

Abstract
SARS-CoV-2 hybrid immunity is more protective than vaccination or prior infection alone. To investigate the kinetics of spike (S) reactive T cells (TS) from SARS-CoV-2 infection through mRNA vaccination in persons with hybrid immunity, we identified the T cell receptor (TCR) sequences of thousands of index TS cells and tracked their frequency in bulk TCRβ repertoires sampled longitudinally from the peripheral blood of recovered COVID-19 patients. Vaccinations led to large expansions in memory TS cell clonotypes, the majority of which were CD8+ T cells, while also eliciting diverse TS cell clonotypes not observed prior to vaccination. TCR sequence similarity clustering identified public CD8+ and CD4+ TCR motifs associated with S-specificity. Synthesis of longitudinal bulk ex vivo single-chain TCRβ repertoires and paired-chain TCRɑβ sequences from droplet sequencing of TS cells provides a roadmap for rapid assessment of T cell responses to vaccines and emerging pathogens.
Keywords: SARS-CoV-2, T cell receptor, mRNA vaccine, immune repertoire, clustering analysis
Hybrid immunity from natural infection and vaccination is more protective against SARS-CoV-2 infection than either alone1–3. Vaccination after SARS-CoV-2 infection increases the abundance of virus-specific memory T cells (Tm cells)4–7 and diversifies the S-specific Tm cell pool8,9, two potential mechanisms by which hybrid immunity is protective against severe disease10, despite antigenic escape from neutralizing antibodies (nAb) by variants of concern (VOC)11. In most persons, the recognition of multiple T cell epitopes12,13 largely preserves T cell responses despite VOC evolution14, although T cell escape has been observed15. To profile the development of hybrid immunity, we collected 259 longitudinal PBMC samples and 46 nasal swabs from 54 persons from COVID-19 convalescence through three mRNA vaccinations.
To investigate the diversity and kinetics of TS cells, we identified T cells that expressed the activation induced markers (AIM) CD69 and CD137 upon S peptide stimulation of PBMCs, defined their CD4+ T cell and CD8+ T cell phenotypes with barcoded mAbs and obtained paired chain TCRαβ by single-cell sequencing (hereafter CD69+CD137+ AIM-scTCRɑβ-seq). We also identified activated S-reactive CD4+ TRB clonotypes from PBMCs that express either one of CD69 or CD137 plus at least one of the AIM CD134 or CD154 by single chain TRB locus sequencing (CD4+AIM-TCRβ-seq). To track TS cells longitudinally we matched the TCRβ sequences from CD69+CD137+ AIM-scTCRɑβ-seq and CD4+ AIM-TCRβ-seq to TRB locus sequences from bulk PBMC (hereafter TCRβ-seq) obtained at 6 intervals between convalescence from SARS-CoV-2 infection and receipt of three mRNA vaccinations. By tracking the frequency of index CD69+CD137+ AIM-scTCRɑβ-seq TS clones within bulk repertoires at all timepoints and matching to pre-vaccine memory subset repertoires, we were able to measure the recruitment and expansion of TS cell clonotypes as hybrid immunity developed. We observed divergent clonotype-level kinetics between S-reactive circulating CD8+ and CD4+ T cells. Sequence-similarity clustering of paired-chain TCRs, identified by CD69+CD137+ AIM-scTCRɑβ-seq, permitted inference of the restricting human leukocyte antigens (HLA) alleles for many S-specific TCR motifs. Taken together, integration of bulk and single-cell TCR sequencing with HLA information provides a broadly applicable roadmap to “de-orphan” pathogen-specific TCR sequences without a priori knowledge of epitope-level specificity.
Results
Vaccination provokes heterogeneous clonal expansions
To investigate heterogeneity in T cell response to vaccination after COVID-19, we performed bulk TCRβ-seq on PBMC from 54 adults (28 females and 26 males, median age 60.3 years, range 31.4–73.5 years) who were infected with SARS-CoV-2 between April-August 2020, when the ancestral-like D614G strain prevailed16. The cohort contained 35 participants with mild or moderate (WHO severity scale 1–2)17 and 19 with severe (WHO severity scale 3–7) COVID-19 (Supplementary Data Table 1)18–21. Blood samples were obtained at six visits. Visit E00 (n = 54) occurred at day 78.5 (median, interquartile range (IQR) 56–105) post-COVID-19 diagnosis. Visit E01 (n = 34, 16 with severe COVID-19) occurred immediately before vaccine dose 1 (BNT162b2 or mRNA-1273), corresponding to day 369 (median, IQR 333–390) post-symptom onset. Visit E02 (n = 52) occurred at day 19.5 (median, IQR 15–24) post-dose 1 and prior to dose 2. Visit E03 (n = 53) occurred at day 24 (median, IQR 20–28) post-dose 2. Visit E04 (n = 7, 2 with severe COVID-19) occurred pre-dose 3, at day 274 (median, IQR 251–308). Visit E05 (n = 44, 15 with severe COVID-19) occurred at day 41.5 (median, IQR 26–69) post-dose 3 (Fig. 1a, Extended Data Fig. 1).
Fig. 1. Vaccines expand previously detected and low abundance clonotypes.
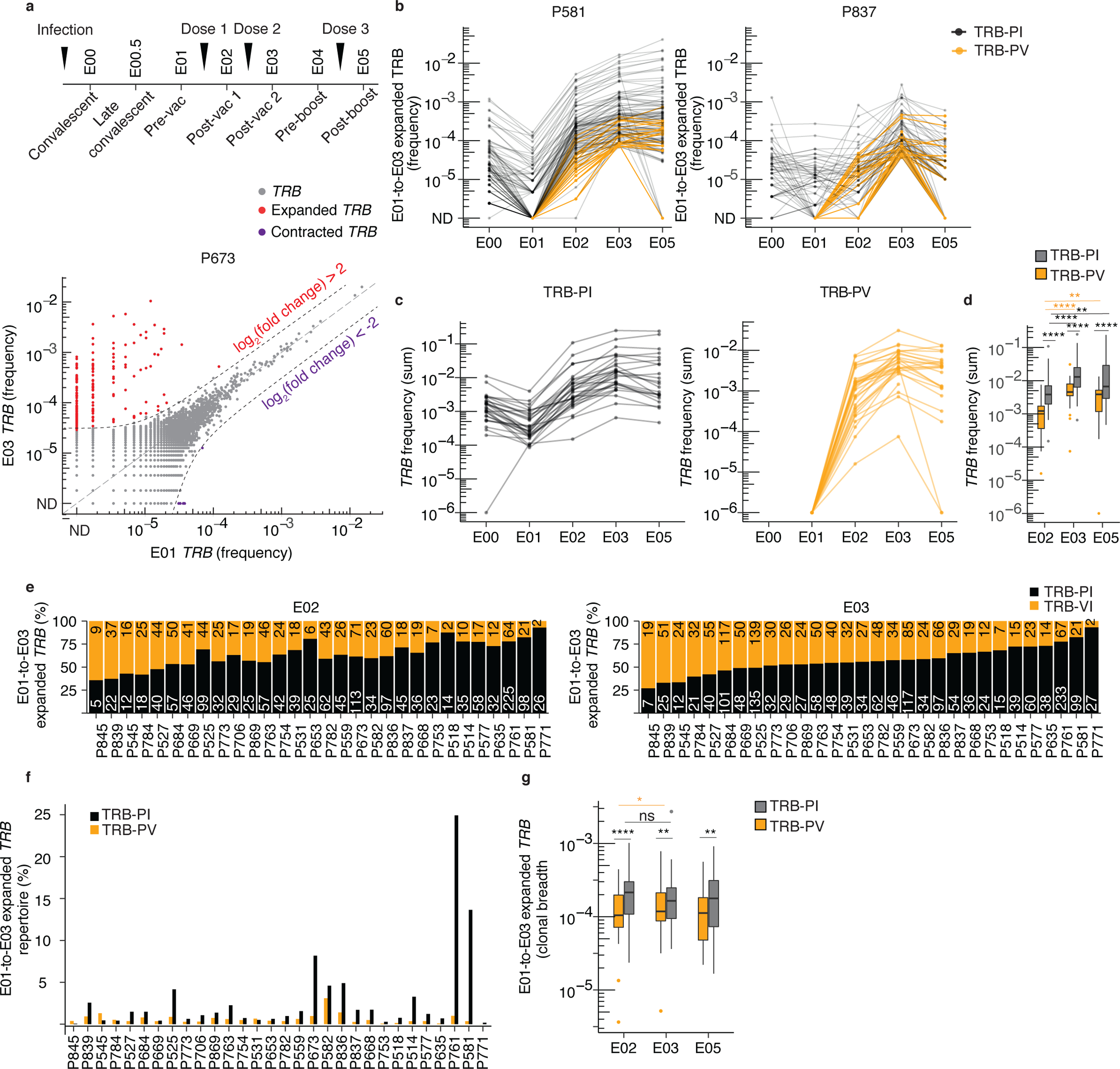
(a) Timing of PBMC sampling and frequency (fraction of the bulk TRB repertoire) of individual clonotypes in E01 vs. E03 in one representative participant (P673). Expanded clonotypes were defined as log2(fold change) > 2 (or < 0.5) and Fisher’s exact test FDR-adjusted p value < 0.05. (b) Frequency of E01-to-E03 expanded clonotypes from E00 through E05 in two representative participants (P581 and P837). (c) Sum of E01-to-E03 expanded TRB clonotype frequency (abundance) per participant detected at E00 and E01 (post-infection TRB clonotypes, TRB-PI) or detected exclusively starting at E02 (post-vaccination TRB clonotypes, TRB-PV, n = 28). (d) Comparison of abundance of E01-to-E03-expanded TRB-PI and TRB-PV at E02 (n = 30), E03 (n = 30) E05 (n = 28) relative to E01. (e) Number of unique E01-to-E03 expanded TRB-PI and TRB-PV clonotypes detected at E02 (post-dose 1) and E03 (post-dose 2), ranked by fraction of repertoire present at E03. (f) Breadth of E01-to-E03-expanded TRB-PI and TRB-PV clonotypes at E02 (n = 30), E03 (n = 30) and E05 (n = 28). (g) The percent of total E01-to-E03-expanded TRB clonotypes detected at E02 (n = 30), E03 (n = 30) and E05 (n = 28).d, f, g, level of statistical significance by Wilcox rank sum test between groups or Wilcoxon signed-rank test within groups between time periods is indicated: ns = not significant, *p<0.05, **p<0.01, ****p<0.0001. Box plots represent median, IQR, and whiskers (1.5 * IQR). All tests are two-sided.
Before evaluating T cell specificity directly, we examined the kinetics of all unique TCRβ sequences (hereafter, TRB clonotypes) detectable in blood. To assess the impact of vaccination on the structure of the T cell repertoire, we compared the frequency of TRB clonotypes across the E01-E05 timepoints by pairwise differential relative abundance in the bulk TRB repertoire at each timepoint in 32 participants (Fig. 1a, Extended Data Fig. 2, Supplementary Data Table 2). Among participants with E01 and E03 repertoires, 21 of 32 had large expansions (1,000-fold or greater) of individual TRB clonotypes at E03 (post-dose 2) compared to E01 (pre-vaccine) (Extended Data Fig. 2). To track the kinetics of potentially vaccine-expanded TRB clonotypes over the course of the study, we identified clones increasing by a log2 fold change >2 and a statistically significant change in frequency between serial visits and between E01 and E03 (hereafter E01-to-E03 expanded), when evaluated by a Fisher’s Exact Test with correction for multiple hypotheses (FDR-adjusted q-value < 0.05) (Fig. 1a, Supplementary Data Tables 3, 4).
When assessing the clonal expansions induced by the first and second mRNA vaccination, a median of 38 (IQR 21–80, n = 28) and 44 (IQR 29–69, n = 32) statistically significant clonal expansions occurred across the intervals E01-to-E02 and E02-to-E03, respectively (Supplementary Data Table 4). Cumulatively, a median of 72 (IQR 51–104) statistically significant E01-to-E03 expanded TRB clonotypes were detected per participant, representing a median of 1.8% (IQR 0.9–3.5%) of the circulating T cell repertoire in each participant (Supplementary Data Table 4). To assess the contribution of T cells established during infection to the vaccine response, we tracked TRB clonotypes that were either detected at E00-E01 (post-infection TRB clonotypes, hereafter TRB-PI) or detected exclusively starting at E02 (post-vaccination TRB clonotypes, TRB-PV) (Fig. 1b). At E02 and E03, the total frequencies of E01-to-E03 expanded TRB clonotypes ranged from 0.001–11% and 1–26%, respectively (Fig. 1c). At E03, the combined abundance of E01-to-E03 expanded TRB-PI represented 0.1% to 25% of each participant’s repertoire while TRB-PV represented 0.01% to 3% (Fig. 1c, d, Extended Data Fig. 3a). TRB-PI were numerically dominant (Fig. 1f); however, the proportion of unique TRB clonotypes was balanced between TRB-PI and TRB-PV (Fig. 1e, g).
The repertoires of participants with E01-to-E03 expanded clones exceeding 5% of their total repertoire at E03 (P673, P582, P836, P761 and P581) were characterized by a strong polyclonal expansion of clonotypes detected post-infection (Fig. 1f). The breadth (i.e., diversity) of TRB-PV increased after dose 2 (E03) compared to dose 1 (E02) (Fig. 1g), indicating that successive doses increased the frequency and diversity of expanded clones. Most E01-to-E03 expanded TRB-PV persisted in participant repertoires through E05, but these clonotypes were not as likely to persist as expanded clonotypes detected post-infection (Extended Data Fig. 3b).
Next, to evaluate the memory phenotype of clonotypes detected before the start of the vaccine series, we performed TCRβ-seq on sorted, pooled CCR7−CD45RA+, CCR7+CD45RA− and CCR7−CD45RA− (CD4+ and CD8+ combined) Tm cells (see Methods) from PBMCs isolated at E00 in 7 participants (median age 66.6, 2 female, 2 with severe COVID-19) (Supplementary Data Fig. 1, Supplementary Data Table 5). Cross-referencing TRB sequences from E00 Tm cells against E01-to-E03 expanded TRB clonotypes indicated that at least 25–84% (median 66%) of vaccine-expanded TRB-PI had a Tm cell phenotype before vaccination (Supplementary Data Fig. 2a, b). Thus, serial bulk PBMC TRB sequencing identified clonotype expansions across each of two mRNA vaccine doses approximately one year after COVID-19, with a substantial contribution of confirmed Tm cells.
Vaccination strongly expands CD8+ T cells in blood
To obtain paired TCRαβ sequences and determine the CD4+ vs. CD8+ T cell phenotype of E01-to-E03 expanded TRB clonotypes, we sorted T cells that co-expressed the activation-induced markers CD69 and CD137 after 18 hours stimulation with peptides spanning the SARS-CoV-2 S protein (hereafter S peptides) from the E03 PBMCs of 17 participants (median age 60.6 years, range 40.9 – 73.5, 4 with severe COVID-19, 6 female) with at least 50 E01-to-E03 expanded TRB clonotypes (Supplementary Data Table 4). Droplet single cell TCR sequencing of CD69+CD137+ double positive T cells (CD69+CD137+ AIM-scTCRɑβ-seq) was then used to define TS TCRs. TRB clonotypes from bulk TCRβ-seq were classified as TS if the frequency of the TRB rearrangement was enriched by CD69+CD137+ AIM-scTCRɑβ-seq compared to unsorted bulk TCRβ-seq at the same time point (see Methods). The clonotype frequency of TS cells in CD69+CD137+ AIM-scTCRɑβ-seq was correlated with the frequency in the bulk repertoires (Methods, Extended Data Fig. 4). We identified 5,733 unique S-activated, paired TCRɑβ clonotypes (Methods, Supplementary Data Table 6), with roughly equal numbers of CD4+ and CD8+ TCRɑβ clonotypes (Supplementary Data Table 7). TCRβ-seq of nasal swabs at E05 (post-dose 3) showed that TS identified by CD69+CD137+ AIM-scTCRɑβ-seq were detected in the nasal mucosa in 13 of 14 participants with CD69+CD137+ AIM-scTCRɑβ-seq data who performed nasal swabs (Extended Data Fig. 5).
There was substantial overlap between CD69+CD137+ AIM-scTCRɑβ-seq clones and E01-to-E03 expanded TRB clonotypes (Fig. 2a, Extended Data Fig. 6). In 12 participants with both datasets (median age 62.2 years, range 47.7 – 73.5, 3 female, 2 with severe COVID-19), a median of 34% (IQR 26–53%) of CD69+CD137+ AIM-scTCRɑβ-seq index TCRs matched E01-to-E03 expanded TRB sequences (Fig. 2b). Despite recovering similar numbers of CD4+ and CD8+ T cell clonotypes, 93% of the robustly E01-to-E03 expanded TRB matching CD69+CD137+ AIM-scTCRɑβ-seq TCRɑβ clonotypes were CD8+ T cells (Fig. 2b). Most CD69+CD137+ AIM-scTCRɑβ-seq-identified S-reactive CD4+ TCRɑβ clonotypes did not expand as markedly in response to vaccination as S-reactive CD8+ TCRɑβ clonotypes (Fig. 2c, e, Extended Data Fig. 6). Comparison of the mean trajectories (Fig. 2d) and median fold expansion (Fig. 2e) of the abundance of the CD69+CD137+ AIM-scTCRɑβ-seq-generated S-reactive clonotypes in all 17 samples indicated greater expansion of CD8+ than CD4+ T cells post-vaccination.
Fig. 2. Longitudinal kinetics of S-reactive clonotypes defined by AIM-scTCRɑβ-seq from post-infection to post-vaccination.
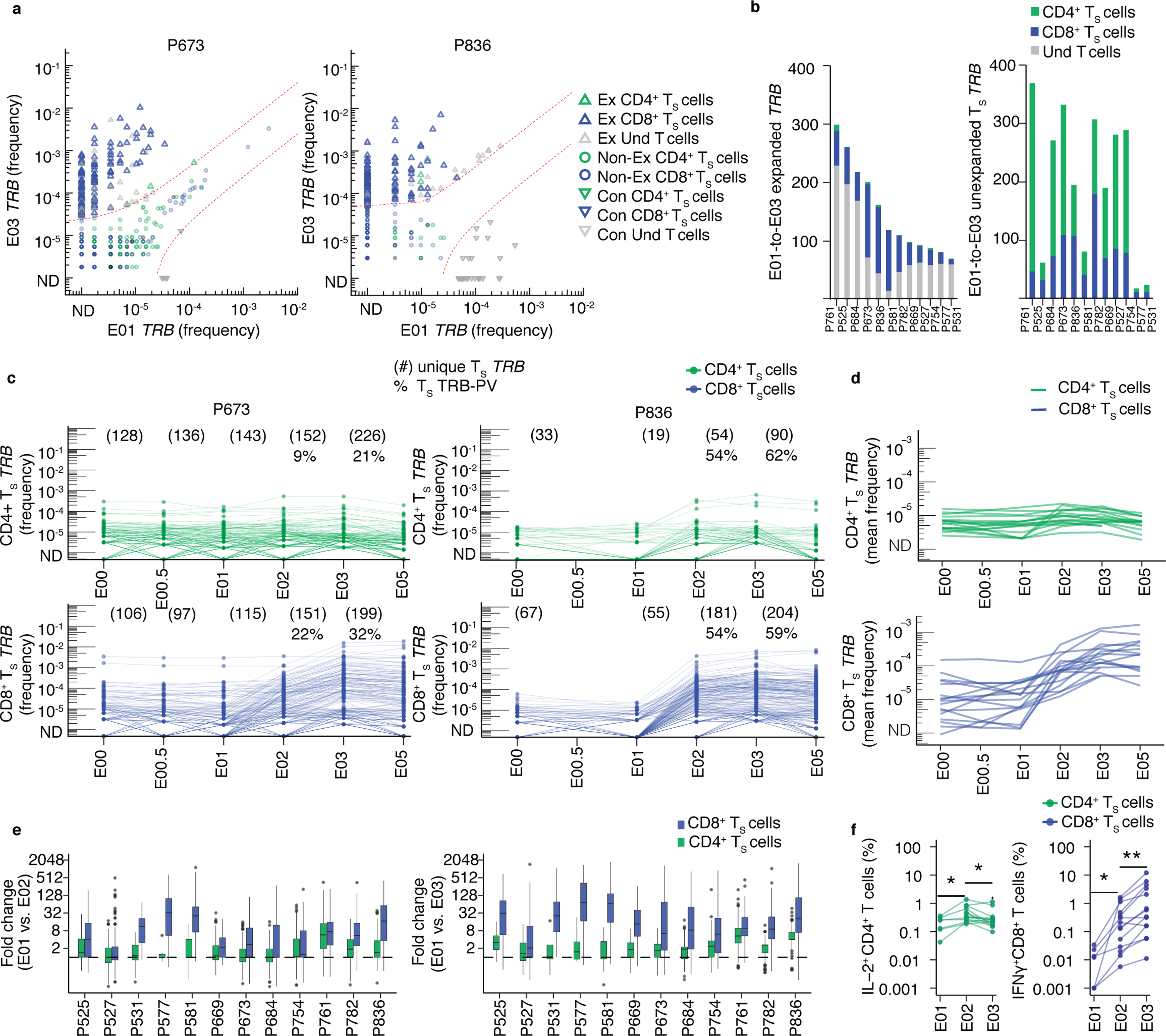
(a) Overlay of TRB sequences from CD69+CD137+ AIM-scTCRɑβ-seq of TS cells onto bulk TRB clonotype frequency at E01 and E03 in two representative participants. E01-to-E03 expanded (Ex), non-expanded (non-Ex) or contracted (Con) TRB clonotypes for TRB sequences matching CD69+CD137+ AIM-scTCRɑβ-seq (TS) and unmatched TRB sequences (undetected, Und) are shown. (b) Numbers of expanded or non-expanded PBMC TRB-defined clonotypes matching CD8+ or CD4+ TS clonotypes in 12 participants with E01-E03 expanded samples. (c) Longitudinal tracking of CD69+CD137+ AIM-scTCRɑβ-seq-identified CD4+ and CD8+ TS TRB clonotype abundance in PBMC of two representative participants. The number of unique CD69+CD137+ AIM-scTCRɑβ-seq TRB clonotypes from E03 detected at each time point are shown. Percentages represent CD69+CD137+ AIM-scTCRɑβ-seq TRB-PV clonotypes for the E02 and E03 timepoints. (d) Mean abundances of S-reactive CD4+ and CD8+ T cell clonotypes identified by CD69+CD137+ AIM-scTCRɑβ-seq in 17 participants at E00 to E05. (e) Distribution of fold-changes (median, box (IQR), and whiskers (1.5 * IQR)) observed for S-reactive CD4+ or CD8+ T cell clonotypes between E01 and E02 and E01 and E03. (f) Intracellular cytokine staining after stimulation of PBMC isolated at E01 (n=7), E02 (n=14) and E03 (n=14) with S peptides. Level of statistical significance by paired, two-sided Wilcoxon signed-rank test is indicated *p<0.05, **p<0.01.
Focusing on CD8+ TS cells, we analyzed the recruitment of rare versus abundant clonotypes by the first two vaccine doses. A median of 50% (IQR 33–61%) of the CD8+ CD69+CD137+ AIM-scTCRɑβ sequences matching bulk TRB clonotypes at E03 were not detected at either E00 or E01 (Extended Data Fig. 7). A median of 20% (IQR 11–32%) of the S-reactive CD8+ TRB clonotypes found at E03 were not detected at E02 (Extended Data Fig. 7), showing that the second dose of the vaccine entrained previously undetected CD8+ TS into the repertoire between E02 and E03 (Fig. 2c, Extended Data Fig. 7, Supplementary Data Fig. 2). Thus, even in persons with an appreciable pre-existing S-reactive CD8+ Tm cell population due to SARS-CoV-2 infection, the second dose of mRNA vaccine expanded pre-existing CD8+ Tm and recruited previously naive or extremely rare CD8+ Tm cell clonotypes, potentially broadening S recognition.
Vaccine kinetics of CD4+ and CD8+ TS cells diverge
To compare CD4+ and CD8+ TS trajectories by an independent method, we performed intracellular cytokine staining in PBMC from E01 (n = 7), E02 (n =14) and E03 (n =14) visits from participants with E03 CD69+CD137+ AIM-scTCRɑβ-seq data using S peptides. We observed that the kinetics of IL-2+CD4+ and IFN-γ+CD8+ T cell responses to S peptide stimulation at E03, relative to E02 and E01, were consistent to those tracked with CD69+CD137+ AIM-scTCRɑβ-seq (Fig. 2d, f, Supplementary Data Table 8). Prior to vaccination, at E01, S-reactive IL-2+CD4+ T cells were detected at higher frequencies (median 0.25%) than S-reactive IFN-γ+CD8+ T cells median (0.012%) (Fig. 2f), The frequency of S-reactive IL-2+CD4+ T cells in blood increased from E01 to E02 (Fig. 2f). The frequency of IFN-γ+CD8+ T cells increased stepwise from the previous visit after each vaccine dose at E02 and E03 in 12 out of 14 participants (Fig. 2f). These data indicated a distinct pattern of expansion in CD4+ T cells and CD8+ T cells in response to the second vaccine dose.
To more closely examine the kinetics of CD4+ and CD8+ CD69+CD137+ AIM-scTCRɑβ-seq clonotypes in response to each vaccine dose, we performed unsupervised clustering of clonotype-level trajectories in 12 participants (as above) with matching bulk TCRβ-seq repertoire sequencing at E00, E01, E02, E03 and E05. Five classifications described the expansion or contraction trajectory of these T cell clonotypes: minimal proliferation (Group 1), proliferation at E02 followed by contraction (Group 2), proliferation at E03 (Group 3), proliferation at E02 without contraction (Group 4) or proliferation at E02 and E03 (Group 5) (Fig. 3a). In all 12 participants, many S-reactive CD8+ clonotypes proliferated at both E02 and E03 (Group 5) (Fig. 3b). In contrast, the majority of S-reactive CD4+ T cell clonotypes had minimal expansion (76% in Group 1) or proliferated at E02 followed by contraction (21% in Group 2), with few showing expansion in response to dose 2 (Groups 3, 4 and 5) (Fig. 3a).
Fig. 3. Trajectories of S-reactive clonotypes defined by AIM-scTCRɑβ-seq differentiate CD4+ and CD8+ T cells.
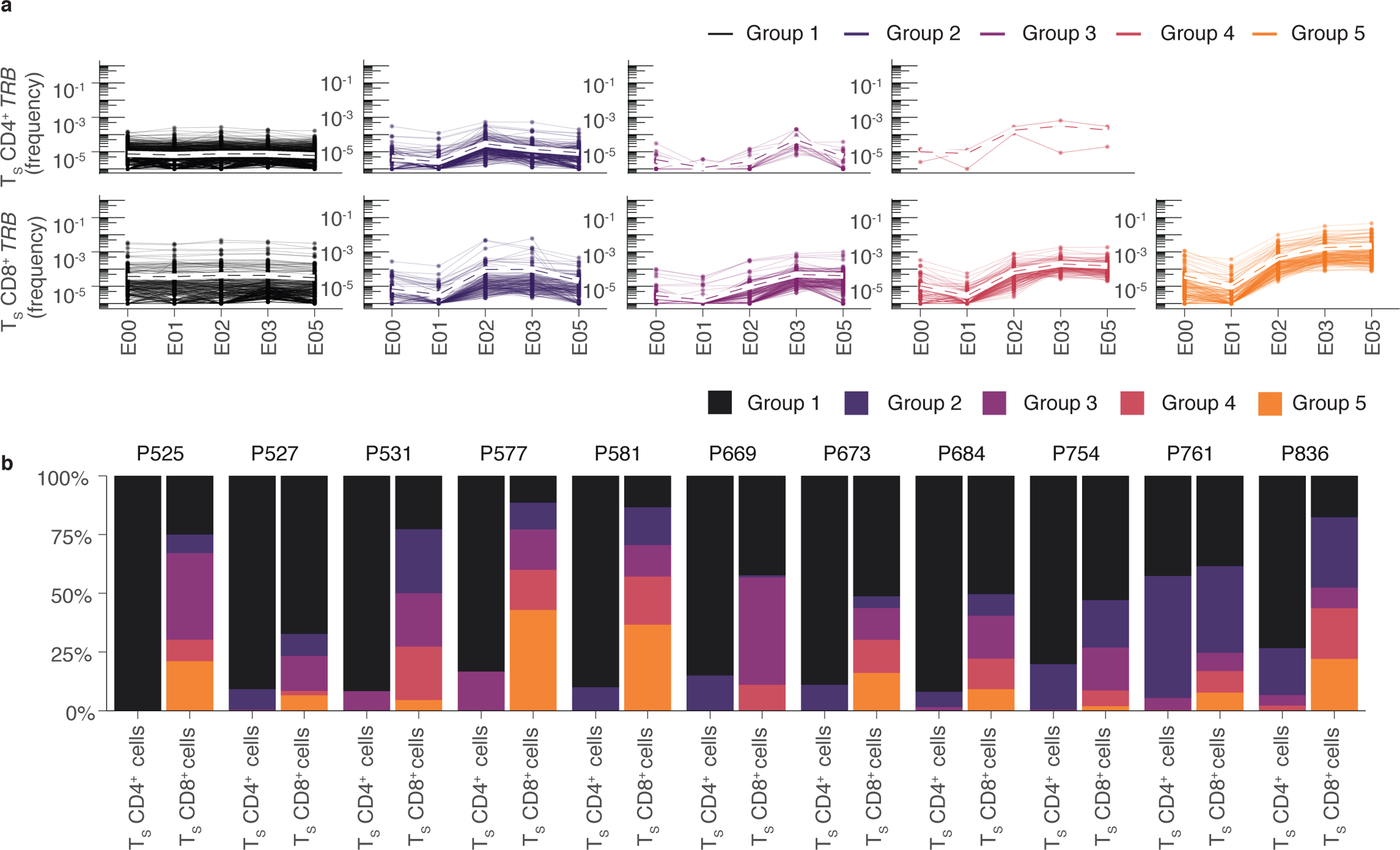
(a) Unsupervised hierarchical clustering of S-reactive clonotypes identified by CD69+CD137+ CD69+CD137+ AIM-scTCRɑβ-seq based on cosine similarity of their E00, E01, E02, E03, E05 log productive frequency showing five major trajectory types: minimal proliferation (Group 1), proliferation at E02 followed by contraction (Group 2), proliferation at E03 (Group 3), proliferation at E02 without contraction (Group 4) or proliferation at E02 and E03 (Group 5). Mean trajectory with each group is shown as a dashed line. (b) The percentage of clonotypes per participant by trajectory group for CD4+ and CD8+ TS cells in the 11 participants with CD69+CD137+ AIM-scTCRɑβ-seq phenotyping and PBMC sampling at all 5 visits (E00-E05).
Activated CD4+ T cells can express diverse combinations of cell surface markers beyond CD69 and CD13722 (Supplementary Data Fig. 3). We therefore performed TCRβ-seq from sorted CD4+ T cells from PBMCs at E03 expressing either CD69 or CD137 and at least one of the activation markers CD134 or CD154 (CD69+(CD134/CD154)+ and CD137+(CD134/CD154)+) after incubation with S peptides (hereafter CD4+AIM-TCRβ-seq) in 7 participants (median age 66.6, 2 female, 2 with severe COVID-19) (Supplementary Data Table 9). CD69+(CD134/CD154)+ and CD137+(CD134/CD154)+CD8+ AIM identified very few CD8+ T cells (Supplementary Data Fig. 4, Supplementary Data Table 10), but CD4+AIM-TCRβ-seq identified nearly twice as many S-reactive CD4+ TRB clonotypes as identified by CD69+CD137+CD4+AIM-TCRβ-seq (Extended Data Fig. 8). However, the expansion kinetics of the S-reactive CD4+ clonotypes identified by CD69+CD137+ AIM-scTCRɑβ-seq and CD4+AIM-TCRβ-seq were similar (Supplementary Data Fig. 5). To further investigate the expansion of CD4 clonotypes from E01 to E03, we sorted bulk CD4+ T cells from both time points and compared clonotype expansion in these cells to that observed in whole PBMC. Few E01-to-E03 expanding clonotypes could be confirmed as CD4+ T cells (Fig. 2, Extended Data Figs. 6, 8, Supplementary Data Fig. 5). Overall, these experiments suggested that at the clonotype level, independent of method, CD4+ TS cells expanded only modestly in our cohort.
Hybrid immunity elicits highly public TS TCR motifs
TCRs that recognize a common ligand often exhibit convergent sequence features23,24 in the CDR3 residues, which directly contact the peptide, and in CDR1 and CDR2, which can also contact peptide-HLA. Using TCRdist – a multi-CDR position-weighted, biochemical distance metric – we computed pairwise sequence divergence amongst the 5,733 unique S-reactive TCRɑβ clonotypes recovered by CD69+CD137+AIM-scTCRɑβ-seq from 17 participants (Methods) to define public TCR clusters24. A similarity graph was constructed from the 1,458 clonotypes that had at least one other similar TCR in the dataset, with edges joining sufficiently similar TCRs (a TCRdist metric ≤100 generally corresponds to similar TRBV and TRAV gene usage and one to four amino acid substitutions or deletions within CDR3ɑ and CDR3β). This identified 284 TCR clusters (Fig. 4a), often characterized by distinct CDR3 motifs, which were consistently expanded by mRNA vaccination from E01 to E03 across participants with a common HLA (Fig. 4). The ten largest clusters (cluster 0 – cluster 9) contained cells from 3–11 participants and between 25–144 unique clonotypes (Fig. 4b–h, Supplementary Data Fig. 6). More than 97% of edges within TCR clusters connected clonotypes with matching CD4+ or CD8+ assignments (Fig. 4a), and many public TCR clusters were formed from groups of persons expressing a shared HLA class I or class II allele (Supplementary Data Fig. 7, Supplementary Data Table 11), suggesting common specificity for a peptide ligand restricted by this allele.
Fig. 4. TCRαβ sequence similarity network shows public CD8+ T cell responses among sequences recovered by AIM-scTCRɑβ-seq.
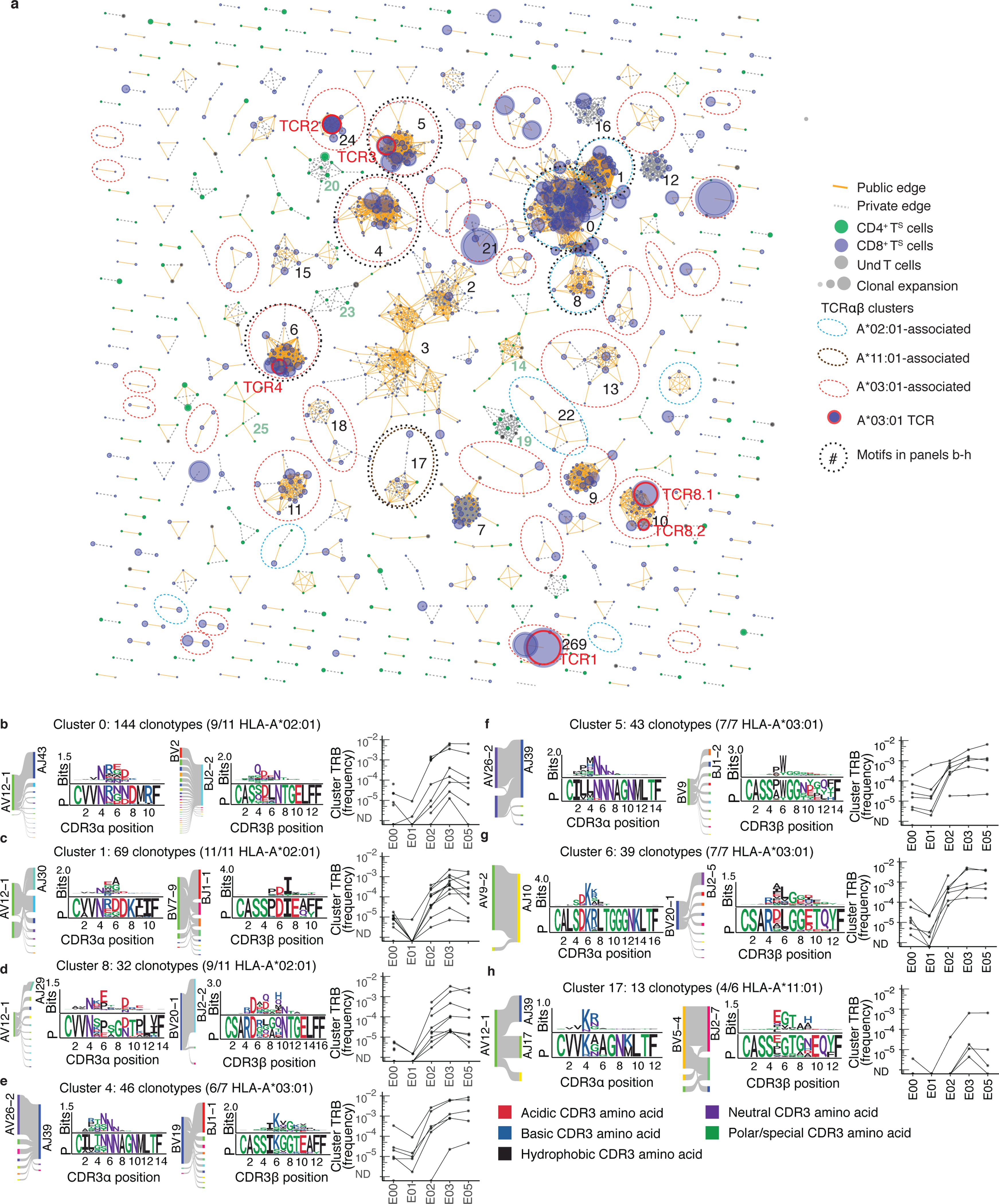
(a) Sequence similarity graph with 1448 paired TCRɑβ clonotypes and 248 convergent CD8+ T cell and CD4+ T cell clusters of two or more S-reactive clonotypes recovered from 17 persons at E03. Edges are formed between similar receptors (TCRdist ≤ 100). Edges indicate connections between TCRs observed in multiple or single participants. Circle size represents the relative frequency of each TCRαβ clonotype. Equivalent TCRαβ amino acid sequences may be included more than once if found in multiple participants. (b-h) Logo plots for representative clusters indicated by integers in (a) with inferred restricting HLA class I alleles (see Methods) and number of participants contributing to each cluster with the matching HLA allele, and graphs showing the sum of clonal frequency of the TRB sequences in each cluster in longitudinal PBMC repertoires for each participant. For each CDR3 motif, the lower sequence logo shows the probability of each amino acid residue at each CDR3 position, and the upper sequence logo depicts the information content in bits comparing the residue usage to a set of randomly selected CDR3 with the same V and J gene usage as the sequence cluster (Methods).
Cluster analyses identified many public TCRαβ paired motifs with matching TRB clonotypes expanded by vaccination, most notably in persons expressing the HLA-A*02:01, A*03:01 or A*11:01 alleles (Fig. 4, Supplementary Data Figs. 6,7). Two large public clusters (cluster 0 and cluster 1) corresponded to previously identified NTGEL-TRBJ2–2 (Fig. 4b) and PDIE (Fig. 4c) motifs, which recognize the HLA-A*02:01-restricted epitope YLQPRTFLL (amino acid 269–277 in S, S269–277)9,14,25–29. While TCRs from cluster 0 and cluster 1 were ubiquitous among HLA-A*02:01 participants post-infection (E00) and post-dose 1 (E02 and later), a related HLA-A*02:01-assigned motif (cluster 8) was only commonly observed post-dose 1 (E02 and later) (Fig. 4d). Cluster 8 TCRs were distinguished by longer CDR3α (14 amino acids) and CDR3β (16 amino acids) and strict TRAV12–1/TRBV29–1 usage compared to cluster 0 TCRs (Fig. 4b, d). Cluster 8 TCRs had a much lower probability of being generated30,31 (median Pgen CDR3α = 5.1×10−9, CDR3β = 9.7×10−11) compared to cluster 0 TCRs (median Pgen CDR3α = 2.2×10−8, CDR3β = 1.5×10−8) (Fig. 4b, d), yet cluster 8 TCRs were expanded from E01 compared to E03 in 8 of 11 HLA-A*02:01 participants (Fig. 4d). This suggests vaccination may diversify the set of circulating TCRs targeting immunodominant epitopes.
We observed highly public HLA-A*03:01-associated TCR clusters, with sequence motifs found in at least 6 of the 7 HLA-A*03:01-expressing participants (Fig. 4e–g). Two large HLA-A*03:01-associated clusters (cluster 4 and cluster 5) shared CDR3α junctions with central NNNAG residues paired with distinct and V-gene biased CDR3β receptor motifs in cluster 4 (TRBV19-dominated) and cluster 5 (TRBV9-dominated) (Fig. 4e, f). Cluster 4 and 5 CDR3β motifs shared similar central junctional residues, such as SIKGG (Fig. 4e) and SPWGG (Fig. 4f), with a hydrophobic residue preceding diglycine residues (Fig. 4e, f). This pattern also appeared within the CDR3β motif of public HLA-A*03:01-associated cluster 6 (Fig. 4g). A prevalent HLA-A*11:01-associated receptor motif was found in 4 of 6 HLA-A*11:01 participants (Fig. 4h). CD69+CD137+ AIM-scTCRɑβ-seq groupings also included two large clusters (cluster 2 and cluster 3) using the TRAV1–2 and TRAJ33 genes (Fig. 4e, f), characteristic of MAIT cells32–34. Clonotypes in these MAIT clusters did not expand after vaccination in most participants (Supplementary Data Fig. 6). Together, this analysis linked clusters of similar TCRs with likely shared peptide reactivity to a potentially restricting HLA allele for experimental investigation.
AIM-scTCRαβ public cluster members are S-specific
To confirm the ligands of representative TCRs assigned to HLA-A*03:01, we expressed six TCRαβ identified with CD69+CD137+ AIM-scTCRɑβ-seq from participant P673 in Jurkat cells that express mNeonGreen after TCR engagement (hereafter Jurkat reporter cells) (Fig. 5a). The TCRs selected were strongly expanded after vaccination (Fig. 5b) and representative of AIM-scTCRαβ clonotypes found in TCR clusters (Fig. 4a).29 All six TCRs showed reactivity with peptide S378–387 (KCYGVSPTKL), an HLA-A*03:01-restricted epitope (Fig. 5c)29. Strong activation of Jurkat reporter cells was observed for each TCR in the presence of artificial antigen presenting cells (aAPC) co-expressing HLA-A*03:01 and full-length S from ancestral strain Wu-1 (Fig. 5d). Control aAPC expressing other HLA-A or B from participant P673 with S, or HLA-A*03:01 alone, did not activate the Jurkat reporter cells (Fig. 5d). While this core epitope is invariant in Omicron variants of SARS-CoV-2, flanking amino acid residues can influence antigen processing35. Near full-length S and relevant peptides from Omicron BA.1, BA.2 or BA.4 SARS-CoV-2 also activated each TCR (Fig. 5d, Supplementary Data Fig. 8). We observed potential differences in the ligand requirements of the TCRs (Fig. 5c, e), indicating that TCRdist may cluster TCRs into functionally meaningful groups. TCR1 and TCR4, from clusters 269 and 6, respectively, recognized the 10-mer peptide S378–387 (KCYGVSPTKL), but not the internal 9-mers (KCYGVSPTK or CYGVSPTKL) (Fig. 5c). In contrast, TCR3 was versatile, and equally recognized the parent 10-mer and each 9-mer (Fig. 5c). TCR2 optimally recognized S378–387 and partially recognized the internal 9-mers (Fig. 5c). Peptide titration confirmed differences in peptide length requirements between TCRs (Fig. 5e, Supplementary Data Table 12).
Fig. 5. Transgenic CD8+ T cell-origin TCRs from 5 public clusters are activated in the context of HLA-A*03:01 and the KCY epitope.
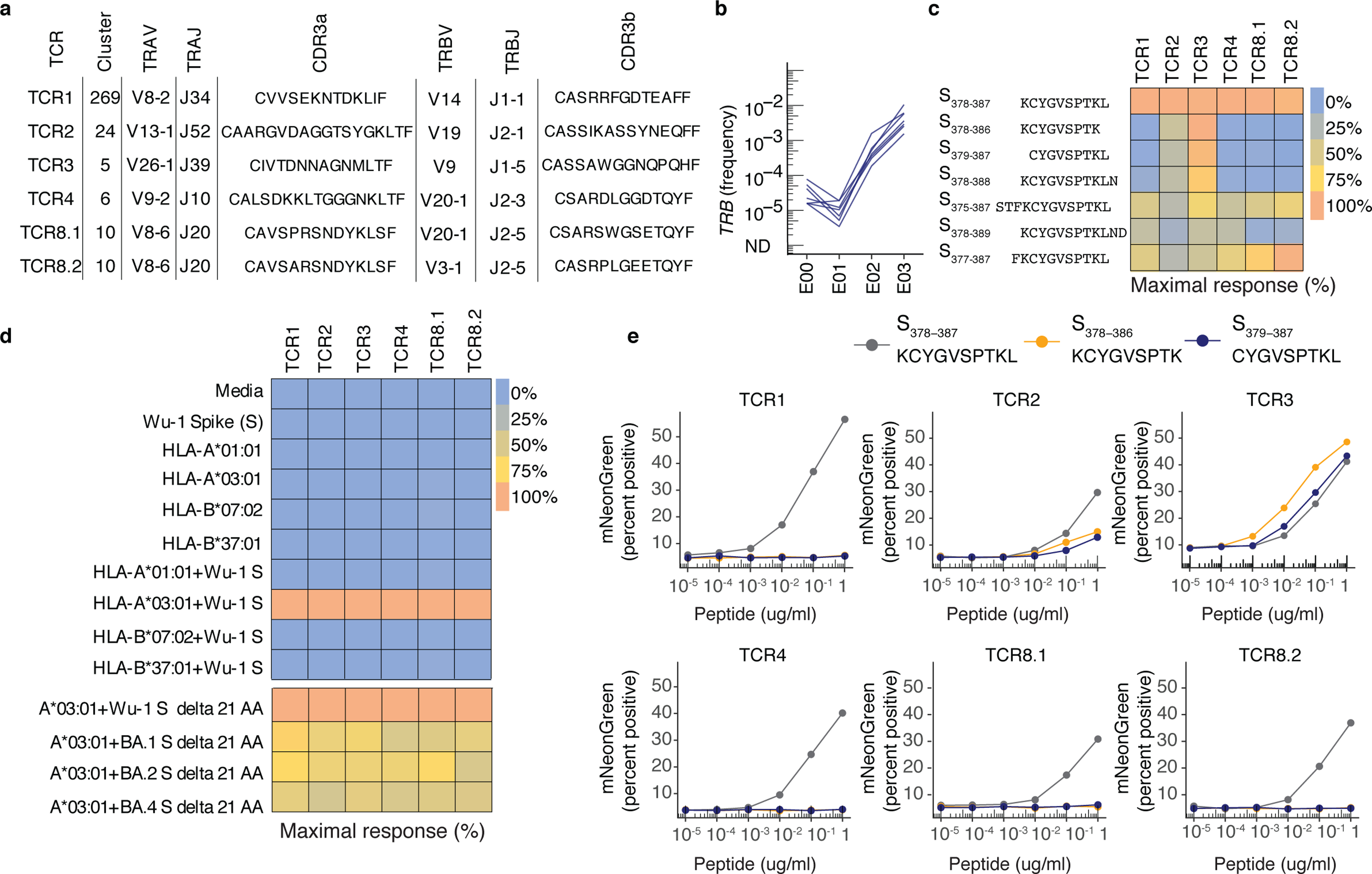
(a) TRA- and TRB-gene usage and CDR3 sequences of transgenic TCRs from five public sequence-similarity clusters expressed in Jurkat reporter cells (Methods). (b) Frequency in PBMC of selected clonotypes in the bulk TCRβ-seq datasets from E00 to E03. (c) Heatmap showing the percent of maximal response for each TCR expressed in the Jurkat reporter cells cultured with HLA-A*03:01-expressing APC exposed to peptides of varying lengths in the region of residues 378–386 of S protein [1μg/mL] within a single experiment. (d) Heatmap showing the percent of maximal response for each TCR expressed in Jurkat reporter cells stimulated with HLA class I-expressing artificial APC with or without co-transfection of full-length S from SARS-CoV-2 strain Wuhan-1 (Wu-1) within a single experiment. Controls include APC treated with media or S alone. The last four rows of the heatmap represent a separate experiment, whereby each TCR was tested with HLA-A*03:01 and Wu-1 S or Omicron variants BA.1, BA.2, and BA.4. (e) Peptide titration (10−5 to 1 μg/mL) with SARS-CoV-2 S 10-mer (KCYGVSPTKL) and internal 9-mers (KCYGVSPTK and CYGVSPTKL) tested with TCR-transduced Jurkat reporter cells cultured with lymphoblastoid cell lines known to express HLA-A*03:01. Y-axis shows percent of reporter cells with mNeonGreen fluorescence.
To confirm that CD69+CD137+ AIM-scTCRɑβ-seq can identify antigen-specific CD4+ T cells, we expressed representative CD4+ TCR clonotypes in autologous polyclonal CD4+ T cells transduced with TCRs of interest (Methods) and tested activation by S protein and peptides (Supplementary Data Fig. 9, Supplementary Data Tables 13, 14). We further queried clusters of CD4+ TS cells recovered by CD69+CD137+ AIM-scTCRɑβ-seq against previously identified TS motifs. CD69+CD137+ AIM-scTCRɑβ-seq clusters of CD4+ TS clonotypes overlapped with published CD69+CD154+CD4+TCR-seq single chain TRA and TRB of CD4+ T cells responsive to mRNA vaccines in SARS-CoV-2-naïve individuals36 (Supplementary Data Fig. 10a). In addition, CD69+CD137+ AIM-scTCRɑβ-seq cluster 49 matched the TRAV35-NYGGSQ motif found enriched in draining lymph nodes of DPA1*01:03/DPB1*04:02 individuals in response to an immunodominant S167–180 class II epitope37 (Supplementary Data Fig. 10b). The CDR3β of cluster 49 TCRs contained the S*RG**GY-TRBJ1–2 motif proposed to commonly pair with the TRAV35-NYGGSQ motif to form S167–180 reactive receptors38. Thus, the publicity of TS TCR motifs, the experimental confirmation of selected CD4+ and CD8+ T cell-origin TCRs and the recovery of TCRs similar to those previously identified as SARS-CoV-2-specific validated the use of AIM to discover antigen specific receptors without prior epitope knowledge.
Severe disease imprints CD4+ TS cells
To further assess the participant level heterogeneity and longitudinal dynamics of SARS-CoV-2-reactive CD4+ T cells, we computed the breadth of clones in E00-E05 samples that matched a diagnostic set of CD4+ T cell-associated TRB sequences reported to be enriched in SARS-CoV-2 convalescent compared to healthy control repertoires39 as a measure of antigen-specific TCR diversity (hereafter ‘diagnostic clonal breadth’). These TRB sequences40–43 were previously assigned to S (n=917) or non-Spike (n=1,564) SARS-CoV-2 antigens42 (Methods). To examine if COVID-19 severity resulted in differential imprinting of the T cell repertoire, we compared the diagnostic breadth of patients with mild/moderate and severe COVID-19 (Fig. 6a, Supplementary Data Table 15). Severe infection was associated with greater overall SARS-CoV-2 specific TRB repertoire diversity after infection18,44. We observed greater diagnostic breadth of S-reactive CD4+ T cells in patients with severe COVID-19 compared to patients with only mild/moderate infection at E00 (0.014 vs 0.006%, Fig. 6a), but this difference was not detected at E01 or E02 (Fig. 6a). Diagnostic S-reactive CD4+ T cell breadth was elevated in participants with severe COVID-19 compared to those with mild/moderate disease at E03 (0.012 vs 0.008%) and E05 (0.009% vs 0.005%) (Fig. 6a). In contrast, the breadth of E01-to-E03 expanded TRB clonotypes at E03 did not correlate with severity of prior infection (Fig. 6b). The diagnostic breadth of S and non-S CD4+ T cells weakly correlated (rank correlation ρ = 0.38, p = 0.019) at E00 (Fig. 6c). The diagnostic breadth of S-reactive CD4+ T cells increased promptly at E02 and slowly declined at E03 and E05, in contrast to the non-S-reactive CD4+ T cell breadth, which remained stable from E01 to E05 after the initial decline from E00 to E01 (Fig. 6d). CMV infection has been reported to impact immune parameters related to SARS-CoV-2 infection or vaccine45,46. We did not observe any associations between imputed CMV infection status47 and parameters of SARS-CoV-2-specific TRB repertoires at E00 (Fig. 6c).
Fig. 6. TRB sequence-defined metrics and nAb associate with disease severity before and after mRNA vaccination.
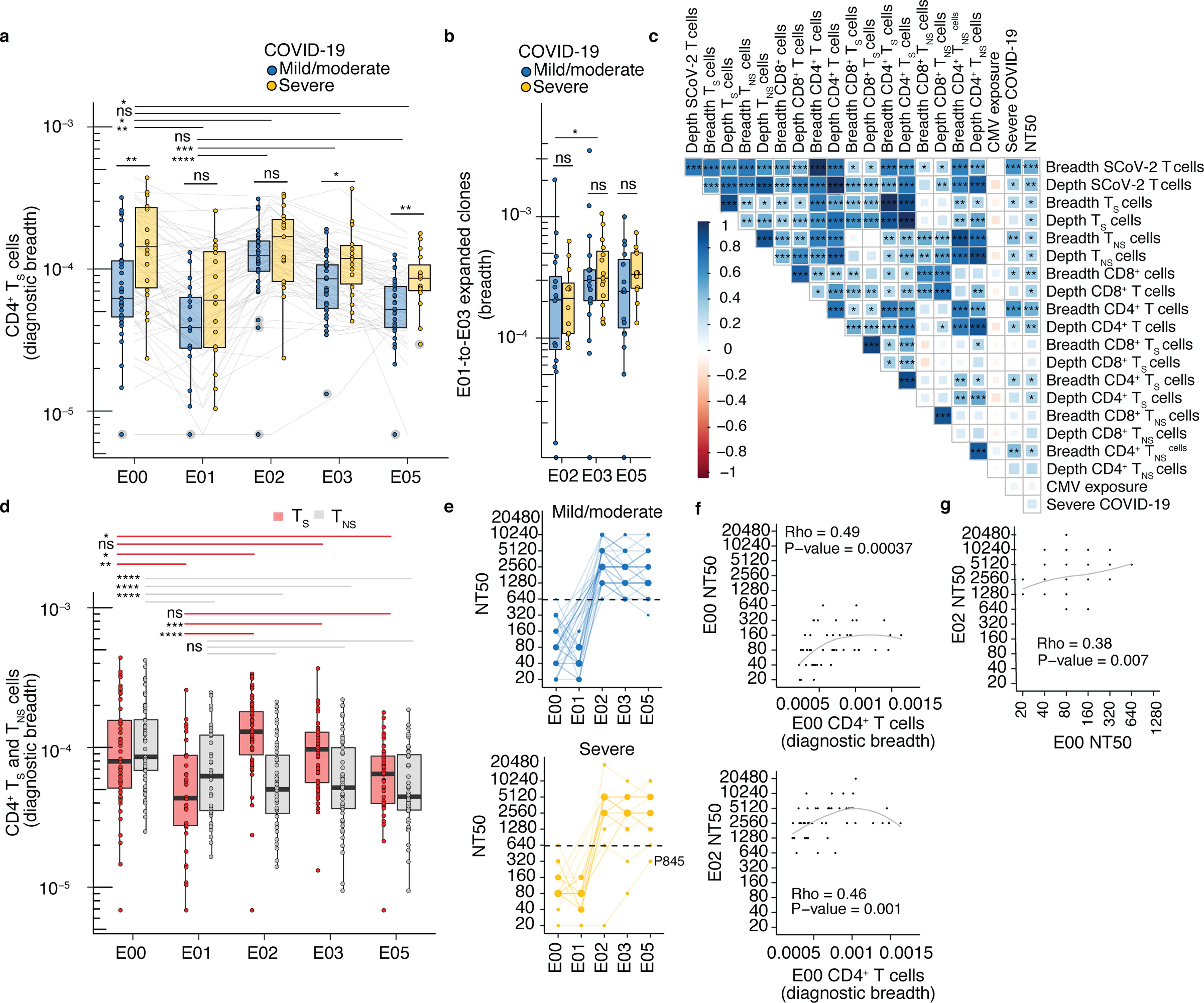
(a) Comparison of diagnostic CD4+ TS breadth (Methods) between participants with mild/moderate COVID-19 at E00 (n=35), E01 (n=18), E02 (n=35), E03 (n=35), E05 (n =29) and severe COVID-19 at E00 (n=19), E01 (n=16), E02 (n=17), E03 (n=18), E05 (n=15)). (b) Breadth of E01-to-E02, E01-E03 and E01-E05 expanded clonotypes in mild/moderate COVID-19 at E02 (n=17), E03 (n=18), E05 (n=16), and severe COVID-19 at E02 (n=12), E03 (n=15), E05 (n=12). (c) Rank correlation of diagnostic sequence-defined metrics (S, spike; NS, non-spike) with severity and neutralization titers (NT50) at E00 (n = 51). CMV exposure was imputed from TRB repertoire (Methods). Shading, strength of correlation (ρ correlation coefficient); asterisks, level of statistical significance. (d) Breadth of inferred CD4+ TS and TNS TRB sequences from PBMC at E00 (n=54), E01 (n=34), E02 (n=52), E03 (n=53), E05 (n=44). (e) NT50 of serum antibodies in mild/moderate (n=35) and severe COVID-19 (n=19) at E00, E01, E02, E03, and E05). (f) Association of CD4+ T cell diagnostic breadth at E00 with nAb NT50 at E00 and E02 (n=52). (g) Association between E00 nAb NT50 and E02 nAb NT50 (n=52) in participants with prior SARS-CoV-2 infection. In (a, b, d), tests between time points are paired Wilcoxon signed-rank and between groups with different severity are unpaired Wilcoxon rank-sum. Median, IQR, and whiskers (1.5 * IQR) are shown. Level of two-sided statistical significance is as indicated: ns = not significant, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
To examine a potential correlation between T cell and humoral responses in this cohort, we measured neutralizing antibody titers (NT50) in all participants. NT50 declined post SARS-CoV-2 infection (median 80, IQR 50–160 at E00; median 60, IQR 40–80 at E01), but was boosted in 51 of 52 participants at E02 (median 2,560, IQR 2,250–5,120, Fig. 6e). In contrast to dose-dependent serial boosting of CD8+ T cells from E02 to E03, little to no further increase in NT50 was observed from E02 to E03 (Fig. 6e). The post-infection diagnostic breadth of CD4+ T cell at E00 was correlated with both NT50 at E00 (ρ = 0.49, p-value 0.00004, Fig. 6f) and after vaccination at E02 (ρ = 0.46, p-value 0.001 Fig. 6f). These data were consistent with CD4+ T cells help to B cells, and possible detection of S-specific circulating T follicular helper-like cells (Tfh cells), which have been positively associated with neutralizing antibody titers48. These results suggested that in hybrid immunity severe disease imprints a more diverse and persistent CD4+ TS cell population, which was maintained after repeated antigen exposures.
Discussion
Here we longitudinally profiled the cellular and humoral immunity in a cohort of adults from post-SARS-CoV-2 infection through 3 doses of mRNA vaccine. We used CD69+CD137+ AIM-scTCRɑβ-seq and CD4+ AIM-TCRβ-seq to obtain index TS clonotypes to track in serial bulk TRB repertoires. This revealed divergent vaccine response kinetics between CD4+ and CD8+ TS cells. In individuals with prior SARS-CoV-2 infection, mRNA vaccines induced profound, albeit variable, expansion of pre-existing circulating Tm cell clones. We observed that the first two doses of the vaccine augmented the S-reactive clonotypic diversity in the blood, consistent with observations in peptide-HLA oligomer-sorted T cells following repeated antigen exposure9. These doses lead to strong expansion in CD8+ TS clonotypes measured by both CD69+CD137+ AIM-scTCRɑβ-seq and intracellular cytokine staining. Rapid expansion of CD8+ T cells after the first mRNA vaccination has also been described in SARS-CoV-2-naive persons25.
In contrast to our results, a study that measured the percentage of AIM+CD4+ TS cells or AIM+CD8+ TS cells (AIM+ defined as 4 of the 5 activation markers CD200, CD154, CD137, CD107a and IFN-γ) in the blood of 11 persons with prior SARS-CoV-2 infection found prominent CD4+ TS cell expansion, but little increase in CD8+ TS cells49. Differences in study design - the cohort was younger (median age 34.7 compared to 60.7 years in our report), with shorter intervals between infection, vaccination and PBMC sampling49, and an emphasis on specificity over sensitivity may account for these divergent conclusions. The CD8+ TS cell selection49 was more restrictive than ours and included CD154, which is seldom expressed on activated CD8+ T cells50,51. The different results could also stem from an incomplete coverage of S protein when measuring CD8+ T cell responses49,52. To increase sensitivity, we used CD69+CD137+CD8+ to identify vaccine-responsive CD8+ TS cells and two methods to identify vaccine-responsive CD4+ TS cells (CD69+CD137+ and (CD69/CD137)+(CD134/CD154)+). We further used intracellular cytokine staining to compare CD4+ and CD8+ TS cells after the first and second vaccine doses. With all approaches, we found robust CD8+ T cell expansion and more modest CD4+ T cell expansion after the second vaccination.
This report builds on prior work describing longitudinal dynamics in TCR repertoires in general, and the response to SARS-CoV-2 infection and vaccination in particular. A foundational study identified vaccine-expanded TRB sequences after yellow fever vaccination in naive persons to link yellow-fever-associated TRB sequences and shared HLA alleles to T cell clones of unknown specificity53,54. Prior work has defined many individual TCRs and TCR clusters in the context of Tm cell contraction after COVID-1955 or SARS-CoV-2 vaccination in naive persons56 by peptide-HLA oligomer-sorted T cells or statistical enrichment versus a control (often historical) population. Our work used AIM with statistical filters to assign S-specificity to individual TCRs. We recovered thousands of paired-chain TCRαβ sequences with S-reactivity to augment public databases57 for characterizing T cell responses to vaccines58,59 and reveal both α-and β-chain sequence features contributing to epitope specificity. We report a large set of experimentally validated public CDR3 sequence motifs for an immunodominant A*03:01-restricted S epitope. Through experimental validation of proposed S-specific TCRs within sequence-similar clusters, we successfully de-orphaned TCR-peptide-HLA ligand pairings without foreknowledge of epitopes or restricting HLA.
Our study has several limitations. We studied heterologous exposures only in the context of infection before vaccination. The order of serial antigen exposures may differentially shape the TCR repertoire. Our study was not structured to detect associations of TCR clonotypes with functional profiles or with COVID-19 severity. Since our subjects were infected early in the pandemic and were generally older (median age 60.6 years), our findings may not directly translate to younger persons. Another limitation is the inability to infer directly whether E01-to-E03 expanded clonotypes that were below the limit of detection at all time points prior to vaccination came from either very rare Tm or naive T cell populations. However, we showed that both doses of mRNA vaccination can expand low-abundance CD8+ T cell clonotypes that were undetectable post SARS-CoV-2 infection. Synthesis of serial bulk TCRβ with single cell TCRɑβ sequencing provides insights into the spectrum of mRNA vaccine potency across doses, and it provides a tool to efficiently focus research attention on antigen specific TCRs for detailed assignment of epitope specificity. Further research is required to determine how the phenotype, durability and distribution of CD4+ and CD8+ T cells elicited by hybrid exposures compare to responses elicited by infection or vaccination alone.
Methods
Participants and specimens.
Persons with a self-reported history of PCR-confirmed COVID-19 were recruited through local advertising from April through August 2020 in the Seattle area for convalescent plasma donation (NCT 04338360, 04344977). Participants provided informed written consent for University of Washington (UW) Institutional Review Board-approved protocol STUDY00004312 “Protocol for the Collection of Laboratory Research Specimens”. Participant data was collected and managed in RedCap, a protected health information-secure data collection and management resource (hosted at UW Institute of Translational Health Sciences, Seattle, WA)60. PBMC were cryopreserved at 5–10 X 106 cells/vial in 10% DMSO, 50% human serum, 40% RPMI-1640 in LN2. Plasma from heparin-anticoagulated blood or serum was frozen at −20°C.
Study cohort.
57 persons agreed to observational PBMC samples from disease convalescence through vaccination. No statistical methods were used to determine sample sizes. Two persons without an E00 or E03 sample were excluded, as was one person who did not receive an mRNA vaccine. Of the remaining 54 participants, 35 participants had mild/moderate and 19 had severe COVID-19 requiring hospitalization and oxygen support. Eight had critical illness requiring ICU admission. All persons attested to a diagnostic PCR sample confirming SARS-CoV-2 infection, 50 could be confirmed. Twenty-eight were female. The median age was 60.3 years, range 31.4–73.5 years. Comorbidities which did not preclude blood or plasma donation included a history of cancer (n=8), heart disease (n=9), renal disease (n=4), hypertension (n=9), hyperlipidemia (n=10), lung disease (n=9), and diabetes (n=9). One participant (P845) was serologically naive prior to vaccination and was excluded from statistical analysis (Supplementary Data Fig. 11). Three participants (P545, P664, and P669) had breakthrough infections between the second and third mRNA vaccine doses and E05 samples were excluded from statistical analyses (Extended Data Fig. 1, Supplementary Data Table 16). Data collection and analysis were not performed blind to the conditions of the experiments.
SARS-CoV-2 and CMV infection status.
Plasma neutralizing antibodies against SARS-CoV-2 strain WA1 were measured by microfluorescence61 and resulted as the reciprocal dilution inhibiting infection by 50% (NT50). Antibodies used in the neutralization assay were an anti-SARS nucleocapsid antibody (1:8000, rabbit monoclonal; Sino Biological #40143-R001, Wayne, PA, USA), Alexa Fluor 594-conjugated goat anti-rabbit IgG secondary (1:2500, Thermo Fisher Scientific #A11037), and Hoechst 33342 nuclear stain (Thermo Fisher Scientific #H3570). Plasma SARS-CoV-2 anti-strain Wu-1 S and anti-N IgG were measured by microbead-based binding assay62 and reported as μg/ml. CMV infection status was imputed from analysis of whole PBMC TRB repertoires from the first available sample from each subject using the CMV classifier tool (Adaptive Biotechnologies, Seattle, WA), an update of published methods47 (Supplementary Data Table 1).
Isolation of memory and naïve T cell subsets.
From the earliest available timepoint (E00), for selected subjects, cryopreserved PBMC (2×107) were thawed and stained with anti-human CCR7-PE (1.25:100, G043H7, BioLegend), anti-human CD95-Pacific Blue (2.5:100, DX2, BioLegend), anti-human CD45RA-APC (1.25:100, HI100, BioLegend), anti-human CD4-BV510 (2.5:100, A161A1, BioLegend), anti-human CD8-FITC (5:100, 3B5, LifeTech), anti-human CD3-ECD (2:100, UCHT1, Becton Dickinson), and 7-actinomycin D (7-AAD, 1:10, Becton Dickinson). Among gated, live CD3+ single cells, single-positive CD4+CD8− and CD4−CD8+ cells were identified and double-negative or double-positive cells were excluded. CD4+CD8− and CD4−CD8+ cells were analyzed separately for CCR7 and CD45RA. CCR7+/CD45RA+ cells were separated from cells expressing one or neither marker. CD45RA-intermediate and CCR7-intermediate cells (<5% of total CD3+ T cells) were excluded. Among pooled CCR7+/CD45RA+ TCM, CCR7−CD45RA− TEMRA, and CCR7−/CD45RA+ TEM cells, the majority (>99%) were CD95+. Among CCR7+CD45+ cells preliminarily assigned as naive, cells with CD95 expression below the lower limit of CD95 expression amongst memory cells were defined as naive. Thus, specificity for memory or naive subsets was emphasized over cell inclusion in the gating scheme. CD4+ and CD8+ cells were pooled for memory and naive TRB repertoire analyses. Representative gating scheme is shown in Supplementary Data Fig. 1, participants and raw sequencing results are shown in Supplementary Data Table 5.
AIM detection and sorting of SARS-CoV-2-reactive T cells.
PBMC were thawed and cultured at 4 X 106 cells/well in 2 mL/well T cell medium (TCM)63 in 2 to 3 wells of a 24-well plates with 1 μg/mL each peptide covering SARS-CoV-2 strain Wu-1 Spike (PM-WCPV-S-1, JPT, Berlin, Germany) in a final concentration of 0.2% DMSO, or DMSO negative control. Peptides were 15 amino acids long with 11 amino acid overlap. After 18 hours, cells were recovered by centrifugation into 50 μL PBS with 1% BSA and incubated with 5 μL TruStainFcX™ blocking reagent (BioLegend) for 10 minutes on ice, followed by addition of 50 μL of a cocktail (Supplementary Data Table 17) of oligonucleotide-labeled mAbs (Totalseq™ C, BioLegend) for 5 minutes on ice. Cells were then stained with anti-CD3-FITC (3:200, SK7, BioLegend), anti-CD69-BV421 (3:100, FN50, BioLegend), anti-CD137-APC (2:10, 4B4–1, Becton Dickinson), and 7-AAD (1:10, Becton Dickinson), washed and resuspended in 1 ml TCM. Live, single, CD3+ cells expressing CD69 and CD137 were sorted (FACSAria II, Becton Dickinson) from S-stimulated PBMC for subsequent CD69+CD137+ AIM-scTCRɑβ-seq (representative gating tree, Supplementary Data Fig. 12a).
To study AIM combinations, we investigated both the abundance and TRB repertoire of CD4 T cells staining for various markers. PBMC were stimulated and stained with anti-CD3-PE (1:200, UCHT1, Biolegend), anti-CD4-APC-H7 (1:40, RPA-T4, Becton Dickinson), anti-CD8-FITC (1:20, 3B5, Life Technologies), anti-CD25-PE-Cy7 (1:50, BC96, BioLegend), anti-CD134-BV480 (1:50, L106, Becton Dickinson), anti-CD69-BV421 (3:100, FN50, BioLegend), anti-CD137-APC (1:5, 4B4–1, Becton Dickinson), anti-CD154-BV711 (1:50, 24–31, BioLegend), and 7-AAD. After washing, data was acquired on a FACSAria III in analytic mode. Gating and Boolean analyses used FlowJo 10.7.1 (Becton Dickinson). Gating schemes are shown in Supplementary Data Fig. 12. We investigated activation of CD4+ T cells by measuring TNF receptor (TNFR) family members CD134 (OX40L), CD137 (4–1BB), and CD154 (CD40L), CD25 (IL-2 receptor subunit), and CD69. We considered five pairs of activation markers: CD69/CD137, CD25/CD134, CD134/CD137, CD134/CD154, and CD137/CD154, and set the sum of cells expressing at least two markers to 100%. For CD8+ T cells, CD69/CD137 detected a median of 85.7% of all activated cells, with CD134 and CD154 showing very little activation (Supplementary Data Figs. 2, 12). For CD4+ T cells, the sensitivity to detect peptide activation using CD69/CD137 was more limited (median 21.1%). However, no AIM molecule pair was consistently the most sensitive. CD69/CD137 showed better sensitivity than combinations of two TNFRs and was used for AIM-scTCRab-seq to identify both CD4+ and CD8+ T cell activation by peptide stimulation. To more formally study the relationship between selection markers and TCR repertoires, we selected seven subjects with PBMC availability at the E03 timepoint (Supplementary Data Table 10). We sequentially sorted live, single, CD3+, CD8− T cells stimulated for 18 hours with S peptides, first selecting all CD69+/CD137+ cells (fraction 1, black gates), and then selecting any residual CD69+ cells expressing CD134 and/or CD154 (fraction 2, blue gates) and finally any residual CD137+ cells expressing CD134 and/or CD154 (fraction 3, red gates) (representative gating scheme, Supplementary Data Fig. 4). Fraction 1 and pooled fractions 2 and 3 were submitted (separately) for bulk TCRβ-seq.
Intracellular cytokine staining.
Cryopreserved peripheral blood mononuclear cells (PBMC) were thawed and rested overnight. PBMC (1 × 106 per well) were stimulated with a SARS-CoV-2 spike overlapping peptide pool (JPT, 1 μg/mL each peptide; 0.4% final DMSO concentration), 0.4% DMSO as a negative control, or PHA-P (Remel, Lenexa, KS, USA; 1.6 μg/mL final) as a positive control, in the presence of anti-CD28 (1:1000, L293, BD Biosciences) and anti-CD49d (1:1000, L25, BD Biosciences) antibodies at 37°C for 6 hours. Brefeldin A (Sigma) was added after 2 hours. Cells were stained with Live/Dead Near IR dye (1:250, Invitrogen), treated with FACS lyse (BD Biosciences) and frozen at −80°C. For staining, the cells were thawed, washed, and permeabilized with Permeabilizing solution 2 (BD Biosciences) then stained with fluorochrome labeled monoclonal antibodies to anti-human CD3-PE-Texas Red (1:40, UCHT, Beckman Coulter), anti-human CD4-BV510 (1:83, SK3, Biolegend), anti-human CD8-PerCP-Cy5.5 (1:33, SK1, BD Biosciences), and the activation markers anti-human CD40L-BV421 (1:167, TRAP1, BD Biosciences), anti-human IFNγ-PE (1:500, 4S.B3, BD Biosciences), anti-human IL2-APC (1:250, MQ1–17H12, BD Biosciences), and anti-human TNFα-FITC (1:400, Mab11, BD Biosciences). Events were recorded with BD Fortessa and analyzed with FlowJo (v10 for Mac; BD).
Bulk TCR sequencing.
Genomic DNA was extracted from frozen PBMC samples using the Qiagen Dneasy Blood Extraction Kit (Qiagen). Immunosequencing of CDR3 regions of TCR-β chains used the ImmunoSEQ™ Assay (Adaptive Biotechnologies, Seattle, WA, USA). Input DNA was amplified in a bias-controlled multiplex PCR, followed by high-throughput sequencing. Sequences were collapsed and filtered to identify and quantitate the absolute abundance of each unique TCR-β CDR3 region for further analysis, as described40,41,43. Blood TRB sequencing recovered medians of 429,661 productive TRB templates and 242,861 unique productive TRB rearrangements (‘clonotypes’) per sample (Supplementary Data Table 2). For analyses of bulk repertoires, the term clonotype is used for a unique TRB sequence: a complementarity determining region 3 (CDR3) nucleotide sequence and associated TRBV and TRBJ genes. These generally distinguish a unique T cell clonotype; occasionally, a single TRB may pair with > 1 TRA in distinct T cell clonotypes. To assist the assessment of expanded or AIM-selected T cell clonotypes as CD4+ T cells, we stained cells with anti-human CD3-PE-Texas Red (1:40, UCHT, Beckman Coulter), anti-human CD4-BV510 (1:83, SK3, Biolegend), and anti-human CD8-PerCP-Cy5.5 (1:33, SK1, BD Biosciences). We then sorted unstimulated live single CD3+CD4+CD8− cells from the E03 post-vaccine timepoint for bulk TCR sequencing (representative gating scheme, Supplementary Data Fig. 4a), selecting the same subjects used to study CD4+ T cell AIM selection conditions and memory and naive cells (cells sorted per subject, Supplementary Data Table 10).
Single cell TCR sequencing.
Sorted single cells were segregated into nanoliter droplets (Chromium Next GEM Single Cell 5’ Kit v2, PN-1000263, Chromium Next GEM Chip K Single Cell Kit, PN-1000286, Chromium Controller, 10X Genomics, Pleasanton, CA, USA). The VDJ and antibody feature barcode libraries were made per user guide (CG000330 Rev C, 10X Genomics). Library quality was measured by TapeStation (Agilent, Santa Clara, CA, USA). Library DNA quantification was measured by Qubit 3.0 Fluorometer (ThermoFisher, Waltham, MA, USA). Sequencing depth was 5,000 paired reads per cell with configuration 26, 10, 10, 150, assuming 10,000 cells per library. Sequencing used the NovaSeq 6000 System (Illumina, San Diego, CA, USA) and SP200 kit (Illumina).
Single-cell VDJseq and feature barcode data analysis and alignment.
Raw sequencing data were processed with the Cell Ranger version 6.1.0 (10X Genomics) pipeline. Demultiplexing from raw .bcl data and conversion to .fastq data used Cell Ranger mkfasq. Surface feature barcode antibody binding analyses used Cell Ranger counts with the reference feature barcode library (Supplementary Data Table 17). Initial TCR VDJ analyses used the Cell Ranger VDJ module and GRCh-Alts-ensembl-5.0.0.
CD4+/CD8+ assignments.
For analyses of single cell data, we computed the percent of UMI counts corresponding to DNA barcodes for CD8 and CD4 assigned to each marker. To assign a phenotype per cell, we computed a score based on the natural logarithm of total CD8 divided total CD4 counts. A score greater than 1 was classified as CD8 and a score less than −1 was classified as CD4. Cells with fewer than 10 UMIs or values between 1 and −1 were considered ambiguous and not assigned a T cell phenotype. When a TCR clonotype (cells with identical TRA and TRB nucleotide sequences) was present in multiple cells, the median score was used to classify that clonotype. For bulk TRB analyses of AIM-selected total CD4+ T cells, we considered TRB sequences that were present and enriched in the flow sorted (bulk) CD4+ T cell population as compared to unselected PBMCs to be CD4+. For bulk CD4+ TRB analyses, TRB sequences that were enriched in the bulk CD4+ T cell population as compared to unselected PBMCs were assigned to be CD4+.
Longitudinal analysis of S-reactive clones.
We tested for enrichment of S-reactive (TS) clones in the AIM assay using a statistical test. The observed frequency of each AIM+ clonotype among the total AIM+ cells was compared with an expectation from a null model based on each clonotype’s frequency in the bulk sequenced repertoire from the same sample. The p-value of the observed counts of each AIM+ clonotype under the null model was computed from the complement of the binomial cumulative distribution function:
From the binomial cumulative distribution function, we computed the chance of observing single cells of a given clonotype in a pool of total AIM+ single cells, with the null success probability equal to the fraction of the matching TRB in the unsorted bulk repertoire. We then applied a multiple hypothesis correction with the Benjamini-Hochberg procedure to compute an FDR-adjusted q-value for each AIM+ clonotype. We designated clonotypes with a q-value < 0.05 as stringently enriched by the AIM sort and thus high confidence S-reactive clones. These highest-confidence S-reactive clonotypes were used for trajectory analysis and estimation of the total fraction of the repertoire composed of S-reactive CD4+ and CD8+ T cells, respectively.
TCR sequence clustering.
To compare and cluster paired TRA/TRB sequences between cells, we first filtered sequences from CD69+CD137+ AIM-scTCRɑβ-seq to those with a matching TRB from the deeply sequenced bulk blood repertoire from the same subject and timepoint. Next, we filtered out sequences with TRB occurring at a lower frequency in the set of clonotypes expressing AIM markers than in the bulk repertoire, clonotypes which had not been enriched by AIM. Non-enriched clonotypes were assumed to have been sorted after bystander activation and were not considered further. Next we computed pairwise dissimilarity between 5,569 subject-unique TRA/TRB clonotypes using the TCRdist distance metric24 as implemented using default parameters in tcrdist3 version 0.2.264. Pruning the pairwise distance matrix to include connections between sequences within 100 TCRdist units, we formed a sequence graph.
When selecting samples to analyze by TCRdist, we restricted analyses to samples to include ≥2 persons with prevalent HLA alleles, such that the subcohort studied included persons with HLA-A*02:01 (n=11), HLA-A*03:01 (n=7), HLA-A*11:01 (n=6), HLA-B*07:02 (n=6), HLA-B*15:01 (n=4), HLA-B*35:01 (n=4), HLA-C*07:02 (n=8), and HLA-C*03:04 (n=3), and HLA-A*24:02 (n=2) (Supplementary Data Tables 1, 2, 11). To examine whether connected components with the graph (i.e., any subgraph where a pair of nodes is connected with each other via an edge path) might recognize an HLA-restricted epitope, we used a graph walking approach to discover minimal sets of feasible HLA alleles that participants shared within closely connected nodes. Briefly, for each node, we sorted nodes in ascending order by TCRdist to all other nodes with its largest connected component. Starting at the closest public node found in another HLA-genotyped participant, we took the intersection of the set of all class I (CD8+ nodes) or class II (CD4+ nodes), before moving on to the next-nearest connected node and taking the next stepwise intersection. If possible, the algorithm continues to narrow the set of feasible presenting HLA alleles to a minimal possible set. We inferred feasible HLA alleles, and in many cases only one allele was shared among closely connected S-reactive TCR sequences. This allele was assigned to the corresponding TCR cluster. Code to assign feasible HLA restriction from TCR sequence similarity analyses/graphs is provided in the Code Availability Statement using custom scripts run in Python version 3.8. Sequence similarity networks were visualized with the Networkx v2.8.6 package65.
TCR motif visualization.
From the weighted sequence similarity graphs formed from all CD69+CD137+ AIM-scTCRɑβ-seq S-reactive clones, we identified clusters of similar sequences using the Louvain community detection algorithm with the communities v3.0.0 package in Python. For each public sequence cluster with sequences donated from 3 or more participants, we depict selected TCR clusters by six graphical elements, with the CDR3α and β junctions on the left and right, respectively (Fig. 3b–h). The lower sequence logo shows the observed position-specific frequency of each amino acid within the TCR cluster, and the upper logo plots represent the position-specific information content in bits (i.e., a signal of selection) compared to CDR3α and β receptors, with the same and -gene usages, randomly sampled from naive repertoires64,66. The Sankey flow diagrams left of the CDR3 motifs show the frequencies of TRAV/TRAJ and TRBV/TRJV gene usages within each cluster. Motifs were aligned and computed in palmotif v0.4, and graphics rendered using ggplot2 and ggseqlogo67 in R version 4.1.2.
TRB repertoire analyses.
Breadth of TRB sequences significantly expanding (or contracting) between serially collected blood specimens was calculated as the number of unique clonotypes meeting significance criteria. In brief, to determine longitudinal persistence and previous detection, TRB were filtered for productive sequences and analyzed at the nucleotide level (CDR3, TRBV, TRBJ). TRB from bulk sequencing data were defined as expanded if their log2 fold change was >2 relative to the E01 timepoint and met a second criteria for a statistically significant change in counts between samples using Fisher’s Exact Test with correction for multiple hypotheses (FDR-adjusted q-value <0.05). Analysis used custom R and Python scripts detailed in Code Availability Statement. CDR3 amino acid sequence with V and J gene usage and HLA restriction (if published) was used to determine whether a clonotype had been shown previously to be associated with a known antigen.
Separately, TRB CDR3 assigned to SARS-CoV-2 were generated by statistically comparing TRB CDR3 sequences from whole blood Immunoseq TRB repertoires between persons with documented SARS-CoV-2 infection and healthy controls (HD). Sequences were assigned as likely to represent CD4+ T cells based on publicity between persons sharing HLA class II alleles, or as likely to represent CD8+ T cells based on publicity between persons sharing HLA class I alleles. Assignments to SARS-CoV-2 S or non-Spike specificity were performed using the output of multiplexed antigen restimulation assays (MIRA)68–70. Briefly, defined SARS-CoV-2 antigens were used to stimulate expanded PBMC from SARS-CoV-2-infected persons and sorted CD4+ or CD8+ T cells expressing activation markers were bulk-sequenced at the TRB locus. Further refinements were performed to exclude non-SARS-CoV-2-specific TRB sequences associated with ubiquitous antigens such as CMV or EBV, or with TRB sequences non-specifically associated with HLA alleles in a cohort of healthy controls18. MIRA-enriched and statistically SARS-CoV-2-associated TRB sequences were co-analyzed to create sets of TRB sequences spanning CDR3 and assigned, when possible, as CD4+ or CD8+, or as S- or non-S-specific. The diagnostic breadth of blood TRB repertoires were calculated as described18,71 and represent the proportion of productive TRB clonotypes present in a repertoire that are assigned as SARS-CoV-2-specific.
CD8+ T cell-origin TCR reporter cell lines.
TCR CDR3 sequences from CD69+CD137+ AIM-scTCRɑβ-seq were integrated into assigned TRA and TRB genomic variable genes using ImMunoGeneTics (IMGT)72. Codon-optimized TCR lentiviral expression constructs (Genscript, Piscataway, NJ, USA) were cloned into pRRLSIN.cPPT.MSCV/GFP.WPRE73. The TRB polypeptide is N-terminal within a fusion protein separated from TRA by a porcine echovirus P2A sequence, with both TCR constant regions of murine origin and extra cysteine residues to promote pairing74. Lentiviral particles were expressed in HEK293 cells (CRL-1573, ATCC, Manassas, VA, USA) by co-transfection with packaging plasmids75. Lentivirus was concentrated (Lenti-X, Takara, San Jose, CA, USA). The NR4A1 mNeonGreen TCR reporter cell line has been described76. In brief, mNeonGreen was integrated in-frame into the NR4A1 locus using CRISPR-induced homology-directed repair into Jurkat E6–1 cells (ATCC). The cells were additionally modified to knock out endogenous TCR expression. CD8αβ were also inserted using CRISPR to create NR4A1_mNeonGreen_035 (Jurkat_035). Jurkat_035 were maintained in TCM. 1 X 106 Jurkat_035 cells were transduced with 200–300 μl of lentiviral stock based on analysis for gag protein (Lenti-X Go Stix, Clontech) for an estimated 5 infectious units per cell. Five days or more after transduction, flow cytometry routinely showed >80% TCR expression as measured with anti-murine TRB clone H57–597-APC (eBioscience, San Diego, CA, USA).
CD8+ T cell-origin TCR specificity, HLA restriction, and dose responsiveness.
To evaluate specificity, Cos-7 cells were transfected as described77 in 96-well flat-bottom plates with HLA class I cDNA, SARS-CoV-2 full length S from strain Wu-1 cloned into pDEST103, HLA and S, or neither. Details of HLA cDNA and S cloning are published19,77–79. HLA and S were sequence-confirmed. At 48 hours, TCR-expressing Jurkat_035 were stained with Cell Trace Violet (Invitrogen, Carlsbad, CA, USA), washed, and added to Cos-7 cells at 3 X 105/well. To determine peptide epitopes TCR-expressing Jurkat_035 were co-cultured with B-LCL (1 X 105 each) and pools of 1 μg/ml each 15 amino acid peptide covering the N or C terminal halves of SARS-CoV-2 Wu-1 S (PM-WCPV-S-1, JPT, Berlin, Germany) in 200 μL TCM in round-bottom plates. Next, HLA-transfected Cos-7 aAPC and 2 X 105/well CTV-labeled Jurkat_035 were incubated with S peptides, 13 AA long with 9 AA overlap covering full-length SARS-CoV-2 strain Wu-1, arrayed in rectangular matrices with row or column pool complexities of 8 to 12 peptides as reported19. Peptides at the intersection of positive rows and columns, or internal shorter peptides, were repeated in follow-up experiments. Subsequent experiments used fresh synthesis of selected single peptides and truncated internal peptides in the vicinity of the active peptide, at 1 μg/mL (Genscript, 70% purity). For some peptide experiments, the APC used were autologous EBV-transformed B-lymphoblastoid cell lines (B-LCL). These were cultured as described80 and CTV-labeled, in which case Jurkat_035 responder wells were not CTV-labeled. To determine dose-responsiveness of confirmed KCY epitope-specific CD8 TCRs, SARS-CoV-2 S 10-mer (KCYGVSPTKL) and internal 9-mers (KCYGVSPTK and CYGVSPTKL) were tested independently against 6 Jurkat-based reporter lines with TCRs recognizing SARS-CoV-2 S in the context of HLA-A*03:01. B-LCL known to express HLA-A*03:01 were used as APC with supplied peptide concentration ranging from 10−5 to 1 μg/mL. To study S variants, Cos-7 aAPC were co-transfected with HLA-A*03:01 and S plasmid constructs representing Wu-1 or SARS-CoV-2 Omicron variants BA.1, BA.2, and BA.4, each with 21 amino acid C-terminal deletions. Regardless of APC and antigen, after 24 hours, cells were analyzed by flow cytometry (LSR II, Becton Dickinson) and the percent of gated Jurkat cells, either CTV-high or CTV-low, expressing mNeonGreen was analyzed.
CD4+ T cell-origin TCR reporter cells
To test the CD4+ T cell-origin candidate TCRs, engineered reporter T cells were generated by transduction of autologous CD4+ T cells using lentiviruses with candidate-paired TCR expression cassettes. Autologous CD4+ T cells were immuno-magnetically enriched (19662, STEMCELL, Vancouver, British Columbia, Canada) per the manufacturer, seeded at 5 × 105/well in 48 well plates in 1 ml TCM, and activated with anti-CD3/anti-CD28 Dynabeads (11131D, Invitrogen) at a bead:cell ratio of 3:1. Recombinant human IL-2 (50 U/ml, Miltenyi, Gaithersburg, MD, USA) was added. After a 24-hour incubation, T cells were transduced with 200–300 μl of concentrated lentiviral stock. On day 4, beads were removed. Cells were further expanded for 10 days in TCM containing recombinant IL-2, washed, and counted. To evaluate specificity of CD4+ T cell-origin TCRs, 105 TCR-transduced reporter cells were cultured in 96-well round bottom plates in 200 μl TCM containing 105 autologous, irradiated PBMC as APC and SARS-CoV-2 S protein (D614G, 10587-CV-100, R&D Systems, Minneapolis, MN, USA) at 0.1 μg/ml. Other stimuli included pooled SARS-CoV-2 S1 or S2 peptide pools from the N or C terminal regions of S, respectively (PM-WCPV-S-1, JPT) at a final concentration of 1 μg/ml each peptide, TCM or 0.1% DMSO as negative controls, or 1.6 μg/ml PHA-P as a positive control. At day 3, supernatants were evaluated for IFN-γ secretion by ELISA.
HLA typing.
HLA class I and II allotypes were determined by next-generation sequencing at Scisco (Seattle, WA, USA)21.
Statistical Testing.
Statistical comparison between paired samples are Wilcoxon signed-rank tests, and comparison between groups are Wilcoxon rank sum tests. Tests use a two-sided alternative hypothesis. Associations among immunological parameters were measured with Spearman rank correlation. Asterisks represent level of statistical significance (ns = not significant, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001). TRB from bulk sequencing data were defined as expanded if their log2 fold change was >2 relative to the E01 timepoint and met a second criteria for a statistically significant change in counts between samples using Fisher’s Exact Test with correction for multiple hypotheses (FDR-adjusted q-value <0.05). Statistical testing was performed in R version 4.1.2. Repertoires were generated from distinct samples collected longitudinally.
Data availability statement
Whole PBMC and nasal TRB repertoires are at https://doi.org/10.5281/zenodo.7698787. Sorted naive and memory PBMC T cell subset TRB repertoires from timepoint E00 and sorted total CD4+ T cells and AIM–sorted CD4+ T cell subsets from timepoint E03 are at https://doi.org/10.5281/zenodo.7686500. Processed single cell CD69+CD137+ AIM-scTCRɑβ-seq and feature barcode oligonucleotide-labeled mAb data are at https://zenodo.org/record/6909380#.Yvqcg3bMKUk. Flow cytometry results from intracellular cytokine staining of CD4+ and CD8+ S-reactive T cells are available at https://doi.org/10.5281/zenodo.8088178. Sequences of CD8+ and CD4+ T cell-origin TCRs expressed in reporter cells are available in Genbank OP245920-OP245935 and OR239787-OR239798, respectively. Reference dataset for CellRanger used was GRCh-Alts-ensembl 5.0.0 and is available at 10xgenomics.com/support/software/cell-ranger/downloads.
Biologic material availability statement
Please contact the corresponding author. Biological materials may be available after IRB and/or technology transfer office approval from sending and receiving institutions and execution of a material transfer agreement (MTA).
Code availability statement
Code used to analyze and present data is based on Python version 3.8 or R version 4.1.2 and is available at https://github.com/kmayerb/NIA34780B. Availability of the tool used to classify PBMC TRB repertoires for evidence of CMV infection is discussed at https://www.immunoseq.com/cmv-classifier/
Extended Data
Extended Data Fig. 1. Schedule of infection, vaccination, and sample collection.
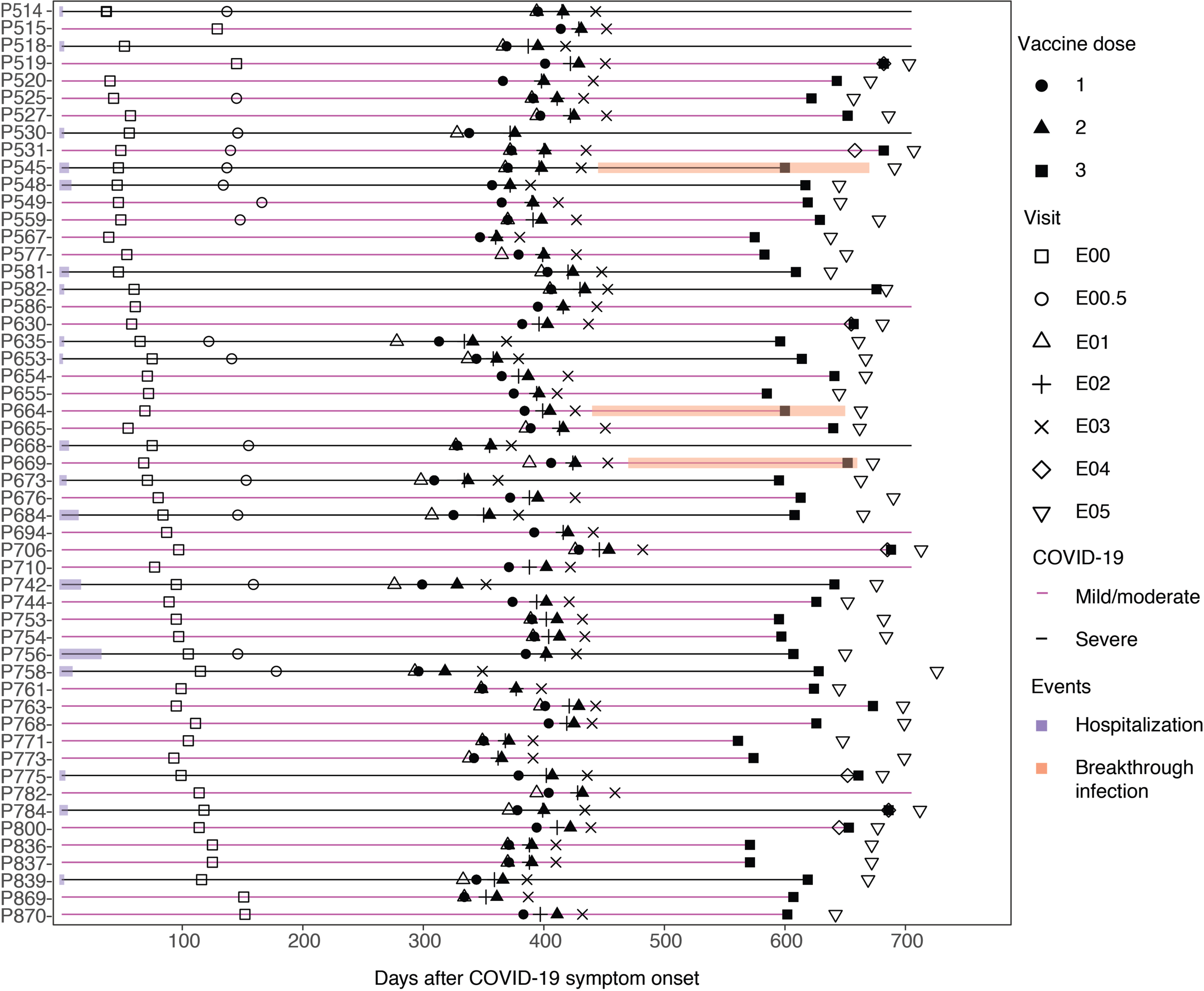
Fifty-three study participants with prior SARS-CoV-2 infection as documented by seropositivity to S and N proteins and participant P845 who was seronegative prior to vaccination received either BNT162 or mRNA-1273 1st dose (closed circle), 2nd dose (closed triangle), and booster (3rd) dose (closed square) on the days after symptom onset as shown. Persons with mild or moderate COVID-19 are shown in magenta, persons with severe COVID-19 in black. Duration of hospitalization in persons with severe COVID-19 is shown in purple. PBMC were obtained at exam visits convalescence (E00, n = 54), late convalescence (E00.5, n = 16), pre-dose 1 (E01, n = 34), post-dose 1 (E02, n = 52), post-dose 2 (E03, n = 53), pre-boost (E04, n = 7), and post-boost (E05, n = 44). 33 persons had samples at E00, E01 and E03, 31 had samples at E00, E01, E02, and E03, and 26 had samples at all of E00, E01, E02, E03, and E05. Three participants were observed to have breakthrough COVID-19 infection based on boosting of anti-nucleocapsid antibody levels at visit E05 (P545, P664, P669), indicated in orange. All participants received primary vaccination but not all received a booster dose.
Extended Data Fig. 2. Primary vaccination led to expansion in specific TCR clonotypes in vaccinated persons.
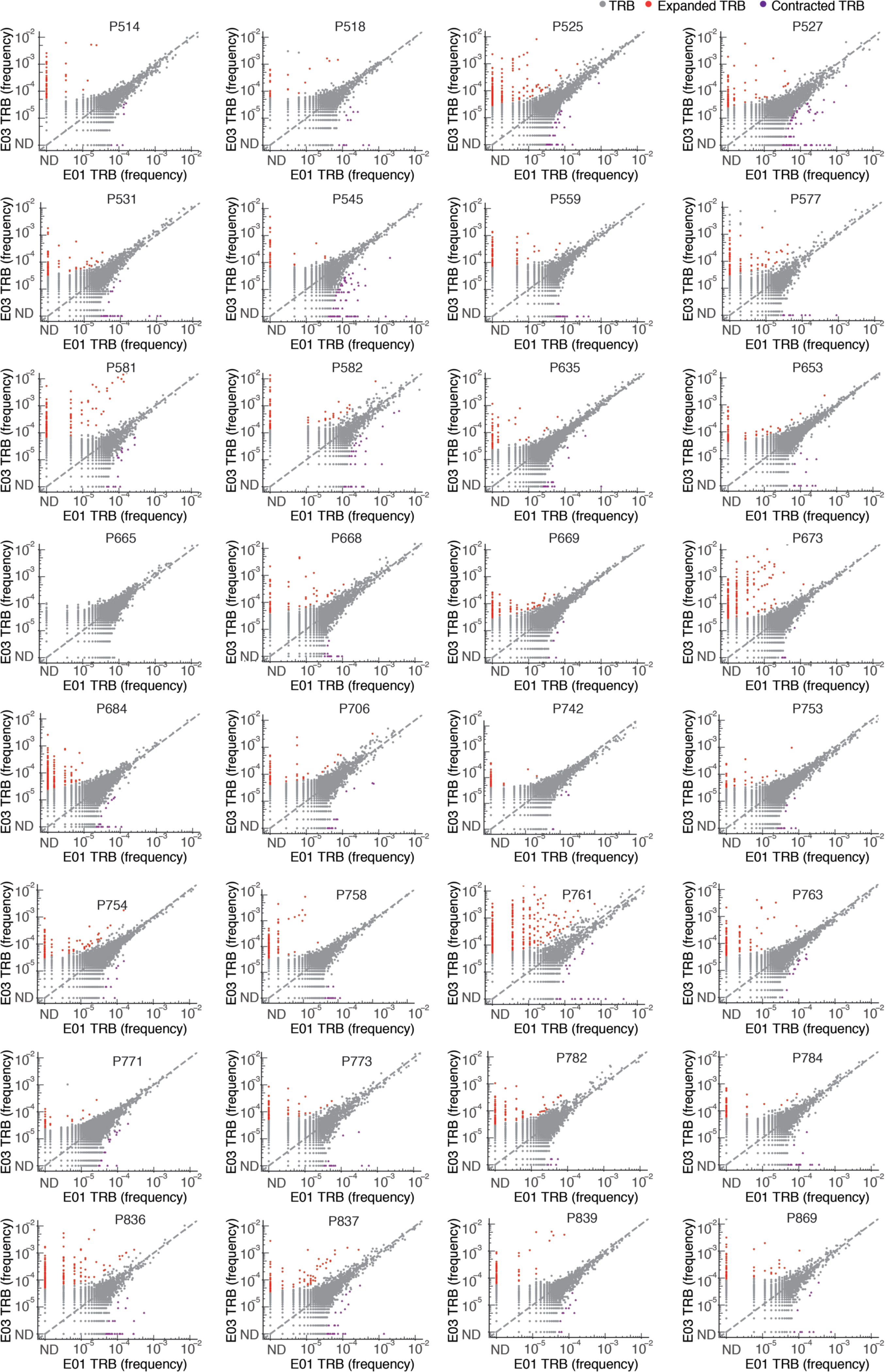
Frequency (% of bulk TRB repertoire) of individual clonotypes in E01 vs. E03 in 32 persons with both samples. Expanded (red) (or contracted, purple) clonotypes were defined as log2(fold change) > 2 (or < 0.5) and Fisher’s exact test FDR-adjusted q value < 0.05. Dotted line indicates y = x. Participant ID at top of each graph. ND = not detected. Serologically-naive Participant P845 is not shown.
Extended Data Fig. 3. Frequency of E01-to-E03 expanded clonotypes from E00 through E05.
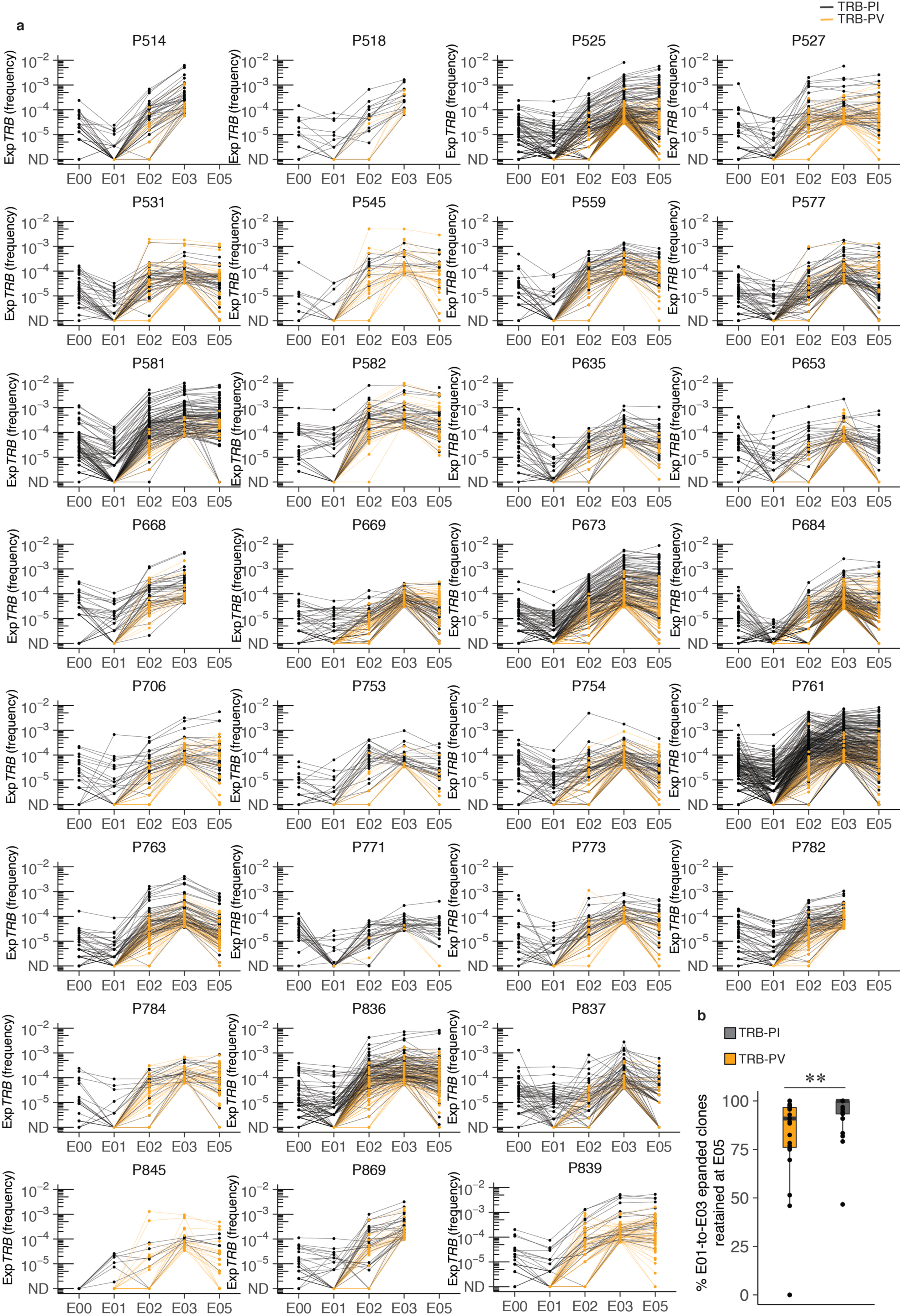
(a) E01-to-E03 expanded clonotype frequency (abundance) over the course of the study. Each line is an individual clonotype. TRB-PI are shown in black, TRB-PV are in orange (n = 30). ND = not detected. (b) Boxplot at lower right shows the percent of TRB-PI and TRB-PV for each participant that were detectable after a 3rd vaccine dose (E05) (n = 26). Median, IQR and whiskers (1.5*IQR) are noted. Comparison between groups is by two-sided Wilcoxon rank sum test, p = 0.0023. Participants P742 and P758 were not sampled at E02 and trajectories are not shown. Participant P665 had no expanded clonotypes and is not shown. Participant P845 was serologically naive at E00. Participants P545 and P669 experienced breakthrough infection between the E03 and the E05 timepoints and so repertoires at E05 represent both repeat natural infection as well as mRNA booster vaccine dose.
Extended Data Fig. 4. AIM-scTCRɑβ-seq enriches a complex set of clonotypes from PBMC after mRNA vaccination of previously SARS-CoV-2 infected persons.
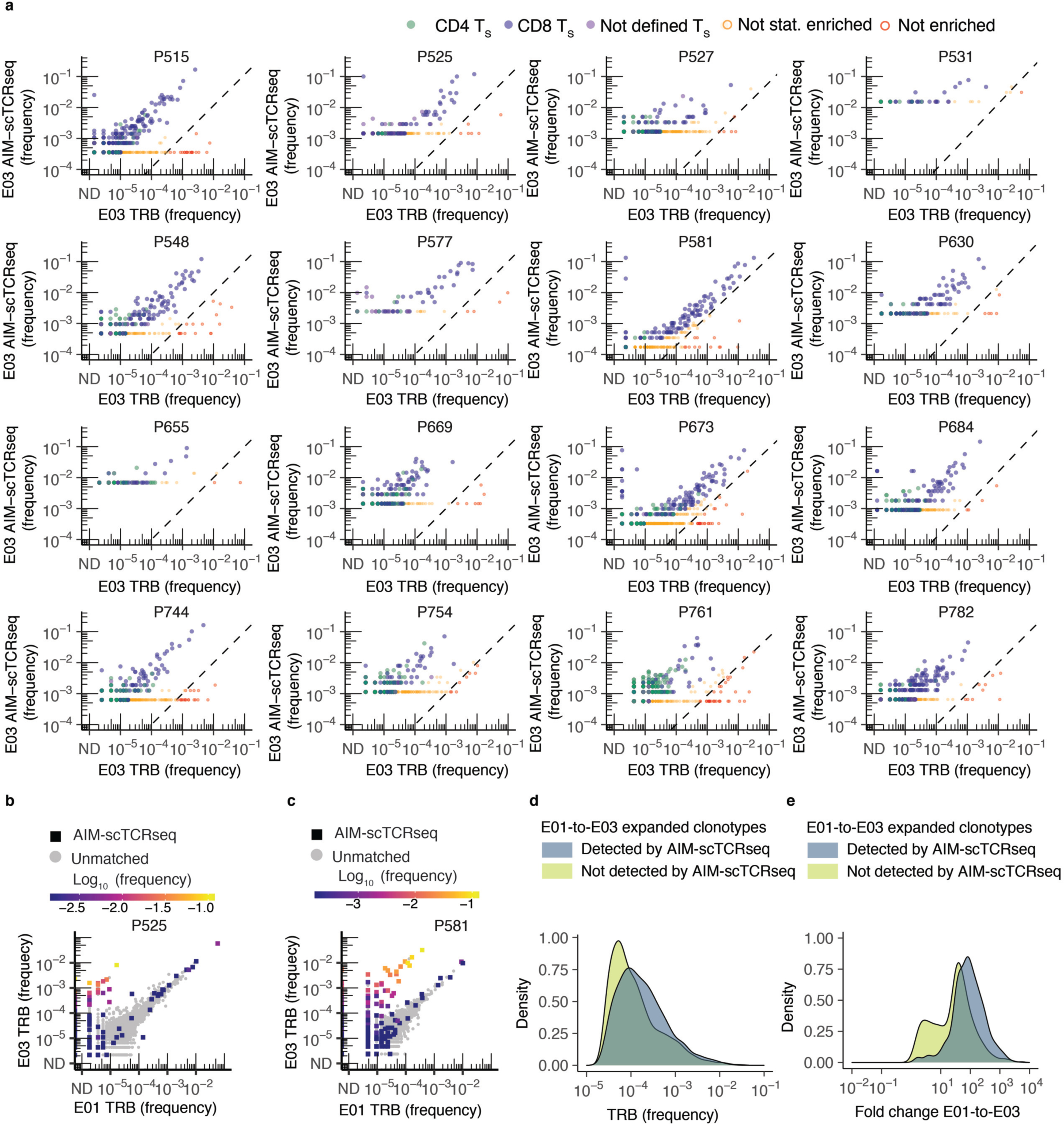
(a) Frequency of clonotypes detected by CD69+CD137+ AIM-scTCRɑβ-seq plotted against the productive frequency of TRB-matched templates in bulk TRB sequencing at E03. Cell types defined by oligonucleotide-labeled mAbs are shown as CD4+ (green), CD8+ (blue), or phenotype not defined (purple) T cells. Clonotype enrichment in CD69+CD137+ AIM-scTCRɑβ-seq was determined by cumulative distribution function (CDF) with false discovery rate (FDR) correction (Methods). Clonotypes that were detected, but not enriched, in CD69+CD137+ AIM-scTCRɑβ-seq are shown in red (n = 180) and were omitted from CDR3 motif discovery analysis. UND indicates clonotypes that could not be assigned a TCRB unambiguously. (b,c) Productive frequency of CD69+CD137+ AIM-scTCRɑβ-seq detected clonotypes in relation to change in productive frequency from E01 to E03 in bulk TCR-β-seq is shown for 2 representative participants, including one with a lower proportion of representation in CD69+CD137+ AIM-scTCRɑβ-seq of their significantly expanded clonotypes (P525, b) and another with a higher proportion of significantly expanded clonotypes also seen by CD69+CD137+ AIM-scTCRɑβ-seq (P581, c). (d,e) Density plots show proportion of unique, expanded clonotypes by frequency at E03 (d) or fold change from E01 to E03 (primary vaccination) (e) by detection in CD69+CD137+ AIM-scTCRɑβ-seq at E03 amongst 12 participants with both paired E01/E03 bulk TCR-β-seq and CD69+CD137+ AIM-scTCRɑβ-seq.
Extended Data Fig. 5. TS from CD69+CD137+ AIM-scTCRɑβ-seq matching TRB clonotypes from nasal swabs in 14 participants at E05.
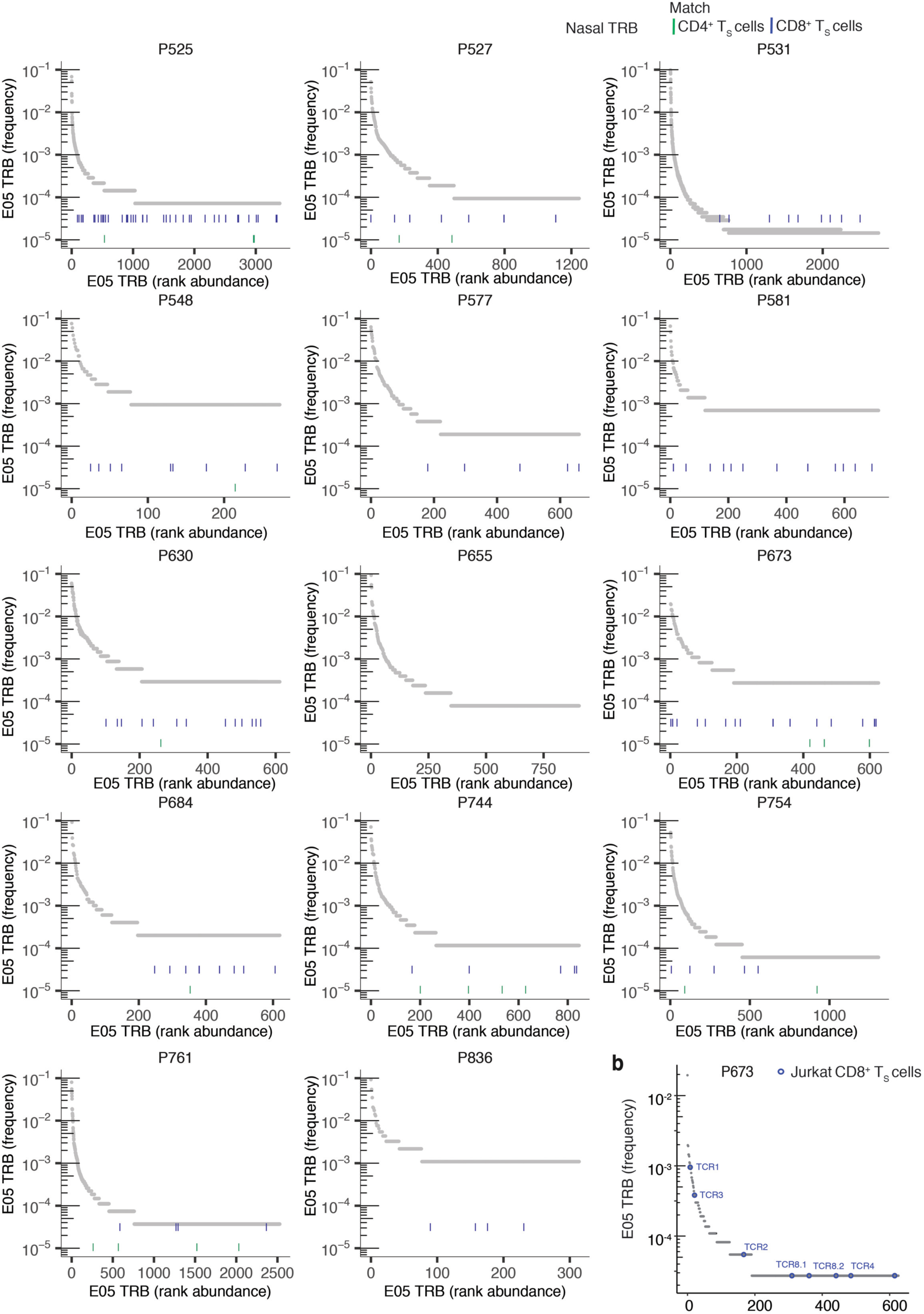
(a) Rank abundance plots of TRB clonotypes in nasal samples in 14 participants, where blue (CD8+) and green (CD4+) dashes indicate rank of clones identified in the same participant’s blood by CD69+CD137+ AIM-scTCRɑβ-seq at E03. (b) In participant P673, rank abundance plot of TRB clonotypes in nasal samples collected at E05. Clones labeled TCR1, TCR2, TCR3, TCR4, TCR8.1 and TCR8.2 indicate TRB clonotypes with exact sequence match to experimentally-confirmed receptors shown to recognize HLA-A*03:01 S epitopes.
Extended Data Fig. 6. TS and vaccine-expanded clonotypes.
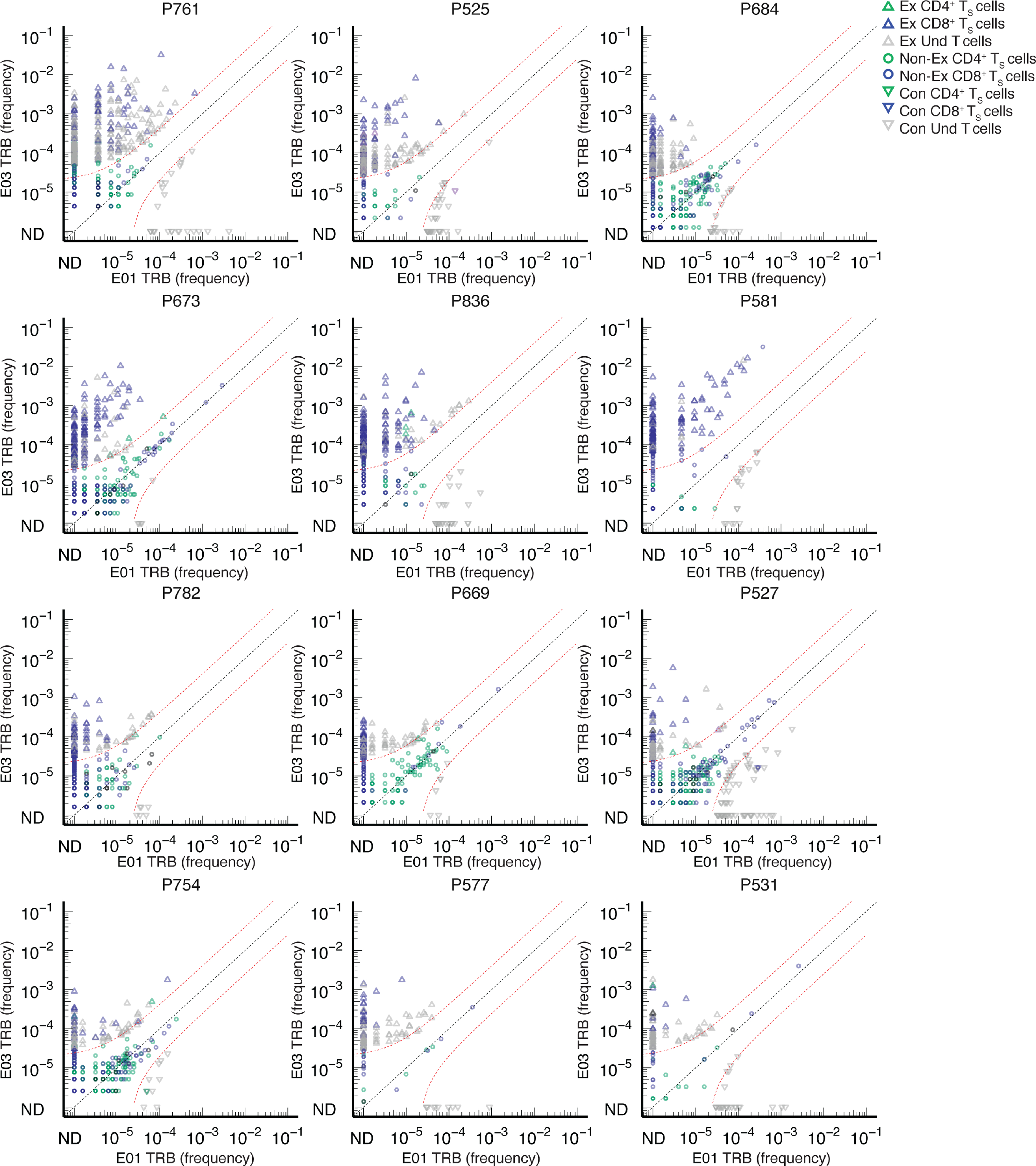
Overlay of TRB sequences from CD69+CD137+ AIM-scTCRɑβ-seq of TS cells onto bulk TRB clonotype frequency at E01 and E03 in two representative participants. E01-to-E03 expanded (Ex) or contracted (Con), TRB clonotypes are shown, with TRB matching an CD69+CD137+ AIM-scTCRɑβ-seq (TS) shown in color and unmatched TRB are shown in gray (Und). Among clonotypes that neither expanded nor contracted (Non-Ex), only CD69+CD137+ AIM-scTCRɑβ-seq TRB-matched clonotypes are shown between red dashed lines. Frequencies of TRB clonotypes pre- and post-vaccine are as resolved by bulk TCR-β-seq. ND = not detected.
Extended Data Fig. 7. Abundance of TS clonotypes by TCRβ-seq over time.
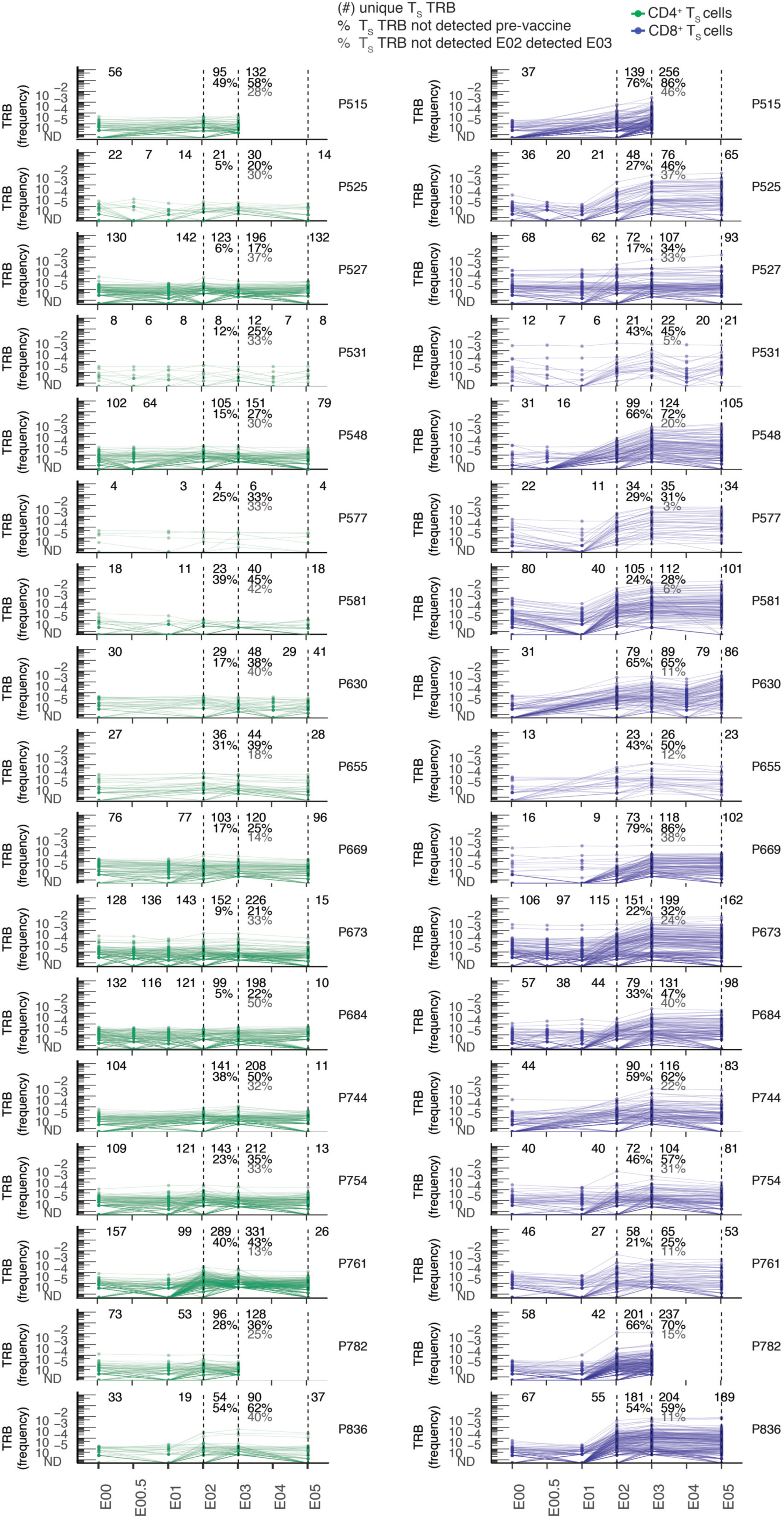
Longitudinal tracking of abundance of CD69+CD137+ AIM-scTCRɑβ-seq-identified CD4+ and CD8+ TS TRB clonotypes in PBMC by TCRβ-seq in all participants with AIM-scTCRɑβ-seq. Numbers in top rows indicate the number of unique CD69+CD137+ AIM-scTCRɑβ-seq TRB clonotypes from E03 detected at each time point. Percentages refer to the fraction of CD69+CD137+ AIM-scTCRɑβ-seq clonotypes detected at the E02 and E03 timepoints, respectively, detected only post-vaccination. Percentages in gray are the fraction of unique clones detected at E03 that are below the level of detection at E02. Not all participants had samples at each time point, indicated by absence of dot symbols at those samples. Participant P669 had SARS-CoV-2 infection between E03 and E05 timepoints and so this E05 repertoire reflects repeat SARS-Co-2 infection and mRNA booster vaccine.
Extended Data Fig. 8. Selection of AIM-scTCRɑβ-seq T cells using CD69, CD137, and CD134/ CD154 marker sets compared to bulk TCRβ-seq and sorted CD4 TCRβ-seq from PBMCs from E01 to E03.
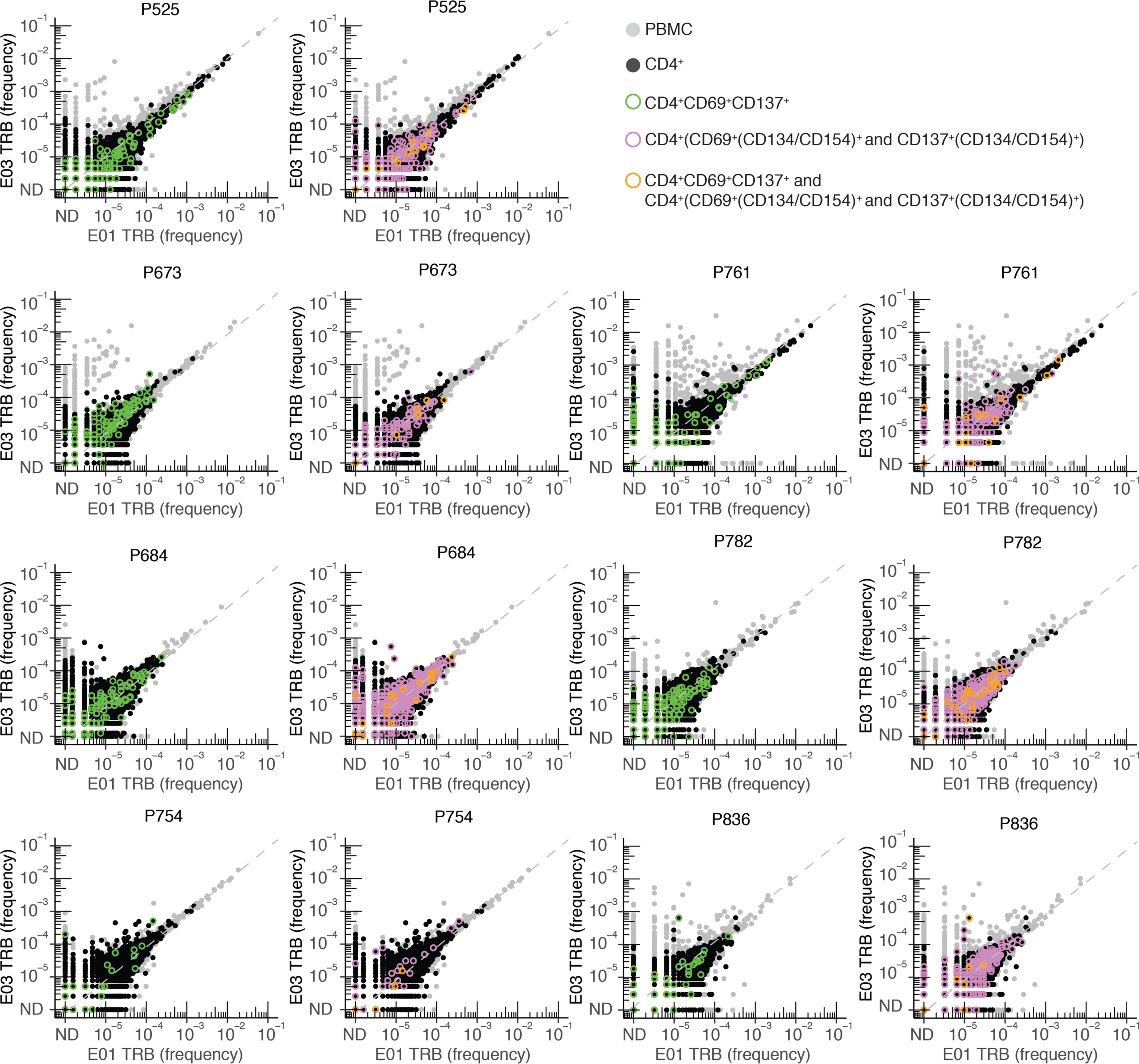
Frequency (% of bulk TRB repertoire) of individual clonotypes in E01 vs. E03 in 7 persons studied by CD69+CD137+ AIM-scTCRɑβ-seq and (CD69/CD137)+(CD134/CD154)+ CD4+ AIM-TCRβ-seq. Dotted line indicates y = x. Participant ID at top of each pair of graphs. ND = not detected. All PBMC are shown. Clonotypes found in the total CD4+ sorted fraction are shown in black. Clonotypes present in the total CD4+ sorted fraction and also enriched in sequential sorting of CD4+CD69+CD137+ (green) cells are overlaid with CD4+CD69+(CD134/CD154)+ and CD4+CD137+(CD134/CD154)+ (pink) cells in the left and right panels, respectively, for seven participants. Clonotypes in all three fractions (total CD4+, CD4+CD69+CD137+, and CD4+CD69+(CD134/CD154)+ and CD4+CD137+(CD134/CD154)+) are shown in orange. Gray shaded clonotypes were not identified as CD4+ by any of these methods.
Supplementary Material
Acknowledgments:
We thank the participants; the Virology Research Clinic, University of Washington for collecting specimens and data; D. Geraghty and C.-W. Pyo at Fred Hutchinson Cancer Center (FHCC) for HLA typing; L. Stamatatos, FHCC for SARS-CoV-2 S protein with stabilizing proline (S2P) substitutions used in antibody assays; J. Bloom, FHCC, for expression plasmids encoding SARS-CoV-2 S from strain Wu-1, or Omicron BA.1, BA.2 and BA.4, with 21 amino acid C-terminal deletions.
Support
US NIH NIAID Contract 75N93019C00063 (DMK). US NIH grants AI163999 (DMK), K08 AI148588 (ESF), R01 AI136514 (KMB, AFG), F30 CA254168 (THP), T32 CA080416 (SJ), and P01 CA225517 (AGC, DMK, LJ, THP, SJ), R01 AI134878 (AMS, ELB and RSG), UM1 AI068614 (AMS, ELB and RSG). Scientific computing infrastructure at Fred Hutchinson Cancer Center was funded by US NIH ORIP grant S10 OD028685 (KMB, AFG, ESF). Bill and Melinda Gates Foundation INV-027499 (AFG, KMB). This project was funded in part with Federal funds from the NIAID, NIH, DHHS, under US NIH Contract No. HHSN272201800013C. BE, EE, and MRH performed this work as employees of Laulima Government Solutions, LLC. MM and EP are subcontractors to Laulima Government Solutions, LLC who performed this work as employees of Tunnell Government Services, Inc.
Footnotes
Conflict of Interest Statements:
HSR and RE are employees of Adaptive Biotechnologies, Inc. B.E., E.E. and M.R.H. performed this work as employees of Laulima Government Solutions. M.M. and E.P. are subcontractors to Laulima Government Solutions; they performed this work as employees of Tunnell Government Services. The other authors have no competing interest. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
REFERENCES:
- 1.Goldberg Y. et al. Protection and waning of natural and hybrid immunity to SARS-CoV-2. N. Engl. J. Med 386, 2201–2212 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Suarez Castillo M, Khaoua H. & Courtejoie N. Vaccine-induced and naturally-acquired protection against Omicron and Delta symptomatic infection and severe COVID-19 outcomes, France, December 2021 to January 2022. Eurosurveillance 27, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Qu P. et al. Durability of booster mRNA vaccine against SARS-CoV-2 BA.2.12.1, BA.4, and BA.5 subvariants. N. Engl. J. Med 1–4 (2022) doi: 10.1056/NEJMc2210546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lim JME et al. SARS-CoV-2 breakthrough infection in vaccinees induces virus-specific nasal-resident CD8+ and CD4+ T cells of broad specificity. J. Exp. Med 219, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peng Y. et al. Broad and strong memory CD4+ and CD8+ T cells induced by SARS-CoV-2 in UK convalescent individuals following COVID-19. Nat. Immunol 21, 1336–1345 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tavukcuoglu E, Horzum U, Cagkan Inkaya A, Unal S. & Esendagli G. Functional responsiveness of memory T cells from COVID-19 patients. Cell. Immunol 365, 104363 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rodda LB et al. Functional SARS-CoV-2-specific immune memory persists after mild COVID-19. Cell 184, 169–183.e17 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dykema AG et al. SARS-CoV-2 vaccination diversifies the CD4+ spike-reactive T cell repertoire in patients with prior SARS-CoV-2 infection. eBioMedicine 80, 1–13 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Minervina AA et al. SARS-CoV-2 antigen exposure history shapes phenotypes and specificity of memory CD8+ T cells. Nat. Immunol 23, 781–790 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kared H. et al. Immune responses in Omicron SARS-CoV-2 breakthrough infection in vaccinated adults. Nat. Commun 13, 1–12 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Altarawneh HN et al. Effects of previous infection and vaccination on symptomatic Omicron infections. N. Engl. J. Med (2022) doi: 10.1056/nejmoa2203965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Keeton R. et al. T cell responses to SARS-CoV-2 spike cross-recognize Omicron. Nature 603, 488–492 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gao Y. et al. Ancestral SARS-CoV-2-specific T cells cross-recognize the Omicron variant. Nat. Med 28, 472–476 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dolton G. et al. Emergence of immune escape at dominant SARS-CoV-2 killer T cell epitope. Cell 185, 2936–2951 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Naranbhai V. et al. T cell reactivity to the SARS-CoV-2 Omicron variant is preserved in most but not all individuals. Cell 185, 1041–1051.e6 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Müller NF et al. Viral genomes reveal patterns of the SARS-CoV-2 outbreak in Washington State. Sci. Transl. Med 13, 1–12 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mueller YM et al. Stratification of hospitalized COVID-19 patients into clinical severity progression groups by immuno-phenotyping and machine learning. Nat. Commun 13, 1–13 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Elyanow R. et al. T cell receptor sequencing identifies prior SARS-CoV-2 infection and correlates with neutralizing antibodies and disease severity. JCI Insight 7, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang JZ et al. Thermodynamically coupled biosensors for detecting neutralizing antibodies against SARS-CoV-2 variants. Nat. Biotechnol 40, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johansson AM et al. Cross-reactive and mono-reactive SARS-CoV-2 CD4+ T cells in prepandemic and COVID-19 convalescent individuals. PLoS Pathog. 17, 1–28 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boonyaratanakornkit J. et al. Clinical, laboratory, and temporal predictors of neutralizing antibodies against SARS-CoV-2 among COVID-19 convalescent plasma donor candidates. J. Clin. Invest 131, 1–9 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reiss S. et al. Comparative analysis of activation induced marker (AIM) assays for sensitive identification of antigen-specific CD4 T cells. PLoS One 12, 1–22 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Glanville J. et al. Identifying specificity groups in the T cell receptor repertoire. Nature 547, 94–98 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dash P. et al. Quantifiable predictive features define epitope-specific T cell receptor repertoires. Nature 547, 89–93 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oberhardt V. et al. Rapid and stable mobilization of CD8+ T cells by SARS-CoV-2 mRNA vaccine. Nature 597, 268–273 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Francis JM et al. Allelic variation in Class I HLA determines pre-existing memory responses to SARS-CoV-2 that shape the CD8 + T cell repertoire upon viral exposure Collection and Processing Team. bioRxiv 3070, 2021.04.29.441258 (2021). [Google Scholar]
- 27.Shomuradova AS et al. SARS-CoV-2 epitopes are recognized by a public and diverse repertoire of human T cell receptors. Immunity 53, 1245–1257.e5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Szeto C. et al. Molecular basis of a dominant SARS-CoV-2 spike-derived epitope presented by HLA-A*02:01 recognised by a public TCR. Cells 10, 1–15 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ferretti AP et al. Unbiased screens show CD8+ T cells of COVID-19 patients recognize shared epitopes in SARS-CoV-2 that largely reside outside the spike protein. Immunity 53, 1095–1107.e3 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Robins HS et al. Overlap and effective size of the human CD8 + T cell receptor repertoire. Immunology 2, 47ra64 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sethna Z, Elhanati Y, Callan CG, Walczak AM & Mora T. OLGA: Fast computation of generation probabilities of B-and T-cell receptor amino acid sequences and motifs. Bioinformatics 35, 2974–2981 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Flament H. et al. Outcome of SARS-CoV-2 infection is linked to MAIT cell activation and cytotoxicity. Nat. Immunol 22, 322–335 (2021). [DOI] [PubMed] [Google Scholar]
- 33.Parrot T. et al. MAIT cell activation and dynamics associated with COVID-19 disease severity. Sci. Immunol 5, 1–14 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boulouis C. et al. MAIT cell compartment characteristics are associated with the immune response magnitude to the BNT162b2 mRNA anti-SARS-CoV-2 vaccine. Mol. Med 28, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Le Gall S, Stamegna P. & Walker BD Portable flanking sequences modulate CTL epitope processing. J. Clin. Invest 117, 3563–3575 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Saggau C. et al. The pre-exposure SARS-CoV-2-specific T cell repertoire determines the quality of the immune response to vaccination. Immunity 1–16 (2022) doi: 10.1016/j.immuni.2022.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mudd PA et al. SARS-CoV-2 mRNA vaccination elicits a robust and persistent T follicular helper cell response in humans. Cell 185, 603–613.e15 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pogorelyy MV et al. Resolving SARS-CoV-2 CD4+ T cell specificity via reverse epitope discovery. Cell Reports Med. 3, 100697 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gittelman RM et al. Longitudinal analysis of T cell receptor repertoires reveals shared patterns of antigen-specific response to SARS-CoV-2 infection. JCI Insight 7, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carlson CS et al. Using synthetic templates to design an unbiased multiplex PCR assay. Nat. Commun 4, 1–9 (2013). [DOI] [PubMed] [Google Scholar]
- 41.Robins HS et al. Comprehensive assessment of T-cell receptor beta-chain diversity in alpha-beta T cells. Blood 114, 4099–107 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu AM et al. Differences in SARS-CoV-2 vaccine response dynamics between class-I- and class-II-specific T-cell receptors in inflammatory bowel disease. Front. Immunol 13, 1–10 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Robins H. et al. Ultra-sensitive detection of rare T cell clones. J. Immunol. Methods 375, 14–19 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu J. et al. T cell receptor β repertoires in patients with COVID-19 reveal disease severity signatures. Front. Immunol 14, 1–16 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pothast CR et al. SARS-CoV-2-specific CD4+ and CD8+ T cell responses can originate from cross-reactive CMV-specific T cells. Elife 11, 1–22 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jo N. et al. Aging and CMV infection affect pre-existing SARS-CoV-2-reactive CD8+ T cells in unexposed individuals. Front. Aging 2, 1–16 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Emerson RO et al. Immunosequencing identifies signatures of cytomegalovirus exposure history and HLA-mediated effects on the T cell repertoire. Nat. Genet 49, 659–665 (2017). [DOI] [PubMed] [Google Scholar]
- 48.Boppana S. et al. SARS-CoV-2-specific circulating T follicular helper cells correlate with neutralizing antibodies and increase during early convalescence. PLoS Pathog. 17, 1–18 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Painter MM et al. Rapid induction of antigen-specific CD4+ T cells is associated with coordinated humoral and cellular immunity to SARS-CoV-2 mRNA vaccination. Immunity 54, 2133–2142.e3 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ngalamika O, Kawimbe M. & Mukasine MC Expression of CD40L on CD4+ T cells distinguishes active versus inactive HIV-associated Kaposi’s Sarcoma. Cancer Treat. Res. Commun 27, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Curato C. et al. Frequencies and TCR Repertoires of Human 2,4,6-Trinitrobenzenesulfonic Acid-specific T Cells. Front. Toxicol 4, 1–13 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sette A. & Crotty S. Adaptive immunity to SARS-CoV-2 and COVID-19. Cell 184, 861–880 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.DeWitt WS et al. Dynamics of the cytotoxic T cell response to a model of acute viral infection. J. Virol 89, 4517–4526 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pogorelyy MV et al. Precise tracking of vaccine-responding T cell clones reveals convergent and personalized response in identical twins. PNAS 115, 12704–12709 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Minervina AA et al. Longitudinal high-throughput tcr repertoire profiling reveals the dynamics of t-cell memory formation after mild covid-19 infection. Elife 10, 1–17 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Meckiff BJ et al. Imbalance of regulatory and cytotoxic SARS-CoV-2-reactive CD4+ T cells in COVID-19. Cell 183, 1340–1353.e16 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Goncharov M. et al. VDJdb in the pandemic era: A compendium of T cell receptors specific for SARS-CoV-2. Nat. Methods 2–4 (2022) doi: 10.1038/s41592-022-01578-0. [DOI] [PubMed] [Google Scholar]
- 58.Alter G. et al. Immunogenicity of Ad26.COV2.S vaccine against SARS-CoV-2 variants in humans. Nature 596, 268–272 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Swanson PA et al. AZD1222/ChAdOx1 nCoV-19 vaccination induces a polyfunctional spike protein-specific TH1 response with a diverse TCR repertoire. Sci. Transl. Med 13, 1–15 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Harris PA et al. Research electronic data capture (REDCap)-A metadata-driven methodology and workflow process for providing translational research informatics support. J. Biomed. Inform 42, 377–381 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bennett RS et al. Scalable, micro-neutralization assay for assessment of SARS-CoV-2 (Covid-19) virus-neutralizing antibodies in human clinical samples. Viruses 13, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sholukh AM et al. Evaluation of cell-based and surrogate SARS-CoV-2 neutralization assays. J. Clin. Microbiol 59, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Koelle DM Expression cloning for the discovery of viral antigens and epitopes recognized by T cells. Methods 29, 213–226 (2003). [DOI] [PubMed] [Google Scholar]
- 64.Mayer-Blackwell K. et al. TCR meta-clonotypes for biomarker discovery with tcrdist3 enabled identification of public, HLA-restricted clusters of SARS-CoV-2 TCRs. Elife 10, 1–32 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hagberg AA, Schult DA & Swart PJ Exploring network structure, dynamics, and function using NetworkX. in Proceedings of the 7th Python in Science Conference (SciPy2008), Gäel Varoquaux, Travis Vaught, and Jarrod Millman (Eds). (Pasadena, CA USA: ) 11–15 (2008). [Google Scholar]
- 66.Britanova OV et al. Age-related decrease in TCR repertoire diversity measured with deep and normalized sequence profiling. J. Immunol 192, 2689–2698 (2014). [DOI] [PubMed] [Google Scholar]
- 67.Wagih O. Ggseqlogo: A versatile R package for drawing sequence logos. Bioinformatics 33, 3645–3647 (2017). [DOI] [PubMed] [Google Scholar]
- 68.Snyder TM et al. Magnitude and dynamics of the T-cell response to SARS-CoV-2 infection at both individual and population levels. medRxiv September, (2020). [Google Scholar]
- 69.Nolan S. et al. A large-scale database of T-cell receptor beta (TCRβ) sequences and binding associations from natural and synthetic exposure to SARS-CoV-2. Res. Sq 1–28 (2020) doi: 10.21203/rs.3.rs-51964/v1. [DOI] [Google Scholar]
- 70.Klinger M. et al. Multiplex identification of antigen-specific T cell receptors using a combination of immune assays and immune receptor sequencing. PLoS One 10, 1–21 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li D. et al. The T-cell clonal response to SARS-CoV-2 vaccination in inflammatory bowel disease patients is augmented by anti-TNF therapy and often deficient in antibody-responders. medRxiv (2021). [Google Scholar]
- 72.Lefranc MP et al. IMGT R, the international ImMunoGeneTics information system R 25 years on. Nucleic Acids Res. 43, D413–D422 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Schmitt TM et al. Generation of higher affinity T cell receptors by antigen-driven differentiation of progenitor t cells in vitro. Nat. Biotechnol 35, 1188–1195 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Linnemann C. et al. High-throughput identification of antigen-specific TCRs by TCR gene capture. Nat. Med 19, 1534–1541 (2013). [DOI] [PubMed] [Google Scholar]
- 75.Jing L. et al. Extensive CD4 and CD8 T cell cross-reactivity between alphaherpesviruses. J. Immunol 196, 2205–2218 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ford ES et al. Expansion of the HSV-2-specific T cell repertoire in skin after immunotherapeutic HSV-2 vaccine. medRxiv 1–48 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jing L. et al. Cross-presentation and genome-wide screening reveal candidate T cells antigens for a herpes simplex virus type 1 vaccine. J. Clin. Invest 122, 654–673 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.van Velzen M. et al. Local CD4 and CD8 T-cell reactivity to HSV-1 antigens documents broad viral protein expression and immune competence in latently infected human trigeminal ganglia. PLoS Pathog. 9, 12–15 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jing L. et al. Prevalent and diverse intratumoral oncoprotein-specific CD8+ T cells within polyomavirus-driven merkel cell carcinomas. Cancer Immunol. Res 8, 648–659 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tigges MA et al. Human CD8+ herpes simplex virus-specific cytotoxic T-lymphocyte clones recognize diverse virion protein antigens. J. Virol 66, 1622–1634 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Whole PBMC and nasal TRB repertoires are at https://doi.org/10.5281/zenodo.7698787. Sorted naive and memory PBMC T cell subset TRB repertoires from timepoint E00 and sorted total CD4+ T cells and AIM–sorted CD4+ T cell subsets from timepoint E03 are at https://doi.org/10.5281/zenodo.7686500. Processed single cell CD69+CD137+ AIM-scTCRɑβ-seq and feature barcode oligonucleotide-labeled mAb data are at https://zenodo.org/record/6909380#.Yvqcg3bMKUk. Flow cytometry results from intracellular cytokine staining of CD4+ and CD8+ S-reactive T cells are available at https://doi.org/10.5281/zenodo.8088178. Sequences of CD8+ and CD4+ T cell-origin TCRs expressed in reporter cells are available in Genbank OP245920-OP245935 and OR239787-OR239798, respectively. Reference dataset for CellRanger used was GRCh-Alts-ensembl 5.0.0 and is available at 10xgenomics.com/support/software/cell-ranger/downloads.