

Abstract

The H1047R mutation of PIK3CA is highly prevalent in breast cancers and other solid tumors. Selectively targeting PI3KαH1047R over PI3KαWT is crucial due to the role that PI3KαWT plays in normal cellular processes, including glucose homeostasis. Currently, only one PI3KαH1047R-selective inhibitor has progressed into clinical trials, while three pan mutant (H1047R, H1047L, H1047Y, E542K, and E545K) selective PI3Kα inhibitors have also reached the clinical stage. Herein, we report the design and discovery of a series of pyridopyrimidinones that inhibit PI3KαH1047R with high selectivity over PI3KαWT, resulting in the discovery of compound 17. When dosed in the HCC1954 tumor model in mice, 17 provided tumor regressions and a clear pharmacodynamic response. X-ray cocrystal structures from several PI3Kα inhibitors were obtained, revealing three distinct binding modes within PI3KαH1047R including a previously reported cryptic pocket in the C-terminus of the kinase domain wherein we observe a ligand-induced interaction with Arg1047.
Introduction
The PI3K (Phosphoinositide 3-Kinase) pathway regulates many important cellular processes including cell growth, survival, and proliferation.1−6 PI3K family members are lipid kinases that phosphorylate specific species of phosphoinositides in cellular membranes and transduce signals in a tightly regulated manner. There are three classes of PI3K proteins (Classes I–III),1−6 of which the Class I PI3K proteins catalyze the phosphorylation of PIP2 (phophoinositol-3,4-diphosphate) to PIP3 (phophoinositol-3,4,5-triphosphate), resulting in the recruitment of AKT to the plasma membrane and eventual phosphorylation of AKT. Class IA PI3Ks, like PI3Kα, are heterodimers comprised of a regulatory subunit (p85α, p85β, p55α, p55γ, or p50α) and a catalytic subunit (p110α, p110β, or p110δ). Activating mutations of the PIK3CA gene that encodes for the p110α isoform (PI3Kα) are highly prevalent in solid tumors and are present in nearly 30% of all breast cancers.7−13 The three most common activating point mutations of PIK3CA occur within the p110α subunit, resulting in PI3KαH1047R, PI3KαE545K, and PI3KαE542K mutant proteins.14 The H1047R mutation accounts for the highest occurrence of somatic PIK3CA mutations in breast cancers,15−17 and additional therapies are needed for these patients.18,19
The high occurrence of these PI3Kα mutant proteins in solid tumors has led to the design of isoform-selective orthosteric inhibitors of PI3Kα,20−23 including the only clinically approved inhibitor of PI3Kα, alpelisib (1).19,22,24,25 However, treatment with alpelisib commonly results in hyperglycemia within patients, a form of on-target toxicity that can result in dose interruption, reduction, or discontinuation.24−26 This on-target toxicity is a consequence of the key role that PI3KαWT plays in the regulation of glucose metabolism,26−28 and results in a limited therapeutic index. For this reason, recent research in this field has focused on the discovery of compounds that selectively inhibit the mutant forms of PI3Kα (i.e., H1047X, E545X, and E542X).
In 2012, Williams and co-workers reported the binding of a PI3Kβ-selective inhibitor, PIK-10830 (2), within the wild-type form of PI3Kα (Figure 1).29 Serendipitously, although PIK-108 displays high selectivity for PI3Kβ (PI3Kβ IC50: 57 nM, PI3Kα IC50: 2600 nM),30 it produced superior cocrystals with PI3Kα when compared to more potent and selective inhibitors of PI3Kα. The cocrystal structure revealed that PIK-108 bound to both the ATP-binding site (Figure 1, cyan) and a second, cryptic site in close proximity to the His1047 residue (Figure 1, magenta). This allosteric binding site provided a potential druggable pocket for a selective inhibitor of PI3KαH1047R.
Figure 1.

Reported cocrystal structure of PIK-108 bound to two sites within PI3KαWT (PDB: 4A55(29)). PIK-108 in cyan is bound within the orthosteric site, and PIK-108 in magenta is binding within the allosteric site near His1047.
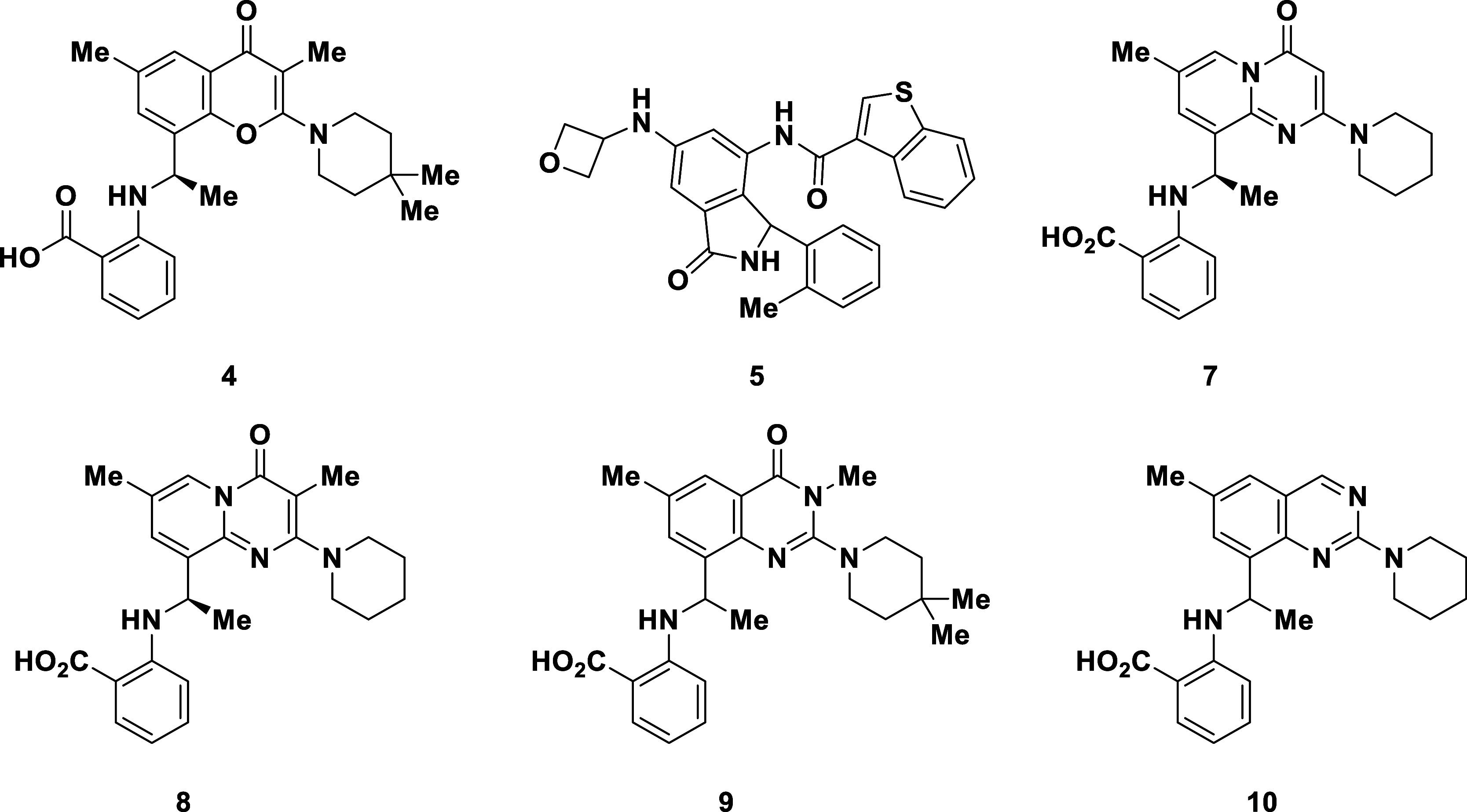
Years later, in 2021, Petra Pharma/Loxo/Eli Lilly published a patent application detailing selective PI3KαH1047R inhibitors (e.g., 3–4, Figure 2) that closely resemble the structure of PIK-108,31 and several additional closely related applications were published shortly thereafter.32−34 Simple removal of the hinge binding motif (morpholine) from the C2-substituent on the chromenone ring in 2 and installation of a carboxylic acid on the phenyl ring to provide compounds like 3 and 4, resulted in potent and selective inhibitors of PI3KαH1047R (13.5-fold selectivity reported for 3 against PI3KαWT).31 Within the same year, Relay Therapeutics35−40 published a patent application describing pan mutant selective PI3Kα inhibitors (e.g., 5),35 which contain no obvious motif for targeting the Arg1047 residue. The patent application from Relay also contained no reported selectivity data against PI3KαWT. While Lilly currently has the only reported PI3KαH1047R selective inhibitor in the clinic (LOXO-783), Relay Therapeutics and Scorpion Therapeutics41−43 are currently in clinical trials with pan mutant selective inhibitors of PI3Kα (Relay: RLY-260839 and RLY-5836; Scorpion: STX-478). Although the structures of STX-478 (6)43 and RLY-260839 were both recently disclosed, the chemical structures and structural details of binding to PI3Kα for LOXO-783 and RLY-5836 have not been released. Herein, we provide crystallographic evidence for three distinct binding modes of PI3Kα inhibitors and detail the discovery of a pyridopyrimidinone series that selectively inhibits the function of PI3KαH1047R.
Figure 2.

Chemical Structures of Alpelisib (1), PIK-108 (2), and representative literature compounds (3–6) from Lilly, Relay, and Scorpion.
Results and Discussion
To provide more insight into the binding modes of these literature compounds, two discrete cocrystal structures of compounds 4 and 5 bound to PI3KαH1047R (Figure 3A,B) were solved (in the presence of the alpelisib derivative A-66,44 PDB: 8V8H and 8V8I, respectively). Interestingly, while 4 binds near the Arg1047 residue in the same allosteric pocket as PIK-108, we observed the binding of compound 5 in a previously unreported cryptic pocket within the protein. This cryptic pocket was recently revealed as the binding site of STX-478 (6)43 and RLY-2608.39
Figure 3.

A. Cut-away view of the discrete cocrystal structure of compound 4 bound to PI3KαH1047R showing the cryptic binding pocket (PDB: 8V8H) B. Discrete cocrystal structure of compound 5 bound to PI3KαH1047R (PDB: 8V8I). C. Co-crystal structure of A-66, 4, and 5 simultaneously bound to PI3KαH1047R (PDB: 8 V8J).
In the initial cocrystal structure of compound 4 bound to PI3KαH1047R (Figure 3A), the benzoic acid appears to form a salt bridge with the Arg1047 residue that likely accounts for the reported selectivity31 against wild-type for this series of chromenone inhibitors. While the carboxylate also makes a hydrogen bonding interaction with Gln981 and the chromenone core participates in a π-stacking interaction with Phe954, no additional significant polar interactions between 4 and the protein are observed. However, the cocrystal structure also revealed an intramolecular hydrogen bond between the anilinic N–H and the carboxylate of 4 (Figure S3).
The pan mutant PI3Kα inhibitor 5 was found to bind in a second cryptic pocket under the activation loop (residues 933–958) of PI3KαH1047R, secluded from any of the common point mutations found in PI3Kα mutant cancers. The cocrystal structure (Figure 3B) revealed that 5 participates in hydrogen bonding interactions with three distinct backbone carbonyls: the first between the benzothiophenyl amide N–H and the backbone carbonyl of Leu911, the second between the anilinic N–H and Gly912, and the final hydrogen bond between the isoindolinone N–H and Asp1018. Additionally, the 2-methylphenyl and benzothiophene moieties share edge-to-face interactions with Phe1002, and the thiophenyl sulfur makes a mildly favorable sigma-hole interaction with the side chain of Thr813.
Based on results from our laboratory and recent reports detailing the cocrystal structures of STX-47843 bound to PI3KαH1047R and RLY-260839 bound to WT PI3Kα, it is clear that compounds like 5, RLY-2608, and STX-478 (6) bind within the same cryptic pocket of PI3Kα. It is difficult to provide justification solely on the basis of the cocomplexed crystal structures for how the binding mode of 5 can provide selectivity against PI3KWT since the oncogenic mutations are relatively distal to the binding pocket. Buckbinder et al. suggest selectivity is the result of altered kinetics of the mutants.43 Varkaris et al. concur and further demonstrate that the altered kinetics are due to mutant perturbed conformations increasing accessibility of the cryptic pocket.39
In our work, further crystallographic studies led to the discovery that under optimized crystallization conditions, a cocomplex crystal structure with A-66, 4, and 5 simultaneously bound to PI3KαH1047R could be obtained (Figure 3C). The interactions observed in the discrete crystal structure of 4 are present in this structure. Similarly, 5 showed no significant changes between the discrete structure and the cocrystal containing all three compounds.
Compounds 4 and 5 were further evaluated in two cellular assay formats, an AlphaLISA assay (measuring phospho-AKT (pAKT) levels) and a viability assay in both the T47D (PI3KαH1047R) and SKBR3 (PI3KαWT) cell lines (Table 1). In these cell-based assays, SKBR3 is a PIK3CA wild-type cell line used as a surrogate model to evaluate the effects of WT PI3Kα inhibition and to determine selectivity for these compounds. Compound 4 gave high potency and selectivity across both cellular assay formats (pAKT T47D IC50: 3 nM, Viability T47D IC50: 160 nM, Table 1) while compound 5 (pAKT T47D IC50: 332 nM, Viability T47D IC50: 488 nM, Table 1) was less potent and selective under the assay conditions.
Table 1. Evaluation of Published Selective PI3Kα Inhibitors and Discovery of a Novel Pyridopyrimidinone Core.
| compound | pAKT AlphaLISA IC50 (nM)a | viability IC50 (nM)b | viability selectivity 1047R/WT IC50 | ||
|---|---|---|---|---|---|
| T47D | SKBR3 | T47D | SKBR3 | ||
| 4 | 3 | 3185 | 160 | 9082 | 57 |
| 5 | 332 | 115 | 488 | 4309 | 9 |
| 7 | 78 | 5894 | 1110 | >10,000 | 9 |
| 8 | 3 | 1465 | 120 | >10,000 | 83 |
| 9 | 179 | >10,000 | 6239 | >10,000 | 2 |
| 10 | 2544 | >10,000 | >10,000 | >10,000 | |
Measured inhibition of pAKT formation using alphaLISA after 24 h of treatment.
Measured decrease in viability using CellTiter-Glo 2.0 (CTG) after 72 h of treatment.
Utilizing a structure-based drug design, our goal was to discover a highly selective inhibitor of PI3KαH1047R. The pyridopyrimidinone 7 was designed and resulted in modest potency in T47D cells (pAKT T47D IC50: 78 nM, Viability T47D IC50: 1110 nM, Table 1). Further installation of a C3-methyl to give 8 significantly increased the potency of this novel scaffold and provided high selectivity against the cells expressing wild-type PI3Kα. The transposition of the pyridone nitrogen to the 3-position of the 6,6-scaffold to give compounds 9 and 10 resulted in a 39-fold decrease or complete loss in potency (viability) when compared to compound 4. An X-ray cocrystal structure of 7 bound to PI3KαH1047R revealed that 7 shares a similar binding mode to chromenone 4 (Figure 4A) and a superposition of the cocomplex structures of compounds 4 and 7, demonstrate that the compounds overlay well (Figure 4B). Further analysis of the structure reveals that the C3-vector could potentially be used to target the His931 residue. Additionally, the C2-substituent protrudes into a large lipophilic portion of the allosteric binding site, providing a potential growth vector for new analogs.
Figure 4.

A. Discrete co-crystal structure of compound 7 (PDB: 8V8V). B. Overlay of compounds 4 (pink) and 7 (magenta) bound to PI3KαH1047R.
While installation of the C3-methyl in compound 8 resulted in a significant increase in potency, this change was met with a decrease in metabolic stability in human hepatocytes (Clint: 340 mL/min/kg, Table 2). Introduction of a fluorine in the C3-position (11) further increased the potency of the scaffold (pAKT T47D IC50: 0.3 nM); however, the fluoro-analog 11 provided less stability than the C3-methyl analog 8 (Clint for 11: 862 mL/min/kg). In an attempt to decrease the metabolism and target the His931 residue, the C3-nitrile analogue 12 was synthesized and provided a compound that was 3-fold more potent and 4-fold more stable in human hepatocytes (Clint for 12: 79 mL/min/kg) than 8. Conversely, replacing the C3-nitrile in 12 with a trifluoromethyl (13) or an amide (14) resulted in a substantial loss in potency.
Table 2. Examination of C3-substitution on the Pyridopyrimidinone Core.

| compound | R | pAKT AlphaLISA IC50 (nM)a | Clint human hepatocytes (mL/min/kg)b | |
|---|---|---|---|---|
| T47D | SKBR3 | |||
| 7 | –H | 78 | 5894 | 181 |
| 8 | –Me | 3 | 1465 | 340 |
| 11 | –F | 0.3 | >10,000 | 862 |
| 12 | –CN | 1 | 4739 | 79 |
| 13 | –CF3 | 28 | >10,000 | N.D. |
| 14 | –CONH2 | >10,000 | >10,000 | N.D. |
Measured inhibition of pAKT formation using alphaLISA after 24 h of treatment.
Intrinsic clearance obtained from scaling in vitro half-lives from pooled hepatocytes.
A cocrystal structure of 12 bound to PI3KαH1047R was obtained and confirmed that the nitrogen of the C3-nitrile participates in a hydrogen bonding interaction with His931 (Figure 5A). As observed in the structure of 7 (Figure 4), the cocrystal structure of 12 and PI3KαH1047R also displays a salt bridge formed between the benzoic acid moiety and Arg1047, further supporting the hypothesis that H1047R selectivity for this chemotype is largely driven by direct interaction with the point mutation. Interestingly, the position of the Arg1047 side chain is in a qualitatively similar position relative to the cryo-EM structure of PI3KαH1047R (Figure 5B)45 suggesting that minimal protein rearrangement is required for engagement of the salt bridge interaction. The binding of 12 displaces the activation loop C-terminus by ∼6 Å at Pro953 compared to that of apo PI3KαH1047R. This, in turn, changes the positioning of the second basic box membrane binding element (residues 948–951),46 which we hypothesize negatively impacts membrane interaction and leads to inhibition of enzymatic activity for PI3KαH1047R. While the C-terminal end of kα11 is displaced ∼2 Å at Asp1045 relative to the apo protein, it is unclear if this impacts the autoinhibitory ordering of the C-terminal tail including the WIF membrane interaction element (residues 1057–1059).47 However, both 12 and the displaced C-terminus of the activation loop sit adjacent to the catalytic HRD element (residues 915–917, Figure 5C). Dynamic changes in HRD could impact phosphoryl transfer, potentially resulting in an additional mechanism of inhibition.
Figure 5.

A. X-ray cocrystal structure of compound 12 bound to PI3KαH1047R (PDB: 8V8U). B. Overlay of the X-ray cocrystal structure of 12 (green protein) with the cryo-EM structure of PI3KαH1047R (gray protein, PDB: 8GUB)45 showing inhibitor-induced perturbation of the activation loop and basic box 2 (blue). Inset shows kα11 helix and similar relative positions of the Arg1047 side chain between apo and liganded structures. C. X-ray cocrystal structure of compound 12 showing proximity of ligand to HRD motif (orange) and activation loop (yellow).
To further decrease the metabolism of these pyridopyrimidinones, designs were focused on finding replacements for the C2-piperidine ring (Table 3). Decreasing the ring size (15) or installing dimethyl groups at the 4-position (16) resulted in compounds with nearly the same potency and stability as parent compound 12. However, substituting the piperidine ring with two fluorines at the 4-position to give 17, decreased the intrinsic clearance in human hepatocytes by 3-fold (Clint for 17: 29 mL/min/kg) while retaining high cellular potency (Viability T47D IC50: 48 nM) when compared to 12. Increased substitution around the 4,4-difluoropiperidine ring to give 18, preserved the potency of the scaffold while slightly decreasing the metabolic stability (Clint for 18: 39 mL/min/kg). Removing the nitrogen from the piperidine ring to give the 4,4-difluorocylcohexane 19 resulted in a significantly decreased intrinsic clearance in human hepatocytes (Clint for 19: 12 mL/min/kg), albeit with a 5-fold loss in potency within the T47D cellular viability assay. Replacing the piperidine ring in 12 with a difluoroazetidine (20) was well-tolerated and provided a similar in vitro profile to the difluoropiperidine 17. Conversely, the installation of the difluoroazaspiroheptane 21 or the thiomorpholine dioxide 22 resulted in analogs that were completely inactive in the T47D viability assay.
Table 3. Compounds Designed to Lower the Clint in Human Hepatocytes.
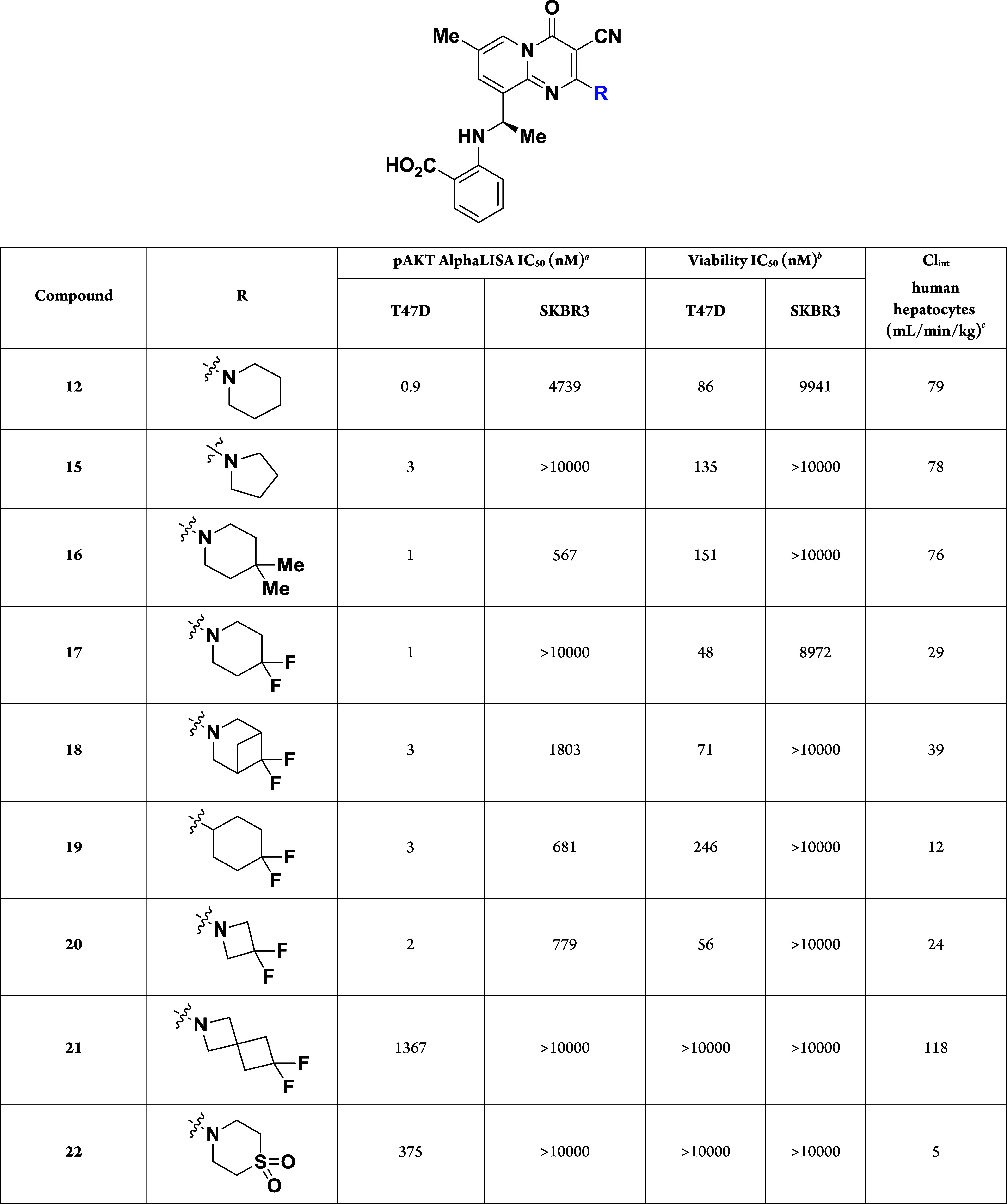
Measured inhibition of pAKT formation using alphaLISA after 24 h of treatment.
Measured decrease in viability using CellTiter-Glo 2.0 (CTG) after 72 h of treatment.
Intrinsic clearance obtained from scaling in vitro half-lives from pooled hepatocytes.
While the difluoropiperidine 17 displayed high cellular potency and moderate stability, it was found that glucuronidation of the benzoic acid was the only discernible metabolite in human hepatocytes (see Figure S1, Supporting Information). In the hopes of minimizing the glucuronidation of this acid, several benzoic acid replacements were explored (23–27, Table 4). Unfortunately, substitution around the benzene ring (23–24, Table 4) or replacement of the ring with a heteroaromatic ring (25–27, Table 4) led to a significant decrease in cellular potency. For this reason, compound 17 was progressed into pharmacokinetic studies within mice, rats, and dogs.
Table 4. Evaluating Analogs to Replace the Benzoic Acid Found in Compound 17.

Measured inhibition of pAKT formation using alphaLISA after 24 h of treatment.
Measured decrease in viability using CellTiter-Glo 2.0 (CTG) after 72 h of treatment.
When administered in rodents, 17 displayed moderate bioavailability (48–57%F, Table 5) with high clearance in mice and moderate clearance in rats, similar to the intrinsic clearance measured in hepatocytes for these species. IV administration of 17 in dogs also resulted in high clearance, correlating well with Clint found in dog hepatocytes. Upon oral dosing of 17 in dogs, the resulting bioavailability was low (7%), indicating that 17 was unsuitable for progression into development. However, the moderate bioavailability in rodents and high selectivity for PI3KαH1047R (see Tables SI-2–SI-4 in Supporting Information) suggested 17 as a compelling tool compound for pharmacodynamic (PD) and efficacy models in mice, while further analogs were designed to address the metabolic stability of the scaffold.
Table 5. Overall Pharmacokinetic (PK) Parameters for Compound 17 Across Species.

| ADME parametersa | mouse | rat | dog |
|---|---|---|---|
| Clint hepatocytes (mL/min/kg) | 304 | 70 | 104 |
| in vivo Cl (mL/min/kg) | 180 | 46 | 64 |
| Vd,ss (L/kg) | 13.6 | 4.8 | 1.5 |
| IV t1/2 (h) | 2 | 3.5 | 0.7 |
| F (%) | 48 | 57 | 7 |
| dose: IV/PO (mg/kg) | 3/100 | 3/100 | 2/10 |
See Supporting Information for details.
Compound 17 was first evaluated in an HCC1954 (ER-/HER2+ human breast tumor cell line with endogenous PI3KαH1047R) mouse xenograft model to assess its pharmacodynamic properties. Modulation of pAKT was monitored after a single dose of 17 via oral or intraperitoneal (IP) administration. The results from the PD study showed that oral dosing of 17 at 100 or 300 mg/kg resulted in negligible inhibition of pAKT (Figure 6), likely due to the low concentrations of 17 at these time points (Table SI-5, Supporting Information). However, administration of 17 via IP injection at either 60 or 120 mg/kg (as a single dose, qd) resulted in a much higher concentration of 17 (Table SI-5), leading to 42 and 81% inhibition of pAKT, respectively, for up to 8 h (Figure 6). When compared to the PI3KαWT inhibitor alpelisib, the 120 mg/kg IP dose of 17 displayed similar inhibition of pAKT at both the 1 and 8 h time points.
Figure 6.

Single Dose Pharmacodynamic Study and a 14-Day TGI Study of Compound 17 in the HCC1954 tumor model. A. pAKT modulation at 1 h post dose B. pAKT modulation at 8 h post dose C. 14-day efficacy study.
Based on the results from the PD study, a 14-day tumor growth inhibition (TGI) study was performed in HCC1954 tumor-bearing mice (Figure 6C). Compound 17 was dosed at IP at either 100 mg/kg qd or 80 mg/kg twice daily (b.i.d.) for 14 days. While the 100 mg/kg (IP, qd) dose resulted in significant tumor growth inhibition (88%), the 80 mg/kg (IP, bid) dosing regimen provided tumor regressions (−34%). To the best of our knowledge, this is the first reported case of an H1047R-selective inhibitor of PI3Kα providing tumor regressions in an HCC1954 tumor model.
Chemistry
Cyclization of the 4H-pyrido[1,2-a]pyrimidin-4-one core (30) was accomplished through the condensation of 2-amino-3-bromo-5-methylpyridine (28) and malonyl dichloride (27, Scheme 1). Next, direct installation of 4,4-difluoropiperidine onto the core was realized using PyBOP for activation of the cyclic amide motif to provide 31.48,49 A Heck reaction of the aryl bromide 31 using butyl vinyl ether followed by acidic hydrolysis provided aryl ketone 32, which was easily brominated using NBS to provide 33. Condensation of the chiral Ellman’s sulfinamide50 onto ketone 33, followed by a highly diastereoselective reduction using Schwartz’s reagent (zirconocene hydrogen chloride) provided the benzylic sulfinamide 34 with >97% de. Installation of the key nitrile at the C3 position was achieved through a palladium-catalyzed cross-coupling with zinc cyanide, and acidic removal of the chiral auxiliary provided the benzylic amine 35. Finally, the synthesis was completed with the installation of the critical benzoic acid motif that provided selectivity for PI3KαH1047R mutant inhibition. Initial efforts were focused on Ullmann–Goldberg type cross-couplings of aryl halides bearing carboxylic acids; however, yields were generally low. A Chan-Evans-Lam coupling using 2-carboxylphenylboronic acid proved fruitful when run in amide-based solvents, and the use of the stronger base DBU in comparison to triethylamine was found to be necessary to achieve higher conversions, providing 17 in 40% isolated yield.51
Scheme 1. Gram-Scale Synthesis of 17.

Reagents and conditions: (a) DCM, 25 °C, 48 h, 61% yield; (b) PyBOP, DIPEA, DMF, 25 °C, 12 h, 38% yield; (c) butyl vinyl ether, P(t-Bu)3Pd G2, N,N-dicyclohexylamine, dioxane, 100 °C, 1 h; (d) HCl (12M), THF, 25 °C, 1 h, 25% yield over 2 steps; (e) NBS, DMF, 25 °C, 2 h, 78% yield; (f) (R)-tert-butylsulfinamide, Ti(OEt)4, THF, 80 °C, 12 h, 80% yield; (g) Cp2ZrHCl, DCM, 25 °C, 1 h, 70% yield; (h) Zn(CN)2, XantPhos Pd G3, DMA, 100 °C 1 h; (i) HCl (4 M in dioxane), dioxane, 25 °C, 1 h, 89% yield over 2 steps; (j) 2-carboxyphenylboronic acid, Cu(OAc)2, DBU, DMF, 25 °C, 12 h, 40% yield.
Conclusions
Our efforts toward a PI3KαH1047R mutant selective inhibitor led to the discovery of a pyridopyrimidinone series that provided compound 17, a potent and selective inhibitor of PI3KαH1047R that provided tumor regressions in the HCC1954 tumor model in mice. Crystallographic studies provided insights into the binding mode of several literature inhibitors of PI3Kα. Additionally, we elucidated several cocrystal structures of the pyridopyrimidinone inhibitors bound within the PI3KαH1047R mutant protein, leading to suggested mechanisms of inhibition and selectivity. Encouraged by the results from the HCC1954 tumor studies, we began further optimization of our scaffold to help increase the metabolic stability of these selective PI3KαH1047R inhibitors. However, increasing competitive activity focused on this chemical scaffold38,52,53 drove us to alternative designs, to be disclosed in due course.
Experimental Section
General Procedures
All final compounds were purified to ≥95% purity by either high-performance liquid chromatography (HPLC) or supercritical liquid chromatography (SFC). Purity was determined by HPLC and additional structural characterization was performed by proton NMR, carbon NMR, and high-resolution mass spectrometry, as described below. All chemicals were purchased from commercial suppliers and used as received unless otherwise indicated. Proton nuclear magnetic resonance (1H NMR) spectra were recorded on Bruker Avance 400 MHz spectrometers. Chemical shifts are expressed in δ ppm and are calibrated to the residual solvent peak: proton (e.g., CDCl3, 7.27 ppm). Coupling constants (J), when given, are reported in hertz. Multiplicities are reported using the following abbreviations: s = singlet, d = doublet, dd = doublet of doublets, t = triplet, q = quartet, m = multiplet (range of multiplets is given), br = broad signal, dt = doublet of triplets. Carbon nuclear magnetic resonance (13C NMR) spectra were recorded using a Bruker Avance HD spectrometer at 100 MHz. Chemical shifts are reported in parts per million (ppm) and are calibrated to the solvent peak: carbon (CDCl3, 77.23 ppm). The purity of the test compounds was determined by high-performance liquid chromatography (HPLC) on an LC-20AB Shimadzu instrument. HPLC conditions were as follows: The purity for test compounds was determined by high-performance liquid chromatography (HPLC) on an LC-20AB Shimadzu instrument. HPLC conditions were as follows: HPLC conditions were as follows: Kinetex EVO C18 3.0 × 50 mm, 2.6 um, 10–80% ACN (0.0375%TFA) in water (0.01875% TFA), 3–10 min runs, flow rate 1.2 mL/min, UV detection (λ = 220, 215, 254 nm), or Kinetex C18 LC column 4.6 × 50 mm, 5 μm, 10–80% ACN (0.0375%TFA) in water (0.01875% TFA), 4–10 min runs, flow rate 1.5 mL/min, UV detection (λ = 220, 215, 254 nm), or XBridge C18, 2.1 × 50 mm, 5 μm, 10–80% ACN in water buffered with 0.025% ammonia, 4–10 min runs, flow rate 0.8 mL/min, UV detection (λ = 220, 215, 254 nm). The mass spectra were obtained using liquid chromatography–mass spectrometry (LC-MS) on an LCMS-2020 Shimadzu instrument using electrospray ionization (ESI). LCMS conditions were as follows: Kinetex EVO C18 30 × 2.1 mm, 5 μm, 5–95% ACN (0.0375% TFA) in water (0.01875% TFA), 1.5 min run, flow rate 1.5 mL/min, UV detection (λ = 220, 254 nm), or Kinetex EVO C18 2.1 × 30 mm, 5 μm, 5–95% ACN in water buffered with 0.025% ammonia, 1.5 min run, flow rate 1.5 mL/min, and UV detection (λ = 220, 254 nm). High-resolution mass measurements were carried out on an Agilent 1290LC and 6530Q-TOF series with ESI. The SFC purity was determined with a Shimadzu LC-30ADsf.
Preparation of Compound 17
9-Bromo-2-hydroxy-7-methyl-4H-pyrido[1,2-a]pyrimidin-4-one (30)
To a solution of 3-bromo-5-methylpyridin-2-amine (300 g, 1.60 mol, 1.0 equiv) in dichloromethane (2.0 L) was added malonyl dichloride (249 g, 1.76 mol, 172 mL, 1.10 equiv), and the mixture was stirred at 25 °C for 48 h. The mixture was then filtered, and the filter cake was washed with dichloromethane (500 mL × 2). The filter cake was further concentrated in vacuo to give compound 30 (269 g, 983 mmol, 61% yield, 93% purity) as a yellow solid. LCMS [M + 3]+= 256.9.
9-Bromo-2-(4,4-difluoropiperidin-1-yl)-7-methyl-4H-pyrido[1,2-a]pyrimidin-4-one (31)
To a solution of 30 (269 g, 983 mmol, 1.00 equiv) in DMF (2.0 L) was added PyBOP (614 g, 1.18 mol, 1.20 equiv), N,N-diisopropylethylamine (508 g, 3.93 mol, 685 mL, 4.00 equiv), and 4,4-difluoropiperidine (131 g, 1.08 mol, 1.10 equiv), and the mixture was left to stir at 25 °C for 12 h. After this time, water (4.0 L) was added to the mixture. The resultant precipitate was filtered, and the filter cake was dried under a vacuum to give 31 (172 g, 377 mmol, 38% yield, 78% purity) as a yellow solid. LCMS [M + 3]+= 360.0.
9-Acetyl-2-(4,4-difluoropiperidin-1-yl)-7-methyl-4H-pyrido[1,2-a]pyrimidin-4-one (32)
A mixture of 31 (167 g, 366 mmol, 1.00 equiv), butyl vinyl ether (110 g, 1.10 mol, 141 mL, 3.00 equiv), dicylcohexylamine (73.0 g, 402 mmol, 80.2 mL, 1.10 equiv), P(t-Bu)3Pd G2 (9.38 g, 18.3 mmol, 0.05 equiv) in dioxane (1.70 L) was degassed and purged with nitrogen 3 times, and then the mixture was stirred at 100 °C for 1 h under a nitrogen atmosphere. The mixture was then cooled to 25 °C, filtered, and concentrated under reduced pressure to give 9-(1-butoxyvinyl)-2-(4,4-difluoropiperidin-1-yl)-7-methyl-4H-pyrido[1,2-a]pyrimidin-4-one (200 g, crude) as a yellow solid, which was without further purification. LCMS [M + 1]+= 378.2. To a solution of 9-(1-butoxyvinyl)-2-(4,4-difluoropiperidin-1-yl)-7-methyl-4H-pyrido[1,2-a]pyrimidin-4-one (200 g, 530 mmol, 1.00 equiv) in tetrahydrofuran (1.5 L) was added hydrochloric acid (12 M, 132 mL, 3.00 equiv), and the mixture was left to stir at 25 °C for 1 h. The reaction mixture was then concentrated under reduced pressure to give a residue. The residue was diluted with dichloromethane (1.0 L) and the pH of the mixture was adjusted (pH ∼8) with sodium bicarbonate (saturated aqueous solution). The mixture was then extracted with dichloromethane(500 mL), and the organic layer was washed with brine (1.0 L), dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure to give a residue. The residue was purified by flash column chromatography (SiO2, petroleum ether/ethyl acetate = 10/1 to 1/1) to give 32 (45.6 g, 134 mmol, 25% yield, 94% purity) as a yellow solid. LCMS [M + 1]+ = 322.1. 1H NMR (400 MHz, CDCl3) δ = 8.85 (d, J = 0.8 Hz, 1H), 7.84 (d, J = 2.0 Hz, 1H), 5.70 (s, 1H), 3.91–3.68 (m, 4H), 2.76 (s, 3H), 2.36 (s, 3H), 2.09–1.96 (m, 4H).
9-Acetyl-3-bromo-2-(4,4-difluoropiperidin-1-yl)-7-methyl-4H-pyrido[1,2-a]pyrimidin-4-one (33)
To a solution of 32 (45.6 g, 134 mmol, 1.00 equiv) in DMF (450 mL) was added NBS (26.1 g, 147 mmol, 1.10 equiv), and the mixture was left to stir at 25 °C for 2 h. The mixture was then diluted with water (900 mL) and filtered, and the filter cake was dried under vacuum to give 33 (47.0 g, 104 mmol, 78% yield, 89% purity) as a yellow solid. LCMS [M + 1]+= 402.0. 1H NMR (400 MHz, CDCl3) δ = 9.00–8.96 (m, 1H), 8.92 (s, 1H), 7.95 (d, J = 2.0 Hz, 1H), 3.83–3.74 (m, 4H), 2.81 (s, 3H), 2.43 (s, 3H), 2.22–2.07 (m, 4H).
(R)-N-((R)-1-(3-Bromo-2-(4,4-difluoropiperidin-1-yl)-7-methyl-4-oxo-4H-pyrido[1,2-a]pyrimidin-9-yl)ethyl)-2-methylpropane-2-sulfinamide (34)
To a solution of 33 (47.0 g, 104 mmol, 1.00 equiv), (R)-2-methyl-2-propanesulfinamide (15.2 g, 125 mmol, 1.20 equiv) in tetrahydrofuran (500 mL) was added titanium(IV) ethoxide (47.5 g, 208 mmol, 43.2 mL, 2.00 equiv) and 1,2-dimethoxyethane (9.39 g, 104 mmol, 10.8 mL, 1.00 equiv), and the mixture was stirred at 80 °C for 12 h. The reaction was then diluted with water (15.0 mL) and ethyl acetate (2.00 L), filtered, and concentrated under reduced pressure to give a residue. The residue was purified by column chromatography (SiO2, petroleum ether/ethyl acetate = 5/1 to 1/1) to give (R)-N-(1-(3-bromo-2-(4,4-difluoropiperidin-1-yl)-7-methyl-4-oxo-4H-pyrido[1,2-a]pyrimidin-9-yl)ethylidene)-2-methylpropane-2-sulfinamide (51.7 g, 83.3 mmol, 80% yield, 81% purity) as a yellow oil. LCMS [M + 1]+ = 505.2. To a solution of (R)-N-(1-(3-bromo-2-(4,4-difluoropiperidin-1-yl)-7-methyl-4-oxo-4H-pyrido[1,2-a]pyrimidin-9-yl)ethylidene)-2-methylpropane-2-sulfinamide (51.7 g, 83.3 mmol, 1.00 equiv) in dichloromethane (500 mL) was added Schwartz’ Reagent (25.8 g, 100 mmol, 1.20 equiv), and the mixture was stirred at 25 °C for 1 h. The mixture was then diluted with dichloromethane (500 mL) and washed with water (500 mL × 2). The organic phase was dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure to give a residue. The residue was purified by column chromatography (SiO2, petroleum ether/ethyl acetate = 10/1 to 1/0) to give 34 (30.0 g, 57.9 mmol, 70% yield, 98% purity) as a yellow solid. LCMS [M + 1]+= 505.2. 1H NMR (400 MHz, CDCl3) δ = 8.74 (s, 1H), 7.60 (d, J = 1.6 Hz, 1H), 4.89 (t, J = 7.2 Hz, 1H), 4.72 (br d, J = 7.6 Hz, 1H), 3.87–3.57 (m, 4H), 2.40 (s, 3H), 2.17 (m, 4H), 1.64 (d, J = 6.8 Hz, 3H), 1.22 (s, 9H).
(R)-9-(1-Aminoethyl)-2-(4,4-difluoropiperidin-1-yl)-7-methyl-4-oxo-4H-pyrido[1,2-a]pyrimidine-3-carbonitrile (35)
A mixture of 34 (20.0 g, 38.6 mmol, 1.00 equiv), zinc cyanide (9.06 g, 77.1 mmol, 4.90 mL, 2.00 equiv), XantPhos Pd G3 (3.66 g, 3.86 mmol, 0.10 equiv) in N,N-dimethylacetamide (200 mL) was degassed and purged with nitrogen 3 times, and then, the mixture was stirred at 100 °C for 1 h under a nitrogen atmosphere. The mixture was then cooled to 25 °C and filtered, and the filter cake was washed with ethyl acetate (600 mL). The filtrate was diluted with water (600 mL) and extracted with ethyl acetate (300.0 mL × 2). The combined organic layers were washed with brine (600 mL × 2), dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure to give (R)-N-((R)-1-(3-cyano-2-(4,4-difluoropiperidin-1-yl)-7-methyl-4-oxo-4H-pyrido[1,2-a]pyrimidin-9-yl)ethyl)-2-methylpropane-2-sulfinamide (23.0 g, crude) as a yellow solid, which was used without further purification. LCMS [M + 1]+ = 452.3. To a solution of (R)-N-((R)-1-(3-cyano-2-(4,4-difluoropiperidin-1-yl)-7-methyl-4-oxo-4H-pyrido[1,2-a]pyrimidin-9-yl)ethyl)-2-methylpropane-2-sulfinamide (23.0 g, 45.5 mmol, 1.00 equiv) in ethyl acetate (230 mL) was added hydrochloric acid (4 M in dioxane, 45.5 mL, 4.00 equiv), and the mixture was left to stir at 25 °C for 1 h. After this time, a precipitate formed, and the precipitate was filtered. The filter cake was dried under vacuum to give 35 (15.6 g, 40.3 mmol, 89% yield, 99% purity, hydrochloric acid salt) as a yellow solid. LCMS [M + 1]+ = 348.3. 1H NMR (400 MHz, DMSO-d6) δ = 8.64 (br s, 4H), 8.17 (d, J = 1.6 Hz, 1H), 5.04–4.87 (m, 1H), 4.07–3.93 (m, 4H), 2.38 (s, 3H), 2.26–2.05 (m, 4H), 1.59 (d, J = 6.8 Hz, 3H).
(R)-2-((1-(3-Cyano-2-(4,4-difluoropiperidin-1-yl)-7-methyl-4-oxo-4H-pyrido[1,2-a]pyrimidin-9-yl)ethyl)amino)benzoic Acid (17)
To a solution of 35 (12.0 g, 31.1 mmol, 1.00 equiv, hydrochloric acid salt) in DMF (100 mL) was added copper acetate (8.47 g, 46.6 mmol, 1.50 equiv), DBU (14.2 g, 93.2 mmol, 14.1 mL, 3.00 equiv), 4 Å molecular sieves (1.00 g), and 2-carboxyphenylboronic acid (7.74 g, 46.6 mmol, 1.50 equiv), and the mixture was left to stir at 25 °C for 12 h. The pH of the mixture was then adjusted to pH ∼5 with hydrochloric acid (2N in water). The solution was extracted with ethyl acetate (250 mL × 2), and the combined organic layers were washed with brine (500 mL), dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. The residue was purified by column chromatography (SiO2, petroleum ether/ethyl acetate = 1/0 to 1/1) to give a yellow solid. The crude product was triturated with methyl tert-butyl ether (100 mL) at 25 °C for 1 h to give (R)-2-((1-(3-cyano-2-(4,4-difluoropiperidin-1-yl)-7-methyl-4-oxo-4H-pyrido[1,2-a]pyrimidin-9-yl)ethyl)amino)benzoic acid (17, 5.77 g, 12.3 mmol, 40% yield, 99% purity) as a yellow solid. 1H NMR (400 MHz, CD3OD) δ = 8.51 (br s, 1H), 7.91 (br d, J = 7.6 Hz, 1H), 7.76 (s, 1H), 7.20 (br t, J = 7.6 Hz, 1H), 6.56 (br t, J = 7.6 Hz, 1H), 6.37 (br d, J = 8.4 Hz, 1H), 5.28 (q, J = 6.4 Hz, 1H), 4.14 (br s, 4H), 2.25 (s, 3H), 2.22–2.10 (m, 4H), 1.64 (br d, J = 6.4 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ = 170.0, 159.6, 159.1, 149.3, 146.9, 138.5, 137.2, 134.5, 131.7, 124.4, 123.8, 117.4, 115.0, 112.1, 110.6, 70.5, 47.3, 43.8, 40.1, 33.5(t, J = 23.0 Hz), 21.5, 17.5 HRMS: Calculated for C24H23F2N5O3 [M + H]+: 468.1842; found 468.1852. [α]D25 – 318.62 (c = 0.93 g, DMSO).
Acknowledgments
We would like to thank the following teams/people for their valuable contributions to this work. The IDSU chemistry team (WuXi AppTec, Wuhan, China): Tao Guo, Feng Zhao, Pan Hu, Xiaodong Xu, Xing Su. CSU chemistry team (WuXi AppTec, Wuhan, China): Rongfeng Zhao, Shaojun Song, Wenbing Ruan, and Wenchao Fei. In vitro ADME and PK team (WuXi Apptec, Shanghai, China): Shiyan Chen, Mengmeng Wei, Binbin Tian, and Yan Yuni. Todd Baumgartner for his assistance with analytical support. The X-ray crystallography work is based upon research conducted at the Northeastern Collaborative Access Team beamlines, which are funded by the National Institute of General Medical Sciences from the U.S. National Institutes of Health (P30 GM124165). The Eiger 16M detector on 24-ID-E is funded by an NIH-ORIP HEI grant (S10OD021527). Part of the research described in this paper was performed using beamline CMCF-ID at the Canadian Light Source, a national research facility of the University of Saskatchewan, which is supported by the Canada Foundation for Innovation (CFI), the Natural Sciences and Engineering Research Council (NSERC), the National Research Council (NRC), the Canadian Institutes of Health Research (CIHR), the Government of Saskatchewan, and the University of Saskatchewan. This research used resources of the National Synchrotron Light Source II, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory under Contract No. DE-SC0012704. This work was carried out with the support of Shanghai Synchrotron Radiation Facility Beamline 19U1. This research used resources from the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. All research described in this manuscript was funded by Mirati Therapeutics. Protein production and structural biology support: Yichen Zhang, Xiaoyun Yang, Ruimei Gao, Xiuhong Zeng, and Zhelin Sun (Viva Biotech, Shanghai, China).
Glossary
Abbreviations
- bid
bis in die (twice a day)
- Cl
clearance
- IV
intravenous
- PI3Kα
phosphoinositide 3-kinase alpha
- PK
pharmacokinetics
- PO
per os (oral)
- qd
quaque die (once daily)
- SAR
structure–activity relationship
- SFC
supercritical fluid chromatography
- t1/2
half-life
- TGI
tumor growth inhibition
- Vd,ss
volume of distribution at steady state
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.4c00078.
Accession Codes
Atomic coordinates for PI3Kα cocrystal structures are available from the RCSB Protein Databank (www.rcsb.org) with accession codes: 8V8H, 8V8I, 8V8J, 8V8U, 8V8V.
Author Contributions
‡ J.K. and S.H. are co-first authors. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. J.M.K. and S.J.H. contributed equally to the preparation of this manuscript.
The authors declare the following competing financial interest(s): All authors of this manuscript are employees of Mirati Therapeutics.
Supplementary Material
References
- Vanhaesebroeck B.; Leevers S. J.; Panayotou G.; Waterfield M. D. Phosphoinositide 3-kinases: a conserved family of signal transducers. Trends Biochem. Sci. 1997, 22 (7), 267–272. 10.1016/S0968-0004(97)01061-X. [DOI] [PubMed] [Google Scholar]
- Leevers S. J.; Vanhaesebroeck B.; Waterfield M. D. Signalling through phosphoinositide 3-kinases: the lipids take centre stage. Curr. Opin. Cell Biol. 1999, 11 (2), 219–225. 10.1016/S0955-0674(99)80029-5. [DOI] [PubMed] [Google Scholar]
- Brazil D. P.; Hemmings B. A. Ten years of protein kinase B signalling: a hard Akt to follow. Trends Biochem. Sci. 2001, 26 (11), 657–664. 10.1016/S0968-0004(01)01958-2. [DOI] [PubMed] [Google Scholar]
- Cantley L. C. The phosphoinositide 3-kinase pathway. Science 2002, 296 (5573), 1655–1657. 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- Okkenhaug K.; Vanhaesebroeck B. PI3K in lymphocyte development, differentiation and activation. Nat. Rev. Immunol. 2003, 3 (4), 317–330. 10.1038/nri1056. [DOI] [PubMed] [Google Scholar]
- Jean S.; Kiger A. A. Classes of phosphoinositide 3-kinases at a glance. J. Cell Sci. 2014, 127 (Pt 5), 923–928. 10.1242/jcs.093773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stemke-Hale K.; Gonzalez-Angulo A. M.; Lluch A.; Neve R. M.; Kuo W. L.; Davies M.; Carey M.; Hu Z.; Guan Y.; Sahin A.; Symmans W. F.; Pusztai L.; Nolden L. K.; Horlings H.; Berns K.; Hung M. C.; van de Vijver M. J.; Valero V.; Gray J. W.; Bernards R.; Mills G. B.; Hennessy B. T. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res. 2008, 68 (15), 6084–6091. 10.1158/0008-5472.CAN-07-6854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuels Y.; Waldman T. Oncogenic mutations of PIK3CA in human cancers. Curr. Top. Microbiol. Immunol. 2010, 347, 21–41. 10.1007/82_2010_68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorpe L. M.; Yuzugullu H.; Zhao J. J. PI3K in cancer: divergent roles of isoforms, modes of activation and therapeutic targeting. Nat. Rev. Cancer 2015, 15 (1), 7–24. 10.1038/nrc3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fruman D. A.; Chiu H.; Hopkins B. D.; Bagrodia S.; Cantley L. C.; Abraham R. T. The PI3K Pathway in Human Disease. Cell 2017, 170 (4), 605–635. 10.1016/j.cell.2017.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.; Nie J.; Ma X.; Wei Y.; Peng Y.; Wei X. Targeting PI3K in cancer: mechanisms and advances in clinical trials. Mol. Cancer 2019, 18 (1), 26. 10.1186/s12943-019-0954-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoxhaj G.; Manning B. D. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2020, 20 (2), 74–88. 10.1038/s41568-019-0216-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhardt K.; Stuckrath K.; Hartung C.; Kaufhold S.; Uleer C.; Hanf V.; Lantzsch T.; Peschel S.; John J.; Pohler M.; Bauer M.; Burrig F. K.; Weigert E.; Buchmann J.; Kantelhardt E. J.; Thomssen C.; Vetter M. PIK3CA-mutations in breast cancer. Breast Cancer Res. Treat. 2022, 196 (3), 483–493. 10.1007/s10549-022-06637-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang N.; Dai Q.; Su X.; Fu J.; Feng X.; Peng J. Role of PI3K/AKT pathway in cancer: the framework of malignant behavior. Mol. Biol. Rep. 2020, 47 (6), 4587–4629. 10.1007/s11033-020-05435-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comprehensive molecular portraits of human breast tumours. Nature 2012, 490 (7418), 61–70. 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis C.; Shah S. P.; Chin S. F.; Turashvili G.; Rueda O. M.; Dunning M. J.; Speed D.; Lynch A. G.; Samarajiwa S.; Yuan Y.; Graf S.; Ha G.; Haffari G.; Bashashati A.; Russell R.; McKinney S.; Langerod A.; Green A.; Provenzano E.; Wishart G.; Pinder S.; Watson P.; Markowetz F.; Murphy L.; Ellis I.; Purushotham A.; Borresen-Dale A. L.; Brenton J. D.; Tavare S.; Caldas C.; Aparicio S. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 2012, 486 (7403), 346–352. 10.1038/nature10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Saez O.; Chic N.; Pascual T.; Adamo B.; Vidal M.; Gonzalez-Farre B.; Sanfeliu E.; Schettini F.; Conte B.; Braso-Maristany F.; Rodriguez A.; Martinez D.; Galvan P.; Rodriguez A. B.; Martinez A.; Munoz M.; Prat A. Frequency and spectrum of PIK3CA somatic mutations in breast cancer. Breast Cancer Res. 2020, 22 (1), 45. 10.1186/s13058-020-01284-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo S.; Loibl S.; von Minckwitz G.; Darb-Esfahani S.; Lederer B.; Denkert C. PIK3CA H1047R Mutation Associated with a Lower Pathological Complete Response Rate in Triple-Negative Breast Cancer Patients Treated with Anthracycline-Taxane-Based Neoadjuvant Chemotherapy. Cancer Res. Treat 2020, 52 (3), 689–696. 10.4143/crt.2019.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andre F.; Ciruelos E.; Rubovszky G.; Campone M.; Loibl S.; Rugo H. S.; Iwata H.; Conte P.; Mayer I. A.; Kaufman B.; Yamashita T.; Lu Y. S.; Inoue K.; Takahashi M.; Papai Z.; Longin A. S.; Mills D.; Wilke C.; Hirawat S.; Juric D.; Alpelisib for PIK3CA-Mutated, Hormone Receptor-Positive Advanced Breast Cancer. N. Engl. J. Med. 2019, 380 (20), 1929–1940. 10.1056/NEJMoa1813904. [DOI] [PubMed] [Google Scholar]
- Zhang M.; Jang H.; Nussinov R. PI3K inhibitors: review and new strategies. Chem. Sci. 2020, 11 (23), 5855–5865. 10.1039/D0SC01676D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ndubaku C. O.; Heffron T. P.; Staben S. T.; Baumgardner M.; Blaquiere N.; Bradley E.; Bull R.; Do S.; Dotson J.; Dudley D.; Edgar K. A.; Friedman L. S.; Goldsmith R.; Heald R. A.; Kolesnikov A.; Lee L.; Lewis C.; Nannini M.; Nonomiya J.; Pang J.; Price S.; Prior W. W.; Salphati L.; Sideris S.; Wallin J. J.; Wang L.; Wei B.; Sampath D.; Olivero A. G. Discovery of 2–3-[2-(1-isopropyl-3-methyl-1H-1,2–4-triazol-5-yl)-5,6-dihydrobenzo[f]imidazo[1,2-d][1,4]oxazepin-9-yl]-1H-pyrazol-1-yl-2-methylpropanamide (GDC-0032): a beta-sparing phosphoinositide 3-kinase inhibitor with high unbound exposure and robust in vivo antitumor activity. J. Med. Chem. 2013, 56 (11), 4597–4610. 10.1021/jm4003632. [DOI] [PubMed] [Google Scholar]
- Fritsch C.; Huang A.; Chatenay-Rivauday C.; Schnell C.; Reddy A.; Liu M.; Kauffmann A.; Guthy D.; Erdmann D.; De Pover A.; Furet P.; Gao H.; Ferretti S.; Wang Y.; Trappe J.; Brachmann S. M.; Maira S. M.; Wilson C.; Boehm M.; Garcia-Echeverria C.; Chene P.; Wiesmann M.; Cozens R.; Lehar J.; Schlegel R.; Caravatti G.; Hofmann F.; Sellers W. R. Characterization of the novel and specific PI3Kalpha inhibitor NVP-BYL719 and development of the patient stratification strategy for clinical trials. Mol. Cancer Ther. 2014, 13 (5), 1117–1129. 10.1158/1535-7163.MCT-13-0865. [DOI] [PubMed] [Google Scholar]
- Hanan E. J.; Braun M. G.; Heald R. A.; MacLeod C.; Chan C.; Clausen S.; Edgar K. A.; Eigenbrot C.; Elliott R.; Endres N.; Friedman L. S.; Gogol E.; Gu X. H.; Thibodeau R. H.; Jackson P. S.; Kiefer J. R.; Knight J. D.; Nannini M.; Narukulla R.; Pace A.; Pang J.; Purkey H. E.; Salphati L.; Sampath D.; Schmidt S.; Sideris S.; Song K.; Sujatha-Bhaskar S.; Ultsch M.; Wallweber H.; Xin J.; Yeap S.; Young A.; Zhong Y.; Staben S. T. Discovery of GDC-0077 (Inavolisib), a Highly Selective Inhibitor and Degrader of Mutant PI3Kalpha. J. Med. Chem. 2022, 65 (24), 16589–16621. 10.1021/acs.jmedchem.2c01422. [DOI] [PubMed] [Google Scholar]
- Mayer I. A.; Prat A.; Egle D.; Blau S.; Fidalgo J. A. P.; Gnant M.; Fasching P. A.; Colleoni M.; Wolff A. C.; Winer E. P.; Singer C. F.; Hurvitz S.; Estevez L. G.; van Dam P. A.; Kummel S.; Mundhenke C.; Holmes F.; Babbar N.; Charbonnier L.; Diaz-Padilla I.; Vogl F. D.; Sellami D.; Arteaga C. L. A Phase II Randomized Study of Neoadjuvant Letrozole Plus Alpelisib for Hormone Receptor-Positive, Human Epidermal Growth Factor Receptor 2-Negative Breast Cancer (NEO-ORB). Clin. Cancer Res. 2019, 25 (10), 2975–2987. 10.1158/1078-0432.CCR-18-3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer I. A.; Abramson V. G.; Formisano L.; Balko J. M.; Estrada M. V.; Sanders M. E.; Juric D.; Solit D.; Berger M. F.; Won H. H.; Li Y.; Cantley L. C.; Winer E.; Arteaga C. L. A Phase Ib Study of Alpelisib (BYL719), a PI3Kalpha-Specific Inhibitor, with Letrozole in ER+/HER2- Metastatic Breast Cancer. Clin. Cancer Res. 2017, 23 (1), 26–34. 10.1158/1078-0432.CCR-16-0134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D.; Weintraub M. A.; Garcia C.; Goncalves M. D.; Sisk A. E.; Casas A.; Harding J. J.; Harnicar S.; Drilon A.; Jhaveri K.; Flory J. H. Characterization, management, and risk factors of hyperglycemia during PI3K or AKT inhibitor treatment. Cancer Med. 2022, 11 (8), 1796–1804. 10.1002/cam4.4579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sopasakis V. R.; Liu P.; Suzuki R.; Kondo T.; Winnay J.; Tran T. T.; Asano T.; Smyth G.; Sajan M. P.; Farese R. V.; Kahn C. R.; Zhao J. J. Specific roles of the p110alpha isoform of phosphatidylinsositol 3-kinase in hepatic insulin signaling and metabolic regulation. Cell Metab. 2010, 11 (3), 220–230. 10.1016/j.cmet.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins B. D.; Goncalves M. D.; Cantley L. C. Insulin-PI3K signalling: an evolutionarily insulated metabolic driver of cancer. Nat. Rev. Endocrinol. 2020, 16 (5), 276–283. 10.1038/s41574-020-0329-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hon W. C.; Berndt A.; Williams R. L. Regulation of lipid binding underlies the activation mechanism of class IA PI3-kinases. Oncogene 2012, 31 (32), 3655–3666. 10.1038/onc.2011.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight Z. A.; Gonzalez B.; Feldman M. E.; Zunder E. R.; Goldenberg D. D.; Williams O.; Loewith R.; Stokoe D.; Balla A.; Toth B.; Balla T.; Weiss W. A.; Williams R. L.; Shokat K. M. A pharmacological map of the PI3-K family defines a role for p110alpha in insulin signaling. Cell 2006, 125 (4), 733–747. 10.1016/j.cell.2006.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson E. D. A.; Sean Douglas; Boyles N. A.; Dahlgren M. K.; Feng S.; Gerasyuto A. I.; Hickey E. R.; Irvin T. C.; Kesicki E. A.; Klippel-Giese A.; Knight J. L.; Kolakowski G. R.; Kumar M.; Long K. F.; Mayne C. G.; McElligott D. L.; McLean J. A.; Puca L.; Ravi K. K.; Severance D. L.; Welch M. B.; Widjaja T.. Preparation of chromenones as allosteric inhibitors of phosphoinositide 3-kinase (PI3K) for the treatment of diseases associated with PI3K modulation. WO2021202964, 2021.
- Anderson E. D. A.; Douglas S.; Boyles N. A.; Chen X.; Dawadi S.; Hickey E. R.; Irvin T. C.; Kesicki E. A.; Knight J. L.; Kolakowski G. R.; Kumar M.; Long K. F.; Mayne C. G.; Picado A.; Pototschnig G. M.; Wang H.-Y.; Welch M. B.; Widjaja T.; Wright N. E.. Allosteric chromenones as inhibitors of phosphoinositide 3-kinase (PI3K) for the treatment of disease and their preparation. WO2022235574, 2022.
- Anderson E. D. A.; Douglas S.; Boyles N. A.; Chen X.; Dawadi S.; Hickey E. R.; Irvin T. C.; Kesicki E. A.; Knight J. L.; Kolakowski G. R.; Kumar M.; Long K. F.; Mayne C. G.; Picado A.; Pototschnig G. M.; Wang H.-Y.; Welch M. B.; Widjaja T.; Wright N. E.. Allosteric chromenones as inhibitors of PI3K for the treatment of disease and their preparation. WO2022235575, 2022.
- Anderson E. D. A.; Douglas S.; Boyles N. A.; Chen X.; Dawadi S.; Hickey E. R.; Irvin T. C.; Kesicki E. A.; Knight J. L.; Kolakowski G. R.; Kumar M.; Long K. F.; Mayne C. G.; McLean J. A.; Pototschnig G. M.; Wang H.-Y.; Welch M. B.; Widjaja T.. Chromenones as allosteric inhibitors of phosphoinositide 3-kinase (PI3K) for the treatment of cancer and their preparation. WO2022251482, 2022.
- Boezio A. D.; Lucian V.; Fridrich C. G.; Gunaydin H.; Kurukulasuriya R.; Lescarbeau A.; Mader M. M.; McLean T. H.; Pierce L. C. T.; Raynor K. D.; Shortsleeves K. C.; Tang Y.; Taylor A. M.; Walters W. P.; Zhang H.; Giordanetto F.; Pechersky Y.; Wang Q.; Atienza B.-J.; Bertrand-Laperle M.; Burnie A. J.; Chen F.; Sampada G.; Shorena G.; Giguere J.-B.; Landagaray E.; Larivee A.; Lepitre T.; Maertens G.; Outin J.; Pal M.; Sturino C.; Tanveer K.; Thorat R.; Vemula N.; Krueger E. B.; Pan Y.; Deninno M. P.; Bousquet Y.; Jobin-Des Lauriers A.; Lee J.; Mohammed T.. Preparation of isoindolinone derivatives as PI3K-α inhibitors and methods of use thereof. WO2021222556, 2021.
- Boezio A. T.; Alexander M.; Gunaydin H.; Zhang H.; Raynor K. D.; Shortsleeves Kelley C.; DiPietro Lucian V.; Pierce L. C. T.; Pabon N.; McLean T. H.; Giordanetto F.; Pechersky Y.; Wang Q.; Larivee A.; Chen F.; Maertens G.; Outin J.; Bertrand Laperle M.; Pal M.; Chitale S.; DeNinno M. P.. Preparation of heterocycles as PI3K-alpha inhibitors and methods of use thereof. WO2023039532, 2023.
- Boezio A. T.; Alexander M.; Fridrich C. G.; Gunaydin H.; Dipietro L. V.; Pierce L. C. T.; Mader M. M.; Kurukulasuriya R.; Mclean T. H.; Pan Y.; Deninno M. P.; Alexandre L.; Burnie A. J.; Medena C.; Maertens G.; Tanveer K.; Pal M.; Mohamed T.; Lepitre T.; Atienza B.-J.; Vemula N.; Gelozia S.. Preparation of heterocycles as PI3Kα inhibitors and methods of use thereof. WO2023288242, 2023.
- Boezio A. T.; Alexander M.; Zhang J.; Shortsleeves K. C.; Pierce L. C. T.; McLean T. H.; Kaplan A.; Madec A.; Hudson Brandi M.; Ma J.; Pan Y.; Maertens G.; Outin J.. PI3K-alpha inhibitors and methods of use thereof. WO2023060262, 2023.
- Varkaris A.; Pazolli E.; Gunaydin H.; Wang Q.; Pierce L.; Boezio A. A.; DiPietro L.; Frost A.; Giordanetto F.; Hamilton E. P.; Harris K.; Holliday M.; Hunter T. L.; Iskandar A.; Ji Y.; Larivee A.; LaRochelle J. R.; Lescarbeau A.; Llambi F.; Lormil B.; Mader M. M.; Mar B. G.; Martin I.; McLean T. H.; Michelsen K.; Pechersky Y.; Puente-Poushnejad E.; Samadani R.; Schram A. M.; Shortsleeves K.; Swaminathan S.; Tajmir S.; Tan G.; Tang Y.; Valverde R.; Wehrenberg B.; Wilbur J.; Williams B. R.; Zeng H.; Walters W. P.; Wolf B. B.; Shaw D. E.; Bergstrom D. A.; Watters J.; Fraser J. S.; Fortin P. D.; Kipp D. R. Discovery and clinical proof-of-concept of RLY-2608, a first-in-class mutant-selective allosteric PI3Ka inhibitor that decouples anti-tumor activity from hyperinsulinemia. Cancer Discovery 2024, 14, 240. 10.1158/2159-8290.CD-23-0944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varkaris A.; Fece de la Cruz F.; Martin E. E.; Norden B. L.; Chevalier N.; Kehlmann A. M.; Leshchiner I.; Barnes H.; Ehnstrom S.; Stavridi A. M.; Yuan X.; Kim J. S.; Ellis H.; Papatheodoridi A.; Gunaydin H.; Danysh B. P.; Parida L.; Sanidas I.; Ji Y.; Lau K.; Wulf G. M.; Bardia A.; Spring L. M.; Isakoff S. J.; Lennerz J. K.; Pierce L.; Pazolli E.; Getz G.; Corcoran R. B.; Juric D. Allosteric PI3K-alpha inhibition overcomes on-target resistance to orthosteric inhibitors mediated by secondary PIK3CA mutations. Cancer Discovery 2024, 14, 227. 10.1158/2159-8290.CD-23-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St. Jean D. Jr.; Cummings M. D.. Substituted urea derivatives as PI3K inhibitors and their preparation and use in the treatment of cancer. WO2022265993, 2022.
- St. Jean D., Jr. Preparation of benzimidazoles and related heterocycles as PI3K isoform alpha inhibitors useful in treatment of cancer. WO2023018636, 2023.
- Buckbinder L.; St Jean D. J.; Tieu T.; Ladd B.; Hilbert B.; Wang W.; Alltucker J. T.; Manimala S.; Kryukov G. V.; Brooijmans N.; Dowdell G.; Jonsson P.; Huff M.; Guzman-Perez A.; Jackson E. L.; Goncalves M. D.; Stuart D. D. STX-478, a Mutant-Selective, Allosteric PI3Ka Inhibitor Spares Metabolic Dysfunction and Improves Therapeutic Response in PI3Ka-Mutant Xenografts. Cancer Discovery 2023, 13, 2432. 10.1158/2159-8290.CD-23-0396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvatti G.; Fairhurst R. A.; Furet P.; Guagnano V.; Imbach P.. Organic Compounds. WO2010029082, 2010.
- Liu X.; Zhou Q.; Hart J. R.; Xu Y.; Yang S.; Yang D.; Vogt P. K.; Wang M. W. Cryo-EM structures of cancer-specific helical and kinase domain mutations of PI3Kalpha. Proc. Natl. Acad. Sci. U.S.A. 2022, 119 (46), e2215621119 10.1073/pnas.2215621119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirola L.; Zvelebil M. J.; Bulgarelli-Leva G.; Van Obberghen E.; Waterfield M. D.; Wymann M. P. Activation loop sequences confer substrate specificity to phosphoinositide 3-kinase alpha (PI3Kalpha). Functions of lipid kinase-deficient PI3Kalpha in signaling. J. Biol. Chem. 2001, 276 (24), 21544–21554. 10.1074/jbc.M011330200. [DOI] [PubMed] [Google Scholar]
- Jenkins M. L.; Ranga-Prasad H.; Parson M. A. H.; Harris N. J.; Rathinaswamy M. K.; Burke J. E. Oncogenic mutations of PIK3CA lead to increased membrane recruitment driven by reorientation of the ABD, p85 and C-terminus. Nat. Commun. 2023, 14 (1), 181. 10.1038/s41467-023-35789-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Z. K.; Wacharasindhu S.; Binnun E.; Mansour T. An efficient direct amination of cyclic amides and cyclic ureas. Org. Lett. 2006, 8 (11), 2425–2428. 10.1021/ol060815y. [DOI] [PubMed] [Google Scholar]
- Wan Z. K.; Wacharasindhu S.; Levins C. G.; Lin M.; Tabei K.; Mansour T. S. The scope and mechanism of phosphonium-mediated S(N)Ar reactions in heterocyclic amides and ureas. J. Org. Chem. 2007, 72 (26), 10194–10210. 10.1021/jo7020373. [DOI] [PubMed] [Google Scholar]
- Ellman J. A.; Owens T. D.; Tang T. P. N-tert-butanesulfinyl imines: versatile intermediates for the asymmetric synthesis of amines. Acc. Chem. Res. 2002, 35 (11), 984–995. 10.1021/ar020066u. [DOI] [PubMed] [Google Scholar]
- West M. J.; Fyfe J. W. B.; Vantourout J. C.; Watson A. J. B. Mechanistic Development and Recent Applications of the Chan-Lam Amination. Chem. Rev. 2019, 119 (24), 12491–12523. 10.1021/acs.chemrev.9b00491. [DOI] [PubMed] [Google Scholar]
- Tan H. Z. L.; Liu B.; Wang Y.; He C.; Liu Q.; Xu H.; Qi Y..; Liu Y.; Lin S.; Wang W.. Compounds As Protein Kinase Inhibitors. WO2023078401, 2023.
- Orr S. T. M. B. B. C.; Do Q.-Q. T.; Lee C. C.-H.; Mochalkin I.; Bunker K. D.; Huang P. Q.. PI3K Inhibitors and Methods of Treating Cancer. WO2023081209, 2023.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.