

Abstract
The transcription factor RUNX2 is a key regulator of chondrocyte phenotype during development, making it an ideal target for prevention of undesirable chondrocyte maturation in cartilage tissue engineering strategies. Here, we engineered an autoregulatory gene circuit (cisCXp-shRunx2) that negatively controls RUNX2 activity in chondrogenic cells via RNA interference initiated by a tunable synthetic Col10a1-like promoter (cisCXp). The cisCXp-shRunx2 gene circuit is designed based on the observation that induced RUNX2 silencing after early chondrogenesis enhances the accumulation of cartilaginous matrix in ATDC5 cells. We show that the cisCXp-shRunx2 initiates RNAi of RUNX2 in maturing chondrocytes in response to the increasing intracellular RUNX2 activity without interfering with early chondrogenesis. The induced loss of RUNX2 activity in turn negatively regulates the gene circuit itself. Moreover, the efficacy of RUNX2 suppression from cisCXp-shRunx2 can be controlled by modifying the sensitivity of cisCXp promoter. Finally, we show the efficacy of inhibiting RUNX2 in preventing matrix loss in human MSC-derived cartilage under conditions that induce chondrocyte hypertrophic differentiation, including inflammation. Overall, our results demonstrated that the negative modulation of RUNX2 activity with our autoregulatory gene circuit enhanced matrix synthesis and resists ECM degradation by reprogrammed MSC-derived chondrocytes in response to the microenvironment of the degenerative joint.
Keywords: cartilage, human mesenchymal stem cells, RUNX2, synthetic gene circuit
Graphical Abstract
A gene circuit was created to induce an intracellular RUNX2-suppressing feedback loop, which abrogates MMP13 activity and enhances matrix accumulation in response to cues that induce chondrocyte hypertrophy.
INTRODUCTION
Runt related transcription factor 2 (RUNX2) has a significant role in musculoskeletal development, including regulation of the chondrocyte phenotype during endochondral ossification(1, 2). RUNX2 expression in chondrocytes promotes hypertrophic maturation and cartilage matrix turnover, in preparation for replacement with bone, while RUNX2 expression is important for initial commitment and differentiation of stem cells into osteoblasts(3). Multiple intracellular pathways, including those mediated by SMADs(4), β-catenin(5), and p38(6) signaling, are involved in hypertrophic maturation of chondrocytes during this process. These pathways are regulated by various extracellular signaling molecules, including bone morphogenetic proteins (BMPs)(7), transforming growth factor beta (TGFβ)(8), parathyroid hormone-related peptide (PTHrP)(9), Indian hedgehog (IHH)(10), fibroblast growth factor (FGF), and insulin like growth factor (IGF)(11) as well as extracellular matrix (ECM) ligands such as hyaluronic acid(12).
Hypertrophic maturation of chondrocytes also poses a significant challenge towards clinical application of tissue engineering-based cartilage repair strategies. During hypertrophy, RUNX2 upregulates expression of matrix metalloproteinases (MMPs) resulting in cartilage matrix catabolism, compromising the integrity of the engineered tissue(13-15). Hence, inhibition of chondrocyte hypertrophy has been a long withstanding goal of cartilage tissue engineering community.
Synthetic regulation of the RUNX2 pathway has long been a target for cartilage tissue engineering, but its multifaceted functions in cell biology have led to mixed results. For example, blocking SMAD1/5/8 after chondrogenic differentiation of mesenchymal stem cells (MSCs) prevents terminal differentiation and mineralization while sustaining production of the articular cartilage structural macromolecules aggrecan and collagen type II(4). Similarly, blocking the Wnt/β-catenin pathway during chondrogenic culture of MSCs suppresses hypertrophy and in vivo mineralization(16). Combined strategies using dynamic loading or hypoxic culture of MSCs in hydrogels containing ECM ligands can also regulate hypertrophic pathways(17, 18). However, it has also been shown that complete inhibition or silencing of these and other RUNX2-related pathways will prevent or significantly delay chondrogenesis(4, 18). While these studies have investigated the role of various pathways that regulate RUNX2 gene expression and activity in MSCs, the direct effect of stage-specific suppression of RUNX2 activity on cartilage tissue engineering outcomes has yet to be explored.
Gene silencing through RNA interference (RNAi) allows straightforward loss-of-function studies in mammalian cells to help understand the molecular mechanisms that underpin cartilage development. A virally introduced inducible short hairpin RNA (shRNA) system provides temporal and reversible control of protein expression so that its activity may be evaluated in vitro over long culture periods, such as those required for tissue engineering. These insights could help us develop targeted silencing protocols in cartilage tissue engineering applications, which has advantages over broad spectrum inhibition of intracellular pathways that drive or respond to RUNX2 activity. However, chondrogenesis and subsequent maturation of MSCs is not a homogenous process. Therefore, inducible RNAi systems cannot be used to address the stage-specific effects of RUNX2 inhibition in cell populations that are at various stages of maturation. Furthermore, RNAi systems cannot be used to study the direct effect of exogenous cues on RUNX2-driven pathways.
Advances in synthetic biology allow us to reprogram cells with new functions by site-specific editing of the genome or combining basic genetic modules with well-characterized functions in novel ways to create gene circuits. The reprogrammed cells are then capable of autonomously detecting and adapting to changes in their environment. The utility of these techniques has been demonstrated in the production of stem cells with autoregulatory resistance to inflammatory stimulus for cartilage tissue engineering(19, 20). Moreover, synthetic gene circuits have been shown to promote stem cell-based cartilage regeneration by preserving circadian clock(21). Autoregulatory gene circuits can be used to evaluate the impact of exogenous cues on RUNX2 activity levels as well as regulate the maturation of chondrogenic cells mediated by RUNX2.
In this study, we sought to engineer an autoregulatory, closed-loop gene circuit to negatively regulate RUNX2 activity in chondrogenic cells. Using an inducible RNAi system(22), the effect of timing of RUNX2 interference on the retention of articular cartilage-specific structural macromolecules was first assessed. Based on the outcomes of these experiments, we then designed an RNA-based regulatory unit containing a partial Col10a1-like promoter region with tunable activity that drives expression of short hairpin RNA (shRNA) for RUNX2. Therefore, we constructed compact synthetic promoters de novo that contain multiple copies of RUNX2 cis-enhancers upstream of the essential regulatory elements to initiate transcription. This design permits tuning of the gene circuit’s sensitivity to intracellular RUNX2 concentration as well as control of RUNX2 activity levels. We characterized our circuits with in vitro specificity and sensitivity assays using the ATDC5 cell line. We then tested the hypothesis that the level of RUNX2 activity can be tuned by varying the number of cis-enhancers incorporated into the promoter and that targeted disruption of RUNX2 activity could increase the production and retention of structural macromolecules in a cell-state-dependent manner. Finally, we demonstrate that the gene circuit resists the expression of hypertrophy markers and subsequent matrix degradation by human mesenchymal stem cell-derived chondrocytes (hMdChs) under hypertrophic and inflammatory stimulus.
METHODS
Experimental Design
Part 1: Design and Assessment of the Inducible shRunx2 System
The experiments to characterize the effect of cell state-specific RUNX2 suppression on chondrogenesis and accumulation of cartilage-specific structural macromolecules are outlined in Fig. 1. ATDC5 cells were first transduced with lentiviruses expressing a TetOn inducible Runx2 shRNA. Stable polyclonal cell populations were then established via antibiotic selection. RUNX2 suppression was then induced using doxycycline at day 0, 4, 7, 14, and 21 during a four-week chondrogenic program in monolayer culture followed by gene and protein expression profiling.
Figure 1.

Experimental design of the inducible shRunx2 study. (a) design of the dox-inducible shRunx2 vector (b) study design where ATDC5 cells were transduced with dox-inducible shRUNX2 vector, selected using Puromycin treatment and then induced to chondrogenesis. RUNX2 suppression was induced from Day 0, 4, 7, 14 or 21 using doxycycline treatment.
Part 2: Design and Assessment of the Autoregulatory shRunx2 Gene Circuit
To enable autoregulatory RUNX2 suppression and characterize its effect on chondrogenesis, we engineered a gene circuit that induces the expression of Runx2 shRNA in response to increasing intracellular concentrations of active RUNX2 using a synthetic Col10a1-like promoter. Chondrogenesis of a polyclonal ATDC5 cell population stably expressing such a gene circuit was evaluated in both 3-week monolayer culture and 5-week pellet culture (Fig. 2).
Figure 2. Experimental design of the autoregulatory shRunx2 gene circuit study.

(a) Design of the autoregulatory shRunx2 gene circuit. (b) ATDC5 cells were transduced with autoregulatory shRunx2 gene circuits, selected using puromycin and then induced to chondrogenesis in monolayer and pellet culture. Runx2 activity was measured using the luciferase reporter throughout chondrogenic culture.
Chondrogenic Cell Cultures
ATDC5 (Sigma; RRID:CVCL_3894) cells were used to model the transition through the different phases of endochondral ossification, as this chondroprogenitor cell line can recapitulate chondrogenic maturation in both monolayer (2D) and pellet (3D) cultures on an accelerated timescale(23-25). After expansion, cells were transduced with the vectors described above. Chondrogenic behavior of these cells and their mineralization in response to exogenous phosphate treatment were carefully characterized in a previous study, which also describes the detailed chondrogenic culture protocol used in this study(26). Briefly, chondrogenesis in 2D cultures of transduced or wild-type cells was initiated by addition of 1% ITS+ Premix (Corning) and 50 μg/ml L-ascorbic acid-2-phosphate upon 100% confluence. In 3D cultures, cell pellets containing 2.5×105 cells were cultured in round-bottomed polypropylene 96-well-plates and media was changed every 2 days for 35 days.
Adult human bone marrow-derived MSCs (hMSCs) were plated at a density of 4,000 cells/cm2 and cultured in media containing (glucose DMEM + 10% FBS (Gibco qualified, lot# 1805387) + 1% antibiotic and antimycotic + 10ng/ml FGF-2 (Shenandoah, Cat# 100-146)). At ~80% confluency, cells were detached from plates with 0.25% trypsin/EDTA and subcultured. Experiments were performed using cells that underwent 12 population doublings (PD) as calculated from the formula PD = 3.32[log (final cell #) − log (0.2x106)]. Pellets of transduced or wild-type cells were formed and cultured for either 21 or 28 days in chondrogenic media containing (DMEM, 4.5 g Glucose/L), 1% (v/v) Antibiotic-Antimycotic, 1% (v/v) ITS+ Premix (corning, cat# 354350), 1% Non-essential Amino Acids (Gibco, 11-140-050), 40 μg/ml L-proline (Sigma cat# P-0380), 50 μg/ml Ascorbic Acid 2-Phosphate, 0.1 μM Dexamethasone, 10 ng/ml TGFβ1 (Shenandoah, cat# 100-39). To induce hypertrophy, pellets were treated with 10nM triiodothyronine (T3) and 10mM beta glycerophosphate (βGP) starting on day 14 of chondrogenic culture, as previously described(27). For the inflammation studies, cell pellets were pre-cultured in chondrogenic media for 21 days. On day 21, the chondrogenic culture media was supplemented with 0.1ng/ml IL-1β (Shenandoah cat#100-167) in absence of TGFβ and dexamethasone to provide inflammatory stimulus.
Lentiviral Vector Design and Synthesis
Doxycycline Inducible shRunx2 Vector
The inducible system that drives RUNX2 knockdown was modified from the pINDUCER lentiviral toolkit originally developed by Meerbrey et al.(22). pINDUCER13 was acquired from Addgene (plasmid # 46936) for further modification as such a lentiviral vector conveniently contains a Tet-on inducible cassette that co-expresses luciferase and shRNA sequence as well as a cDNA sequence of a constitutively active puromycin resistance gene (Fig 3a). Three shRNA sequences targeting murine/human RUNX2 from RNAi Codex(28) were screened for RUNX2 knockdown via western blotting and shRunx2 sequence #294717 (5'–TGCTGTTGACAGT-GAGCGCCGAATGGCAGCACGCTATTAATAGTGAAGCCACAGATGTATTAATAGCGT-GCTGCCATTCGATGCCTACTGCCTCGGA–3') was further selected for downstream vector synthesis.
Figure 3. Construction of the TetOn inducible shRunx2 vectors and autoregulatory cisCXp-shRunx2 gene circuits.

(a) The shRunx2 sequence #294717 was inserted into the pINDUCER vector to produce the inducible TetOn-Luc-shRunx2 vector. (b) The inducible promoter was replaced with an engineered promoter containing 1-, 2-, or 3 copies of the 150bp Runx2 cis-enhancer upstream of Col10a1 basal promoter, creating the ncisCXp-shRunx2 gene circuits. This gene circuit creates a synthetic intracellular feedback loop that drives expression of the short hairpin RNA in response to Runx2 binding to the engineered promoter. The activity of the gene circuit is monitored using a luciferase reporter. TRE2, tetracycline response element 2 promoter; Luc, luciferase; Ubc, Ubiquitin C promoter; rtTA3, reverse tetracycline-controlled transactivator 3; Puro, puromycin resistance sequence. Diagram of pINDUCER13 is adapted from Meerbrey et al19.
TetOn-Luc-shRunx2 inducible vectors were created by sub-cloning PCR-amplified shRunx2 #294717 into TetOn-inducible cassette of pINDUCER13 via XhoI/EcoRI sites. In addition, a vector containing a scrambled shRunx2 sequence (TetOn-Luc-scramble) was synthesized to serve as experimental controls (Table S1). Polyclonal populations were established by combining selected cells from two independent transduction experiments (n=3 technical replicates from two viral batches) to evaluate the reproducibility of the system. Expression of short hairpin sequences was induced by the addition of (0.5μg/ml) of doxycycline (Dox) beginning on the day indicated and continued to the end of culture.
Autoregulatory cisCXp-shRunx2 gene circuit
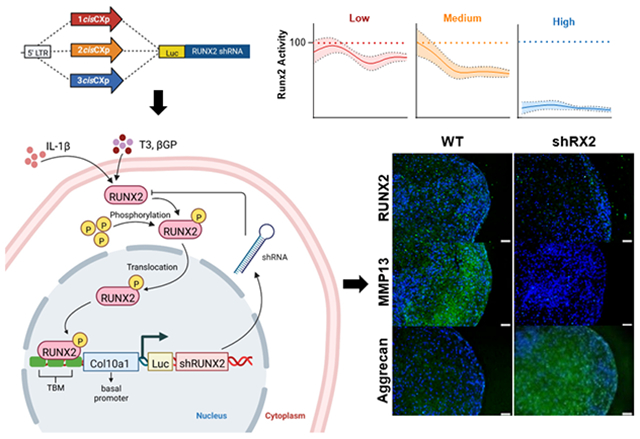
The autoregulatory cisCXp-shRunx2 gene circuits rely on a Col10a1-like promoter that was first reported by Zheng et al.(29) to drive RUNX2 knockdown via expression of the shRunx2 sequence. Zheng et al.(29) demonstrated that the Runx2-driven transcriptional activity of the Col10a1 basal (−220 to 110 bp) promoter can be regulated by varying the number of Runx2 binding sites (cis enhancers; −4296 to −4147 bp) incorporated upstream of the promoter. Therefore, we first designed the 1cisCXp promoter which consists of one cis enhancer upstream of the Col10a1 basal promoter and then increased the number of cis enhancers incorporated into the promoter (Fig. 3b). All sequences (1cisCXp, 2cisCXp, and 3cisCXp) were acquired from IDT technology. To create the 1cisCXp-shRunx2 vector, the 1cisCXp promoter was PCR amplified using corresponding primers (Table S2) created so that both 5'- and 3'-ends of the reaction products have an overlap of >30 nucleotides with the backbone of TetOn-Luc-shRunx2 vector after the TRE2 promoter was removed by restriction digestion with NheI/AgeI. The linearized Luc-shRunx2 vector and 1cisCXp promoter were then assembled using the Gibson Assembly Kit (NEB) according to the manufacturer’s instructions. A conceptual schematic of the gene circuits’ function is shown in Fig. 4.
Figure 4. Schematic of the intracellular synthetic negative-feedback regulation of RUNX2.

Maturation signals like T3, βGP and IL-1β upregulate RUNX2 expression which binds to the transcription factor binding motifs (TBM) in the gene circuit to induce the expression of shRUNX2, thereby establishing a negative-feedback loop that suppresses RUNX2 activity, allowing MdChs to resist maturation during neo-cartilage formation.
To create 2cisCXp-shRunx2, PCR amplification of both the cis-enhancer and 1cisCXp promoter was performed using the corresponding primers (Table S3). Each primer was designed with an overlap of >30 nucleotides between the reaction products to support the sequential assembly of linearized Luc-shRunx2 vector, cis enhancer, and 1cisCXp. Similarly, 3cisCXp-shRunx2 vectors were assembled from the three components described above as well as an additional PCR product that inserts an extra copy of cis enhancer in between the first cis enhancer and 1cisCXp promoter. Correct cloning was confirmed by Sanger sequencing at University of Michigan Sequencing core.
Lentiviral Transduction and Stable Selection
To establish ATDC5 cell lines that express the vectors of interest, lentiviral supernatants of the chosen plasmids were produced by the University of Michigan Vector Core. Twenty-four hours prior to transduction, cells were plated in individual wells of a 6-well plate at a density of 10,000 cell/cm2. Proliferating cells were transduced at multiplicity of infection (MOI) = 1 with lentiviral supernatant of the TetOn-shRunx2, the ncisCXp-Luc-shRunx2, or their corresponding scramble control gene circuits in the absence of serum for 48 hours. Polyclonal populations of cells stably expressing the chosen vectors were subsequently selected with continuous treatment of puromycin (2μg/ml) for 10-14 days for ATDC5 cells.
For human MSCs (hMSCs), 10,000 cells/cm2 were plated in individual wells of 6-well plate and transduced at MOI = 5 with lentiviral supernatant of cisCXp-Luc-shRUNX2 gene circuits for 24 hours. Cells stably expressing the vectors were selected by treatment with 1μg/ml puromycin for 4 days.
For both cell types, cells were subcultured for one additional passage prior to each experiment as described above.
Luciferase Assay
Luciferase activity of 2D and 3D cultures in multi-well plates was measured once daily beginning on day -1 or 0. Thirty minutes prior to each measurement, concentrated D-luciferin stock (1mg/ml) was added to existing culture media to achieve a final concentration of 150 μg/mL and gently mixed. Cultures containing luciferin were then incubated at 37°C for 30 minutes before measured using SYNERGY H1 microplate reader (BioTek). Each sample was measured three times (1 second/read) and the means were normalized to the day -1 values and expressed as relative luminescence units (RLU). Media was replaced in measured cultures immediately after reading.
Biochemical Assays
To assess the accumulation of cartilage-specific matrix in both 2D and 3D cultures, the sGAG levels were quantified using the DMMB assay, as previously described(26, 30). Specifically, differentiated cell masses from 2D cultures or pellets from 3D cultures were washed with ice-cold PBS before being digested with 1mg/ml proteinase K in 200-500μl ammonium acetate (0.1M) at 50°C for 16 hours. sGAG content in the digested lysate of each sample was subsequently determined by comparing its DMMB reading to a standard curve. The DNA content of each sample was measured using Hoechst 33258 dye (Sigma) to normalize its corresponding sGAG content(31).
Gene Expression
Analysis of gene expression was performed as previously described(26, 30). Total RNA of both 2D and 3D cultures were extracted using TRI Reagent® RT (Molecular Research Center). Cell masses from each well of 12-well plate were collected as one 2D sample. Four pellets were combined to generate 1 3D sample. Extracted RNA were reverse-transcribed into cDNA using High-Capacity cDNA Reverse Transcription Kit (Life Technologies). Based on previous reports on the instability of references genes in tissue engineered products(32), the average value for 2 reference genes (Hprt and Ppia) were used to normalize the expression levels of target genes. The efficiency of the primers was verified to be >85% for all genes. Relative expression levels were calculated as , in which , at the time-point of interest, at day 0, and is the average value of two reference genes. The forward and reverse primer sequences for all genes are listed in Table S2.
Histology
Monolayer cultures were washed twice with PBS and fixed in 70% ethanol at room temperature for one hour. Fixed pellets were washed with 70% ethanol, embedded in paraffin wax, and then 7 μm sections were taken. Sections were deparaffinized in xylene, rehydrated in increasing dilutions of ethanol. Both 2D cultures and 3D sections were stained with Alcian blue (1% in 3% Acetic Acid, Poly Scientific) for 30 minutes, counter-stained with Fast Nuclear Red, and then imaged using bright field microscopy.
Immunohistochemistry
The pellet sections were deparaffinized in xylene and rehydrated in successive dilutions of ethanol. Slides were then blocked in 3% H2O2 for 10 minutes. This was followed by Antigen retrieval with heated Retrievagen A (BD Biosciences, Cat# 550524) for 20 minutes. Once cooled to room temperature, the samples were then permeabilized using 0.5% TBS-TX100, blocked with 10% Goat serum, 1% BSA in 0.1% TBST for 1 hour at room temperature, and then incubated overnight in either RUNX2 (ABclonal Cat# A2851; 1:100 dilution), COLX (ABclonal Cat# A6889; 1:100 dilution), MMP13 (ABclonal Cat# A16920; 1:200 dilution) or ACAN (ABclonal Cat# A8536; 1:500 dilution) primary antibody solution in 10% Goat serum in 0.1% TBS-TX100 at 4°C. After washing, samples were incubated with Goat Anti-Rabbit IgG (H+L) Alexa Fluor 488 secondary antibody (Invitrogen, Cat#A11034; 1:500 dilution in 10% Goat serum in 0.1% TBS-TX100) followed by 300nM DAPI solution. Lastly, the slides were cover-slipped with anti-fade mounting media.
Immunoblotting
Whole cell extracts of 2D cultures from one well of a 6-well plate were prepared using RIPA Lysis and Extraction Buffer (Thermo Scientific) supplemented with protease inhibitor cocktail (Sigma) using the Micro Tube Homogenizer (Thermo Scientific). Homogenized lysate was rotated at 4 °C for one hour and centrifuged at 12,000g for 10 minutes to remove cellular debris. Total protein content within each sample was determined with the Pierce BCA Protein Assay Kit (Pierce). Proteins (5-15 μg) were separated on a 10% NuPAGE Bis-Tris Protein Gel and then transferred to a polyvinylidene difluoride membrane (Millipore). Membranes were blocked with 5% BSA made up in Tris-buffered saline-Tween 20 (TBS-T, 0.1% Tween 20) for 1 hour at room temperature. Following blocking, membranes were incubated in TBS-T, 0.1% Tween 20 and 5% BSA overnight at 4 °C with anti-Runx2 antibody (RUNX2 D1H7 Rabbit mAb, Cell Signaling Technology, 84866 1:2000) and anti-β-actin antibody (Rabbit Anti-beta Actin, Abcam, ab119716, 1:5000). Secondary incubation was performed at room temperature for 1 hour using WesternSure® Goat anti-Rabbit HRP (LiCor, 926-80011 1:20000). Positive staining was visualized using the LiCor WesternSure® PREMIUM Chemiluminescent Substrate and quantified using the LiCor Image Studio. Densitometry was quantified using Licor software. To control for the loading of total protein into the gel, RUNX2 protein content was normalized by β-actin.
Statistics
Statistical analyses were performed in GraphPad Prism version 10 for Windows (GraphPad Software, La Jolla California USA) using one or two-way analysis of variance followed by Tukey’s post-hoc test. P-values less than 0.05 were considered statistically significant. All plots represent the mean ± the standard deviation.
RESULTS
Part 1: Assessment of the Inducible shRunx2 System
Constitutively Silencing of RUNX2 Inhibits Chondrogenesis
To examine the effect of constitutive RUNX2 silencing on in vitro differentiation of chondroprogenitors, we induced chondrogenesis in monolayer cultures of ATDC5 cells stably expressing TetOn-shRunx2 (shRunx2 cells) in presence of doxycycline (Dox) and evaluated sGAG accumulation and expression levels of chondrogenic markers after 14 days. In cultures treated with Dox, RUNX2 protein was depleted in shRunx2 cells by day 7 and remained downregulated through day 14 (Figs. 5a and S6). There was also a significant decrease in sGAG accumulation by shRunx2 cells by day 14, while the scramble controls had successfully formed Alcian blue-positive nodules (Figs. 5b and c). The expression of mRNA for Acan and Col2a1 was also significantly inhibited with Dox treatment of shRunx2 cells (Fig. 5d), suggesting that constitutive RUNX2 suppression interfered with early chondrogenesis.
Figure 5. Effects of constitutive RUNX2 silencing on early chondrogenesis.

ATDC5 cells expressing the TetOn-Luc-shRunx2 (shRx2) or the TetOn-Luc-scrambled (Scr) gene circuits were differentiated in the presence (+Dox) or absence (−Dox) of 0.5 μg/ml doxycycline for 14 days. (a) Western blot analysis of RUNX2 and β-actin at day 7 and 14 demonstrating silencing of Runx2. Alcian blue staining, (b) and % sGAG accumulation relative to samples at 7 days without Dox (c) at day 14. (d) Fold change in expression of aggrecan and collagen type II genes on day 14. Polyclonal populations were established by combining selected cells from two independent transduction experiments (two viral batches, n=3). All data represented as mean ± SD. Significance values are indicated in the plots. Scale bar: 200μm.
Stage-dependent Effect of RUNX2 Silencing on Chondrogenesis
RUNX2 suppression in chondroprogenitors as they mature through the stages of endochondral ossification was examined by adding Dox continuously starting on the following time-points of culture that were found to correlate with specific stages of chondrocyte maturation, as determined in preliminary studies (Fig. 6c and Fig. S1): minimum RUNX2 protein expression (D4), just prior to rapid increase in RUNX2 protein expression (D7), peak gene expression of Col10a1 (D14)(28), and the peak of RUNX2 protein expression (D21). These were compared to cultures that received no Dox or were treated with Dox from day 0 (D0). By day 28, Dox induction in monolayer suppressed RUNX2 protein levels by 20-60% in all cultures containing the TetOn-Luc-shRunx2 gene circuits compared to controls that did not receive Dox (Fig. 6c). This effect depended on the time-point at which Dox treatment, and therefore gene circuit activation, started. At this point, the expression of RUNX2 protein in the TetOn-Luc-scrambled gene circuits was ~97% of controls that did not receive Dox. When RUNX2 suppression was induced from Day 4, 7, and 14, sGAG accumulation was increased by 1.9-fold, 2.14-fold, and 1.75-fold respectively on day 28 relative to scramble controls and the no Dox group (Fig. 6d). There was no difference between the scramble and no Dox groups.
Figure 6. Effects of RUNX2 silencing throughout chondrogenesis.

(a) The time-course of cell-state transitions and relative Runx2 protein expression/activity in monolayer cultures of naïve ATDC5 cells as determined in preliminary experiments. These are also the points at which Runx2 suppression was induced in TetOn-Luc-shRunx2 (shRx2)- or TetOn-Luc-scrambled (Scr)-expressing ATDC5 cells with the addition of doxycycline (Dox). (b) Runx2 protein band intensity normalized by β-actin during ATDC5 chondrogenesis. The plot shows the percent increase in protein expression at each time-point indicated compared to just prior to induction of chondrogenesis (day −1) during normal chondrogenic differentiation of ATCD5 cells. (c) Runx2 protein expression in ATDC5 cells containing either the TetOn-shRunx2 or TetOn-scrambled vectors in response to Dox treatment starting on Day 0, 4, 7, 14, or 21 of chondrogenic differentiation relative to the No Dox controls. (d) Fold change in sGAG accumulation normalized to DNA content after 14 and 28 days in culture. The data is presented relative to D14 No Dox group as mean ± SD. Significant difference between TetOn shRunx2 and scrambled groups at each time point is indicated in the plots. (e) The effects of delayed RUNX2 silencing on gene expression of chondrogenic markers and matrix degrading enzymes are shown. Quantification of mRNA expression of Acn, Col2a1, Col10a1, Adamts4, Adamts5, and Mmp13 at day 14 and 28 in cells with inducible expression of shRUNX2 beginning on D4 or D7. Polyclonal populations were established by combining selected cells from two independent transduction experiments (two viral batches, n = 3 technical replicates). Data represented as mean ± SD. Significant differences between day 14 and day 28 are as indicated in the plots.
We next examined the effect of Runx2 suppression beginning on D4 or D7 on genes associated with chondrocyte hypertrophy, ECM production, and ECM turnover (Fig 6e). Runx2 suppression reduced expression of the hypertrophy marker Col10a1 by 2-fold at 14 days of culture. This level was maintained through day 28. Expression of Mmp13, another significant marker of hypertrophy, was downregulated 5-20-fold by Runx2 suppression independent of when Dox treatment began. When RUNX2 was suppressed beginning on day 4 or day 7, gene expression of the chondrogenic ECM markers aggrecan and collagen II were not significantly reduced compared to controls that did not receive Dox. Runx2 suppression beginning at day 4 had no effect on Adamts5 gene expression while delaying suppression until day 7 lead to increased expression of this metalloproteinase. Suppression beginning at either time point had no effect on Adamts4 expression.
This inducible study indicates that suppression of RUNX2 at specific points of chondrocyte maturation has a differential impact on sGAG accrual of cartilage-specific structural macromolecules and regulation of metalloproteinases expression. Therefore, persistent and broad-spectrum silencing of RUNX2 could have deleterious effects on chondrogenesis and sGAG production in heterogeneous populations of chondroprogenitors at various stages of maturation. This makes it difficult to use inducible silencing to optimally target its suppression. Next, we engineered an autoregulatory genetic modification that would allow the cells to independently regulate their response to exogenous cues that raise intracellular RUNX2 levels.
Part 2: Design and Assessment of the Autoregulatory shRunx2 Gene Circuit
Engineering of a Synthetic Col10a1-like Promoter
To engineer a RUNX2-responsive promoter that is specific to chondrogenic cells, we assembled the 150bp RUNX2-binding cis-enhancer of the Col10a1 promoter upstream of a truncated basal sequence from the same promoter (ncisCXp; Fig. 3). To demonstrate that this promoter is only active in chondrogenic cells undergoing hypertrophic maturation, a promoter containing 1 cis enhancer (1cisCXp) was used to drive the expression of luciferase or enhanced green fluorescent protein (eGFP; Fig. 7a). Luciferase activity mirrored the time-course of Col10a1 gene expression (Fig. 7b) in chondrogenic ATDC5 cells: minimal until day 6 followed by a rapid increase to maximum level at day 10 and sustained thereafter (Fig. 7c). eGFP fluorescence was only seen in cartilaginous nodules starting on day 7 (Fig. 7d). Little eGFP fluorescence was observed in cells that were not chondrogenically induced or ones that had not yet formed cartilaginous nodules. Western blot analysis on day 14 showed that eGFP protein was solely expressed in cells that were exposed to chondrogenic stimuli (Figs. 7e and S8). Taken together, these data illustrate that the 1cisCXp promoter has phenotype-specificity, that its transcriptional activity is limited to differentiated ATDC5 cells, and that activity of the gene circuit increases as these cells transition to the hypertrophic phenotype.
Figure 7. Specificity of the gene circuit.

(a) Diagrams of 1cisCXp-Luc and 1cisCXp-eGFP vectors used to evaluate the activity and chondrogenic specificity, respectively, of the engineered Col10a1-like promoter. (b) Quantification of mRNA expression of Col10a1 in non-transduced ATDC5 monolayer cultures over 21 days of chondrogenic differentiation (two independent experiments, n ≥ 5). (c) Luciferase activity from ATDC5 cultures expressing 1cis-Luc over 21 days of differentiation relative to day -1 (three independent experiments, n = 6). (d) Bright field and fluorescent imaging of ATDC5 cells transduced with 1cisCXp-eGFP showing that GFP is localized to cartilage nodules of chondrogenic cultures at the onset of Col10a1 mRNA upregulation (day 7), demonstrating the specificity of the gene circuit activity to chondrogenic cultures. (e) Western blot of eGFP and β-actin (internal control) levels after 14-day chondrogenic cultures in non-transduced ATDC5 cells compared to cells transduced with 1cisCXp-eGFP, also demonstrating specificity of the gene circuit activity. Scale bar: 200μm. eGFP – enhanced green fluorescent protein; RLU – relative luminescence units.
Col10a1-like promoter can be engineered to tune the activity of RUNX2 during chondrogenesis
To determine whether the Col10a1-like promoter could be used to provide self-regulated silencing of RUNX2 and to test the hypothesis that the level of RUNX2 suppression could be tuned by varying the number of cis-enhancers, we monitored the activity of cisCXp-shRunx2 gene circuits during the chondrogenesis of polyclonal ATDC5 cell populations stably expressing these vectors (Fig. 3). During the 21-day monolayer chondrogenic differentiation assay, luciferase activity was used as a surrogate measurement of promoter activation. Since the scrambled control gene circuits cannot suppression Runx2 activity, the difference in luminescence between cells containing cisCXp-shRunx2 gene circuits and those containing cisCXp-scrambled gene circuits is indicative of the level of intracellular Runx2 suppression.
Luminescence was minimal in cells expressing the 1cis-shRunx2 vector during the first 6 days, similar to cells transduced with 1cis-scramble vector. From day 7, shRunx2 containing cells exhibited a significantly lower level of luminescence than the scramble controls (Fig. 8a and b). While the total luminescence from both groups fluctuated throughout further differentiation, a stable relative decrease in Runx2 activity of 18.1 ± 5.1% compared to scrambled controls was reached under the 1cis-shRunx2 vector. Similarly, gene circuits containing the 2cisCXp promoter exhibited the same level of minimal activity as 1cisCXp during the first three days of chondrogenic differentiation. Thereafter, the 2cisCXp promoter drove the total gene circuit activity in 2cis-shRunx2 cultures to equilibrate at a level that was 30.4 ± 4.2% lower than the scrambled controls. The addition of a third cis-enhancer exhibited a more prominent reduction (77.5 ± 2.6%) in gene circuit activity starting at the onset of chondrogenesis. Gene expression of Col10a1 was decreased (Fig. 8c) and sGAG content increased (Fig. 8d) in the 3cis-shRunx2 expressing cultures at D14. Alcian blue stained sections of ATDC5 pellet cultures showed an increase in the staining area of cartilage nodules. RUNX2 protein expression decreased with increasing numbers of cis-enhancers compared to the scramble control (Fig. 8e).
Figure 8. Activity of RUNX2 Suppressing Gene Circuits and Their Effect on Chondrogenesis.

(a) Luminescence measured over 21-day chondrogenic cultures expressing 1cisCXp-shRunx2/scramble, 2cisCXp-shRunx2/scramble, 3cisCXp-shRunx2/scramble as a measure of RUNX2 and gene circuit activity in ATDC5 cells. (b) Relative activity of each gene circuit was calculated by normalizing to activity of corresponding scramble controls at each time point. (c) Quantification of mRNA expression (relative to day 0 and the housekeeping genes Hprt and Ppia) of the RUNX2 target gene, Col10a1, at day 14. (d) Fold change in sGAG accumulation normalized to DNA content at days 14 and 28. (e) Staining for sGAG and Runx2 in ATDC5 pellet cultures after 28 days of culture. Data represented as mean ± SD (n=6 technical replicates from two independent transduction experiments). Scale bar: 200μm. RLU – relative luminescence units.
RUNX2 suppressing gene circuits attenuate hypertrophy in MSC-derived chondrocytes
Once we verified the functionality of the gene circuit in the ATDC5 cell line, we wanted to verify its functionality during cartilage formation during chondrogenesis and neo-cartilage formation of human mesenchymal stem cells (hMSCs). To challenge the gene circuit, cells were treated with T3 and β-GP—stimuli known to induce RUNX2 activity and hypertrophy in primary and hMSC-derived chondrocytes (hMdChs). Alcian blue-positive sGAG was observed in all pellets created from hMdChs expressing the 1cisCXp-shRunx2 and 31cisCXp-shRunx2 gene circuits after 28-days in chondrogenic culture. The peripheral regions of the pellets of the modified cells had more intense Alcian blue staining, accompanied by less hypertrophy-like cell morphology compared to MdChs derived from WT hMSCs (Fig S3). In response to hypertrophy-inducing stimuli, hMdChs reprogrammed with cisCXp-shRunx2 gene circuits accumulated more sGAGs compared to both WT and scramble controls (Fig. 9). The 1- and 3cisCXp-shRunx2 gene circuits also suppressed mineral deposition under hypertrophic conditions (Fig. 9b). Furthermore, RUNX2 protein expression and that of its target genes, MMP13 and collagen X were downregulated and aggrecan upregulated by both the 1- and 3cisCXp-shRunx2 gene circuits (Fig. S5). Taken together, this data indicates that RUNX2 suppressing gene circuits are successfully able to prevent hypertrophic maturation of hMdChs and increase their sGAG accumulation in vitro.
Figure 9. RUNX2 Suppressing Gene Circuits attenuate hypertrophy in human MSC-derived chondrocytes.

(a) sGAG accumulation by hMdChs at day 21 after 7 days of hypertrophic induction with T3 and β-glycerophosphate (n ≤ 4 technical replicates). (b) Alcian blue staining for sGAG and alizarin red staining for calcium deposition in untreated and T3 + βGP treated hMdCh WT and reprogrammed cells. Scale bar: 50μm.
RUNX2 suppressing gene circuits mitigate IL-1β-induced sGAG degradation in hMdChs
RUNX2 activity is also upregulated in hMdChs in response to inflammatory stimulus(33-35). Therefore, we evaluated the effect of RUNX2 suppression on hMdChs response to IL-1β treatment using the 3cis-shRUNX2 gene circuit to induce maximum RUNX2 suppression in hMdChs. Within 6 hours of the addition of IL-1β to hMdCh cultures, the levels of luminescence, and thus gene circuit activity, were significantly elevated in 3cis-scramble pellets and remained unchanged in 3cis-shRUNX2 pellets (Fig. 10a). There was higher retention of sGAG in 3cis-shRUNX2 modified hMdCh pellets compared to WT and scrambled controls (Fig. 10b). Immunofluorescence analysis revealed lower expression of hypertrophic proteins RUNX2, COLX and MMP13 as well as elevated expression of aggrecan and more intense sGAG staining in 3cis-shRUNX2 modified hMdCh pellets (Fig. 10c). Taken together, these results show that RUNX2 suppression in hMdCh pellets partially rescues hMdChs from IL-1β induced sGAG degradation, potentially by reducing the expression of matrix degrading enzymes like MMP13 that are downstream targets of RUNX2 activity.
Figure 10. Effect of RUNX2-suppressing gene circuits on MdCh response to IL-1β.

(a) Luminescence measured in pellet cultures of hMSC expressing 3cisCXp-shRunx2/scramble, after 6 hours of 0.1ng/ml IL-1β treatment (n=3 technical replicates). (b) sGAG accumulation normalized to DNA content and (c) Alcian blue staining for sGAG and immunofluorescence staining for hypertrophic (RUNX2, COLX, MMP13) and chondrogenic marker (ACAN) in WT and 3cisCXp-shRunx2/scramble MdCh pellets treated with 0.1ng/ml IL-1β. Scale bar 50μm. RLU – relative luminescence units.
DISCUSSION
Here, we demonstrate that the loss of RUNX2 function during chondrogenesis of mesenchymal progenitors can elicit distinct cellular responses at different stages of differentiation. Using a 2D ATDC5 model with inducible shRunx2 expression, we show that RNAi of Runx2 in the undifferentiated cells inhibits mesenchymal proliferation and differentiation into chondrocytes. However, induced RNAi of Runx2 after the pre-chondrogenic proliferation phase or early chondrogenesis enhances the accumulation of cartilaginous matrix structural macromolecules. We further demonstrate that the synthetic Col10a1-like promoters can initiate RNAi of Runx2 in chondrocytes that are transitioning to the pre-hypertrophic phenotype in response to the increasing intracellular RUNX2 activity without interfering with early chondrogenesis. The induced loss of RUNX2 function in turn negatively regulates the activity of the cis-shRunx2 gene circuit to resist upregulation of RUNX2 during hypertrophy and maturation-associated matrix degradation. Together, our findings highlighted three key features of the cis-shRunx2 gene circuit: 1) phenotype-specific activation of RNAi, 2) closed-looped, intracellular negative-feedback regulation of RUNX2 activity, and 3) tunable levels of RUNX2 repression. We show that these features of the gene circuits can enhance accumulation of cartilaginous matrix by MdChs during neo-cartilage formation under conditions that drive loss of cartilage tissue functionality in the injured joint, namely hypertrophy and inflammation. Overall, we demonstrate the application of synthetic biology tools like synthetic gene circuits to improve cartilage tissue engineering outcomes.
RUNX2 is essential to chondrogenesis and plays different roles at different stages.
Unlike the well-established role of RUNX2 in driving chondrocyte maturation, its involvement in early events of chondrogenesis remains unclear. Progenitor cells within the pre-chondrogenic condensation first proliferate before committing to chondrogenic differentiation(36-38) and Runx2 expression has been detected within these condensations(13-15). Recently, Dexheimer et al. showed that this transient phase of proliferation is required for in vitro chondrogenesis of human MSCs after cells are condensed into a micromass pellet(39, 40). In our study, when chondrogenesis is induced in confluent monolayer ATDC5 cultures, differentiation initiates with a similar phase of proliferation that overrides contact inhibition during the first 4 days, which does not occur under the RNAi of Runx2 using the inducible system (Fig S2). In accordance with our observations, Akiyama et al. also showed that introduction of the dominant negative form of RUNX2 in ATDC5 cells inhibits cellular condensation and subsequent chondrogenic differentiation(41). Similar suppression of chondrogenesis is also observed when Zfp521, an inhibitor of RUNX2, is overexpressed in ADTC5 cells(40). However, it is worth noting that Runx2−/− mice do form cartilaginous skeleton(3, 42). Nonetheless, prior in vitro studies and ours show that constitutively active RNAi of Runx2 is not suitable cartilage tissue engineering possibly due to the redundant functions of other RUNX proteins and the complexity of the in vivo system compared to cells in isolation under static, in vitro cultures.
As chondrogenesis proceeds, the role of RUNX2 changes(43). Delayed RUNX2 silencing in chondroprogenitors after the pre-chondrogenic proliferation phase does not interfere with their further chondrogenic progression; instead, it increases the amount of sGAG accumulated by these differentiated chondrocytes and decreases the gene expression of MMP13. After pre-chondrogenic proliferation in vivo, Nkx3.2-mediated repression of Runx2 promotes early chondrogenesis by activating Sox9(44). These findings are analogous to our observation of the transient upregulation of RUNX2 prior to the elevated levels of Col2a1 and Acan expression (Fig S1). They also likely explain why loss of RUNX2 function after day 4 no longer blocks chondrogenesis in ATDC5 cells. As in growth plate chondrocytes(45), the low protein expression of RUNX2 is not permanent in the ATDC5 model. Quickly following the upregulation of Col2a1 and Acan, protein expression of RUNX2 rose and drove differentiated chondrocytes to the pre-hypertrophic and then hypertrophic phenotypes. As expected, induced RUNX2 silencing at this stage of chondrocyte maturation decreased mRNA levels of both Col10a1 and Mmp13. Since both aggrecan and type II collagen are degradation targets of MMP13(46-48), this outcome is consistent with our observation that RUNX2 silencing leads to an increased level of sGAG accumulation under hypertrophic as well as inflammatory environments.
An important observation from the inducible study is that RNAi of Runx2 in chondrocytes that are transitioning to the pre-hypertrophic phenotype maximizes the accumulation of sGAG since it is a net function of production and turnover. Therefore, maximizing the accumulation of cartilaginous matrix via RNAi of Runx2 requires balancing the inhibition of degradation and increasing ECM production by optimizing the timing and dosing of shRunx2 expression. While RUNX2 silencing is desirable for reducing MMP13-mediated matrix degradation, we also noticed that it also downregulated the gene expression of Acan and Col2a1 to varying levels. While the relationship RUNX2 activity and transcription of Acan and Col2a1 remains to be clarified, RUNX2, together with RUNX1, has been shown to induce the expression of Sox5 and Sox6, which further control the induction of Col2a1(49). Additionally, RUNX2 is a common target of TGF-β1 and BMP-2, both of which are frequently used to induce chondrogenesis of MSCs(50). These studies, together with our results, suggest that RUNX2 activity may also contribute to the production of aggrecan and type II collagen. Although we cannot easily decouple the regulation of early chondrogenic markers and matrix-degrading enzymes by RUNX2, the differential expression profiles of early and late chondrogenic markers allow us to optimize the temporal activation of shRunx2 expression to maximize the accumulation of matrix during chondrogenesis. Identifying such an optimal time is critical because the premature loss of RUNX2 function reduces the production of aggrecan and type II collagen while the belated silencing permits uncontrolled matrix degradation.
The cisCXp-shRunx2 gene circuit is designed to enable autonomous RUNX2 suppression in heterogeneous stem cell population in preparation for unknown environmental cues.
The cisCXp-shRunx2 gene circuit relies on its synthetic Col10a1-like promoter to induce hypertrophy-specific RUNX2 silencing. The specific expression of Col10a1 in pre-hypertrophic and hypertrophic chondrocytes requires the binding of RUNX2 in addition to the recruitment of the general transcription factors near the transcription start site(29, 51, 52), which usually reside within the basal region of mammalian polymerase II promoters. The 330-bp Col10a1 basal promoter we incorporated contains a highly conserved sequence that precisely describes the transcription start site of Col10a1 across species(29), providing the DNA template that supports the assembly of the RNA polymerase II transcription initiation complex. Meanwhile, the two putative tandem-repeat within the 150-bp cis-enhancer ensures direct binding of RUNX2(29, 53). As a result of the cooperative actions of these two regulatory elements, all three versions of cis promoter are sufficient to direct hypertrophy-specific transcription resembling the endogenous Col10a1 promoter in differentiating chondrocytes. Critically, the suppression of early chondrogenesis can be avoided as the loss of RUNX2 function does not occur until progenitors fully differentiate into chondrocytes and transition to pre-hypertrophy.
The cisCXp-shRunx2 gene circuits establish closed-loop intracellular negative-feedback regulation of RUNX2, allowing differentiating chondrocytes to dynamically resist maturation in response to exogenous cues.
The negative feedback motif of cisCXp-shRunx2 utilizes RUNX2 activity as the central signal. In chondrocytes that are transitioning to hypertrophy, the cisCXp promoter initiates the production of shRunx2 that downregulates RUNX2, which in turn decreases the transcriptional activity of the cisCXp promoter. The negative feedback regulation of chondrocyte maturation also occurs naturally, often limited by the range of paracrine signaling (e.g., PTHrP/IHH feedback loop)(54-56). Free from the dependence on paracrine signaling, cisCXp-shRunx2 gene circuits allow chondrocytes to resist maturation based their internal tendency to undergo hypertrophy, measured by RUNX2 activity. As a gatekeeper, RUNX2 mediates the signaling of many molecular and biophysical cues in chondrocytes(5, 57, 58). Therefore, by inputting the intracellular RUNX2 activity as the signal for the negative-feedback regulation, cells expressing cisCXp-shRunx2 can dynamically adjust the level of silencing required to maintain their steady states (levels of RUNX2 suppression) without needing to address the specific pathways underlying hypertrophy stimuli, such as thyroid hormone T3 and beta glycerophosphate or inflammatory factors like IL-1β (Fig. 4). Finally, design of cisCXp promoter allows cisCXp-shRunx2 gene circuits to provide a tunable level of RUNX2 suppression. We observed that increasing the number of cis-enhancers in the gene circuit progressively enhanced RUNX2 silencing from 18% to nearly 80%. Increasing the number of cis-enhancers also led to faster equilibrium of the activity of the gene circuits.
CONCLUSION
Overall, our circuit design is modular with a straightforward construction, which allows for simple testing of various combinations of circuit components and outputs. This flexible design enables circuits to be optimized for a variety of environmental conditions. Our results demonstrate that this approach could be a viable alternative to strategies that use broad spectrum spatial/temporal regulation of exogenous cues or constitutive or transient silencing of intracellular messengers. However, the behavior of cisCXp-shRunx2 or similar gene circuits need to be further investigated in long-term in vitro and in vivo studies. Future studies in the gene circuit design should focus on fine tuning promoter activity to specific cell states, reducing off target effects, and targeting transcriptional and translational regulators of cell function.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Ciara Davis, John Braford, Meghan Burns, Hannah Floyd, Samad Emory, and Sunny Karnan for their assistance in analysis of the data and Colleen Flanagan for her editorial assistance. Supported by the ANRF Award No. 310 047420, Orthopaedic Research and Education Foundation Award No. 651100, and NIH-NIAMS Award No. 1R21AR07401101. Human MSCs were provided by the Case Western Reserve University Center for Multimodal Evaluation of Engineered Cartilage – NIH-NIBIB 1P41EB021911.
Footnotes
COMPETING INTERESTS
The authors declare no Competing Financial or Non-Financial Interests.
DATA AVAILABILITY
The data in the paper is available upon reasonable request.
REFERENCES
- 1.Nakashima K, and de Crombrugghe B (2003) Transcriptional mechanisms in osteoblast differentiation and bone formation. Trends Genet 19, 458–466 [DOI] [PubMed] [Google Scholar]
- 2.Chen H, Ghori-Javed FY, Rashid H, Adhami MD, Serra R, Gutierrez SE, and Javed A (2014) Runx2 regulates endochondral ossification through control of chondrocyte proliferation and differentiation. J Bone Miner Res 29, 2653–2665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Komori T, Yagi H, Nomura S, Yamaguchi A, Sasaki K, Deguchi K, Shimizu Y, Bronson RT, Gao YH, Inada M, Sato M, Okamoto R, Kitamura Y, Yoshiki S, and Kishimoto T (1997) Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell 89, 755–764 [DOI] [PubMed] [Google Scholar]
- 4.Hellingman CA, Davidson EN, Koevoet W, Vitters EL, van den Berg WB, van Osch GJ, and van der Kraan PM (2011) Smad signaling determines chondrogenic differentiation of bone-marrow-derived mesenchymal stem cells: inhibition of Smad1/5/8P prevents terminal differentiation and calcification. Tissue Eng Part A 17, 1157–1167 [DOI] [PubMed] [Google Scholar]
- 5.Dong YF, Soung do Y, Schwarz EM, O'Keefe RJ, and Drissi H (2006) Wnt induction of chondrocyte hypertrophy through the Runx2 transcription factor. J Cell Physiol 208, 77–86 [DOI] [PubMed] [Google Scholar]
- 6.Stanton LA, Sabari S, Sampaio AV, Underhill TM, and Beier F (2004) p38 MAP kinase signalling is required for hypertrophic chondrocyte differentiation. Biochem J 378, 53–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shu B, Zhang M, Xie R, Wang M, Jin H, Hou W, Tang D, Harris SE, Mishina Y, O'Keefe RJ, Hilton MJ, Wang Y, and Chen D (2011) BMP2, but not BMP4, is crucial for chondrocyte proliferation and maturation during endochondral bone development. J Cell Sci 124, 3428–3440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Keller B, Yang T, Chen Y, Munivez E, Bertin T, Zabel B, and Lee B (2011) Interaction of TGFbeta and BMP signaling pathways during chondrogenesis. PLoS One 6, e16421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guo J, Chung UI, Yang D, Karsenty G, Bringhurst FR, and Kronenberg HM (2006) PTH/PTHrP receptor delays chondrocyte hypertrophy via both Runx2-dependent and -independent pathways. Dev Biol 292, 116–128 [DOI] [PubMed] [Google Scholar]
- 10.Mak KK, Kronenberg HM, Chuang PT, Mackem S, and Yang Y (2008) Indian hedgehog signals independently of PTHrP to promote chondrocyte hypertrophy. Development 135, 1947–1956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang J, Zhou J, and Bondy CA (1999) Igf1 promotes longitudinal bone growth by insulin-like actions augmenting chondrocyte hypertrophy. FASEB J 13, 1985–1990 [DOI] [PubMed] [Google Scholar]
- 12.Zhu M, Feng Q, Sun Y, Li G, and Bian L (2017) Effect of cartilaginous matrix components on the chondrogenesis and hypertrophy of mesenchymal stem cells in hyaluronic acid hydrogels. J Biomed Mater Res B Appl Biomater 105, 2292–2300 [DOI] [PubMed] [Google Scholar]
- 13.Inada M, Yasui T, Nomura S, Miyake S, Deguchi K, Himeno M, Sato M, Yamagiwa H, Kimura T, Yasui N, Ochi T, Endo N, Kitamura Y, Kishimoto T, and Komori T (1999) Maturational disturbance of chondrocytes in Cbfa1-deficient mice. Dev Dyn 214, 279–290 [DOI] [PubMed] [Google Scholar]
- 14.Stricker S, Fundele R, Vortkamp A, and Mundlos S (2002) Role of Runx genes in chondrocyte differentiation. Dev Biol 245, 95–108 [DOI] [PubMed] [Google Scholar]
- 15.Kim IS, Otto F, Zabel B, and Mundlos S (1999) Regulation of chondrocyte differentiation by Cbfa1. Mech Dev 80, 159–170 [DOI] [PubMed] [Google Scholar]
- 16.Deng Y, Lei G, Lin Z, Yang Y, Lin H, and Tuan RS (2019) Engineering hyaline cartilage from mesenchymal stem cells with low hypertrophy potential via modulation of culture conditions and Wnt/beta-catenin pathway. Biomaterials 192, 569–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Foyt DA, Taheem DK, Ferreira SA, Norman MDA, Petzold J, Jell G, Grigoriadis AE, and Gentleman E (2019) Hypoxia impacts human MSC response to substrate stiffness during chondrogenic differentiation. Acta Biomater 89, 73–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aisenbrey EA, and Bryant SJ (2018) A MMP7-sensitive photoclickable biomimetic hydrogel for MSC encapsulation towards engineering human cartilage. J Biomed Mater Res A 106, 2344–2355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brunger JM, Zutshi A, Willard VP, Gersbach CA, and Guilak F (2017) Genome Engineering of Stem Cells for Autonomously Regulated, Closed-Loop Delivery of Biologic Drugs. Stem Cell Reports 8, 1202–1213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brunger JM, Zutshi A, Willard VP, Gersbach CA, and Guilak F (2017) CRISPR/Cas9 Editing of Murine Induced Pluripotent Stem Cells for Engineering Inflammation-Resistant Tissues. Arthritis Rheumatol 69, 1111–1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pferdehirt L, Damato AR, Dudek M, Meng QJ, Herzog ED, and Guilak F (2022) Synthetic gene circuits for preventing disruption of the circadian clock due to interleukin-1-induced inflammation. Sci Adv 8, eabj8892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meerbrey KL, Hu G, Kessler JD, Roarty K, Li MZ, Fang JE, Herschkowitz JI, Burrows AE, Ciccia A, Sun T, Schmitt EM, Bernardi RJ, Fu X, Bland CS, Cooper TA, Schiff R, Rosen JM, Westbrook TF, and Elledge SJ (2011) The pINDUCER lentiviral toolkit for inducible RNA interference in vitro and in vivo. Proc Natl Acad Sci U S A 108, 3665–3670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Atsumi T, Miwa Y, Kimata K, and Ikawa Y (1990) A chondrogenic cell line derived from a differentiating culture of AT805 teratocarcinoma cells. Cell Differ Dev 30, 109–116 [DOI] [PubMed] [Google Scholar]
- 24.Shukunami C, Ishizeki K, Atsumi T, Ohta Y, Suzuki F, and Hiraki Y (1997) Cellular hypertrophy and calcification of embryonal carcinoma-derived chondrogenic cell line ATDC5 in vitro. J Bone Miner Res 12, 1174–1188 [DOI] [PubMed] [Google Scholar]
- 25.Shukunami C, Shigeno C, Atsumi T, Ishizeki K, Suzuki F, and Hiraki Y (1996) Chondrogenic differentiation of clonal mouse embryonic cell line ATDC5 in vitro: differentiation-dependent gene expression of parathyroid hormone (PTH)/PTH-related peptide receptor. J Cell Biol 133, 457–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu B, Durisin EK, Decker JT, Ural EE, Shea LD, and Coleman RM (2017) Phosphate regulates chondrogenesis in a biphasic and maturation-dependent manner. Differentiation 95, 54–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mueller MB, and Tuan RS (2008) Functional characterization of hypertrophy in chondrogenesis of human mesenchymal stem cells. Arthritis Rheum 58, 1377–1388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Olson A, Sheth N, Lee JS, Hannon G, and Sachidanandam R (2006) RNAi Codex: a portal/database for short-hairpin RNA (shRNA) gene-silencing constructs. Nucleic Acids Res 34, D153–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zheng Q, Keller B, Zhou G, Napierala D, Chen Y, Zabel B, Parker AE, and Lee B (2009) Localization of the cis-enhancer element for mouse type X collagen expression in hypertrophic chondrocytes in vivo. J Bone Miner Res 24, 1022–1032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carrion B, Souzanchi MF, Wang VT, Tiruchinapally G, Shikanov A, Putnam AJ, and Coleman RM (2016) The Synergistic Effects of Matrix Stiffness and Composition on the Response of Chondroprogenitor Cells in a 3D Precondensation Microenvironment. Adv Healthc Mater 5, 1192–1202 [DOI] [PubMed] [Google Scholar]
- 31.Kim YJ, Sah RL, Doong JY, and Grodzinsky AJ (1988) Fluorometric assay of DNA in cartilage explants using Hoechst 33258. Anal Biochem 174, 168–176 [DOI] [PubMed] [Google Scholar]
- 32.Rauh J, Jacobi A, and Stiehler M (2015) Identification of stable reference genes for gene expression analysis of three-dimensional cultivated human bone marrow-derived mesenchymal stromal cells for bone tissue engineering. Tissue Eng Part C Methods 21, 192–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takahashi N, Rieneck K, van der Kraan PM, van Beuningen HM, Vitters EL, Bendtzen K, and van den Berg WB (2005) Elucidation of IL-1/TGF-beta interactions in mouse chondrocyte cell line by genome-wide gene expression. Osteoarthritis Cartilage 13, 426–438 [DOI] [PubMed] [Google Scholar]
- 34.Wehling N, Palmer GD, Pilapil C, Liu F, Wells JW, Muller PE, Evans CH, and Porter RM (2009) Interleukin-1beta and tumor necrosis factor alpha inhibit chondrogenesis by human mesenchymal stem cells through NF-kappaB-dependent pathways. Arthritis Rheum 60, 801–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gouze JN, Bianchi A, Becuwe P, Dauca M, Netter P, Magdalou J, Terlain B, and Bordji K (2002) Glucosamine modulates IL-1-induced activation of rat chondrocytes at a receptor level, and by inhibiting the NF-kappa B pathway. FEBS Lett 510, 166–170 [DOI] [PubMed] [Google Scholar]
- 36.Murtaugh LC, Chyung JH, and Lassar AB (1999) Sonic hedgehog promotes somitic chondrogenesis by altering the cellular response to BMP signaling. Genes Dev 13, 225–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Murtaugh LC, Zeng L, Chyung JH, and Lassar AB (2001) The chick transcriptional repressor Nkx3.2 acts downstream of Shh to promote BMP-dependent axial chondrogenesis. Dev Cell 1, 411–422 [DOI] [PubMed] [Google Scholar]
- 38.Marcelle C, Ahlgren S, and Bronner-Fraser M (1999) In vivo regulation of somite differentiation and proliferation by Sonic Hedgehog. Dev Biol 214, 277–287 [DOI] [PubMed] [Google Scholar]
- 39.Dexheimer V, Frank S, and Richter W (2012) Proliferation as a requirement for in vitro chondrogenesis of human mesenchymal stem cells. Stem Cells Dev 21, 2160–2169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Correa D, Hesse E, Seriwatanachai D, Kiviranta R, Saito H, Yamana K, Neff L, Atfi A, Coillard L, Sitara D, Maeda Y, Warming S, Jenkins NA, Copeland NG, Horne WC, Lanske B, and Baron R (2010) Zfp521 is a target gene and key effector of parathyroid hormone-related peptide signaling in growth plate chondrocytes. Dev Cell 19, 533–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Akiyama H, Kanno T, Ito H, Terry A, Neil J, Ito Y, and Nakamura T (1999) Positive and negative regulation of chondrogenesis by splice variants of PEBP2alphaA/CBFalpha1 in clonal mouse EC cells, ATDC5. J Cell Physiol 181, 169–178 [DOI] [PubMed] [Google Scholar]
- 42.Otto F, Thornell AP, Crompton T, Denzel A, Gilmour KC, Rosewell IR, Stamp GW, Beddington RS, Mundlos S, Olsen BR, Selby PB, and Owen MJ (1997) Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell 89, 765–771 [DOI] [PubMed] [Google Scholar]
- 43.Komori T. (2002) Runx2, a multifunctional transcription factor in skeletal development. J Cell Biochem 87, 1–8 [DOI] [PubMed] [Google Scholar]
- 44.Lengner CJ, Hassan MQ, Serra RW, Lepper C, van Wijnen AJ, Stein JL, Lian JB, and Stein GS (2005) Nkx3.2-mediated repression of Runx2 promotes chondrogenic differentiation. J Biol Chem 280, 15872–15879 [DOI] [PubMed] [Google Scholar]
- 45.Tchetina E, Mwale F, and Poole AR (2003) Distinct phases of coordinated early and late gene expression in growth plate chondrocytes in relationship to cell proliferation, matrix assembly, remodeling, and cell differentiation. J Bone Miner Res 18, 844–851 [DOI] [PubMed] [Google Scholar]
- 46.Fosang AJ, Last K, Knauper V, Murphy G, and Neame PJ (1996) Degradation of cartilage aggrecan by collagenase-3 (MMP-13). FEBS Lett 380, 17–20 [DOI] [PubMed] [Google Scholar]
- 47.Liao L, Zhang S, Gu J, Takarada T, Yoneda Y, Huang J, Zhao L, Oh CD, Li J, Wang B, Wang M, and Chen D (2017) Deletion of Runx2 in Articular Chondrocytes Decelerates the Progression of DMM-Induced Osteoarthritis in Adult Mice. Sci Rep 7, 2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mitchell PG, Magna HA, Reeves LM, Lopresti-Morrow LL, Yocum SA, Rosner PJ, Geoghegan KF, and Hambor JE (1996) Cloning, expression, and type II collagenolytic activity of matrix metalloproteinase-13 from human osteoarthritic cartilage. J Clin Invest 97, 761–768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kimura A, Inose H, Yano F, Fujita K, Ikeda T, Sato S, Iwasaki M, Jinno T, Ae K, Fukumoto S, Takeuchi Y, Itoh H, Imamura T, Kawaguchi H, Chung UI, Martin JF, Iseki S, Shinomiya K, and Takeda S (2010) Runx1 and Runx2 cooperate during sternal morphogenesis. Development 137, 1159–1167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee KS, Kim HJ, Li QL, Chi XZ, Ueta C, Komori T, Wozney JM, Kim EG, Choi JY, Ryoo HM, and Bae SC (2000) Runx2 is a common target of transforming growth factor beta1 and bone morphogenetic protein 2, and cooperation between Runx2 and Smad5 induces osteoblast-specific gene expression in the pluripotent mesenchymal precursor cell line C2C12. Mol Cell Biol 20, 8783–8792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Arnosti DN, and Kulkarni MM (2005) Transcriptional enhancers: Intelligent enhanceosomes or flexible billboards? J Cell Biochem 94, 890–898 [DOI] [PubMed] [Google Scholar]
- 52.Harafuji N, Keys DN, and Levine M (2002) Genome-wide identification of tissue-specific enhancers in the Ciona tadpole. Proc Natl Acad Sci U S A 99, 6802–6805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li F, Lu Y, Ding M, Napierala D, Abbassi S, Chen Y, Duan X, Wang S, Lee B, and Zheng Q (2011) Runx2 contributes to murine Col10a1 gene regulation through direct interaction with its cis-enhancer. J Bone Miner Res 26, 2899–2910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kronenberg HM (2003) Developmental regulation of the growth plate. Nature 423, 332–336 [DOI] [PubMed] [Google Scholar]
- 55.Schipani E, Lanske B, Hunzelman J, Luz A, Kovacs CS, Lee K, Pirro A, Kronenberg HM, and Juppner H (1997) Targeted expression of constitutively active receptors for parathyroid hormone and parathyroid hormone-related peptide delays endochondral bone formation and rescues mice that lack parathyroid hormone-related peptide. Proc Natl Acad Sci U S A 94, 13689–13694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Weir EC, Philbrick WM, Amling M, Neff LA, Baron R, and Broadus AE (1996) Targeted overexpression of parathyroid hormone-related peptide in chondrocytes causes chondrodysplasia and delayed endochondral bone formation. Proc Natl Acad Sci U S A 93, 10240–10245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Leboy P, Grasso-Knight G, D'Angelo M, Volk SW, Lian JV, Drissi H, Stein GS, and Adams SL (2001) Smad-Runx interactions during chondrocyte maturation. J Bone Joint Surg Am 83-A Suppl 1, S15–22 [PubMed] [Google Scholar]
- 58.Yoshida CA, Yamamoto H, Fujita T, Furuichi T, Ito K, Inoue K, Yamana K, Zanma A, Takada K, Ito Y, and Komori T (2004) Runx2 and Runx3 are essential for chondrocyte maturation, and Runx2 regulates limb growth through induction of Indian hedgehog. Genes Dev 18, 952–963 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data in the paper is available upon reasonable request.