

Abstract
Understanding allosteric regulation of proteins is fundamental to our study of protein structure and function. Moreover, allosteric binding pockets have become a major target of drug discovery efforts in recent years. However, even though the function of almost every protein can be influenced by allostery, it remains a challenge to discover, rationalise and validate putative allosteric binding pockets. This review examines how the discovery and analysis of putative allosteric binding sites has been influenced by the availability of centralised facilities for crystallographic fragment screening, along with newly developed computational methods for modelling low occupancy features. We discuss the experimental parameters required for success, and how new methods could influence the field in the future. Finally, we reflect on the general problem of how to translate these findings into actual ligand development programs.
Introduction
The concept of allostery is fundamental to our understanding of biomolecular reactions. It means that structural perturbations away from the active site, can lead to modulation of protein function. The field was initially defined by the KNF (Koshland-Nemethy-Filmer) [1] and MWC (Monod-Wyman-Changeux) [2] models which were derived from oligomeric proteins where allosteric regulation was described through conformational changes between well-defined structural states. But the concept was considerably refined with the introduction of the energy landscape model [3] and the realisation that allostery can manifest itself without discernible conformational changes [4]. This has eventually resulted in a unified view of allostery [5,6] where proteins are now seen as an ensemble of structures which populate different conformations with different energies [3]. Nevertheless, allostery remains challenging to rationalise, because structural changes are often minute and differences in free energy may be dominated by changes in entropy related to protein, solvent, or ligand dynamics [4,7].
Allostery is not only fundamental to our understanding of protein structure and function, but also highly relevant to drug discovery. Extracellular targets like G-protein-coupled receptors (GPCRs) have a long history of allosteric modulators [8], and there is new attention on exploiting allostery in intracellular targets like kinases [9] or GTPases [10]. Small molecules that bind in allosteric binding pockets open up new possibilities to target proteins involved in disease, especially those that have previously been deemed “undruggable”. Allosteric compounds may present an alternative targeting strategy for targets that have been avoided because their active sites are too conserved: allosteric binding pockets tend to be less conserved, making them more tractable for developing target-specific compounds [11].
Recent successes also demonstrate how allosteric molecules can circumvent other problems of designing small molecules against orthosteric sites [12]. For example, Ras has been a posterchild for “undruggable” proteins because it has high affinity at its active site for its highly abundant natural ligands (GTP/GDP). Recently efforts identified molecules that bind allosteric binding pockets achieving reasonable potency either through covalent linkage to a nearby mutant cysteine [13] or by targeting the switch I/II pocket which was previously deemed undruggable [14]. A second example where allosteric modulation is a better strategy comes from the family of protein tyrosine phosphatases (PTPs). Their active sites are generally highly positively charged, to accommodate the negative charge on the phosphotyrosine. Consequently, in vitro inhibitors of phosphatases are generally highly charged as well, meaning they cannot cross cell membranes. A clever screening strategy was employed on SHP2 PTP, which counter-selected against hits that were likely to target the orthosteric site, to yield a molecule that inhibits allosterically by “gluing” the regulatory SH2 domains over the active site [15]. Molecules designed to bind this site, and another allosteric pocket [16] have favourable drug-like properties, avoiding the problems that have faced PTP inhibitors in the past. Thus, although allosteric pockets may generally be smaller than orthosteric pockets, this does not prevent their drugability [14].
Despite the practical and theoretical importance of allosteric regulation, it remains a major challenge to describe the energy landscape of proteins, as well as to predict sites and investigate the effect of modulating agents. Most computational and experimental methods interrogate only one aspect and invariably require orthogonal experiments [17]. Instead, crystallography has the potential to provide comprehensive insights from a single approach, now that fragment screening directly in crystals has become routinely accessible. This review examines how the directly observed map of protein-ligand interactions across the entire protein surface might allow allosteric opportunities to be directly inferred, and indeed be more thoroughly probed through chemical elaboration of fragments hits to achieve binding potency; and what the future holds for this approach.
Crystal packing does not preclude studying protein dynamics
Protein crystals tend to be seen as static entities and therefore unsuitable for studying allosteric events, given that these involve conformational changes. However, it has been known for a long time that proteins remain catalytically active within crystals [18], despite the environment within a crystal lattice having little resemblance to the actual cellular neighbourhood of a protein. Proteins are highly dynamic molecules, but even though the lattice restrains their accessible conformational space (figure 1), they retain a remarkable degree of plasticity [19] and often display surprisingly large-scale conformational changes during ligand binding [20] (supplementary movie 1). The accessible conformational space depends on how the protein has packed in a given crystal form, and analysis of multiple crystal forms gives a more complete picture of the conformational landscape [21].
Figure 1.
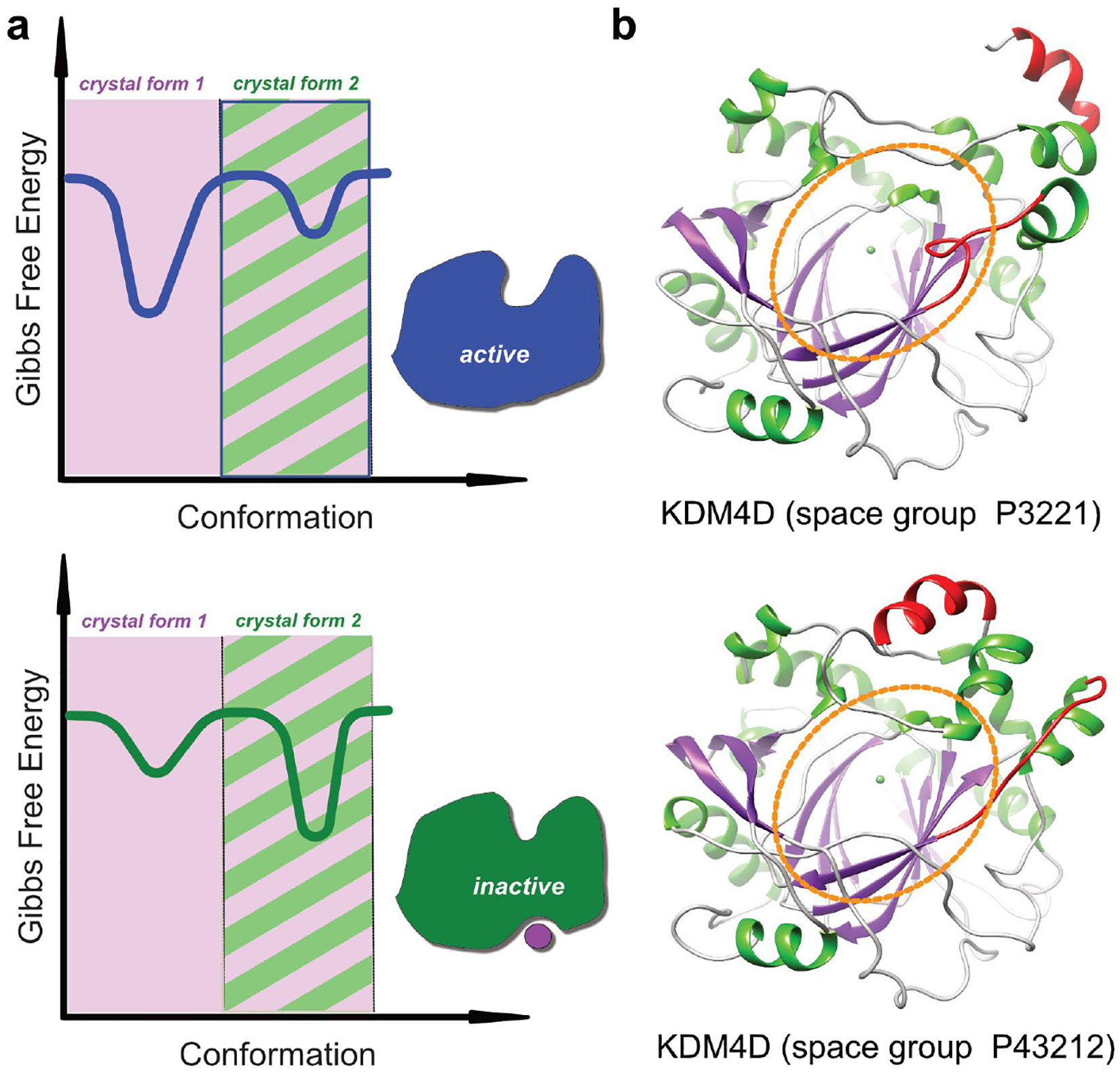
Accessible conformational landscape in protein crystals. (a) The plots show a schematic representation of the energy landscape. Binding of an allosteric effector molecule leads to a conformational change which shifts the energy landscape from the active (top) to the inactive form (bottom). The shaded regions indicate the accessible conformational space in two different crystal forms. In the example, crystal form 1 can accommodate conformational changes which accompany binding of an allosteric effector, whereas crystal from 2 can only accommodate one conformation. (b) Structure of human KDM4D in two different crystal lattices. KDM4D is a histone demethylase and the region encircled in orange indicates the active site pocket. The protein conformation crystallized in space group P3221 is identical with a structure of KDM4D in complex with H3K9Me3, whereas the conformation crystallized in space group P43212 has a distorted active site pocket and appears to represent an inactive conformation. Hence, any ligands that are able to stabilize this unusual conformation, whether by binding to the active site or to any other conformation-specific pocket, should in principle be able to serve as inhibitors. The conformation observed in space group P43212 is stabilized through a loop which is part of the active site (coloured in red), but which is involved in crystal contacts and which therefore does not allow conformational changes that would lead to a properly formed active site pocket.
This means there is in fact a vast need to get better at discovering multiple crystal forms for any crystallizing protein, because the energy landscape model highlights that unexpected conformations cannot simply be dismissed as “crystallographic artefacts”, since crystal contacts are weak. Instead, it is far more instructive to think of crystals as capturing (purifying) low-energy states of the protein, even ones that are not highly populated under some physiological conditions (figure 1b). Any such conformation is thus relevant for study and targeting with small molecules. Different crystal forms are most often obtained by chance and it is still not possible to produce crystal forms on demand, but productive approaches include matrix seeding [22], changing expression constructs [23] or surface mutations [24,25].
Recently, new methods have been devised which explicitly take advantage of the structural plasticity of proteins in crystals to study allostery. Ranganathan and co-workers monitored the structural changes caused by an electric field through time-dependent Laue diffraction experiments [26], whereas Fraser and co-workers have established multi-temperature crystallography as a means to reliably map allosteric networks by defining and explicitly modeling structural elements that populate different conformations as the data collection temperature is shifted in steps from the standard 100K to 273–310K [27].
Crystallographic fragment screens modulate the energy landscape
The use of X-ray crystallography to probe the binding characteristics of the entire protein surface is not new, and several elegant studies in the 90s mapped surface interactions by immersing cross-linked protein crystals in pure organic solvents [28,29].
At around the same time, fragment-based lead discovery (FBLD) emerged as an alternative to high-throughput screening (HTS) for inhibitor discovery [30]. The basic concept is simple: chemical starting points for inhibitor development are identified by screening simple, fragment-like compounds, rather than complex, lead-like molecules. Fragment libraries can be orders of magnitude smaller than HTS libraries, because their low molecular weight and consequential low molecular complexity means they sample chemical space more efficiently [31]. The drawback is that fragments bind weakly, so the screening technique and validation cascade are crucial [32], and accordingly many methodologies have been explored and refined over the years [33]; but the earliest approach [34], of soaking compounds into crystals to directly observe binding structurally, has remained relevant throughout.
The primary aim of most screening campaigns is to find starting points for chemical probe or drug discovery projects, which therefore tend to focus on hits found in well-characterised, orthosteric sites. Nevertheless, screening by X-ray crystallography routinely finds fragments bound all over the protein surface (Figure 3) [11,35,36] and fragment binding to such secondary pockets can lead to rearrangements around the site of interaction which can result in subtle perturbations in distant parts of the structure (figure 2). However, to consider a site truly allosteric requires orthogonal experiments to establish whether modulating them has measureable biochemical effects, and therefore they are here denoted “putative allosteric” pockets to highlight the tentative nature of these non-orthosteric binding events. While binding is often too weak to give a measurable signal in biophysical or biochemical assays, these direct observations can provide a blueprint for the analysis of allosteric networks [37]. In other cases, fragment binding can uncover conformational states which deviate considerably from the average [20]. Hence, a global analysis of the individual snapshots obtained from a fragment screen can give a surprisingly dynamic picture of the energy landscape which resemble trajectories from MD simulations (supplementary movies 2 & 3).
Figure 3.
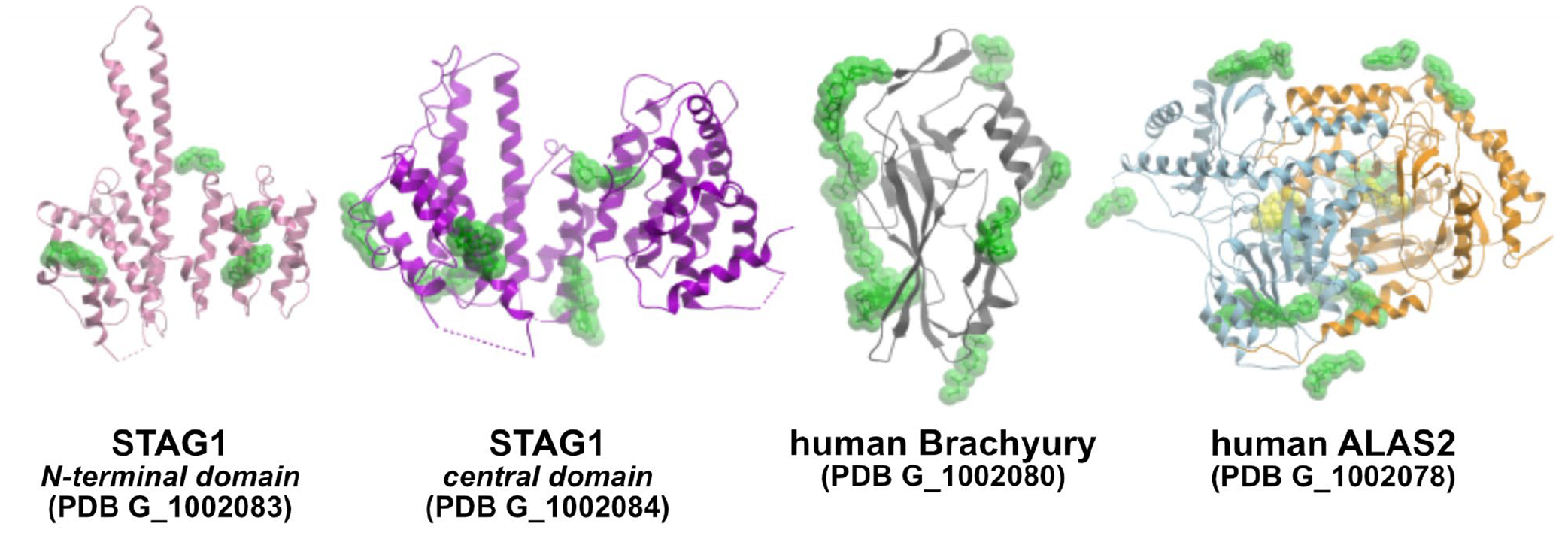
Crystallographic fragment screening comprehensively explores the binding surface of proteins. Overlay of fragment hits from four recent screening campaigns conducted at the DLS XChem facility by SGC Oxford. The structures are available from the PDB under the provided group deposition ID. Additional information about the targets and fragment hits can be found at the SGC website (https://www.thesgc.org/tep). Results from all SGC Oxford fragment campaigns can be accessed via Fragalysis (https://fragalysis.diamond.ac.uk).
Figure 2.
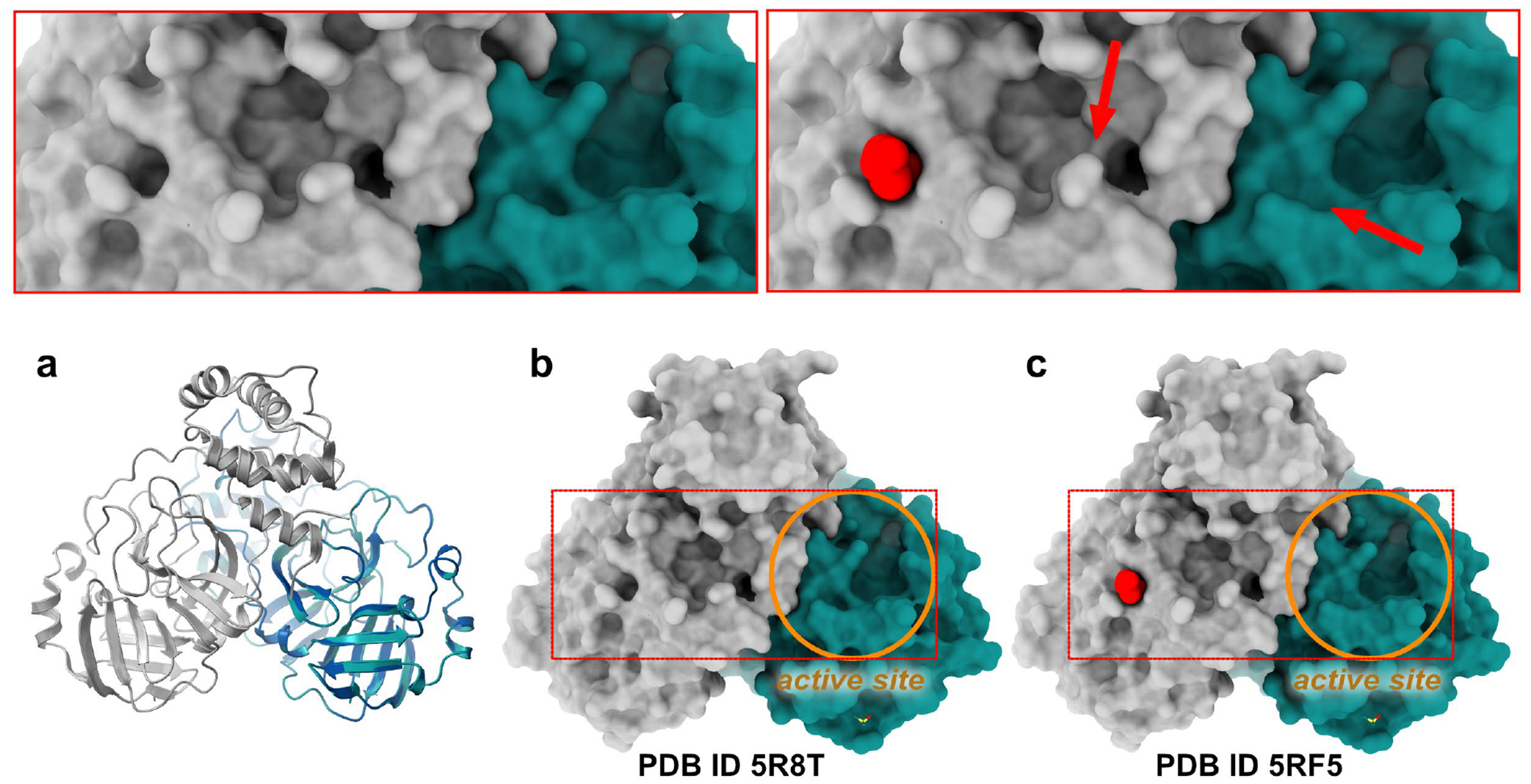
Fragment binding modulates the surface topography of proteins. The figure shows a comparison of the ground-state structure of the SARS-Cov-2 main protease (Mpro) (PDB ID 5R8T) and in complex with a fragment (PDB ID 5RF5). The protein crystallised with one molecule in the asymmetric unit, but forms a dimer in solution which is present as a crystallographic dimer in the crystal. (a) Superposition of the two proteins shows no discernible differences in the main-chain conformation of the two structures. (b,c) Surface representation of ligand-free and fragment-bound Mpro. The active site pocket of the protein of one subunit is indicated with an orange circle, and the fragment (red) binds on the back side of the other subunit. The insets on the top provide a magnified view of the affected region and highlight subtle conformational changes which are caused by fragment binding and which seem to propagate all the way to the active site of the adjacent protomer. In this case, the difficulty remains establishing whether these are merely baseline structural fluctuations, or instead the result of true allosteric signalling.
X-ray crystallography is now a routine method for fragment screening
While fragments are excellent probes for characterizing the binding landscape of proteins, not only are their interactions weak so that binding is difficult to detect [38], but also to associate them with putative allosteric pockets requires direct 3-dimensional readout from X-ray crystallography or NMR spectroscopy. For a long time, crystallography as primary screening method remained comparatively rare, given the technical difficulty to all but a few well-optimised organisations [39,40]. However, the last decade has seen vast technological advances regarding beam intensity, beamline instrumentation, robotics and software which all have resulted in a step-change in terms of productivity [41]. This has led to the development of dedicated, crystallographic screening centres, most notably at the Diamond Light Source (XChem), ESRF/EMBL Grenoble, BESSY (HZB) and MAX IV (FragMAX). These publicly accessible facilities provide platforms which enable screening of hundreds of fragment molecules per day [42]. Availability of these centres has turned crystallography into one of the most popular fragment screening methods [43].
An further major but rarely discussed advantage of the approach is the existence of a well-established data dissemination platform, namely the Protein Data Bank (PDB) [44] (Figure 3), providing a vast and freely-available resource of data for meta-analyses [45]. Other platforms exist, such as the Fragalysis Cloud (https://fragalysis.diamond.ac.uk) [46] that we developed to serve data from all our fragment campaigns.
New algorithms enable identification of low-occupancy conformations
Structural models resulting from X-ray crystallography represent an average of the billions of molecules in a crystal. As a consequence, less-populated, high-energy, conformational states, tend to be ‘blurred out’, even though these minor states remain present in electron density maps [47] and indeed frequently make map interpretation difficult. To make that process less subjective, methods like RINGER [47] and qFit [48] were devised to algorithmically sample electron density maps and thereby expose hidden alternative conformations (figure 4b). Alternatively, ensemble refinement, where local molecular vibrations are sampled by molecular-dynamics (MD) simulations constrained by agreement to the experimental data, can be used to better model the inherent dynamics of protein molecules [49] (figure 4a).
Figure 4.
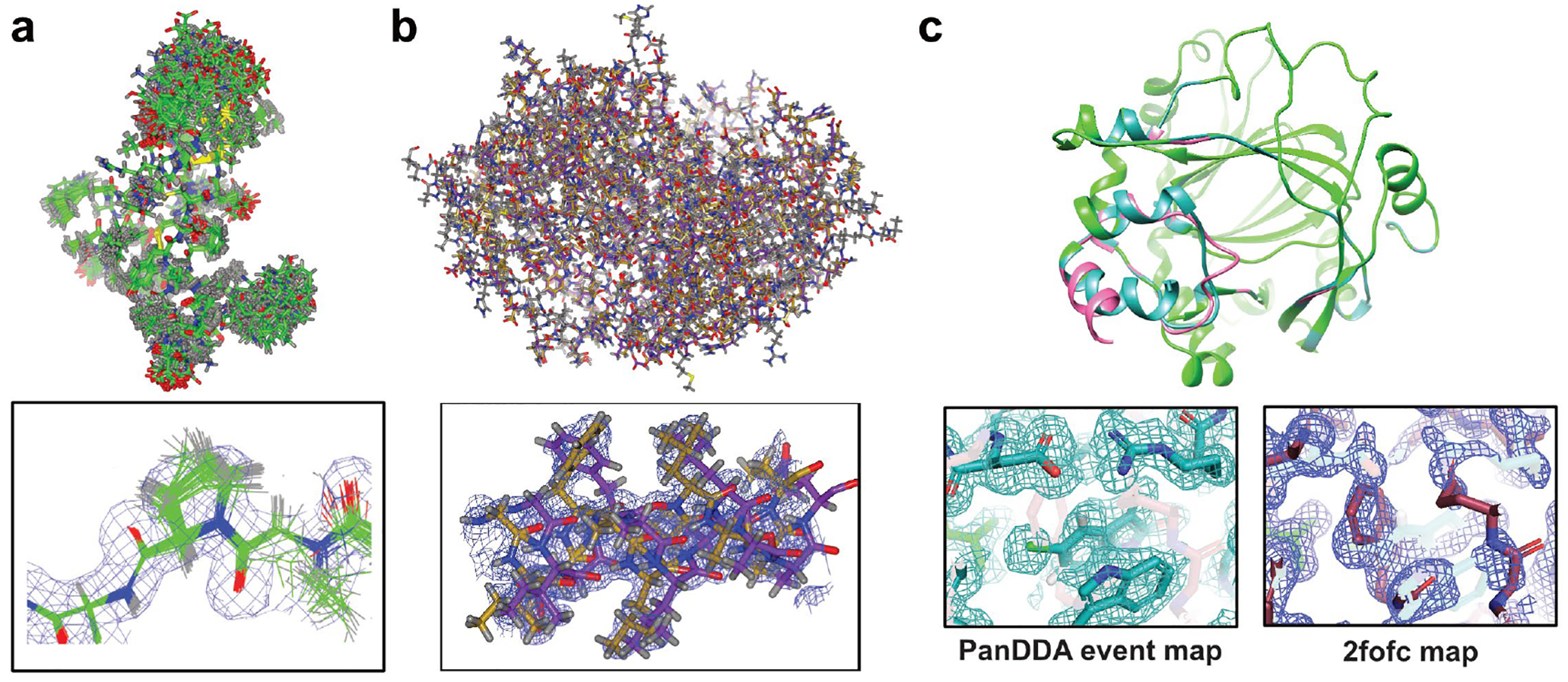
Strategies to model hidden alternative conformations in X-ray crystal structures. (a) MD-driven ensemble refinement (PDB ID 3CA7). (b) Algorithmic sampling of electron density maps with qFit (PDB ID 6NI9). (c) Multi-crystal PanDDA method for extracting low-occupancy conformations (PDB ID 5PHL).
These methods are helpful in finding allosteric networks, but do not directly reveal the ligand-bound conformation on its own, which is problematic for weakly binding fragment which do not bind to a majority of protein molecules in a crystal. The PanDDA (Pan-Dataset Density Analysis) algorithm addresses this by taking advantage of the large number of datasets collected during a fragment campaign to detect partial-occupancy ligands that are not visible in normal crystallographic maps [50]. PanDDA uses a statistical analysis to identify bound ligands and resulting conformational changes, and then generates an “event map” for the bound state of the crystal. The event map approximates what would be observed if the ligand was bound at full occupancy, and is generated by subtracting the unbound fraction of the crystal from the partial-occupancy dataset. The observed structural changes are often imperceptible in standard crystallographic maps and therefore facilitate identification of binding pockets and provide a direct readout of the effect of modulating agents (figure 4c).
Routine room-temperature crystallography opens up new possibilities to study allostery
For decades, routine crystallographic data collection has relied on frozen crystals at 100K, because cryogenic conditions increase crystal lifetime by mitigating radiation damage, and consequently, sample handling and logistics are highly optimized for cryo-crystallography, both at synchrotron sources and home labs [51]. However, cryogenic temperatures tend to mask alternate conformational states that are only accessible at ambient temperatures and therefore bias our conclusions [52,53]. Room temperature data collection is thus of great scientific interest, but until recently has been very difficult to perform, and certainly not routinely.
This is changing fast: the development of new light sources and sample handling procedures has brought a resurgence of room-temperature crystallography. X-ray free-electron lasers (XFELs) can produce radiation-damage free room temperature structures [54,55], but remain difficult to access. Instead, the intensity of state-of-the-art synchrotron sources, combined with ultra-fast detectors, enables many similar experiments [51,56]. Alternatively, dedicated end-stations for in-situ experiments considerably extend experiments that can be made with standard crystal plates [57–59]. These developments will powerfully complement established multi-temperature experiments, which enable mapping of allosteric networks in proteins [37]. Combined information on bound ligands at physiologically relevant temperatures and knowledge of the underlying “connecting rods” will greatly facilitate discovery of allosteric drugs.
Outlook
The self-evident power of structure-based mapping of protein surfaces is tempered by the remaining challenge of assessment biochemical or biological relevance of these pockets. Developing high-affinity small molecule ligands is even more difficult for these pockets than for orthosteric sites, especially if a very specific modulation of protein function is required, so for the time being, complementary approaches will remain vital. Disulphide tethering and mutational studies are established methods for functional validation of putative allosteric pockets [13,60] and the availability of cheap synthetic genes combined with high-throughput purification simplifies their application. Covalent fragments offer an attractive possibility for developing site-specific binders due to their unmatched potency, selectivity, and duration of action [61] and work on new warheads will greatly expand the scope of the method beyond cysteine residues [62]. An interesting new approach comes from the PROTACs field (proteolysis-targeting chimeras) [63]: these bivalent compounds mediate interactions between a target protein and E3 ligases, thereby labelling the protein for cellular degradation by the proteasome. Substrate degradation seems to be driven by cooperative protein-protein recognition, rather than target binding affinity and therefore even suboptimal ligands can end up potent enough when complemented with the right ligase [64]. This conceivably puts many putative pockets in reach of the technique, either for studying in vivo through actual degradation behaviour, or in vitro by using the cooperatively achieved potency to study enzyme or other kinetics.
Crystallographic fragment screens can routinely identify putative allosteric pockets, but they may contain additional, still unrealised insights. Typically, less than 10% of all collected datasets of a screening campaign have fragments bound, so there is a large number of structures available that should allow us to determine crystal form dependent, baseline conformational states. This would enable us to determine whether the observed, subtle perturbations of fragments bound to putative allosteric sites are indeed caused through allosteric communication or are still within expected conformational fluctuations (figure 2).
A key open question in the fragment field is whether crystallographically observed but weakly binding fragments are useful starting points for compound elaboration; this question is even more important for probing putative allosteric sites where the biochemical effect may not even be known, so that only biophysical binding can be assessed. As the fragment field in general develops new technologies and methods for rapid fragment elaboration to address this challenge [65,66], it will directly advance the discovery of allosteric drugging opportunities too.
The advent of 4th generation synchrotron facilities will further increase throughput, enable new experiments and ultimately lead to many more and diverse fragment screening campaigns. In the longer term, this will be complemented by cryo-EM for systems that are not suitable for crystallographic analysis [67]. A big challenge for the coming years will be how to transform the wealth of information from protein-fragment structures and multi-dataset experiments into novel biological and medical insights. We still lack the necessary infrastructure for data presentation and analysis in order to quickly devise new hypotheses and turn them into orthogonal experiments. The sheer amount of available data will require a concerted, interdisciplinary effort in order to harness the full potential of the method. Technical approaches like the Fragalysis cloud computing platform will help tackle these challenges, and initiatives like it will allow not only discovering true allosteric sites, but lead to an increasingly thorough understanding of the structural principles governing allosteric regulation.
Supplementary Material
Acknowledgements
The SGC is a registered charity (number 1097737) that receives funds from AbbVie, Bayer Pharma AG, Boehringer Ingelheim, Canada Foundation for Innovation, Eshelman Institute for Innovation, Genome Canada, Innovative Medicines Initiative (EU/EFPIA) [ULTRA-DD grant no. 115766], Janssen, Merck KGaA Darmstadt Germany, MSD, Novartis Pharma AG, Ontario Ministry of Economic Development and Innovation, Pfizer, São Paulo Research Foundation-FAPESP, Takeda, and Wellcome [106169/ZZ14/Z].
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Koshland DE, Nemethy G, Filmer D: Comparison of Experimental Binding Data and Theoretical Models in Proteins Containing Subunits. Biochemistry 1966, 5:365–385. [DOI] [PubMed] [Google Scholar]
- 2.Monod J, Wyman J, Changeux JP: On Nature of Allosteric Transitions - a Plausible Model. Journal of Molecular Biology 1965, 12:88–118. [DOI] [PubMed] [Google Scholar]
- 3.Frauenfelder H, Sligar SG, Wolynes PG: The Energy Landscapes and Motions of Proteins. Science 1991, 254:1598–1603. [DOI] [PubMed] [Google Scholar]
- 4.Cooper A, Dryden DTF: Allostery without Conformational Change - a Plausible Model. European Biophysics Journal with Biophysics Letters 1984, 11:103–109. [DOI] [PubMed] [Google Scholar]
- 5.Hilser VJ, Wrabl JO, Motlagh HN: Structural and Energetic Basis of Allostery. Annual Review of Biophysics, Vol 41 2012, 41:585–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Motlagh HN, Wrabl JO, Li J, Hilser VJ: The ensemble nature of allostery. Nature 2014, 508:331–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tzeng SR, Kalodimos CG: The role of slow and fast protein motions in allosteric interactions. Biophys Rev 2015, 7:251–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thal DM, Glukhova A, Sexton PM, Christopoulos A: Structural insights into G-protein-coupled receptor allostery. Nature 2018, 559:45–53. [DOI] [PubMed] [Google Scholar]; •• Comprehensive review on the atomic details of allosteric transitions that govern GPCR biology.
- 9.Lu SY, Qiu YR, Ni D, He XH, Pu J, Zhang J: Emergence of allosteric drug-resistance mutations: new challenges for allosteric drug discovery. Drug Discovery Today 2020, 25:177–184. [DOI] [PubMed] [Google Scholar]
- 10.Gray JL, von Delft F, Brennan PE: Targeting the Small GTPase Superfamily through Their Regulatory Proteins. Angewandte Chemie-International Edition 2020, 59:6342–6366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ludlow RF, Verdonk ML, Saini HK, Tickle IJ, Jhoti H: Detection of secondary binding sites in proteins using fragment screening. Proceedings of the National Academy of Sciences of the United States of America 2015, 112:15910–15915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guarnera E, Berezovsky IN: Allosteric sites: remote control in regulation of protein activity. Current Opinion in Structural Biology 2016, 37:1–8. [DOI] [PubMed] [Google Scholar]
- 13.Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM: K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013, 503:548–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kessler D, Gmachl M, Mantoulidis A, Martin LJ, Zoephel A, Mayer M, Gollner A, Covini D, Fischer S, Gerstberger T, et al. : Drugging an undruggable pocket on KRAS. Proceedings of the National Academy of Sciences of the United States of America 2019, 116:15823–15829. [DOI] [PMC free article] [PubMed] [Google Scholar]; • Recent study which gives detailed account of the structure-based development process of an allosterically binding chemical probe for KRAS.
- 15.Chen YN, LaMarche MJ, Chan HM, Fekkes P, Garcia-Fortanet J, Acker MG, Antonakos B, Chen CH, Chen Z, Cooke VG, et al. : Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature 2016, 535:148–152. [DOI] [PubMed] [Google Scholar]
- 16.Fodor M, Price E, Wang P, Lu H, Argintaru A, Chen Z, Glick M, Hao HX, Kato M, Koenig R, et al. : Dual Allosteric Inhibition of SHP2 Phosphatase. ACS Chem Biol 2018, 13:647–656. [DOI] [PubMed] [Google Scholar]
- 17.Lu SY, Ji MF, Ni D, Zhang J: Discovery of hidden allosteric sites as novel targets for allosteric drug design. Drug Discovery Today 2018, 23:359–365. [DOI] [PubMed] [Google Scholar]
- 18.Doscher MS, Richards FM: Activity of an Enzyme in Crystalline State - Ribonuclease S. Journal of Biological Chemistry 1963, 238:2399–2406. [Google Scholar]
- 19.Forneris F, Burnley BT, Gros P: Ensemble refinement shows conformational flexibility in crystal structures of human complement factor D. Acta Crystallographica Section D-Structural Biology 2014, 70:733–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pearce NM, Bradley AR, Krojer T, Marsden BD, Deane CM, Von Delft F: Partial-occupancy binders identified by the Pan-Dataset Density Analysis method offer new chemical opportunities and reveal cryptic binding sites. Structural Dynamics-Us 2017, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tyka MD, Keedy DA, Andre I, DiMaio F, Song YF, Richardson DC, Richardson JS, Baker D: Alternate States of Proteins Revealed by Detailed Energy Landscape Mapping. Journal of Molecular Biology 2011, 405:607–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stewart PDS, Kolek SA, Briggs RA, Chayen NE, Baldock PFM: Random Microseeding: A Theoretical and Practical Exploration of Seed Stability and Seeding Techniques for Successful Protein Crystallization. Crystal Growth & Design 2011, 11:3432–3441. [Google Scholar]
- 23.Gileadi O, Burgess-Brown NA, Colebrook SM, Berridge G, Savitsky P, Smee CE, Loppnau P, Johansson C, Salah E, Pantic NH: High throughput production of recombinant human proteins for crystallography. Methods Mol Biol 2008, 426:221–246. [DOI] [PubMed] [Google Scholar]
- 24.Collins PM, Douangamath A, Talon R, Dias A, Brandao-Neto J, Krojer T, von Delft F: Achieving a Good Crystal System for Crystallographic X-Ray Fragment Screening. Modern Approaches in Drug Discovery 2018, 610:251–264. [DOI] [PubMed] [Google Scholar]
- 25.Derewenda ZS: Rational protein crystallization by mutational surface engineering. Structure 2004, 12:529–535. [DOI] [PubMed] [Google Scholar]
- 26.Hekstra DR, White KI, Socolich MA, Henning RW, Srajer V, Ranganathan R: Electric-field-stimulated protein mechanics. Nature 2016, 540:400–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Keedy DA, Kenner LR, Warkentin M, Woldeyes RA, Hopkins JB, Thompson MC, Brewster AS, Van Benschoten AH, Baxter EL, Uervirojnangkoorn M, et al. : Mapping the conformational landscape of a dynamic enzyme by multitemperature and XFEL crystallography. Elife 2015, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Allen KN, Bellamacina CR, Ding XC, Jeffery CJ, Mattos C, Petsko GA, Ringe D: An experimental approach to mapping the binding surfaces of crystalline proteins. Journal of Physical Chemistry 1996, 100:2605–2611. [Google Scholar]
- 29.Fitzpatrick PA, Steinmetz ACU, Ringe D, Klibanov AM: Enzyme Crystal-Structure in a Neat Organic-Solvent. Proceedings of the National Academy of Sciences of the United States of America 1993, 90:8653–8657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Erlanson DA, Fesik SW, Hubbard RE, Jahnke W, Jhoti H: Twenty years on: the impact of fragments on drug discovery. Nature Reviews Drug Discovery 2016, 15:605–619. [DOI] [PubMed] [Google Scholar]
- 31.Hall RJ, Mortenson PN, Murray CW: Efficient exploration of chemical space by fragment-based screening. Progress in Biophysics & Molecular Biology 2014, 116:82–91. [DOI] [PubMed] [Google Scholar]
- 32.Mashalidis EH, Sledz P, Lang S, Abell C: A three-stage biophysical screening cascade for fragment-based drug discovery. Nature Protocols 2013, 8:2309–2324. [DOI] [PubMed] [Google Scholar]
- 33.Osborne J, Panova S, Rapti M, Urushima T, Jhoti H: Fragments: where are we now? Biochemical Society Transactions 2020, 48:271–280. [DOI] [PubMed] [Google Scholar]
- 34.Hol WGJ: Protein Crystallography and Drug Design. Arzneimittelforschung-Drug Research 1989, 39–2:1016–1019. [DOI] [PubMed] [Google Scholar]
- 35.Bauman JD, Patel D, Dharia C, Fromer MW, Ahmed S, Frenkel Y, Vijayan RSK, Eck JT, Ho WC, Das K, et al. : Detecting Allosteric Sites of HIV-1 Reverse Transcriptase by X-ray Crystallographic Fragment Screening. Journal of Medicinal Chemistry 2013, 56:2738–2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Douangamath A, Fearon D, Gehrtz P, Krojer T, Lukacik P, Owen CD, Resnick E, Strain-Damerell C, aimon A, Ábrányi-Balogh P, et al. : Crystallographic and electrophilic fragment screening of the SARS-CoV-2 main protease. bioRxiv 2020, 10.1101/2020.05.27.118117:2020.2005.2027.118117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Keedy DA, Hill ZB, Biel JT, Kang E, Rettenmaier TJ, Brandao-Neto J, Pearce NM, von Delft F, Wells JA, Fraser JS: An expanded allosteric network in PTP1B by multitemperature crystallography, fragment screening, and covalent tethering. Elife 2018, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Seminal study which combines multi-temperature crystallography and fragment screening to illuminate the allosteric networks governing the protein tyrosine phosphatase PTP1B.
- 38.Murray CW, Rees DC: The rise of fragment-based drug discovery. Nature Chemistry 2009, 1:187–192. [DOI] [PubMed] [Google Scholar]
- 39.Price AJ, Howard S, Cons BD: Fragment-based drug discovery and its application to challenging drug targets. Structure-Based Drug Design: Insights from Academia and Industry 2017, 61:475–484. [DOI] [PubMed] [Google Scholar]
- 40.Spurlino JC: Fragment Screening Purely with Protein Crystallography. Fragment-Based Drug Design: Tools, Practical Approaches, and Examples 2011, 493:321–356. [DOI] [PubMed] [Google Scholar]
- 41.Owen RL, Juanhuix J, Fuchs M: Current advances in synchrotron radiation instrumentation for MX experiments. Archives of Biochemistry and Biophysics 2016, 602:21–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schiebel J, Krimmer SG, Rower K, Knorlein A, Wang XJ, Park AY, Stieler M, Ehrmann FR, Fu K, Radeva N, et al. : High-Throughput Crystallography: Reliable and Efficient Identification of Fragment Hits. Structure 2016, 24:1398–1409. [DOI] [PubMed] [Google Scholar]; • This study gives a detailed overview of the parameters influencing hit rates of a crystallographic fragment screen on the aspartic protease endothiapepsin.
- 43.Erlanson DA: Poll results: affiliation and fragment-finding methods in 2019. Edited by; 2019. [Google Scholar]
- 44.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE: The Protein Data Bank. Nucleic Acids Research 2000, 28:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Giordanetto F, Jin CT, Willmore L, Feher M, Shaw DE: Fragment Hits: What do They Look Like and How do They Bind? Journal of Medicinal Chemistry 2019, 62:3381–3394. [DOI] [PMC free article] [PubMed] [Google Scholar]; • Systematic study analysing binding properties of 489 protein-fragment structures in the PDB.
- 46.Fragalysis on World Wide Web URL: https://diamondlightsource.atlassian.net/wiki/spaces/FRAG/overview
- 47.Lang PT, Ng HL, Fraser JS, Corn JE, Echols N, Sales M, Holton JM, Alber T: Automated electron-density sampling reveals widespread conformational polymorphism in proteins. Protein Science 2010, 19:1420–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Keedy DA, Fraser JS, van den Bedem H: Exposing Hidden Alternative Backbone Conformations in X-ray Crystallography Using qFit. Plos Computational Biology 2015, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Burnley BT, Afonine PV, Adams PD, Gros P: Modelling dynamics in protein crystal structures by ensemble refinement. Elife 2012, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pearce NM, Krojer T, Bradley AR, Collins P, Nowak RP, Talon R, Marsden BD, Kelm S, Shi JY, Deane CM, et al. : A multi-crystal method for extracting obscured crystallographic states from conventionally uninterpretable electron density. Nature Communications 2017, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Description of the PanDDA algorithm which utilises the large number of ligand-free datasets collected during crystallographic fragment screening in order to detect weakly bound ligands.
- 51.Grimes JM, Hall DR, Ashton AW, Evans G, Owen RL, Wagner A, McAuley KE, von Delft F, Orville AM, Sorensen T, et al. : Where is crystallography going? Acta Crystallographica Section D-Structural Biology 2018, 74:152–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fraser JS, Clarkson MW, Degnan SC, Erion R, Kern D, Alber T: Hidden alternative structures of proline isomerase essential for catalysis. Nature 2009, 462:669–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fraser JS, van den Bedem H, Samelson AJ, Lang PT, Holton JM, Echols N, Alber T: Accessing protein conformational ensembles using room-temperature X-ray crystallography. Proceedings of the National Academy of Sciences of the United States of America 2011, 108:16247–16252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Barends TRM, Foucar L, Botha S, Doak RB, Shoeman RL, Nass K, Koglin JE, Williams GJ, Boutet S, Messerschmidt M, et al. : De novo protein crystal structure determination from X-ray free-electron laser data. Nature 2014, 505:244–247. [DOI] [PubMed] [Google Scholar]
- 55.Moreno-Chicano T, Ebrahim A, Axford D, Appleby MV, Beale JH, Chaplin AK, Duyvesteyn HME, Ghiladi RA, Owada S, Sherrell DA, et al. : High-throughput structures of protein-ligand complexes at room temperature using serial femtosecond crystallography. Iucrj 2019, 6:1074–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Weinert T, Olieric N, Cheng R, Brunle S, James D, Ozerov D, Gashi D, Vera L, Marsh M, Jaeger K, et al. : Serial millisecond crystallography for routine room-temperature structure determination at synchrotrons. Nature Communications 2017, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bingel-Erlenmeyer R, Olieric V, Grimshaw JPA, Gabadinho J, Wang X, Ebner SG, Isenegger A, Schneider R, Schneider J, Glettig W, et al. : SLS Crystallization Platform at Beamline X06DA-A Fully Automated Pipeline Enabling in Situ X-ray Diffraction Screening. Crystal Growth & Design 2011, 11:916–923. [Google Scholar]
- 58.le Maire A, Gelin M, Pochet S, Hoh F, Pirocchi M, Guichou JF, Ferrer JL, Labesse G: In-plate protein crystallization, in situ ligand soaking and X-ray diffraction. Acta Crystallographica Section D-Structural Biology 2011, 67:747–755. [DOI] [PubMed] [Google Scholar]
- 59.Sanchez-Weatherby J, Sandy J, Mikolajek H, Lobley CMC, Mazzorana M, Kelly J, Preece G, Littlewood R, Sorensen TLM: VMXi: a fully automated, fully remote, high-flux insitu macromolecular crystallography beamline. Journal of Synchrotron Radiation 2019, 26:291–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Erlanson DA, Wells JA, Braisted AC: Tethering: Fragment-based drug discovery. Annual Review of Biophysics and Biomolecular Structure 2004, 33:199–223. [DOI] [PubMed] [Google Scholar]
- 61.Resnick E, Bradley A, Gan JR, Douangamath A, Krojer T, Sethi R, Geurink PP, Aimon A, Amitai G, Bellini D, et al. : Rapid Covalent-Probe Discovery by Electrophile-Fragment Screening. Journal of the American Chemical Society 2019, 141:8951–8968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gehringer M, Laufer SA: Emerging and Re-Emerging Warheads for Targeted Covalent Inhibitors: Applications in Medicinal Chemistry and Chemical Biology. Journal of Medicinal Chemistry 2019, 62:5673–5724. [DOI] [PubMed] [Google Scholar]
- 63.Lu J, Qian YM, Altieri M, Dong HQ, Wang J, Raina K, Hines J, Winkler JD, Crew AP, Coleman K, et al. : Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chemistry & Biology 2015, 22:755–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gadd MS, Testa A, Lucas X, Chan KH, Chen WZ, Lamont DJ, Zengerle M, Ciulli A: Structural basis of PROTAC cooperative recognition for selective protein degradation. Nature Chemical Biology 2017, 13:514–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bentley MR, Ilyichova OV, Wang GQ, Williams ML, Sharma G, Alwan WS, Whitehouse RL, Mohanty B, Scammells PJ, Heras B, et al. : Rapid Elaboration of Fragments into Leads by X-ray Crystallographic Screening of Parallel Chemical Libraries (REFiL(x)). Journal of Medicinal Chemistry 2020, 63:6863–6875. [DOI] [PubMed] [Google Scholar]
- 66.Cox OB, Krojer T, Collins P, Monteiro O, Talon R, Bradley A, Fedorov O, Amin J, Marsden BD, Spencer J, et al. : A poised fragment library enables rapid synthetic expansion yielding the first reported inhibitors of PHIP(2), an atypical bromodomain. Chemical Science 2016, 7:2322–2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Saur M, Hartshorn MJ, Dong J, Reeks J, Bunkoczi G, Jhoti H, Williams PA: Fragment-based drug discovery using cryo-EM. Drug Discovery Today 2020, 25:485–490. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.