

Abstract
Spermatogenesis is critical to sexual reproduction yet evolves rapidly in many organisms. High-throughput single-cell transcriptomics promises unparalleled insight into this important process but understanding can be impeded in nonmodel systems by a lack of known genes that can reliably demarcate biologically meaningful cell populations. Tribolium castaneum, the red flour beetle, lacks known markers for spermatogenesis found in insect species like Drosophila melanogaster. Using single-cell sequencing data collected from adult beetle testes, we implement a strategy for elucidating biologically meaningful cell populations by using transient expression stage identification markers, weighted principal component clustering, and SNP-based haploid/diploid phasing. We identify populations that correspond to observable points in sperm differentiation and find species specific markers for each stage. Our results indicate that molecular pathways underlying spermatogenesis in Coleoptera are substantially diverged from those in Diptera. We also show that most genes on the X chromosome experience meiotic sex chromosome inactivation. Temporal expression of Drosophila MSL complex homologs coupled with spatial analysis of potential chromatin entry sites further suggests that the dosage compensation machinery may mediate escape from meiotic sex chromosome inactivation and postmeiotic reactivation of the X chromosome.
Keywords: computational biology, meiotic sex chromosome inactivation, scRNA-seq, spermatogenesis, Tribolium castaneum
Significance.
Sperm development is critical to sexual reproduction yet evolves rapidly in many organisms. Our study presents the first gene expression study of beetle sperm development at single-cell resolution. We develop a new computational approach that phases cells into diploid (premeiotic) and haploid (postmeiotic) cells, which provides an independent test of the computationally derived cell type identifications based on coexpressed genes alone. We find that the genetic pathways underlying beetle spermatogenesis are substantially diverged from previously studied model organisms. The work provides an atlas of genes that may be used to identify specific cell populations and sheds new light on how differentiated sex chromosomes are regulated throughout meiosis.
Introduction
The process of spermatogenesis evolves rapidly at both the anatomical (Fitzpatrick et al. 2022) and molecular levels (Murat et al. 2023) despite having a core function that is essential for sexual reproduction across metazoans. The rapid pace of evolution is likely driven by several factors including sexual selection and genetic conflict imposed by selfish genetic elements (reviewed in Kleene 2005). Additionally, in many male heterogametic species the X and Y chromosomes lack homology across much or all of their length, which imposes challenges for gene regulation and meiotic segregation (Blackmon and Demuth 2015, 2014). In many species, the inability to pair by homology induces meiotic sex chromosome inactivation (MSCI). MSCI occurs when unsynapsed X and Y chromosomes are silenced during the meiotic stages of spermatogenesis and is thought to be related to a phylogenetically widely conserved surveillance system that protects against aneuploid gametes by condensing unsynapsed chromatin (MSUC) (Turner et al. 2005). This has been observed in insects through high-throughput single-cell transcriptome (scRNA-Seq) experiments of both Drosophila melanogaster and Anopheles gambiae testes (Page et al. 2023; Raz et al. 2023a, 2023b). This type of analysis provides high-level detail of cell-specific differentiation in tissue maps but is limited by the need for quality molecular evidence usually associated with well-studied model organisms (Alfieri et al. 2022). Challenges arise when characterizing sequenced cells from nontraditional model organisms such as the red flour beetle (Tribolium castaneum), which share little homology to better studied species.
Here, we investigate gene expression at single-cell resolution throughout T. castaneum spermatogenesis. Tribolium, like many beetles in the suborder Polyphaga, possesses an unconventional mechanism of meiosis wherein the X and Y chromosomes are asynaptic. Early in meiosis they form an “XY parachute”, pairing at a distance by only protein scaffolding that seems to ensure proper segregation (Blackmon and Demuth 2014). This unique feature of beetle spermatogenesis makes them an excellent system to test evolutionary hypotheses about MSCI origins while gaining deeper insights into sex chromosome regulation and chromatin dynamics. Moreover, because T. castaneum is an important stored product pest, understanding gene regulation throughout spermatogenesis may provide additional molecular targets for development of sterile insect technique; one approach to population control that has proven to be safer for humans than traditional pesticide treatment but requires detailed understanding of the insect reproductive system.
The molecular basis of spermatogenesis is comparatively well studied in model organisms like Drosophila (Demarco et al. 2014; Siddall and Hime 2017). However, it has not been as thoroughly investigated in T. castaneum, and there are many genomic differences between Drosophila and Tribolium that could negatively affect homologous marker gene discovery (Pointer et al. 2021). While there are few studies regarding the molecular basis for spermatogenesis in beetles, the cell biology of Tribolium sperm development is relatively well characterized (Dias et al. 2012, 2015; Fishman et al. 2017). The teste organ is oval shaped (Fig. 1a and b) with the germinal stem cell (GSC) hub that contains premeiotic sperm cells located at the distal end of the organ (I), and the seminiferous tubule (III) connected to the proximal end (II) where mature spermatozoa exit the tissue. In the middle of the organ, cyst-cell encased, meiotic spermatogonia undergo successive rounds of mitotic and meiotic division, transitioning from round-shaped, diploid spermatocytes to spindly, haploid, spermatids (Fig. 1c). Independent fluorescent microscopy studies have implicated the roles of Transformer 2 (Tra2), Anastral spindle 1 (Ana1), β2-tubulin, Rad50, and Enolase in beetle sperm development (Fig. 1d) (Shukla and Palli 2013, 2014; Fishman et al. 2017; Khan et al. 2021). Expression of β2-tubulin and Rad50 occur in premeiotic spermatocytes then decrease while Enolase is expressed (Fig. 1d). Expression of β2-tubulin is observed during the transition between spermatids and spermatozoa, when nuclei become more elliptical and a neck is formed to separate the head and body of the sperm cell.
Fig. 1.

Extraction and single-cell sequencing of testes from four adult beetles. a and b) Microscope pictures of adult beetle testes dissected. a) Stereomicroscope image of dissected beetle testes in water. b) DAPI stained slide squash of adult beetle testes. I, Distal end of one teste organ. II, Proximal end of same teste organ; and III, Seminary tubule. c) Magnified views of cell populations for GSC (upper panel) and spermatocytes and spermatids (lower panel). d) Diagram of beetle testes with estimated expression levels of previously validated markers; β2-tubulin (TC009035), Rad50 (TC006703), and Enolase (TC011729). Estimated expression patterns were derived through examination of promoter driven GFP expression in Khan et al., (2021). Numbers represent the number of cells at each stage of mitotic/meiotic division. Spermatids are represented with 29 cells even though there is no division due to each cyst encompassing 2 bundles of antiparallel sperm. The distal end of testes is at the bottom of diagram and proximal end is at the top. e) Expression of previously validated markers for spermatogenic staging across all cells. f) Expression of homologs to tissue markers used to stage spermatogenesis in D. melanogaster and A. gambiae (Table 1). Expression of genes displayed as rows of heatmap with cell expression in each column. g) UMAP projection of weighted principal components showing stage specific clusters. h–j) Expression of previously validated stage specific markers in cells displayed as UMAP projection. k) Switch-like mechanics of β2-tubulin, Rad50, and Enolase for determining pseudotime status of each cell.
Tribolium castaneum uniquely shows interesting dynamics that differentiate its sperm cell development from the sperm cell development found in other insect species. Cyst cells in Tribolium encase two antiparallel packets of elongating cells leading to each cyst containing twice the number of flagellated sperms per straw, which has been suggested to develop due to higher competition between males (Fishman et al. 2017). Analyses of sex chromosome gene expression in Tribolium suggest that dosage compensation follows a pattern similar to that of Drosophila, although the molecular mechanisms are less well understood (Prince et al. 2010; Mahajan and Bachtrog 2015; Whittle et al. 2020). Furthermore, bulk RNA-seq from male gonads show a similar pattern to Drosophila wherein X-linked genes appear to be suppressed but not fully silenced (Whittle et al. 2020). However, the status of dosage compensation and MSCI in germline tissues remains unresolved because previous studies could not differentiate expression from somatic cells that were included in the gonad samples. To better understand sex chromosome regulation in the Tribolium male germline, we employ high-throughput single-cell transcriptome sequencing from testes-derived heterogeneous cell populations and present an atlas of gene expression throughout spermatogenesis. To compensate for the lack of markers for cell type identification and clustering, we develop novel strategies of using nonmarker evidence for sperm stage determination.
Results
Spermatogenesis Differentiation Marker Expression
Pooled single-cell RNA-sequencing (scRNA-seq) from 4 individuals yielded 1,662,669 cells expressing 16,592 genes. Filtering for lowly expressed genes and cells, doublets, and high mitochondrial expression resulted in 14,443 cells expressing 10,817 features (supplementary fig. S1a and b, Supplementary Material online). We observed that low variation in gene expression between potential cell groups resulted in poor spatial subdivision within principal component analysis (PCA) and uniform manifold projection (UMAP) (supplementary fig. S1c and d, Supplementary Material online). Nongermline or cyst somatic cells were predicted and filtered using common cell line markers (supplementary fig. S1e and f, Supplementary Material online) leaving 10,587 potential germ line cells and 3,856 possible cyst cells. Due to limited markers for cyst identification, we included cyst cells for further analyses.
We corroborate from previous reports that β2-tubulin, Rad-50, and Enolase display switch-like patterns of expression across cells, indicating their potential utility for staging cells in spermatogenic time (Fig. 1e, supplementary fig. S1g, Supplementary Material online). These experimentally validated markers demonstrated more temporal coherence than beetle homologs of Drosophila markers used to previously stage Drosophila testes scRNA-seq data (Fig. 1f, Table 1) (Raz et al. 2023a, 2023b). Similarly, UMAP projections derived from principal components of the 400 most variably expressed genes did not result in identifiable features of spermatogenesis (i.e. recognizable clusters) or linear relationships to development (supplementary fig. S1h, Supplementary Material online). This is a common problem in scRNA seq experiments of nonmodel species that lack identifiable markers of cell type and low variability in gene expression between cells (Alfieri et al. 2022).
Table 1.
Markers previously used to identify sperm differentiation in insect species
| Marker | Cell type | Drosophila gene | Tribolium gene | Average expression | Source |
|---|---|---|---|---|---|
| stg | GSC | FBgn0003525 | TcasGA2-TC001885 | 0 | Fly |
| vasa | GSC | FBgn0283442 | – | – | Fly |
| esg | GSC | FBgn0287768 | TcasGA2-TC014474 | 0 | Fly |
| zpg | GSC | FBgn0024177 | TcasGA2-TC005460 | 0 | Fly |
| squid | GSC | FBgn0263396 | TcasGA2-TC000928 | 0.3 | Mosquito |
| geminin | GSC | FBgn0033081 | TcasGA2-TC006282 | 0.08 | Mosquito |
| RacGAP1 | GSC | FBgn0045843 | TcasGA2-TC010549 | 0.19 | Mosquito |
| bam | Spermatogonia | FBgn0000158 | – | – | Fly |
| aub | Spermatogonia | FBgn0000146 | TcasGA2-TC008711 | 0.09 | Fly |
| kmg | Spermatogonia | FBgn0032473 | TcasGA2-TC014806 | 0 | Fly |
| Rbp4 | Primary Spermatocyte | FBgn0010258 | – | – | Fly |
| aly | Primary Spermatocyte | FBgn0004372 | – | – | Fly |
| can | Primary Spermatocyte | FBgn0011569 | TcasGA2-TC011780 | 0.6 | Fly |
| sa | Primary Spermatocyte | FBgn0002842 | – | – | Fly |
| CycB | Secondary Spermatocyte | FBgn0000405 | TcasGA2-TC014544 | 0.53 | Fly |
| β2-tub | Secondary Spermatocyte | FBgn0003889 | TcasGA2-TC009035 | 23 | Mosquito |
| fzo | Secondary Spermatocyte | FBgn0011596 | – | – | Fly |
| Dic61B | Secondary Spermatocyte | FBgn0263988 | TcasGA2-TC034331 | 0 | Fly |
| elf4E-5 | Spermatid | … | – | – | Mosquito |
| Protamine | Spermatid | FBgn0013300 | TcasGA2-TC007828 | 17 | Mosquito |
| Dnah-3 | Spermatid | FBgn0035581 | TcasGA2-TC005485 | 0.21 | Mosquito |
| p-cup | Spermatid | FBgn0030840 | TcasGA2-TC007034 | 0.8 | Mosquito |
| Pp2C1 | Elongating spermatid | FBgn0022768 | TcasGA2-TC034727 | 0.08 | Fly |
| CG6701 | Elongating spermatid | FBgn0033889 | TcasGA2-TC008350 | 3.9 | Fly |
| Parp16 | Elongating spermatid | FBgn0034129 | TcasGA2-TC010849 | 0.003 | Fly |
| Glut3 | Elongating spermatid | FBgn0015230 | – | – | Fly |
| tj | Hub Cyst Cell | FBgn0000964 | TcasGA2-TC033194 | 0 | Fly |
| CG3902 | Hub Cyst Cell | FBgn0036824 | TcasGA2-TC005596 | 0.007 | Fly |
| piwi | Hub Cyst Cell | FBgn0004872 | TcasGA2-TC008711 | 0.09 | Fly |
| Amph | Hub Cyst Cell | FBgn0027356 | TcasGA2-TC032117 | 0.24 | Fly |
| eya | Cyst cell | FBgn0000320 | TcasGA2-TC008985 | 0.09 | Fly |
| Akr1B | Cyst cell | FBgn0086254 | TcasGA2-TC004881 | 2.9 | Fly |
| geko | Cyst cell | FBgn0020300 | TcasGA2-TC031823 | 0 | Fly |
| so | Elongating Cyst cells | FBgn0003460 | TcasGA2-TC030468 | 0.19 | Fly |
| Nep4 | Elongating Cyst cells | FBgn0038818 | TcasGA2-TC006174 | 0.024 | Fly |
| Fas3 | Cyst marker | FBgn0000636 | TcasGA2-TC014942 | 0 | Fly |
Sources of marker genes from previous scRNA-seq studies in Drosophila and Anopheles (Page et al. 2023; Raz et al. 2023a, 2023b). Average expression refers to normalized mean expression across cells for each marker.
To resolve cell types in the absence of marker homology with other model systems, we used weighted principal components generated from markers independently shown in other studies to be differentially expressed in germline cell types (supplementary table S1, Supplementary Material online) to generate UMAP projections and Leiden clusters. Weighted principal components yielded a higher degree of separation between cell type clusters (Fig. 1g, supplementary fig. S1i, Supplementary Material online). We also observed a greater linear relationship in UMAP space between our three marker genes (Fig. 1h–j) which we confirmed by pseudotime analysis using Ouija (Fig. 1k). We also found that marker expression independently matched the experimentally observed spatial expression (Fig. 1d, Khan et al. 2021), with early expression switch behavior on the left side of the UMAP space and a similar pattern of expression occurring as cells move to the right. This could indicate that the UMAP space is temporally divided from early stage germ cells (left) to later stage matured spermatozoa (right). This marker gene expression was repeated in spatially separated cell groupings indicating redundant clustering's and tell us nothing of other relevant cell populations like cyst cells. Ouija pseudotime also displayed higher linearity in UMAP projection from weighted principal components than by those using variable genes alone (supplementary fig. S1j, Supplementary Material online).
Tribolium Lacks Classical Marker Genes for Spermatogenesis
We found little correlation between Drosophila or Anopheles markers of spermatogenesis and marker expression in Tribolium (Table 1; supplementary fig. S1k–m, Supplementary Material online) (Page et al. 2023; Raz et al. 2023a, 2023b). This is because essential markers for sperm staging in the two dipterans such as vasa, fuzzy onions (fzo), or bag of marbles (bam) (Raz et al. 2023a, 2023b) do not seem to play the same role in beetle spermatogenesis. While interesting from an evolutionary perspective, the lack of functional homology provided a challenge for clustering cells by spermatogenic stage. To address this, we chose to look at markers of stage specific cell transition that were not specific to previously discovered experimental evidence in other species. Because spermatogenesis is a dynamic process in which cells at different stages of spermatogenesis are likely to be found at different states of mitotic and meiotic division, we decided to use a classifier of mitotic cycle stage to help identify cluster identity. Homologs of genes used to phase mitotic cycle in Drosophila were variably expressed across clusters (Fig. 2a and b).We found that predicted clusters lined up with markers of mitotic and meiotic activity (Fig. 2c) and predicted mitotic phase, with G2M being highest at early mitotic division in Germline Stem Cells (GSC) and spermatogonia and the end of meiosis II in primary spermatocytes.
Fig. 2.
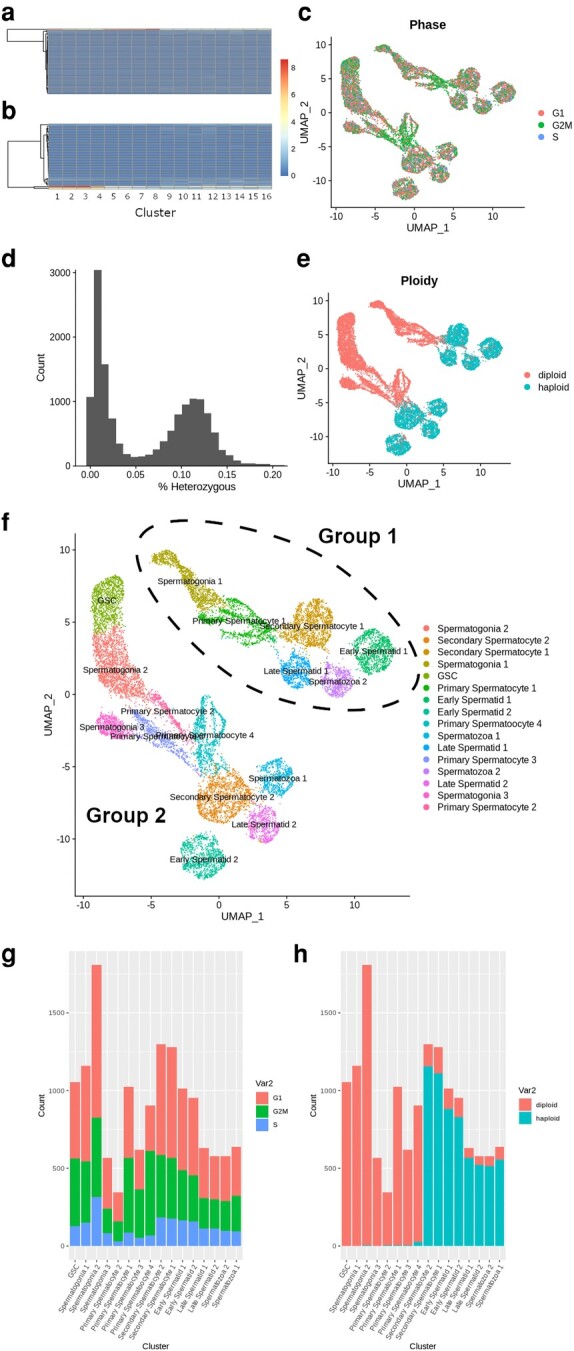
Identification of cluster cell types using phasing evidence. a and b) Expression of a) G2M and b) S phase specific expression markers as rows across cell clusters in columns. c) Estimated mitotic phase of each cell displayed in UMAP projection from homologous Drosophila markers. d) Histogram showing the distribution of cells as a function of their percent heterozygosity. Heterozygosity estimated from SNPs in sequenced reads may not represent actual allele status due to low sequencing coverage and variable gene expression. e) Ploidy status of each cell as inferred from cells < 5% heterozygous across alleles. f) Inferred cell type using multiple evidences. Parallel clusters independently separated during UMAP construction represent 2 distinct groups labeled group 1 and group 2. g and h) Stacked barplots showing the total numbers of g) G1, G2M, and S and h) haploid and diploid phased cells in each cluster.
We can also identify clusters through their ploidy state, as cells that have recently undergone Meiosis I telophase division will be found at the secondary spermatocyte stage. We further attempted to computationally validate our cell clustering by differentiating postmeiotic, haploid cells, from meiotic and premeiotic, diploid cell populations. By predicting SNPs from sequenced RNA, we were able to predict the heterozygous content of each cell at all alleles (Fig. 2d). Because haploid cells are in theory hemizygous, which would appear in the data as high homozygosity, we used a cutoff of 2.5% heterozygosity to phase diploid cells (Fig. 2e) which represented secondary spermatocytes, spermatids and spermatozoan cells in our data. In our dataset we found 8,283 diploid cells and 6,180 haploid cells. Using this information, rudimentary labels were given to clusters based on relative values of mitotic, ploidy, and temporal data (Fig. 2f). While there was still some mixture of cell types likely left over, the number of clusters matched with our expected number of germline cell types, the proportion of cells at mitotic stages matched with mitotically active cell types (Fig. 2g), and transition to diploid occurred immediately in almost 90% of cells within the cluster determined to be secondary spermatocytes (Fig. 2h), which is expected to occur following telophase of meiosis I. Cluster identities were refined and confirmed through differential expression of markers important to stage dependent activity (supplementary table S2, Supplementary Material online).
While clusters evenly represented stages of spermatogenesis, we observed two distinct groups of clusters, which we refer to as group 1 and group 2. We found that there were 7,659 cells in group 2 and 6,804 cells in group 1 and there was proportional representation of both clusters and sequenced individuals in both groups (supplementary fig. S2a, Supplementary Material online, supplementary table S3, Supplementary Material online). The relative proportion of cell count in each cell type matched roughly with the cell density of regions of the testes measured by microscopy (supplementary fig. S2b, Supplementary Material online).
Nonclassical Markers of Sperm Differentiation in Tribolium
While nonexpression markers (e.g. ploidy) can confirm rough temporal division in spermatogenesis, we also employed additional methods that provide more context in our analysis. Specifically, the pseudotime of our validated marker genes lined up perfectly with UMAP projections and predicted clusters (Fig. 3a and b). We also observed linear relationships in the UMAP projections when plotted with pseudotime and RNA velocity values (Fig. 3c and d); however, the RNA velocity displayed recursive patterns that pseudotime did not. We have confirmed, however, that the velocity equation itself does not derive a linear relationship from the specific markers that have been shown to stage spermatogenesis in beetles (supplementary fig. S2c, Supplementary Material online).
Fig. 3.
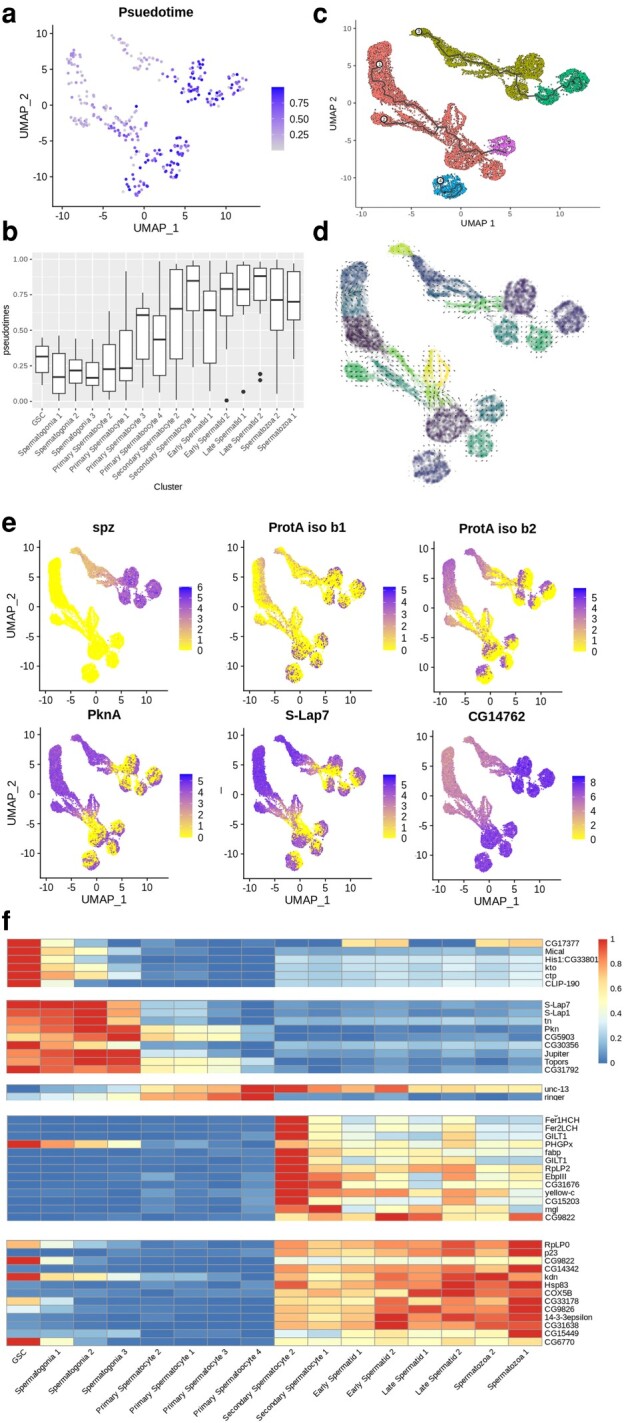
Differentiation trajectory and marker expression of beetle testes. a and b) The linear development of sperm cells as inferred by ouija pseudotime analysis of β2-tubulin, Rad50, and Enolase is consistent with labeled clusters. Reported as per cell expression on a) UMAP projection and b) per cluster boxplot. c and d) Trajectories are also reported as determined by c) monocle v3 and d) SCvelo RNA velocity. Cells are colored by their cluster identification. e) Markers that show some differentiation and linear relationships between or within clusters are reported as expressions of each cell in UMAP projection. f) Markers identified from monocle-derived gene expression markers and represented as normalized average expression values across clusters on a heatmap.
Due to the lack of differentiation markers in beetles that have been identified in other species, it was important to find potential unique markers that could be used to predict cell types in beetle spermatogenesis. Some markers that have been shown through molecular evidence to correlate with stages of spermatogenesis in Tribolium, such as Tra2 and Ana1, were lowly expressed in the single-cell data (supplementary fig. S2d and e, Supplementary Material online) (Shukla and Palli 2013; Fishman et al. 2017).
A majority of the highest expressed genes did not show variable patterns of expression across cell clusters, but a few genes were identified that did. The most striking expression pattern we discovered was that of the Tribolium homolog of Spaetzle (spz), which was expressed in group 1 clusters but not group 2 (Fig. 3e). Two isoforms of Protamine A (ProtA) exhibited strange expression patterns with isoform b1 showing variable and polar expression within all clusters and groups, while b2 only showed variable expression within secondary spermatocytes, spermatids, and spermatozoa (Fig. 3e). These split cluster expression patterns were not opposite between the two isoforms, but we did see opposing expression from isoform b2 in protein kinase A (PknA) (Fig. 3e). We also saw similar expression patterns between Sperm-Luecylaminopeptidase 7 (S-Lap7) and ProtA isoform b1 (Fig. 3e). The Tribolium homolog of CG17377 has an interesting pattern as well and may be a general marker of meiosis as it increases until secondary spermatocytes (Fig. 3e). CG17816 is known for negative regulation of microtubule binding and is found to increase expression as it goes into meiosis II (supplementary fig. S2f, Supplementary Material online) (Rodrigues et al. 2022). Unc-13 is interesting as it shows an opposite pattern of expression to that of Drosophila where it builds up to secondary spermatocytes and then decreases in expression in later stages (supplementary fig. S2g, Supplementary Material online).
Using monocle3, we were able to predict modules of gene coexpression that correlated with stages of spermatogenesis (supplementary fig. S2h, Supplementary Material online). This allowed us to find markers that could be used to stage cell differentiation in beetle spermatogenesis (Fig. 3f). Due to expression of Akb1r, we determined that there was likely equal contamination of cyst cells in germline clusters. While we have little evidence that Akb1r is a marker for cyst cells in beetles we did observe higher median expression of Akb1r in diploid cells than haploid; however, the gene would require further validation to confirm that it is a cyst cell marker in beetle (supplementary fig. S2i and j, Supplementary Material online). We did confirm that the increased weighting of Akb1r in principal components was still not enough to accurately separate cyst and germ clusters and so was not considered in downstream analysis (supplementary fig. S2k, Supplementary Material online). Similarly, we determined that ProtA, while showing interesting expression dynamics, could not be used to correctly stage cyst cells (supplementary fig. S2l, Supplementary Material online).
Preference Toward Female Meiotic Pathways Throughout Spermatogenesis
To better understand the unique dynamics of beetle spermatogenesis, we have performed cell level gene set enrichment analysis (GSEA) on our dataset. We found several patterns of expression for certain Drosophila gene ontology (GO) biological processes (BP) terms (supplementary fig. S3a, Supplementary Material online). There is a stronger enrichment of genes with annotation for female meiotic pathways in contrast to male meiotic pathways (Fig. 4a). Spermatogenesis and sperm storage terms increased during spermatogenesis in beetles, but we observed low enrichment of spermatid development genes in late-stage sperm cells (supplementary fig. S3b, Supplementary Material online). While there was little enrichment of terms for oocyte development, “oocyte dorsal ventral axis specification” was highly enriched, especially in group 1 clusters (supplementary fig. S3c, Supplementary Material online). Other highly enriched female specific pathways included “imaginal disc derived female genitalia development” and “female gamete generation” (supplementary fig. S3d, Supplementary Material online). Along with the mitotic phasing of our data, we were also encouraged to see enrichment of “regulation of exit from mitosis” genes be high in GSC and spermatogonia and decrease over time (supplementary fig. S3e, Supplementary Material online). We also saw similar patterns in GO terms related to germline maintenance (supplementary fig. S3f, Supplementary Material online).
Fig. 4.

Pathway analysis of cluster expressions. a and b) Average enrichment scores per cluster for GO biological process terms related to a) meiosis and b) chromosome. Reported values are averaged across per cell enrichment for each term. c) Average expression of genes contained within relevant terms. d) Expression of female bias genes by cluster by chromosome.
Surprisingly, there was significant enrichment of “dosage compensation by hyperactivation of the X chromosome (GO:0009047)” which we found highly correlated to female meiotic expression (Fig. 4b and c). Overall, expression of genes related to female meiosis was greater than those associated with male meiosis (supplementary fig. S3g and h, Supplementary Material online). When we looked at genes previously associated with male- or female-biased expression (Prince et al. 2010), we found that more male-biased genes are expressed in our dataset overall with 500 out of 1,311 male-biased genes being expressed and only 13 out of 1,763 female-biased genes being expressed. We also found that male-biased genes are more strongly expressed in all clusters (supplementary fig. S4a and b, Supplementary Material online). However, the female-biased genes that were expressed had a significant increase in autosomal expression immediately after meiosis I in secondary spermatocytes as opposed to the male biased genes that saw comparable expression in autosomal chromosomes throughout all stages of spermatogenesis (Fig. 4d, supplementary fig. S4c, Supplementary Material online). In both male- and female-biased genes, we do not see a large change in expression of X-chromosomal genes at any stage, which could be either due to total MSCI or a lack of nonautosomal sex-biased genes (<5% of both male- and female-biased genes).
Incomplete X Inactivation During Meiotic Phase Change due to Dosage Compensation Activity
Due to previous studies showing interesting dynamics for X chromosome hyperactivation in somatic tissues and possible MSCI during beetle spermatogenesis (Prince et al. 2010), and given the enrichment of hyperactivation pathways that we observed, we further investigated patterns of X-linked gene expression across cell clusters. In premeiotic and meiotic cell populations, we observed significantly higher silencing of X-linked genes when compared to autosomal genes than has been found in other species (supplementary table S4, Supplementary Material online). However, the mean expression of genes across the X chromosome shows selective upregulation at various points during late sperm development (Fig. 5a). This appears to correlate with specific regions of the X chromosome that peak in log expression at secondary spermatocyte development (Fig. 5b, supplementary fig. S3d, Supplementary Material online).
Fig. 5.

X inactivation and MSCI of adult beetle testes. a) Boxplot showing average expression of each gene across all chromosomes, the X chromosome, and autosomes by cluster. b) Percent of cells expressing X chromosome genes above an average of 0.25 normalized expression. c) Expression of Unr (TC002472) per cell displayed on UMAP projection. d) Expression of msl1-3 per cluster. e) Density plot showing motif enrichment of DCC CES sites on the X chromosome plotted over mean expression data of X chromosome genes in “Secondary Spermatocyte 1” cluster cells. Dashed line represents density of highly expressed X-chromosomal genes.
A motif enrichment analysis of upstream promoter regions of the genes that spike in expression through secondary spermatocyte development revealed a motif common to more than half of upregulated genes (supplementary fig. S4e, Supplementary Material online). Expression of genes associated with the Drosophila dosage compensation complex also increase in expression at similar stages, with expression of Upstream of N-Ras (Unr) and male sex lethal 3 (msl-3) increasing and decreasing in expression, respectively, through early stages of spermatogenesis (Fig. 5c and d), with msl-3 expression rebounding at the secondary spermatocyte stage which is after meiosis I and when we see a peak in X-reactivation. It is unknown if roX1/2 expression exists in beetles due to limited annotation of lncRNAs or if it is also expressed at the same stages. Using previously identified chromatin entry sites (CES) for msl in Drosophila, we predicted DCC occupancy on the X chromosome (Fig. 5e, Witt et al. 2021). CES like mut1-3 and CES11D1 showed high linear correlation (P < 0.005) with expression at later stages of sperm development (supplementary fig. S4f, Supplementary Material online).
Discussion
A comprehensive analysis of the beetle adult testes through a scRNA-seq atlas provides deep insight into mechanisms of beetle sexual development. In this paper, we address a key issue that has prevented similar analyses in nonmodel organisms like T. castaneum, which is the lack of homologous markers used for clustering cells. We show that weighted PCA and nonexpression markers can be used to drastically improve variance in dimensionality reduction and properly stage clusters by cell type. Development of these novel methodologies are important as many scRNA-seq analyses rely on the underlying algorithms of clustering packages to form clusters that represent the biological ground truth even though most cannot accurately do this (Krzak et al. 2019). We have in our analysis a logical progression of cell differentiation from GSC to Spermatozoa that closely matches the haploid distribution and pseudotime derived from previously validated markers which we know to be accurate to the cell stage. This allowed us to identify new markers that can be used to identify sperm stages in development. We must acknowledge that even though we saw high consistency among our techniques for cell identification, the results of these new techniques have not yet been validated by microscopy. For example, we find many homologous markers for mitotic phase prediction that show similar low expression across cell clusters. This may reflect a lack of conserved gene function in Tribolium, but may be a limitation of scRNA-Seq’s ability to quantify moderate and lowly expressed genes, which could obscure proper cell classification. That said, the results of the techniques we used matched not only each other but also reflect the evidence from the three expression markers that were experimentally validated in previous Tribolium work.
This atlas gives us the ability to directly compare to other studies in Drosophila and Anopheles (Page et al. 2023; Raz et al. 2023a, 2023b). We did not observe separate clusters for germline and cyst cells, likely due to a lack of known beetle markers for cyst cells for the weighted principal components; however, the mosquito study also did not identify separate clusters for cyst cells. ProtA and Ak1br might be candidates for germ vs. cyst markers as ProtA replaces histones during sperm development and Ak1br is highly expressed in cyst cells in Drosophila (Okada 2022; Raz et al. 2023a, 2023b). The fact that Tribolium was missing or lacked expression of key Drosophila spermatogenesis genes like fzo, escargo (esg), and traffic jam (tj) made the overall analysis difficult but highlights the rapid evolutionary dynamics of male gamete development. Many of these genes are shared in mosquitoes which, like fruit flies, belong to the order Diptera (Page et al. 2023). The fact that these genes are not expressed in Coleoptera spermatogenesis suggests that critical genes for Drosophila, and other insect sperm development, may not actually be reflective of the larger class and phylum. Surprisingly, many enriched pathways and expressed genes are associated with female meiotic pathway annotations in other species, indicating that there may be less divergent meiotic pathways between sexes in Coleoptera. A big surprise was the impact that spz had on cluster assignment even though its role in the toll receptor pathway does not lend to any effect on sperm development. We do note that it is shown to be upregulated in male gonadal tissues in Drosophila (flybase; https://flybase.org/) and could be an aspect of asymmetrical development of the teste organs.
One key aspect of our study is the dynamics of MSCI and X-linked expression that we demonstrate in beetle spermatogenesis. We observed that expression of X chromosome genes is already extremely depressed in GSC and rises in expression until secondary spermatocytes. The main difference with other species here is the scale, as we only observed an X-Autosome expression ratio of 0.2%, while other insect species note minima never crossing below 20% (Page et al. 2023; Raz et al. 2023a, 2023b). This implies an overall suppression of the X chromosome and MSCI more absolute than other species and localized entirely to meiotic cells unlike what has been observed in Drosophila (Prince et al. 2010; Meiklejohn et al. 2011; Mikhaylova and Nurminsky 2011; Vibranovski et al. 2012; Landeen et al. 2016; Mahadevaraju et al. 2021; Witt et al. 2021).
Where it has been studied in detail, MSCI is critical for fertility; however, its purpose and the evolutionary factors influencing its origins remain unclear. One hypothesis is that MSCI is driven by sexual antagonism and X inactivation, the so-called “SAXI hypothesis” (Wu and Xu 2003). Since X chromosomes spend 2/3 of their time in females, the X chromosome may accumulate female-beneficial genes even at the expense of those genes' effects in males. Consequently, MSCI may arise to protect male gametogenesis from these sexually antagonistic effects by silencing X-linked genes during spermatogenesis. Our data are consistent with one aspect of the SAXI hypothesis, that the X chromosome will become demasculinized as male-biased and/or testes expressed genes are translocated to autosomes to avoid inactivation (Vibranovski et al. 2009). We found more expression of female-biased genes (186) on the X chromosome than male-biased genes (1). However, SAXI also predicts that there should be a greater concentration of the very early spermatogenic genes on the X chromosome than on autosomes, which we do not see. Overall X expression increases in later stages of Tribolium spermatogenesis including expression of both male- and female-biased genes. We observed site specific X-linked expression increase over time, which combined with the correlation of DCC CES enrichment at comparative stages of spermatogenesis, leads us to believe that there is selective upregulation of the X chromosome that may be mediated by the dosage compensation machinery and be necessary for premeiotic sperm development (Alekseyenko et al. 2008; Landeen et al. 2016).
Alternative hypotheses for the origins of MSCI suggest that it may be a form of host genome defense, protecting against selfish genetic elements, suppressing sex ratio distorters, and/or preventing nonhomologous recombination between the X and Y chromosomes (Hamilton 1967; McKee and Handel 1993; Namekawa and Lee 2009; Meiklejohn and Tao 2010). This line of reasoning follows from MSCI's ancestral origin being MSUC and fits neatly with our observation of early and extreme X chromosome coupled with the unique meiotic mechanism present in Tribolium. Like many species in the Coleopteran subphylum Polyphaga, Tribolium has a unique form of meiosis wherein the X and Y pair at a distance, held together by a protein scaffold that ensures proper meiotic segregation without synaptic pairing. This silencing more closely resembles MSUC than MSCI and the results within this study may be indicative of the differences behind these two related processes. To our knowledge, Tribolium is the first species where gene expression in this type of meiosis has been explored. Based on our results, we might expect MSUC to be active as early as Anaphase I in spermatogenesis (Dutrillaux and Dutrillaux 2010). We believe the insights we find here can serve as the basis for using Tribolium and potentially other polyphagan beetles as an evolutionary model for MSCI development and chromatin dynamics and regulation. Future investigation into the dynamics of sperm cell differentiation within this clade will allow researchers to make comparative analysis to find the molecular origin behind this pervasive biological phenomena.
Methods
Preparation and Sequencing of Testis Single-cell RNA-seq Libraries
Testes from four male beetles were dissected in cold aerated phosphate buffered salin (PBS). The resulting eight testes were placed in 200 mL of lysis buffer (100 mL 0.5% Trypsin EDTA + 100 mL of 4 mg/mL collagenase) in preparation for single-cell dissociation as previously described (Mahadevaraju et al. 2021). The samples were incubated in the lysis buffer for 15 min in ice with gentle mixing every 30 s using a pipette. After incubation, we added 20 µL of 1% fetal bovine serum to stop the enzymatic activity by gentle pipetting. The sample was filtered through a 40 mm cell strainer coated with 5 µL of 0.04% bovine serum albumin (BSA) solution followed by a 5 min centrifugation at 2,000 rpm (rcf 325 x g) and 4 °C. The resulting cell pellet was resuspended in 15 µL of 0.04% BSA solution before further processing. For cell counting, 5 µL of the single-cell suspension were mixed with 5 µL of the trypan blue dye, and the total cell number and the ratio between live and dead cells were analyzed using an automated cell counter (Bio-Rad Automated Cell Counter TC20). This method yielded high numbers of single cells (∼5 million live cells) with an average of 70% to 75% viability. We then submitted cells to the UT Southwestern genomics core for library preparation with the 10× Genomics chromium 3′ kit v3 chemistry. Libraries were then sequenced using 150 bp PE on Illumina NovaSeq at the North Texas Genome Center (Arlington, TX). Unused testis from similar aged male beetles was prepared as a squash slide by pipetting onto a glass slide containing 45% acetic acid and using force to affix a glass coverslip after a 30-min stain in 5 µg/mL Hoechst 33342 (Invitrogen Cat. No. H3570). Images were taken on a Zeiss Imager A1 with fluorescence and then raw images were analyzed for cell density using ImageJ.
Sequencing Data Processing
Illumina sequencing data was filtered and aligned to Tcas5.2 (Herndon et al. 2020) using cell ranger 7.1.0. Cell ranger output was uploaded to R v4.2.3 (R Core Team 2017) through Seurat v4.1.0 (Hao et al. 2021) into a Seurat object containing 1,662,669 cells with expression data for 16,592 genes. Cells were filtered for low expression, doublets, and high mitochondrial content by setting a cutoff of nFeatures > 100 and < 3,000, and percent mitochondrial features < 5%, resulting in 14,443 cells expressing 10,817 genes. Expression data were normalized using “vst” and likely nongermline or cyst cells were filtered by cells expressing marker genes greater than 0.1 normalized expression (supplementary fig. S1a and b, Supplementary Material online).
Marker Identification and Gene Annotation
Tcas5.2 gene protein sequences were aligned to Drosophila r6.52 (dos Santos et al. 2015) using blastp 2.5.0 with gap penalty of 5 and word size of 3. Previously documented markers for β2-tubulin, Rad50, and Enolase were found at loci “TcasGA2-TC009035”, “TcasGA2-TC006703”, and “TcasGA2-TC011729”, respectively. Drosophila and Anopheles (Page et al. 2023; Raz et al. 2023a, 2023b) markers that have been used to identify cell types of the testes were queried through blast annotation (Table 1). Cyst/germline markers in Drosophila were extracted from previous scRNA-seq data using a Wilcoxon ranked sum test and markers differentially expressed with a P < 0.001 that were represented in more than 10% of cells were used for additional analysis. Nonpertinent sex organ tissue cells such as epithelial, muscle, and neuronal cells were removed through positive detection of expression of the following markers: “shg”, “Mhc”, “Prm”, “atilla”, “para”, “ems”, “Sox100B”, and “Lsp2”.
Weighted Principal Components and Clustering
To increase the variance of differentiation markers, we applied weighted PCA (Delchambre 2015), weighting the selected markers (supplementary table S1, Supplementary Material online) 10× against the 400 most variable genes. After generating acceptable UMAP projections, we used Seurat native functions to cluster cells with resolutions from 0.25 to 4. Cell types were determined from a combination of staging markers and differential expression markers identified through the findallmarkers function (supplementary table S2, Supplementary Material online).
Trajectory Analysis, coexpression and Gene set Enrichment
Sequencing data were subjected to different forms of trajectory analysis. β2-tubulin, Rad50, and Enolase were used to generate pseudotime estimates on 300 randomly sampled cells using Ouija (Campbell and Yau 2019). Alignment bam files were used to generate RNA velocity estimates through SCVelo then plotted in Python v3.7.12 using loom files extracted through R. Seurat data were imported into monocle3 (Trapnell et al. 2014) cell datasets and used to generate pseudotime estimates of cell differentiation. Using monocle3 functions, we calculated modules of similarly expressing genes using the K-nearest neighbor graph method. To determine patterns of expression related to function, we turned to GSEA using a gene set constructed from Drosophila biological process GO annotations (https://maayanlab.cloud/FlyEnrichr/#stats, downloaded 2023 June 21). GSEA on individual cells was conducted using the escape package (Borcherding et al. 2021) in R with 1,000 groups and a minimum size of 5. Median enrichment scores were determined per cluster and displayed for most variably enriched terms as well as terms that contained “meiosis”, “mitosis”, “male”, “female”, “sperm”, “cyst”, “germ”, and “chromosome”.
Ploidy Analysis
To determine the fractions of cells that were haploid and diploid in our dataset, we sought to estimate the level of heterozygosity in samples through single nucleotide polymorphism (SNP) analysis of the single reads. This can be represented with the equation:
where H is the percent heterozygosity for each cell, a is the total number of alleles for which expression was detected in each cell, R is the number of reads mapped to the reference allele, and C is the total number of reads at each allele. We predicted SNPs using bcftools mpileup (Li 2011) to get biallelic markers. Using the program scAlleleCount (https://github.com/barkasn/scAlleleCount), we calculated for each cell the number of reads that mapped to either allele. We then calculated the frequency of reads at each allele site to determine the heterozygosity of each allele in every cell. This allowed us to determine the percent heterozygosity across all detected alleles for each cell. Hierarchical clustering of allele states was used to demultiplex pooled samples and resulted in 4 unique populations with approximately equivalent cell representation (∼4,000 cells per individual) which were found to be equally distributed between clusters and showed no bias in the clustering process.
Motif Enrichment
Previously identified msl CES motifs (Alekseyenko et al. 2008) were predicted on the X chromosome using the Fimo tool in the meme-suite (https://meme-suite.org/meme/index.html). Novel motifs were identified from 1,000 bp regions upstream of highly expressed X-linked genes. Distance was calculated from the start of each motif to the start of each gene and the shortest distance was tested for linear correlation with expression of each gene using a three-way ANOVA with TukeyHSD.
Supplementary Material
Supplementary material is available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
We would like to thank Heath Blackmon for graciously providing us the protocol for testes dissection using the Tribolium beetles. We thank the Oliver lab (National Institute of Diabetes and Digestive and Kidney Diseases) for providing us prepublication protocols for preparing single-cell suspension from Drosophila testes and the Betran lab (University of Texas at Arlington) for graciously guiding us with the use the fluorescence microscopy. We are grateful to B. Margabandhu and Chris, who helped to fine-tune the protocol for the testes dissections in T. castaneum. We would also like to thank Avishek Das for contributing code and data processing work for this analysis. This work was funded by the Phi Sigma Biology Graduate Student Society at the University of Texas at Arlington, the University of Texas System Rising STARs Award and the CPRIT First Time Faculty Award. We thank the Texas Advanced Computing Cluster for the use of their high-performance computing resources.
Contributor Information
Michael Robben, Department of Computer Science and Engineering, University of Texas at Arlington, Arlington, TX 76019, USA.
Balan Ramesh, Department of Biology, University of Texas at Arlington, Arlington, TX 76019, USA.
Shana Pau, Department of Biology, University of Texas at Arlington, Arlington, TX 76019, USA.
Demetra Meletis, Department of Biology, University of Texas at Arlington, Arlington, TX 76019, USA.
Jacob Luber, Department of Computer Science and Engineering, University of Texas at Arlington, Arlington, TX 76019, USA.
Jeffery Demuth, Department of Biology, University of Texas at Arlington, Arlington, TX 76019, USA.
Author Contributions
The experiment was designed by M.R., B.R., J.L., and J.D. Material was collected, imaged, and sequenced by B.R. and S.P. Computational and statistical analysis was performed by M.R. and D.M. Manuscript was prepared by M.R., J.L., and J.D.
Data Availability
All code used to generate images and analysis in the manuscript can be found hosted on github: https://github.com/RobbenLab/Testes_scRNAseq. Sequencing and count data have been uploaded for public access at NCBI SRA and GEO databases under the bioproject accession number PRJNA994887.
Literature Cited
- Alekseyenko AA, Peng S, Larschan E, Gorchakov AA, Lee O-K, Kharchenko P, McGrath SD, Wang CI, Mardis ER, Park PJ, et al. A sequence motif within chromatin entry sites directs MSL establishment on the Drosophila X chromosome. Cell. 2008:134(4):599–609. 10.1016/j.cell.2008.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfieri JM, Wang G, Jonika MM, Gill CA, Blackmon H, Athrey GN. A primer for single-cell sequencing in non-model organisms. Genes (Basel). 2022:13(2):380. 10.3390/genes13020380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackmon H, Demuth JP. Estimating tempo and mode of Y chromosome turnover: explaining Y chromosome loss with the fragile Y hypothesis. Genetics. 2014:197(2):561–572. 10.1534/genetics.114.164269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackmon H, Demuth JP. The fragile Y hypothesis: Y chromosome aneuploidy as a selective pressure in sex chromosome and meiotic mechanism evolution. Bioessays. 2015:37(9):942–950. 10.1002/bies.201500040. [DOI] [PubMed] [Google Scholar]
- Borcherding N, Vishwakarma A, Voigt AP, Bellizzi A, Kaplan J, Nepple K, Salem AK, Jenkins RW, Zakharia Y, Zhang W. Mapping the immune environment in clear cell renal carcinoma by single-cell genomics. Commun Biol. 2021:4(1):122. 10.1038/s42003-020-01625-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell KR, Yau C. A descriptive marker gene approach to single-cell pseudotime inference. Bioinformatics. 2019:35(1):28–35. 10.1093/bioinformatics/bty498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delchambre L. Weighted principal component analysis: a weighted covariance eigendecomposition approach. Mon Not R Astron Soc. 2015:446(4):3545–3555. 10.1093/mnras/stu2219. [DOI] [Google Scholar]
- Demarco RS, Eikenes ÅH, Haglund K, Jones DL. Investigating spermatogenesis in Drosophila melanogaster. Methods. 2014:68(1):218–227. 10.1016/j.ymeth.2014.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias G, Lino-Neto J, Mercati D, Dallai R. The sperm ultrastructure and spermiogenesis of Tribolium castaneum (Coleoptera: Tenebrionidae) with evidence of cyst degeneration. Micron. 2015:73:21–27. 10.1016/j.micron.2015.03.003. [DOI] [PubMed] [Google Scholar]
- Dias G, Yotoko KSC, Gomes LF, Lino-Neto J. Uncommon formation of two antiparallel sperm bundles per cyst in tenebrionid beetles (Coleoptera). Naturwissenschaften. 2012:99(9):773–777. 10.1007/s00114-012-0949-6. [DOI] [PubMed] [Google Scholar]
- dos Santos G, Schroeder AJ, Goodman JL, Strelets VB, Crosby MA, Thurmond J, Emmert DB, Gelbart WM; FlyBase Consortium . FlyBase: introduction of the Drosophila melanogaster release 6 reference genome assembly and large-scale migration of genome annotations. Nucleic Acids Res. 2015:43(D1):D690–D697. 10.1093/nar/gku1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutrillaux AM, Dutrillaux B. Y-chromosome disomy and trisomy in Scarabaeid and Cerambycid beetles. Cytogenet Genome Res. 2010:132(3):195–202. 10.1159/000321569. [DOI] [PubMed] [Google Scholar]
- Fishman EL, Jo K, Ha A, Royfman R, Zinn A, Krishnamurthy M, Avidor-Reiss T. Atypical centrioles are present in Tribolium sperm. Open Biol. 2017:7(3):160334. 10.1098/rsob.160334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzpatrick JL, Kahrl AF, Snook RR. SpermTree, a species-level database of sperm morphology spanning the animal tree of life. Sci Data. 2022:9(1):30. 10.1038/s41597-022-01131-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton WD. Extraordinary sex ratios: a sex-ratio theory for sex linkage and inbreeding has new implications in cytogenetics and entomology. Science. 1967:156(3774):477–488. [DOI] [PubMed] [Google Scholar]
- Hao Y, Hao S, Andersen-Nissen E, Mauck WM 3rd, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, et al. Integrated analysis of multimodal single-cell data. Cell. 2021:184(13):3573–3587.e29. 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herndon N, Shelton J, Gerischer L, Ioannidis P, Ninova M, Dönitz J, Waterhouse RM, Liang C, Damm C, Siemanowski J, et al. Enhanced genome assembly and a new official gene set for Tribolium castaneum. BMC Genomics. 2020:21(1):47. 10.1186/s12864-019-6394-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan SA, Jakes E, Myles KM, Adelman ZN. The β2Tubulin, Rad50-ATPase and enolase cis-regulatory regions mediate male germline expression in Tribolium castaneum. Sci Rep. 2021:11(1):18131. 10.1038/s41598-021-97443-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleene KC. Sexual selection, genetic conflict, selfish genes, and the atypical patterns of gene expression in spermatogenic cells. Dev Biol. 2005:277(1):16–26. 10.1016/j.ydbio.2004.09.031. [DOI] [PubMed] [Google Scholar]
- Krzak M, Raykov Y, Boukouvalas A, Cutillo L, Angelini C. Benchmark and parameter sensitivity analysis of single-cell RNA sequencing clustering methods. Front Genet. 2019:10:1253. 10.3389/fgene.2019.01253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landeen EL, Muirhead CA, Wright L, Meiklejohn CD, Presgraves DC. Sex chromosome-wide transcriptional suppression and compensatory cis-regulatory evolution mediate gene expression in the Drosophila male germline. PLoS Biol. 2016:14(7):e1002499. 10.1371/journal.pbio.1002499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011:27(21):2987–2993. 10.1093/bioinformatics/btr509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahadevaraju S, Fear JM, Akeju M, Galletta BJ, Pinheiro MMLS, Avelino CC, Cabral-de-Mello DC, Conlon K, Dell’Orso S, Demere Z, et al. Dynamic sex chromosome expression in Drosophila male germ cells. Nat Commun. 2021:12(1):892. 10.1038/s41467-021-20897-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahajan S, Bachtrog D. Partial dosage compensation in Strepsiptera, a sister group of beetles. Genome Biol Evol. 2015:7(2):591–600. 10.1093/gbe/evv008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKee BD, Handel MA. Sex chromosomes, recombination, and chromatin conformation. Chromosoma. 1993:102(2):71–80. 10.1007/BF00356023. [DOI] [PubMed] [Google Scholar]
- Meiklejohn CD, Landeen EL, Cook JM, Kingan SB, Presgraves DC. Sex chromosome-specific regulation in the Drosophila male germline but little evidence for chromosomal dosage compensation or meiotic inactivation. PLoS Biol. 2011:9(8):e1001126. 10.1371/journal.pbio.1001126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meiklejohn CD, Tao Y. Genetic conflict and sex chromosome evolution. Trends Ecol Evol. 2010:25(4):215–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikhaylova LM, Nurminsky DI. Lack of global meiotic sex chromosome inactivation, and paucity of tissue-specific gene expression on the Drosophila X chromosome. BMC Biol. 2011:9(1):29. 10.1186/1741-7007-9-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murat F, Mbengue N, Winge SB, Trefzer T, Leushkin E, Sepp M, Cardoso-Moreira M, Schmidt J, Schneider C, Mößinger K, et al. The molecular evolution of spermatogenesis across mammals. Nature. 2023:613(7943):308–316. 10.1038/s41586-022-05547-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namekawa SH, Lee JT. XY and ZW: is meiotic sex chromosome inactivation the rule in evolution? PLoS Genet. 2009:5(5):e1000493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada Y. Sperm chromatin condensation: epigenetic mechanisms to compact the genome and spatiotemporal regulation from inside and outside the nucleus. Genes Genet Syst. 2022:97(1):41–53. 10.1266/ggs.21-00065. [DOI] [PubMed] [Google Scholar]
- Page N, Taxiarchi C, Tonge D, Chesters E, Kuburic J, Game L, Nolan T, Galizi R. Single-cell profiling of mosquito spermatogenesis defines the onset of meiotic silencing and pre-meiotic overexpression of the X chromosome. Commun Biol. 2023;6(1):850. 10.1038/s42003-023-05224-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pointer MD, Gage MJG, Spurgin LG. Tribolium beetles as a model system in evolution and ecology. Heredity (Edinb). 2021:126(6):869–883. 10.1038/s41437-021-00420-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prince EG, Kirkland D, Demuth JP. Hyperexpression of the X chromosome in both sexes results in extensive female bias of X-linked genes in the flour beetle. Genome Biol Evol. 2010:2:336–346. 10.1093/gbe/evq024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raz AA, Vida GS, Stern SR, Mahadevaraju S, Fingerhut JM, Viveiros JM, Pal S, Grey JR, Grace MR, Berry CW, et al. ASAP single cell dataset visualization. 2023b. https://asap.epfl.ch/projects/ASAP24 [dataset]. [DOI] [PMC free article] [PubMed]
- Raz AA, Vida GS, Stern SR, Mahadevaraju S, Fingerhut JM, Viveiros JM, Pal S, Grey JR, Grace MR, Berry CW, et al. Emergent dynamics of adult stem cell lineages from single nucleus and single cell RNA-Seq of Drosophila testes. Elife. 2023a:12:e82201. 10.7554/eLife.82201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team . R: a language and environment for statistical computing. 2017. https://www.R-project.org/.
- Rodrigues LR, Zwoinska MK, Wiberg RAW, Snook RR. The genetic basis and adult reproductive consequences of developmental thermal plasticity. J Anim Ecol. 2022:91(6):1119–1134. 10.1111/1365-2656.13664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla JN, Palli SR. Tribolium castaneum transformer-2 regulates sex determination and development in both males and females. Insect Biochem Mol Biol. 2013:43(12):1125–1132. 10.1016/j.ibmb.2013.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla JN, Palli SR. Production of all female progeny: evidence for the presence of the male sex determination factor on the Y chromosome. J Exp Biol. 2014:217(Pt 10):1653–1655. 10.1242/jeb.100438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddall NA, Hime GR. A Drosophila toolkit for defining gene function in spermatogenesis. Reproduction. 2017:153(4):R121–R132. 10.1530/REP-16-0347. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, Rinn JL. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol. 2014:32(4):381–386. 10.1038/nbt.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner JMA, Mahadevaiah SK, Fernandez-Capetillo O, Nussenzweig A, Xu X, Deng C-X, Burgoyne PS. Silencing of unsynapsed meiotic chromosomes in the mouse. Nat Genet. 2005:37(1):41–47. 10.1038/ng1484. [DOI] [PubMed] [Google Scholar]
- Vibranovski MD, Lopes HF, Karr TL, Long M. Stage-specific expression profiling of Drosophila spermatogenesis suggests that meiotic sex chromosome inactivation drives genomic relocation of testis-expressed genes. PLoS Genet. 2009:5(11):e1000731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vibranovski MD, Zhang YE, Kemkemer C, Lopes HF, Karr TL, Long M. Re-analysis of the larval testis data on meiotic sex chromosome inactivation revealed evidence for tissue-specific gene expression related to the Drosophila X chromosome. BMC Biol. 2012:10(1):49. author reply 50. 10.1186/1741-7007-10-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittle CA, Kulkarni A, Extavour CG. Absence of a faster-X effect in beetles (Tribolium, Coleoptera). G3. 2020:10(3):1125–1136. 10.1534/g3.120.401074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witt E, Shao Z, Hu C, Krause HM, Zhao L. Single-cell RNA-sequencing reveals pre-meiotic X-chromosome dosage compensation in Drosophila testis. PLoS Genet. 2021:17(8):e1009728. 10.1371/journal.pgen.1009728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CI, Xu EY. Sexual antagonism and X inactivation—the SAXI hypothesis. Trends Genet. 2003:19(5):243–247. 10.1016/S0168-9525(03)00058-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All code used to generate images and analysis in the manuscript can be found hosted on github: https://github.com/RobbenLab/Testes_scRNAseq. Sequencing and count data have been uploaded for public access at NCBI SRA and GEO databases under the bioproject accession number PRJNA994887.