

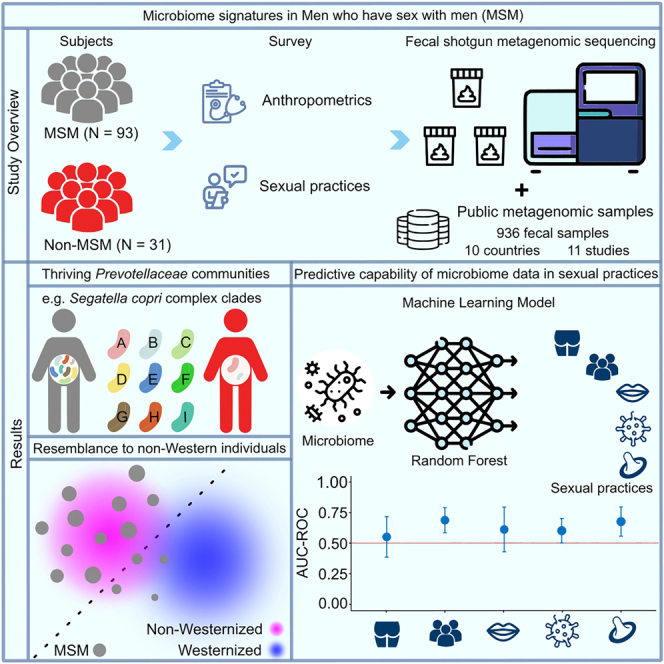
Summary
The human gut microbiota is influenced by various factors, including health status and environmental conditions, yet considerable inter-individual differences remain unexplained. Previous studies identified that the gut microbiota of men who have sex with men (MSM) is distinct from that of non-MSM. Here, we reveal through species-level microbiota analysis using shotgun metagenomics that the gut microbiota of many MSM with Western origin resembles gut microbial communities of non-Westernized populations. Specifically, MSM gut microbiomes are frequently dominated by members of the Prevotellaceae family, including co-colonization of species from the Segatella copri complex and unknown Prevotellaceae members. Questionnaire-based analysis exploring inter-individual differences in MSM links specific sexual practices to microbiota composition. Moreover, machine learning identifies microbial features associated with sexual activities in MSM. Together, this study shows associations of sexual activities with gut microbiome alterations in MSM, which may have a large impact on population-based microbiota studies.
Keywords: MSM, gut microbiome, Prevotella, Segatella, sexual orientation, RAI, oral sex, sex partner, non-Westernized microbiota
Graphical abstract
Highlights
-
•
Men who have sex with men (MSM) harbor a distinctive gut microbiota
-
•
Dominant members include the Segatella copri complex and other Prevotellaceae species
-
•
The MSM gut microbiome resembles that of non-Westernized populations
-
•
Sexual practices are associated with a non-Westernized gut microbiome in MSM
Huang et al. analyze the gut microbiota composition of men who have sex with men (MSM) using shotgun metagenomics. Characterization of microbiome architecture identifies an enrichment of Prevotellaceae including the Segatella copri complex in MSM. Integration of questionnaire-collected data reveals associations for sexual practices in shaping community structure and their similarity to non-Westernized communities.
Introduction
The human microbiota is shaped by diverse factors including geographical and ethnic origin, socioeconomic status, genetics, health status, diet, and medications; yet, much of the interindividual variability of the human gut microbiota is currently unexplained.1,2 This suggests that so-far-unknown factors not routinely recorded in large population-based cross-sectional studies may affect the gut microbiota. One of these factors that has been considered is sexual orientation, as men who have sex with men (MSM) have been characterized by a distinctive gut microbiota, characterized by the depletion of Bacteroides and the expansion of the genus Segatella/Prevotella.3,4,5 Notably, earlier studies had suggested HIV infection as a strong driver of gut microbiota alterations toward a Segatella/Prevotella-rich gut microbiome,6 but subsequent studies revealed that these alterations can be explained by an enrichment of men who have sex with men (MSM) in HIV-positive cohorts.3,4,7 While MSM exhibit a Segatella/Prevotella-rich gut microbiome independently of HIV status, more recent studies have revealed that HIV infection and HIV disease progression have been associated with independent changes in the gut microbiota.8,9
The gut microbiome signature of MSM is reminiscent of the initially reported human Prevotella enterotype, which has been associated with inflammatory diseases and insulin resistance in Westernized (Wes) populations.10,11,12 However, the increased prevalence and/or increased relative abundance of Segatella/Prevotella spp. was also associated with health-promoting diets, i.e., a plant-based diet, and beneficial outcomes after dietary interventions.13,14,15,16,17 Moreover, conflicting results were reported for potential detrimental roles of Segatella/Prevotella spp. on human health, with associations varying between studies.18,19 Interestingly, in a global context, the expansion of the genus Segatella/Prevotella and the depletion of Bacteroides were also observed in non-Westernized (non-Wes) healthy individuals following traditional lifestyles and typically consuming high-fiber and low-fat diets.18,20,21
Shotgun metagenome sequencing and advances in bioinformatic analyses have recently uncovered that the family Prevotellaceae, the genera Prevotella and Segatella, and its most prevalent species in humans, Segatella copri (formerly Prevotella copri), display a much larger taxonomic and functional diversity compared to what has been initially suggested based on 16S rRNA gene sequencing studies.5,18 Specifically, through combining cultivation- and metagenomics-based approaches, we recently revealed that the species S. copri can be subdivided into at least 13 different clades, which represent different species with distinct functional profiles.22
Here, we performed shotgun metagenomc sequencing for a cohort of 124 individuals including MSM and non-MSM and characterized their gut microbiota at the species level. Moreover, we studied the MSM microbiota alteration in the global context using publicly available datasets representing typical Wes and non-Wes populations and those from China but in urbanized communities. With the analysis of clinical and self-reported data together, we were able to identify an association for sexual activities to MSM microbiome composition and diversification. These data and analyses provide an insight into a previously under-appreciated role of sexual practices in shaping the human gut microbiota.
Results
Increased diversity and altered gut microbiome architecture at the species level in MSM
Insights into the gut microbiome structure of MSM have been mostly generated using 16S rRNA gene sequencing,3,4,23,24 which lacks taxonomic precision. Therefore, we performed SMS from fecal samples of a total of 124 subjects (MSM n = 93, non-MSM n = 31; Tables 1 and S1), resulting in, on average, 25 M (with a minimum of 14 M) quality-controlled paired-end reads per sample (average: 6.3 Gbp/sample; Figure S1A; Table S1). Microbial community composition with relative abundance quantification was analyzed using a reference-based approach, namely MetaPhlAn 4.25 MetaPhlAn 4 identifies species-level genome bins (SGBs), which are clusters of genomes spanning the typical diversity of bacterial species and can include previously cultured (known SGBs [kSGBs]) or can be composed of solely unknown metagenome-assembled genomes (unknown SGBs [uSGBs]).21 Within the 124 subjects, we identified a total of 1,675 SGBs including 1,124 kSGBs and 551 uSGBs.
Table 1.
Subject characteristics at study entry (N = 124)
| Anthropometric and clinical parameter | MSM (%) | Non-MSM (%) | p |
|---|---|---|---|
| Age | 1.0000 | ||
| <45 years | 45 (48) | 14 (45) | – |
| >45 years | 48 (52) | 17 (55) | – |
| HIV infection | 1.0000 | ||
| Positive | 79 (85) | 26 (84) | – |
| Negative | 14 (15) | 5 (16) | – |
| Ethnicity | 0.0065 | ||
| White | 92 (99) | 26 (84) | – |
| Others | 1 (1) | 5 (16) | – |
| Antibiotics treatment in the last 6 months | 1.0000 | ||
| Yes | 26 (28) | 5 (16) | – |
| No | 66 (71) | 26 (84) | – |
| N/A | 1 (1) | – | – |
| BMI classification | 0.4212 | ||
| Normal | 48 (52) | 12 (39) | – |
| Overweight | 37 (40) | 11 (35) | – |
| Obese class I | 5 (5) | 5 (16) | – |
| Obese class II | 1 (1) | 1 (3) | – |
| Obese class III | 1 (1) | 0 (0) | – |
| Underweight | 1 (1) | 2 (7) | – |
Differences between MSM (n = 93) and non-MSM (n = 31) for each parameter were evaluated using Fisher’s exact test with correction of Benjamini-Hochberg method. Values are given as counts with percentages in parentheses. The cohort consists of 119 male and 5 female subjects.
N/A, not available; BMI classification, body mass index class based on WHO recommendations. See also Table S1.
Comparison of alpha diversity using the Shannon index revealed a higher microbiome diversity in MSM than in non-MSM (p = 0.028, Wilcoxon rank-sum test; adjusted p value [padj] = 0.017, adjusted by fixed-effect linear models for confounding effects including age, ethnicity, antibiotics use, BMI, and HIV infection (see STAR Methods; Figure 1A). To study the differences in the microbiome of MSM and non-MSM, we calculated beta diversity using the Bray-Curtis distance, performed a permutational multivariate analysis of variance (PERMANOVA) controlled for confounders (age, ethnicity, antibiotics use, BMI, and HIV infection), and confirmed a species-level microbiome differentiation between MSM and non-MSM (PERMANOVA, R2 = 0.042 and padj < 0.001; Figure 1B). Additional analysis for subjects stratified according to major covariate groups such as age, BMI, HIV status, or recent antibiotic intake confirmed our observation (Figure S1B). Examining the effect of immunological parameters such as the counts and frequency of CD4+ T cells as well as the CD4/CD8 T cell ratio in 103 HIV-infected individuals further confirmed that the association of the gut microbiome change with MSM was significant, independently of HIV infection (Table 2; Figure S1C). Taken together, our species-level microbiome diversity analysis identified a significantly altered microbiome structure in MSM compared to non-MSM, which is in line with earlier 16S rRNA gene sequencing-based microbiome studies.3,24,26 These results corroborate that sexual practices or lifestyle are relevant factors affecting the gut microbiome community composition, regardless of previously reported variables.3,27,28,29,30
Figure 1.

Gut microbiome community architecture in MSM and non-MSM
(A) Alpha diversity analysis of MSM compared to non-MSM (adjusted by fixed-effect linear models,31 Shannon diversity padj = 0.017, richness padj = 0.237).
(B) Principal-coordinate analysis (PCoA) based on microbial abundance profiled using MetaPhlAn 425 between MSM and non-MSM samples (PERMANOVA, R2 = 0.042 and padj< 0.001). Marginal density plots show distributions of samples along each axis. MSM N = 93, non-MSM N = 31.
Table 2.
Immunological measures for HIV-infected subjects (N = 103)
| Immunological measures | MSM | Non-MSM | p |
|---|---|---|---|
| CD4 cell counts | 680.92 ± 282.36 (92–1,640) | 683.19 ± 267.24 (149–1,310) | 0.74 |
| CD4/CD8 ratio | 0.86 ± 0.50 (0.2–2.24) | 0.92 ± 0.42 (0.08–1.77) | 0.73 |
| CD3/CD4 (%) | 31.58 ± 8.95 (15.1–54) | 32.85 ± 10.13 (6.82–49.8) | 0.73 |
Differences between MSM (N = 77) and non-MSM (N = 26) with HIV infection were evaluated for each measure using Wilcoxon rank-sum test with correction of Benjamini-Hochberg method. Values are given as mean ± standard deviation with ranges in parentheses. All HIV-infected individuals were treated with antiretroviral therapy. See also Table S1.
Prevotellaceae-dominated communities in MSM comprise Segatella copri complex members and unknown species
A high Segatella/Prevotella abundance has been reported as the most distinct feature of the gut microbiome of MSM in Wes populations.3,4,32 We next aimed to explore Segatella/Prevotella communities at the species level, specifically whether Prevotellaceae expansion is driven by a single species or rather a diversification of the family. Therefore, we compared the richness, abundance, and prevalence of all species belonging to the Prevotellaceae family between MSM and non-MSM. Notably, the average number of Prevotellaceae species was three times higher in the microbiome of MSM compared to non-MSM (Wilcoxon rank-sum test p = 1.65e−06; covariates adjusted by fixed-effect linear model padj = 1.63e−04; Figure 2A). For comparing Prevotellaceae community diversity, we estimated the Shannon index specifically for members of Prevotellaceae family, confirming a higher alpha diversity in the MSM gut microbiome compared to that in non-MSM (Wilcoxon rank-sum test p = 1.35e−06; covariates adjusted by fixed-effect linear model padj = 2.69e−06; Figure 2A). We also observed a significant separation between MSM and non-MSM samples based on a Prevotellaceae-centric beta diversity analysis (covariate-controlled PERMANOVA padj < 0.001; Figure 2B). Of 47 Prevotellaceae species identified in this study (kSGBs n = 28, uSGBs n= 19), when only considering species with a minimum relative abundance of 0.1 at the 90th percentile of all samples, 16 were differentially enriched for MSM compared to non-MSM (Bonferroni-corrected Fisher’s exact test false discovery rate [FDR] < 0.05; Figures 2C and S2; Table S2). Unknown Prevotellaceae members (uSGBs) were similarly overrepresented in the gut microbiome of MSM displaying an elevated abundance and prevalence compared to non-MSM (Figures 2C and S2). Strikingly, members belonging to the S. copri species complex were the most common and abundant Prevotellaceae members in MSM, with 70% of MSM carrying at least two S. copri clades compared to only about 30% of non-MSM (Figures 2C and 2D). Moreover, the co-occurrence of strains from multiple S. copri clades, a feature previously observed in typical non-Wes microbiomes,18 was consistently higher in MSM than in non-MSM (Wilcoxon rank-sum test p = 0.001; Figure 2D).
Figure 2.

The Prevotellaceae community structure in MSM and non-MSM
(A) Richness and Shannon diversity of Prevotellaceae species members in MSM and non-MSM (adjusted by fixed-effect linear models,31 richness padj = 1.63e−4, Shannon diversity padj = 2.69e−6).
(B) PCoA based on log2-transformed abundances of Prevotellaceae members of MSM and non-MSM (PERMANOVA, R2 = 0.034 and padj< 0.001). Marginal density plots show distributions of samples along each axis.
(C) Log2-transformed abundances of Prevotellaceae members with a minimum abundance of 0.1 at 90th percentile in all subjects. S. copri complex clades are highlighted in red, and uSGBs are denoted in blue. Asterisk signs indicate species differentially enriched in MSM relative to non-MSM.
(D) Percentage of individuals harboring multiple S. copri complex clades (p = 0.001; Wilcoxon rank-sum test). Only samples carrying Prevotellaceae members were considered (MSM n = 84, non-MSM n = 24).
The MSM gut microbiota resembles a microbial diversity present in non-Wes microbiomes
To further compare the MSM microbiota structure in the global context, we utilized publicly available and curated shotgun metagenomic datasets from 936 healthy adult individuals representing typical Wes (n = 93) and non-Wes (n = 481) populations across nine countries as well as those from a recently Wes (or urbanized) population in China (China [urban], n = 66)18,21,33,34,35,36,37,38,39,40,41 (Table S3; see STAR Methods). These samples were profiled for quantitative taxonomic composition using MetaPhlAn 4 in the same way as samples sequenced in this study (see STAR Methods). The family Bacteroidaceae is ubiquitous in the gut microbiome of Wes populations, with Bacteroides being the most abundant genus,14,42 while Prevotellaceae/Segatella/Prevotella represent a major part in non-Wes populations.18,21,43,44,45,46 In MSM, both Bacteroidaceae and Bacteroides were significantly lower in abundance compared to both the Wes and urban Chinese populations (Bonferroni-corrected Fisher’s exact test, to Wes population, FDRs = 6.33e−13 and = 8.33e−25, respectively; to urban Chinese population, FDRs = 0.02 and = 3.51e−09, respectively) but were significantly more abundant relative to non-Wes samples (FDRs = 6.73e−11 and 9.19e−10, respectively, Figure 3A). In contrast, Prevotellaceae and Segatella/Prevotella abundances were significantly higher in MSM compared to both Wes subjects (FDRs = 7.15e−19 and 4.46e−18) and urban Chinese subjects (FDRs = 3.75e−07 and 1.32e−05) but were not different from non-Wes subjects (FDRs = 0.25 and 0.52; Figure 3A). We further extended the comparison from Prevotellaceae to the whole microbiome community, which revealed that overall MSM microbiomes were characterized by signatures that are distinct from both Wes and non-Wes samples (PERMANOVA p < 0.001; Figure 3B). Expectedly, urban Chinese samples clustered with those from Wes populations, suggesting that modern lifestyles exerted a converging effect of shaping the gut microbiome composition in geographically different populations. However, with only a few MSM scattering between the non-Wes and Wes samples in principal-coordinate analysis (n = 12 overlapped with hyperplane), a large proportion of MSM samples clustered with the non-Wes samples (n = 54 falling on the non-Wes side), and a smaller number of samples grouped with the Wes samples (n = 27 falling on the Wes side, Figure 3B). This reveals that most MSM microbiomes bear remarkable similarity to those of the non-Wes subjects, particularly for Prevotellaceae/Segatella/Prevotella accumulation and Bacteroidaceae/Bacteroides depletion, compared to the Wes subjects. Accordingly, of the top 50 abundant species in each population, MSM and non-Wes individuals shared 33 species, compared to only 15 that were shared between MSM and Wes individuals as well as 15 between MSM and urban Chinese individuals (Figure 3C). Of note, 92 out of 93 MSM in this study have European ancestry, and all have long resided in Germany, a typical Wes country.
Figure 3.

Gut microbiome community of MSM in the global context
(A) Differential abundance of Bacteroidaceae and Prevotellaceae families and Bacteroides and Segatella/Prevotella genera between MSM (n = 93), Westernized (n = 481), non-Westernized (n = 389), and urban Chinese (n= 66) samples (Wilcoxon rank-sum test with FDR correction).
(B) PCoA based on MetaPhlAn 425 microbial abundances (log2 transformed) (PERMANOVA, p < 0.001). The hyperplane drawing a boundary between Westernized and non-Westernized individuals was generated using support vector machine (SVM).
(C) Venn diagram of the number of species shared and distinct between MSM and Westernized, non-Westernized, and urban Chinese individuals, respectively. Only the 50 most abundant species were considered based on the 90th percentile in each population.
(D) Presence-absence heatmap (blue, present; white, absent) for differentially enriched species (Fisher’s exact test, FDR correction). For each comparison, only 15 species were displayed (a complete list is included in Table S3). Circles illustrate the transmissibility47 of each enriched species and ranges from 0 (low) to 1 (high) with circle size increasing proportional to the value. Country abbreviations: MDG, Madagascar; TZA, Tanzania; GHA, Ghana; ETH, Ethiopia; FJI, Fiji; JPN, Japan; ITA, Italy; SWE, Sweden; USA, United States; CHN, China.
See also Table S3.
Yet, there were other microbial traits likely to differentiate MSM from both the Wes and non-Wes individuals. Specifically, we found 216 and 187 species enriched in MSM samples relative to Wes or non-Wes samples, respectively (Table S3). In addition to multiple S. copri complex species and Prevotella members such as Prevotella pectinovora (FDR = 6.2e−28, to Wes), microbes enriched in MSM relative to both the Wes and non-Wes individuals included Megasphaera elsdenii (FDRs = 3.6e−05 and 1.7e−04, to Wes and non-Wes, respectively), Megasphaera hexanoica (FDRs = 8.7e−17 and 1.8e−19), and Paraprevotella clara (FDRs = 3.4e−12 and 9.1e−41; Figure 3D; Table S3). Interestingly, these species were previously identified to be preferentially transferred during fecal microbiota transplantation (FMT), suggesting they are engraftment-amenable genera.48 Intriguingly, a subtype of eukaryotic parasite, Blastocystis subtype 1 (FDR = 4.2e−03), which was reported as a widespread colonizer in non-Wes populations,49 was also found enriched in MSM relative to Wes subjects (Figure 3D; Table S3). On the other hand, 94 species were more abundant in Wes and 357 in non-Wes samples compared to MSM (Table S3). Wes-population-enriched species included Akkermansia muciniphila (FDR = 3.3e−03), Streptococcus infantis (FDR = 1.7e−02), and Erysipelatoclostridium ramosum (FDR = 5.0e−04) (Figure 3D; Table S3). Non-Wes-population-enriched species were represented by Klebsiella pneumoniae (FDR = 5.9e−14), Veillonella parvula (FDR = 1.7e−04), Veillonella tobetsuensis (FDR = 2.7e−05), and Haemophilus parainfluenzae (FDR = 3.5e−12) (Figure 3D; Table S3). Notably, these species belong to the least likely engrafted genera during FMT.50
Assessing engraftment rate and horizontal transmissibility of MSM-enriched species
Engraftment probabilities of microbes during FMT are influenced by complex factors such as antibiotic pre-conditioning and microbial properties compared to transmission and engraftment in non-medical settings, e.g., between familial members or partners.47,50 Hence, to examine if microbial inter-host transfer capability was related to MSM enrichment/depletion, we first assessed the engraftment rate and horizontal transmissibility of microbial species differentially abundant in MSM samples compared to non-MSM samples, utilizing previously quantified gut microbial engraftment rate and household transmissibility data47,50 (see STAR Methods). Shortly, these recent studies contributed methodological developments to the strain-level profiling of the microbiome, including the establishment of operational species-specific strain boundaries, and then quantified frequency of engraftment of microbial species upon FMT50 (engraftment rate) and of transmission within households47 (horizontal transmissibility). Overall, we found that there was no significant difference in both engraftment rate and horizontal transmissibility between MSM and non-MSM samples (Figure S3A; Table S3). Likewise, species associated with MSM and non-MSM appeared to have similar carriage patterns of engraftment-/transmission-related phenotypes including spore formation, aerotolerance, motility, and Gram-staining (Figure S3B; Table S3).
Next, we compared the engraftment rate and horizontal transmissibility of microbial species that were enriched in MSM with those enriched in the Wes and the non-Wes subjects. We observed a slightly higher mean transmissibility in MSM-enriched species when comparing MSM- to Wes-enriched microbes (Figure S3A; Table S3). As for the engraftment rate, a similar trend was observed but without significance (Figure S3A; Table S3). Analyzing engraftment-/transmission-related phenotypes also in this case did not yield a general difference between species enriched in MSM and those enriched in Wes subjects (Figure S3C; Table S3). However, compared to species enriched in MSM, those enriched in the non-Wes subjects were more likely to be Gram-negative—generally more resilient to sanitizers and disinfectants—which were expected to have enhanced gut microbial transmissibility due to longer persistence outside the human body47,51,52 (FDR = 0.02; Figure S3D; Table S3).
Sexual practices associated with the change of gut microbiome of MSM toward non-Westernization
Sexual practices can increase transmission and incidence of microbial infections, e.g., HIV, viral hepatitis, and syphilis.53,54,55 However, the influence of sexual practices on the gut microbiome remains underexplored, especially in MSM.56,57 Hence, we explored this link in a subset of our cohort (N = 52 MSM) surveyed for sexually transmitted infections (STIs) and sexual practices including receptive anal intercourse (RAI), number of sexual partners (in the last 12 months), oral sex, and condom use (Table S1). Alpha diversity was not generally associated with sexual practices, as only “condom use” reached statistical significance for Shannon diversity (padj = 0.023) and observed richness (padj = 0.016) after accounting for confounding factors (Figure S4A). Specifically, individuals who practiced condomless RAI were characterized by a higher alpha diversity of the microbiota than individuals who sometimes or always used condoms (Figure S4A). For beta diversity, a significant difference in the whole microbiome composition was found associated with “oral sex” and “number of partners” (covariate-controlled PERMANOVA padj < 0.05; Figure S4B).
As summary metrics like alpha/beta diversity and richness may not integratively reflect microbial features related to sexual practices, a random-forest-based machine learning approach was next utilized to evaluate the prediction power provided by microbiota-derived information for sexual practices. In our case, we aimed to estimate how much accuracy can be achieved to classify a sample into the corresponding categories of sexual practices (e.g., having >3 or 0–3 partners) solely based on the gut microbiome taxonomic composition. Notably, microbiome-based prediction capability was observed for all sexual practices (Figure 4A), and it reached the highest performance in identifying individuals who have either >3 or 0–3 partners (area under the receiver operating characteristic curve 0.70) (Figure 4B). Intriguingly, the number of sexual partners was the only factor linked to the relative abundance of members belonging to both Prevotellaceae and Segatella/Prevotella, with statistically higher abundances in those who had >3 sexual partners during the last 12 months (Figure S4C).
Figure 4.

Associations between gut microbiome alteration and sexual practices in MSM
(A) Association analysis between the whole microbiome composition and sexual practices assessed by machine learning prediction. The approach was based on a 20-repeated 3-fold-stratified cross-validation using random forest approach, given species-level relative abundances as input.
(B) ROC (receiver operating characteristic) curve of the learning model based on the species-level relative abundances of individuals corresponding to having 0–3 and >3 partners (in the last 12 months) using a random-forest-based approach.
(C) Species-level taxonomic biomarkers identified using LEfSe58 to associate with behaviors having >3 (and 0–3) sexual partners. Only taxonomically known species are displayed (refer to Table S4 for a complete list).
(D) UpSet plot showing overlaps of taxonomic biomarkers identified using LEfSe for sexual practices classified as risk increasing and reducing. Numbers above bars indicate the number of biomarkers, solid dots denote sexual practices, biomarkers shared by multiple practices are denoted by connected dots, and biomarkers exclusively associated with a single sexual practice are denoted by single dots.
(E) The number of taxonomic biomarkers shared by the minimum number of practice combinations in risk-increasing and -reducing categories.
(F) Boxplots showing the transmissibility (left) and engraftment rate (right) of taxonomic biomarkers associated with risk-increasing and -reducing practices.
(G) Closeness of the gut microbiome to Westernized and non-Westernized populations with respect to sexual practices and STIs. Circles show the AUC-ROC mean and error bars represent the standard deviation. Asterisk denotes the statistical significance (∗p < 0.05) by Wilcoxon rank-sum test.
Risky sexual activities such as condomless intercourse and multiple sex partners were thought to increase pathogen exchange between individuals to increase infection incidences; thus, we hypothesize they might contribute to shaping a distinctive gut microbiome in MSM as well. We next sought to identify microbial biomarkers associated with sexual practices using linear discriminant analysis effect size (LEfSe).58 For comparison, we divided sexual practices into risk-increasing and risk-reducing categories in analogy to their STI risk (see STAR Methods). Of note, we generally observed a higher number of microbial biomarkers associated with the risk-increasing behaviors, e.g., not using condoms during RAI, in comparison to risk-reducing behaviors, e.g., always using condoms during RAI (Figure 4D; Table S4). This suggests that risk-increasing activities introduced more microbial features differentiating gut microbiome structure compared to risk-reducing ones. Along these lines, having >3 sexual partners was linked to a co-existence of multiple Segatella/Prevotella members including S. copri clades B and C as well as P. pectinovora, which were mutually enriched for subjects with >3 partners (Figure 4C). Notably, the majority of identified biomarkers for specific sexual practices were exclusively associated with the specific practice, however, we noted a few biomarkers that were common to multiple practices in both risk-increasing and risk-reducing categories, respectively (Figure 4D). For example, uSGB2230 (Rikenellaceae spp.) was an overlapping biomarker across anal intercourse receivers, oral sex practitioners, STI carriers (in the last 24 months), and those not using condoms during RAI (Figure 4D; Table S4). By contrast, kSGB5075 (Lachnospira pectinoschiza) was mutually associated with those who always used condoms during RAI and had 0–3 sexual partners (in the last 12 months) (Figure 4D; Table S4). Overall, it appeared that risk-increasing behaviors share a higher number of microbial biomarkers than risk-reducing behaviors (Figures 4D and 4E; Table S4).
Next, we analyzed whether the biomarkers associated with risk-increasing practices are overall more transmissible or likely to engraft in the recipient’s gut than those with risk-reducing practices (see STAR Methods). We did not observe either a significant difference in horizontal transmissibility (Wilcoxon rank-sum test p = 0.1616) or transmission-related properties including being spore forming, anaerobic, motile, or Gram-negative (Fisher’s exact test FDR >0.05; Table S4). Counterintuitively (as pathogens tend to display more aggressive colonization strategies59), species associated with risk-reducing practices seemed to show a higher engraftment rate than those associated with risk-increasing ones (Wilcoxon rank-sum test p = 0.0001; Figure 4F).
Finally, we assessed links between the sexual practices of MSM living and raised in a Wes society and Westernization or non-Westernization of the gut microbiome. We quantified the similarity of MSM compared to Wes and non-Wes populations (see Figure 3B) based on principal coordinates calculated using Bray-Curtis distances of species-level relative abundances (see STAR Methods). Interestingly, risk-increasing behaviors, e.g., practicing oral sex, having >3 sexual partners, and not using condoms during RAI, were linked to a microbiome composition significantly closer to that of non-Wes subjects (Figure 4G; practicing oral sex p = 0.01, having >3 sexual partners p = 0.02, condomless [during RAI] p = 0.02). This suggests that specific sexual practices were associated with distinctly different microbiota in MSM that shared considerable commonality with the gut microbial composition of non-Wes individuals.
Enrichment of undescribed microbial species in the gut microbiome of MSM
The above reference-based approach allowed us to easily analyze our cohort in the context of numerous previous studies, yet this approach is limited by the existing databases, which are most likely from populations dominated by non-MSM. Thus, to characterize the microbial species that were not yet classified with described species-level taxonomy, we performed de novo metagenomic assembly on all samples in this study (see STAR Methods). Based on commonly used quality criteria,21,60 we recovered medium quality (50% < completeness ≤90% and contamination <5%) and high quality (completeness >90% and contamination <5%) genomes for a total of 6,065 putative genomes representing 765 species (Figure 5A). 183 genomes (=strains) belonged to 72 previously undescribed species, as they lacked a characterized species-level taxonomy assignment by the GTDB-Tk database61 (Table S5). These previously undescribed species were distributed across nearly all phyla represented by all genomes reconstructed in this study with the exception of Cyanobacteria, Elusimicrobiota, Methanobacteriota, and Myxococcota (Figure 5A; Table S5). Their strain genomes were significantly more prevalent in MSM samples than non-MSM samples, with 86% of MSM carrying at least one previously undescribed strain genome compared to 55% in non-MSM (Figure 5A; Table S5; Fisher’s exact test p = 3.5e−04). To estimate the read abundance of strains from previously undescribed species in MSM and non-MSM samples, we sought to align the reads of each metagenomic sample against 183 previously undescribed strain sequences. Because some strains were underrepresented in both MSM and non-MSM samples, for stringency, we only considered 130 strain sequences, which attracted, on average, >0.001% of total reads per sample from both MSM and non-MSM samples (see STAR Methods). This resulted in a considerably larger proportion of previously undescribed strain-specific reads contributed by MSM samples relative to non-MSM samples (Figure 5B; padj = 0.041; adjusted by fixed-effect linear models for confounding effects reported as above). Focusing on single, previously undescribed strains, 76% (99/130) of strain genomes appeared to attract more reads from MSM samples compared to non-MSM samples (Figure 5C). The de novo genome reconstruction complements the reference-based analysis, revealing that the MSM gut microbiome contains more frequently than the non-MSM gut microbiota not-yet-characterized bacterial species.
Figure 5.

Quantification of previously undescribed microbial species in the gut microbiome of MSM and non-MSM
(A) GTDB-Tk taxonomy structure of 765 species represented by 6,065 putative genomes reconstructed by de novo metagenomic assembly (only top seven phyla are highlighted in colors). The binary color of mustard and teal denote whether a species has been previously described or not, respectively. The gradient of magenta indicates the prevalence of species-assigned genomes in MSM and non-MSM (the color gradient was rescaled by arcsine-square root transformation for enhancing readability). Heights of the outmost histograms are in proportion to the number of genomes corresponding to the assigned species. Genome prevalence is defined as the number of species-assigned genomes divided by the number of individuals of MSM and non-MSM in percentage, respectively.
(B) The distribution of read abundance of previously undescribed strains in the metagenomic samples of MSM and non-MSM (adjusted by fixed-effect linear models,31 mean difference padj = 0.041).
(C) Read abundance stratified by strains, averaged over 93 MSM and 31 non-MSM, respectively. The read abundance of previously undescribed strains is defined as the percentage of reads mapped to previously undescribed strains to total reads in a metagenomic sample.
See also Table S5.
Discussion
Here, we describe on the species level a distinct gut microbiota with an elevated alpha diversity in MSM compared to their non-MSM counterparts regardless of anthropometric (e.g., BMI) and clinical factors (e.g., HIV infection) (Figures 1 and S1), which corroborates previous 16S rRNA gene sequencing studies performed using lower taxonomic resolution.3,4,7 Enabled by deeply sequenced metagenomes, we also identified more not-yet-characterized microbial species in the gut microbiome of MSM relative to non-MSM (Figure 5). Specifically, we uncovered thriving Prevotellaceae communities in the MSM gut microbiome including co-existing S. copri complex clades and unknown species members (Figure 2). Distinctive species features identified in this study, as well as community-level alteration patterns in MSM, are consistent with previous reports3,4,7 and implicate that identifying as MSM is a relevant factor for explaining inter-individual variability in the human gut microbiome. Notably, in another study, we recently observed a strong association of S. copri species complex members with the male gender in a large cross-sectional analysis of more than 4,000 healthy Wes individuals from 24 studies.22 Importantly, as sexual orientation is not recorded in these and many other microbiota studies, it may be an underlying confounder that is currently largely overlooked, as MSM are estimated to constitute up to 6% of the male population.62
Despite the reported association of Prevotellaceae enrichment with fiber-rich diets in Wes individuals,14,35 the increase in both prevalence and relative abundance of S. copri complex members and Prevotellaceae in MSM appears independent of diet, as only two were on a high-fiber diet based on our dietary survey in 52 participants (Table S1). Two other studies in MSM also failed to link dietary patterns and Prevotella abundance in MSM.3,4 The Prevotellaceae enrichment in MSM has been hypothesized to be related to rectal mucosal injury, as functionalities involved in injury repair were indirectly predicted based on a small group of HIV-negative MSM engaging in condomless RAI.57 Nonetheless, mechanisms underlying the Prevotellaceae expansion in MSM should be further explored regarding functional potentials, particularly in the context of rectal immune environment and mucosal inflammation.
Analyzing the gut microbiome of MSM combining publicly available metagenomic datasets covering many regions of the world (Table S3) led to the observation that the MSM microbiomes frequently bear remarkable resemblance to non-Wes microbiomes (Figure 3). This observation is in line with a similar observation by Lozupone and colleagues6: that the gut microbiome of HIV-infected persons in the US resembled that of healthy agrarians in Malawi and Venezuela, largely based on the Prevotella enrichment characteristics shared between two populations. Nonetheless, consistent with other studies,3,4,7,57 our results revealed an increased abundance and diversity of Segatella/Prevotella in MSM compared to non-MSM, independently of HIV infection (Figure 2). Moreover, the reminiscent pattern of MSM and non-Wes microbiomes was featured by numerous abundant and prevalent species beyond Segatella/Prevotella members. Because the HIV-infected subjects in many enrollment settings are predominantly MSM, comparison of the gut microbiota of HIV-infected populations with other cohorts without taking into account their MSM status might have been confounded in prior studies. Although a clear effect of Westernization on MSM gut microbiome alteration was apparent (Figure 3), owing to our approach such as selecting samples from healthy adults of different cohorts (Table S3), common confounding factors like batch effects should be better technically controlled in future studies when integrating large public datasets.
Kelly and colleagues reported for a small group of HIV-negative MSM57 that sexual practices such as condomless RAI and enema use are linked to an aberrant rectal mucosal environment concerning both immune context and microbiome composition. Mechanical damage of the mucosa and nutrients in seminal fluid might contribute to shifts of the microbiota composition toward microbes that are better adapted to the altered rectal environment. However, we found that sexual practices more than RAI exerted profound effects in differentiating the gut microbiota of MSM and driving the similarity to non-Wes microbiomes (Figure 4). Our observation of distinctive microbial features for different sexual activities suggests that alteration is an additive process driven by combinations of specific sexual practices (Figure 4). While potential confounding effects (e.g., age and BMI) were not included in the machine learning and LEfSe analysis for sexual activities due to the technique limitation to include multiple parameters, association analysis of each sexual activity and confounders revealed that age and BMI showed insignificant confounding effects (Table S4). However, HIV status and antibiotics treatment might still be linked with STI and oral sex, respectively, for which confounding effects can be better controlled with larger recruitment with improved match for covariables in the future.
As high-risk behaviors for acquiring STIs are associated with more diverse and non-Wes gut microbiota, we hypothesized that those behaviors and sexual practices also facilitate the transmission of gut bacteria. Therefore, we attempted to establish the transmission-based mechanism to explain the alteration in the MSM gut microbiome, utilizing species-resolved transmissibility scores and engraftment rates from two recent large-scale studies.47,50 While the MSM gut microbiome is rich in species with high transmissibility, e.g., S. copri, we observed only a slightly increased mean transmissibility and insignificantly different engraftment rate for species that were differentially abundant in MSM (Figure S3; Table S3). This may be partially due to limited transmission data available for MSM-associated species, with a large proportion currently lacking quantitative transmissibility scores or engraftment rates (Table S3). Moreover, the transmission measures were estimated in a global approach independent of potential transfer routes and therefore may not accurately reflect the transmission probabilities in the MSM population. For instance, specific sexual practices and higher numbers of sexual partners probably overcome transmission barriers measured in other settings, such as mother-infant transmission. For transmission quantification per se, the approach was based on species-level marker genes instead of whole-genome sequences and genetic identity without directionality inference, which might be potential caveats in the case of limited strain genetic variation, although it allowed inferring transmissibility and engraftment metrics for a much larger number of species. Since the transmission of microbes is a highly complex dynamic affected vastly by factors beyond a species’ intrinsic biology, e.g., endogenous colonization resistance,47,63 clearly more effort is required to determine biological features involved in the gut microbial transmission, to establish transmission dynamics in different scenarios, and to provide insight on the transmission routes and mechanisms.
Along these lines, our cross-sectional cohort identified a strong influence of sexual practices, especially higher numbers of sexual partners, on gut microbiota composition, yet larger cohorts comprising samples from multiple body sites (e.g., oral cavity, penis, and gut) collected at different time points from participants sharing sexual interaction networks are required to reconstruct a more detailed alteration trajectory and transmission landscape between MSM. While our study focused on MSM, it is intriguing to speculate to what degree sexual practices and numbers of sexual partners affect microbiota structure and diversity in the general population and in women who have sex with women.
Limitations of the study
We acknowledge that the self-report questionnaire might introduce biases if participants fail to reliably respond to questions. Although common anthropometric and clinical effects were taken into account in analyzing samples generated in this study, batch effects should be considered when including published samples that were generated from other studies using different methods. In addition, an increasing effort in expanding the dataset would be appreciated, particularly for enhancing the robustness of machine learning and transmissibility analysis.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Fecal samples | This study | N/A |
| Critical commercial assays | ||
| Stratec (now Invitek) Stool Collection Tubes | Stratec (now Invitek), Berlin, Germany | 1038111200 |
| Stool DNA Stabilizer PSP Spin Stool DNA | Stratec (now Invitek), Berlin, Germany | 1038120200 |
| NEBNext Ultra DNA Library Prep Kit for Illumina | New England Biolabs, Massachusetts, USA | E7645L |
| Deposited data | ||
| Raw sequencing data | This study | PRJNA947377 |
| Metagenome-assembled genomes | This study | PRJNA947377 |
| Software and algorithms | ||
| BBMap | sourceforge.net/projects/bbmap/ | https://jgi.doe.gov/data-and-tools/software-tools/bbtools/bb-tools-user-guide/bbmap-guide/ |
| MetaPhlAn 4.0 | Blanco-Miguez et al.25 | https://github.com/biobakery/MetaPhlAn |
| GTDB-Tk v1.5.0 | Chaumeil et al.61 | https://github.com/ecogenomics/gtdbtk |
| metaWRAP v1.3.2 | Uritskiy et al.64 | https://github.com/bxlab/metaWRAP |
| VAMB v3.0.2 | Nissen et al.65 | https://github.com/RasmussenLab/vamb |
| DAS Tool v1.1.1 | Sieber et al.66 | https://github.com/cmks/DAS_Tool |
| CheckM v1.0.7 | Parks et al.67 | https://ecogenomics.github.io/CheckM/ |
| Bowtie2 v2.3.5.1 | Langmead and Salzberg68 | https://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| CMSeq v1.0.0 | Pasolli et al.21 | https://github.com/SegataLab/cmseq |
| GraPhlAn | Asnicar et al.69 | https://github.com/biobakery/graphlan |
| PhyloPhlAn 3.0 | Asnicar et al.70 | https://github.com/biobakery/phylophlan |
| iTOL v6.0 | Letunic and Bork71 | https://itol.embl.de/ |
| python package hclust2 | N/A | https://github.com/SegataLab/hclust2 |
| python package scikit-bio v0.5.7 | N/A | http://scikit-bio.org/ |
| python package scikit-learn v1.1.2 | Pedregosa et al.72 | https://scikit-learn.org/ |
| python package matplotlib v3.5.0 | Hunter73 | https://matplotlib.org/ |
| python package scipy v1.7.3 | Virtanen et al.74 | https://scipy.org/ |
| python package seaborn v0.11.2 | Waskom75 | https://seaborn.pydata.org/ |
| python package pandas v1.3.5 | McKinney76 | https://pandas.pydata.org/ |
| python package statsmodels v0.12.1 | Seabold and Perktold77 | https://www.statsmodels.org/ |
| python package numpy v1.21.2 | Harris et al.78 | https://numpy.org/ |
| LEfSe v1.1.2 | Segata et al.58 | https://huttenhower.sph.harvard.edu/lefse/ |
| R package ggplot2 v3.3.6 | Wickham79 | https://ggplot2.tidyverse.org/ |
| R package ggpubr v0.4.0 | N/A | https://rpkgs.datanovia.com/ggpubr |
| R package ComplexHeatmap v2.8.0 | Gu80 | https://bioconductor.org/packages/ComplexHeatmap/ |
| R package mia v1.0.8 | N/A | https://github.com/microbiome/mia |
| R package lfe v2.8.8 | Gaure31 | https://github.com/sgaure/lfe |
| R package vegan v2.6.2 | Oksanen et al.81 | https://github.com/vegandevs/vegan |
Resource availability
Lead contact
Further information and requests for resources should be requested from the Lead Contact, Prof. Dr. Till Strowig (Till.Strowig@helmholtz-hzi.de).
Materials availability
This study did not generate new unique reagents.
Data and code availability
-
•
Shotgun metagenomic reads, detailed descriptions for each sample and metagenome-assembled genomes in this study are available under NCBI BioProject: PRJNA947377
-
•
Codes and corresponding tutorials for reproducing results in this study are avaialble at: https://github.com/KunDHuang/KunDH-2024-CRM-MSM_metagenomics
-
•
Any additional information required to reanalyze the data reported in this work is available from the lead contact upon request.
Experimental model and study participant details
This study recruited patients from two cohorts treated in the Clinic of Dermatology, Department of Venerology of the University Hospital Essen, Germany. Patients under 18 years of age were excluded for both cohorts. One cohort comprised 57 patients living with HIV of the HIV Heart study group, including 41 MSM and 16 non-MSM. The HIV-heart study is a prospective study assessing the incidence, prevalence and clinical course of cardiovascular diseases in HIV infected patients and is registered under Clinical Trials.gov (NCT01119729). The cohort was initiated for assessing differences in the gut microbiome between people living with HIV linked to coronary heart disease. All patients were treated with antiretroviral therapy. Both groups, with and without coronary heart disease were matched for gender, age, diabetes mellitus and chronic inflammatory bowel disease. The entire characteristics of the cohort is described elsewhere.82 A second cohort included 67 patients (48 HIV positive and 19 HIV negative individuals, comprising 52 MSM and 15 non-MSM), who additionally completed a questionnaire assessing information about diet, BMI, practice of RAI, use of condoms when practicing RAI, number of sexual partners within last 12 months and STIs of the gut or other gut inflammatory diseases within last 24 months to gain insights into risk factors linked to Prevotellaceae-richness of the gut microbiome (Table S1). Human sample and data collections have been performed in agreement with the guidelines of the Ethics Committee of the Medical Faculty of the University of Duisburg-Essen (permit No. 14-5874-BO and 18-8409-BO) and the European Data Protection Laws (Europäische Datenschutz-Grundverordnung DSGVO). All subjects enrolled have signed a letter of informed consent in accordance with the World Medical Association Declaration of Helsinki (Version 2013).
Method details
DNA extraction, library preparation and sequencing
DNA was extracted using the PSP Spin Stool DNA Extraction Kit (Stratec, now Invitek, Berlin, Germany) according to the manufacturer’s protocol “stool samples from difficult to lyse bacteria”. Libraries were constructed using the NEBNext Ultra DNA Library Prep Kit and sequenced on NovaSeq S4 PE150 platform with a target depth of 35,000,000 reads/sample.
Profiling species-level microbial community
For all samples in this study the metagenomic reads were first quality-checked and processed using BBMap (sourceforge.net/projects/bbmap/) with the Ensembl masked human genome GRCh38 and phiX. Afterward, we performed profiling of the species-level microbial community on the pre-processed reads by running MetaPhlAn 425 using default settings to identify microbial species and estimate the corresponding relative abundances in the metagenomic samples. To compare the MSM microbiome architecture with that of the global population, we downloaded 936 publicly available shotgun metagenomes (Table S3) spanning ten countries and different lifestyles, and subsequently profiled the species-level microbial community as described above. In each profile, we only considered species with a minimum relative abundance of 0.0001 because the precision of taxonomy identification might be compromised in the case of low-abundance members. All external metagenomes were from adult individuals including males and females who were healthy defined as free of self-reported diseases and medical intervention in order to represent general populations. Each of them was characterized by whether they were from a Westernized or non-Westernized or urban Chinese population (Table S3). The term “non-Westernized” describes a population practicing a traditional lifestyle relating to factors such as diet, hygiene, and with limited access to modern medical healthcare and pharmaceuticals (e.g., antibiotics) as previously described.18,20,21,83 Briefly, Westernization is a complex process with incremental urbanization that has been undergoing for centuries. During the process, lifestyles have changed profoundly, including but not limited to, transition from autarchic means of producing food to controlled food production chain, increased hygiene and accessibility to modern medicals, introduction of food sterilization, increased exposure to pollutants, and switch from high-fiber simple diets to high-fat high-protein processed foods. All these factors are thought to play a pivotal part in shaping the human gut microbiome structure. The concept of “Westernization” and “non-Westernization” was adopted here to demarcate populations by combining at least the majority of the factors as discussed above. Although this defines very heterogeneous populations, significance of MSM status to the gut microbiome change could be depicted in a much broader context encompassing many factors. Noteworthily, to complement the binary demarcation, we also included two Chinese cities - GuangDong and TangShan - which are located in Southern China and have been experiencing rapid urbanization in the last decade. They were used to represent populations from non-Western but relatively urbanized communities.
Alpha and beta diversity calculations
Alpha diversities were calculated using Shannon index estimated by R package mia (https://github.com/microbiome/mia) and beta diversities were calculated using Bray-Curtis dissimilarity based on species-level relative abundances, followed by a principal coordinates analysis (PCoA) for the visualization (Python package scikit-bio v0.5.7; http://scikit-bio.org/). To rule out confounding effects, the significance of differences in alpha diversity was additionally measured using fixed effects linear models31 by accounting for co-variables, including age, ethnicity, antibiotics use, BMI and HIV status. Likewise, the R package vegan v2.6.281 (https://github.com/vegandevs/vegan) was used to perform covariate-controlled PERMANOVA tests for beta diversities.
Prevotellaceae community comparison in MSM and non-MSM individuals
To compare the Prevotellaceae community structure in MSM and non-MSM gut microbiomes, we extracted Prevotellaceae members’ abundances from MetaPhlAn 4 outputs and removed samples with no Prevotellaceae members being identified. Afterward, Prevotellaceae community’s richness was calculated for the kept samples in this study, and the beta diversity of Prevotellaceae community between MSM and non-MSM individuals was estimated using extracted Prevotellaceae members’ log2-transformed abundances. Besides analyzing the whole Prevotellaceae community, we also analyzed Segatella copri complex clades due to their likely association with MSM behavior.19 In particular, we quantified the co-existing pattern of multiple S. copri complex clades in the MSM and non-MSM subjects using the method as described elsewhere.18 The Prevotellaceae members’ log2-transformed abundances were visualized in heatmap using ComplexHeatmap80 for only those with a minimum abundance of 0.1 at 90th percentile in all kept samples. To present the phylogenetic structure of all Prevotellaceae members identified in this study, we considered one representative genome for each of the 47 Prevotellaceae SGBs. The phylogenetic tree was built using PhyloPhlAn (v. 3.0)70 and by considering the 400 universal marker genes available in PhyloPhlAn. iTOL tool71 (v. 6.0) was then used to integrate the information about a member’s prevalence in MSM/non-MSM and if it belongs to kSGB or uSGB in the tree.71
Analyzing MSM microbiome architecture in a global context
To contextualize the MSM microbiome structure in the global population, we used MetaPhlAn 4 outputs to generate a matrix including microbial abundances from 93 MSM individuals of this study and 936 downloaded metagenomic samples which include typical Westernized, non-Westernized societies, and urban Chinese communities whose lifestyles are transitioning into more modern ones18,21,33,34,35,36,37,38,39,40,41 (Table S3). Individual abundance profiles were merged into one matrix using merge_metaphlan_tables.py, a utility script in MetaPhlAn 4. Afterward, beta diversity based on log2-transformed relative abundances was calculated on the merged matrix as described above in order to understand the overall difference between MSM microbial community and that of global populations. Afterward, a hyperlane was created using Support Vector Machine (SVM) to draw a boundary between Westernized and non-Westernized individuals (a python package scikit-learn v1.1.2; https://scikit-learn.org/). For identifying the top 50 abundant species in the population of MSM, Westernized and non-Westernized and urban Chinese individuals respectively, we used a python package hclust2 (https://github.com/SegataLab/hclust2) with parameter setting of --fperc 90 --ftop 50 --f_dist_f correlation --s_dist_f braycurtis -s --slinkage complete. To further understand the difference at the single species level, we used the merged matrix for identifying single species which are significantly enriched in MSM individuals, Westernized or non-Westernized (MSM vs. Westernized, MSM vs. Non-Westernized, Westernized vs. MSM and Non-Westernized vs. MSM) using Bonferroni-corrected Fisher’s exact test as described elsewhere.18,60
Assessing engraftment rate, horizontal transmissibility and relevant biological properties
To examine the horizontal transmissibility, engraftment rate and transmission-related properties including spore formation, anaerobic growth, motility and Gram-negative staining, we retrieved the per-species basis measures from two integrative multi-cohort studies by Valles-Colomer et al., and Ianiro et al.,47,50 respectively. Briefly, Valles-Colomer and colleagues first aggregated a multi-cohort dataset comprising 9,715 human metagenomic samples with curated metadata on subject identifiers, time points, participant’s age, gender, delivery mode, family identifiers, family relationships, two zygosity and age at which twins moved apart, village, and country. Species-level profiling was performed for all samples using MetaPhlAn 425 based on the SGB database same as for the 124 samples from this study. Strain-level profiling was subsequently performed for detected SGBs (complying with SGB selection criteria reported elsewhere47) using StrainPhlAn4,25,84 which estimates phylogenetic distances between strains from different samples for each SGB. For inferring horizontal transmissibility, Valles-Colomer and colleagues used StrainPhlAn profiles of 3,192 samples whose subjects shared a household, and calculated the species horizontal transmissibility following strain-sharing-inference pipeline (https://github.com/biobakery/MetaPhlAn/wiki/Strain-Sharing-Inference). In the original study, transmission-related properties were predicted for all SGBs using Traitar (v 1.1.12)85 on the 50% core genes. On the other hand, Ianiro and colleagues quantified engraftment rate using 1,371 fecal metagenomic samples specific to FMT settings, which mainly reflects the extent of vertical microbiome transmission between donors and recipients. Microbiome species profiles and strain profiles were generated in the same way as described above, which were subsequently used to quantify FMT engraftment rate following the strain-sharing-pipeline (https://github.com/SegataLab/Strain-sharing-pipeline).
As microbial species identified in this study were based on the same SGB database as for those from two large studies, we next annotated the differentially enriched species in this study for their corresponding horizontal transmissibility, engraftment rate and transmission-related properties using the quantifications from Valles-Colomer et al., and Ianiro et al., . For biological properties involved in microbial transmission, we used Fisher’s exact test to examine if any of the properties was differentially present in species sets that were enriched in MSM relative to non-MSM, Westernized and non-Westernized individuals, respectively. Afterward, Benjamini-Hochberg methods were used for correcting multiple hypotheses comparison.
Analysis of associations between sexual practices and microbiome composition
To analyze links between sexual practices and gut microbiome characteristics in MSM, we generated six datasets merging MetaPhlAn 4 species-level relative abundances, respectively, each represented by RAI (Yes and No), sexually transmitted infections (Positive and Negative), oral sex (Yes and No), number of partners (0–3 and >3) and condom use during RAI (Always and No). These datasets were then used for assessing the association between sexual practices and microbiome composition based on machine learning, for identifying practice-associated taxonomic biomarker and for estimating microbiome closeness for sexual practices to Westernization and non-Westernization as elaborated below.
Sexual practice-microbiome association analysis using random forest-based machine learning approach
All learning experiments depended on random forest implemented by a python package scikit-learn v1.1.212,86 as it has been shown to reach overall a better performance for microbiome data compared to other approaches.87,88 We created a learning model using 1,000 estimator trees and Shannon entropy to evaluate the quality for each node splitting of a tree. In the hyperparameter setting, we assigned at least one sample per leaf and 30% of features for a tree as suggested elsewhere.89,90 The prediction capability for each dataset was evaluated by stratified 3-fold cross-validation to ensure that each partition has enough and balanced binary cases. The procedure of dataset folding and model evaluation was repeated 20 times with a result of an average over 60 validation folds per practice.
Identifying taxonomic biomarkers
On a per-dataset basis, linear discriminant analysis effect size (LEfSe) with default parameter settings58 was used to identify microbial features that were statistically different between sexual practices (e.g., RAI: Yes vs. RAI: No) and to estimate their effect size represented by logarithmic LDA score. Features with a logarithmic LDA score over 2.0 were considered significantly discriminant.
Grouping sexual practices based on risk analogous to STI risk
Risk-increasing category was represented by receiving anal intercourse (RAI: Yes), having >3 sexual partners (#partners: >3), practicing oral sex (Oral sex: Yes), diagnosed with STI in the last 24 months (STI: Positive) or not using condom during RAI (Condom use (during RAI): No). By contrast, risk-reducing category included not receiving anal intercourse (RAI: No), having 0–3 sexual partners (#partners: 0–3), no oral sex (Oral sex: No), free of STI in the last 24 months (STI: Negative) or always using condom during RAI (Condom use (during RAI): Always).
Estimating microbiome similarity to Westernized and non-Westernized microbiomes
We measured the closeness of each sexual practice in MSM to the Westernized and non-Westernized based on the MSM gut microbiome community composition in a global context (Figure 3B). Firstly, species-level relative abundances of 870 publicly available samples (Westernized N = 481; non-Westernized N = 389) were merged with the dataset of each sexual practice, resulting in six expanded datasets additionally containing global samples characterized by Westernization and non-Westernization. Secondly, Bray-Curtis distances on a per-dataset basis were measured, based on which principal coordinates were estimated for each sample, with only the first two coordinates (PC1 and PC2) being selected to determine a sample’s placement. We finally calculated the coordinates distance between each practice-characterized sample and the centroid of the Westernized and non-Westernized samples respectively. The closeness was defined as the complement of the distance (e.g., closeness = 1 - distance).
Metagenomic assembly and estimation of previously undescribed microbial strains
In order to complement the reference-based analysis, metagenomic assembly was performed in order to reconstruct putative strain genomes following the metaWRAP pipeline64 (version 1.3.2). Briefly, for each of 124 metagenomic samples from this study, reads were first assembled into contigs by a de novo metagenome assembler metaSpades91 whose performance has been reported to outstand among other similar approaches.21,92,93 Afterward, contigs were binned using five different binning tools including metabat1,94 metabat295 and maxbin296 which have been implemented in metaWRAP pipeline already and two external methods (VAMB65 and DAStool66). The genome bins generated by five approaches were further consolidated using bin_refinement module in the metaWRAP pipeline for producing putative genomes. The putative genomes were assessed for completeness, contamination and strain heterogeneity using CheckM67 (version 1.0.7; lineage specific workflow). We retained only medium-quality (MQ) genomes (50% < completeness ≤90% and contamination ≤5%) and high-quality genomes (completeness >90% and contamination ≤5%) for the analysis to follow as suggested in the previous studies.21,60 Finally, GTDB-Tk61 (v.1.5.0; classify_wf module; default settings) was used to assign species-level taxonomy to qualified genomes; genomes lacking known species-level taxonomy were identified as previously undescribed microbial strains. The taxonomy structure of genomes was visualized using GraPhlAn.69
To estimate the read abundance of previously undescribed microbial strains in the metagenomic samples, we aligned metagenomic reads for each sample in this study against genome sequences of previously undescribed strains using bowtie268 (v2.3.5.1; --end-to-end --no-unal -U -S) to generate read alignments in bam files. The resulting bam files, based on each previously undescribed strain sequence, were further cleaned using a python package CMSeq (https://github.com/SegataLab/cmseq) with following criteria: (1) reads alignment quality ≥ 30, (2) reads coverage depth ≥ 5-folds, (3) minimum identity of reads ≥ 96%, (4) aligned read length ≥ 90 nt, (5) minimum dominant allele frequency ≥ 50%, (6) previously undescribed strain sequence coverage breadth ≥ 80%. The read abundance of previously undescribed strains for each sample was then calculated by dividing the filtered aligned reads by the total number of reads of a metagenomic sample. This approach has been validated by other studies for retaining strain-specific reads.18,93,97 Strains were not considered in reads alignment estimation if the percentage of aligned reads account for, on average, <0.001% of total reads per sample.
Quantification and statistical analysis
All statistical significance for mean difference between categories was assessed using the two-tailed Wilcoxon rank-sum test. Statistical significance for multivariate analysis was performed using PERMANOVA test controlled for confounding factors including ethnicity, antibiotics use, BMI, and HIV infection. Statistical significance for enrichment analysis was performed using Fisher’s exact test corrected with the Bonferroni method for multiple hypothesis testing. Associations with transmission-related properties were tested using Fisher’s exact test followed by Benjamini-Hochberg correction. Tests were considered as significant if p values <0.05 (Padj < 0.05 in the case of adjusting for confounding effects or FDR <0.05 in the case of multiple hypothesis correction).
Acknowledgments
We acknowledge and appreciate those participants whose clinical, anthropometric, and microbiome data were analyzed for this research. We thank members of the T.S. laboratory, especially Caroline Taouk, for the discussion and critical reading of the manuscript and Pavaret Sivapornnukul for assistance with metagenomic data pre-processing. We thank the high-performance computing team at Helmholtz Center for Infection Research for consistent technical support. This research was supported by the association of “Förderverein des HZI” (Singh-Chhatwal Postdoctoral Fellowship to K.D.H.). Funding was provided by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy - EXC 2155 - project number 390874280.
Author contributions
Conceptualization, K.D.H., T.S., and J.K.; resources, K.D.H., S.E., L.A., A.B., N.S., T.S., and J.K.; data curation, K.D.H., L.A., and A.B.; methodology, K.D.H., T.S., and J.K.; investigation, K.D.H., L.A., E.J.C.G., T.-R.L., R.d.O., A.B.-M., M.V.-C., I.R., E.P., J.B., N.S., S.E., T.S., and J.K.; writing – original draft, K.D.H., T.S., and J.K.; writing – review & editing, K.D.H., L.A., E.J.C.G., M.V.-C., T.S., and J.K.; formal analysis, K.D.H., L.A., T.-R.L., A.B.-M., I.R., and E.P.; visualization, K.D.H. and E.P.; supervision, K.D.H., T.S., and J.K.
Declaration of interests
The authors declare no competing interests.
Published: February 15, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2024.101426.
Contributor Information
Till Strowig, Email: till.strowig@helmholtz-hzi.de.
Jan Kehrmann, Email: jan.kehrmann@uk-essen.de.
Supplemental information
The overall information of each project in the publicly available datasets utilized in this study and samples were from healthy adults (18–65 years) (Tab 1). Sample-wise characterization by geography and Westernization for each project in the dataset (Tab 2). Species enriched in MSM individuals relative to the Westernized (Tab 3). Species enriched in MSM individuals relative to the non-Westernized (Tab 4). Species enriched in the Westernized populations compared to MSM individuals (Tab 5). Species enriched in the non-Westernized populations compared to MSM individuals (Tab 6). Species differentially abundant in MSM and non-MSM samples (Tab 7). The False Discovery Rates (FDRs) were estimated by Fisher’s exact test corrected for multiple hypotheses using Bonferroni method. FDRs lower than 0.05 were considered significant. Horizontal transmissibility measures, engraftment rates and phenotypes involved in microbial transmission (including spore former, anaerobic growth, motility and Gram-negative staining) were retrieved from other two studies [S4],[S5]. Horizontal transmissibility and engraftment rate are within a range of 0–1. For phenotypes (spore former, anaerobic growth, motility and Gram-negative staining), 1 indicates presence and 0 indicates absence. Missing values were supplemented if not publicly available.
The significance of a biomarker for each sexual practice was indicated by effect size measures (LDA score (log 10)). Horizontal transmissibility measures, engraftment rates and phenotypes involved in microbial transmission (including spore formation, anaerobic growth, motility and Gram-negative staining) were retrieved from other two studies [S4],[S5]. For phenotypes (spore formation, anaerobic growth, motility and Gram-negative staining), 1 indicates presence and 0 indicates absence. Missing values were supplemented if not publicly available (Tab 1). Association significance of sexual practices and confounding effects, assessed by Fisher’s exact test with Benjamini-Hochberg method for multiple comparison correction. Values indicate FDRs of the association between a sexual practice and a confounding effect. Association is considered significant when FDR <0.05 (Tab 2).
The genome prevalence in MSM/non-MSM was quantified as, for each species, dividing the total number of genomes by the number of MSM and non-MSM individuals, respectively.
References
- 1.Rothschild D., Weissbrod O., Barkan E., Kurilshikov A., Korem T., Zeevi D., Costea P.I., Godneva A., Kalka I.N., Bar N., et al. Environment dominates over host genetics in shaping human gut microbiota. Nature. 2018;555:210–215. doi: 10.1038/nature25973. [DOI] [PubMed] [Google Scholar]
- 2.Scepanovic P., Hodel F., Mondot S., Partula V., Byrd A., Hammer C., Alanio C., Bergstedt J., Patin E., Touvier M., et al. A comprehensive assessment of demographic, environmental, and host genetic associations with gut microbiome diversity in healthy individuals. Microbiome. 2019;7:130. doi: 10.1186/s40168-019-0747-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Noguera-Julian M., Rocafort M., Guillén Y., Rivera J., Casadellà M., Nowak P., Hildebrand F., Zeller G., Parera M., Bellido R., et al. Gut Microbiota Linked to Sexual Preference and HIV Infection. EBioMedicine. 2016;5:135–146. doi: 10.1016/j.ebiom.2016.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Armstrong A.J.S., Shaffer M., Nusbacher N.M., Griesmer C., Fiorillo S., Schneider J.M., Preston Neff C., Li S.X., Fontenot A.P., Campbell T., et al. An exploration of Prevotella-rich microbiomes in HIV and men who have sex with men. Microbiome. 2018;6:198. doi: 10.1186/s40168-018-0580-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hitch T.C.A., Bisdorf K., Afrizal A., Riedel T., Overmann J., Strowig T., Clavel T. A taxonomic note on the genus Prevotella: Description of four novel genera and emended description of the genera Hallella and Xylanibacter. Syst. Appl. Microbiol. 2022;45 doi: 10.1016/j.syapm.2022.126354. [DOI] [PubMed] [Google Scholar]
- 6.Lozupone C.A., Li M., Campbell T.B., Flores S.C., Linderman D., Gebert M.J., Knight R., Fontenot A.P., Palmer B.E. Alterations in the gut microbiota associated with HIV-1 infection. Cell Host Microbe. 2013;14:329–339. doi: 10.1016/j.chom.2013.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vujkovic-Cvijin I., Sortino O., Verheij E., Sklar J., Wit F.W., Kootstra N.A., Sellers B., Brenchley J.M., Ananworanich J., Loeff M.S.v.d., et al. HIV-associated gut dysbiosis is independent of sexual practice and correlates with noncommunicable diseases. Nat. Commun. 2020;11:2448. doi: 10.1038/s41467-020-16222-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen Y., Lin H., Cole M., Morris A., Martinson J., Mckay H., Mimiaga M., Margolick J., Fitch A., Methe B., et al. Signature changes in gut microbiome are associated with increased susceptibility to HIV-1 infection in MSM. Microbiome. 2021;9:237. doi: 10.1186/s40168-021-01168-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vujkovic-Cvijin I., Dunham R.M., Iwai S., Maher M.C., Albright R.G., Broadhurst M.J., Hernandez R.D., Lederman M.M., Huang Y., Somsouk M., et al. Dysbiosis of the gut microbiota is associated with HIV disease progression and tryptophan catabolism. Sci. Transl. Med. 2013;5 doi: 10.1126/scitranslmed.3006438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dillon S.M., Lee E.J., Kotter C.V., Austin G.L., Dong Z., Hecht D.K., Gianella S., Siewe B., Smith D.M., Landay A.L., et al. An altered intestinal mucosal microbiome in HIV-1 infection is associated with mucosal and systemic immune activation and endotoxemia. Mucosal Immunol. 2014;7:983–994. doi: 10.1038/mi.2013.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scher J.U., Sczesnak A., Longman R.S., Segata N., Ubeda C., Bielski C., Rostron T., Cerundolo V., Pamer E.G., Abramson S.B., et al. Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. Elife. 2013;2 doi: 10.7554/eLife.01202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pedersen H.K., Gudmundsdottir V., Nielsen H.B., Hyotylainen T., Nielsen T., Jensen B.A.H., Forslund K., Hildebrand F., Prifti E., Falony G., et al. Human gut microbes impact host serum metabolome and insulin sensitivity. Nature. 2016;535:376–381. doi: 10.1038/nature18646. [DOI] [PubMed] [Google Scholar]
- 13.David L.A., Maurice C.F., Carmody R.N., Gootenberg D.B., Button J.E., Wolfe B.E., Ling A.V., Devlin A.S., Varma Y., Fischbach M.A., et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2014;505:559–563. doi: 10.1038/nature12820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu G.D., Chen J., Hoffmann C., Bittinger K., Chen Y.-Y., Keilbaugh S.A., Bewtra M., Knights D., Walters W.A., Knight R., et al. Linking long-term dietary patterns with gut microbial enterotypes. Science. 2011;334:105–108. doi: 10.1126/science.1208344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kovatcheva-Datchary P., Nilsson A., Akrami R., Lee Y.S., De Vadder F., Arora T., Hallen A., Martens E., Björck I., Bäckhed F. Dietary Fiber-Induced Improvement in Glucose Metabolism Is Associated with Increased Abundance of Prevotella. Cell Metab. 2015;22:971–982. doi: 10.1016/j.cmet.2015.10.001. [DOI] [PubMed] [Google Scholar]
- 16.Roager H.M., Vogt J.K., Kristensen M., Hansen L.B.S., Ibrügger S., Mærkedahl R.B., Bahl M.I., Lind M.V., Nielsen R.L., Frøkiær H., et al. Whole grain-rich diet reduces body weight and systemic low-grade inflammation without inducing major changes of the gut microbiome: a randomised cross-over trial. Gut. 2019;68:83–93. doi: 10.1136/gutjnl-2017-314786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vitaglione P., Mennella I., Ferracane R., Rivellese A.A., Giacco R., Ercolini D., Gibbons S.M., La Storia A., Gilbert J.A., Jonnalagadda S., et al. Whole-grain wheat consumption reduces inflammation in a randomized controlled trial on overweight and obese subjects with unhealthy dietary and lifestyle behaviors: role of polyphenols bound to cereal dietary fiber. Am. J. Clin. Nutr. 2015;101:251–261. doi: 10.3945/ajcn.114.088120. [DOI] [PubMed] [Google Scholar]
- 18.Tett A., Huang K.D., Asnicar F., Fehlner-Peach H., Pasolli E., Karcher N., Armanini F., Manghi P., Bonham K., Zolfo M., et al. The Prevotella copri Complex Comprises Four Distinct Clades Underrepresented in Westernized Populations. Cell Host Microbe. 2019;26:666–679.e7. doi: 10.1016/j.chom.2019.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Iljazovic A., Amend L., Galvez E.J.C., de Oliveira R., Strowig T. Modulation of inflammatory responses by gastrointestinal Prevotella spp. - From associations to functional studies. Int. J. Med. Microbiol. 2021;311 doi: 10.1016/j.ijmm.2021.151472. [DOI] [PubMed] [Google Scholar]
- 20.Maixner F., Sarhan M.S., Huang K.D., Tett A., Schoenafinger A., Zingale S., Blanco-Míguez A., Manghi P., Cemper-Kiesslich J., Rosendahl W., et al. Hallstatt miners consumed blue cheese and beer during the Iron Age and retained a non-Westernized gut microbiome until the Baroque period. Curr. Biol. 2021;31:5149–5162.e6. doi: 10.1016/j.cub.2021.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pasolli E., Asnicar F., Manara S., Zolfo M., Karcher N., Armanini F., Beghini F., Manghi P., Tett A., Ghensi P., et al. Extensive Unexplored Human Microbiome Diversity Revealed by Over 150,000 Genomes from Metagenomes Spanning Age, Geography, and Lifestyle. Cell. 2019;176:649–662.e20. doi: 10.1016/j.cell.2019.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blanco-Míguez A., Gálvez E.J.C., Pasolli E., De Filippis F., Amend L., Huang K.D., Manghi P., Lesker T.-R., Riedel T., Cova L., et al. Extension of the Segatella copri complex to 13 species with distinct large extrachromosomal elements and associations with host conditions. Cell Host Microbe. 2023;31:1804–1819.e9. doi: 10.1016/j.chom.2023.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cook R.R., Fulcher J.A., Tobin N.H., Li F., Lee D., Javanbakht M., Brookmeyer R., Shoptaw S., Bolan R., Aldrovandi G.M., Gorbach P.M. Effects of HIV viremia on the gastrointestinal microbiome of young MSM. AIDS. 2019;33:793–804. doi: 10.1097/QAD.0000000000002132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li S.X., Sen S., Schneider J.M., Xiong K.-N., Nusbacher N.M., Moreno-Huizar N., Shaffer M., Armstrong A.J.S., Severs E., Kuhn K., et al. Gut microbiota from high-risk men who have sex with men drive immune activation in gnotobiotic mice and in vitro HIV infection. PLoS Pathog. 2019;15 doi: 10.1371/journal.ppat.1007611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blanco-Míguez A., Beghini F., Cumbo F., McIver L.J., Thompson K.N., Zolfo M., Manghi P., Dubois L., Huang K.D., Thomas A.M., et al. Extending and improving metagenomic taxonomic profiling with uncharacterized species using MetaPhlAn 4. Nat. Biotechnol. 2023;41:1633–1644. doi: 10.1038/s41587-023-01688-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tuddenham S., Koay W.L., Sears C. HIV, Sexual Orientation, and Gut Microbiome Interactions. Dig. Dis. Sci. 2020;65:800–817. doi: 10.1007/s10620-020-06110-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Deschasaux M., Bouter K.E., Prodan A., Levin E., Groen A.K., Herrema H., Tremaroli V., Bakker G.J., Attaye I., Pinto-Sietsma S.-J., et al. Depicting the composition of gut microbiota in a population with varied ethnic origins but shared geography. Nat. Med. 2018;24:1526–1531. doi: 10.1038/s41591-018-0160-1. [DOI] [PubMed] [Google Scholar]
- 28.Maier L., Goemans C.V., Wirbel J., Kuhn M., Eberl C., Pruteanu M., Müller P., Garcia-Santamarina S., Cacace E., Zhang B., et al. Unravelling the collateral damage of antibiotics on gut bacteria. Nature. 2021;599:120–124. doi: 10.1038/s41586-021-03986-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goodrich J.K., Waters J.L., Poole A.C., Sutter J.L., Koren O., Blekhman R., Beaumont M., Van Treuren W., Knight R., Bell J.T., et al. Human genetics shape the gut microbiome. Cell. 2014;159:789–799. doi: 10.1016/j.cell.2014.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Loftfield E., Herzig K.-H., Caporaso J.G., Derkach A., Wan Y., Byrd D.A., Vogtmann E., Männikkö M., Karhunen V., Knight R., et al. Association of Body Mass Index with Fecal Microbial Diversity and Metabolites in the Northern Finland Birth Cohort. Cancer Epidemiol. Biomarkers Prev. 2020;29:2289–2299. doi: 10.1158/1055-9965.EPI-20-0824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gaure S. OLS with multiple high dimensional category variables. Comput. Stat. Data Anal. 2013;66:8–18. [Google Scholar]
- 32.Fulcher J.A., Li F., Tobin N.H., Zabih S., Elliott J., Clark J.L., D’Aquila R., Mustanski B., Kipke M.D., Shoptaw S., et al. Gut dysbiosis and inflammatory blood markers precede HIV with limited changes after early seroconversion. EBioMedicine. 2022;84 doi: 10.1016/j.ebiom.2022.104286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bäckhed F., Roswall J., Peng Y., Feng Q., Jia H., Kovatcheva-Datchary P., Li Y., Xia Y., Xie H., Zhong H., et al. Dynamics and Stabilization of the Human Gut Microbiome during the First Year of Life. Cell Host Microbe. 2015;17 doi: 10.1016/j.chom.2015.04.004. 852–703. [DOI] [PubMed] [Google Scholar]
- 34.Brito I.L., Yilmaz S., Huang K., Xu L., Jupiter S.D., Jenkins A.P., Naisilisili W., Tamminen M., Smillie C.S., Wortman J.R., et al. Mobile genes in the human microbiome are structured from global to individual scales. Nature. 2016;535:435–439. doi: 10.1038/nature18927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.De Filippis F., Pasolli E., Tett A., Tarallo S., Naccarati A., De Angelis M., Neviani E., Cocolin L., Gobbetti M., Segata N., Ercolini D. Distinct Genetic and Functional Traits of Human Intestinal Prevotella copri Strains Are Associated with Different Habitual Diets. Cell Host Microbe. 2019;25:444–453.e3. doi: 10.1016/j.chom.2019.01.004. [DOI] [PubMed] [Google Scholar]
- 36.Human Microbiome Project Consortium Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rampelli S., Schnorr S.L., Consolandi C., Turroni S., Severgnini M., Peano C., Brigidi P., Crittenden A.N., Henry A.G., Candela M. Metagenome Sequencing of the Hadza Hunter-Gatherer Gut Microbiota. Curr. Biol. 2015;25:1682–1693. doi: 10.1016/j.cub.2015.04.055. [DOI] [PubMed] [Google Scholar]
- 38.Smits S.A., Leach J., Sonnenburg E.D., Gonzalez C.G., Lichtman J.S., Reid G., Knight R., Manjurano A., Changalucha J., Elias J.E., et al. Seasonal cycling in the gut microbiome of the Hadza hunter-gatherers of Tanzania. Science. 2017;357:802–806. doi: 10.1126/science.aan4834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yachida S., Mizutani S., Shiroma H., Shiba S., Nakajima T., Sakamoto T., Watanabe H., Masuda K., Nishimoto Y., Kubo M., et al. Metagenomic and metabolomic analyses reveal distinct stage-specific phenotypes of the gut microbiota in colorectal cancer. Nat. Med. 2019;25:968–976. doi: 10.1038/s41591-019-0458-7. [DOI] [PubMed] [Google Scholar]
- 40.He Q., Gao Y., Jie Z., Yu X., Laursen J.M., Xiao L., Li Y., Li L., Zhang F., Feng Q., et al. Two distinct metacommunities characterize the gut microbiota in Crohn’s disease patients. GigaScience. 2017;6:1–11. doi: 10.1093/gigascience/gix050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li J., Zhao F., Wang Y., Chen J., Tao J., Tian G., Wu S., Liu W., Cui Q., Geng B., et al. Gut microbiota dysbiosis contributes to the development of hypertension. Microbiome. 2017;5:14. doi: 10.1186/s40168-016-0222-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Arumugam M., Raes J., Pelletier E., Le Paslier D., Yamada T., Mende D.R., Fernandes G.R., Tap J., Bruls T., Batto J.-M., et al. Enterotypes of the human gut microbiome. Nature. 2011;473:174–180. doi: 10.1038/nature09944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.De Filippo C., Cavalieri D., Di Paola M., Ramazzotti M., Poullet J.B., Massart S., Collini S., Pieraccini G., Lionetti P. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Natl. Acad. Sci. USA. 2010;107:14691–14696. doi: 10.1073/pnas.1005963107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yatsunenko T., Rey F.E., Manary M.J., Trehan I., Dominguez-Bello M.G., Contreras M., Magris M., Hidalgo G., Baldassano R.N., Anokhin A.P., et al. Human gut microbiome viewed across age and geography. Nature. 2012;486:222–227. doi: 10.1038/nature11053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Obregon-Tito A.J., Tito R.Y., Metcalf J., Sankaranarayanan K., Clemente J.C., Ursell L.K., Zech Xu Z., Van Treuren W., Knight R., Gaffney P.M., et al. Subsistence strategies in traditional societies distinguish gut microbiomes. Nat. Commun. 2015;6:6505. doi: 10.1038/ncomms7505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schnorr S.L., Candela M., Rampelli S., Centanni M., Consolandi C., Basaglia G., Turroni S., Biagi E., Peano C., Severgnini M., et al. Gut microbiome of the Hadza hunter-gatherers. Nat. Commun. 2014;5:3654. doi: 10.1038/ncomms4654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Valles-Colomer M., Blanco-Míguez A., Manghi P., Asnicar F., Dubois L., Golzato D., Armanini F., Cumbo F., Huang K.D., Manara S., et al. The person-to-person transmission landscape of the gut and oral microbiomes. Nature. 2023;614:125–135. doi: 10.1038/s41586-022-05620-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Podlesny D., Durdevic M., Paramsothy S., Kaakoush N.O., Högenauer C., Gorkiewicz G., Walter J., Fricke W.F. Identification of clinical and ecological determinants of strain engraftment after fecal microbiota transplantation using metagenomics. Cell Rep. Med. 2022;3 doi: 10.1016/j.xcrm.2022.100711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Beghini F., Pasolli E., Truong T.D., Putignani L., Cacciò S.M., Segata N. Large-scale comparative metagenomics of Blastocystis, a common member of the human gut microbiome. ISME J. 2017;11:2848–2863. doi: 10.1038/ismej.2017.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ianiro G., Punčochář M., Karcher N., Porcari S., Armanini F., Asnicar F., Beghini F., Blanco-Míguez A., Cumbo F., Manghi P., et al. Variability of strain engraftment and predictability of microbiome composition after fecal microbiota transplantation across different diseases. Nat. Med. 2022;28:1913–1923. doi: 10.1038/s41591-022-01964-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mahnert A., Moissl-Eichinger C., Zojer M., Bogumil D., Mizrahi I., Rattei T., Martinez J.L., Berg G. Man-made microbial resistances in built environments. Nat. Commun. 2019;10:968. doi: 10.1038/s41467-019-08864-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wickham G. An investigation into the relative resistances of common bacterial pathogens to quaternary ammonium cation disinfectants. Bioscience Horizons. 2017;10:hzx008. [Google Scholar]
- 53.Dosekun O., Fox J. An overview of the relative risks of different sexual behaviours on HIV transmission. Curr. Opin. HIV AIDS. 2010;5:291–297. doi: 10.1097/COH.0b013e32833a88a3. [DOI] [PubMed] [Google Scholar]
- 54.Szmuness W., Much I., Prince A.M., Hoofnagle J.H., Cherubin C.E., Harley E.J., Block G.H. On the role of sexual behavior in the spread of hepatitis B infection. Ann. Intern. Med. 1975;83:489–495. doi: 10.7326/0003-4819-83-4-489. [DOI] [PubMed] [Google Scholar]
- 55.Holtgrave D.R., Crosby R.A. Social determinants of tuberculosis case rates in the United States. Am. J. Prev. Med. 2004;26:159–162. doi: 10.1016/j.amepre.2003.10.014. [DOI] [PubMed] [Google Scholar]
- 56.Pescatore N.A., Pollak R., Kraft C.S., Mulle J.G., Kelley C.F. Short Communication: Anatomic Site of Sampling and the Rectal Mucosal Microbiota in HIV Negative Men Who Have Sex with Men Engaging in Condomless Receptive Anal Intercourse. AIDS Res. Hum. Retroviruses. 2018;34:277–281. doi: 10.1089/aid.2017.0206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kelley C.F., Kraft C.S., de Man T.J., Duphare C., Lee H.-W., Yang J., Easley K.A., Tharp G.K., Mulligan M.J., Sullivan P.S., et al. The rectal mucosa and condomless receptive anal intercourse in HIV-negative MSM: implications for HIV transmission and prevention. Mucosal Immunol. 2017;10:996–1007. doi: 10.1038/mi.2016.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Segata N., Izard J., Waldron L., Gevers D., Miropolsky L., Garrett W.S., Huttenhower C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12:R60. doi: 10.1186/gb-2011-12-6-r60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Browne H.P., Neville B.A., Forster S.C., Lawley T.D. Transmission of the gut microbiota: spreading of health. Nat. Rev. Microbiol. 2017;15:531–543. doi: 10.1038/nrmicro.2017.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wibowo M.C., Yang Z., Borry M., Hübner A., Huang K.D., Tierney B.T., Zimmerman S., Barajas-Olmos F., Contreras-Cubas C., García-Ortiz H., et al. Reconstruction of ancient microbial genomes from the human gut. Nature. 2021;594:234–239. doi: 10.1038/s41586-021-03532-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chaumeil P.-A., Mussig A.J., Hugenholtz P., Parks D.H. GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics. 2019;36:1925–1927. doi: 10.1093/bioinformatics/btz848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Grey J.A., Bernstein K.T., Sullivan P.S., Purcell D.W., Chesson H.W., Gift T.L., Rosenberg E.S. Estimating the Population Sizes of Men Who Have Sex With Men in US States and Counties Using Data From the American Community Survey. JMIR Public Health Surveill. 2016;2:e14. doi: 10.2196/publichealth.5365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sarkar A., Harty S., Johnson K.V.-A., Moeller A.H., Archie E.A., Schell L.D., Carmody R.N., Clutton-Brock T.H., Dunbar R.I.M., Burnet P.W.J. Microbial transmission in animal social networks and the social microbiome. Nat. Ecol. Evol. 2020;4:1020–1035. doi: 10.1038/s41559-020-1220-8. [DOI] [PubMed] [Google Scholar]
- 64.Uritskiy G.V., DiRuggiero J., Taylor J. MetaWRAP-a flexible pipeline for genome-resolved metagenomic data analysis. Microbiome. 2018;6:158. doi: 10.1186/s40168-018-0541-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nissen J.N., Johansen J., Allesøe R.L., Sønderby C.K., Armenteros J.J.A., Grønbech C.H., Jensen L.J., Nielsen H.B., Petersen T.N., Winther O., Rasmussen S. Improved metagenome binning and assembly using deep variational autoencoders. Nat. Biotechnol. 2021;39:555–560. doi: 10.1038/s41587-020-00777-4. [DOI] [PubMed] [Google Scholar]
- 66.Sieber C.M.K., Probst A.J., Sharrar A., Thomas B.C., Hess M., Tringe S.G., Banfield J.F. Recovery of genomes from metagenomes via a dereplication, aggregation and scoring strategy. Nat. Microbiol. 2018;3:836–843. doi: 10.1038/s41564-018-0171-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Parks D.H., Imelfort M., Skennerton C.T., Hugenholtz P., Tyson G.W. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015;25:1043–1055. doi: 10.1101/gr.186072.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Asnicar F., Weingart G., Tickle T.L., Huttenhower C., Segata N. Compact graphical representation of phylogenetic data and metadata with GraPhlAn. PeerJ. 2015;3 doi: 10.7717/peerj.1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Asnicar F., Thomas A.M., Beghini F., Mengoni C., Manara S., Manghi P., Zhu Q., Bolzan M., Cumbo F., May U., et al. Precise phylogenetic analysis of microbial isolates and genomes from metagenomes using PhyloPhlAn 3.0. Nat. Commun. 2020;11:2500. doi: 10.1038/s41467-020-16366-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Letunic I., Bork P. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021;49:W293–W296. doi: 10.1093/nar/gkab301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pedregosa F., Varoquaux G., Gramfort A., Michel V., Thirion B., Grisel O., Blondel M., Prettenhofer P., Weiss R., Dubourg V., et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011;12:2825–2830. [Google Scholar]
- 73.Hunter J.D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007;9:90–95. [Google Scholar]
- 74.Virtanen P., Gommers R., Oliphant T.E., Haberland M., Reddy T., Cournapeau D., Burovski E., Peterson P., Weckesser W., Bright J., et al. SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat. Methods. 2020;17:261–272. doi: 10.1038/s41592-019-0686-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Waskom M. seaborn: statistical data visualization. J. Open Source Softw. 2021;6:3021. [Google Scholar]
- 76.McKinney W. Proceedings of the 9th Python in Science Conference (SciPy) 2010. Data Structures for Statistical Computing in Python. [DOI] [Google Scholar]
- 77.Seabold S., Perktold J. Proceedings of the 9th Python in Science Conference (SciPy) 2010. Statsmodels: Econometric and statistical modeling with python. [DOI] [Google Scholar]
- 78.Harris C.R., Millman K.J., Van Der Walt S.J., Gommers R., Virtanen P., Cournapeau D., Wieser E., Taylor J., Berg S., Smith N.J., et al. Array programming with NumPy. Nature. 2020;585:357–362. doi: 10.1038/s41586-020-2649-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wickham H. Springer International Publishing; 2016. ggplot2: Elegant Graphics for Data Analysis. [Google Scholar]
- 80.Gu Z. Complex heatmap visualization. iMeta. 2022;1 doi: 10.1002/imt2.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Oksanen J., Kindt R., Legendre P., O’Hara B. The vegan package. Community Ecol. Package. 2007;10:617. [Google Scholar]
- 82.Kehrmann J., Menzel J., Saeedghalati M., Obeid R., Schulze C., Holzendorf V., Farahpour F., Reinsch N., Klein-Hitpass L., Streeck H., et al. Gut Microbiota in Human Immunodeficiency Virus-Infected Individuals Linked to Coronary Heart Disease. J. Infect. Dis. 2019;219:497–508. doi: 10.1093/infdis/jiy524. [DOI] [PubMed] [Google Scholar]
- 83.Brewster R., Tamburini F.B., Asiimwe E., Oduaran O., Hazelhurst S., Bhatt A.S. Surveying Gut Microbiome Research in Africans: Toward Improved Diversity and Representation. Trends Microbiol. 2019;27:824–835. doi: 10.1016/j.tim.2019.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Beghini F., McIver L.J., Blanco-Míguez A., Dubois L., Asnicar F., Maharjan S., Mailyan A., Manghi P., Scholz M., Thomas A.M., et al. Integrating taxonomic, functional, and strain-level profiling of diverse microbial communities with bioBakery 3. Elife. 2021;10 doi: 10.7554/eLife.65088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Weimann A., Mooren K., Frank J., Pope P.B., Bremges A., McHardy A.C. From Genomes to Phenotypes: Traitar, the Microbial Trait Analyzer. mSystems. 2016;1 doi: 10.1128/mSystems.00101-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Breiman L. Random Forests. Mach. Learn. 2001;45:5–32. [Google Scholar]
- 87.Pasolli E., Truong D.T., Malik F., Waldron L., Segata N. Machine Learning Meta-analysis of Large Metagenomic Datasets: Tools and Biological Insights. PLoS Comput. Biol. 2016;12 doi: 10.1371/journal.pcbi.1004977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Curry K.D., Nute M.G., Treangen T.J. It takes guts to learn: machine learning techniques for disease detection from the gut microbiome. Emerg. Top. Life Sci. 2021;5:815–827. doi: 10.1042/ETLS20210213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Thomas A.M., Manghi P., Asnicar F., Pasolli E., Armanini F., Zolfo M., Beghini F., Manara S., Karcher N., Pozzi C., et al. Metagenomic analysis of colorectal cancer datasets identifies cross-cohort microbial diagnostic signatures and a link with choline degradation. Nat. Med. 2019;25:667–678. doi: 10.1038/s41591-019-0405-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hastie T., Hastie T., Tibshirani R., Friedman J. Springer; 2009. The Elements of Statistical Learning. [Google Scholar]
- 91.Nurk S., Meleshko D., Korobeynikov A., Pevzner P.A. metaSPAdes: a new versatile metagenomic assembler. Genome Res. 2017;27:824–834. doi: 10.1101/gr.213959.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Quince C., Walker A.W., Simpson J.T., Loman N.J., Segata N. Shotgun metagenomics, from sampling to analysis. Nat. Biotechnol. 2017;35:833–844. doi: 10.1038/nbt.3935. [DOI] [PubMed] [Google Scholar]
- 93.Manara S., Selma-Royo M., Huang K.D., Asnicar F., Armanini F., Blanco-Miguez A., Cumbo F., Golzato D., Manghi P., Pinto F., et al. Maternal and food microbial sources shape the infant microbiome of a rural Ethiopian population. Curr. Biol. 2023;33:1939–1950.e4. doi: 10.1016/j.cub.2023.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kang D.D., Froula J., Egan R., Wang Z. MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ. 2015;3:e1165. doi: 10.7717/peerj.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kang D.D., Li F., Kirton E., Thomas A., Egan R., An H., Wang Z. MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ. 2019;7 doi: 10.7717/peerj.7359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wu Y.-W., Simmons B.A., Singer S.W. MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics. 2016;32:605–607. doi: 10.1093/bioinformatics/btv638. [DOI] [PubMed] [Google Scholar]
- 97.Granehäll L., Huang K.D., Tett A., Manghi P., Paladin A., O’Sullivan N., Rota-Stabelli O., Segata N., Zink A., Maixner F. Metagenomic analysis of ancient dental calculus reveals unexplored diversity of oral archaeal Methanobrevibacter. Microbiome. 2021;9:197. doi: 10.1186/s40168-021-01132-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The overall information of each project in the publicly available datasets utilized in this study and samples were from healthy adults (18–65 years) (Tab 1). Sample-wise characterization by geography and Westernization for each project in the dataset (Tab 2). Species enriched in MSM individuals relative to the Westernized (Tab 3). Species enriched in MSM individuals relative to the non-Westernized (Tab 4). Species enriched in the Westernized populations compared to MSM individuals (Tab 5). Species enriched in the non-Westernized populations compared to MSM individuals (Tab 6). Species differentially abundant in MSM and non-MSM samples (Tab 7). The False Discovery Rates (FDRs) were estimated by Fisher’s exact test corrected for multiple hypotheses using Bonferroni method. FDRs lower than 0.05 were considered significant. Horizontal transmissibility measures, engraftment rates and phenotypes involved in microbial transmission (including spore former, anaerobic growth, motility and Gram-negative staining) were retrieved from other two studies [S4],[S5]. Horizontal transmissibility and engraftment rate are within a range of 0–1. For phenotypes (spore former, anaerobic growth, motility and Gram-negative staining), 1 indicates presence and 0 indicates absence. Missing values were supplemented if not publicly available.
The significance of a biomarker for each sexual practice was indicated by effect size measures (LDA score (log 10)). Horizontal transmissibility measures, engraftment rates and phenotypes involved in microbial transmission (including spore formation, anaerobic growth, motility and Gram-negative staining) were retrieved from other two studies [S4],[S5]. For phenotypes (spore formation, anaerobic growth, motility and Gram-negative staining), 1 indicates presence and 0 indicates absence. Missing values were supplemented if not publicly available (Tab 1). Association significance of sexual practices and confounding effects, assessed by Fisher’s exact test with Benjamini-Hochberg method for multiple comparison correction. Values indicate FDRs of the association between a sexual practice and a confounding effect. Association is considered significant when FDR <0.05 (Tab 2).
The genome prevalence in MSM/non-MSM was quantified as, for each species, dividing the total number of genomes by the number of MSM and non-MSM individuals, respectively.
Data Availability Statement
-
•
Shotgun metagenomic reads, detailed descriptions for each sample and metagenome-assembled genomes in this study are available under NCBI BioProject: PRJNA947377
-
•
Codes and corresponding tutorials for reproducing results in this study are avaialble at: https://github.com/KunDHuang/KunDH-2024-CRM-MSM_metagenomics
-
•
Any additional information required to reanalyze the data reported in this work is available from the lead contact upon request.