since several SNPs associated with allergic diseases are clustered around the Ets1-SE element, we postulated that compromised Th1 differentiation in Ets1-SE−/− mice can lead to enhanced allergic responses in vivo. Hence, we challenged wildtype and Ets1-SE−/− mice with House Dust Mite (HDM) extracts for 5 consecutive days after an initial exposure and quantified immune cell infiltration and type 2 cytokine production in the lungs 17 days after the initial exposure (Figure 3I). We found a significantly higher number of infiltrating CD4+ and CD8+ T cells in the lungs of Ets1-SE−/− animals compared to wildtype counterparts (Figure 3J). Notably, we observed dramatically increased numbers of eosinophils and Th2 cells in HDM-challenged Ets1-SE−/− mice, a key signature of over-active type 2 responses (Figure 3J). Moreover, Ets1-SE−/− mice also showed a significantly higher number of IL-5+ producing Th2 cells during HDM-induced allergic airway inflammatory responses (Figure 3K). We did not observe any major differences in the frequencies or numbers of FoxP3+ CD4+ T cells, T-bet+ FoxP3+ CD4+ T cells, or IFNγ-producing Th1 cells (Figure S2C–D). These results suggest that in the context of HDM-induced allergic airway inflammatory responses, Ets1-SE−/− CD4+ T cells might have an increased intrinsic capacity to differentiate into Th2 cells. However, we cannot fully rule out the possibility that the Ets1-SE has an important role in other yet-to-be-identified immune cells that are critical for the expansion and maintenance of Th2 cells during allergic airway inflammatory responses.
Transcriptional outputs of Th1 cells depend on Ets1-SE
Considering the strong in vivo phenotype in the absence of Ets1-SE, we next measured changes in transcriptional outputs of CD4+ T helper cells using RNA-seq. At the bulk level, Ets1 expression was reduced by ~27% in Ets1-SE−/− Th1 cells but Fli1 expression remained intact (Figure 4A). Moreover, the expression of 33 genes was reduced while the expression of 51 genes increased in Ets1-SE−/− Th1 cells (Figure 4B, Table S2). Strikingly, Ets1-SE deletion had a specific effect on the Th1 gene expression program, with Th1 signature genes, such as Ifng, selectively and significantly demonstrating reduced expression in Ets1-SE−/− Th1 cells compared with wildtype counterparts (Figure 4B) while Th2 signature genes were selectively upregulated in Ets1-SE−/− cells polarized under Th1 condition (Figure S3A). Unlike the Th1-specific effect of Ets1-SE, deletion of this regulatory element did not change the transcriptional landscape of Th2 cells (Figure S3B). Of note, the effect of Ets1-SE deletion on the expression level of Ets1 and the number of differentially expressed genes was more pronounced in naïve CD4+ T cells (Figures. 4A and S3C–D). The Ets1-SE-deregulated genes in naïve T cells were enriched with interferon-associated gene ontology, for example Ifit3, implying the role of Ets1-SE in controlling the baseline expression of interferon genes (Figure S3F). Notably, Ets1-SE did not control the expression of cytokine receptors such as Il12ra, Il4ra, or Il5ra in naïve T cells (Table S3). Together, the partial reduction in Ets1 expression may cause a significant and specific decrease in the Th1-associated gene expression program.
Figure 4. Reorganization of the multi-enhancer hub in the absence of the Ets1-SE element.
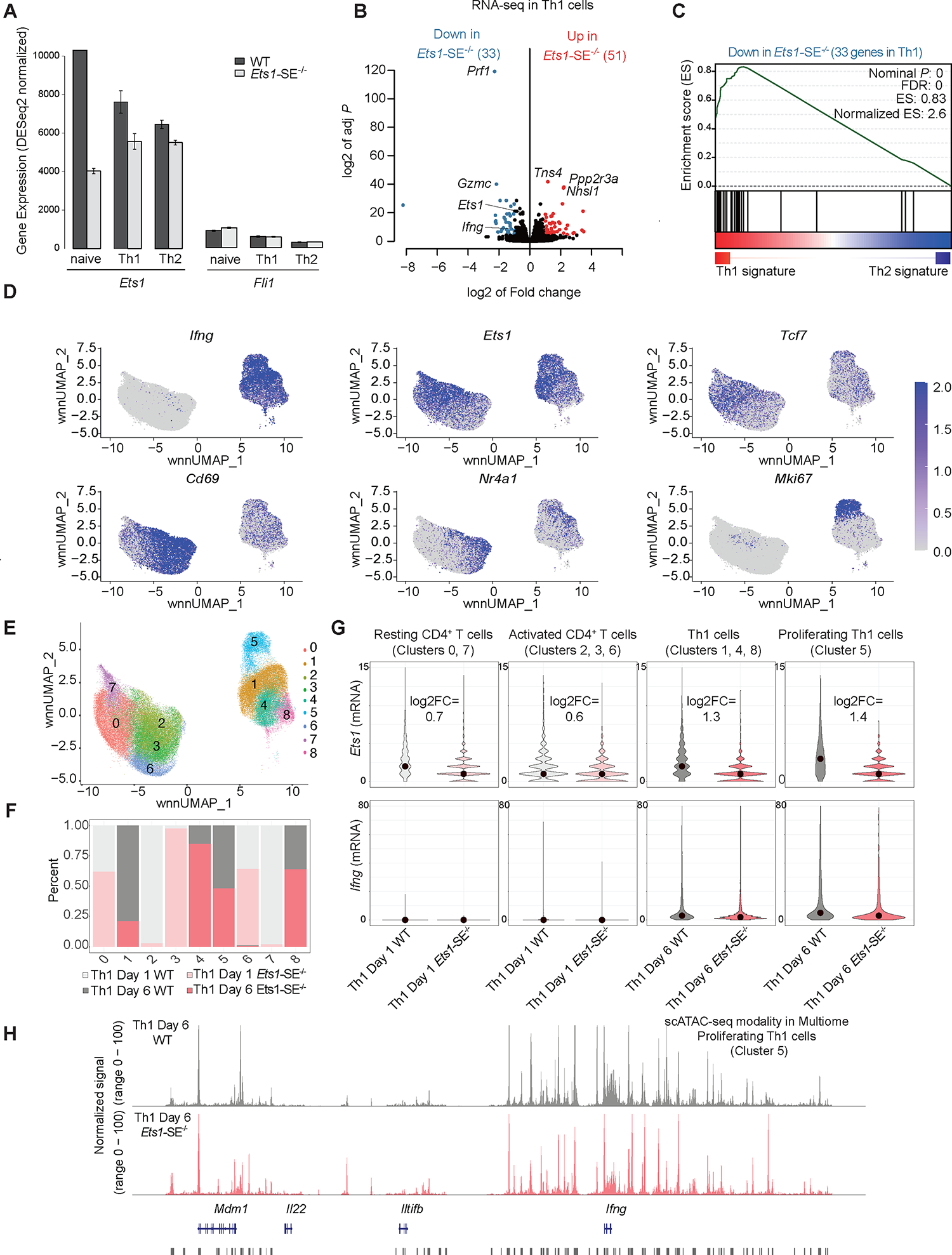
A, Barplot demonstrates normalized mRNA levels of Ets1 and Fli1 using bulk RNA-seq experiments in wildtype and Ets1-SE−/− naïve CD4+ and in vitro polarized Th1 and Th2 cells performed in three replicates.
B, Volcano plot demonstrates differential expression analysis of bulk RNA-seq experiments in wildtype and Ets1-SE−/− in vitro Th1 polarized cells studied at day 6. Three replicates were used to perform DESeq2 analysis and |log2FC|>1, adjusted P <0.05 were used to determine differentially expressed genes.
C, Pre-ranked Gene Set Enrichment Analysis (GSEA) depicts the enrichment of gene-set for downregulated genes in Ets1-SE−/− compared with wildtype Th1 cells. The pre-ranked genes were defined based on DESeq2 analysis of wildtype Th1 and Th2 cells.
D,E, Weighted nearest neighbor UMAP (wnnUMAP) projection, which uses both gene expression and chromatin accessibility measurements for dimensionality reduction and clustering of single cell multiomics analysis on wildtype and Ets1-SE−/− day 1 and day 6 Th1 cells using technical replicates, showing mRNA level of Ifng, Ets1, Tcf7, Cd69, Nr4a1, and Mki67 in each cell. e, cluster numbers across wnnUMAP.
F, Barplots showing the composition of each of the 9 clusters including proportion and contribution from each of the four conditions: wildtype and Ets1-SE−/− Th1 after day 1 and day 6 of in vitro cluster. Biological replicates were performed and cells were pooled.
G, Violin plots showing the expression levels of Ets1 and Ifng across individual wildtype and Ets1-SE−/− cells within clusters annotated as resting CD4+ T cells (clusters 0,7), activated CD4+ T cells (clusters 2, 3, 6), Th1 cells (clusters 1, 4, 8) and proliferating Th1 cells (cluster 5).
H, Representative pseudo-bulk ATAC-seq tracks from scATAC-seq modality in multiome of proliferating Th1 cells (cluster 5), showing comparable chromatin accessibility between wildtype and Ets1-SE−/− Th1 cells.
The chromatin accessibility landscape of Th1 cells does not depend on the Ets1 level
We next examined the chromatin accessibility landscape of Th1 cells using bulk ATAC-seq and found only a few genomic elements to be differentially accessible between wildtype and Ets1-SE−/− Th1 cells (Figure S4A). Similarly, a small number of genomic regions demonstrated significant alterations in histone acetylation as measured by H3K27ac CUT&RUN44 in Ets1-SE−/− compared to wildtype Th1 cells (Figure S4B). Hence, we did not find strong evidence for the active enhancer landscape of Th1 cells to be dependent on Ets1-SE using these bulk measurements. Examining binding of transcription factors within accessible chromatin regions of Ets1-SE suggested STAT1, STAT4, STAT3, STAT5a, and T-bet as potential upstream regulators of this locus in response to changes in the cytokine environment (Figure S4C). In line with transcriptional profiling, the chromatin accessibility landscapes of wildtype and Ets1-SE−/− Th2 cells were virtually indistinguishable (Figure S4D). Notably, the Ets1-SE deletion led to major changes on the chromatin accessibility landscape of naïve T cells (Figure S4E–I). We also found that the Ets1-SE-dependent open chromatin regions in naïve T cells had a low level of accessibility in differentiated Th1 cells, suggesting a distinct effect of Ets1-SE on the naïve state (Figure S4G–I). Of note, the noncoding RNA Gm27162 demonstrated its highest expression and strongest chromatin accessibility in naïve and Th1 cells (Figure S4J–K). Altogether, the analysis of bulk data suggests that the Th1-specific enhancer landscape was largely independent of the Ets1-SE element.
Single-cell multiomic profiling reveals that Ets1-SE deletion impairs high Ets1-expressing cells
Although bulk RNA-seq measurements implied a link between Ets1-SE, the Ets1 expression level, and transcriptional outputs of Th1 cells, it remained unclear whether the disruption of Ets1-SE can (a) reduce the Ets1 expression level on a per-cell basis and thus leading to a uniform reduction across individual cells or (b) reduce the frequency of high Ets1 expressing cells. We next used single-cell multiomics profiling45 and generated joint single-cell RNA- and single-cell ATAC-seq measurements in CD4+ T cells from wildtype and Ets1-SE−/− mice at two different dynamic time points (days 1 and 6) after Th1 polarization in vitro. After quality control filtering (Figure S5A), we obtained a total of ~80,000 cells with comparable contributions from wildtype and Ets1-SE−/− mice using two technical replicates. We detected 9 distinct clusters using the weighted nearest neighbor analysis45, which incorporated both gene expression and chromatin accessibility measurements for the dimensionality reduction and clustering analysis (Figures 4D–E and S5B). Relying on marker genes, we assigned clusters to four different CD4+ T cell states: (1) the resting state (Ccr7 and Tcf7 expressing cells in clusters 0, 7), (2) the activated state (Nrf4a1 and Cd69 expressing cells in clusters 2, 3, 6), (3) the Th1 state (Ifng and Tbx21 expressing cells in clusters 1, 4, 8) and (4) the proliferating Th1 state (Ifng and Mki67 expressing cells in cluster 5) (Figures 4D–E and S5B). Cells from both wildtype and Ets1-SE−/− T cells comprised these clusters with different frequencies (Figure 4F).
We next assessed the extent of variation in the Ets1 mRNA level across individual cells. Focusing first on wildtype CD4+ T cells revealed that the frequency of high Ets1 expressing cells was the largest in cells associated with the resting state and the Ifng producing Th1 state (Figure 4G, gray violin plots). High Ets1 expressing cells were not frequently detected in wildtype activated T cells representing low Ifng expression cells 1 day after Th1 polarization. This reduction in frequencies of high Ets1 expressing cells in the activated state is in line with previous studies suggesting the downregulation of Ets1 level by T cell activation46. Strikingly, the dynamic increase of the Ets1 level during Th1 polarization was impaired in Ets1-SE−/− cells (Figure 4G, red/pink violin plots). Collectively, the distributions of Ets1 expression were comparable in individual cells from Ets1-SE−/− mice grouped into all four states (Figure 4G, red/pink violin plots). Quantitatively, the largest difference for Ets1 expression between wildtype and Ets1-SE−/− cells occurred in proliferating Th1 cells, suggesting ~60% reduction in the Ets1 level after Ets1-SE deletion. The simultaneous measurement of chromatin accessibility in the multiome assay across these clusters corroborated findings based on bulk ATAC-seq (Figure 4H). Together, our single-cell multiome profiling measurements suggest that deletion of Ets1-SE within the multi-enhancer hub impairs the ability of CD4+ T cells to express high levels of Ets1 in response to Th1 stimulation.
ETS1 dosage controls Th1 differentiation
We next assessed whether the partial reduction of Ets1 was responsible for the compromised Th1 differentiation using two complementary dosage experiments. First, we used Ets1 heterozygous mice (Ets1fl/+Cd4cre) that harbor a 50% reduction of Ets1 transcript and protein levels in CD4+ T cells. Second, we overexpressed the ETS1 protein in Ets1-SE−/− Th1 cells that carry a partial reduction of Ets1 as compared to wildtype cells. Thus, using these Ets1 dosage experiments, if the Ets1 expression level controls Th1 differentiation through the Ets1-SE element (in cis), we expect that cells carrying one copy of the Ets1 gene, but an intact Ets1-SE region, would also fail to differentiate effectively towards the Th1 program. Moreover, we expect that the reconstitution of ETS1 in Ets1-SE−/− Th1 cells to the wildtype level can rescue optimal Th1 differentiation. We observed a significant loss of IFNγ expressing cells upon in vitro Th1 polarization of naïve CD4+ cells from Ets1fl/+Cd4cre mice when compared with wildtype counterparts (Figure 5A). Furthermore, the reconstitution of ETS1 expression into Ets1-SE−/− cells using retroviral transduction during in vitro Th1 polarization significantly increased the frequency of IFNγ expressing cells compared to empty-vector transduced cells (Figure 5B–C). Transcriptional profiling of Ets1-SE−/− Th1 cells with the ectopic expression of ETS1 also confirmed selective rescue of Th1-signature genes (Figures 5D and S5C–D). Together, Ets1-SE acts in cis, tuning a precise expression level of Ets1 which is required for optimal Th1 differentiation.
Figure 5: ETS1 dosage controls Th1 differentiation.
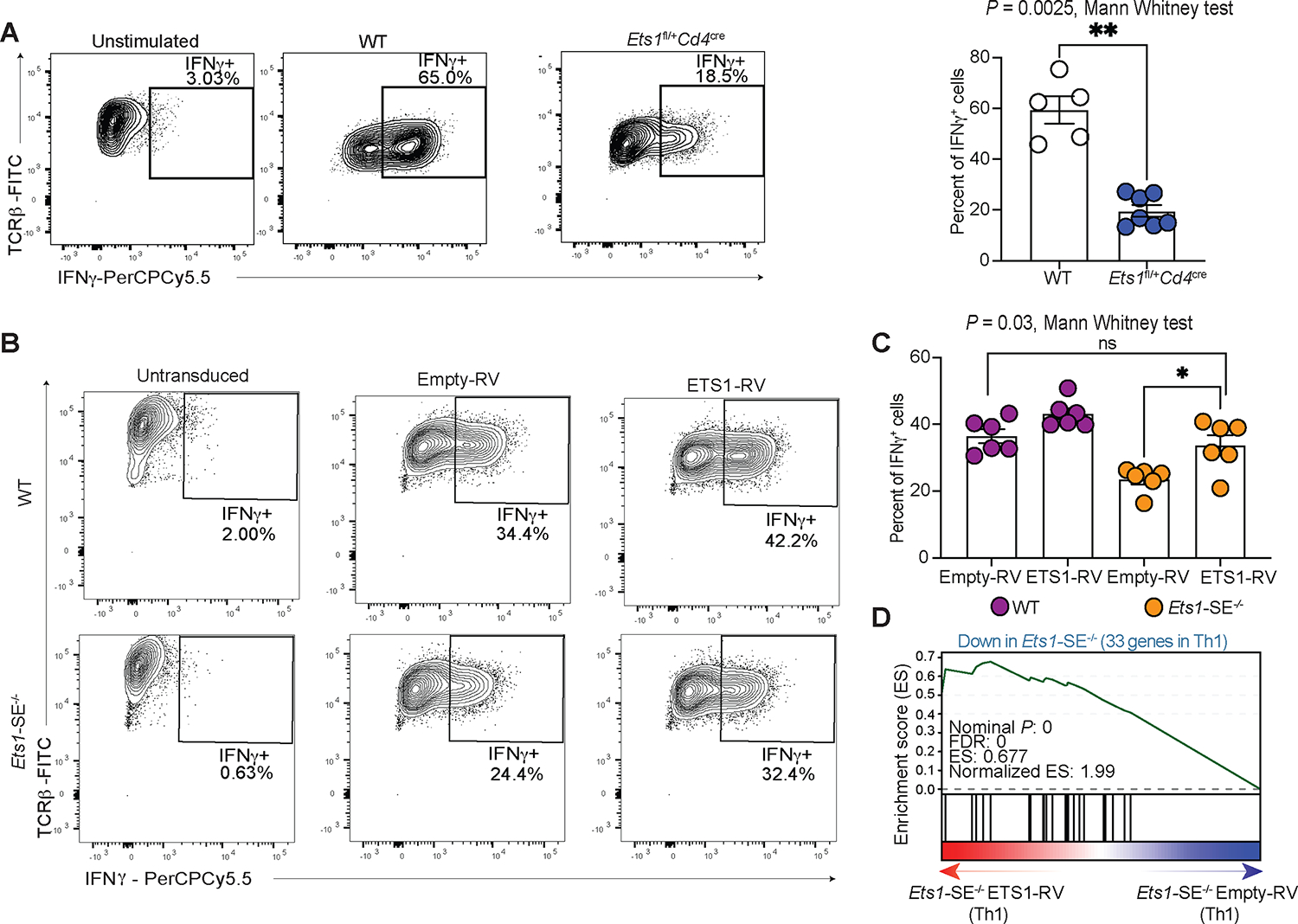
A, Flow cytometry results demonstrating frequency of IFNγ expressing cells under unstimulated (left) and in vitro Th1 polarization for 6 days in wildtype (middle) and Ets1 heterozygous mice (Ets1fl/+Cd4cre) (right). The right panel shows bar plot of average results from two independent experiments. Each dot represents an individual mouse. Statistical significance was evaluated using non-parametric Mann-Whitney test. ** P = 0.0025.
B, Flow cytometry results demonstrating frequency of IFNγ expressing cells when naïve CD4+ T cells were retrovirally transduced with empty vector (Empty-RV) or ETS1 expressing vector (ETS1-RV) and were polarized under in vitro Th1-differentiating conditions for 6 days. The panel shows representative contour plots, C, panel shows bar plot of average results from two independent experiments. Each dot represents an individual mouse. Significance was tested using non-parametric Mann-Whitney test. * P = 0.03.
D, Pre-ranked Gene Set Enrichment Analysis (GSEA) depicts the enrichment of the downregulated genes in Ets1-SE−/− compared with wildtype Th1 cells as gene-set. The pre-ranked genes based on DESeq2 analysis of Ets1-SE−/− cells transduced with empty-vector and ETS1-expressing vector. Three technical replicates were used for DESeq2 analysis.
Ets1 level controls the 3D genome topology of Th1 cells
To assess whether Ets1-SE is required for the spatial localization of enhancers to Th1 signature genes, we created unbiased maps of long-range interactions in CD4+ T helper cells using ultra-deep Hi-C47,48. Focusing on the genome organization of the Ets1 locus in wildtype T cells, we found that the Ets1-SE demonstrated the strongest interaction with the Ets1 gene locus in naïve and CD4+ Th1 cells, a trend which is consistent with the expression level of Ets1 in these T cell subsets (Figure S5E–F). Comparing wildtype and Ets1-SE−/− Th1 cells revealed that compartments and topologically associating domains (TADs) were independent of Ets1-SE (Figure S5G–H). However, we observed extensive weakening of long-range interactions in Ets1-SE−/− as compared to wildtype Th1 cells (Figure S6A). Genes associated with Th1 biology including Ifng and Il1r-Il18r were located within loop domains with reduced interaction in Ets1-SE−/− compared with wildtype cells, suggesting the specific effect of Ets1-SE on Th1-specific genome topology (Figure S6B–C). Consistent with gene expression and chromatin accessibility measurements, the effect of the Ets1-SE deletion on long-range interactions was less pronounced in Th2 cells compared with the effect of this regulatory element on Th1 cells (Figure S6D).
To dissect whether changes in genome topology in Ets1-SE−/− Th1 cells were mediated by a reduction in the level of Ets1 expression, we also measured long-range interactions using Hi-C in Ets1 heterozygous (Ets1fl/+Cd4cre) Th1 cells, where the Ets1-SE sequence is intact. Remarkably, we detected an extensive similarity between Ets1-SE−/− and Ets1fl/+Cd4cre Th1 cells: 4,726 loops including interactions at the Ets1 locus were weaker in both Ets1-SE−/− and Ets1fl/+Cd4cre Th1 cells compared with wildtype counterparts (Figure 6A–C). The weaker interactions in Ets1-SE−/− Th1 cells at Ets1 locus were consistent with the Oligopaint results in DP T cells, demonstrating an increased spatial distance among regulatory elements in Ets1-SE−/− cells (Figure 1F–H). Thus, a precise level of Ets1 expression controlled by the Ets1-SE element is required for the Th1-associated genome topology.
Figure 6: Ets1 level controls the 3D genome topology of Th1 cells in a CTCF-dependent manner.
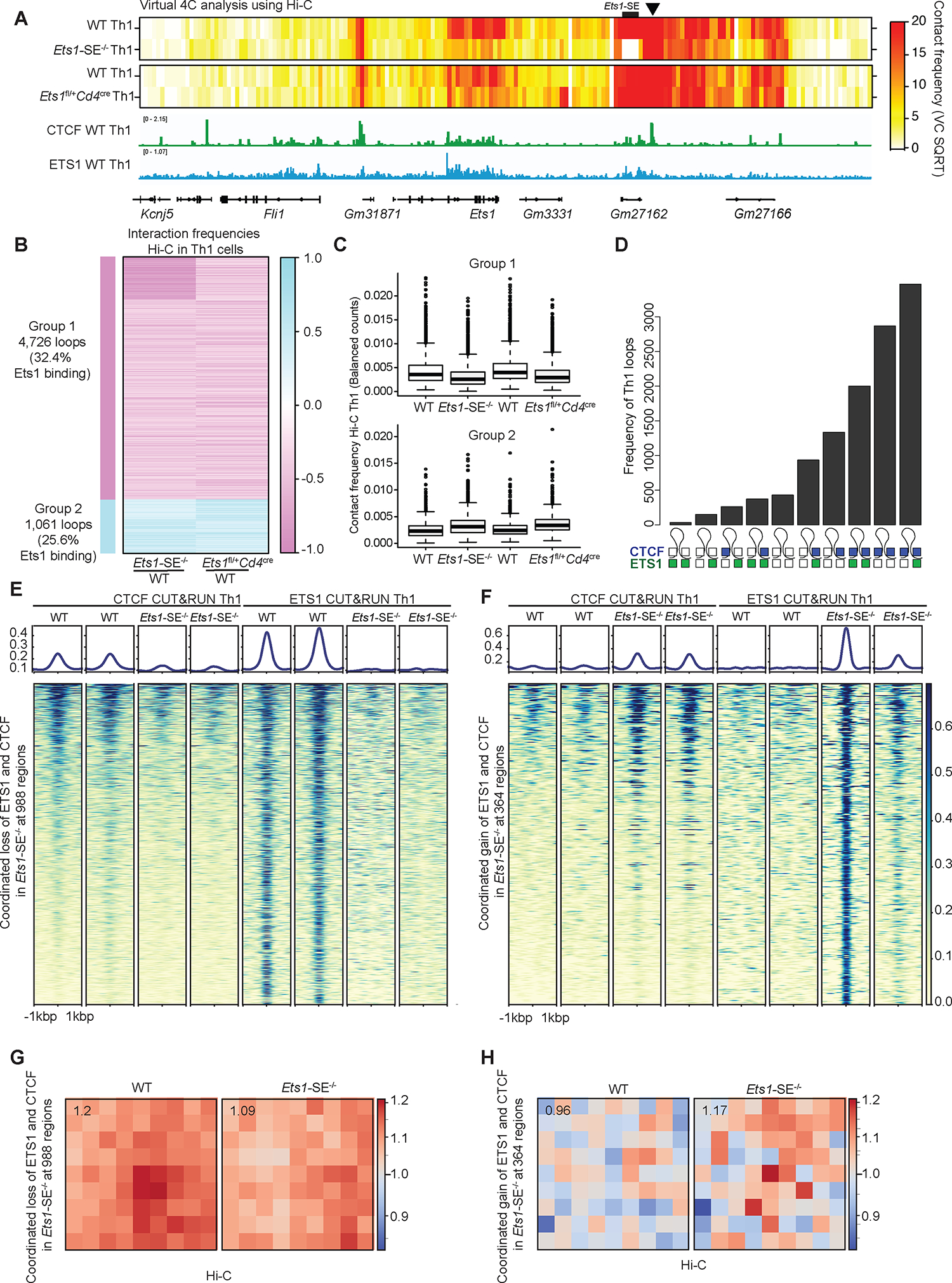
A, Heatmap of contact frequencies from virtual 4C analysis of Hi-C data generated from wildtype, Ets1-SE−/− and Ets1fl/+Cd4cre Th1 cells, using the immediate downstream locus of Ets1-SE as the 4C-anchor (chr9:32,940,001–32,945,000) (marked with black arrow). The genome browser view of the corresponding locus is shown along with CTCF and ETS1 binding tracks generated from CUT&RUN on wildtype Th1 cells.
B, Heatmap showing log2 fold change in interaction frequencies from Hi-C in Ets1-SE−/− and Ets1fl/+Cd4cre Th1 cells, compared to their respective matched wildtype controls. Group 1 which includes 4,726 loops demonstrates weaker interactions in in Ets1-SE−/− and Ets1fl/+Cd4cre Th1 cells, compared to their respective matched wildtype controls. Group 2 which includes 1,061 loops demonstrates stronger interactions in in Ets1-SE−/− and Ets1fl/+Cd4cre Th1 cells, compared to their respective matched wildtype controls. The percentage of loops with ETS1 binding based on CUT&RUN in wildtype Th1 cells is also provided. Each row represents an individual interaction or a loop.
C, Boxplot showing average interaction frequencies from Hi-C in Ets1-SE−/− and Ets1fl/+Cd4cre Th1 cells, and their respective matched wildtype controls, using group 1 and group 2 loops.
D, UpSet plot demonstrates the number of Th1-associated loops with different binding patterns of CTCF and ETS1 across two loop anchors. A filled square represents bound, and an open square represents no binding. Blue and green squares represent CTCF and ETS1 binding, respectively. Bedtools intersect was used to define overlapping peaks with loop anchors detected in Th1 cells.
E,F, Heatmap demonstrates CTCF and ETS1 occupancy levels at genome regions where both proteins are lost (f) and gained (g) in Ets1-SE−/− compared with wildtype Th1 cells. DESeq2 was used to define co-bound regions by ETS1 and CTCF measured by CUT&RUN with differential occupancy between wildtype and Ets1-SE−/− Th1 cells using P<0.05 & |logFC|>0.5.
G,H, Loop pileups of long-range interactions anchored at genomic regions described in (e) and (f) with lost (g) and gained (h) occupancy generated by coolpuppy. Hi-C data in wildtype and Ets1-SE−/− Th1 cells were used. The number in the top left corner represents the average intensity in 3×3 box at the center.
ETS1 and CTCF co-bound sites sensitive to the Ets1 expression level shape the 3D genome topology of Th1 cells
We next assessed whether the partial reduction of Ets1 could modify the genome-wide occupancy of ETS1 protein and if there was any direct evidence for ETS1 binding affecting Th1-associated long-range interactions. We therefore mapped the global binding pattern of ETS1 protein in biological replicates of Th1 cells using CUT&RUN44 (Figure S6E). Overall, we detected more than 27,000 genomic regions bound by ETS1 in wildtype Th1 cells enriched with the canonical ETS recognition motif and found ~33% occupancy at promoter regions, suggesting the high quality of our CUT&RUN measurements (Figure S6E–F, Table S4). Considering the unexpected effect of the Ets1 expression level on the 3D genome organization of Th1 cells (Figure 6A–C), we next measured the genome-wide occupancy of CTCF using CUT&RUN and found that more than 50% of ETS1 binding events co-occurred at CTCF-bound sites in Th1 cells (Figure S6G). Moreover, ETS1 and CTCF co-binding was detected at a significant proportion of Th1-associated loop anchors, suggesting the potential cooperation of ETS1 and CTCF in establishing 3D genome topology of Th1 cells (Figure 6D). Although it has been reported that the complete deletion of Ets1 in CD4+ T cells reduces the expression of CTCF49, the CTCF mRNA level was intact in Ets1-SE−/− Th1 cells (Table S2A). To further understand how the reduced expression of Ets1 weakened long-range interactions in Ets1-SE−/− Th1 cells, we compared ETS1 and CTCF occupancies in wildtype and Ets1-SE−/− Th1 cells. While the majority of ETS1 and CTCF binding events were not sensitive to the Ets1 expression level, 998 genomic regions demonstrated a coordinated loss of both ETS1 and CTCF binding in Ets1-SE−/− compared to wildtype Th1 cells (Figure 6E, Table S5). In addition, 364 genomic regions demonstrated a coordinated gain of both proteins in Ets1-SE−/− compared to wildtype Th1 cells (Figure 6F). The subset of ETS1-CTCF co-occupied regions with a coordinated loss of both proteins in Ets1-SE−/− Th1 cells demonstrated lower ETS1 and CTCF occupancy compared to the majority of unaffected binding events in wildtype Th1 cells, implying the overall sensitivity of these loci to the Ets1 expression level (Figure S6H). Remarkably, the ETS-RUNX cooperative binding motif, which has been characterized as the T cell activation signature of ETS1 binding17,50, was enriched at genomic regions which were sensitive to the Ets1 level (Figure S6I–J). STAT2 and Tbox motifs which are recognition sites for Th1-specific transcription factors T-bet and STAT2 were also enriched at these ETS1-CTCF lost sites, suggesting the potential cooperation of ETS1 with these proteins (Figure S6I–J). Remarkably, genomic regions whose ETS1 and CTCF co-occupancy were sensitive to the Ets1 level demonstrated altered long-range interactions in Ets1-SE−/− Th1 cells (Figure 6G–H). These results indicate that a precise level of ETS1 protein was required for CTCF occupancy and the 3D long-range interactions for Th1 regulatory elements. Altogether, the hyperconnectivity of the Ets1-Fli1 locus controlled the expression level of Ets1 which was dispensable for the active enhancer signature but required for the Th1-specific DNA folding through recruitment of CTCF. This specific deficit in genome folding due to a partial reduction of Ets1 led to compromised Th1 differentiation and allergic responses in vivo.
Discussion
Here, we interrogated the functional relevance of a frequently reported 3D structure referred to as a multi-enhancer hub in the context of T cell differentiation. Our systematic prioritization of 3D genome interactions in mouse T cells ranked the Ets1-Fli1 region as the second most hyperconnected locus after the Bcl11b region with an unusual degree of enhancer connectivity. This multi-enhancer locus was conserved in human T cells and represented a hotspot for SNPs associated with allergic diseases. To better understand the functional relevance of the hyperconnectivity of this genetic hotspot in T cell biology, we generated a new mouse strain and deleted a noncoding regulatory element homologous to the allergy-associated polymorphic region in the human genome. While T cell development remained intact, Th1 differentiation was compromised after the genetic deletion of this regulatory element. Detailed mechanistic investigation demonstrated the link between the hyperconnectivity of the Ets1-Fli1 locus, Ets1 expression level, CTCF recruitment, and long-range interactions required for the Th1 gene expression program. Although it has been shown that the complete ablation of Ets1 can lead to changes in CTCF recruitment49, our study is the first demonstration of the sensitivity of genome organization to the Ets1 expression level as a mechanism for predisposition to immune-mediated diseases. Related to our findings, it has been known that the graded expression of interferon regulatory factor-4 (IRF-4) coordinates isotype switching with plasma cell differentiation51. We speculate that IRF-4 or other transcription factors may follow a similar dose-dependent mechanism and reorganize the genome by interacting with CTCF at specific genome regions.
Since the completion of the Human Genome Project, research on the genetic basis of Mendelian traits has been extremely successful. In most cases, the rare single-gene disorders masquerade a multifactorial trait in their clinical phenotype52, but detailed clinical examination and studying ‘human allelic knockouts’ have guided to one gene responsible for the disease in those particular families. Thus, genetic ablation of an individual gene in rodents became a powerful tool to dissect the molecular processes of such monogenic diseases. Despite the success of genetic strategies in Mendelian traits, human genetics has been less successful in dissecting complex conditions which are in fact more commonly found across populations. Unlike Mendelian traits that can be modeled as gene knockouts, complex traits might be caused by single-nucleotide variants disrupting transcriptional enhancers. However, the link between sequence variation, cellular context, 3D genome folding, and the extent of change in gene expression in most complex diseases remains largely unknown. Here we presented a mouse model, which was inspired by the mathematical analysis of genome organization data, reporting how noncoding elements can control the precise dosage of a key transcription factor through the formation of a multi-enhancer hub. It is worth mentioning that the heterozygous alleles of transcription factors such as HNF1A53 or FoxA254 can impact target genes, leading to developmental defects. Yet, our study highlights dosage control through regulatory elements of the transcription factor gene, not the coding region of the transcription factor gene itself. Considering that individuals carrying risk factors for genetic predispositions to common diseases are far from gene-knockout mouse models, we reason that comprehensive strategies following the integrative approaches used in this study can shed light on molecular mechanisms through which single-nucleotide variants can affect the gene expression programs sometimes in subtle ways which can lead to substantial clinical phenotypes.
Limitation of the Study
As of today, we are limited in Oligopaint experiments to select three genomic loci, but chromatin tracing experiments55 for the entire multi-enhancer hub could allow us to better understand the dynamics of multi-enhancer interactions at the single-cell resolution. Combining RNA FISH and chromatin tracing such as optical reconstruction of chromatin architecture (ORCA)55 can enable the investigation of gene dosage and chromatin conformation across individual alleles.
STAR★ Methods
Resource availability
Lead contact
Golnaz Vahedi (vahedi@pennmedicine.upenn.edu)
Materials availability
This study did not generate new unique reagents.
Data and code availability
The accession number for the HiC, CUT&RUN, ATAC-seq and RNA-seq reported in this study is NCBI GEO: GSE211178.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice:
The Ets1-SE−/− mice were generated using the CRISPR/Cas9 system as previously described88. The sgRNA sequences 5’ GGACGTTGTGCACCTAGGATTGG and 3’ ATAAACGTCAATAATGGTATAGG were used for the generation of knockout mice and allowed the deletion of the following DNA regions referred to as Ets1-SE: chr9:32,928,966–32,904,069 (mm10 genome). Ets1-SE−/− mice were backcrossed onto the C57BL/6 background for at least three generations to control for potential off-target effects. Rag1−/− mice (B6.129S7-Rag1tm1Mom/J; Strain #:002216) were purchased from the Jackson Laboratory. Ets1fl/fl Cd4cre mice were generated by Dr. Barbara Kee’s laboratory at the University of Chicago by insertion of loxP sites flanking exons 7 and 8 of the Ets1 locus. Ets1fl/+ mice were then crossed to mice expressing the Cre recombinase under the Cd4 promoter (Cd4cre) to generate Cd4cre Ets1fl/+ mice. Spleens and lymph nodes (LNs) from these mice were used for in vitro polarization assays. All mice were bred and maintained under pathogen-free conditions at an American Association for the Accreditation of Laboratory Animal Care accredited animal facility at the University of Pennsylvania. Mice were housed in accordance with the procedures outlined in the Guide for the Care and Use of Laboratory Animals under an animal study proposal approved by an institutional Animal Care and Use Committee. All experiments were performed using 6- to 12-weeks old age and sex matched mice and using both males and females.
Tissue and cell Preparation:
Different organs were extracted from the mice for T cell phenotyping study, including thymus, lungs, liver, spleen, lymph nodes and bone marrow. All red blood cells were lysed using ACK lysis buffer (GIBCO). Single-cell suspensions from spleen, thymus and lymph nodes were isolated by physical dissociation of tissues and filtered through a 70μm cell strainer (Falcon) in RPMI-1640 media (Invitrogen) containing 1% fetal bovine serum (FBS) (Fisher Scientific). For bone marrow cell isolation, femurs were collected and crushed using a mortar and pestle. Lungs were isolated, minced with scissors, and digested in PBS containing FBS (2%), Collagenase D (1mg/ml), DNase I (0.2mg/ml) for 35 minutes at 37°C with shaking at 200 RPM. The digested lungs were then passed through a 70μm cell strainer. Mice were perfused with 10ml PBS and then transferred to DMEM on ice. The liver was then removed from media and mechanically dissociated using a tissue grinder, then filtered through a 100μm cell strainer. To pellet hepatocytes, the digested livers were centrifuged at 20g for 5min at 4°C. Leukocytes were then purified over an 80/40% Percoll gradient.
CD4+ T cell isolation and polarization.
Splenocytes were isolated from mouse spleen and lymph nodes. Spleen cells were subjected to ACK lysis buffer (Gibco, Invitrogen) to remove red blood cells. Naïve CD4+ T cells were enriched using negative selection beads (STEMCELL, Cat# 19765) following manufacturer’s recommendations. Purity of naïve cells were assessed using flow cytometry and found to be >=90% pure. Cells were cultured in RPMI 1640 medium (Invitrogen), supplemented with 10% fetal bovine serum (Fisher Scientific), 1mM sodium pyruvate (Gibco), 1% non-essential amino acids (Gibco), 1X GlutaMAX (Gibco), 1% HEPES (Gibco), 1% penicillin-streptomycin and 0.1% 2-Mercaptoethanol (Gibco). For in vitro polarization, flat bottom 96-well plates were coated with 2 μg/mL of anti-CD3 (Clone: 145–2C11, BioLegend, Cat#100302) in PBS overnight at 4°C or 4 hrs at 37°C. For Th1 polarization, 0.2×106 naïve CD4+ T cells were cultured in presence of 10 ng/mL of recombinant IL-12 (BioLegend, Cat#577002), 1 ng/mL of recombinant IL-2 (BioLegend, Cat#575402) and 1 μg /mL of soluble anti-CD28 (Clone: 37.51, BD Biosciences, Cat# 553294) for 6 days at 37°C. Th2 polarization was induced by cultivating 0.2×106 naïve CD4+ T cells in complete RPMI supplemented with 10 μg /mL of anti-IFNγ (Clone: XMG1.2, BioXCell, Cat#BE0055), 50 ng/mL of recombinant IL-4 (BioLegend, Cat# 574302) and 1 μg/mL of soluble anti-CD28 for 6 days at 37°C. For Th17 polarization, 0.2×106 naïve CD4+ T cells were cultured in complete RPMI containing 10 μg/mL of anti-IFNγ, 10 μg/mL of anti-IL-4 (Clone: 11B11, BioXCell, Cat# BE0045), 2 ng/mL of recombinant TGFβ (BioLegend, Cat#763102), and 20 ng/mL of recombinant IL-6 (BioLegend, Cat# 575702) and 1 μg/mL of soluble anti-CD28 for 6 days at 37°C. For induced Treg (iTreg) polarization, 0.2×106 naïve CD4+ T cells were cultured in complete RPMI containing 10 μg/mL of anti-IFNγ (Clone: XMG1.2, BioXCell, Cat#BE0055), 10 μg/mL of anti-IL-4 (Clone: 11B11, BioXCell, Cat# BE0045), 3 ng/mL of recombinant TGFβ (BioLegend, Cat#763102), and 5 ng/mL of recombinant IL-2 (BioLegend, Cat# 575702) and 2 μg/mL of soluble anti-CD28 for 6 days at 37°C. At day 3 post stimulation, half of the medium was removed and replaced by fresh media containing 2X cytokines concentration of Th1, Th2 or Th17-related polarizing medium.
ETS-1 retroviral transduction experiments:
cDNA encoding the short isoform of ETS-1 (p54) was cloned into pENTR vector and then into the destination vector MSCV-IRES-Thy1.1 DEST (Addgene: 17442) using Gateway cloning strategy (Gateway Clonase II, Invitrogen). To generate retroviral particles for Ets1-overexpression, 293T cells were purchased from ATCC. Briefly, HEK-293T cells were maintained in high glucose DMEM medium 1X with L-Glutamine (Invitrogen), supplemented with 100 U/mL penicillin and 100 mg/mL streptomycin (Gibco) with 10% FBS. Cells were maintained at low passage number (< 12), at 70–80% confluency, and were grown at 37°C and 5% CO2. Retroviral vectors were packaged in HEK 293T cells. Briefly, 4×106 HEK 293T cells were plated in 5 ml media in 10 cm dishes on the day prior to transfection. During transfection, 15 μg of MSCV-Thy1.1-Ets1 plasmid was co-transfected with packaging plasmid, 15 μg of pCL-Eco, using Lipofectamine 3000 (Invitrogen). The MSCV-Thy1.1-EV (Empty vector) plasmid was also similarly transfected. The cells were returned to the incubator for 6 hours. Subsequently, the medium was changed to fresh media. Virions were collected 24 and 48 hrs after transfection, snap-frozen, and stored at −80°C for future use.
CD4+ T cell transduction:
Primary naïve CD4+ T cells were transduced by addition of virions to culture media supplemented with polybrene (Sigma-Aldrich, cat# H9268) at 4 μg/mL final concentration, followed by centrifugation at 32°C for 1 hour at 3000 rpm. Cells were returned to incubator at 37°C for at least 4 hrs. Subsequently, the medium was changed to fresh culture media supplemented with Th1 polarizing cytokines. Transduction efficiency was checked next day and frequency of polarizing cells was checked on day 6 after transduction.
Antibodies, Flow cytometry, and cell sorting.
All antibodies were diluted in FACS buffer (PBS + 2% FBS + 2mM EDTA) and used to stain single cell suspensions for 30 minutes at 4°C. First, dead cells were stained by incubation of cell suspension in Viability Dye (eFluor780, eFluor506, or Aqua) diluted in PBS for 10mins at 4°C. Then after a wash with PBS, cells were staining with surface antibodies diluted in FACS buffer (PBS + 2% FBS + 2mM EDTA) and washed with FACS buffer. Cells were then either fixed for intracellular staining using the Foxp3 staining buffer (eBioscience) or were fixed with 2% PFA. For intracellular staining, fixed cells were washed with permwash buffer (eBioscience) and incubated with intracellular antibodies diluted in permwash buffer for 30 mins or left overnight. Cells were washed with permwash buffer and resuspended in FACS buffer with the addition of 123count eBeads (ThermoFischer Scientific, ref:01–1234-42) following manufacturer’s recommendations for cell counting. For cell sorting, cells were stained at a concentration of 100×106 cells/ml in FACS buffer (PBS + 2% FBS + 2mM EDTA) for 30 mins at 4°C then filtered through a 70μm filter prior acquisition and sort using either a 100μm or 70μm nozzle on a BD FACS Aria II SORP under aseptic conditions. Cells were sorted in FACS buffer containing 10% of FBS.
Flow Cytometry on in vitro polarized cells
For flow cytometry, in vitro polarized cells were stimulated with PMA, Ionomycin and Golgi Plug for 4 hrs at 37C. Subsequently, they were stained with a viability dye (1:500 L/D Aqua), and extracellular stains with anti-mouse CD4-APC (1:400, clone RM4–5, BioLegend Cat#100516), anti-mouse TCRb chain-FITC (1:400, clone H57–597, BD Biosciences Cat#553170). Cell were then fixed using Foxp3/ Transcription factor Staining buffer Set (eBioscience, Thermo Cat# 00–5523-00) followed by intra-cellular stains with anti-mouse IFNγ-PerCP/Cyanine5.5 (1:200, clone XMG1.2, BioLegend Cat#505821), T-bet monoclonal antibody-PE-Cyanine 7 (1:200, clone 4B10, eBioscience, Cat# 25–5825-80), anti-mouse IL-13-PE eFluor 610 (clone eBio13A, Fisher Cat# 61–7133-80), anti-GATA3 BV711 (BD Biosciences Cat# 565449), anti-mouse IL-17 PE (clone TC11–18H10.1, BioLegend, Cat# 506903), anti-mouse RORgt BV421 (BD Biosciences Cat#562894). Cells were washed and resuspended in PBS for flow cytometry. Data were collected on an LSR II running DIVA software (BD Biosciences) and were analysed using FlowJo software v10.8.1.
In vivo House Dust Mite (HDM) extract exposure model:
HDM extracts (Dermatophagoides pteronyssinus extracts; Greer Laboratories lot #361863, 385930, 387032) were used to induce allergic airway inflammation, following the protocol described previously89. Briefly, mice were sensitized intranasally with 20 μg HDM extracts on day 0 and subsequently challenged intranasally with 10 μg/mouse per day on days 7–13. Three days after the last challenge, mice were anesthetized and used either for immune cell quantification in the lung parenchyma or ex vivo restimulation. For lung parenchyma experiments, lungs were isolated, minced with scissors, and digested in PBS containing FBS (2%), Collagenase D (1mg/ml), DNase I (0.2mg/ml) for 35 minutes at 37°C with shaking at 200 RPM. The digested lungs were then passed through a 70μm cell strainer. Cells were resuspended in PBS then split in order to perform lung parenchyma cell infiltration or ex vivo restimulation. For ex vivo restimulation, cells were plated into round-bottom 96 well plates and stimulated in complete RPMI media containing PMA (Sigma; final concentration 100ng/mL) and Ionomycin (Sigma; final concentration 10ng/mL) and GolgiPlug (1X - BD Bioscience). Cytokines and transcription factor expression were measured by intracellular staining using the “Foxp3 staining buffer” (Ebioscience).
Induction and Evaluation of colitis:
CD4+ T cells were enriched from spleen and lymph node cell suspensions using a cocktail of biotinylated antibodies containing anti-CD8 (53–6.7), anti-CD19 (6D5), anti-BB20 (RA3–6B2), anti-Gr1 (RB6–8C5), anti-TCRγδ (GL3), anti-CD11c (N418), anti-I-A/I-E (M5), anti-CD25 (PC61) all purchased from Biolegend. Splenocytes were incubated for 30mins at 4°C with the previously mentioned mAbs, washed with PBS then incubated with streptavidin magnetic beads for 20mins at 4°C. Negative fraction containing CD4 T cell was then stained with Viability Dye-APC-eF780, anti-TCRβ-APC (H57–597), anti-CD4-FITC (GK1.5), anti-CD8-PerCP-Cy5.5 (53–6.7) and anti-CD45RB-PE (C363–16A) mAbs. Live, TCRβ+, CD4+, CD45RBhigh colitogenic T cells were then separated by fluorescent cell sorting using a BD FACS Aria II SORP with a purity over 90%. Colitis was induced in 9- to 11-week-old Rag1−/− mice by retro-orbital injection of 1×106 naive CD4+CD45RBhigh from WT or Ets1-SE−/− colitogenic CD4+ T cells in 100 μl of PBS. Weight loss of Ets1-SE−/− or WT-injected Rag1−/− was recorded every week for 6 to 7 weeks prior to mice euthanasia. Cell infiltration characterization, restimulation and macroscopic scoring was performed on week 6 to 7.
Macroscopic analysis:
Macroscopic colonic tissue damage was evaluated by the Comparative Pathology Core (CPC) at the Veterinary school of the University of Pennsylvania using a scale described in Figure 3E. Colonic tissue specimens were excised 2 cm proximal to the cecum and immediately transferred into 10% formaldehyde to be embedded in paraffin. Colonic sections were then stained with hematoxylin and eosin by the Comparative Pathology Core (CPC) at the Veterinary school of the University of Pennsylvania. Each slide was scored (blind readings) by a single pathologist. Colon length was measured using a ruler right after mice euthanasia (Figure S2A)
Colon Lamina propria (cLP) harvest digestion and cell infiltration phenotyping.
Colons were excised and fat was removed using forceps, unrolled, and measured using a ruler. Colons were flushed with cold PBS to remove feces then were opened lengthwise and tissues were shaken in a petri dish containing cold PBS to remove any remaining feces. Colons were washed 2 times 10mins at 180 RPM and 37°C in PBS containing FBS (2%), HEPES (20mM) and EDTA (10mM). Colons were thoroughly washed 2 times with ice cold PBS and minced into 1 cm pieces using scissors. The minced colons were then digested PBS containing FBS (2%), HEPES (20mM), Collagenase D (1mg/ml), DNAse I (0.2mg/mL), and Dispase (0.1 U/ml). The digested colons were filtered through a 100μm cell strainer and pelleted. Cells were then enriched over an 80/40% Percoll gradient prior to staining or ex vivo restimulation. For cytokines production by colonic CD4+ T cells, cells were plated in a round bottom 96-well plate in complete RPMI medium containing PMA (Sigma; final concentration 100ng/mL) and Ionomycin (Sigma; final concentration 10ng/mL) and GolgiPlug (1X - BD Bioscience) and incubated for 4 hours at 37°C 5% CO2. Cells were then harvested and used for subsequent flow cytometry staining and analysis.
Genomics and sequencing experiments
RNA-seq
Around 100,000 cells were washed once with 1x PBS before resuspending pellet in 350 μL Buffer RLT Plus (QIAGEN) with 1% 2-Mercaptoethanol (Sigma), vortexed briefly, and stored at −80°C. Subsequently, total RNA was isolated using the RNeasy Plus Micro Kit (QIAGEN). RNA integrity numbers were determined using a TapeStation 2200 (Agilent), and all samples used for RNA-seq library preparation had RIN numbers greater than 9. Libraries were prepared using the SMARTer® Stranded Total RNA-seq Kit v2- Pico Input Mammalian kit (Takara). Two technical replicates were generated for each experiment. Libraries were validated for quality and size distribution using a TapeStation 2200 (Agilent). Libraries were paired-end sequenced (38bp+38bp) on a NextSeq 550 (Illumina) or 61bp+61bp on Novaseq 6000 (Illumina).
ATAC-seq
ATAC-seq was performed as previously described with minor modifications10. Fifty thousand cells were pelleted at 550 g and washed with 50 μL ice-cold 1x PBS, followed by treatment with 50 μL lysis buffer (10 mM Tris-HCl [pH 7.4], 10 mM NaCl, 3 mM MgCl2, 0.1% IGEPAL CA-630). Nuclei pellets were resuspended in 50 μL transposition reaction containing 2.5 μL Tn5 transposase (FC-121–1030; Illumina). The reaction was incubated in a 37°C heat block for 45 min. Tagmented DNA was purified using a MinElute Reaction Cleanup Kit (QIAGEN) and amplified with varying cycles, depending on the side reaction results. Libraries were purified using a QIAQuick PCR Purification Kit (QIAGEN). Libraries were validated for quality and size distribution using a TapeStation 2200 (Agilent). Libraries were paired-end sequenced (38bp+38bp) on a NextSeq 550 (Illumina) or 61bp+61bp on Novaseq 6000 (Illumina).
Hi-C
Hi-C libraries were generated on 0.5 – 1×106 cells using with Arima-HiC+ kit (Arima Genomics) and Accel-NGS @S Plus DNA Library kit (21024 Swift Biosciences), according to the manufacturer’s recommendations. Libraries were validated for quality and size distribution using Qubit dsDNA HS Assay Kit (Invitrogen, cat# Q32851) and TapeStation 2200 (Agilent). Libraries were paired-end sequenced (66bp+66bp) on NovaSeq 6000 (Illumina).
CUT&RUN
CUT&RUN was performed on in vitro polarized Th1 cells using CUTANA™ ChIC/CUT&RUN Kit (EpiCypher, Cat#14–1048), using manufacturer’s recommendation. Briefly, 4 × 105 live cells were sorted out and nuclei were extracted, washed, and allowed to adsorb onto activated ConA beads. Cells were then resuspended in recommended buffer, 0.5 mg of antibody was added, mixed well, and allowed to incubate at 4°C overnight on a nutator. Recommended antibodies were used, including anti-H3K27ac (Acetyl-Histone H3 (Lys27) (D5E4) XP® Rabbit mAb, CST, Cat #8173S), anti-ETS-1 (C-20, SantaCruz, Cat# sc-350X) and anti-CTCF (Millipore, Cat# 07–729), along with positive and negative controls. Subsequently, the reactions were washed with cell permeabilization buffer and incubated with pAG-MNase, and the DNA was isolated for the antibody-bound regions. At least two biological replicates were generated for each experiment. Library preparation was carried out using NEBNext Ultra II DNA Library Prep Kit for Illumina (NEB) and were paired-end sequenced (38bp+38bp) on a NextSeq 550 (Illumina) or 61bp+61bp on Novaseq 6000 (Illumina).
Oligopaint FISH probe generation
The OligoMiner pipeline was used to design Oligopaint libraries as performed earlier10. 42bp probes were designed to a 50 kbp region at a density of approximately 5 probes per kilobase for the Ets1 locus using the GRCm38.87 genome.
Oligopaint FISH hybridization
Thymocytes were isolated by dissociating the mouse thymus through a 70 mM filter (Falcon) in RPMI (Corning). Cells were washed with PBS and filtered again. Following this, the cells were diluted to 4 million cells per mL, and 80uL of diluted cells were added to polysine microscope slides (Thermo Scientific, cat#P4981–001) using silicone isolators (Electron Microscopy Sciences, cat #70339–05). Cells adhered to the slides for 1 hour at room temperature inside humidified chambers. Cells were then briefly washed in 1XPBS, fixed in 4% formaldehyde (Fisher Scientific, cat#PI28908) in PBS for 10 min, and then washed in 1XPBS. Slides were stored temporarily in 1xPBS at 4°C or used immediately for DNA FISH. Cells were permeabilized in 0.5% Triton in PBS for 15 min and dehydrated with an ethanol row of 70%, 90%, and 100% ethanol for 2 min each. After allowing the slides to dry for 3–5 minutes, cells were washed in 2XSSCT/50% formamide (0.3M NaCl, 0.03M sodium citrate, 0.1% Tween-20) at room temperature for 5 minutes, 2.5 min at 92°C in 2XSSCT/50% formamide, and 20 min at 60°C in 2XSSCT/50% formamide. For primary probe hybridization, slides were cooled down to room temperature, and cells were immersed in hybridization buffer (10% dextran sulfate, 50% formamide, 4% PVSA, 5.6 mM dNTPs, and 10ug of RNase A) containing 50 pmol of primary Oligopaint probes, covered with a coverslip (Fisher Scientific, cat#12–548-5M), and sealed with no-wrinkle rubber cement (Elmer’s). Cells were denatured for 2.5 min at 92°C on top of a heated block, followed by hybridization at 37°C in a humidified chamber for ~16 hrs. Coverslips were then carefully removed using a razor blade, and cells were washed for 15 min in 2XSSCT at 60°C, followed by 10 min wash at room temperature in 2XSSCT shaking at 75 rpm and another 10 min wash at room temperature in 0.2XSSCT. After allowing the slides to air-dry, cells were immersed in secondary hybridization buffer (10% dextran sulfate, 10% formamide, and 4% PVSA) with 2pmol bridges and 10pmol of secondary probes (Alexa-488, Atto-565, and Alexa-647), covered with a coverslip (Fisher Scientific, cat#12–548-5M), and sealed with no-wrinkle rubber cement (Elmer’s). Slides were then incubated in the dark in a humidified chamber for 2 hrs at room temperature. Coverslips were then carefully removed using a razor blade, and slides were briefly washed in 2XSSCT at room temperature, followed by a 5 min wash in 2XSSCT at 60°C, a 5 min wash in 2XSSCT with DAPI (0.1 μg/mL), and a 5 min wash in 0.2XSSC. Slide were held in 2XSSC before mounting with Slowfade Gold Antifade Reagent (Invitrogen by Thermo Fisher Scientific, cat#S36936) and sealing with Sally Hansen’s “dries instantly top coat”.
scRNA- and scATAC-seq library generation
The Chromium Next GEM Single Cell Multiome ATAC + Gene Expression kit (10x Genomics) was used to generate single cell multiome data. Around 1×106 live Th1 cells were sorted, on day 1 and day 6 of in vitro Th1 polarization of wildtype and Ets1-SE−/− cells. Nuclei isolation was performed based on the manufacturer’s instruction (CG000365) with the following modifications. A diluted lysis buffer was made using 10 mM Tris-HCl (pH 7.4), 10 mM NaCl, 3 mM MgCl2, 1% BSA, 1 mM DTT, 1 U/μl RNase inhibitor, and nuclease-free water. Then, 200uL lysis (CG000368) and 400uL diluted lysis buffers were mixed to make the final diluted lysis buffer. Cells were incubated in 100uL final diluted lysis on ice for 5 min. Around 10,000 cells were targeted for recovery per genotype and per time point, and done in replicates. The libraries were generated based on the manufacturer’s instruction. All libraries were validated for quality and size distribution using a TapeStation 2200 (Agilent) and quantified using Kapa (Illumina). The scATAC and gene expression libraries were pooled separately and paired-end sequencing (ATAC: Read 1: 50 cycles, i7 Index: 8 cycles, i5 index: 24 cycles, and Read 2: 49 cycle; Gene Expression: Read 1: 28 cycles, i7 Index: 10 cycles, i5 index: 10 cycles, and Read 2: 90 cycle) was performed on the NovaSeq6000 (Illumina).
Genomics Data Analysis
HiChIP data processing and 3D clique analysis:
H3K27ac HiChIP measurements in mouse DP T cells were generated in our previous study10. Data analysis was performed as previously described: Raw reads for each HiChIP sample were processed with HiC-Pro (version v2.5.0)67 to obtain putative interactions with default parameters except LIGATION_SITE = GATCGATC and GENOME_FRAGMENT generated for MboI restriction enzyme. Valid pairs (VI), self-circle (SC) and dangling-end (DE) interactions in cis were used as input for significant interaction calling in ‘.bedpe’ format. Mango (version 1.2.0) (Phanstiel et al., 2015) step 4 identified putative significant interaction anchors by MACS peak calling with MACS_qvalue = 0.05 and MACS_shiftsize = 75. Mango step 5 identified significant interactions with default parameters except maxinteractingdist = 2000000 and MHT = found. Two biological repeats for each strain were processed and only significant interactions with PETs >= 2 reproduced in both replicates were used for further analysis. For each library, each significant interaction was normalized to contacts per hundred million, i.e., divided by the number of interactions in the Mango input.bedpe file and multiplied by 1E8. Mango outputs of two biological replicates where two anchors were within 5kbp were called reproducible interactions in DP T cells.
3D clique analysis:
3D clique analysis was performed following the same procedure as reported earlier9,10. An undirect graph of regulatory interactions was constructed for reproducible interactions with at least one H3K27ac peak at one anchor. In this graph, each vertex was an enhancer or a promoter and each edge was a significant and reproducible enhancer-enhancer, enhancer-promoter, or promoter-promoter interaction. “3D Cliques” were defined by spectral clustering of the regulatory graph interactions using cluster_louvain function in igraph R package with default parameters. A 3D clique connectivity was defined as the number of edges connecting vertices within the clique. The connectivity of cliques was ranked in ascending order and plotted against the rank. The cutoff for hyperconnected 3D cliques was set to the elbow of the curve and a tangent line at the cutoff was shown. Super-enhancers were defined using H3K27ac ChIP-seq in DP T cells as described earlier36. Annotation of noncoding RNA was performed using gencode.vM10.annotation.gtf file. Architectural stripes were defined using Stripenn 14 using Hi-C measurements in DP T cells. Odds ratio analysis was performed to evaluate the significance of enrichment of super-enhancers, ncRNA and architectural stripes.
Hi-C data analysis
Hi-C alignment:
We used the ArimaHiC protocol to generate our Hi-C libraries following the manufacturer’s recommendations and processed the data with HiC-Pro using parameters “LIGATION_SITE =GAATAATC,GAATACTC,GAATAGTC,GAATATTC,GAATGATC,GACTAATC,GACTACTC,GACTAGTC,GACTATTC,GACTGATC,GAGTAATC,GAGTACTC,GAGTAGTC,GAGTATTC,GAGTGATC,GATCAATC,GATCACTC,GATCAGTC,GATCATTC,GATCGATC,GATTAATC,GATTACTC,GATTAGTC,GATTATTC,GATTGATC” and GENOME_FRAGMENT file was generated using “digest_genome.py -r ĜATC GÂATC GÂTTC GÂCTC GÂGTC”. ValidPairs generated by HiC-Pro were further converted to cool and hic files.
Compartment analysis:
The principal component analysis (PCA) was performed with 50 kbp resolution for both wild type and Ets1-SE−/− Th1 Hi-C data. To generate the PC1 plot in the Figure S6a, a customized script (cool_compartment.py) utilizing cooltools was used.
TAD analysis:
TAD coordinates were estimated using two Perl scripts named ‘matrix2insulation.pl’ and ‘insulation2tads.pl’ from the cworld-Dekker Github page for both wild type and Ets1-SE−/− Th1 Hi-C data. The overlap of TAD boundaries was evaluated using the findOverlaps function in an R package called GenomicRanges.
Loop analysis:
Loops were called using Mustache77 from both wild type and Ets1-SE−/−Th1 and Th2 Hi-C with 5kbp resolutions. Loops with FDR < 0.1 were used for further analysis. The scatter plot for loop intensity of wild type and Ets1-SE−/− mice was generated, and the loops with intensity higher than |WT/ Ets1-SE−/− |>0.5 was highlighted.
Triangle heatmaps:
Triangle heatmaps for 3D chromatin conformation data and corresponding tracks were generated using Sushi R package (version 1.28.0)
CUT&RUN data analysis:
The FASTQ files of CUT&RUN experiments were aligned to the bam file using BWA (version 0.7.17-r1188). In this process, minor chromosomes such as mitochondrial chromosome or chrY were removed using samtools (version 1.11). Next, duplicated reads were removed using Picard (version 2.26.7) and then the bam files were indexed using samtools. BigWig files were generated using bamCoverage (version 3.3.2) with parameters ‘normalizedUsing=CPM, binsize=30, smoothLength=300, p=5, extendReads=200’. For peak calling, macs2 (version 2.1.4) was used with following commands: ‘macs2 callpeak -t input_file -c control -g mm -n output_path –nomodel -f BAMPE -B –keep-dup all –broad –broad-cutoff 0.25 -q 0.25’. For the background (control), the bam file of IgG CUT&RUN data was used. CUT&RUN peaks from two conditions and both replicates were merged and the number of fragments in each peak were counted with bedtools. The count data of each peak was then fed to DESeq2 for differential analysis.
Integration of CUT&RUN data with 3D loop analysis
Loop classification:
The loops called from the wild type Th1 Hi-C with 5kb resolution were classified based on the occupancy of CTCF and/or ETS1. First, the CTCF and ETS1 peaks from replicates were combined, respectively. Next, both ends of each loop (loop anchors) were extended by ±5 kbp and then the occupancy of CTCF or ETS1 peaks within loop anchors was examined using the R package named GenomicRanges.
DESeq2 analysis and pileup plot:
To identify CTCF and ETS1 peaks that were lost or gained in a coordinated manner in Ets1-SE−/− Th1 cells, DESeq2 was performed by (1) combining CTCF and ETS1 peaks across all replicates, (2) counting reads across this union of peaks, and (3) comparing wildtype and Ets1-SE−/− Th1 cells samples regardless of the protein occupancy. As a result, 988 and 364 co-bound CTCF-ETS1 peaks were detected to be co-lost and co-gained, respectively (|log2 fold change| > 1 and p-value<0.05). 1D heatmaps using deeptools or pileup plots for 3D interactions using coolpuppy examined the 1D and 3D features of these peak sets. For coolpup.py, a parameter –pad=50 was used. Local pileup analysis at different sets of peaks was done with coolpup.py 63 using parameters “--pad 250 --local”. The average interaction of the upright corner of pileup plots is quantified with custom python script, by parsing the results from coolpup.py. Interactions between regions in bedfiles were done with parameters “--mindist 200000 --maxdist 2000000”. Pileup of loops was also plot with coolpup.py. To do multiple pileup analysis parallelly, we used GNU parallel to run the shell script.
Deeptools analysis of CUT&RUN and ATAC-seq data:
The differentially gained or lost sites in Naive CD4+ T, Th1 and Th2 cells were obtained using DESeq2 (|log2 fold change| > 1 and adjusted p-value<0.05) and then combined using bedtools sort and merge function. Next, deeptools plot was generated with computeMatrix function using following parameters: reference-point –referencePoint center -a 2000 -b 2000. The heatmap was generated with the ‘plotHeatmap’ function with the following parameters: ‘–heatmapHeight 15 –averageTypeSummaryPlot median –colorMap Greys’. In comparison of Naive CD4 T and Th1 cells, we performed DESeq2 for Naive CD4 T and Th1 cells (Ets1-SE+/+). Then, Naive CD4 T-specific (log2 fold change > 1 and adjusted P-value < 0.05), Th1-specific (log2 fold change < −1 and adjusted P-value < 0.05) and non-significant (adjusted P-value > 0.05) peaks were obtained and fed to computeMatrix/plotHeatmap function.
RNA-seq data analysis:
The FASTQ files of RNA-seq experiments were aligned and further counted using STAR 2.7.7a with parameters ‘--outSAMtype BAM SortedByCoordinate --outWigType wiggle read1_5p --outWigStrand Stranded --outWigNorm RPM --quantMode GeneCounts’. Next, DESeq2 was performed to identify differentially expressed genes (|log2 fold change|>1 and adjusted p-value < 0.05).
ATAC-seq data analysis:
The alignment process of ATAC-seq data is identical to that of CUT&RUN data except MACS2 parameters which is as follows: ‘macs2 callpeak -t input_file -g mm -n output_path –nomodel -f BAM -B –keep-dup all –broad –broad-cutoff 0.1 -q 0.1’. ATAC-seq peaks from two conditions and both replicates were merged and the number of fragments in each peak were counted with bedtools. The count data of each peak was then fed to DESeq2 for differential analysis.
scRNA- and scATAC-seq data preprocessing and quality control
The cellranger-arc mkfastq from Cell Ranger ARC pipeline (version 2.0.0) was used to demultiplex samples. Demultiplexed scRNA- and scATAC-seq fastq files were inputted into the cellranger-arc count pipeline from 10x Genomics to generate barcoded count matrices of gene expression and ATAC data. cellranger-arc aggr was used to aggregate samples across all conditions. For the aggregate of all samples, count matrices were loaded in Seurat pipeline and selected for barcodes that appeared in both the scRNA-seq and scATAC-seq datasets. scRNA-seq data from nuclei remaining after quality control filtering were analyzed using Seurat (version 4.3). Gene expression counts were normalized using the SCT function. Graph-based clustering was then performed on the data using the top 20 principal components at a resolution of 0.5. Cluster identities were manually annotated based on the expression of genes from published scRNA-seq studies of T cell differentiation. Marker genes for each cluster were additionally identified using the FindAllMarkers function with a minimum fraction of 0.5 and a log2 fold change of 1. Clusters expressing low RNA count were removed from further analysis. scATAC-seq data were analyzed using Signac (V 1.9.0) based on barcoded cell type identities from scRNA-seq. Chromatin accessibility peaks on chromosomes 1–22 and X and outside of blacklist regions were then called using the MACS2 with scATAC peaks for all cells.
GSEA and Gene Ontology analysis:
Pre-ranked Gene Set Enrichment Analysis (GSEA) was used for enrichment analyses. Gene-sets were provided by DESeq2 for downregulated and upregulated genes in Ets1-SE−/− compared with wildtype Th1 cells. Pre-ranked genes based on DESeq analysis of across different conditions were used. Metascape using ImmuneSigDB was used for gene-ontology analysis.
ImmGen gene expression data processing:
Gene Skyline feature for RNA-seq data provided by ImmGen was used to profile Ets1 and Fli1 expression levels across various cell types.
Motif analysis:
To find the conserved motifs from the lost or gained ATAC-seq/Hi-C peaks(=loops) in Ets1-SE−/− mice, a function named ‘findMotifsGenome.pl’ in Homer program (version 4.11) was utilized. For ATAC-seq data of naive CD4 T cells, gained and lost peaks in Ets1-SE−/− mice were determined by DESeq2 results such that |log2 fold change| > 1 and adjusted P-value < 0.05. Here, the peaks that were not significantly changed (adjusted P-value>0.8) were used as background. The loops gained and lost in Ets1-SE were searched from Hi-C data of Th1 cells. Here, gained and lost loops were defined as those |fold change|>0.5, and the loops showing minute changes (|fold change| < 0.1) were considered as background. These loop coordinates were then fed to ‘bedtools intersect (version 2.30.0)’ to obtain the coordinates overlapping with CUT&RUN peaks of ETS1.The output consists of (1) consensus motifs (known motifs) and (2) de novo motifs conserved in the input sequences (homer motifs). Here, we reported the latter one.
Oligopaint FISH imaging and analysis
Imaging was carried out on a Bruker Vutara VXL in the Widefield imaging modality, which has an imaging field-of-view (FOV) of 200 μm × 200 μm. The VXL has a 60X silicon oil immersion objective with a 1.3 numerical aperture (NA). Z-stack size was maximum 20 μm with a Z-step size of 150 nm. Analysis was carried out on the raw images in a semi-automated manner on a cell-by-cell basis as describe in Raj. et al. 2008 (https://github.com/arjunrajlaboratory/rajlabimagetools). Briefly, the DAPI signal was used for manual nuclei segmentation of DP T cells. The exact numbers of nuclei analyzed per genotype are as follows: 561 nuclei for wildtype and 727 nuclei for the cells from Ets1-SE−/−. One mouse per genotype was used. Spots for each of the 3 channels (Alexa-488, Atto-565, and Alexa-647) were individually detected using a linear filter approximately conforming to a Laplacian convolved with a Gaussian. For each spot, the brightest z slice was used as the z coordinate. Centroid positions for each spot in xy were found by fitting a Gaussian. X, Y, and Z coordinates were extracted, and pairwise Euclidean distances between nearest neighbors were calculated. The spatial distance of two probes in each cell were determined by taking a minimum from all pair-wise distances of corresponding probes. The Kolmogorov-Smirnov test was used to compare differences in the cumulative distributions, and the Mann-Whitney test was used to compare differences in the medians. In addition, the proportion of cells having probe distance less than threshold was examined for 3D clique dynamics analysis.
Representative Image processing
Imaging was carried out on a Leica Multiphoton Confocal using a 63X oil immersion objective with a 2.0 zoom factor, a pixel size of 58.77 nm × 58.77 nm, and Z-stack size of 15 μm with a Z-step size of 300 nm. Z-stacks were maximally projected. Each cell, allele, and locus for each strain were individually processed using ImageJ via adjusting the brightness/contrast/minimum/maximum, as well as smoothing.
Ets1+/− mice data analysis
Virtual 4C analysis
The Virtual 4C analysis evaluates the DNA interaction that takes place between a genomic anchor and the loci located in its proximity. Here, the direct downstream of Gm27162 was set as an anchor, and its normalized contact frequencies (VC_SQRT) with the Ets1-Fli1 region (chr9:32325001–33205000) were measured using Hi-C data of Ets1-SE−/−, and Ets1fl/+Cd4cre Th1 cells and their wildtype counterparts. Instead of the commonly used line plots, virtual 4C plot here is represented by a heatmap to effectively compare the contact frequencies from four datasets.
Loop analysis
From the Hi-C data of Ets1-SE−/− naïve CD4+ T cells, Ets1-SE−/− Th1 cells, Ets1fl/+Cd4cre Th1 cells and their wildtype counterparts, 11031, 13164, 17790, 15642, 18077 and 17818 long-range interactions were detected using Mustache (version 1.2.7, FDR<0.1). The loops from six datasets were then merged into 67119 loops using sort and uniq command in Linux to collect as many loops as possible for testing. Next, by utilizing the balancing in the python cooler package, the normalized contact frequencies were calculated for Ets1-SE−/− and Ets1fl/+Cd4cre Th1 cells and their wildtype counterparts. Upon calculating the fold change, we found 5,787 loops that exhibited simultaneous alterations with Ets1 dose reduction (|log2FC|>0.25).
ChIP-Atlas
ChIP-Atlas is a comprehensive database that collects public ChIP-seq, DNase-seq, ATAC-seq and Bisulfite-seq data. It provides an integrative analysis such as peak browser, TF target gene search, colocalization and enrichment analysis. In this study, peak browser was utilized to search for the transcription factors bound on Gm27162 locus in Th1 cells. Experiment type, cell type class, threshold for significance and cell type were set as ChIP:TFs and others, blood, 50 and Th1 cells, respectively. The bed file was further processed with awk to include TFs bound on Gm27162 region.
Allergy-associated SNPs enrichment
A list of 58 SNPs within Ets1-Fli1 region was manually collected. The orthologous human coordinate of Gm27162 (chr11:128303536–128330986) included 9 SNPs in total. Among them, four were related to allergy/asthma. P-value and odds ratio were estimated by Fisher exact test using fisher.test function in R stats package.
Statistical analysis:
For all experiments, the difference between two groups was calculated using unpaired t-test (Mann and Whitney) Prism 10 GraphPad Software. ANOVA and Bonferroni test were used for multiple comparisons (ns = not significant, * = p ≤ 0.05, ** = p ≤ 0.01, *** = p ≤ 0.0005, **** = p ≤ 0.0001). The Mann and Whitney unpaired t-test was used for comparisons of two conditions within one group. One-way ANOVA was used to compare more than two conditions for one group. The Two-way ANOVA was used to compare two conditions across multiple groups. All graphs show the mean and the standard error of the mean (SEM). One- and Two-way ANOVA were corrected for multiple comparison using Bonferroni correction.
Supplementary Material
Key resources table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-mouse CD4 (Clone: RM4–5) APC | BioLegend | Cat# 100516; RRID: AB_312719 |
| Anti-mouse CD4 (Clone: GK1.5) FITC | BioLegend | Cat# 10040;, RRID: AB_312690 |
| Anti-mouse CD4 (Clone: RM4–5) BV510 | BioLegend | Cat# 100559; RRID: AB_2562608 |
| Anti-mouse CD4 (Clone: RM4–5) BV711 | BioLegend | Cat# 100549; RRID: AB_11219396 |
| Anti-mouse CD4 (Clone: RM4–5) BV785 | BioLegend | Cat# 100552; RRID: AB_2563053 |
| Anti-mouse CD5 (Clone: 57–7.3) FITC | BioLegend | Cat# 100606; RRID: AB_312735 |
| Anti-mouse CD8 (Clone: 53–6.7) PE-eFluor 610 | Thermo Fisher Scientific | Cat# 61–0081-80; RRID: AB_2574523 |
| Anti-mouse CD8 (Clone:53–6.7) BV785 | BioLegend | Cat# 100749; RRID: AB_11218801 |
| Anti-Mouse CD8 (Clone: 53.6–7) PerCP-Cy5.5 | BioLegend | Cat# 100733; RRID: AB_2075239 |
| Anti-mouse CD8 (Clone:53–6.7) BV711 | BioLegend | Cat# 100744; RRID: AB_2562609 |
| Anti-mouse CD11b (Clone: M1/70) AF700 | BioLegend | Cat# 101222; RRID: AB_493705 |
| Anti-mouse CD11c (Clone: N418) BV785 | BioLegend | Cat# 117335; RRID: AB_11219204 |
| Anti-mouse CD24 (Clone : M1/69) Pe/Cy7 | BioLegend | Cat# 101821; RRID: AB_756047 |
| Anti-mouse CD25 (Clone: PC61) PerCP-Cy5.5 | BioLegend | Cat# 102030; RRID: AB_893288 |
| Anti-mouse CD25 (Clone: PC61) Pacific Blue | BioLegend | Cat# 102021; RRID: AB_493642 |
| Anti-mouse CD44 (Clone: IM7) BV785 | BioLegend | Cat# 103059; RRID: AB_2571953 |
| Anti-mouse CD44 (Clone: IM7) AF700 | BioLegend | Cat# 103026; RRID: AB_493713 |
| Anti-Mouse CD44 (Clone: IM7) PerCP-Cy5.5 | BioLegend | Cat# 103035; RRID: AB_10639933 |
| Anti-mouse CD45 (Clone: 30-F11) AF700 | BioLegend | Cat# 103128; RRID: AB_493715 |
| Anti-mouse CD45 (Clone: 30-F11) FITC | BioLegend | Cat# 103108; RRID: AB_312973 |
| Anti-mouse CD45RB (Clone: C363–16A) PE | BioLegend | Cat# 103307; RRID: AB_313014 |
| Anti-mouse CD62L (Clone: MEL-14) BV421 | BioLegend | Cat# 104435; RRID: AB_10900082 |
| Anti-mouse CD90.2 (Clone: 53–2.1) AF700 | BioLegend | Cat# 140324; RRID: AB_2566740 |
| Anti-rat CD90/mouse CD90.1 (Thy-1.1) (Clone: OX-7) APC/Fire™ | BioLegend | Cat# 202543; RRID: AB_2650816 |
| Anti-mouse/pig CD117 (Clone: 2B8) APC-eF780 | ThermoFisher Scientific | Cat# 47–1171-82; RRID: AB_1272177 |
| Anti-mouse CD193 (CCR3) (Clone: J07325) APC | BioLegend | Cat# 144511; RRID: AB_2565737 |
| Anti-mouse TCRb (Clone: H57–597) FITC | BD Biosciences | Cat# 553170; RRID: AB_394682 |
| Anti-mouse TCRb (Clone: H57–597) APC | BioLegend | Cat# 109212; RRID: AB_313435 |
| Anti-mouse TCRb (Clone: H57–597) PE | BioLegend | Cat# 109207; RRID: AB_313430 |
| Anti-mouse TCRb (Clone: H57–597) BB700 | BD Biosciences | Cat# 745846; RRID: AB_2743291 |
| Anti-mouse TCRb (Clone: H57–597) PE/Cy7 | BioLegend | Cat# 109222; RRID: AB_893625 |
| Anti-mouse IFNγ (Clone: XMG1.2) PerCP/Cyanine5.5 | BioLegend | Cat# 505821; RRID: AB_961361 |
| Anti-mouse IFNγ (Clone: XMG1.2) - APC | BD Biosciences | Cat# 554413; RRID: AB_398551 |
| Anti-mouse IFNγ (Clone: XMG1.2) - AF700 | BD Biosciences | Cat# 557998; RRID: AB_396979 |
| Anti-mouse IL-13 Monoclonal Antibody (Clone: eBio13A) PE-eFluor™ 610 | Thermo Fisher Scientific | Cat# 61–7133-80; RRID: AB_2574653 |
| Anti-mouse IL-13 (Clone: eBio13A) efluor 450 | Thermo Fisher Scientific | Cat# 48–7133-80; RRID: AB_11218502 |
| Anti-mouse IL-13 (Clone: eBio13A) Alexa488 (FITC) | Thermo Fisher Scientific | Cat# 53–7133-82; RRID: AB_2016708 |
| Anti-mouse IL-17A (Clone: TC11–18H10.1) PE | BioLegend | Cat# 506903; RRID: AB_315463 |
| Anti-mouse IL-17A (Clone: TC11–18H10.1) BV785 | BioLegend | Cat# 506928; RRID: AB_2629787 |
| Anti-mouse IL-4 (Clone: 11B11) PE | BioLegend | Cat# 504103; RRID: AB_315317 |
| Anti-mouse IL-5 (Clone: TRFK5) PE | ThermoFisher Scientific | Cat# 12–7052-81; RRID: AB_763588 |
| Anti-mouse IL-5 (Clone: TRFK5) APC | BioLegend | Cat# 504305; RRID: AB_315329 |
| Anti-mouse Granzyme B (Clone: GB11) BV421 | BioLegend | Cat# 515407; RRID: AB_2562195 |
| T-bet Monoclonal Antibody (eBio4B10 (4B10)), PE-Cyanine7 | eBioscience (Thermo Fisher Scientific) | Cat# 25–5825-80; RRID: AB_11041809 |
| Anti-mouse/Human T-bet (Clone: 4B10) PE | eBioscience (Thermo Fisher Scientific) | Cat# 12–5825-80; RRID: AB_925762 |
| Anti-mouse/Human T-bet (Clone: 4B10) BV711 | BioLegend | Cat# 644819; RRID: AB_11218985 |
| Anti-mouse/Human T-bet (Clone: 4B10) BV421 | BioLegend | Cat# 644815; RRID: AB_10896427 |
| Anti-mouse/Human T-bet (Clone: 4B10) PeCy7 | Thermo Fisher Scientific | Cat# 25–5825-82; RRID: AB_11042699 |
| Anti-Mouse RORγt Antibody (Clone: Q31–378) BV421 | BD Biosciences | Cat# 562894; RRID: AB_2687545 |
| Anti-mouse RORγt (Clone: B2D) PE | Thermo Fisher Scientific | Cat# 12–6981-82; RRID: AB_10807092 |
| Anti-mouse RORγt (Clone: B2D) PE-eFluor 610 | Thermo Fisher Scientific | Cat# 61–6981-80; RRID: AB_2574649 |
| Anti-mouse Ly6G (Clone: 1A8) FITC | BioLegend | Cat# 127606; RRID: AB_1236494 |
| Anti-mouse Eomes (Clone: Dan11mag) APC | ThermoFisher Scientific | Cat# 17–4875-82; RRID: AB_2866428 |
| Anti-mouse GATA3 (Clone: L50–823) BV711 | BD Biosciences | Cat# 565449; RRID: AB_2739242 |
| Anti-mouse GATA3 (Clone: L50–823) PeCy7 | BD Biosciences | Cat# 560405; RRID: AB_1645544 |
| Anti-mouse FoxP3 (Clone: MF-14) BV421 | BioLegend | Cat# 126410; RRID: AB_2105047 |
| Anti-mouse FoxP3 (Clone: MF-14) PE | BioLegend | Cat# 126403; RRID: AB_1089118 |
| Anti-mouse NK1.1 (Clone: PK136) BV711 | BioLegend | Cat# 108745; RRID: AB_2563286 |
| Anti-mouse NK1.1 (Clone: PK136) FITC | BioLegend | Cat# 108705; RRID: AB_313392 |
| Anti-Mouse CD1d-PE (PBS57 - NIH Core) | NIH Tetramer Core Facility | Item # 14461; RRID: N/A |
| Anti-mouse SiglecF (Clone: S17007L) PE | BioLegend | Cat# 155505; RRID: AB_2750234 |
| Anti-mouse Siglec-F (Clone: E50–2440) PE-CF594 | BD Biosciences | Cat# 562757; RRID: AB_2687994 |
| Anti-mouse T1/ST2 (Clone: DIH9) PeCy7 | BioLegend | Cat# 145304; RRID: AB_2561915 |
| Anti-CD3ε Armenian Hamster Monoclonal Antibody (LEAF - Clone: 145–2C11) | BioLegend | Cat# 100302; RRID: AB_312667 |
| Anti-CD28 Syrian Hamster Monoclonal Antibody (Clone: 37.51) |
BD Biosciences | Cat# 553294; RRID: AB_394763 |
| InVivoMab anti-mouse IFNγ (Clone: XMG1.2) | Bio X Cell | Cat# BE0055; RRID: AB_1107694 |
| InVivoMab anti-mouse IL-4 (Clone: 11B11) | Bio X Cell | Cat# BE0045; RRID: AB_1107707 |
| Anti-CTCF | Millipore | Cat# 07–729; RRID: AB_441965 |
| Anti-Acetyl-Histone H3 (Lys27) (D5E4) | Cell Signaling Technology | Cat# 8173; RRID: AB_10949503 |
| Anti-Ets1 (C-20) | Santa Cruz Biotechnology | Cat# sc-350; RRID: AB_2100688 |
| Anti-CD8 (biotin) (clone: 53–6.7) | BioLegend | Cat# 100704; RRID: AB_312743 |
| Anti-CD19 (biotin) (clone: 6D5) | BioLegend | Cat# 115504; RRID: AB_313639 |
| Anti-CD45R (biotin) (clone: RA3–6B2 | BioLegend | Cat# 103204; RRID: AB_312989 |
| Anti-Gr1 (biotin) (clone: RB6–8C5) | BioLegend | Cat# 108403; RRID: AB_313368 |
| Anti-TCRg/d (biotin) (clone: GL3) | BioLegend | Cat# 118103; RRID: AB_313827 |
| Anti-CD11c (biotin) (clone: N418) | BioLegend | Cat# 117303; RRID: AB_313772 |
| Anti-I-A/I-E (biotin) (clone: M5) | BioLegend | Cat# 107604; RRID: AB_313319 |
| Anti-CD25 (biotin) (clone: PC61) | BioLegend | Cat# 102003; RRID: AB_312852 |
| Anti-NK1.1 (biotin) (clone:PK136) | BioLegend | Cat# 108703; RRID:AB_313390 |
| Chemicals, peptides, and recombinant proteins | ||
| Recombinant IL-2 (mouse) | BioLegend | Cat# 575402 |
| Recombinant IL-12 (mouse) | BioLegend | Cat# 577002 |
| Recombinant TGFb (mouse) | BioLegend | Cat# 763102 |
| Recombinant IL-4 (mouse) | BioLegend | Cat# 574302 |
| Recombinant IL-6 (mouse) | BioLegend | Cat# 575702 |
| Viability Dye LIVE/DEAD™ Fixable Aqua Dead Cell Stain Kit | ThermoFisher Scientific | Cat# L34957 |
| Fixable Viability Dye eFluor™ 506 | ThermoFisher Scientific | Cat# 65–0866-14 |
| Fixable Viability Dye eFluor™ 520 | ThermoFisher Scientific | Cat# 65–0867-14 |
| Fixable Viability Dye eFluor™ 780 | ThermoFisher Scientific | Cat# 50–112-9035 |
| CellTrace™ Violet Cell Proliferation Kit, for flow cytometry | ThermoFisher Scientific | Cat# C34557 |
| HDM extracts (Dermatophagoides pteronyssinus extracts) | Fischer Scientific (Greer Laboratories) | Cat# NC9756554 (lot: 361863, 385930, 387032) |
| Phorbol 12-myristate 13-acetate (PMA) | Sigma- Aldrich | Cat# P8139–1MG |
| Ionomycin calcium salt from Streptomyces conglobatus | Sigma- Aldrich | Cat# I0634–1MG |
| GolgiPlug Protein Transport Inhibitor | BD Biosciences | Cat# 555029 |
| RPMI 1640 medium | Invitrogen | Cat# 11875085 |
| Fetal Bovine Serum | Sigma- Aldrich | Cat# F2442 |
| GemCell™ - Fetal Bovine Serum | Gemini Bio | Cat# 100–500, Lot# A03HOOK |
| 2-bMercaptoethanol | Sigma | Cat# M6250–10ML |
| ACK Lysing Buffer | ThermoFisher Scientific | Cat# A1049201 |
| Deoxyribonuclease I from bovine pancreas | Sigma-Aldrich | Cat# D5025 |
| Collagenase D | Roche | Cat# 11088866001 |
| Dispase (5U/ml) | STEMCELL Technologies | Cat# 07913 |
| HEPES | ThermoFisher | Cat#15630080 |
| Non-Essential Amino Acids | ThermoFisher | Cat# 11140050 |
| Penicillin-Streptomycin | ThermoFisher | Cat# 15140122 |
| Sodium Pyruvate | ThermoFisher | Cat# 11360070 |
| L-Glutamine (200 mM) | ThermoFisher | Cat# 25030081 |
| Percoll® Density Gradient Media | VWR | Cat# 17–0891-01 |
| Formaldehyde solution 16% | Thermo | Cat# PI28908 |
| cOmplete™, EDTA-free Protease Inhibitor Cocktail | Roche | Cat# 11873580001 |
| Qubit dsDNA HS Assay Kit | Invitrogen | Cat# Q32851 |
| Phusion PCR Master Mix | NEB | Cat# M0531 |
| Nextera XT Index Kit | Illumina | Cat# FC-131–1001 |
| SPRIselect | Beckman Coulter | Cat# B2331 |
| Polybrene | Sigma | Cat# H9268 |
| Lipofectamine 3000 | Thermo | Cat# L3000008 |
| Chloroquine | Sigma | Cat# C6628–25G |
| Secondary Oligopaint probe: /5Alex647N/ TGATCGACCACGGCCAAGACGGAGAG CGTGTG/ 3AlexF647N/ | https://www.pnas.org/doi/full/10.1073/pnas.1714530115 | N/A |
| Secondary Oligopaint probe: /5ATTO565N/ ACACN/ACCTTGCACGTCGTGGACCTCC TGCGCTA/ 3ATTO565N/ | https://www.pnas.org/doi/full/10.1073/pnas.1714530115 | N/A |
| Secondary Oligopaint probe: /5Alex488N/ CACAN/ACGCTCTTCCGTTCTATGCGAC GTCGGTG/ 3AlexF488N/ | https://www.pnas.org/doi/full/10.1073/pnas.1714530115 | N/A |
| Polysine microscope slides | Thermo Scientific | Cat# P4981–001 |
| Silicone isolators | Electron Microscopy Sciences | Cat# 70339–05 |
| Ethanol | Decon Laboratories | Cat# 2716 |
| Dimethylformamide | Sigma-Aldrich | Cat# D4551 |
| Polyvinylsulfonic acid (PVSA) | Sigma Aldrich | Cat# 278424 |
| Coverslips | Fisher Scientific | Cat# 12–548-5M |
| Nowrinkle rubber cement | Elmer’s | N/A |
| Slowfade Gold Antifade Reagent | Invitrogen by Thermo Fisher Scientific | Cat# S36936 |
| Dries instantly top coat | Sally Hansen’s | Cat# 45114 |
| Fixation/Permeabilization Solution Kit | BD Biosciences | Cat#554714 |
| Foxp3/ Transcription Factor Staining Buffer Set | Thermo | Cat# 00–5523-00 |
| Critical commercial assays | ||
| CUTANA™ ChIC/CUT&RUN Kit | EpiCypher | Cat# 14–1048 |
| Tn5 Transposase | Illumina | Cat# FC-121–1030 |
| SMARTer Stranded Total RNA-Seq Kit – Pico Input Mammalian kit | Takara | Cat# 635006 |
| NEBNext Ultra II DNA Library Prep Kit for Illumina | NEB | Cat# E7645S |
| D1000 ScreenTape | Agilent | Cat# 5067–5582 |
| D1000 Reagents | Agilent | Cat# 5067–5583 |
| High Sensitivity D1000 ScreenTape | Agilent | Cat# 5067–5584 |
| High Sensitivity D1000 Reagents | Agilent | Cat# 5067–5585 |
| Genomic DNA ScreenTape | Agilent | Cat# 5067–5365 |
| Genomic DNA Reagents | Agilent | Cat# 5067–5366 |
| RNA ScreenTape | Agilent | Cat# 5067–5576 |
| RNA ScreenTape Ladder | Agilent | Cat# 5067–5578 |
| RNA ScreenTape Sample Buffer | Agilent | Cat# 5067–5577 |
| Arima HiC kit | Arima Genomics | N/A |
| Accel-NGS 2S Plus DNA Library Kit | Swift Biosciences | Cat# 21024 |
| MinElute Reaction Cleanup Kit | QIAGEN | Cat# 28204 |
| QIAQuick PCR Purification Kit | QIAGEN | Cat# 28104 |
| RNeasy Plus Micro Kit | QIAGEN | Cat# 74034 |
| EasySep™ Mouse Naïve CD4+ T Cell Isolation Kit | StemCell | Cat# 19765 |
| Chromium Next GEM Single Cell Multiome ATAC + Gene Expression Reagent Bundle, 16 rxns | 10X Genomics | PN-1000283 |
| Chromium Next GEM Chip J Single Cell Kit, 48 rxns | 10X Genomics | PN-1000234 |
| Single Index Kit N Set A, 96 rxns | 10X Genomics | PN-1000212 |
| Dual Index Kit TT Set A, 96 rxns | 10X Genomics | PN-1000215 |
| Deposited data | ||
| HiC, CUT&RUN, ATAC-seq and RNA-seq | This study | GEO: GSE211178 |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6J | Jackson Laboratories | RRID:IMSR_JAX:000664 |
| Mouse: C57BL/6J-Ets1-SE−/− | This study | N/A |
| Mouse: CD4-cre (B6.Cg-Tg(Cd4-cre)1Cwi/BfluJ) | The Jackson Laboratory | Cat#022071; RRID:IMSR_JAX:022071 |
| Mouse: Ets1fl/fl | Gift from Barbara L. Kee | N/A |
| Mouse: Rag1−/−mice (B6.129S7-Rag1tm1Mom/J) | Jackson Laboratories | Strain #:002216, RRID:IMSR_JAX:002216 |
| Recombinant DNA | ||
| Plasmid: MSCV-IRES-Thy1.1 DEST | Addgene | Cat# 17442 |
| Plasmid: pCL-Eco | (Naviaux et. al., 1996) | N/A |
| Software and algorithms | ||
| R 4.1.1 | R Core Team58 | https://www.R-project.org/ |
| Python 3.8 | Van Rossum & Drake59 | https://peps.python.org/pep-0569/ |
| FastQC v0.11.9 | Andrews60 | http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| Trim_Galore v0.6.6 | Krueger61 | https://github.com/FelixKrueger/TrimGalore |
| BWA v0.7.17-r1188 | Li & Durbin 62 | https://bio-bwa.sourceforge.net |
| Picard v2.26.7 | Broad Institute63 | http://broadinstitute.github.io/picard/ |
| MACS2 v2.1.4 | Zhang et al.64 | https://pypi.org/project/MACS2/2.1.4/ |
| bamCoverage v3.3.2 | Ramirez et al.65 | https://deeptools.readthedocs.io/en/develop/content/tools/bamCoverage.html |
| STAR v2.7.7a | Dobin et al.66 | https://github.com/alexdobin/STAR |
| HiC-Pro v2.8 | Servant et al.67 | https://github.com/nservant/HiC-Pro/ |
| Homer | Heinz, Benner, Spann, Bertolino et al.68 | http://homer.ucsd.edu/homer/ |
| DESeq2 v1.34.0 | Love, Huber & Anders 69 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| bedtools v2.30.0 | Quinlan & Hall 70 | https://bedtools.readthedocs.io/en/latest/content/installation.html |
| GSEA v4.3.2 | Subramanian et al.71 | https://www.gsea-msigdb.org/gsea/index.jsp |
| Mango v1.2.0 | Phanstiel et al. 72 | https://github.com/dphansti/mango |
| igraph v1.3.5 | Csardi & Nepusz73 | https://igraph.org |
| Seurat v4.3.0 | Hao et al.45 | https://satijalab.org/seurat/ |
| Integrative Genome Browser v2.9.13 | Robinson et al.74 | https://software.broadinstitute.org/software/igv/ |
| Cell Ranger-ARC v2.0.0 | https://support.10xgenomics.com/single-cell-multiome-atac-gex/software/pipelines/latest/what-is-cell-ranger-arc | |
| Sushi v1.32.0 | Phanstiel75 | http://bioconductor.riken.jp/packages/3.1/bioc/html/Sushi.html |
| RajLabImageTool v1.0 | Raj et al.76 | https://github.com/arjunrajlaboratory/rajlabimagetools |
| Mustache v1.2.7 | Ardakany et al.77 | https://github.com/ay-lab/mustache |
| Deeptools v3.5.1 | Ramirez et al.65 | https://deeptools.readthedocs.io/en/develop/ |
| Oligominer | Beliveau et al.41 | https://github.com/beliveau-lab/OligoMiner |
| Cooltools | Open2C et al.78 | https://github.com/open2c/cooltools |
| Cool_compartment.py | Wang, Chandra, Goldman, Yoon & Ferrari et al. 79 | https://github.com/VahediLab/TCF13D_code/blob/master/Figure4/cool_compartment.py |
| GenomicRanges v1.46.1 | Lawrence et al.80 | https://bioconductor.org/packages/release/bioc/html/GenomicRanges.html |
| Matrix2insulation.pl | Crane et al.81 | https://github.com/dekkerlab/cworld-dekker/commit/09dd804f8cf9f7522351b97d6b22296b75d2d7f8 |
| Insulation2tads.pl | Crane et al.81 | https://github.com/dekkerlab/cworld-dekker/blob/master/scripts/perl/insulation2tads.pl |
| Stripenn v1.1.65.15 | Yoon et al.14 | https://github.com/ysora/stripenn |
| Coolpup.py v0.9.5 | Flyamer et al.82 | https://github.com/open2c/coolpuppy |
| Signac v1.9.0 | Stuart et al.83 | https://cran.r-project.org/web/packages/Signac/ |
| Samtools 1.11 | LI et al.84 | https://samtools.sourceforge.net |
| ImmGen | The Immunological Genome Project85 | https://www.immgen.org |
| ImageJ | Schneider et al.86 | https://imagej.net |
| ChIP-Atlas | Zou et al.87 | https://chip-atlas.org |
| FlowJo v10.8.2 | TreeStar | https://www.flowjo.com/solutions/flowjo/downloads |
| GraphPad Prism 9 Version 9.5.1 (528), January 24, 2023 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
Acknowledgments
We thank helpful discussions with Atishay Jay and other members of the Vahedi lab in addition to Robert B. Faryabi, Yeqiao Zhou, Kenneth Zaret, Rajan Jain, Sara Cherry, Arjun Raj, and E. John Wherry. We would like to thank Behdad Afzali and his lab for sharing optimizations of the CUT&RUN protocol in T helper cells. We also thank the CRISPR/Cas9 Mouse Targeting Core and Transgenic & Chimeric Mouse Facility for generating the knockout mouse strains. The work in this manuscript was supported by the Burroughs Welcome Fund, the Chang Zuckerberg Initiative Award, and the NIH grant R01AI168240 (J. H-M. and G.V.); W. W. Smith Charitable Trust, the Sloan Foundation, and the NIH grants UC4 DK112217, U01 DK112217, R01 HL145754, U01 DK127768, U01 DA052715 (G.V); the PEW Charitable Trust and the NIH grant R01 HL136572 (J. H-M.); and the NIH grant R01 AI106352 (B. L. K.).
Inclusion and Diversity
We support inclusive, diverse, and equitable conduct of research.
Footnotes
Declaration of Interests
The authors declare no competing interests.
References
- 1.Phanstiel DH et al. Static and Dynamic DNA Loops form AP-1-Bound Activation Hubs during Macrophage Development. Mol Cell 67, 1037–1048 e6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lim B & Levine MS Enhancer-promoter communication: hubs or loops? Curr Opin Genet Dev 67, 5–9 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jiang T et al. Identification of multi-loci hubs from 4C-seq demonstrates the functional importance of simultaneous interactions. Nucleic Acids Res 44, 8714–8725 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tsai A, Alves MR & Crocker J Multi-enhancer transcriptional hubs confer phenotypic robustness. Elife 8(2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Allahyar A et al. Enhancer hubs and loop collisions identified from single-allele topologies. Nat Genet 50, 1151–1160 (2018). [DOI] [PubMed] [Google Scholar]
- 6.Monahan K et al. Cooperative interactions enable singular olfactory receptor expression in mouse olfactory neurons. Elife 6(2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Madsen JGS et al. Highly interconnected enhancer communities control lineage-determining genes in human mesenchymal stem cells. Nat Genet 52, 1227–1238 (2020). [DOI] [PubMed] [Google Scholar]
- 8.Levo M et al. Transcriptional coupling of distant regulatory genes in living embryos. Nature 605, 754–760 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Petrovic J et al. Oncogenic Notch Promotes Long-Range Regulatory Interactions within Hyperconnected 3D Cliques. Mol Cell 73, 1174–1190 e12 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fasolino M et al. Genetic Variation in Type 1 Diabetes Reconfigures the 3D Chromatin Organization of T Cells and Alters Gene Expression. Immunity 52, 257–274 e11 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Link VM et al. Analysis of Genetically Diverse Macrophages Reveals Local and Domain-wide Mechanisms that Control Transcription Factor Binding and Function. Cell 173, 1796–1809 e17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beagrie RA et al. Complex multi-enhancer contacts captured by genome architecture mapping. Nature 543, 519–524 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boudaoud I et al. Connected Gene Communities Underlie Transcriptional Changes in Cornelia de Lange Syndrome. Genetics 207, 139–151 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yoon S, Chandra A & Vahedi G Stripenn detects architectural stripes from chromatin conformation data using computer vision. Nat Commun 13, 1602 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vian L et al. The Energetics and Physiological Impact of Cohesin Extrusion. Cell 173, 1165–1178 e20 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wei GH et al. Genome-wide analysis of ETS-family DNA-binding in vitro and in vivo. EMBO J 29, 2147–60 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hollenhorst PC, McIntosh LP & Graves BJ Genomic and biochemical insights into the specificity of ETS transcription factors. Annu Rev Biochem 80, 437–71 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nye JA, Petersen JM, Gunther CV, Jonsen MD & Graves BJ Interaction of murine ets-1 with GGA-binding sites establishes the ETS domain as a new DNA-binding motif. Genes Dev 6, 975–90 (1992). [DOI] [PubMed] [Google Scholar]
- 19.Dittmer J The biology of the Ets1 proto-oncogene. Mol Cancer 2, 29 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kola I et al. The Ets1 transcription factor is widely expressed during murine embryo development and is associated with mesodermal cells involved in morphogenetic processes such as organ formation. Proc Natl Acad Sci U S A 90, 7588–92 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maroulakou IG, Papas TS & Green JE Differential expression of ets-1 and ets-2 proto-oncogenes during murine embryogenesis. Oncogene 9, 1551–65 (1994). [PubMed] [Google Scholar]
- 22.Kim CJ et al. The Transcription Factor Ets1 Suppresses T Follicular Helper Type 2 Cell Differentiation to Halt the Onset of Systemic Lupus Erythematosus. Immunity 49, 1034–1048 e8 (2018). [DOI] [PubMed] [Google Scholar]
- 23.Lee CG et al. Ets1 suppresses atopic dermatitis by suppressing pathogenic T cell responses. JCI Insight 4(2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Walunas TL, Wang B, Wang CR & Leiden JM Cutting edge: the Ets1 transcription factor is required for the development of NK T cells in mice. J Immunol 164, 2857–60 (2000). [DOI] [PubMed] [Google Scholar]
- 25.Muthusamy N, Barton K & Leiden JM Defective activation and survival of T cells lacking the Ets-1 transcription factor. Nature 377, 639–42 (1995). [DOI] [PubMed] [Google Scholar]
- 26.Ramirez K et al. Gene deregulation and chronic activation in natural killer cells deficient in the transcription factor ETS1. Immunity 36, 921–32 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zook EC et al. The ETS1 transcription factor is required for the development and cytokine-induced expansion of ILC2. J Exp Med 213, 687–96 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen Z et al. In vivo CD8(+) T cell CRISPR screening reveals control by Fli1 in infection and cancer. Cell 184, 1262–1280 e22 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hinds DA et al. A genome-wide association meta-analysis of self-reported allergy identifies shared and allergy-specific susceptibility loci. Nat Genet 45, 907–11 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Paternoster L et al. Multi-ancestry genome-wide association study of 21,000 cases and 95,000 controls identifies new risk loci for atopic dermatitis. Nat Genet 47, 1449–1456 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.O’Shea J & Paul WE Mechanisms underlying lineage commitment and plasticity of helper CD4 + T cells. Science 327, 1098–1102 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ouyang W et al. Inhibition of Th1 development mediated by GATA-3 through an IL-4-independent mechanism. Immunity 9, 745–55 (1998). [DOI] [PubMed] [Google Scholar]
- 33.Kanhere A et al. T-bet and GATA3 orchestrate Th1 and Th2 differentiation through lineage-specific targeting of distal regulatory elements. Nat Commun 3, 1268 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fang R et al. Mapping of long-range chromatin interactions by proximity ligation-assisted ChIP-seq. Cell Res 26, 1345–1348 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mumbach MR et al. HiChIP: efficient and sensitive analysis of protein-directed genome architecture. Nat Methods 13, 919–922 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vahedi G et al. Super-enhancers delineate disease-associated regulatory nodes in T cells. Nature 520, 558–62 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Whyte WA et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153, 307–19 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li L, Leid M & Rothenberg EV An early T cell lineage commitment checkpoint dependent on the transcription factor Bcl11b. Science 329, 89–93 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Isoda T et al. Non-coding Transcription Instructs Chromatin Folding and Compartmentalization to Dictate Enhancer-Promoter Communication and T Cell Fate. Cell 171, 103–119 e18 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Beliveau BJ et al. Versatile design and synthesis platform for visualizing genomes with Oligopaint FISH probes. Proc Natl Acad Sci U S A 109, 21301–6 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Beliveau BJ et al. OligoMiner provides a rapid, flexible environment for the design of genome-scale oligonucleotide in situ hybridization probes. Proc Natl Acad Sci U S A 115, E2183–E2192 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kanno Y, Vahedi G, Hirahara K, Singleton K & O’Shea JJ Transcriptional and epigenetic control of T helper cell specification: molecular mechanisms underlying commitment and plasticity. Annu Rev Immunol 30, 707–31 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Powrie F, Leach MW, Mauze S, Caddle LB & Coffman RL Phenotypically distinct subsets of CD4+ T cells induce or protect from chronic intestinal inflammation in C. B-17 scid mice. Int Immunol 5, 1461–71 (1993). [DOI] [PubMed] [Google Scholar]
- 44.Skene PJ & Henikoff S An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. Elife 6(2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hao Y et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587 e29 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bhat NK et al. Reciprocal expression of human ETS1 and ETS2 genes during T-cell activation: regulatory role for the protooncogene ETS1. Proc Natl Acad Sci U S A 87, 3723–7 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lieberman-Aiden E et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–93 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rao SS et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–80 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pham D et al. Batf Pioneers the Reorganization of Chromatin in Developing Effector T Cells via Ets1-Dependent Recruitment of Ctcf. Cell Rep 29, 1203–1220 e7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhao JY, Osipovich O, Koues OI, Majumder K & Oltz EM Activation of Mouse Tcrb: Uncoupling RUNX1 Function from Its Cooperative Binding with ETS1. J Immunol 199, 1131–1141 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sciammas R et al. Graded expression of interferon regulatory factor-4 coordinates isotype switching with plasma cell differentiation. Immunity 25, 225–36 (2006). [DOI] [PubMed] [Google Scholar]
- 52.Peltonen L, Perola M, Naukkarinen J & Palotie A Lessons from studying monogenic disease for common disease. Hum Mol Genet 15 Spec No 1, R67–74 (2006). [DOI] [PubMed] [Google Scholar]
- 53.Nkonge KM, Nkonge DK & Nkonge TN The epidemiology, molecular pathogenesis, diagnosis, and treatment of maturity-onset diabetes of the young (MODY). Clin Diabetes Endocrinol 6, 20 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kittappa R, Chang WW, Awatramani RB & McKay RD The foxa2 gene controls the birth and spontaneous degeneration of dopamine neurons in old age. PLoS Biol 5, e325 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mateo LJ et al. Visualizing DNA folding and RNA in embryos at single-cell resolution. Nature 568, 49–54 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhou Y et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun 10, 1523 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Beliveau BJ et al. Single-molecule super-resolution imaging of chromosomes and in situ haplotype visualization using Oligopaint FISH probes. Nat Commun 6, 7147 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Team RCR: A language and environment for statistical computing. (R Foundation for Statistical Computing, Vienna, Austria, 2021). [Google Scholar]
- 59.Van Rossum G & Drake FL Python 3 Reference Manual, (CreateSpace, Scotts Valley, CA, USA, 2009). [Google Scholar]
- 60.Andrews S FastQC: A Quality Control Tool for High Throughput Sequence Data. (2010). [Google Scholar]
- 61.Krueger F Trimgalore. (2021). [Google Scholar]
- 62.Li H & Durbin R Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–60 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Institute, B. Picard toolkit. (Broad Institute, GitHub repository, 2021). [Google Scholar]
- 64.Zhang Y et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ramirez F, Dundar F, Diehl S, Gruning BA & Manke T deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res 42, W187–91 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Servant N et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol 16, 259 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Heinz S et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38, 576–89 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Quinlan AR & Hall IM BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–2 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Subramanian A et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–50 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Phanstiel DH, Boyle AP, Heidari N & Snyder MP Mango: a bias-correcting ChIA-PET analysis pipeline. Bioinformatics 31, 3092–8 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Csardi G & Nepusz T The igraph software package for complex network research. InterJournal Complex Systems, 1695 (2006). [Google Scholar]
- 74.Robinson JT et al. Integrative genomics viewer. Nat Biotechnol 29, 24–6 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Phanstiel DH Sushi: Tools for visualizing genomics data. (2021). [Google Scholar]
- 76.Raj A, van den Bogaard P, Rifkin SA, van Oudenaarden A & Tyagi S Imaging individual mRNA molecules using multiple singly labeled probes. Nat Methods 5, 877–9 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Roayaei Ardakany A, Gezer HT, Lonardi S & Ay F Mustache: multi-scale detection of chromatin loops from Hi-C and Micro-C maps using scale-space representation. Genome Biol 21, 256 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Open2C et al. Cooltools: enabling high-resolution Hi-C analysis in Python. BioRxiv (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wang W et al. TCF-1 promotes chromatin interactions across topologically associating domains in T cell progenitors. Nat Immunol 23, 1052–1062 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lawrence M et al. Software for computing and annotating genomic ranges. PLoS Comput Biol 9, e1003118 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Crane E et al. Condensin-driven remodelling of X chromosome topology during dosage compensation. Nature 523, 240–4 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Flyamer IM, Illingworth RS & Bickmore WA Coolpup.py: versatile pile-up analysis of Hi-C data. Bioinformatics 36, 2980–2985 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Stuart T, Srivastava A, Madad S, Lareau CA & Satija R Single-cell chromatin state analysis with Signac. Nat Methods 18, 1333–1341 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Li H et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–9 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Aguilar SV et al. ImmGen at 15. Nature Immunology 21, 700–703 (2020). [DOI] [PubMed] [Google Scholar]
- 86.Schneider CA, Rasband WS & Eliceiri KW NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9, 671–5 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zou Z, Ohta T, Miura F & Oki S ChIP-Atlas 2021 update: a data-mining suite for exploring epigenomic landscapes by fully integrating ChIP-seq, ATAC-seq and Bisulfite-seq data. Nucleic Acids Res 50, W175–W182 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Henao-Mejia J et al. Generation of Genetically Modified Mice Using the CRISPR-Cas9 Genome-Editing System. Cold Spring Harb Protoc 2016, pdb prot090704 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Laffont S et al. Androgen signaling negatively controls group 2 innate lymphoid cells. J Exp Med 214, 1581–1592 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the HiC, CUT&RUN, ATAC-seq and RNA-seq reported in this study is NCBI GEO: GSE211178.