

Abstract
Key clinical message
Pseudohypoaldosteronism (PHA) carries a good prognosis if treated early and appropriately, but some cases can have life‐threatening events. We underscored the need to consider secondary PHA as one of the differential diagnoses of hyponatremia and hyperkalemia in infancy.
Abstract
Pseudohypoaldosteronism (PHA) type 1 has two classifications; the primary type, caused by genetic abnormalities that develop during neonatal and infancy periods, and the secondary type, caused by urinary tract malformation and urinary tract infection. Secondary PHA, if treated early and appropriately, has a good prognosis; however, some cases can present life‐threatening events. Therefore, early diagnosis is crucial. We present a case of early infancy secondary PHA presented with marked hyponatremia and poor weight gain. The patient's growth and development improved with secondary PHA treatment. Here, were demonstrated the value of prompt action against infection and electrolyte imbalance and the importance of imaging for diagnosis, and underscore the need to consider secondary PHA as a differential diagnoses of hyponatremia and hyperkalemia in infancy. However further studies, including basic research, to elucidate the diseases pathology is warranted.
Keywords: children, poor weight gain, pseudohypoaldosteronism, urinary tract infection, urinary tract malformation
INTRODUCTION
Pseudohypoaldosteronism (PHA) has no abnormalities in aldosterone secretion and kidney spherical function; however, causes sodium reabsorption disorders due to a decrease in response to mineral corticoids in distal tubes. It is a rare hereditary disease that presents symptomatic and metabolic acidosis, and is divided into Type I with recognizable salt loss and Type II with hypertension and no salt loss. 1 PHA type 1 is common in boys during early infant and presents hyponatremia, hyperkalemia, metabolic acidosis, and hypersecretion of aldosterone. PHA type 1 has two classifications: primary type, caused by genetic abnormalities that develop during neonatal and infancy periods and secondary type, caused by urinary tract malformation (UTM) and urinary tract infection (UTI). 2 , 3 Although PHA is considered to have a good prognosis if treated early and appropriately, some cases can have life‐threatening events, such as cardiac arrest. 4 We reported a case of secondary PHA presenting with marked hyponatremia and a chief complaint of poor weight gain.
CASE HISTORY/EXAMINATION
A 9‐month‐old boy with an unremarkable birth history presented with failure to thrive.
He was delivered at 39 weeks and 3 days, with a birth weight at 3360 g. There were no abnormalities on tandem mass screening. Cervical determination was not possible at 4 months, but he was able to roll over at 5 months and could sit up at 9 months. His poor weight gain was noted at 3 months of age. At 7 months after birth, he was noted to be vomiting once a day. The patient had been followed‐up for poor weight gain of unknown cause.
METHODS
Nine months after birth, he was admitted at another hospital because for frequent vomiting and persisted poor weight gain. Physical measurements were height 70 cm (−0.7 standard deviation (SD)) and weight 7300 g (−1.6 SD) and vital signs were body temperature 37.5°C, blood pressure 76/40 mmHg, pulse 148/min, and respiratory rate 42/min.
The patient's anterior fontanels had caved in. He took gasping breaths; however, chest auscultation revealed no riles. His heartbeat was normal and had no abnormalities in the abdomen. His muscle tone had decreased and the extremities were cyanotic.
Edema was not observed on the external surface, and a decrease in skin turgor was observed. He had no vulvar abnormalities or pigmentation.
Investigation revealed serum levels of sodium at 103 mmol/L (normal 137–147 mmol/L), and potassium at 5.88 mmol/L (normal 3.70–4.90 mmol/L). He had nonanionic gap metabolic acidosis and transtubular potassium gradient of 3.67 (normal 8–9), which indicated reduced urinary potassium excretion. The urine sodium was <20 mmol/L. Correction of hyponatremia and dehydration treatment were started with physiological saline infusion. Infusion of 130 mEq/L of saline partially corrected the hyponatremia. Urinalysis revealed leukocytes and nitrites. Extended‐spectrum beta‐lactamase (ESBL) Escherichia coli were isolated from the urine culture. Abdominal ultrasound revealed Grade 1 hydronephrosis and ureteral dilatation on the right. (Figure 1) The blood tests ruled out renal failure (i.e., no increase in the urea nitrogen or creatinine level) and hypertrophic pyloric stenosis (i.e., no evidence of hypokalemia or hypochloremic metabolic alkalosis). Abdominal ultrasound showed UTM and UTI; therefore, secondary pseudohypoaldosteronism was suspected. The levels of 21‐hydroxylase and cortisol were normal. Aldosterone was elevated at 20,000 pg/mL (normal 4–80 pg/dL). Plasma renin activity (PRA) was elevated at >20 ng/mL/h (normal 0.2–4.1 ng/mL/h), implying tubular unresponsiveness. Adrenal insufficiency was ruled out by 17‐OHP, adrenocorticotropic hormone (ACTH), and cortisol levels.
FIGURE 1.
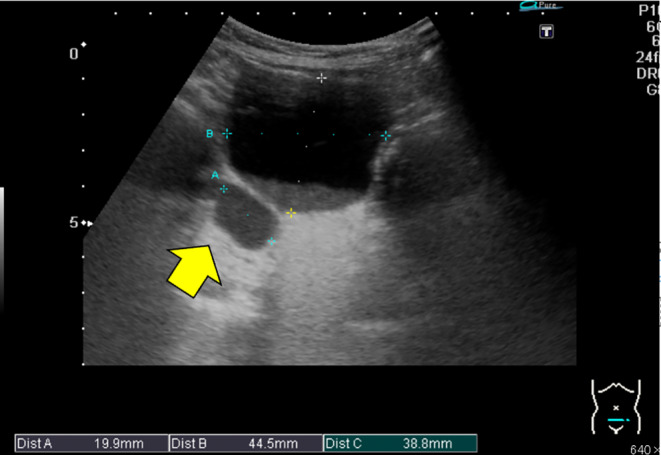
Abdominal ultrasound. Arrow: A luminal structure was observed on the right dorsal side of the bladder.
On the seventh day of illness, there were improvements in the serum levels of sodium to 140 mEq/L and potassium to 3.9 mEq/L, but he was transferred to our hospital, because of marked edema and difficult placement of intravenous access. Despite the absence of fever at the time of admission, intravenous infusion of meropenem (200 mg every 12 h) was started for the recent urine isolate of ESBL E. coli and because of the increased leukocyte count and C‐reactive protein. On the fourteenth day, aldosterone level decreased to 207 pg/mL, PRA decreased to 5.6 ng/mL/h, and the edema improved.
Magnetic resonance urography (MRU), diuretic renogram, and voiding cystography (VCG) were performed to evaluate for UTMs. MRU showed a duplicated right collecting system with an upper pole ectopic ureter that extended to the bladder neck (Figure 2). VCG did not show regurgitation in the ureter or any abnormality in the lower urethra. On diuretic renogram (99mTc DTPA MAG 3), the left kidney had a normal renogram pattern but showed mild functional impairment, compared with the right kidney, while the right kidney had ureteral dilatation and mild hypoexcretion pattern (Figure 3). We diagnosed secondary PHA based on the findings of hyponatremia, hyperkalemia, and renal UTM, accompanied by high PRA and high blood aldosterone levels.
FIGURE 2.
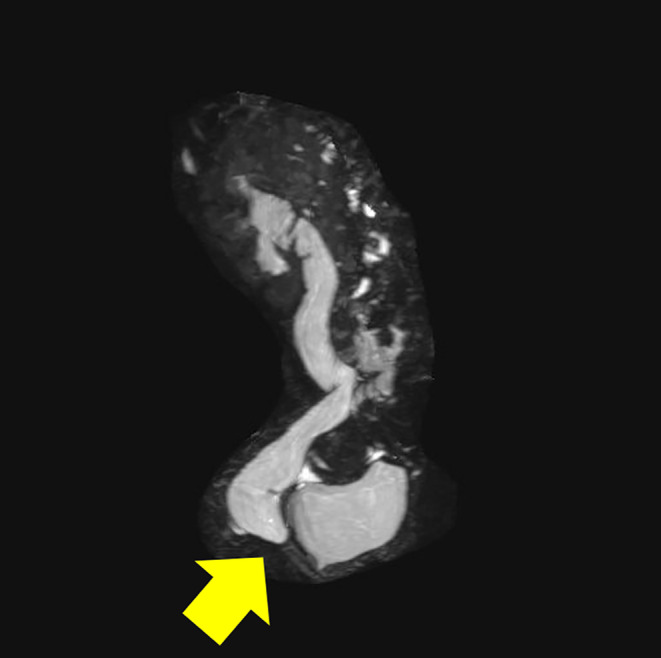
Magnetic resonance urography (MRU). MRU showed a duplicated right collecting system with an upper pole ectopic ureter that extended to the bladder neck. Arrow: Right vesicoureteral junction stenosis was observed.
FIGURE 3.
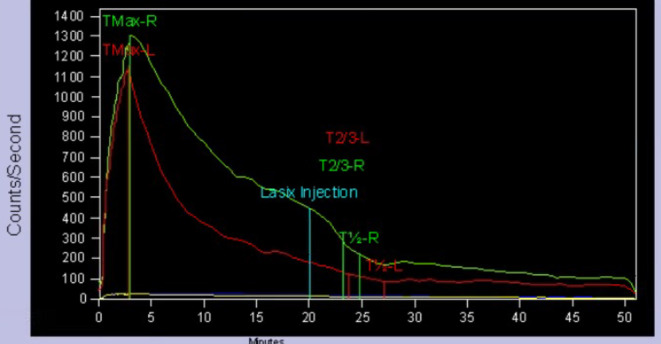
Diuretic renogram (99mTc DTPA MAG 3). No deterioration of both renal function, pattern of right renal ureter dilatation, and mildly decreased excretion.
Figure 4 shows the clinical course. Oral administration of sulfamethoxazole/ trimethoprim (ST) was started on the sixteenth day to prevent UTI; however ST caused drug‐induced eczema, and, thus, had to use cefaclor for half a year. The patient's subsequent course was uneventful. He was able to crawl at 11 months, sit up at 1 year and 1 month, and walk at 1 year and 4 months of age. At the age of 2 years, the renin level was 4.4 ng/mL/h, and the aldosterone level was 171 pg/mL, which confirmed normalization. At the age of 4 years, he was growing well, with height of 101 cm (−0.1 SD), weight of 16.2 kg (+0.2 SD), and normal neurodevelopment (Figure 5).
FIGURE 4.
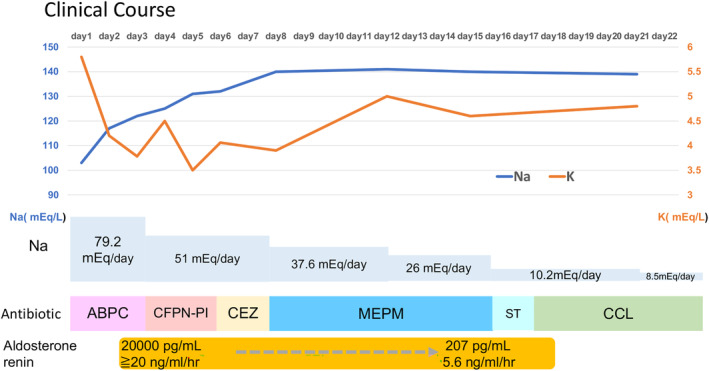
Clinical course. ABPC, ampicillin sodium; CCL, cefaclor; CEZ, cefazolin; CFPN‐PI, cefcapene pivoxil; MEPM, meropenem; ST, sulfamethoxazole‐trimethoprim.
FIGURE 5.
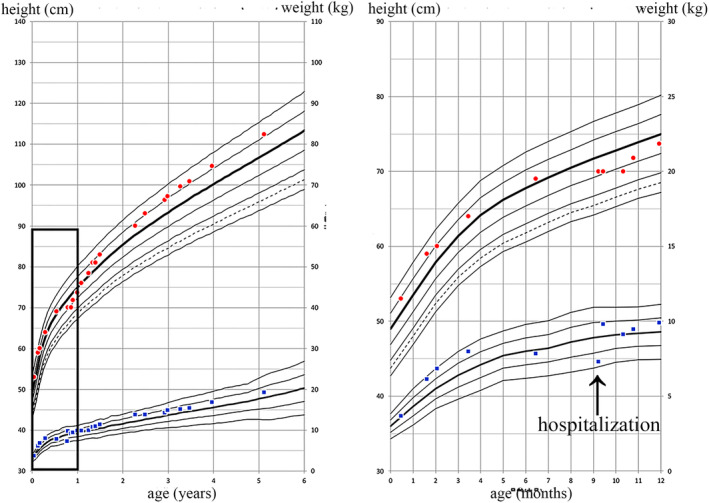
Growth curves. In this case, growth and development improved with treatment. The figure on the right shows details of the box in the figure on the left.
CONCLUSION AND RESULTS
Secondary PHA should be kept in mind as one of the differential diagnoses of hyponatremia and hyperkalemia in infancy and can be diagnosed by imaging. Secondary PHA had been thought to be caused by immaturity of the renal tubules, UTM, and UTI, but its pathophysiology remains unclear. In the future, further progress in elucidating the pathology, including basic research, is required.
DISCUSSION
Secondary PHA associated with UTI and UTM is most common in infants under 7 months of age 5 and is characterized by diminished action of aldosterone receptors secondary to inflammation and pressure damages that are associated with the immaturity of the renal tubules. Urinary toxins and hyperaldosteronism had been thought to cause downregulation of aldosterone receptors, 6 but the details of the pathogenic mechanism remain unknown. Unlike primary PHA, secondary PHA does not demonstrate genetic mutations in the mineralocorticoid receptor (MR) or epithelial sodium channel (ENaC). 7 Symptoms are nonspecific and include poor oral intake, poor weight gain, vomiting, diarrhea, and dehydration. Secondary PHA causes hyponatremia in infancy and is particularly important to differentiate from congenital adrenal hyperplasia (CAH), 6 which has the same pattern of electrolyte abnormalities and may have similar time of onset and clinical symptoms. Urinalysis and imaging of the urinary system are useful in differentiating secondary PHA from CAH, but the diagnosis of secondary PHA requires confirmation of elevated renin and aldosterone levels and normal ACTH and cortisol levels.
In our case, there were no physical findings that strongly suggested CAH, an increase in vitality was observed after the start of fluid replacement, and UTM was confirmed by ultrasound; therefore, glucocorticoids were not administered. Some reports mentioned that patients should be treated with glucocorticoids until endocrine test results are available. 8 , 9 Since the first report on secondary PHA by G.W. Moll et al in 1982, over 100 cases have been reported worldwide. 10 Approximately 90% of secondary PHA develops before 3 months of age. After 3 months of age, secondary PHA develops less frequently and rarely leads to electrolyte abnormalities. 11 Judging from the growth curve in our case, the onset was presumed to have occurred around 4–5 months after birth; notably, the serum sodium level was 103 mEq/L and led to marked hyponatremia. Although the neurological course in this patient was not poor, it should be noted that secondary PHA may develop severe hyponatremia even at 9 months after birth, as in our case. PHA has a wide range of clinical manifestations, the most prominent of which are gastrointestinal, including eating disorders, vomiting, bloating, and diarrhea.
Although secondary PHA is generally considered to have a favorable prognosis, there had been reported cases that developed serious conditions, such as convulsion and disturbance in consciousness secondary to hyponatremia, fatal arrhythmia secondary to hyperkalemia, and circulatory failure and shock. 8 Therefore, early detection is important. Secondary PHA should be considered as one of the differential diagnoses not only in early infancy, as in our case, but also in cases with hyponatremia and hyperkalemia, for which urinalysis and urinary system evaluation should be performed.
Although the etiology of secondary PHA remains unclear, three factors, including age, urinary tract obstruction, and UTI, may be the key in developing secondary PHA. Secondary PHA is more common in infants under 6 months of age, and studies have validated that this may be associated with immature renal tubules. 2 , 12 , 13 For children with UTM, urinary tract obstruction, or vesicoureteral reflux, increased intrarenal pressure causes downregulation of aldosterone receptors. In addition, immature renal tubules and UTM with children lead to a surge in the intrarenal synthesis of various cytokines, such as tumor necrosis factor‐β1 and tumor necrosis factor‐α; the former may block the action of aldosterone. 14 , 15 In children with UTI, internal and external toxins not only directly damage the aldosterone receptors, but they also stimulate the immune system to produce inflammatory factors, such as interleukin‐1, thromboxane, and natriuretic peptides. 16 , 17 Although basic studies on secondary PHA are few, a transient decrease in lymphocyte MR expression is one of the mechanisms of aldosterone resistance in patients with secondary PHA. Examining the genetic polymorphisms in MRs and ENaCs is needed in order to understand the pathogenesis of secondary PHA, but there had been no reports on comprehensive genetic analyses. 5 The pathophysiology of secondary PHA remains largely unknown, and advances in pathophysiological analysis, including basic research, are required.
AUTHOR CONTRIBUTIONS
Keisuke Goshima: Data curation; writing – original draft. Hiroshi Tamura: Conceptualization; data curation; formal analysis; writing – review and editing. Yuko Hidaka: Data curation. Keishiro Furuie: Data curation. Shohei Kuraoka: Data curation; formal analysis.
FUNDING INFORMATION
The authors received no financial support for the research, authorship, and/or publication of this article.
CONFLICT OF INTEREST STATEMENT
The authors have no conflict of interest to declare.
ETHICS STATEMENT
All the procedures performed in studies involving human participants were in accordance with the ethical standards of the Institutional Committee and the 1964 Declaration of Helsinki and its later amendments or comparable ethical standards (64th WMA General Assembly, Fortaleza, Brazil, October 2013). Informed consent for examinations and to publish their cases, including images was obtained from patients parents and/or their family members.
CONSENT
Written informed consent was obtained from his parents for the publication of this case report.
ACKNOWLEDGMENTS
We would like to thank his parents for their participation in this study.
Goshima K, Tamura H, Hidaka Y, Furuie K, Kuraoka S. A case of secondary pseudohypoaldosteronism that presented as poor weight gain. Clin Case Rep. 2024;12:e8722. doi: 10.1002/ccr3.8722
DATA AVAILABILITY STATEMENT
All data generated or analyzed during this study are included in this article. Further enquiries can be directed to the corresponding author.
REFERENCES
- 1. Kaneko K. Pseudohypoaldosteronism Type 1. Jpn J Nephrol. 2011;53(2):150‐154. [PubMed] [Google Scholar]
- 2. Kaninde A, Grace ML, Joyce C, et al. The incidence of transient infantile pseudohypoaldosteronism in Ireland: a prospective study. Acta Paediatr. 2021;110(4):1257‐1263. [DOI] [PubMed] [Google Scholar]
- 3. Delforage X, Kongolo G, Cauliez A, et al. Transient pseudohypoaldosteronism: a potentially severe condition affecting infants with urinary tract malformation. J Pediatr Urol. 2019;15(3):265.e1‐265.e7. [DOI] [PubMed] [Google Scholar]
- 4. Attia NA, Marzouk YI. Pseudohypoaldosteronism in a neonate presenting as life‐threatening hyperkalemia. Case Rep Endocrinol. 2016;6(384):697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gunay F, Siklar Z, Berberoglu M. Difficulties in the diagnosis and management of eight infants with secondary pseudohypoaldosteronism. Turk J Pediatr. 2022;64(3):490‐499. [DOI] [PubMed] [Google Scholar]
- 6. Bogdanovic R, Stajic N, Putnik J, Paripovic A. Transient type 1 pseudohypoaldosteronism: report on an eight‐patient series and literature review. Pediatr Nephrol. 2009;24:2167‐2175. [DOI] [PubMed] [Google Scholar]
- 7. Belot A, Ranchin B, Fichtner C, et al. Pseudohypoaldosteronisms, report on a 10‐patient series. Nephrol Dial Transplant. 2008;23:1636‐1641. [DOI] [PubMed] [Google Scholar]
- 8. Thies KC, Boos K, Müller‐Deile K, Ohrdorf W, Beushausen T, Townsend P. Ventricular flutter in a neonate‐severe electrolyte imbalance caused by urinary tract infection in the presence of urinary tract malformation. J Emerg Med. 2000;18:47‐50. [DOI] [PubMed] [Google Scholar]
- 9. Tuoheti Y, Zheng Y, Lu Y, Li M, Jin Y. Transient pseudohypoaldosteronism in infancy mainly manifested as poor appetite and vomiting: two case reports and review of the literature. Front Pediatr. 2022;10:896647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Moll GW Jr, Rich BH, Roseufield RL. Apparent pseudohy‐ poaldosteronism with unilateral obstructive uropathy in an infant. Clin Res. 1982;30:799A. [Google Scholar]
- 11. Watanabe T. Reversible secondary pseudohypoaldosteronism. Pediatr Nephrol. 2003;18:486. [DOI] [PubMed] [Google Scholar]
- 12. Kibe T, Sobajima T, Yoshimura A, Uno Y, Wada N, Ueta I. Secondary pseudohypoaldosteronism causing cardiopulmonary arrest and cholelithiasis. Pediatr Int. 2014;56:270‐272. [DOI] [PubMed] [Google Scholar]
- 13. Martinerie L, Pussard E, Foix‐L'Hélias L, et al. Physiological partial aldosterone resistance in human newborns. Pediatr Res. 2009;66:323‐328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Korkut S, Akin L, Hatipoglu N, et al. A potential serious complication in infants with congenital obstructive uropathy: secondary pseudohypoaldosteronism. J Pak Med Assoc. 2019;69:108‐112. [PubMed] [Google Scholar]
- 15. Abraham M‐B, Larkins N, Choong C‐S, Shetty V‐B. Transient pseudohypoaldosteronism in infancy secondary to urinary tract infection. J Paediatr Child Health. 2017;53:458‐463. [DOI] [PubMed] [Google Scholar]
- 16. Zhu Y, Kuang X‐Y, Kang Y‐L, et al. Clinical analysis of an infant case with transient pseudohypoaldosteronism‐salt‐losing crisis. J Clin Pediatr Dent. 2019;37:601‐604. [Google Scholar]
- 17. Bülchmann G, Schuster T, Heger A, Kuhnle U, Joppich I, Schmidt H. Transient pseudohypoaldosteronism secondary to posterior urethral valves–a case report and review of the literature. Eur J Pediatr Surg. 2001;11:277‐279. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analyzed during this study are included in this article. Further enquiries can be directed to the corresponding author.