

Abstract
Genome-wide hypertranscription is common in human cancer and predicts poor prognosis. To understand how hypertranscription might drive cancer, we applied our FFPE-CUTAC method for mapping RNA Polymerase II (RNAPII) genome-wide in formalin-fixed paraffin-embedded (FFPE) sections. We demonstrate global RNAPII elevations in mouse gliomas and assorted human tumors in small clinical samples and discover regional elevations corresponding to de novo HER2 amplifications punctuated by likely selective sweeps. RNAPII occupancy at replication-coupled histone genes correlated with WHO grade in meningiomas, accurately predicted rapid recurrence, and corresponded to whole-arm chromosome losses. Elevated RNAPII at histone genes in meningiomas and diverse breast cancers is consistent with histone production being rate-limiting for S-phase progression and histone gene hypertranscription driving overproliferation and aneuploidy in cancer, with general implications for precision oncology.
Keywords: Gene Regulation, Epigenomics, HER2 amplification, Mitochondrial DNA, Meningioma, Whole-arm aneuploidy, Centromeres
Introduction
Upregulation or amplification of oncogenic transcription factors is common in cancer. For example, misregulation of the MYC transcription factor has been observed in most human cancers (1), though exactly how increased MYC binding to gene regulatory elements drives cancer has been controversial (2, 3). More generally, a global increase in transcriptional output, or hypertranscription, is associated with poor prognosis (4, 5). However, it is difficult to reconcile promiscuous incremental increases in expression of thousands of genes with the presumed direct action of oncogenic transcription factors in activating expression of target genes to drive tissue-specific malignancies. Alternatively, hypertranscription in cancer may be relevant only to the subset of genes producing protein products that are rate-limiting for proliferation. For example, the multi-subunit enzyme, ribonucleotide reductase (RNR) – which is required for converting RNA bases to DNA bases – is rate-limiting for DNA synthesis, and RNR activity is a target of widely used anti-cancer catalytic inhibitors (6). Similarly, chromatin components such as histones are rate-limiting for proliferation (7–9), insofar as all newly synthesized DNA must be packaged into nucleosomes every cell cycle, and the 64 genes encoding all 5 histone subunits must produce ~5% of a human cell’s total protein during S-phase (10). RNAPII is so abundant over histone genes during replication that S-phase-specific global depletion of transcription has been observed in mouse embryonic stem cells (9). Replication-coupled (RC) histone mRNAs are not polyadenylated and so are essentially absent from RNA-seq libraries, and the possibility that they drive over-proliferation has been overlooked.
To explore hypertranscription in cancer proliferation, we apply Cleavage Under Targeted Accessible Chromatin (CUTAC) in mouse and human formalin-fixed paraffin-embedded (FFPE) tumor samples (11). We document hypertranscription using antibodies to RNA Polymerase II (RNAPII) in three of four mouse brain tumors and three of seven diverse human tumors. In two other human tumors our method discovered amplifications of the HER2 gene region (12) and likely selective sweeps (13) around other genes nearby. We also found that RNAPII hypertranscription was especially prominent at RC histone genes, leading us to ask whether hypertranscription in cancer is an adaptation to produce more histones at S-phase for more rapid replication. As a critical test of our hypothesis, we performed FFPE-CUTAC on an unselected set of human meningiomas, which are mostly benign human brain tumors that infrequently recur as malignant tumors. We found that FFPE-CUTAC RNAPII occupancy at the 64 RC histone genes predicted WHO grade better than other biomarkers, with similar results for malignant breast tumors. Integration of FFPE-CUTAC data with existing RNA-seq data from 1298 meningiomas accurately separated malignant from benign tumors in predicting recurrence. We also observed striking correlations between levels of RNAPII at histone genes and the number whole-arm chromosome losses in both meningiomas and breast tumors. Successful prediction of tumor aggressiveness based on RNAPII occupancy at histone genes establishes an unanticipated cancer driver paradigm that may also lead to the generation of whole-arm aneuploidies, while opening up diagnostic possibilities not previously considered.
Results
RNAPII Hypertranscription varies between tumors
We recently developed FFPE-CUTAC to directly map RNAPII in fixed tissue samples (11). This provides a DNA-based method for measuring gene expression, instead of RNA-based methods that are limited by RNA instability and variable transcript half-lives. We assessed RNAPII across candidate cis-regulatory elements (cCREs) defined by the ENCODE project, which includes genes and regulatory elements. We compared normal mouse brain tissue to adjacent tumors induced by different transgene drivers: a ZFTA-RELA (RELA) transcription factor gene fusion driving an ependymoma (14), a YAP1-FAM118b (YAP1) transcriptional co-activator gene fusion driving an ependymoma (15), and overexpression of the PDGFB tyrosine-kinase receptor ligand driving a glioma (16). We observed that significantly upregulated cCREs were more frequent than downregulated cCREs (fig. S1). To sensitively detect RNAPII hypertranscription (Fig. 1A), where the absolute change is important, we first counted the number of mapped fragments spanning each base-pair in a cCRE scaled to the mouse genome coverage and averaged the normalized counts over that cCRE. We then plotted Tumor minus Normal (T-N) counts on the y-axis versus the average RNAPII signal (17) on a log10 scale on the x-axis for clarity. This revealed clear hypertranscription (Tumor >> Normal RNAPII) for the RELA tumor (Fig. 1B). Two PDGFB-driven tumors differed in hypertranscription, high in PDGFB-1 (Fig. 1C) and very low in PDGFB-2 (Fig. 1D), whereas the YAP1 tumor showed weak hypotranscription (Fig. 1E). To determine whether hypertranscription is specific to any particular class of regulatory element(s), we divided the data into the five ENCODE-annotated cCRE categories: Promoters (24,114), H3K4me3-marked cCREs (10,538), Proximal Enhancers (108,474), Distal Enhancers (211,185) and CTCF cCREs (24,072). We observed that the five RNAPII hypertranscription profiles are highly consistent with one another (fig. S2), which suggests that RNAPII abundance differences between tumors and normal brains affect all regulatory element classes.
Figure 1: RNAPII-Ser5p FFPE-CUTAC directly maps hypertranscription.

(A) Model for hypertranscription in cancer: Paused RNA Polymerase II (RNAPII) at active gene regulatory elements such as promoters and enhancers increases on average over the cell cycle, resulting in a net proportional gain in RNAPII occupancy across the genome. Using RNAPII FFPE-CUTAC we can map hypertranscription genome-wide using three complementary approaches: 1) Genome-scaled Tumor (T) minus Normal (N) counts at cCREs, 2) T − N at replication-coupled histone genes and 3) SEACR Tumor peak calls using Normal as the background control. (B-E) T − N versus log(T + N)/2 plots showing hypertranscription mapped over the 343,731 annotated mouse cCREs for tumor and normal sections dissected post-tagmentation from a 10 micron FFPE slice from each of four different paraffin blocks. Hypertranscription of a cCRE is defined as the excess of RNAPII-Ser5p in the indicated tumor over normal (Tumor minus Normal in normalized count units for Mm10-mapped fragments pooled from the same slide). (F-M) All fragments were pooled from four slides from the same paraffin block and the number of fragments equalized between tumor and normal for each of the seven cancers. T − N versus log(T + N)/2 plots showing hypertranscription mapped over the 984,834 annotated mouse cCREs for tumor and matched normal sections from 5 micron FFPE slices. Max Diffs displays the Tumor minus Normal maximum of the seven samples for each cCRE. (N-O) For each of the indicated tumors, tracks are shown for 50-kb regions around the #1-ranked cCRE based on Tumor (dark red) and Normal (blue) counts. Raw data tracks were group-autoscaled together for tumor (red) and normal (blue), where SEACR Tumor peak calls (green) use Normal as the negative control. Gene annotations and cCREs (black rectangles) are shown at the top.
To expand on our findings of RNAPII hypertranscription based on transgene-driven mouse brain tumors to a diverse sample of naturally occurring cancers, we obtained 5-μm FFPE sections on slides prepared from paraffin blocks of anonymous human tumor and matched normal sections from the same patient (fig. S3). We performed FFPE-CUTAC and rank-ordered each pair by Tumor minus Normal (T-N) differences to test for RNAPII hypertranscription based on the 984,834 ENCODE-annotated human cCREs. We observed clear hypertranscription in five of the seven tumors (breast, colon, liver, rectum and stomach) and for the composite of all samples (Fig. 1F–M, fig. S4). In contrast, the kidney and lung tumor samples tested showed essentially no hypertranscription, which implies that hypertranscription is a common, but not a defining feature of cancer (2, 4).
We also applied a computational method independent of annotations to assess RNAPII hypertranscription in FFPE-CUTAC profiles of patient samples. The SEACR (Sparse Enrichment Analysis for CUT&RUN) tool is specifically designed for low read-count data (18). To customize SEACR for hypertranscription in cancer, we replaced the background control with the normal sample in each pair, merged fragment data, removed duplicates and equalized read numbers for our seven human Tumor/Normal pairs. SEACR reported a median of 4,483 peaks that were elevated in tumors, whereas when Tumor and Normal were exchanged only a median of 15 peaks elevated in normal tissue were identified, demonstrating that RNAPII hypertranscription is more common than RNAPII hypotranscription in these cancer samples.
We next asked whether SEACR Tumor-versus-Normal peak calls corresponded to the 100 top cCREs ranked by T-N in the overall list representing all seven human tumors. Remarkably, all 100 cCREs at least partially overlapped one or more SEACR Tumor/Normal peak calls, and in addition, the large majority of the 100 top-ranked cCREs intersected with overlapping SEACR peak calls from multiple Tumor/Normal pairs (Table S1). Each of the #1-ranked cCREs in the breast, colon, liver, lung and rectum tumor samples respectively intersected MSL1, RFFL, PABPC1, CLTC and SERINC5 genes and overlapped SEACR peak calls in 4–5 of the 7 tumors (Fig. 1N–O, fig. S5). On average, the same cCRE overlapped SEACR/Normal peak calls in 3.7 of the 7 tumors (Table S1). No SEACR peaks were observed for the kidney sample, as expected given the lack of detectable RNAPII hypertranscription over cCREs. We conclude that the most strongly RNAPII-hypertranscribed regulatory elements tend to be strongly hypertranscribed in multiple human cancers of different types, including liver cancers from different individuals (fig. S6).
Interestingly, we observed a much lower level of mitochondrial DNA (mtDNA) in most tumor samples than in their matched normal samples for both mouse and human (fig. S7A–B), suggesting that these tumors contain fewer mitochondria. To test this interpretation, we mined publicly available ATAC-seq data from both the TCGA and ENCODE projects and observed similar reductions in cancer samples (fig. S7C–D). Such reductions in mtDNA have been reported based on whole-genome sequencing (19).
HER2 amplifications with linkage disequilibrium in human tumors
All but one of the top 25 cCREs are located on Chromosome 17 (Table S1). Eight of these cCREs are within Chr17q12 and 13 are within Chr17q21, each spanning a few hundred kilobases in length in the breast tumor sample not seen in the normal tissue (Fig. 2A–B, fig. S8). For the colon tumor sample, a broad region of RNAPII enrichment is sharply defined within Chr17q21. High RNAPII occupancy over the cCREs in Chr17q12–21 can account for most of the RNAPII hypertranscription signal in the breast and colon samples, centered over the ERBB2 gene (fig. S9). ERBB2 encodes Human Epidermal Growth Factor Receptor 2 (HER2), and is commonly amplified in breast and other tumors and is a target of therapy (12). As our measures of RNAPII hypertranscription are normalized with respect to the human genome coverage, amplification of a region will appear as a proportional increase in the level of FFPE-CUTAC signal, so that we can interpret regional RNAPII hypertranscription in the breast and colon tumor samples as revealing amplification events distinct from the global upregulation that defines hypertranscription.
Figure 2: RNAPII levels identify likely HER2 amplifications and regions of linkage disequilibrium.

(A-B) Normalized count tracks and SEACR peak calls for the 250-kb region on Chromosome 17q21 (A) and for the 250-kb 17q12 region (B) amplified in the breast tumor but evidently not in the colon tumor. Tracks were group-autoscaled together for Tumor (red) and Normal (blue), where SEACR Tumor peak calls (green) use Normal as the negative control. Broad regions of prominent RNAPII hypertranscription indicate likely HER2 amplifications in both tumors. (C-D) Bins of 1-kb were tiled over each 1 Mb region centered on the highest peak in Chr17q21 (C) corresponding to the ERBB2 promoter in Chr17q21, and the RFFL promoter in Chr17q12 (D), and count density within each bin is plotted with curve-fitting and smoothing. Each of the six summits in the Breast tumor sample is centered over the promoter peak indicated by an arrow. (E-H) Same as (C-D) for top-ranked loci outside of the HER2 amplicon. (I) Individual broad summits in (D-E) were zoomed-in and rescaled on the x-axis centered over the indicated promoter peak and superimposed over data tracks scaled to the height of the central peak. (J) Data tracks for the CCNK promoter region, where the normalized count increase in the Breast tumor relative to normal over the 10-kb region shown is 5.4-fold and for Colon is 2.1-fold. The range for the other five tumors is 0.9–2.5.
To confirm that the broad regions of RNAPII enrichment around the ERBB2 promoter correspond to HER2 amplifications in the Breast and Colon patient samples, we applied SEACR broad-peak calling, which densely tiled a ~150 kb region centered over the ERBB2 promoter (Fig. 2A–B). To ascertain whether dense tiling using SEACR can detect amplification events, we called SEACR broad peaks on our published K562 RNAPII-Ser5p CUTAC datasets (20, 21). When we examined Chromosome 22q, we observed a single region heavily tiled with broad SEACR peaks corresponding to an annotated amplification specific for K562 cells. Zooming in revealed that the end of the densely tiled region corresponded precisely to the t(9;22)(q34;q11) translocation breakpoint of BCR-ABL in BCR (fig. S10A–B), which we confirmed by observing a broad SEACR peak on the ABL1 side of the translocation breakpoint (fig. S10C). Thus, our approach using RNAPII-Ser5p CUTAC and SEACR can identify and precisely map regional amplifications such as are found in tumors with BCR-ABL and HER2 amplifications.
To delineate possible RNAPII hypertranscription features within Chr17q12 and Chr17q21, we binned successive 1-kb tiles over each 1 Mb region centered on the highest peak, corresponding to the ERBB2 promoter in Chr17q21 and the RFFL promoter in Chr17q12, and plotted count density within each bin with curve-fitting and smoothing. Remarkably, multiple broad summits appeared in both breast and colon tumor-versus-normal tracks, and the six summits in the breast tumor sample accounted for the six highest ranked Chr17 promoter peaks (Fig. 2C–D). We similarly plotted count densities of the four highest ranking cCREs outside of Chr17q12–21 (Table S1), but tumor peaks in these regions were at least an order-of-magnitude lower than the ERBB2 peaks in the breast and colon tumor samples (Fig. 2E–H). Of the six summits in the breast tumor sample, ERBB2 and MSL1 also appeared in the colon tumor sample, whereas no other samples showed prominent summits above normal in Chr17q12–21 (Fig. 2C–D). MSL1 encodes a subunit of a histone H4-lysine-16 acetyltransferase complex required for upregulation of the mammalian X chromosome (22).
We next superimposed each of the six summits in the Chr17q12–21 region in the breast tumor sample over the genomic tracks on expanded scales for clarity, centered over the highest promoter peak in the region (Fig. 2I). For ERBB2, the ~100 kb broad summit is almost precisely centered over the ~1 kb wide ERBB2 promoter peak. Although the other summits are less broad, each is similarly centered over a promoter peak. Insofar as there are multiple summits much broader than the promoter peaks that they are centered over, our results are inconsistent with independent upregulation of promoters over the HER2-amplified regions. Rather, it appears that a HER2 amplification event was followed by clonal selection for broad regions around ERBB2 and other loci within each amplicon, consistent with the observation of clonally heterogeneous HER2 amplifications in primary breast tumors by whole-genome sequencing (23). Clonal selection may be driven by selective sweeps (24) following amplification events that generate extrachromosomal DNA in double-minute acentric chromosomes, which partition unequally during each cell division (13, 25, 26). Such copy number gains within a tumor can result in intra-tumor heterogeneity (26, 27) and are potential factors for resistance to therapy (28). FFPE-CUTAC thus potentially provides a general diagnostic strategy for detection and analysis of amplifications and clonal selection during cancer progression and therapeutic treatment.
One of the summits in the breast tumor sample absent from the colon tumor sample corresponds to the bidirectional promoters of MED1 and CDK12, both of which have been shown to functionally cooperate with co-amplified ERBB2 in aggressive breast cancer (29, 30). MED1 encodes a subunit of the 26-subunit Mediator complex, which regulates RNAPII pause release, and CDK12 is the catalytic subunit of the CDK12/Cyclin K kinase heterodimer complex, which phosphorylates RNAPII for productive transcriptional elongation (31, 32). We wondered whether the co-amplification of these RNAPII regulators might contribute to hypertranscription in this tumor. As Cyclin K is the regulatory subunit of the CDK12 kinase, we would expect that the CCNK gene that encodes Cyclin K would be strongly upregulated in the breast tumor but not necessarily in the colon tumor. Indeed, we saw a 5.4-fold increase in RNAPII-S5p over the CCNK promoter in the breast tumor relative to adjacent normal tissue, whereas in the colon tumor there is a 2.1-fold increase (Fig. 2J), consistent with RNAPII hypertranscription directly driven in part by CDK12 amplification in this particular patient’s tumor.
RNAPII over histone genes predicts aggressiveness in meningiomas and breast tumors
To evaluate how effectively FFPE-CUTAC can resolve differences between the seven tumor samples, we constructed a cCRE-based UMAP including all 114 individual human datasets with >100,000 mapped fragments (median 925,820). Whereas normal samples produced mixed clusters, tumor samples formed tight homogeneous clusters separated by tissue type (Fig. 3A). This implies that paused RNAPII at regulatory elements is more discriminating between tumors than between the tissues that the tumors emerge from. Relatively few samples and shallow sequencing depths were needed for tight clustering; for example, the stomach tumor cluster comprised four samples from four different experiments with a median of ~470,000 mapped fragments (Fig. 3B). The colon and breast tumor samples, which share HER2 amplifications, clustered immediately adjacent to one another.
Figure 3: RNAPII over histone genes correlates with aggressiveness in meningiomas (A-F) and breast tumors (G-I).

(A) UMAP of 114 human tumor samples and replicates including those from 30 meningiomas. HCC: Hepatocellular carcinoma; ICC: Intrahepatic cholangiocarcinoma. (B) Same as (A) colored for sequencing depth and indicating homogeneous tumor clusters. (C) UMAP of RNAPII FFPE-CUTAC data based on 500-bp bins over the human genome. (D) Same as (C) for cCREs. (E) Same as (C) for the 64 Replication-Coupled (RC) histone genes. (F) WHO grade correlates best with RNAPII occupancy over histone genes. (G) Same as (A) excluding meningiomas but including samples and replicates from 13 additional breast tumors. (H) Same as (E) for breast tumor samples and replicates, colored for tumor type. (I) Same as (H) colored for tumor percentage.
To verify that these differences in global upregulation of cCREs are related to tumor growth, we examined the profiles of the RC histone genes. As these exceptionally S-phase-dependent histone loci are expressed in proportion to the amount of replicated DNA (7), we wondered whether cancer cell hypertranscription functions to increase engaged RNAPII at these loci to load up on histones at S-phase for more rapid cell proliferation. For both the 64 mouse and the 64 human annotated replication-coupled genes we observed differences between tumor samples consistent with RNAPII hypertranscription at cCREs differing between samples (fig. S11). We reasoned that if histone production at S-phase is rate-limiting for proliferation, then we would observe a correlation between cancer aggressiveness and RNAPII, specifically over histone genes. As a test of this hypothesis, we applied FFPE-CUTAC to 4-micron FFPEs from 30 meningioma patients and constructed a UMAP that also included the Tumor-Normal pairs described above. Using RNAPII abundance within 500-bp bins for UMAP construction, we found that the meningiomas clustered separately from other tumors and from normal, and using RNAPII abundance on the cCREs, they formed a single tight cluster (Fig. 3C). When we performed the same UMAP construction using only RNAPII abundance over the RC histone genes, we found that all tumors regardless of type clustered together in a cline that overlapped normal at one end (Fig. 3D). This suggested that the RNAPII at histone genes can distinguish cancer from normal but is insensitive to cancer type differences. In a double-blind test of whether the cline overlapping normal in 2D UMAP space is an indication of tumor aggressiveness, we rank-ordered the 15 samples with WHO grades based on distance from normal samples for RNAPII occupancy in 500-bp bins, cCREs, histone genes or ribosomal protein genes (Fig. 3E). Best performance was for the RC histone genes (r=0.57, p<0.05, Fig. 3F), consistent with our hypothesis that high RNAPII levels at histone genes drive proliferation in cancer.
To determine whether RC histone genes can predict cancer aggressiveness in an invasive tumor type, we performed FFPE-CUTAC on a set of 10-μm breast tumor FFPEs from 13 patients representing three major subtypes. When we included these samples with the 7 diverse tumor and normal samples (Fig. 1F–L) and used cCREs for UMAP construction, we observed tight clustering strictly according to tumor type (Fig. 3G). However, when we used RC histone genes for UMAP construction, we observed a single large cluster at one end of a cline that overlapped normal at the other end (Fig. 3H–I). In total, 24 of the 26 individually amplified samples were entirely within the single large tumor-only cluster. We conclude that RC histone genes alone can predict cancer aggressiveness in both non-invasive meningiomas and multiple invasive breast cancer subtypes.
RNAPII over histone genes accurately predicts rapid recurrence in meningiomas
WHO grade is a coarse predictor of recurrence (33), so to predict rapid recurrence for each tumor sample, we integrated FFPE-CUTAC data with RNA-seq data to use nearest neighbors on a UMAP constructed from available RNA-seq data. We defined a gene as spanning from the 3’-most transcript end through the 5’-most end, stopping when either end of the next gene or LINE element is reached. Based on normalized counts over each RefSeq-annotated human gene, we successfully integrated the public frozen meningioma RNA-seq samples with our 30 meningioma FFPE-CUTAC samples (fig. S12). Remarkably, 17 of 19 matching samples from the same meningioma patient showed near-coincidence of frozen RNA-seq and FFPE-CUTAC.
To predict patient clinical outcomes, we employed a data-driven strategy to classify FFPE-CUTAC samples with overall RNAPII signal at histone genes and leveraged the top 20 shared nearest neighbors of RNA-seq samples to obtain the meningioma patient recurrence information (fig. S13A). Indeed, we found a highly significant association between high histone signals in FFPE-CUTAC samples and rapid patient recurrence (fig. S13B). Using only the normalized FFPE-CUTAC counts at the 64 histone genes, we could distinctly separate the five most rapidly recurring from the 25 other tumors with high significance (p<10−8, Fig. 4A–B left). This observation aligns with the generally low recurrence rate of meningiomas, which are predominantly benign (34). In contrast, levels of RNAPII over Ribosomal Protein genes failed to significantly separate rapidly recurring from benign, regardless of the thresholds applied to designate high Ribosomal Protein gene signal samples (Fig. 4A–B, fig. S13C). We also applied the same logic to determine whether reduced mtDNA is a feature of malignant meningioma but observed no significant difference (Fig. 4A–B, fig. S13D). Although we did observe significant separation of rapidly recurring from benign using Chr22q, the most frequently lost whole-arm (33) (Fig. 4A–B right), and significant separation for Chr1q gains and Chr6p losses (fig. S13E–F), the levels of significance were much lower than for separation using RNAPII at the RC histone genes. Our finding that high levels of RNAPII over RC histone genes accurately predicts poor outcomes in meningioma could imply a causal basis.
Figure 4: RNAPII over histone genes uniquely predicts recurrence in meningiomas.

(A) UMAPs of FFPE-CUTAC and frozen RNA-seq meningioma patient samples grouped by different biomarkers. From the left to right: RNAPII over RC histone genes; RNAPII over ribosomal protein genes; Percent of Chromosome M (chrM = Mitochondrial DNA); Chr22q loss. UMAPs are based on integrating FFPE-CUTAC (circles) with frozen-RNA-seq (triangles) samples (fig. S10) using the canonical correlation analysis. For RC histone and ribosomal protein genes, points are colored by high (red) or low (blue for histone genes and purple for ribosomal protein genes) RNAPII enrichment over the corresponding genes for FFPE-CUTAC samples or their shared nearest neighbor RNA-seq samples. For chrM, points are colored by low (red) or high (dark grey) fractions of Chromosome M fragments, with the hypothesis that a low chrM fraction predicts malignant. For chr22q loss, the FFPE-CUTAC samples with chr22q loss and their RNA-seq samples are colored red, while the ones with chr22q intact are colored green. (B) Kaplan-Meier (KM) plots of recurrence-free rate as a function of time in months based on each grouping strategy using the biomarkers shown in (C). For fair comparisons between Histone genes, Ribosomal Protein genes and chrM, the same number of samples, i.e., the top five patients, are selected in the malignant group. The fraction of the chrM predictor has settings (6 to 18 samples in the malignant group) with small p values. However, the KM curve order is in the wrong direction for the hypothesis that a low fraction of chrM predicts fast recurrence.
RNAPII over histone genes predicts whole-arm chromosome losses
As a positive control for separation of malignant from benign, we counted total aneuploidies from RNA-seq data and observed best separation for five malignant and 25 benign tumors, closely matching our prediction based on RNAPII at histone genes over the range of 3–7 predicted malignant (Figure 5A). This correspondence led us to ask whether there is a relationship between overproduction of histones and total aneuploidy by plotting RNAPII levels over the histone genes for each patient as a function of the total number of whole-arm chromosome gains or losses. We observed a weak non-significant positive Spearman correlation with gains (p<0.2), but a highly significant correlation with losses (p<0.006) (Fig. 5B). To test whether this excess of losses over gains applied to all chromosome arms, we asked whether for each patient, there was a net increase or decrease in the level of RNAPII over histone genes, where whole-arm gains would show a negative correlation with the number of cCREs lacking RNAPII occupancy (no signal over the cCRE) and whole-arm losses would show a positive correlation. Indeed, 38/39 autosomal arms showed a net positive correlation for the meningioma patient population (Fig. 5C, fig. S14A), suggesting that RNAPII at histone genes predicts whole-arm losses in meningioma. We confirmed this excess of whole-arm losses for the breast cancer samples, where we also observed excesses of losses over gains for 38/39 autosomal whole-arm aneuploids (Fig. 5C, fig. S14B), as expected if overexpression of histones drives both over-proliferation and whole-arm chromosome losses in cancer.
Figure 5: RNAPII over histone genes predicts whole-arm chromosome losses.

(A) P-values from the log-rank test for the KM separation of malignant from benign for all thresholds of the 30 meningioma patients and the corresponding 36 FFPE-CUTAC samples for Histone genes (blue), Ribosomal protein genes (purple) and % mitochondrial DNA (grey) (Fig. 4), also including the combined aneuploidy data for all 39 chromosome arms (gold). (B) Scatterplots and Spearman correlations between RNAPII over histone genes and number of whole-arm gains (upper panel) and losses (lower panel) for each of the 30 meningioma patient samples. (C) Spearman correlations between the signal for RNAPII over histone genes and null RNAPII occupancies at cCREs for all 39 autosomal arms for Meningiomas (upper panel) and Breast tumors (lower panel). This summarizes correlation coefficients and significances on the correlation scatterplot in fig. S14. A negative correlation is expected if gains are more frequent than losses (i.e., chromosome arm gain will lead to a lower percentage of cCREs regions with zero RNAPII signals) with increasing RC histone gene signal and a positive correlation if losses are more frequent than gains. The error bar describing the standard deviation of the Spearman correlation coefficient is based on 1000 bootstrap estimation. The significance level is indicated by the stars on the top of each bar with *: p-value < 0.05, **: p-value <0.01, and ***: p-value < 0.001.
Mitotic chromosome segregation errors do not account for loss-over-gain biases
The bias of whole-arm losses over gains in breast tumors is far more significant than in meningiomas (Fig. 5C), presumably because all of the breast tumors but only ~1/4 of the meningiomas are malignant. Bias in favor of loss over gain is counter-intuitive in that a 50% loss should be less fit than a 50% gain, as autosomal trisomies occur in ~0.3% of newborns, but no autosomal monosomies survive to term. Whole-chromosome trisomies result from mitotic segregation errors, such as merotelic attachments, extra centrosomes, unattached kinetochores, loss of the spindle assembly checkpoint or cohesion defects. However, whole-arm aneuploid chromosomes are generated by an initial centromere break, and we wondered whether mitotic segregation errors involving broken centromeres could account for the striking biases in favor of losses over gains that we observed. To test this possibility, we took advantage of the two classes of human chromosomes based on the position of the centromere. Whole-arm gains and losses produced by centromere breaks are far more frequent than focal aneuploidies in cancer (35), and using TCGA data we find that whole-chromosome aneuploids comprise only ~15% of the total (Fig. 6A). Metacentric chromosomes have two euchromatic arms and so require a centromere break to generate a whole-arm SCNA (Fig. 6B). There are five human acrocentric chromosomes (13, 14, 15, 21 and 22), which have similar kinetochore conformations as metacentrics (36), but only a single euchromatic arm. Acrocentrics that are gained or lost by mitotic error will be effectively indistinguishable from those that have undergone a centromere break event. Acrocentric short arms comprise only redundant ribosomal DNA genes and other tandem repeats, and single short-arm gains or losses are not detected in genomic studies. Whole-chromosome meiotic or mitotic segregation errors occur, but centromere position does not predict frequency, based on pre-meiotic mitoses during human oocyte maturation (78% metacentrics expected, 83% observed, n = 52) (37). Thus, if mitotic errors are essential for the generation or perpetuation of a significant number of whole-arm SCNAs, then there should be an excess of acrocentrics (both those with and those without centromere breakpoints) relative to metacentrics (all of which must have centromere breakpoints). To distinguish these models, we have analyzed whole-genome sequencing data from TCGA for acrocentrics and metacentrics from 10,674 patients based on allele-specific copy number segmentation analysis across 33 cancer types. However, we observed no significant difference between acrocentrics and metacentrics in the frequency of whole-arm SCNA gains or losses or both (Fig. 6C, fig. S15). We conclude that centromere breaks are sufficient to generate the excess of whole-arm losses that we observed.
Figure 6: Acrocentric and metacentric whole-arm SCNAs are recovered at similar frequencies in cancer.
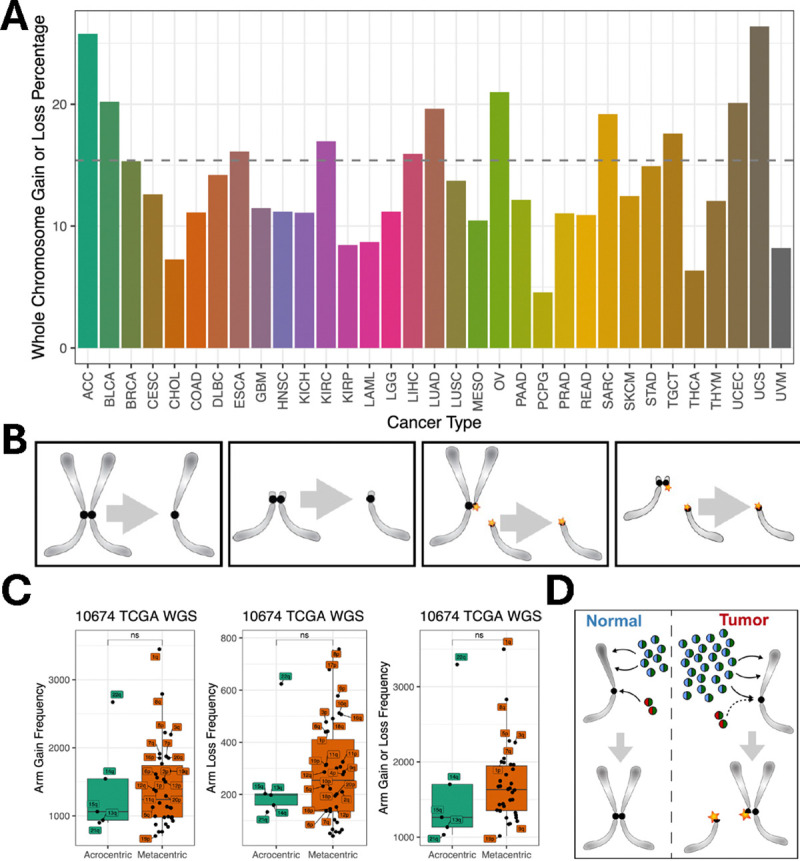
(A) Left to right: A metacentric segregation; An acrocentric segregation; A metacentric segregation following a centromere break; An acrocentric segregation following a centromere break. (B) Summary of whole-genome sequencing data from the Cancer Genome Atlas (TCGA, https://portal.gdc.cancer.gov/) representing 10,674 cancer patients for 33 cancer types (fig. S15). For 32 of 33 cancer types, no significant differences are seen between acrocentrics and metacentrics in the frequencies of whole-arm SCNA gains, losses or both gains and losses (Wilcoxon rank test). Each dot represents a different autosomal chromosome arm (5 acrocentric long arms and 17 metacentrics). (C) We inferred the chromosome arm gain or loss using allele-specific copy number analysis of tumors (ASCAT) (56) profiles for each patient. We summed the major and minor alleles of each segment and took the minimum and maximum of the copy number across segments on each chromosome arm. Any increases in the minimal allelic copy number from the diploid copy number of 2 indicate an arm gain. Similarly, any decreases in the maximum allelic copy number from the diploid copy number of 2 indicate a whole-arm loss for the corresponding autosomal arm. (D) Model for induction of centromere breaks and aneuploidy by histone gene hypertranscription. Histone H3|H4 dimers (blue-green spheres) are deposited along chromosome arms during DNA replication for chromatin duplication, while CENPA|H4 dimers (red-green spheres) are deposited at centromeres and maintain centromere identity. H3|H4 and CENPA|H4 histone dimers are preferentially deposited (black arrows) at the CAF1 and HJURP assembly factors, respectively. However, the excess H3|H4 dimers produced by hypertranscription in cancer cells compete with CENPA|H4 for centromere assembly, reducing centromere function and creating DNA breaks (45, 46). Segregation of broken chromosomes leads to whole-arm aneuploidies.
Discussion
We have demonstrated that elevated RNAPII over genes and regulatory elements is a direct measure of hypertranscription in diverse human cancers and identifies and precisely maps amplifications and selective sweeps in small clinical samples. Likewise, we observed a close correspondence between high RNAPII at the 64 replication-coupled histone genes consistent with cytological evidence of exceptionally high levels of RNAPII at mouse RC histone genes (9) and both RNAPII (7, 8) and Myc (38) at Drosophila histone locus bodies exclusively during S-phase. This led us to hypothesize that the single functional role of hypertranscription in cancer is to produce enough histones to keep up with the requirement for packaging new DNA in cancer cells to proliferate faster than normal cells. We confirmed this prediction by performing FFPE-CUTAC on a set of 30 human meningiomas and showing that RNAPII at the 64 RC Histone genes, which comprise only 1/100,000th of the human genome, successfully estimated WHO grade and accurately predicted rapid recurrence, which corresponds to elevated expression of proliferation genes (33). In sharp contrast, predictions based on RNAPII at Ribosomal Protein genes or mitochondrial DNA abundance failed. Although meningiomas are not invasive, elevated RNAPII at RC histone genes was also observed in invasive breast tumors. The ability of RNAPII FFPE-CUTAC at only the 64 human RC genes to predict aggressiveness in a common intracranial tumor and in multiple breast cancer subtypes implies that a rapid PCR assay for RC histone gene RNAPII or transcription (39) may become an inexpensive general cancer diagnostic tool, revolutionizing precision oncology.
We also found that levels of RNAPII at the 64 RC histone genes correlated with total aneuploidies, which are present in nearly all meningioma patient samples (33), consistent with the occurrence of ~90% whole-arm imbalances in pan-cancer TCGA data (35). Intriguingly, whole-arm losses were observed to be in excess of gains for 38 of the 39 autosomal arms for both meningiomas and breast tumors representing multiple subtypes. Analysis of TCGA data uncovered no evidence that mitotic segregation errors could account for biases of losses over gains. To explain how overproduction of histones might account for this striking whole-arm loss bias in patient tumors, we propose that excess H3 histones compete with CENP-A histones at S-phase for nucleosome assembly at centromeres (40, 41) (Fig. 6D). Production of S-phase histones is tightly regulated (42–44) and overproduction in cancer and displacement of CENP-A nucleosomes are known to result in the generation of DNA–RNA hybrids, likely due to transcription–replication conflicts causing delayed DNA replication, centromere breakage and loss of whole chromosome arms (45, 46). Thus, RNAPII excess over histone genes at S-phase provides a mechanistic basis for understanding not only how cancer cells can proliferate faster than their neighbors but also how the same process might generate centromeric breaks that result in whole-arm imbalances that drive most cancers.
Supplementary Material
Acknowledgements
We thank Christine Codomo and Terri Bryson for technical assistance, the Fred Hutch Genomics Shared Resource for sequencing and data processing and the Fred Hutch Experimental Histopathology Shared Resource for FFPE processing and analysis, Oregon Health Sciences University Knight Biolibrary for breast tumor FFPEs, and Ryan Corces for advice on ATAC-seq data availability. This work was supported by the Howard Hughes Medical Institute (S.H.), National Institutes of Health grant HG012797 (Y.Z.), and grant # T32CA009515 from the National Cancer Institute (R.M.P.).
Footnotes
Competing interests
S.H. is an inventor in a USPTO patent application filed by the Fred Hutchinson Cancer Center pertaining to CUTAC and FFPE-CUTAC (application number 63/505,964). The remaining authors declare no competing interests.
Data and Materials Availability
The sequencing data generated in this study have been deposited in the NCBI GEO database under accession code GSE261351. The raw sequencing data generated from the University of Washington samples are not made available due to data privacy laws. Processed FFPE-CUTAC data for these samples have been deposited on Zenodo and can be accessed with the identifier https://doi.org/10.5281/zenodo.13138686. Custom scripts used in this study are available from GitHub: https://github.com/Henikoff/FFPE.
References
- 1.Dong Y., Tu R., Liu H., Qing G., Regulation of cancer cell metabolism: oncogenic MYC in the driver’s seat. Signal transduction and targeted therapy 5, 124 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Patange S. et al. , MYC amplifies gene expression through global changes in transcription factor dynamics. Cell Rep 38, 110292 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pellanda P. et al. , Integrated requirement of non-specific and sequence-specific DNA binding in Myc-driven transcription. EMBO J 40, e105464 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zatzman M. et al. , Widespread hypertranscription in aggressive human cancers. Sci Adv 8, eabn0238 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cao S. et al. , Estimation of tumor cell total mRNA expression in 15 cancer types predicts disease progression. Nat. Biotechnol. 40, 1624 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huff S. E., Winter J. M., Dealwis C. G., Inhibitors of the Cancer Target Ribonucleotide Reductase, Past and Present. Biomolecules 12, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lu F. et al. , Integrator-mediated clustering of poised RNA polymerase II synchronizes histone transcription. bioRxiv DOI: 10.1101/2023.10.07.561364, (2024). [DOI] [Google Scholar]
- 8.Huang S. K., Whitney P. H., Dutta S., Shvartsman S. Y., Rushlow C. A., Spatial organization of transcribing loci during early genome activation in Drosophila. Curr. Biol. 31, 5102 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mahat D. B. et al. , Single-cell nascent RNA sequencing unveils coordinated global transcription. Nature 631, 216 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Milo R., Phillips R., Cell Biology by the numbers. (Garland, 2016). [Google Scholar]
- 11.Henikoff S. et al. , Epigenomic analysis of Formalin-fixed paraffin-embedded samples by CUT&Tag. Nat Commun 14, 5930 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang H. et al. , HER2 evaluation for clinical decision making in human solid tumours: pearls and pitfalls. Histopathology 10.1111/his.15170, (2024). [DOI] [PubMed] [Google Scholar]
- 13.Yan X., Mischel P., Chang H., Extrachromosomal DNA in cancer. Nat Rev Cancer 24, 261 (2024). [DOI] [PubMed] [Google Scholar]
- 14.Ozawa T. et al. , A De Novo Mouse Model of C11orf95-RELA Fusion-Driven Ependymoma Identifies Driver Functions in Addition to NF-kappaB. Cell Rep 23, 3787 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Szulzewsky F. et al. , Comparison of tumor-associated YAP1 fusions identifies a recurrent set of functions critical for oncogenesis. Genes Dev. 34, 1051 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dai C. et al. , PDGF autocrine stimulation dedifferentiates cultured astrocytes and induces oligodendrogliomas and oligoastrocytomas from neural progenitors and astrocytes in vivo. Genes Dev. 15, 1913 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Altman D. G., Bland J. M., Measurement in medicine: the analysis of method comparison studies. The Statistician 32, 307 (1983). [Google Scholar]
- 18.Meers M. P., Tenenbaum D., Henikoff S., Peak calling by Sparse Enrichment Analysis for CUT&RUN chromatin profiling. Epigenetics Chromatin 12, 42 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reznik E. et al. , Mitochondrial DNA copy number variation across human cancers. eLife 5, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Henikoff S., Henikoff J. G., Ahmad K., Simplified Epigenome Profiling Using Antibody-tethered Tagmentation. bio-protocol 11, e4043 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Janssens D. H. et al. , CUT&Tag2for1: a modified method for simultaneous profiling of the accessible and silenced regulome in single cells. Genome Biol 23, 81 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Deng X. et al. , Mammalian X upregulation is associated with enhanced transcription initiation, RNA half-life, and MOF-mediated H4K16 acetylation. Dev Cell 25, 55 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fan Y. et al. , Characteristics of DNA macro-alterations in breast cancer with liver metastasis before treatment. BMC Genomics 24, 391 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang D. et al. , Lineage tracing reveals the phylodynamics, plasticity, and paths of tumor evolution. Cell 185, 1905 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kaufman R. J., Brown P. C., Schimke R. T., Amplified dihydrofolate reductase genes in unstably methotrexate-resistant cells are associated with double minute chromosomes. Proc. Natl. Acad. Sci. U. S. A. 76, 5669 (1979). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nowell P. C., The clonal evolution of tumor cell populations. Science 194, 23 (1976). [DOI] [PubMed] [Google Scholar]
- 27.Black J. R. M., McGranahan N., Genetic and non-genetic clonal diversity in cancer evolution. Nat Rev Cancer 21, 379 (2021). [DOI] [PubMed] [Google Scholar]
- 28.Schaff D. L., Fasse A. J., White P. E., Vander Velde R. J., Shaffer S. M., Clonal differences underlie variable responses to sequential and prolonged treatment. Cell systems 15, 213 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Forster-Sack M. et al. , ERBB2-amplified lobular breast carcinoma exhibits concomitant CDK12 co-amplification associated with poor prognostic features. The journal of pathology. Clinical research 10, e12362 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marotta M. et al. , Palindromic amplification of the ERBB2 oncogene in primary HER2-positive breast tumors. Sci Rep 7, 41921 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bartkowiak B. et al. , CDK12 is a transcription elongation-associated CTD kinase, the metazoan ortholog of yeast Ctk1. Genes Dev. 24, 2303 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tellier M. et al. , CDK12 globally stimulates RNA polymerase II transcription elongation and carboxyl-terminal domain phosphorylation. Nucleic Acids Res 48, 7712 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thirimanne H. N. et al. , Meningioma transcriptomic landscape demonstrates novel subtypes with regional associated biology and patient outcome. Cell genomics 4, 100566 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goebel S., Mehdorn H. M., Development of anxiety and depression in patients with benign intracranial meningiomas: a prospective long-term study. Support. Care Cancer 21, 1365 (2013). [DOI] [PubMed] [Google Scholar]
- 35.Shih J. et al. , Cancer aneuploidies are shaped primarily by effects on tumour fitness. Nature 619, 793 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sacristan C. et al. , Vertebrate centromeres in mitosis are functionally bipartite structures stabilized by cohesin. Cell 187, 3006 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ghevaria H. et al. , Next Generation Sequencing Detects Premeiotic Errors in Human Oocytes. International journal of molecular sciences 23, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Daneshvar K., Khan A., Goodliffe J. M., Myc localizes to histone locus bodies during replication in Drosophila. PLoS One 6, e23928 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sun R., Qi H., Dynamic expression of combinatorial replication-dependent histone variant genes during mouse spermatogenesis. Gene expression patterns : GEP 14, 30 (2014). [DOI] [PubMed] [Google Scholar]
- 40.Chen C. C. et al. , Establishment of Centromeric Chromatin by the CENP-A Assembly Factor CAL1 Requires FACT-Mediated Transcription. Dev Cell 34, 73 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Blower M. D., Sullivan B. A., Karpen G. H., Conserved organization of centromeric chromatin in flies and humans. Developmental Cell 2, 319 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marzluff W. F., Duronio R. J., Histone mRNA expression: multiple levels of cell cycle regulation and important developmental consequences. Curr. Opin. Cell Biol. 14, 692 (2002). [DOI] [PubMed] [Google Scholar]
- 43.Feser J. et al. , Elevated histone expression promotes life span extension. Mol. Cell 39, 724 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hu Z. et al. , Nucleosome loss leads to global transcriptional up-regulation and genomic instability during yeast aging. Genes Dev. 28, 396 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Giunta S. et al. , CENP-A chromatin prevents replication stress at centromeres to avoid structural aneuploidy. Proc. Natl. Acad. Sci. U. S. A. 118, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Scelfo A. et al. , Specialized replication mechanisms maintain genome stability at human centromeres. Mol. Cell 84, 1003 (2024). [DOI] [PubMed] [Google Scholar]
- 47.Hambardzumyan D., Amankulor N. M., Helmy K. Y., Becher O. J., Holland E. C., Modeling Adult Gliomas Using RCAS/t-va Technology. Translational oncology 2, 89 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hinrichs A. S. et al. , The UCSC Genome Browser Database: update 2006. Nucleic Acids Res 34, D590 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Quinlan A. R., BEDTools: The Swiss-Army Tool for Genome Feature Analysis. Current protocols in bioinformatics 47, 11 12 1 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Martin M., Cutadapt Removes Adapter Sequences From High-Throughput Sequencing Reads. EMBnet.journal 17 DOI: 10.14806/ej.17.1.200, (2010). [DOI] [Google Scholar]
- 51.Langmead B., Salzberg S. L., Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Danecek P. et al. , Twelve years of SAMtools and BCFtools. Gigascience 10, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stuart T. et al. , Comprehensive Integration of Single-Cell Data. Cell 177, 1888 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Serin Harmanci A., Harmanci A. O., Zhou X., CaSpER identifies and visualizes CNV events by integrative analysis of single-cell or bulk RNA-sequencing data. Nat Commun 11, 89 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Clark V. E. et al. , Recurrent somatic mutations in POLR2A define a distinct subset of meningiomas. Nat. Genet. 48, 1253 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ross E. M., Haase K., Van Loo P., Markowetz F., Allele-specific multi-sample copy number segmentation in ASCAT. Bioinformatics 37, 1909 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The sequencing data generated in this study have been deposited in the NCBI GEO database under accession code GSE261351. The raw sequencing data generated from the University of Washington samples are not made available due to data privacy laws. Processed FFPE-CUTAC data for these samples have been deposited on Zenodo and can be accessed with the identifier https://doi.org/10.5281/zenodo.13138686. Custom scripts used in this study are available from GitHub: https://github.com/Henikoff/FFPE.