

Abstract
Targeted protein degradation approaches have been widely used for degrading oncogenic proteins, providing a potentially promising therapeutic strategy for cancer treatment. However, approaches to targeting tumor suppressor proteins are very limited, and only a few agonists have been developed to date. Here, we report the development of a platform termed TF-DUBTAC, which links a DNA oligonucleotide to a covalent ligand of the deubiquitinase OTUB1 via a click reaction, to selectively stabilize tumor suppressor transcription factors. We developed three series of TF-DUBTACs, namely, FOXO-DUBTAC, p53-DUBTAC, and IRF-DUBTAC, which stabilize FOXO3A, p53, and IRF3 in cells, respectively, in an OTUB1-dependent manner. These results suggest that TF-DUBTAC is a generalizable platform to achieve selective stabilization of tumor suppressor transcription factors as a therapeutic means to suppress tumorigenesis.
Graphical Abstract

INTRODUCTION
Tumorigenesis is initiated by either induction/activation of oncoproteins or repression/inactivation of tumor suppressor proteins. Genetic alterations, including amplification or mutation of oncogenes, and deletion or mutation of tumor suppressor genes, have been wildly detected as drivers in tumorigenesis and cancer development.1,2 Small-molecule degraders, such as molecular glues and proteolysis targeting chimeras (PROTACs), have been developed for targeting oncoproteins for proteasomal degradation. However, only a few agonists have been developed for tumor suppressors, including PPARγ,3–5 AMPK (A7696626 and compound 9917), and PP2A (iHAP8). A recent study has described the identification of EN5239 as a covalent small-molecule ligand of OTUB1, one of OTU family members of deubiquitinases (DUBs).10,11 EN523 covalently modifies an allosteric cysteine residue, Cys23, rather than the catalytic Cys91 residue of OTUB1 and thus does not interrupt its DUB enzymatic function.9 On the basis of this discovery, a DUB-targeting chimera (DUBTAC) has been developed to stabilize CTFR as a potential treatment of cystic fibrosis.9
The DNA-binding motifs for approximately 90% of over 1500 transcription factors (TFs) have been well defined by either experimental12–15 or theoretical methods.16 We recently developed a TF-PROTAC platform that targets oncogenic TFs for proteasomal degradation by using TF-specific DNA-binding motifs.17 Herein, we report the development of a generalizable platform termed TF-DUBTAC to selectively stabilize tumor suppressor TFs. Through a series of alkylene linkers to bridge the OTUB1 ligand EN523 and bicyclononyne (BCN), we synthesized a panel of clickable DUB binders (DUBL-X-BCN 1–10). These BCN-linked OTUB1 binders were conjugated onto the 5′-terminus of azide-modified DNA oligomers (N3-ODN) via a copper-free strain-promoted azide–alkyne cycloaddition (SPAAC) reaction (Figures 1 and S1), resulting in TF-DUBTACs for stabilizing tumor suppressor TFs in cells (Figure 1).
Figure 1.

Schematic diagram of the TF-DUBTAC platform. The BCN-linked OTUB1 binder [DUBL-X-BCN, X = (CH2)n, n = 2–11] was conjugated onto an azide-modified DNA oligomer (N3-ODN) via a copper-free SPAAC reaction, resulting in a TF-DUBTAC that recruits the DUB OTUB1 to remove polyubiquitin chain from the targeted TF.
RESULTS AND DISCUSSION
Development of FOXO-DUBTACs to Stabilize FOXO3A Tumor Suppressor Protein.
We chose three tumor suppressor TFs, namely FOXO3A, p53, and IRF3, with well-defined DNA-binding motifs to experimentally test the feasibility of the TF-DUBTAC approach. FOXO3A is a member of FOXO family TFs, all of which have similar DNA-binding motifs, also known as forkhead consensus binding sites (Figure S2A).18,19 As a downstream mediator of several oncogenic pathways, such as Akt18 and IκB,20 FOXO3A plays a tumor suppressive role. Thus, we chose FOXO3A as the first target for our TF-DUBTAC platform. We designed and synthesized a single-strand DNA oligonucleotide (5′-CTATGTAAACAACTTTGTTGTTTACATAG-3′, FOXO-ODN) that contains a FOXO consensus binding sequence (GTAAACA)21 and forms a double-strand hairpin structure via intra-dimerization (Figure 2A). Moreover, the biotin-modified FOXO-ODN (Biotin-FOXO-ODN, Figure 2A) was also synthesized to test the binding and specificity of FOXO-ODN to FOXO family members. As expected, biotin-FOXO-ODN bound both FOXO3A and FOXO1 to a similar extent, and this binding could be blocked by an excess amount of free FOXO-ODN (Figures 2B and S2B–D).
Figure 2.
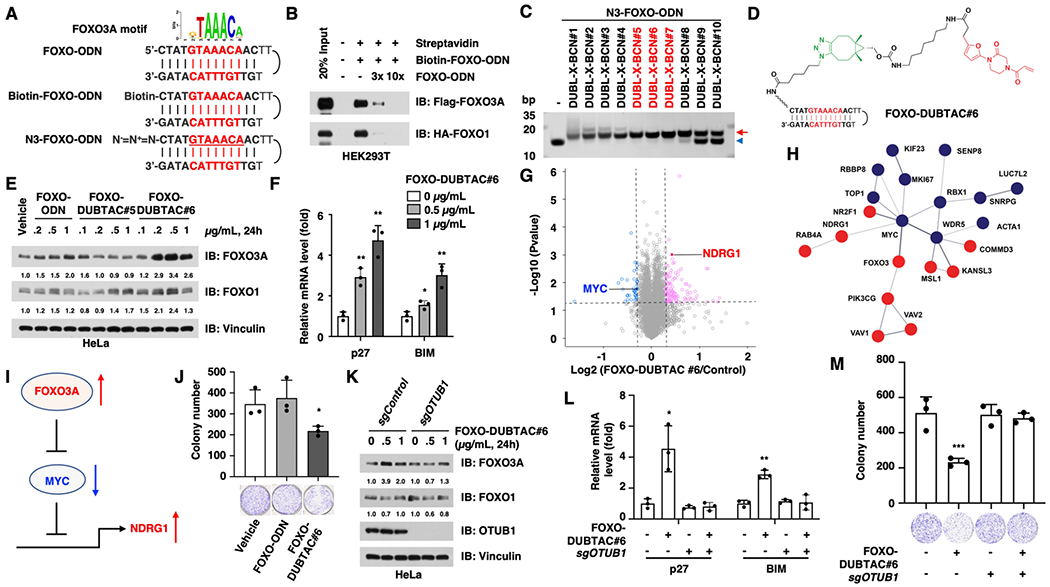
FOXO-DUBTAC #6 stabilizes FOXO3A in an OTUB1-dependent manner. (A) Schematic diagram for the FOXO3A motif and FOXO-ODN. (B) FOXO-ODN binds both FOXO3A and its close family member FOXO1, and the binding can be antagonized by free FOXO-ODN. Cell lysates with Flag-FOXO3A or HA-FOXO1 protein were extracted from HEK293T cells that expressed indicated constructs for 48 h. (C) Linkers with six–eight methylene groups are optimal for the in vitro SPAAC reactions between BCN-modified OTUB1 ligands and N3-FOXO-ODN to form FOXO-DUBTACs. The SPAAC reaction products were separated by 20% native polyacrylamide gel electrophoresis (PAGE). The red arrow indicates the click reaction products, FOXO-DUBTACs, and the blue arrowhead indicates N3-FOXO-ODN. (D) Chemical structure of FOXO-DUBTAC #6. (E) FOXO-DUBTAC #6 stabilizes FOXO3A protein in HeLa cells. HeLa cells were treated with the indicated concentrations of FOXO-ODN, FOXO-DUBTAC #5, or #6 for 24 h, followed by immunoblot analysis. (F) FOXO-DUBTAC #6 treatment increases the expression of p27 and BIM. *, p < 0.05; **, p < 0.01. (G) FOXO-DUBTAC #6 treatment changes the proteome profile of HeLa cells. (H) Enrichment of the MYC-related protein network in FOXO-DUBTAC #6-treated cells. Red indicates increased proteins, and blue indicates decreased proteins. (I) Schematic diagram to show that FOXO3A represses the MYC expression and function, resulting in increase in NDRG1. (J) FOXO-DUBTAC #6 suppresses the tumorigenesis of HeLa cells. HeLa cells were treated with 1 μ/mL FOXO-DUBTAC #6, followed by assessment in a colony formation assay. *, p < 0.05. (K) FOXO-DUBTAC #6 increases FOXO3A protein abundance in OTUB1+/+ cells but not in OTUB1−/− cells. HeLa cells were infected with the sgControl or sgOTUB1 virus to knock out endogenous OTUB1 and selected with puromycin for 72 h, followed by the treatment with FOXO-DUBTAC #6 for 24 h and were further analyzed with immunoblotting. (L) FOXO-DUBTAC #6 treatment increases the expression of p27 and BIM in an OTUB1-dependent manner. *, p < 0.05; **, p < 0.01. (M) FOXO-DUBTAC #6 treatment represses colony formation in an OTUB1-dependent manner. ***, p < 0.001.
The azide-modified FOXO-ODN (N3-FOXO-ODN) was also synthesized for the purpose of conjugating with a DUB ligand via a SPAAC reaction (Figures 1 and 2A). To this end, we synthesized a series of compounds through attaching bicyclo[6.1.0]nonyne (BCN) to EN523, a covalent ligand of OTUB1, with various alkylene linkers [hereafter termed DUBL-X-BCN, X = (CH2)n, n = 2–11, Figure 1 and Scheme S1]. In vitro SPAAC reactions between N3-FOXO-ODN and these DUBL-X-BCNs (#1–10) were performed using an excess amount of DUBL-X-BCNs (10-fold) under physiological conditions (phosphate buffered saline, PBS buffer, 37 °C), to ensure a relatively high yield of TF-DUBTACs (Figure 2C). We found that DUBL-X-BCNs #5–7, which contain six–eight methylene groups as the linkers, respectively, are optimal for the SPAAC reaction to form FOXO-DUBTACs. However, the compounds with a longer linker (such as DUBL-X-BCNs #9 and #10) displayed drastically reduced efficiency in the click reaction to generate FOXO-DUBTACs, while shorter linkers (in DUBL-X-BCNs #1–4) mildly disrupted the intra-molecular dimerization, potentially reducing the binding of these FOXO-DUBTACs to FOXO3A (Figures 2C and S2E). Thus, we chose FOXO-DUBTACs #5 and #6 for further experiments in cells.
Notably, we found that FOXO-DUBTAC #6 (Figure 2D) was effective in stabilizing FOXO3A in a concentration-dependent manner in HeLa cells (0.2–1 μg/mL, Figures 2E, S2F,G). Furthermore, the protein level of FOXO1, a close family member of FOXO3A, was not significantly elevated by the treatment with FOXO-DUBTAC #6 even at 1 μg/mL (Figure 2E), although FOXO-ODN displayed comparable binding to both FOXO3A and FOXO1 (Figures 2B and S2B–D), indicating that FOXO-DUBTAC #6 preferentially stabilizes FOXO3A. This selectivity might be due to the following: (1) the potential difference in FOXO3A and FOXO1 protein structures outside of the DNA-binding pocket may impact the formation of the OTUB1-DUBTAC-FOXO1 ternary complex and/or (2) the distance between OTUB1 and polyubiquitin chain(s) on FOXO1 may not be optimal for its removal. In contrast, the OTUB1 ligands have no effect on the stability of FOXO3A (Figure S2H). In keeping with stabilization results, FOXO-DUBTAC #6 was capable of inducing the interaction between OTUB1 and FOXO3A (Figure S2I).
Furthermore, we measured the expression level of known downstream targets of FOXO3A, including p2722 and BIM,23 both of which were elevated by FOXO-DUBTAC #6 in a dose-dependent manner (Figure 2F). To determine the efficiency and specificity of the FOXO-DUBTAC, we further performed the unbiased mass spectrum (MS) analysis to quantify the proteomics of FOXO-DUBTAC-treated and control cells. Notably, we found 106 elevated and 36 decreased proteins among a total of 6333 detected proteins (Figure 2G). Although the level of FOXO3A itself was under the detective limit, we noticed an enrichment of MYC-related protein networks, with a reduction in the MYC expression and an increase in NDRG1 level, one of the most important MYC downstream genes (Figure 2G,H). MYC is a well-defined FOXO3A downstream protein, and FOXO3A suppresses MYC at transcriptional,24 translational,25 and post-translational levels.26 Moreover, FOXO3A also antagonizes the function of MYC through directly binding to the promoters of MYC target genes.27,28 WDR5 recruits MYC onto the chromatin and is essential for the oncogenic function of MYC.29 MYC forms a complex with TOP1 on chromatin to favor transcriptions.30 Thus, the reduction of WRD5 and TOP1 might be an indirect effect through MYC. Moreover, MYC suppresses the transcription of a series of genes, including NDRG1, a cancer metastasis repressor,31 and FOXO-DUBTAC treatment led to an increase in the NDRG1 level, which could also be due to the reduction of MYC. These results indicated that the FOXO-DUBTAC stabilizes FOXO3A, which suppresses the transcription and function of MYC, unleashing the repression of NDRG1 by MYC (Figure 2H,I). In line with its effect on increasing the FOXO3A protein level and repressing MYC-driven transcription, FOXO-DUBTAC #6 suppressed tumorigenesis of HeLa cells in vitro in a colony formation assay (Figure 2J).
To determine the dependence of DUBTACs on OTUB1, we further depleted endogenous OTUB1 using CRISPR/Cas9 in HeLa cells and found that FOXO-DUBTAC #6 increased the protein level of FOXO3A in OTUB1+/+ cells but not in OTUB1−/− cells (Figures 2K, S2J). Similarly, FOXO-DUBTAC #6 increased the transcription level of p27 and BIM only in OTUB1+/+ cells but not in OTUB1−/− cells (Figure 2L). Consistent with these findings, FOXO-DUBTAC #6 suppressed tumorigenesis of wild-type HeLa cells but not OTUB1-KO cells (Figure 2M). These results indicated that the FOXO3A stabilization induced by FOXO-DUBTAC #6 is dependent on OTUB1.
Development of p53-DUBTACs to Stabilize p53 Tumor Suppressor Protein.
To explore whether other tumor suppressors with known DNA-binding motifs can be targeted by this TF-DUBTAC approach, we next focused on p53, one of the most important tumor suppressors, as a TF-DUBTAC target. The tumor suppressive role of p53 largely depends on its DNA binding and subsequent transcription activation of downstream targets that suppress cell proliferation.32 Tumor-driven p53 mutations are usually located in its DNA-binding domain.33,34 Wild-type p53 protein binds a DNA consensus sequence consisting of 2 tandem repeats of a 10 base-pair motif 5′-PuPuPuC(A/T)(T/A)GPyPyPy-3′ (Pu stands for purine and Py for pyrimidine) separated by 0–13 base pairs.35,36 Thus, we synthesized a double-strand p53-ODN, with a sense strand of 5′-AGACATGCCTAGACATGCCT-3′ and the azide-modified oligomer N3-p53-ODN by attaching an azide group onto the 5′ end of the sense strand (Figure 3A).
Figure 3.

p53-DUBTAC #6 and p53-DUBTAC #7 stabilize p53 in an OTUB1-dependent manner. (A) Schematic diagram for the p53 motif, p53-ODN, biotin-modified p53-ODN (biotin-p53-ODN), and azide-modified p53-ODN (N3-p53-ODN). (B) Biotin-p53-ODN binds Flag-tagged p53. The cell lysates with p53 protein were extracted from HEK293T cells that expressed the Flag-p53 construct for 48 h. (C) Chemical structure of p53-DUBTAC #6. (D) p53-DUBTACs #5, #6, and #7 (1 μ/mL, 24 h) increase the p53 protein level in HeLa cells. (E) p53-DUBTACs #5, #6, and #7 (0.1, 0.2, and 0.4 μ/mL, 24 h) increase the p53 protein level in HeLa cells. Vehicle stands for the treatment with transfection reagents, and control stands for the treatment without transfection reagents. (F) p53-DUBTAC #6 treatment changes the proteome profile of HeLa cells. (G) Enrichment of the AURKA-related protein network in p53-DUBTAC #6-treated cells. Red indicates increased proteins, and blue indicates decreased proteins. (H) p53-DUBTACs #6 and #7 (1 μ/mL, 24 h) increase the p53 protein level in an OTUB1-dependent manner in HeLa cells. HeLa cells infected with either the sgControl or sgOTUB1 virus and selected with puromycin, followed by treatment with the indicated compound for 24 h and immunoblotting analysis of p53 protein abundance.
The streptavidin pulldown study showed dose-dependent interaction between p53 and biotin-p53-ODN (Figure 3B), suggesting that the designed p53-ODN can bind p53. Furthermore, in vitro SPAAC reactions between the N3-p53-ODN and a series of BCN-containing compounds DUBL-X-BCNs (#1–9) resulted in the formation of p53-DUBTACs, as evident by a shift in native PAGE gel (Figure S3A). Based on the SPAAC reaction efficiency, we chose three p53-DUBTACs (#5, #6, and #7, Figures 3C, S3B,C) for further evaluation of stabilizing and increasing the p53 protein level. p53-DUBTACs #6 and #7 significantly increased the p53 protein levels in HeLa cells (over threefold of increase, 1 μg/mL, 24 h, Figures 3D,E,S3D,E), while p53-DUBTAC #5 had a relative weaker effect. On the other hand, DUBL-X-BCNs #5–7 or p53-ODN had minimal effects on the p53 protein level (Figure 3D,E, S2H), thereby excluding the possibility of an indirect effect from the ligands or oligomer. Furthermore, p53-DUBTACs #6 and #7 were capable of guiding the interaction between OTUB1 and p53 (Figure S3F).
Through an unbiased MS-based proteomic analysis, we identified 76 elevated and 33 decreased proteins among the 6333 quantified proteins in p53-DUBTAC-treated HeLa cells in comparison to control cells (Figure 3F). Similar to FOXO3A, p53 was also below the detection limit. Several ARUKA-related proteins were found among the proteins with a reduced expression, while the level of several other p53-related proteins was increased, such as KAT5 (Figure 3F,G). ARUKA has been determined as a p53 partner, and p53 suppresses the AURKA function, either by transcription regulation,37 direct inhibition through protein–protein interaction,38,39 or posttranslational regulation of ARUKA stability.40 KAT5, also known as Tip60, is another p53-binding protein that promotes p53 acetylation,41,42 and is essential for p53 signaling through direct association on the promoters of target genes.41 These results indicate that the p53-DUBTAC has a preference to affect p53-related proteins, partially through suppressing AURKA. Furthermore, p53-DUBTACs were incapable of increasing the p53 protein levels in OTUB1-knockout cells (Figure 3H), indicating that the p53 stabilization induced by these p53-DUBTACs is OTUB1-dependent.
Development of IRF-DUBTACs to Stabilize IRF3 Protein.
We further evaluated this TF-DUBTAC strategy by targeting IRF TF family members, which have a conserved DNA-binding motif (Figure 4). IRF TF family members are responsible for the transcription of genes involved in inflammation and anti-bacteria and anti-virus protective pathways, such as interferons and interleukins.43,44 All IRF family members bind DNA with a consensus motif of AANNGAAA (N stands for any nucleotide, Figure S4A).45,46 Thus, we designed and synthesized a single-strand oligomer with two GAAA core sequences (5′-GAAACTGAAACTTTTAGTTTCAGTTTC-3′, IRF-ODN) and the corresponding biotin-IRF-ODN and N3-IRF-ODN (Figure 4A). We found that biotin-IRF-ODN binds multiple IRF family members we tested except IRF9 (Figures 4B and S4B). In vitro click reactions between N3-IRF3-ODN and DUBL-X-BCNs #1–10 showed the same trend as N3-FOXO-ODN, with DUBL-X-BCNs #5, 6, and 7 exhibiting the best click efficiency in forming IRF-DUBTACs (Figure 4C). Subsequent cellular experiments validated that IRF-DUBTAC #7 was most effective in stabilizing IRF3, while other IRF-DUBTACs had some but less effect (Figures 4D, S4C,D). Taken together, these results suggest that TF-DUBTACs could be a general platform for stabilizing TFs with known DNA-binding motifs.
Figure 4.

IRF-DUBTAC #7 stabilizes IRF3 in HeLa cells. (A) Schematic diagram for the IRF3 motif, IRF-ODN, biotin-modified IRF-ODN (biotin-IRF-ODN), and azide-modified IRF-ODN (N3-IRF-ODN). (B) Biotin-IRF-ODN binds multiple IRF family members, except IRF9. The cell lysates were extracted from HEK293T cells that overexpressed indicated Flag-IRF constructs. (C) DUBL-X-BCNs #5–7 are most effective in forming IRF-DUBTACs via in vitro SPAAC reactions with N3-IRF-ODN. The SPAAC reaction products were separated by 20% native PAGE. The red arrow indicates the click reaction products, IRF-DUBTACs, and the blue arrowhead indicates IRF-ODN. (D) IRF-DUBTAC #7 (2 μ/mL, 24 h) is most effective in increasing the IRF3 protein level in HeLa cells. Vehicle stands for the treatment with transfection reagents.
CONCLUSIONS
In summary, we designed, synthesized, and characterized proof-of-concept TF-DUBTACs that target and stabilize FOXO3A, p53, and IRF3 in cells. Our lead TF-DUBTACs, FOXO-DUBTAC #6, p53-DUBTACs #6 and #7, and IRF-DUBTAC #7, effectively stabilized FOXO3A, p53, and IRF3 tumor suppressor TFs, respectively, in an OTUB1-dependent manner. Collectively, our results suggest that this TF-DUBTAC technology likely provides a universal strategy for targeting TFs with a tumor suppressor role, most of which are still “undruggable” due to the lack of small-molecule activators. Together with our previously reported TF-PROTAC technology,17 which is useful for targeting oncogenic TFs, this TF-DUBTAC platform could potentially target most tumor suppressive TFs with known DNA-binding motifs as therapeutic means to treat cancers. To be noted, compared to the high specificity of reported PROTACs, these prototype oligomer-based TF-DUBTACs have relatively lower specificity, which leads to not only the stabilization of their direct targeted TFs but also some other non-specific proteins. The relatively lower specificity of TF-DUBTACs might be ascribed to two reasons: (1) many TFs have similar DNA-binding motifs, and high dose of TF-DUBTACs might bind to these non-specific TFs; (2) the TF of interest forms a complex with other proteins, and TF-DUBTACs might also remove the polyubiquitin chain from those binding partner proteins, thus leading to the stabilization of these non-target proteins. Thus, this TF-DUBTAC platform warrants further optimization to achieve higher specificity. To overcome these potential limitations, TF-DUBTACs with longer flank sequences of DNA oligomers might improve the binding specificity to the target protein. Moreover, small-molecule agonists or antagonists might provide higher specificity, but there are few such molecules for TFs to date. On the other hand, the DNA oligomer itself or small-molecule antagonists might have a decoy effect to trap TFs that restrict their downstream gene transcription. Therefore, it is important to monitor the effect of TF-DUBTACs carefully to avoid the potential inhibitory effect on its target.
EXPERIMENTAL METHODS
General Chemistry Methods.
All commercially available solvents and reagents were used as received without further purification. Final compounds for biological evaluation were purified with preparative high-performance liquid chromatography (HPLC) on an Agilent Prep 1200 series with the UV detector set to 254/220 nm with solvent A (0.08% ammonium bicarbonate in water) and solvent B (acetonitrile) as eluents with a flow rate of 40 mL/min at rt. Purities of the final compounds were determined by HPLC and were greater than 95%. HPLC conditions to evaluate purity were as follows: an Agilent 1200 series system, 2.1 mm × 150 mm Zorbax 300SB-C18 5 μm column, 1–99% gradient of 0.1% trifluoroacetic acid in water, and 0.1% trifluoroacetic acid in acetonitrile at a flow rate of 0.4 mL/min; high-resolution mass spectrometry data was acquired on an Agilent G1969A API-TOF with an electrospray ionization source. Nuclear magnetic resonance (NMR) spectra were recorded on either an AVANCE NEO 600 MHz or 500 MHz spectrometer. Proton and carbon NMR spectra are reported in parts per million (ppm) on the δ scale.
Oligomer Synthesis.
The single-strand oligonucleotides containing the FOXO3A-binding motif, namely, FOXO-ODN, were synthesized based on the reported FOXO3A DNA-binding consensus. The sequence of 29-mer FOXO-ODN is 5′- CTATGTAAACAACTTTGTTGTTTACATAG-3′, in which the core consensus is underlined. The single-strand IRF-ODN was synthesized based on the reported IRF3 DNA-binding consensus with two repeats of the core consensus, and the sequence is 5′-GAAACTGAAACTTTTAGTTTCAGTTTC-3′. The double-strand p53-ODN was synthesized based on the well-defined p53 DNA-binding consensus with 2 repeats of a 10 base-pair motif. The sense strand of p53-ODN is 5′-AGACATGCCTAGACATGCCT-3′, and the antisense strand is 5′-AGGCATGTCTAGGCATGTCT-3′. Moreover, the biotin modification of ODNs was synthesized by adding a biotin to the 5′ end of the single-strand ODN or the 5′ end of the sense strand for the double-strand ODN using a spacer of NH2–(CH2)2–O–(CH2)2–OH. The azide modification of ODNs was synthesized by incorporating an azide group on the same site as that of biotin through the 5′ amino modifier C6. All oligomers were synthesized by Integrated DNA Technologies, Inc. The unmodified oligomers were purified by standard desalting, while both biotin- and azide-modified oligomers were purified by HPLC. The single FOXO-ODN and IRF-ODN were annealed by heating to 95 °C for 5 min, followed by cooling down to room temperature at 5 °C per min. Similarly, the sense and antisense p53 oligomers were mixed in a 1:1 ratio and annealed before use.
In Vitro Copper-Free SPAAC Reaction.
For incorporation of OTUB1 ligands onto the 5′ end of oligomers, BCN-modified OTUB1 ligands (DUBL-X-BCN) were incubated with an azide-modified oligomer at 37 °C for 24 h. The reaction mixtures were further purified using a Nucleotide Removal Kit (QIAGEN) to remove extra DUB ligands and salts in the reaction mixture, followed by annealing as described above.
Native DNA PAGE.
Oligomers and SPAAC reaction products were separated by 20% native PAGE as previous described.17 Briefly, 0.5 μg of oligomers was separated by native PAGE at 100 V for 1 h, followed by incubation in 0.2% EtBr solution in 1× Tris–boric acid–ethyl-enediaminetetraacetic acid (EDTA) (TBE, pH 8.3) buffer that consisted of 89 mM Tris, 89 mM boric acid, and 2 mM EDTA. The native gels contain 20% acrylamide and 2.5% glycerol, 0.075% ammonium persulfate, and 0.05% tetramethylethylenediamine in 0.5× TBE buffer. Finally, the gels were imaged with UV illumination with the ChemiDoc Touch Imaging System (Bio-Rad).
Cell Culture and Treatment.
Human embryonic kidney 293T (HEK293T) and HeLa cells were maintained in Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum, 100 units/mL penicillin, and 100 μg/mL streptomycin. For the ectopic expression of TFs, Flag-FOXO3A, HA-FOXO1, Flag-p53, and Flag-IRF2, 3, 4, 5, 6, 8, and 9 were transfected into HEK293T cells and harvested for lysis 48 h after transfection. For TF-DUBTAC treatment, HeLa cells in a six-well plate were transfected with individual TF-DUBTACs for 24 h, followed by harvest for western blot analysis. For depletion of endogenous OTUB1, the sgRNA for OTUB1 (5′-TATCAACAGAAGATCAAGGT-3′) was synthesized and inserted into a lenti-CRISPR-V2 construct as previously described.47 The sgOTUB1 lentivirus was generated in HEK293T cells as previously described48,49 for infection of HeLa cells overnight, followed by selection with puromycin for 72 h.
Streptavidin–Biotin Pulldown Assay.
Streptavidin pulldown assay for biotin-ODN with respective TFs was performed as previously described.17 HEK293T cells that ectopically expressed TFs were lysed by EBC lysis buffer (50 mM Tris pH 7.5, 120 mM NaCl, 0.5% NP-40) supplemented with protease inhibitors (Pierce) and phosphatase inhibitors (phosphatase inhibitor cocktail set I and II, Calbiochem). The protein concentrations of the lysates were measured using the Bio-Rad protein assay reagent on a Beckman Coulter DU-800 spectrophotometer. The cell lysates (1 mg) were further incubated with biotin-ODN together with/without excess free ODN (3- or 10-fold) at 4 °C for 3 h, followed by adding 10 μL of streptavidin agarose beads (Thermo Fisher) for another 1 h. The beads were washed four times with NETN buffer (100 mM NaCl, 20 mM Tris-Cl, pH 8.0, 0.5 mM EDTA, 0.5% NP-40), boiled in sodium dodecyl sulfate (SDS) loading buffer, and further separated by 10% SDS-PAGE and blotted with the individual antibody.
Western Blot Assay.
Cells were lysed in EBC buffer supplemented with the protease inhibitor cocktail and phosphatase inhibitors, and the protein concentrations were measured as described above.17 The lysates (60 μg protein) were then resolved by 10% SDS-PAGE at 130 V for 80 min and immunoblotted with indicated antibodies at 4 °C overnight, washed four times with Tris-buffered saline with 0.1% Tween-20 (TBST), incubated with the secondary antibody for 1 h at room temperature, and then washed four times with TBST buffer. Anti-FOXO3A (#12829, 1:1000), FOXO1 (#2880, 1:1000), OTUB1 (#3783, 1:1000), and IRF3 (#11904, 1:1000) antibodies were purchased from Cell Signaling Technology. The anti-HA antibody (clone 16B12, #901513, 1:1000) was purchased from BioLegend. The anti-FLAG (F3165, 1:5000), anti-vinculin antibody (V-4505, 1:50,000), peroxidase-conjugated anti-mouse secondary antibody (A-4416, 1:3000), and peroxidase-conjugated anti-rabbit secondary antibody (A-4914, 1:3000) were purchased from Sigma. The anti-p53 antibody (clone DO-1, sc-126, 1:1000) was purchased from Santa Cruz. All primary antibodies were diluted in 1% bovine serum albumin in TBST buffer, and secondary antibodies were diluted in 5% non-fat milk in TBST buffer. All western blot assays were repeated at least twice. The protein quantification in immunoblot assay was performed using Quantity One software (Bio-Rad).
RT-qPCR.
Total RNAs were extracted using a Qiagen RNeasy mini kit (Qiagen. #74106) and reversed transcripted into cDNA using iScript Reverse Transcription Supermix (Bio-Rad, # 1708841). Reverse transcription-quantitative polymerase chain reaction (RT-qPCR)was performed with SYBR Select Master Mix (Thermo Fisher, #4472908) using primers as mentioned below: p27-forward: AACGTGCGAGTGTCTAACGG, p27-reverse: CCCTCTAGGGGTTTGTGATTCT, BIM-forward: TAAGTTCTGAGTGTGACCGAGA, BIM-reverse: GCTCTGTCTGTAGGGAGGTAGG, GAPDH-forward: TTGAGGTCAATGAAGGGGTC, and GAPDH-reverse: GAAGGTGAAGGTCGGAGTCA.
Proteomics Sample Preparation.
The cell pellets were resuspended in 8 M urea and 50 mM Tris-HCl pH 8.0, reduced with dithiothreitol (5 mM final) for 30 min at room temperature, and alkylated with iodoacetamide (15 mM final) for 45 min in the dark at room temperature. Samples were diluted fourfold with 25 mM Tris-HCl pH 8.0 and 1 mM CaCl2 and digested with trypsin at the 1:100 (w/w, trypsin/protein) ratio overnight at room temperature. There were two biological samples under each condition and total eight samples. Peptides were cleaned using homemade C18 stage tips, and the concentration was determined (peptide assay, Thermo 23275). 25 μg of each was used for labeling with isobaric stable tandem mass tags (TMT10-126, 127C, 128N, 129N, 129C, 130N, 130C, and 131, Thermo Fisher Scientific, San Jose, CA) following manufacturer’s instructions. The mixture of labeled peptides was fractionated into 22 fractions on a C18 stage tip with buffer 10 mM trimethylammonium bicarbonate, pH 8.5 containing 5 to 50% acetonitrile.
Mass Spectrometry Analysis.
Dried peptides were dissolved in 0.1% formic acid and 2% acetonitrile. 0.5 μg of peptides of each fraction was analyzed on a Q-Exactive HF-X coupled with an Easy nanoLC 1200 (Thermo Fisher Scientific, San Jose, CA). Peptides were loaded onto a nanoEase MZ HSS T3 Column (100 Å, 1.8 μm, 75 μm × 250 mm, Waters). Analytical separation of all peptides was achieved with a 110 min gradient. A linear gradient of 5 to 10% buffer B over 5 min, 10 to 31% buffer B over 70 min, and 31 to 75% buffer B over 15 min was executed at a 250 nL/min flow rate followed by a ramp to 100% B in 1 min and 19 min wash with 100% B, where buffer A was aqueous 0.1% formic acid, and buffer B was 80% acetonitrile and 0.1% formic acid. MS experiments were also carried out in a data-dependent mode with full MS (externally calibrated to a resolution of 60,000 at m/z 200) followed by high-energy collision-activated dissociation MS/MS of the top 10 most intense ions with a resolution of 45,000 at m/z 200. High-energy collision-activated dissociation MS/MS was used to dissociate peptides at a normalized collision energy of 32 eV in the presence of nitrogen bath gas atoms. Dynamic exclusion was 45 s.
Raw Proteomics Data Processing and Analysis.
Peptide identification and quantification with TMT reporter ions were performed using the MaxQuant software version 1.6.10.43 (Max Planck Institute, Germany). Protein database searches were performed against the UniProt human protein sequence database (UP000005640). A false discovery rate for both peptide-spectrum match and protein assignment was set at 1%. Search parameters included up to two missed cleavages at Lys/Arg on the sequence, oxidation of methionine, and protein N-terminal acetylation as a dynamic modification. Carbamidomethylation of cysteine residues was considered as a static modification. Peptide identifications are reported by filtering of reverse and contaminant entries and assigning them to their leading razor protein. Data processing and statistical analysis were performed on Perseus (version 1.6.10.50). Protein quantitation was performed on biological replicates, and a two-sample t-test statistic was used with a p-value of 5% to report statistically significant protein abundance fold changes.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by the NIH grants R35CA253027 (W.W.) and P30CA196521 (J.J.) and an endowed professorship from the Icahn School of Medicine at Mount Sinai (to J.J.). This work utilized the NMR Spectrometer Systems at Mount Sinai acquired with funding from National Institutes of Health SIG grants 1S10OD025132 and 1S10OD028504.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.2c04824.
Compound synthesis and experimental details (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/jacs.2c04824
The authors declare the following competing financial interest(s): W.W. is a co-founder and stockholder of the Rekindle Therapeutics. J.J. is a co-founder and equity shareholder in Cullgen, Inc. and a consultant for Cullgen, Inc., EpiCypher, Inc., and Accent Therapeutics, Inc. The Jin laboratory received re-search funds from Celgene Corporation, Levo Therapeutics, Cullgen, Inc. and Cullinan Oncology. All other authors declare no competing interests.
Contributor Information
Jing Liu, Department of Pathology, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, Massachusetts 02215, United States.
Xufen Yu, Mount Sinai Center for Therapeutics Discovery, Departments of Pharmacological Sciences and Oncological Sciences, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States.
He Chen, Mount Sinai Center for Therapeutics Discovery, Departments of Pharmacological Sciences and Oncological Sciences, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States.
H. Ümit Kaniskan, Mount Sinai Center for Therapeutics Discovery, Departments of Pharmacological Sciences and Oncological Sciences, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States.
Ling Xie, Department of Biochemistry and Biophysics, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States.
Xian Chen, Department of Biochemistry and Biophysics, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States.
Jian Jin, Mount Sinai Center for Therapeutics Discovery, Departments of Pharmacological Sciences and Oncological Sciences, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States.
Wenyi Wei, Department of Pathology, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, Massachusetts 02215, United States.
REFERENCES
- (1).Vogelstein B; Kinzler KW Cancer genes and the pathways they control. Nat. Med 2004, 10, 789–799. [DOI] [PubMed] [Google Scholar]
- (2).Hanahan D; Weinberg RA Hallmarks of cancer: the next generation. Cell 2011, 144, 646–674. [DOI] [PubMed] [Google Scholar]
- (3).Lehmann JM; Moore LB; Smith-Oliver TA; Wilkison WO; Willson TM; Kliewer SA An Antidiabetic Thiazolidinedione Is a High Affinity Ligand for Peroxisome Proliferator-activated Receptor γ (PPARγ). J. Biol. Chem 1995, 270, 12953–12956. [DOI] [PubMed] [Google Scholar]
- (4).Willson TM; Cobb JE; Cowan DJ; Wiethe RW; Correa ID; Prakash SR; Beck KD; Moore LB; Kliewer SA; Lehmann JM The Structure–Activity Relationship between Peroxisome Proliferator-Activated Receptor γ Agonism and the Antihyperglycemic Activity of Thiazolidinediones. J. Med. Chem 1996, 39, 665–668. [DOI] [PubMed] [Google Scholar]
- (5).Momose Y; Maekawa T; Yamano T; Kawada M; Odaka H; Ikeda H; Sohda T Novel 5-substituted 2,4-thiazolidinedione and 2,4-oxazolidinedione derivatives as insulin sensitizers with antidiabetic activities. J. Med. Chem 2002, 45, 1518–1534. [DOI] [PubMed] [Google Scholar]
- (6).Cool B; Zinker B; Chiou W; Kifle L; Cao N; Perham M; Dickinson R; Adler A; Gagne G; Iyengar R; Zhao G; Marsh K; Kym P; Jung P; Camp HS; Frevert E Identification and characterization of a small molecule AMPK activator that treats key components of type 2 diabetes and the metabolic syndrome. Cell Metab. 2006, 3, 403–416. [DOI] [PubMed] [Google Scholar]
- (7).Lai Y-C; Kviklyte S; Vertommen D; Lantier L; Foretz M; Viollet B; Hallén S; Rider MH A small-molecule benzimidazole derivative that potently activates AMPK to increase glucose transport in skeletal muscle: comparison with effects of contraction and other AMPK activators. Biochem. J 2014, 460, 363–375. [DOI] [PubMed] [Google Scholar]
- (8).Morita K; He S; Nowak RP; Wang J; Zimmerman MW; Fu C; Durbin AD; Martel MW; Prutsch N; Gray NS; Fischer ES; Look AT Allosteric Activators of Protein Phosphatase 2A Display Broad Antitumor Activity Mediated by Dephosphorylation of MYBL2. Cell 2020, 181, 702–715. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- (9).Henning NJ; Boike L; Spradlin JN; Ward CC; Liu G; Zhang E; Belcher BP; Brittain SM; Hesse MJ; Dovala D; McGregor LM; Valdez Misiolek R; Plasschaert LW; Rowlands DJ; Wang F; Frank AO; Fuller D; Estes AR; Randal KL; Panidapu A; McKenna JM; Tallarico JA; Schirle M; Nomura DK Deubiquitinase-targeting chimeras for targeted protein stabilization. Nat. Chem. Biol 2022, 18, 412–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Mevissen TET; Hospenthal MK; Geurink PP; Elliott PR; Akutsu M; Arnaudo N; Ekkebus R; Kulathu Y; Wauer T; El Oualid F; Freund SMV; Ovaa H; Komander D OTU deubiquitinases reveal mechanisms of linkage specificity and enable ubiquitin chain restriction analysis. Cell 2013, 154, 169–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Komander D; Clague MJ; Urbé S Breaking the chains: structure and function of the deubiquitinases. Nat. Rev. Mol. Cell Biol 2009, 10, 550–563. [DOI] [PubMed] [Google Scholar]
- (12).Berger MF; Philippakis AA; Qureshi AM; He FS; Estep PW 3rd; Bulyk ML Compact, universal DNA microarrays to comprehensively determine transcription-factor binding site specificities. Nat. Biotechnol 2006, 24, 1429–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Weirauch MT; Yang A; Albu M; Cote AG; Montenegro-Montero A; Drewe P; Najafabadi HS; Lambert SA; Mann I; Cook K; Zheng H; Goity A; van Bakel H; Lozano J-C; Galli M; Lewsey MG; Huang E; Mukherjee T; Chen X; Reece-Hoyes JS; Govindarajan S; Shaulsky G; Walhout AJM; Bouget F-Y; Ratsch G; Larrondo LF; Ecker JR; Hughes TR Determination and inference of eukaryotic transcription factor sequence specificity. Cell 2014, 158, 1431–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Jolma A; Yan J; Whitington T; Toivonen J; Nitta KR; Rastas P; Morgunova E; Enge M; Taipale M; Wei G; Palin K; Vaquerizas JM; Vincentelli R; Luscombe NM; Hughes TR; Lemaire P; Ukkonen E; Kivioja T; Taipale J DNA-binding specificities of human transcription factors. Cell 2013, 152, 327–339. [DOI] [PubMed] [Google Scholar]
- (15).Gerstein MB; Kundaje A; Hariharan M; Landt SG; Yan K-K; Cheng C; Mu XJ; Khurana E; Rozowsky J; Alexander R; Min R; Alves P; Abyzov A; Addleman N; Bhardwaj N; Boyle AP; Cayting P; Charos A; Chen DZ; Cheng Y; Clarke D; Eastman C; Euskirchen G; Frietze S; Fu Y; Gertz J; Grubert F; Harmanci A; Jain P; Kasowski M; Lacroute P; Leng J; Lian J; Monahan H; O’Geen H; Ouyang Z; Partridge EC; Patacsil D; Pauli F; Raha D; Ramirez L; Reddy TE; Reed B; Shi M; Slifer T; Wang J; Wu L; Yang X; Yip KY; Zilberman-Schapira G; Batzoglou S; Sidow A; Farnham PJ; Myers RM; Weissman SM; Snyder M Architecture of the human regulatory network derived from ENCODE data. Nature 2012, 489, 91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Pujato M; Kieken F; Skiles AA; Tapinos N; Fiser A Prediction of DNA binding motifs from 3D models of transcription factors; identifying TLX3 regulated genes. Nucleic Acids Res. 2014, 42, 13500–13512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Liu J; Chen H; Kaniskan HÜ; Xie L; Chen X; Jin J; Wei W TF-PROTACs Enable Targeted Degradation of Transcription Factors. J. Am. Chem. Soc 2021, 143, 8902–8910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Brunet A; Bonni A; Zigmond MJ; Lin MZ; Juo P; Hu LS; Anderson MJ; Arden KC; Blenis J; Greenberg ME Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 1999, 96, 857–868. [DOI] [PubMed] [Google Scholar]
- (19).Tsai K-L; Sun Y-J; Huang C-Y; Yang J-Y; Hung M-C; Hsiao C-D Crystal structure of the human FOXO3a-DBD/DNA complex suggests the effects of post-translational modification. Nucleic Acids Res. 2007, 35, 6984–6994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Hu MC-T; Lee D-F; Xia W; Golfman LS; Ou-Yang F; Yang J-Y; Zou Y; Bao S; Hanada N; Saso H; Kobayashi R; Hung M-C IκB Kinase Promotes Tumorigenesis through Inhibition of Forkhead FOXO3a. Cell 2004, 117, 225–237. [DOI] [PubMed] [Google Scholar]
- (21).Pierrou S; Hellqvist M; Samuelsson L; Enerbäck S; Carlsson P Cloning and characterization of seven human forkhead proteins: binding site specificity and DNA bending. EMBO J. 1994, 13, 5002–5012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Dijkers PF; Medema RH; Pals C; Banerji L; Thomas NSB; Lam EW-F; Burgering BMT; Raaijmakers JAM; Lammers J-WJ; Koenderman L; Coffer PJ Forkhead Transcription Factor FKHR-L1 Modulates Cytokine-Dependent Transcriptional Regulation of p27 KIP1. Mol. Cell. Biol 2000, 20, 9138–9148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Sunters A; Fernández de Mattos S; Stahl M; Brosens JJ; Zoumpoulidou G; Saunders CA; Coffer PJ; Medema RH; Coombes RC; Lam EW-F FoxO3a transcriptional regulation of Bim controls apoptosis in paclitaxel-treated breast cancer cell lines. J. Biol. Chem 2003, 278, 49795–49805. [DOI] [PubMed] [Google Scholar]
- (24).Yamashita S; Ogawa K; Ikei T; Fujiki T; Katakura Y FOXO3a potentiates hTERT gene expression by activating c-MYC and extends the replicative life-span of human fibroblast. PLoS One 2014, 9, No. e101864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Kress TR; Cannell IG; Brenkman AB; Samans B; Gaestel M; Roepman P; Burgering BM; Bushell M; Rosenwald A; Eilers M The MK5/PRAK kinase and Myc form a negative feedback loop that is disrupted during colorectal tumorigenesis. Mol. Cell 2011, 41, 445–457. [DOI] [PubMed] [Google Scholar]
- (26).Ferber EC; Peck B; Delpuech O; Bell GP; East P; Schulze A FOXO3a regulates reactive oxygen metabolism by inhibiting mitochondrial gene expression. Cell Death Differ. 2012, 19, 968–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Bouchard C; Marquardt J; Brás A; Medema RH; Eilers M Myc-induced proliferation and transformation require Akt-mediated phosphorylation of FoxO proteins. EMBO J. 2004, 23, 2830–2840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Jensen KS; Binderup T; Jensen KT; Therkelsen I; Borup R; Nilsson E; Multhaupt H; Bouchard C; Quistorff B; Kjaer A; Landberg G; Staller P FoxO3A promotes metabolic adaptation to hypoxia by antagonizing Myc function. EMBO J. 2011, 30,4554–4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Thomas LR; Adams CM; Wang J; Weissmiller AM; Creighton J; Lorey SL; Liu Q; Fesik SW; Eischen CM; Tansey WP Interaction of the oncoprotein transcription factor MYC with its chromatin cofactor WDR5 is essential for tumor maintenance. Proc. Natl. Acad. Sci. U.S.A 2019, 116, 25260–25268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Das SK; Kuzin V; Cameron DP; Sanford S; Jha RK; Nie Z; Rosello MT; Holewinski R; Andresson T; Wisniewski J; Natsume T; Price DH; Lewis BA; Kouzine F; Levens D; Baranello L MYC assembles and stimulates topoisomerases 1 and 2 in a “topoisome”. Mol. Cell 2022, 82, 140–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Zhang J; Chen S; Zhang W; Zhang J; Liu X; Shi H; Che H; Wang W; Li F; Yao L Human differentiation-related gene NDRG1 is a Myc downstream-regulated gene that is repressed by Myc on the core promoter region. Gene 2008, 417, 5–12. [DOI] [PubMed] [Google Scholar]
- (32).Vogelstein B; Kinzler KW p53 function and dysfunction. Cell 1992, 70, 523–526. [DOI] [PubMed] [Google Scholar]
- (33).Pavletich NP; Chambers KA; Pabo CO The DNA-binding domain of p53 contains the four conserved regions and the major mutation hot spots. Genes Dev. 1993, 7, 2556–2564. [DOI] [PubMed] [Google Scholar]
- (34).Bargonetti J; Manfredi JJ; Chen X; Marshak DR; Prives C A proteolytic fragment from the central region of p53 has marked sequence-specific DNA-binding activity when generated from wild-type but not from oncogenic mutant p53 protein. Genes Dev. 1993, 7, 2565–2574. [DOI] [PubMed] [Google Scholar]
- (35).Wang Y; Schwedes JF; Parks D; Mann K; Tegtmeyer P Interaction of p53 with its consensus DNA-binding site. Mol. Cell. Biol 1995, 15, 2157–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).El-Deiry WS; Kern SE; Pietenpol JA; Kinzler KW; Vogelstein B Definition of a consensus binding site for p53. Nat. Genet 1992, 1, 45–49. [DOI] [PubMed] [Google Scholar]
- (37).Nikulenkov F; Spinnler C; Li H; Tonelli C; Shi Y; Turunen M; Kivioja T; Ignatiev I; Kel A; Taipale J; Selivanova G Insights into p53 transcriptional function via genome-wide chromatin occupancy and gene expression analysis. Cell Death Differ. 2012, 19, 1992–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Chen S-S; Chang PC; Cheng YW; Tang FM; Lin YS Suppression of the STK15 oncogenic activity requires a transactivation-independent p53 function. EMBO J. 2002, 21, 4491–4499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Eyers PA; Maller JL Regulation of Xenopus Aurora A activation by TPX2. J. Biol. Chem 2004, 279, 9008–9015. [DOI] [PubMed] [Google Scholar]
- (40).Wu C-C; Yang T-Y; Yu C-TR; Phan L; Ivan C; Sood AK; Hsu S-L; Lee M-H p53 negatively regulates Aurora A via both transcriptional and posttranslational regulation. Cell Cycle 2012, 11, 3433–3442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Tang Y; Luo J; Zhang W; Gu W Tip60-dependent acetylation of p53 modulates the decision between cell-cycle arrest and apoptosis. Mol. Cell 2006, 24, 827–839. [DOI] [PubMed] [Google Scholar]
- (42).Sykes SM; Mellert HS; Holbert MA; Li K; Marmorstein R; Lane WS; McMahon SB Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Mol. Cell 2006, 24, 841–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Yanai H; Negishi H; Taniguchi T The IRF family of transcription factors: Inception, impact and implications in oncogenesis. Oncoimmunology 2012, 1, 1376–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Honda K; Taniguchi T IRFs: master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat. Rev. Immunol 2006, 6, 644–658. [DOI] [PubMed] [Google Scholar]
- (45).Panne D; Maniatis T; Harrison SC An Atomic Model of the Interferon-β Enhanceosome. Cell 2007, 129, 1111–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Panne D; Maniatis T; Harrison SC Crystal structure of ATF-2/c-Jun and IRF-3 bound to the interferon-β enhancer. EMBO J. 2004, 23, 4384–4393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Sanjana NE; Shalem O; Zhang F Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 2014, 11, 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Liu J; Chen H; Ma L; He Z; Wang D; Liu Y; Lin Q; Zhang T; Gray N; Kaniskan HÜ; Jin J; Wei W Light-induced control of protein destruction by opto-PROTAC. Sci. Adv 2020, 6, No. eaay5154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Liu J; Chen H; Liu Y; Shen Y; Meng F; Kaniskan HÜ; Jin J; Wei W Cancer Selective Target Degradation by Folate-Caged PROTACs. J. Am. Chem. Soc 2021, 143, 7380–7387. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.