

Summary
Tissue regeneration following an injury requires dynamic cell-state transitions that allow for establishing the cell identities required for the restoration of tissue homeostasis and function. Here, we present a biochemical intervention that induces an intermediate cell state mirroring a transition identified during normal differentiation of myoblasts and other multipotent and pluripotent cells to mature cells. When applied in somatic differentiated cells, the intervention, composed of one-carbon metabolites, reduces some dedifferentiation markers without losing the lineage identity, thus inducing limited reprogramming into a more flexible cell state. Moreover, the intervention enabled accelerated repair after muscle injury in young and aged mice. Overall, our study uncovers a conserved biochemical transitional phase that enhances cellular plasticity in vivo and hints at potential and scalable biochemical interventions of use in regenerative medicine and rejuvenation interventions that may be more tractable than genetic ones.
Keywords: one-carbon metabolism, small molecules, reprogramming, myogenic lineage, regeneration, histone acetylation
Graphical abstract
Highlights
-
•
Early cell transitions in differentiation include metabolites, supporting identity changes
-
•
Cell-transition biochemicals can be leveraged to induce plasticity
-
•
1C-metabolite supplementation streamlines cell-identity changes in vitro
-
•
1C-metabolite in vivo administration impacts acetylation genes, aiding muscle regeneration
Hernandez-Benitez et al. identify a metabolomic wave conserved in the early transition of cells differentiating in vitro, and they leverage this finding to customize an in vivo supplementation that facilitates the transition of cell phenotypes when needed, like in regeneration after an injury.
Introduction
The dynamics and nature of cell-state transitions that lead to the establishment of cell identities, and ultimately tissue and organ function, have usually been explored during embryogenesis as well as during regeneration. Skeletal muscle tissue differentiation is among the best-studied cell-fate transition models. During physiological differentiation, a stem cell pool gives rise to muscle progenitors (or myoblasts [MBs]), which differentiate into myofibers. Complementarily, the de novo formation of myofibers takes place during the regeneration process after injury and is also mediated by muscle progenitors. Thus, and together with its in vivo accessibility, muscle represents an excellent experimental setting to uncover factors underlying cell-identity changes in both physiological and post-injury contexts.
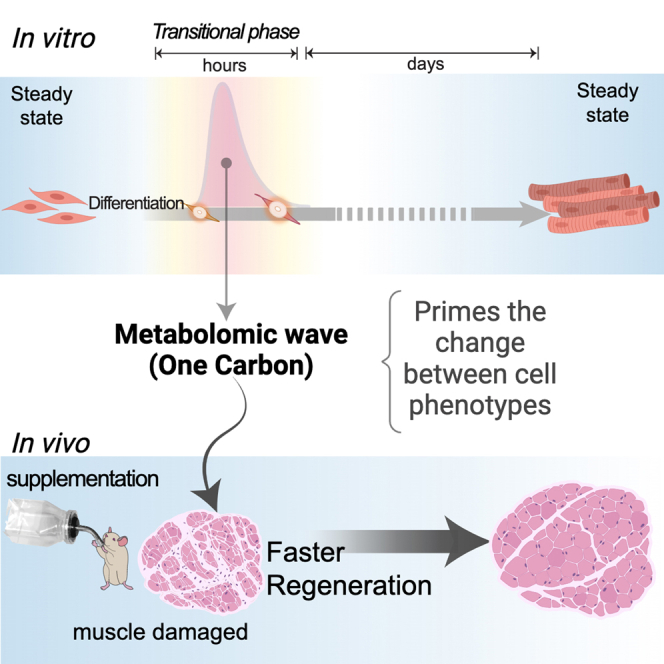
Pioneering studies have revealed the distinctive omics signatures displayed by the steady states held by MBs and myofibers.1,2 The changes between those cell identities are perfectly orchestrated during normal differentiation through a sequence of intermediate transitions that can be followed at different molecular levels.3 The in vitro differentiation of MBs into myofibers takes at least 1 week, and available studies have explored their transitional states on a scale of days.4 Thus, the nature of the earliest time windows associated with (or driving) the first steps critical for the activation of the transition of one cell program to another remains to be revealed. We thus hypothesized that tracking the immediate status of transitional states in differentiation could offer prospective functional candidates to best mirror an environment prone to cell-identity flexibility. With this in mind, we focused on and studied the biochemical shifts taking place in the early-intermediate states of MB differentiation. This approach enabled us to uncover the compounds coupled to the early transitions of programs driving changes in cell phenotypes. Among them, a biochemical wave of one-carbon (1C) metabolites was detected at the initiation of MB differentiation, but it was also shared by different cell types undergoing differentiation. Subsequently, we unveil that by supplementing a few metabolites relevant to the 1C network, we can induce limited plasticity in mature differentiated cells in vitro. Moreover, this intervention enabled accelerated repair after muscle injury in young and aged mice. Together, our results help elucidate the role of the metabolome during the earliest stages of cell differentiation, in addition to their established role in bioenergetics,5 exposing the sufficiency of specific biochemicals to induce cell plasticity and moderately reprogram cell fates.
Results
Early transient states from MB differentiation reveal an upsurge of a specific biochemical niche
We characterized the early transcriptomic transitions from MBs into myofibers in vitro to detect the molecular clues that allow the switch between the two cell identities (Figure 1A). To ensure the selection of an appropriate time frame, we first detected early expression changes in some markers of the progenitor and differentiated states of muscle cells by reverse-transcription polymerase chain reaction (RT-PCR) (Figure S1A). MBs exhibited changes in gene expression as early as 1–3 h after differentiation induction, with characteristic markers of differentiation detected after 6–12 h. Based on that gene expression track, we selected appropriate time points to perform transcriptomics. We identified significant differences in global gene expression after 3 h of differentiation induction (Figure 1B). Then, we utilized Gene Ontology analysis for insights into the molecular niche of those early transitional states. Particularly, in an early time after the induction of differentiation, between 3 and 6 h, we detected a significant enrichment of pathways associated with metabolic processes, including biosynthetic and regulatory ones (Figure 1C). This observation suggested that metabolic clues may have a crucial role in preparing the intracellular niche for achieving the turning off and on of programs during cell-identity changes. Therefore, we proceeded with untargeted metabolomic analyses in an equivalent time frame.
Figure 1.

Identification of biochemical transitional states between myoblasts (MBs) and myofibers
(A–C) Transcriptomic analysis during the early transitional states between MBs and myofibers in vitro. (A) The timing of potential interest to identify the switching between transcriptional programs of two cell phenotypes. (B) Principal-component analysis (PCA) of transcriptomics at the indicated time points. (C) Gene enrichment analyses. Transcriptomic profiles were obtained by bulk RNA-seq; each sample is represented by single dots (n = 3).
(D–F) Metabolomic transitions between MBs and myofibers in vitro. (D) Analysis of fingerprint of the immediate populations differentiated from MBs represented in PCA. Overlapped in yellow over the PCA are the intermediate points during differentiation. Each dot represents a replicate (n = 5). (E) A schematic representation of patterns identified regarding the abundance of individual metabolites over time. The bell-shaped pattern represents an early wave of metabolites that temporally fit with the intermediate transitional populations differentiated from MBs. Enrichment analysis of the bell-patterned metabolites MBs is in (F). Analyses derived from n = 5 per time collected and per cell type.
(G) Intersections of the metabolic pathways prevalent at the initial, transitional, and terminal states of three progenitor cell types. MBs, neural stem cells (NSCs), and mesenchymal stem cells (MSCs) undergoing differentiation into myofibers, astrocytes, and chondrocytes, respectively. Venn diagrams show the intersection of the top 12 pathways derived from the enrichment analyses of reductive, bell-shaped, and cumulative-patterned metabolites corresponding to initial, transitional, and terminal states for each cell type (left, middle, and right, respectively). See the list of pathways in Table S1. Illustration tracks the change in the progression of differentiation (top horizontal gray arrow). A small inset at the top left of each Venn diagram exemplifies the type of pattern that produces the associated enrichment results. Note the commonalities derived from enrichments of the early-wave bell pattern in the central intersection in the middle image.
Enrichments were obtained by MetaboAnalyst, only with metabolites having identifiers in the Human Metabolome Database (HMDB ID). Enrichments derive from data of n = 5 per time point and per cell type.
We used an ultrahigh-performance liquid chromatography-tandem mass spectrometry platform to comprehensively identify and quantify metabolites. Changes in the abundance of each identified metabolite were assessed, and similarities between samples were computed using principal-component analyses (PCAs). We observed that the metabolome of MBs was well separated from that of their earlier differentiated counterparts (Figure 1D). Next, to identify the biochemicals essential in the intermediate transitions, we applied recurrent pattern classification (RPC), which traces the relative mean abundances of compounds over time (Figure 1E; RPC algorithms in Data S1). RPC enabled us to visualize and identify recurrent patterns of metabolite abundance, even those exhibiting subtle increases in specific windows, allowing us to classify them into cumulative, reductive, U-shaped, or bell waves and exclude them from those that did not change over time (Data S1). Of note, the bell-wave pattern concurred with the frame pointed out by the initial transcriptomics (Figures 1A–1C and S1D). Enrichment analysis of the bell pattern showed pathways such as the Warburg effect, glycolysis and gluconeogenesis, 1C-related pathways, and lipid metabolic pathways (Figure 1F).
We next analyzed additional cell types undergoing differentiation to identify chemicals that may have a broader effect on supporting early transitional niches in cells other than MBs. Thus, we screened by RT-PCR some markers on neural stem cells (NSCs) and mesenchymal stem cells (MSCs) differentiating into astrocytes and chondrocytes, respectively (Figures S1B and S1C). We then profiled the metabolomics at the appropriate times and applied the RPC strategy. In the same way, as with MBs, we detected a bell-wave pattern overlapping the early-intermediate transition (Figures S1E and S1F). Of note, we confirmed the outcome of our classification by K-means clustering and silhouette analyses (Figures S1G and S1H). Subsequently, we performed a comparative analysis on the enriched pathways. Considering the three multipotent models tested (MBs/myofibers, NSCs/astrocytes, and MSCs/chondrocytes), we found more commonalities at the intersections of transient states (bell patterns) than at other intersections (Figure 1G, compare middle vs. right or left; Table S1). Overall, this may suggest the existence of a general fingerprint supporting the partial plasticity of intermediate states.
Transient states from multipotent and pluripotent cells share a fingerprint marked by 1C metabolites
After comparing metabolomic transitions, we found methionine metabolism and spermidine/spermine biosynthesis to be the pathways shared at the bell-pattern intersection of Venn diagrams (Figure 1G, middle). Of note, these pathways are interconnected in the 1C metabolism network. We thus explored in detail the relevant shifts in metabolites after inducing differentiation in MBs, NSCs, and MSCs (Tables S2–S4), with a subsequent focus on the early-transient state (3–6 h), corroborating the significant trends of increases in several 1C metabolites (Figure S2A).
Next, we wondered whether cells with higher levels of differentiation potential, such as pluripotent cells, could have similar readouts. We thus utilized ZHBTc4 embryonic stem cells (ESCs), a well-characterized line that efficiently changes its identity toward a trophoblast phenotype,6 to measure the relative levels of two central 1C metabolites, methionine and S-adenosylmethionine (SAM). We found that both metabolites increased during the intermediate state identified by crucial trophoblasts markers (Figure S2B). Likewise, we explored the early transitional state during human ESC differentiation into cardiomyocytes. Consistently, we corroborated an upsurge of methionine and SAM during the early-intermediate state (Figure S2C). Thus far, our results suggest that biochemicals from the 1C network represent potential metabolites that mark the intermediate flexibility not only of MBs but also of other multipotent and pluripotent cells.
A synthetic 1C niche induces a discrete progenitor-like profile in myofibers
The transitional 1C state might mirror the metabolomic requirements of any other cells confronting a change in their identity. Therefore, we treated differentiated myofibers with the a priori-selected biochemicals that are relevant for the 1C network, including methionine, threonine, glycine, putrescine, SAM, and cysteine (refer to STAR Methods for details on the stability and permeability of chemicals used). These molecules were selected as representatives of the 1C network prevalent across the transitional states of the several cell types presented above (Figure 2A). Because of the type of metabolites involved, we named the cocktail 1C-metabolite induction medium (1C-MIM). To get a proper customization dose effect, we exposed MB cultures to a range of concentrations of each metabolite and quantified viable ATP-metabolic active cells (Figure S3A). Orthogonally, we tested the effects of different concentrations of single and combinations of metabolites in astrocyte cultures (Figures S3B and S3C). Once the concentrations to compose 1C-MIM were chosen, a battery of control parameters were performed. Those included the viability of cultures exposed to individual metabolites and the cocktail at different times, the measurement of pH, and the levels of proteasome activity, which were tracked to verify that the effects were not due to a general loss of fitness (Figures S3D–S3H).
Figure 2.

Mirroring the biochemical transitional niche evokes the reduction of differentiation marks on mature cells
(A) Schematic representation of the supplementation of metabolites to partially imitate a 1C (one-carbon) wave with 1C-MIM (metabolite induction medium) over a terminally differentiated steady state. Note that the 1C-MIM represents an artificial intervention to be added in mature cells that do not have a 1C wave.
(B) Effects on the supplementation of 1C-MIM over the differentiation markers of myofibers Myfh11 (on the left) and Mef2c (on the right). Expression evaluated by RT-PCR. Bars represent the gene expression levels on differentiated control myofibers or those exposed for 3 days to the supplementation. Top left insets show the levels observed during differentiation. The gene expression of genes of interest was normalized with the geometric mean of at least two housekeeping genes (from Actb, Gapdh, and Nat1) and then normalized vs. control condition. Each dot represents an independent sample, n ≥ 3, with means ± SEM. The differentiation level at 72 h (bars control) is normalized to 100% to evaluate the reduction induced by 1C-MIM, where differences compared to control are significant at ∗∗∗p < 0.001.
(C and D) Effects on the supplementation of 1C-MIM over the progenitor markers of myofibers Pax7 and MyoD. Protein expression levels were evaluated by immunofluorescence in myofibers exposed to different concentrations of serum; differences compared to control are significant at ∗p < 0.05, ∗∗p < 0.005, and ∗∗∗p < 0.001.
(E) Representative images of in vitro control myofibers and those 3 days after the treatment with 1C-MIM. Note: a fresh feeding was done to improve the quality of the photograph; scale bar: 50 μm.
(F–J) Transcriptional characterization of the identity acquired by myofibers after 1C-MIM supplementation. (F) PCA map of control untreated myofibers, myofibers treated with a scramble combination of metabolites non-related directly to 1C (see main text for details), and 1C-MIM-treated myofibers. Transcriptomic profile obtained by bulk RNA-seq. (G) Hierarchical cluster analysis (HCA) performed using a Euclidean distance metric comparing gene expression profiles; red to blue color gradient indicates higher to lower expression. (H) Gene set enrichment analyses of the DEGs between 1C-MIM myofibers vs. the rest (control and scramble); to the left are enrichments for pathways, and to the right is enrichment for ontologies. (I) Volcano plot representations of differentially expressed genes (DEGs) at the comparison myofibers control to 1C-MIM. DEGs are represented by dots (adjusted p value [p-adj.] < 0.01). Blue dots indicate significantly upregulated and downregulated genes. Gray dots indicate not significant genes. (J) Enrichment analysis by gProfiler of DEGs included 471 downregulated and 351 upregulated considering p-adj. < 0.01.
See also Figures S2–S4 and Tables S2–S4.
Next, we explored whether the artificial niche created by 1C-MIM could drive discrete reprogramming in differentiated myofibers. This was based on the premise that the intermediate transition between MBs to myofibers has an intracellular niche briefly populated by 1C-related metabolites (Figure 2A). We found that myofibers exposed to 1C-MIM exhibited downregulation of the gene expression of mature cell markers Myh11 and Mef2c and recovered the protein expression of Pax7 associated with the progenitor state (Figures 2B–2D). Because we observed a change in morphology, we validated that all cell cultures lysed for downstream analyses had at least 85% cell viability at the time of collection, eliminating the possibility that reductions in expression were derived only from mortality (Figures 2E and S3D). MB/myofiber differentiation depends on serum levels; therefore, we tested 1C-MIM at two different serum concentrations, 2% and 20% (Figures 2C and 2D). We also tested the 1C-MIM cocktail without serum in other cell types (astrocytes and chondrocytes and in additional cell types from mice and humans; see comments about this small screening in the discussion); in all these conditions, we confirmed effects like those observed in myofibers independent of serum presence (Figure S4). Of note, current strategies handled by conventional reprogramming require reduced serum or serum-free media.7,8 Therefore, our results suggest that 1C-MIM induces in differentiated somatic cells some attributes of the corresponding progenitor state.
1C-metabolite supplementation modulates cell cycle and favors reprogramming responsiveness in vitro
To explore the action of 1C-MIM on myofibers, we used the in vitro model of C2C12. Briefly, C2C12 were differentiated by serum reduction and then exposed for 3 days to 1C-MIM or a scramble metabolite combination (mix of six metabolites non-directly related to 1C metabolism). Then, we performed RNA transcriptomics analysis. The scramble combination presented a similar profile of gene expression to the control of myofibers untreated (Figures 2F and 2G). Orthogonal results by RT-PCR in astrocytes confirmed that applying the scramble combination did not repress the expression of differentiation markers, nor did it increase the progenitor markers (Figures S3E–S3F). The differential gene expression analysis from 1C-MIM-treated myofibers vs. the rest (control and scramble) enriched cell-cycle-related pathways and cytoskeleton ontologies (Figure 2H). The 1C-MIM-treated myofibers exhibited 822 differentially expressed genes (DEGs) to their parental untreated control (Figure 2I). Among the top upregulated genes by 1C-MIM was Mmp13, a contributor for skeletal muscle regeneration and critical for MB migration,9 several metalloproteinases, kinases, and genes from fetal muscle development like Stc110 (Figures 2I and 2J).
Similar deductions were derived from the transcriptomics on 1C-MIM-treated astrocytes, where the profile was closer to the progenitor NSCs and exhibited regulation of cell cycle, and cell-migration-related processes were differentially regulated between astrocytes and MIM astrocytes (Figures 3A–3C). Of note, we validated the acquisition of an intermediate-like state in the 1C-MIM-treated astrocytes. This was done by comparing the DEGs in 1C-MIM-treated astrocytes to the intermediate-NSC-like state formerly reported for the partially reprogramed astrocytes11 and evidenced that both sets share 446 genes that, in congruence with the above results, functionally enrich cell-cycle processes (Figures S5A–S5E). Overall, the treatment of in vitro cells with 1C-MIM modulates the cell cycle, which, in agreement with the literature, is one of the immediate responses to the change of cellular states during cell-fate specification and reprogramming.12,13
Figure 3.

Biochemical transitional niche streamlines transdifferentiation events between cell types
(A–C) Transcriptional characterization of the identity acquired by astrocytes after 1C-MIM supplementation. (A) PCA map of control astrocytes, 1C-MIM-astrocytes, and NSCs based on the gene-normalized expression level. The transcriptomic profile was obtained by bulk RNA-seq; each sample is represented by single dots (n = 3). (B) HCA performed using a Euclidean distance metric comparing gene expression profiles; magenta to green color gradient indicates higher to lower expression Z score and K-means analysis of DEGs classified in four clusters with the indicated number of genes. (C) Gene Ontology-enriched categories in each identified cluster from (B).
(D–G) Functional assay for the conversion of MSCs into MBs accelerated by 1C-MIM. Schematic representation of the pretreatment with 1C-MIM after the infection with AAV-DJ-MyoD-2A-GFP and times for evaluation in (D). Evaluation of cell number and size of population after 4 days of 1C-MIM or control treatments in (E), where MIM-6 contains methionine, glycine, putrescine, cysteine, and S-adenosylmethionine, while MIM-4 contains the former composition minus cysteine and minus S-adenosylmethionine. (F) Representative images of transdifferentiated cells, in green the GFP expression (infected cells that harbor MyoD), and in red MF20 (transdifferentiated cells expressing antigen myosin heavy chain [MyH1E]) evaluated at day 8 post-infection. Scale bar: 50 μm. (G) Percentages of MF20+ cells over GFP expressed cells obtained by direct counting of fluorescents cells. Significant differences indicated as ∗∗p<0.005 and ∗∗∗ p<0.001.
Dots represent each sample, n ≥ 3, and bars the average ± SEM.
See also Figure S5.
To explore whether the 1C-MIM-induced state may increase responsiveness to specific differentiation factors, we infected mouse MSCs with MyoD, a well-characterized myogenic transcription factor, and evaluated the conversion efficiency toward myofibers when combined with 1C-MIM (Figure 3D). In this model, MyoD expression is proportional to the linked GPF+ signal, whereas successful transdifferentiation is measured thereafter by MF20, which stains differentiated muscle fibers. We observed that 1C-MIM not only increased the efficiency of conversion up to 5-fold but also accelerated the cell conversion, as observed by the earlier detection of MF20+ cells on day 4, when the infection alone still did not show any converted cells (Figures 3D–3G). Interestingly, more GFP+ cells over the same number of cells were observed on day 8 post-infection, which may reflect an additional influence of the cocktail in the viral transduction, independently of the efficiency of transdifferentiation (which has been normalized to GFP). Of note, no significant differences after the 4 days of supplementation (and before transduction) were observed between control and supplemented cells (Figure 3E); particularly, on the combination of 4 metabolites, more impact in transdifferentiation was detected (Figures 3F and 3G). Also, we challenged the supplementation in an MB-independent model. We treated human fibroblasts with neurogenin (NGN1/2) with or without 1C-MIM and evaluated the efficiency of neuronal conversion (Figure S5F). We found that 1C-MIM caused a reduction in the fibroblast marker CD44 and increased neural marker expression (Figures S5G–S5J). Overall, the above results demonstrate that exposing cells to a biochemical niche abundant in 1C metabolites favors the niche that facilitates traits for plasticity.
In vivo 1C-metabolite intervention accelerates muscle recovery after injury in young and old mice
Next, to validate the potential translatability of 1C-MIM, we performed an in vivo intervention. We chose the model of muscle degeneration by cardiotoxin (CTX), which injures muscular fibers with subsequent activation of the repair mechanism involving sequential transformative cell events.14 We performed an in vivo intervention using 87 wild-type mice (see allocated groups in Figure S6A). Because this intervention consisted of a drinking supplementation, we measured the drinking volumes, observing just a slight trend of an increase in the drinking of 1C-MIM water compared to control standard water (Figure S6B). The drinking supplementation did not induce weight changes (Figure S6C). In addition, a scramble drinking control composed of six metabolites not directly related to 1C metabolism and an intervention of 3 month glycine supplementation were included (glycine, as an amino acid part of 1C and with reported effects in muscle building15). Neither scramble nor glycine altered the volume of drinking or weight (Figures S6B and S6C).
As part of the in vivo validation, we compared the motor capacity in the intervened young and old mice. As the first step, we preconditioned the mice to the open field test (by placing the mice in the area for 1 h and 2–3 times at least 1 week before inducing the injury) as the last habituation the day before CTX injection (Figure 4A). Next, we confirmed that CTX injury impaired the mice’s movement, as observed by a reduction in motor responses in terms of velocity and jumps, together with an increase in resting time, measured 5 h after recovering from anesthesia (Figure S6D). Interestingly, only 24 h after injury, we observed an increase in ambulatory distance and, consequently, a decrease in the resting time in those mice supplemented with 1C-MIM (Figures 4B and 4C). Only a trend in the improvement of other parameters like the number of jumps and ambulatory time was detected: 59% and 61% of the variation of the relative number of jumps and ambulatory time accounted for the regeneration progression, while the velocity variation is not accounted for that recovery (Figure 4D). Of note, the resting time was the parameter that showed the most relevant difference between groups after CTX injury (i.e., mice rest for more time), and it was also compared to the glycine intervention and to the scramble intervention to test specificity. In summary, by comparison with the other control supplementations, a faster motor recovery was detected in mice supplemented with 1C-MIM; in second place was the group supplemented with glycine; and control-untreated and scramble-supplemented mice were behind, with no difference between them (Figure 4C). Overall, mice reduce the resting time (likewise increase ambulatory distance) as muscle regeneration (recovery) progresses.
Figure 4.

In vivo 1C-MIM intervention accelerates motor recovery from CTX injury
(A) Experimental design. The drinking intervention is maintained during all times, including 3 months before the CTX injection and 8 days of recovery after CTX. The day of CTX injection is marked in the center as “0,” followed by 8 days of recovery. The green bar before CTX marks the period of behavioral preconditioning of mice to the open field test (OFT), and the orange bar after CTX marks the tracking of recovery by the OFT.
(B) Representative tracks of behavioral motor test performed in control and 1C-MIM-supplemented mice 24 h after CTX injury.
(C) Kinetics of resting, evaluated by OFT in CTX-injured mice. The resting time was recorded at 1, 5, and 8 days after cardiotoxin injury during a 60 min session. Preconditioning was performed in all animals 1 week before injury and considered as net 100% for subsequent analyses.
(D) Kinetics of jumps, velocity, and ambulatory time, evaluated by OFT in CTX-injured mice. These were recorded 1, 4 , 5 , and 7 days after cardiotoxin injury during a 60 min session. Preconditioning was performed in all animals 1 week before the injury.
Total intervention n = 87 mice. Pair comparisons at indicated groups are in Figure S6A.
See also Figures S5 and S6.
Next, we analyzed what happened at the tissue level. Our observations corroborated the improvement seen in 1C-MIM-supplemented mice. After an injury, the recovery of muscle architecture is concurrent with the appearance of centrally nucleated regenerating fibers; thus, we wondered whether 1C-MIM benefits the appearance rate of central-nucleated fibers. We observed a 2.4-fold increase in central-nucleated fibers on day 2 in 1C-MIM-supplemented mice (i.e., meeting a number of center-nucleated fibers usually observed in controls after day 7), suggesting that the repair occurs faster in 1C-MIM animals (Figures 5A and 5B). This effect occurs despite having started from a comparable degree of damage with CTX injection (Figure S7A).
Figure 5.

Muscle repair is accelerated after the intervention with 1C metabolites in vivo
(A) Analysis of the number of centrally nucleated cells expressed as “number of fibers/field” in control and cardiotoxin-injected muscle at day 2 in control and 1C-MIM-supplemented mice. Random fields were assessed per condition.
(B) Representative H&E staining of tibialis anterior (TA) 7 days after cardiotoxin injection in control and 1C-MIM-treated mice. Scale bar: 50 μm.
(C) Representative immunostaining of Pax7+ cells in TA 7 days after cardiotoxin injection in control and 1C-MIM-treated mice. White arrows indicate double-positive DAPI+Pax7+. Scale bar: 15 μm.
(D) Quantification of Pax7+ cells per field in the indicated group comparisons.
(E–H) Distribution of the fibers according to their size in TA muscle 7 days after cardiotoxin injection. Comparison between control vs. 1C-MIM intervention in the indicated groups. Insets on the right of each image represent averages ± SD. Note that as the curve displaces to the right, it indicates an advance in the muscle regeneration (because of the bigger size of myofibers as these recover). (H) Comparisons of the scramble supplementation of six metabolites unrelated to 1C metabolism; only glycine supplementation; the conditions control (plain water) and 1C-MIM, all were supplemented for 3 months.
Total intervention n = 87 mice. Significant differences indicated as ∗p<0.05, ∗∗p<0.005, and ∗∗∗p<0.001. Pair comparisons at indicated groups are in Figure S6A.
Previous studies have shown that activated satellite cells (SCs) are prominent on day 7—after CTX injury—and reduced beyond 11 days.14 We observed that, indeed, 1C-MIM enhances this activation, as indicated by a significant increase in the levels of Pax7+ cells. These results were consistently obtained in independent treated groups, including young—male or female—and aged mice (Figures 5C and 5D). Moreover, during the regeneration process, the myofibers from 1C-MIM-supplemented mice exhibited increased cross-sectional areas compared to their untreated counterparts, indicating that this intervention accelerates recovery (Figures 5E–5G). The 1C-MIM intervention did not impact the myofiber size in control animals without injury (Figure S7B). Of note, the scramble 3 month intervention did not impact the myofiber size (Figure 5H), whereas the 3 month glycine supplementation had an effect, increasing the size as reported by others,15 but the effect of 1C-MIM supplementation was still superior (Figure 5H).
Finally, to get mechanistic insights of 1C-MIM in muscle tissue, we measured DNA methylation age in mice quadriceps (without injury). We found that 1C-MIM muscle exhibited a discrete but significant reduction in age according to Horvath’s muscle clock (Figure S7C). This suggests that rejuvenation by DNA methylation plays a minor role. Because in the earlier in vitro models analyzed (both myofibers and astrocytes), modulations in the cell cycle occurred after the addition of 1C-MIM, we measured the Ki67+ cells in mice muscle over the different supplementations, and only 1C-MIM—but not scramble, glycine, or control—had a significant increase in Ki67+ cells (Figure 6A). To distinguish whether 1C-MIM influences proliferating MBs or quiescent SCs, we performed coimmunostaining on isolated single myofibers using MyoD and Pax7. We found that 1C-MIM increased the number of proliferating SCs (Pax7+MyoD+) and the number of quiescent SCs (Pax7+MyoD−) by 1.96- and 1.7-fold, respectively (Figure 6B). Next, we performed RNA transcriptomics, and PCA showed a clear separation of the control muscle from the 1C-MIM-supplemented mice (Figure 6C). DEG analysis (cutoff value of log2 fold change: −0.5,+0.5 and adjusted p < 0.01) showed 789 genes, from which 67% were downregulated and associated with immune response and collagen formation (Figures 6D–6F). Particularly, significant downregulation of the pro-tumorigenic gene Saa3, the pro-dystrophy gene Timp1, and the glycoprotein involved in inflammation and cell death Clu and downregulation of metallothioneins Mt1 and Mt2, the abrogation of which has been associated with increases in myotube size and muscle strength, were observed.16 No significant changes were found in senescent markers (Figure S7D). Conversely, supplemented 1C-MIM muscle, particularly, showed upregulation of genes such as Lep (a gene known for modulating the expression of metabolic and myokine genes), Gdf5 (an inhibitor of muscle atrophy), several cadherins, Efcab6 (related to the recruitment of the histone deacetylase complex), and Hdac3 (histone deacetylase that regulates skeletal muscle fuel metabolism). Correspondingly, gene set enrichment analysis revealed that 1C-MIM intervention was positively correlated with epigenetic regulation of gene expression, chromatin-modifying enzymes, and histone acetylation (Figure S7E). The upregulated DEG enriched for regulation of transcriptional activity by acetylation, suggesting this as the via for modulating cell cycle (Figure 6F). Orthogonal readouts obtained in astrocytes treated with 1C-MIM corroborate this regulation, as we measured the changes in the relative abundance of histone modifications induced by 1C-MIM (including acetylation, methylation, and unmarked histones). No relevant changes in methylation marks were observed, but 1C-MIM increased global acetylation marks, an event supporting the acquisition of chromatin plasticity (Figures S7E and 6G).
Figure 6.

Modifications in acetylation and modulations on cell cycle drive the acceleration of regeneration by 1C-MIM in muscle in vivo
(A) Proliferation quantified by immunofluorescence and Ki67 detection.
(B) Primary myofibers were placed in vitro and treated with or without 1C-MIM for precise evaluation of double markers in nuclei. On the right, there is a summary table indicating the number of nuclear markers in single myofibers for control and 1C-MIM-treated double-positive and the ratio of Pax7+ vs. MyoD+ or MyoD– in myofibers exposed to the indicated conditions.
(C–F) Transcriptomic analysis of 1C-MIM quadriceps extracted from old mice after 3 months of supplementation. (C) PCA. (D) Heatmap with the total of DEGs. (E) Respective volcano plots showing DEGs as blue dots DEG, not significant genes as gray dots, and genes of interest highlighted as red dots (p-adj. <0.01). (F) Enrichment analysis of the DEGs with p-adj. < 0.01.
(G) Orthogonal readouts on the histone modifications detected by mass spectrometry on control astrocytes and 1C-MIM-supplemented astrocytes.
(H) Suggested mechanism of 1C-MIM for eliciting acceleration of the cell transition on regeneration. Several metabolic sensors respond to incoming metabolites, creating modulation feedback in the niche. Particularly, an increase in deacetylation will evoke relaxed chromatin, facilitating the expression of new genes associated with specific cell programs required for a cell in a given time (e.g., during regeneration). Thus, preloading the cells with specific metabolites may have prepared them for the rapid gene activation required for any identity transition.
Total intervention n = 87 mice. Pair comparisons at indicated groups are in Figure S6A. See also Figure S7.
Significant differences as ∗p<0.05, ∗∗p<0.005, and ∗∗∗p<0.001.
Overall, our results show that our 1C-MIM intervention in vivo accelerates the regeneration in mouse muscle, emulating the intermediate flexibility observed in the transitional state of differentiating MBs, therefore not impacting the lineage identity. Moreover, this intervention favors reprogramming transitions that enhance the myogenic potential after an injury event, thereby accelerating the recovery of a healthy state in damaged muscle.
Discussion
This study shows that a biochemical intervention, mirroring physiological transitional cell states captured during in vitro MB differentiation, may improve tissue regeneration and function upon injury in both young and old mice. This proposition is supported by several in vitro (including but not limited to MB/myofibers) and in vivo experimental models and may take place by a discrete reprogramming process mediated by acetylation without a loss of lineage identity.
Previously, several studies have explored in-depth cell transition identities (see, for instance, Bracha et al.4 and Peng et al.17). Knowledge about the biochemical demands on intermediate identity transitions is scarce. Thus, we aimed to capture the initial stages at which cells start to lose their steady state and transition to an intermediate, not-well-defined labile state that subsequently resolves in various differentiation paths within a particular lineage. In this context, through a time-dependent metabolomic approach, we identified a subset of biochemicals to formulate a supplementation we called 1C-MIM. This set of metabolites partially mimics the early-intermediate fingerprint characterized by a transient wave of metabolites belonging to the 1C network. Interestingly, we found similar metabolomic waves at the initial stages of differentiation in three multipotent models, as well as indicators of these metabolomic waves in the transient states derived from pluripotent cells. Of note, the similarities described occurred despite differences in the initial multipotent profiles, their supporting culture media, and time span differentiation disparities between cell models. This observation suggests that a 1C wave could be a potential conserved event for cells starting any identity transition. 1C metabolites may play a role in various contexts involving changes in cell identity and cell plasticity. We hypothesize that a rapid increase in 1C metabolites may prime the intracellular environment for the deactivation or activation of transcriptional networks, such as pathways that modulate the cell cycle of potential progenitors and provide substrates that enable specific epigenetic modulations,18,19,20 thus favoring a state in which cells are prone to make a fate decision. Indeed, supplying 1C-associated metabolites to fully differentiated cells, both in vivo and in vitro, resulted in the acquisition of some traits of a progenitor-like state.
The loss of some mature markers without the loss of lineage identity observed after 1C-MIM supplementation and the higher responsiveness to differentiation signals resemble cell dynamic attributes previously reported in cells undergoing partial reprogramming with conventional Yamanaka factors.21,22,23,24,25 This partially reprogrammed cell state has been defined as an intermediate transitional state where cells are able to retain their lineage identity, and at the same time, they are highly responsive to specific differentiation factors.26 In our experimental designs, we could define whether the 1C-MIM effects were because of selection for remaining undifferentiated cells or reversion in the identity of myotubes. This could be assessed by looking at single isolated myofibers from mouse muscle, treated by the 1C cocktail and processed by immunocytochemistry (Figure 6B). Under these conditions, 1C-MIM myotubes showed the acquisition of some nuclei positive for Pax7 (both with and without MyoD). Since this experiment was performed with single myotubes isolated and subprocessed in low density, it is thus possible to propose the partial reversion of their identity. While this does not occur homogeneously in all myotubes, this validates the multinuclear persistence of myotubes, with some nuclei acquiring the Pax7 signal. As a disclaimer, the same validation is difficult to confirm in C2C12 cultures; this is due to the nature of the differentiation of that artificial model, where some progenitor cells may remain. Nonetheless, the observations from in-vivo-isolated myofibers are enough to support a partial reversion. On the other side, intriguing transcriptomic similarities were observed between astrocytes treated with 1C-MIM and the reported intermediate reprogrammed state for astrocytes.11 We also observed that, in a transdifferentiation paradigm, cells are more receptive to changing their identity after 1C-MIM supplementation, as observed over the classic transcription factor-induced reprogramming, such as the generation of myofibers after transduction with MyoD.27,28 While future expert testing in vivo is needed for challenging tissues like the brain, we consider the coincidences between the supplementation of 1C-MIM and pioneer observations from our lab (and others) about partial-reprogramming promoting muscle repair to be exciting.24,25,29
Addressing muscle regeneration using metabolites as a therapeutic approach has been extensively addressed; nonetheless, the explanations about the positive outcomes are still limited and usually consider either amino acids as merely building blocks or point to some elusive antioxidant effects.15,30,31 Our finding about the upsurge of 1C-related biochemicals during the initiation of change of identity in normal differentiation, which boosts an environment prone to starting a cell cycle mediated by acetylation regulation, may explain the underlying process behind the positive effects observed by others in muscle supplemented particularly with amino acids like glycine or methionine. This opens a different perspective to designing more efficient interventions. Here, our results suggest that a 1C biochemical intervention is sufficient to partially elevate the plasticity of the epigenetic status. The fast capacity of metabolites for changing the niche of cells potentially relies upon several metabolic sensors, which may represent the link to our biochemical intervention; therefore, the regulation of relevant modulators’ acetylation like Hdac3, Hdac4, and Efcab6 in skeletal muscle (Figure 6E) may prime the environment for the deactivation/activation of transcriptional networks, thus favoring a state in which cells are prone to make a fate decision, facilitating the transitions needed during a regeneration process.18,19,20
While here, the 1C-MIM intervention was optimized for muscle cells (and to a lesser degree for astrocytes in culture), we emphasize the necessity of tailoring doses and type 1C metabolites further to the cell type. Only a few relevant markers of progenitor and differentiated states from several cell types were selected to observe the likelihood of having similar outcomes as those observed over myofibers (Figure S4). Larger screenings in expression markers should be done before achieving conclusions about the 1C-MIM effects in other cell types. The induction of an artificial elevation of metabolites with 1C-MIM in muscle cells can be attributed specifically to the metabolite mix 1C-MIM because a scramble metabolite combination did not reproduce the same outcome either in vitro or in vivo, providing robustness for the intervention in muscle lineage without disregarding the need of optimization for other tissues/cell types. For example, we observed that the addition of SAM or cysteine into neuronal cultures evoked cell death; thus, for testing in neurons, we removed both metabolites from the cocktail (Figure S4D). Moreover, the effects of the same cocktail in the different cell types tested in this study may also reflect the proper nature of the starting somatic population. For example, the treatment of bona fide sorted astrocytes with 1C-MIM may not exclude the additional plasticity of glial cells compared to myofibers in vitro.32 Conversely, we tentatively assume that, regarding the metabolomic dynamics of the 1C wave found in early differentiation in MBs/myofibers, NSC/astrocytes, and MSC/chondrocytes (Figure 1), future studies may corroborate our findings and extend the commonality to more cell types. Last, but not the less, other metabolomic patterns could be explored in the future, like the U-shape that mostly reflects the biochemicals reduced during the early intermediate transition, which seems to be cell-type specific. While several details concerning how the application of 1C-MIM regulates other cell types remain to be elucidated, our observations already point to this group of effectors as targets that may lead to the development of approaches toward priming cell plasticity and reprogramming cell fate. Since chemical inductions usually offer tighter control in a stepwise modality than the direct overexpression of genes,8,33,34,35,36 an intervention leveraging endogenous transitional cell niches may provide a powerful and clinically translatable strategy for redirecting phenotypes and restoring cell and tissue function during disease and aging.
Limitations of the study
This study took metabolomics dynamics of three cell types in vitro to obtain the signature during early transitions in their normal change of phenotype, and we proposed that the finding of the 1C network observed in those could be more general and that more metabolomics studies in other cell types should be done to validate that proposition. Metabolomics studies provide a snapshot of molecules, but it is important to consider that fast degradation and subtle changes in collection protocols can impact several molecules, therefore limiting the recognition of metabolites that may also have an early role in cell transitions and that are not pointed out in the current study. Finally, the use of the 1C-MIM cocktail in vivo requires further concentration standardization if it is aimed to be applied over other tissues than muscle and other species than mice.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-Nestin | Millipore | Cat# MAB353, RRID:AB_94911 |
| anti-Pax7 | DSHB | Cat# AB_528428, RRID:AB_528428 |
| anti-MF20 | DSHB | Cat# AB_2147781, RRID:AB_2147781 |
| anti-MyoD | Santa Cruz | Cat# sc-377460, RRID:AB_2813894 |
| anti-Ki67 | Cell Signaling | Cat# 12202, RRID:AB_2620142 |
| Donkey anti-Rabbit IgG (H + L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 568 | Thermo Fisher Scientific | Cat# A10042, RRID:AB_2534017 |
| Goat anti-Mouse IgG2a Cross-Adsorbed Secondary Antibody, Alexa Fluor 568 | Thermo Fisher Scientific | Cat# A21134, RRID:AB_2535773 |
| Donkey anti-Rabbit IgG (H + L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Thermo Fisher Scientific | Cat# A21206, RRID:AB_2535792 |
| Goat anti-Chicken IgY (H + L) Secondary Antibody, Alexa Fluor 488 | Thermo Fisher Scientific | Cat# A11039, RRID:AB_2534096 |
| H3K27me3 antibody | Active Motif | Cat# 39155, RRID:AB_2561020 |
| Chemicals, peptides, and recombinant proteins | ||
| rhEFG | Thermo Fisher Scientific | Cat# PH G0311 |
| Fetal Bovine Serum | Thermo Fisher Scientific | Cat# 16000-044 |
| Maxima H Minus cDNA Synthesis Master MIX | Thermo Fisher Scientific | Cat# M1662 |
| Lipofectamine 2000 | Thermo Fisher Scientific | Cat# 11668019 |
| SB 431542 | Tocris | Cat# 1614 |
| SsoAdvanced Universal SYBR ∗Green Supermix | BIORAD | Cat# 1725274 |
| Astrocyte Growth Supplement | ScienCell | Cat# 1852 |
| L-Methionine | Sigma | Cat# M5308 |
| L-Threonine | Sigma | Cat# T8441 |
| Glycine | Sigma | Cat# G5417 |
| Putrescine dihydrochloride | Sigma | Cat# P5780 |
| L-cysteine | Sigma | Cat# C7477 |
| S-adenosylmethionine tosylate | Cayman Chemical | Cat# 16376 |
| L-Arginine | Sigma | Cat# A8094 |
| Creatine | Sigma | Cat# C0780 |
| D-Fructose | Sigma | Cat# F0127 |
| L-Histidine | Sigma | Cat# H5659 |
| L-Leucine | Sigma | Cat# L8912 |
| L-Valine | Sigma | Cat# V0513 |
| Taurine | Sigma | Cat# T8691 |
| r-bFGF | Joint Protein Central | Cat# BBI-EXP-002 |
| Y27632 | Reagents Direct | Cat# 53-B85-50 |
| LDN193189 | MILTENYI BIOTEC | Cat# 130-106-540 |
| cAMP | Cayman Chemical | Cat# 18820 |
| CHIR99021 | Selleck | Cat# S2924 |
| IWP4 | Stemgent | Cat# 04-0036 |
| B-27 Supplement | Life Technologies | Cat# 17504-044 |
| N2 Supplement | Life Technologies | Cat# 17502048 |
| Cardiotoxin | Latoxan S.A.S. | Cat# L8102 |
| AAV-MyoD-2A-GFP | VectorBiolabs | Cat#230116 |
| Critical commercial assays | ||
| STEMPro Chondrogenesis Differentiation Kit | Thermo Fisher Scientific | Cat# A10069 |
| RNeasy Plus Mini kit | QIAGEN | Cat# 74106 |
| Anti-GLAST (ACSA-1) Microbead Kit | MILTENYI BIOTEC | Cat# 130-095-825 |
| Magnetic plate | OZ Biosciences | Cat# MF10000 |
| Combimag | OZ Biosciences | Cat# CM20200 |
| Methionine Assay Kit (Fluorometric) | ABCAM | Cat# ab234041 |
| S-Adenosylmethionine (SAM) ELISA Kit | Cell Biolabs | Cat# STA-672 |
| Experimental models: Cell lines | ||
| Mouse Adipose-Derived Mesenchymal Stem Cells OriCell Strain | Cyagen | Cat# C57BL/6 |
| Myoblasts C2C12 | ATCC | Cat# CRL 1772 |
| Mouse embryonic stem cells: ZHBTcH4 ESCs | Laboratory of Dr. Hitoshi Niwa | Laboratory of Dr. Hitoshi Niwa |
| Human BJ-fibroblasts | ATCC | Cat# CRL-2522 |
| Human ESC H1 | WiCell | Cat#WA01 |
| Experimental models: Organisms/strains | ||
| Mouse ICR (CD-1®) outbred mice | Envigo | Cat# Hsd:ICR (CD-1®) |
| Mouse C57BL inbred mice | Jackson | JAX® Mice |
| Oligonucleotides | ||
| See Table S5 for list of primers | Eton Bioscience | |
| Software and algorithms | ||
| ImageJ | Open Source | https://imagej.nih.gov/ij/ |
| Metabolon Portal ® | Metabolon® | https://portal.metabolon.com/en/login |
| Metaboanalyst | Xia et al., 200937 | https://www.metaboanalyst.ca/ |
| ArrayStudio | Omicsoft | http://omicsoft.com/software/ArrayStudioLauncher/publish.htm |
| Basepair | Basepair | https://www.basepairtech.com/ |
| Interactivenn | Heberle et al., 201538 | http://www.interactivenn.net/ |
| Enrichr | Chen et al., 201339 | https://maayanlab.cloud/Enrichr/ |
| gProfiler | Raudvere et al., 201940 | https://biit.cs.ut.ee/gprofiler/gost |
| Graph Pad Prism8 | © 2018 GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| Raw graphs software | Mauri et al., 201741 | https://rawgraphs.io/ |
| Adobe Illustrator | © 2020 Adobe | https://www.adobe.com/ |
| ToppGene Suite | Cincinnati Children’s Hospital Medical Center | https://toppgene.cchmc.org/ |
| Gene Set Enrichment Analysis (GSEA) software | Subramanian et al., 200542 | https://www.gsea-msigdb.org/gsea/index.jsp |
| Activity Monitor Software | Med Associates Inc. | https://med-associates.com/product/activity-monitor-7-software/ |
| Deposited Data | ||
| Metabolomics | Mendeley | Mendeley Data:https://doi.org/10.17632/ghjgddwfzz.1 |
| RPC strategy | Mendeley | Mendeley Data: https://doi.org/10.17632/zgftdrtrxx.1 |
| Transcriptomics: Myoblast differentiation and Neural stem cell transitions | Gene Expression Omnibus | Database GEO: GSE155193 and GSE145897 |
| Transcriptomics: Effect of metabolite intervention in mice muscle gene expression | Gene Expression Omnibus | Database GEO: GSE229533 |
| Transcriptomics: Effect of metabolites supplementation on C2C12 gene expression | Gene Expression Omnibus | Database GEO: GSE229534 |
Resource and availability
Lead contact
Further information and requests for resources and reagents should be directed and will be fulfilled by the lead contact Juan Carlos Belmonte (jcbelmonte@altoslabs.com).
Materials availability
This study did not generate unique reagents. A patent application has been filed related to mixture 1C-MIM. Individual reagents to generate 1C-MIM are not exclusive and available elsewhere.
Data and code availability
Metabolomics.
-
•
Data depository, Mendeley Data : https://doi.org/10.17632/ghjgddwfzz.1.
-
•
RPC strategy for metabolomics, https://doi.org/10.17632/zgftdrtrxx.1.
Transcriptomics.
-
•
Data depositories: assigned GEO accession numbers as appended below: ∗ Database GEO: GSE155193 for myoblast differentiation. ∗ Database GEO: GSE145897 for neural stem cell transitions
-
•
Database GEO: GSE229533 Effect of metabolite intervention in mice muscle gene expression
-
•
Database GEO: GSE229534 Effect of metabolites supplementation on C2C12 gene expression
No original code was generated in this study.
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
Method details
Animals
ICR mice were purchased from Envigo and the colony was maintained and expanded at Salk Institute animal facility. C57BL mice were purchased from The Jackson Laboratory and the colony was maintained and expanded at both Salk Institute and Altos Labs animal facilities. Mice were utilized for in vivo interventions and primary culture assays. For in vivo interventions, the gender and age of animals are detailed according to the study sections. All animal handling was approved by the IACUC committee to conform to regulatory standards.
In vitro cell lines and primary cultures
All cell cultures below described were maintained under standard incubation conditions: 37°C in 5% CO2, 95% humidified air. All cell cultures were authenticated by periodic PCR checks against mycoplasma. Viability and cell number were determined as required by trypan blue and the TC10 Automated Cell Counter (BioRad), New-Bauer chamber, or DeNovix automatic system, according to availability. To ensure the collection of good fractions of viable cells for downstream analysis, the viability of samples was recorded. Most viability readouts were assessed by trypan blue at the time of sample collection. The confirmation of viability was originally needed to account for the strong impact that dead cells can have on RNAseq readouts. In most of the cases, cells were seeded in a 6-well-plates format. On the day of collection, several sterile 15mL tubes were placed on ice (to avoid degradation during the time needed to collect all samples). The original medium from each well was collected into their respective sterile tube (to avoid washing out the dead cells, and truly determine potential toxicity by the treatment). Then 500μL/well of TrypLE was added and plates incubated for 5 min at 37°C. Once cells were detached, the TrypLE-solution (with cells in it) was collected into their respective tube. One additional wash with 1mL of PBS per well was performed and the wash was collected in their respective tube. Tubes were centrifuged at 300 g × 5 min, and the supernatant was discarded. Each pellet was resuspended with 1mL medium and cells were counted using trypan blue.
Myoblast-cell line culture and differentiation
Myoblasts (C2C12 from ATCC, Cat. CRL 1772) were cultured in Myoblast medium (DMEM with 20% FBS) up to approximately 50% confluency. Cells were detached for passaging using TrypLE according to growth status. For differentiation, when cultures became fully confluent, were washed with PBS, and the above Myoblast medium was replaced by Myofiber differentiation media (DMEM, 2% FBS). Cell morphology was monitored using an IX51 inverted microscope (Olympus).
Primary myoblasts
Myoblasts were isolated from 5-week-old female mice. The hindlimb skeletal muscles were minced and digested in type-I collagenase (Worthington Biochem Cat. LS004194) and dispase B mixture. The digestion was stopped with F-10 Ham’s medium containing 20% FBS, and the cells were filtered from debris, centrifuged, and cultured in growth media (F-10 Ham’s media, 17% FBS, 4 ng/mL bFGF and 1% penicillin-streptomycin) on uncoated dishes for three days when 5 mL growth media were added each day. Then the supernatant was collected, centrifuged, and resuspended with 0.25% trypsin. After washing off the trypsin, primary myoblasts were seeded on collagen-coated dishes, and the growth medium was changed every two days.
Primary NSCs
NSCs were derived from murine embryonic cortex at 14.5 embryonic days (vaginal plug considered 0.5-day). A single-cell suspension was seeded in anti-adherent solution-treated dishes with Neurobasal medium supplemented with 1X B27 (Life Technologies, Cat. 17504-044) and rh-EGF (Thermo Fisher Scientific, Cat. PH G0311) and hFGF2 (Joint Protein Central, Cat. BBI-EXP-002) (20 ng/mL). Primary neurospheres appeared after 5–6 days of culture and were used only during the first 10-passages. Cultures for NSCs were seeded at 200,000 cells/mL and maintained on standard conditions.
Astrocyte cultures
Astrocytes were derived from cortical tissues postnatal or differentiated from dissociated neurospheres after a second or third passage, generated as described before. For postnatal astrocytes, cortices were isolated from P4 mice pups to get single cells, and cells were sorted with Anti-GLAST (ACSA-1) (MILTENYI BIOTEC, Cat. 130-095-825). Microbead kit according to manufacturer instructions. Cells were plated over pre-treated Poly-L-Lysine plates or Poly-D-Lysine glass-coverslips using Astrocyte Differentiation Medium (DMEM-F12, 10% FBS, 1X B27, and 1X Glutamax). The medium was replaced every other day until day-8 when astrocytes reached a mature phenotype corroborated by immunocytochemistry.
Neuronal cultures
Primary neurons were obtained from the cortex of E14.5 mice brains. Brain dissection was performed in a cold solution of 2% glucose in PBS. Then, tissue was trypsinized, and the suspension was transferred across a 40μm cell strainer to get a single-cell suspension. Cells were plated in a ratio of 800,000 cells per each 22mm poly-D-lysine coverslip with Neuron differentiation medium (Neurobasal media, 1X B27, and 1X Glutamax) and maintained on standard conditions. The half volume of culture media was replaced every other day. Note: In previous studies, we tracked the disappearance of the proliferative neuronal progenitors present in the primary culture by 10μM EdU-pulses every day after plating, and we found that 5-day after seeding, the percentage of EdU+ cells was reduced to basal levels. At this time point, we consider neurons as post-mitotic.
Primary culture of chondrocytes
Primary chondrocytes isolated from femoral and tibial condyles of 5 days old mice were cultured overnight at a density of 7x10ˆ3 cells/cmˆ2. Chondrocyte medium contains DMEM plus 10% FBS, and 1X Glutamax. The media was replaced the following day with fresh media. Cultures were fed every other day.
MSC-cell line culture and chondrocyte differentiation
MSCs from Cyagen (OriCell Strain C57BL/6 Mouse Adipose-Derived Mesenchymal Stem Cells, Cat. C57BL/6) were thawed in StemXVivo medium and expanded using MSC-Maintenance Medium (alpha-MEM, 10% FBS, and 1X Glutamax). MSCs were differentiated into chondrocytes by using StemPro Chondrogenesis Differentiation Kit (Thermo Fisher Scientific, cat. A10069) by the 3D-culture system during the needed times (as indicated in the corresponding figure legends). Briefly, 2.5 x 10ˆ6 MSCs were resuspended in Chondrocyte differentiation medium and pellet down in 15 mL polypropylene tubes, then, the caps were loosened, and the tubes placed on a rack and incubated in standard conditions. Half of the media was replaced every other day.
Mouse embryonic stem cell culture and trophoblast differentiation
ZHBTcH4 ESCs (Laboratory of Dr. Hitoshi Niwa) were cultured on gelatin-coated plates via standard medium containing fetal bovine serum and LIF. For trophoblast differentiation, ZHBTcH4 ESCs were cultured in the presence of 2 μg/mL Dox to repress Oct-3/4 expression.
Human embryonic stem culture and cardiomyocyte differentiation
Human embryonic stem cell (hESC) H1 line was obtained from WiCell Research and maintained on feeder-free plates pre-coated with Matrigel (BD Biosciences) using mTeSRTM1 media (STEMCELL Technologies Inc.). The cardiomyocyte differentiation was derived from hESCs. In short, dissociated hESCs were seeded on Matrigel pre-coated dishes in mTeSR media in the presence of ROCK inhibitor Y-27632 (Reagents Direct,Cat. 53-B85-50). A GSK inhibitor, CHIR (Selleck, Cat. S2924) was applied for 24 h, followed by addition of the canonical Wnt-signaling inhibitor, IWP4 (Stemgent, Cat. 04–0036). During differentiation, cells were cultured in RPMI medium plus B-27 Supplement.
Human derived-ESC astrocytes
To get neural precursor cells (NPCs), human ESCs (Cat#WA01) were dissociated into single cells by accutase, seeded at 20,000 cells/cmˆ2 density on matrigel-coated plates and cultured in mTESR1 medium containing 1:100 of Rock inhibitor overnight. Next day, the medium was switched to N2B27-medium (DMEM/F12, 1X N2, 1X B27, 1X Glutamax, 1X NEAA, β-mercaptoethanol [1:1000], and 25 μg/mL insulin), supplemented with the small molecules SB431542 (Tocris, Cat. 1614) 10μM and LDN193189 (Miltenyl Biotec, Cat. 130-106-540) 1μM. Medium was changed daily until day-8 when SB431542 and LDN193189 were withdrawn. On day-14, cells were dissociated and further maintained at high density, grown on matrigel in NPC-medium (DMEM/F12, 1X N2, 1X B27, and 20 ng/mL bFGF), and split every week. Second, for NPC-Astrocyte differentiation, NPCs were plated at 15,000 cells/cmˆ2 density on matrigel-coated plates in NPC-medium containing 1:100 of Rock inhibitor. Following day after seeding, NPC-medium was switched to astrocyte medium (2% FBS and astrocyte growth supplement). Cells were fed every 2-day for 30-day. Cultures were passaged at a 90–95% confluency.
Human BJ-fibroblasts
BJ skin fibroblast cells were obtained from ATCC (Cat. 27CRL-2522) and grown in DMEM supplemented with 10% FBS, 1X Glutamax, and 1X MEM-NEAA.
Metabolome analysis
Sample collections. Cells and media were collected according to the required time as NSCs, MSCs, and Myoblasts or during their respective differentiation conditions. Each sample was derived from a cell pellet of 100μL-mass-volume measured by the Eppendorf microtube scale. After the indicated time, cell metabolism was stopped by placing cells on an ice-bed, where cells were collected. Cells were scraped from wells, centrifuged 300g, 5min, at 4°C, and pellets were flash-frozen in LN2 until further processing by Metabolon company.
Processing of samples at Metabolon
Briefly, samples were homogenized and subjected to methanol extraction then split into aliquots for analysis by ultrahigh performance liquid chromatography/mass spectrometry (UHPLC/MS) in the positive (two methods) and negative (two methods) mode. Metabolites were then identified by an automated comparison of ion features to a reference library of chemical standards followed by visual inspection for quality control. For statistical analyses and data display, any missing values are assumed to be below the limits of detection. Statistics was performed in ArrayStudio (Omicsoft) or "R" to compare data between experimental groups; p < 0.05 is considered significant and 0.05 < p < 0.10 to be trending. An estimate of the false discovery rate (Q-value) is also calculated to take into account the multiple comparisons that normally occur in metabolomic-based studies, with q < 0.05 used as an indication of high confidence.
Determination of metabolomic patterns. The relative intracellular abundance of metabolites was estimated as Scaled Intensity, where each value is normalized by Bradford protein concentration before being considered (n = 5 biological replicates). Then, the calculated averages of the scaled intensity of 5 replicates per condition were plotted (on axes 'y') against a continuous scale of time (on axes 'x') according to the collection times determined for each cell type. Metabolites were classified and sorted according to fixed parameters by the Recurrent Pattern Classification Strategy, provided in Data S1, as standalone zip file. Note regarding the comparison of metabolomic bell-patterns: Briefly, the comparison was performed in three different progenitor cell types when those cells were exposed to differentiation conditions. Considering the variation of time span in which each cell type reaches full differentiation (being longer for the differentiation from MSC into chondrocytes and shorter for the NSC and MBs models), this makes the early intermediate transition also slightly different. Therefore, to be able to select a comparable time point between the three cell types, we selected 6h, as it is a balanced time for including the three cell types.
Hierarchical cluster analysis (HCA), K-means clustering, and silhouette analysis
Data used correspond to the values of Bradford-normalized median-Scaled value of each metabolite from 5 biological replicates. Those analyses were conducted using the MeV software. For the clustering analysis of the metabolites profile, the k-means algorithm was performed in R. Briefly, normalized metabolite values for each cell sample were analyzed using the silhouette method to determine the optimal number of clusters. Then, the K-means algorithm was performed with the optimal number of clusters to group the metabolites based on the patterns in metabolite expression levels.
Enrichment analyses. The analyses were performed using the Enrichment Analysis tool of MetaboAnalyst, where we used only the metabolites recognized by the Human Metabolome DataBase (HMDB IDs), with the library Pathway-associated metabolite sets (SMPDB). Consideration: a fraction of metabolites recognized in the Metabolon database are novel and do not have correspondent identifier in the Human Metabolome DataBase (HMDB IDs). The representations were obtained using Excel, GraphPad Prism 8 software, RAW graphs software, and Adobe Illustrator.
Concentration tests of components of one-carbon-metabolite induction medium (1C-MIM)
Cocktail’s customization was performed in myoblasts C2C12 and primary cultures of astrocytes.
For astrocytes, cultures tested individual metabolites at different concentrations in a serum-free medium (base media, BM). BM contained equivalent volumes of DMEM-F12 and Neurobasal, plus the supplements N2 (Life Technologies, Cat. 17502048) and B27 at 1X. Only for customization purposes, we observed the relative gene expression of Gfap, primary marker of astrocytes, against the exposure of a range of concentrations of each of the components for 1C-MIM. The tracking of this gene expression was performed only to observe trends to select an appropriate concentration, i.e., not lethal and with a discernible effect in gene expression compared with controls. S-adenosylmethionine (SAMe, Cayman Chemical, Cat. 16376) at millimolar concentrations was lethal for astrocytes, which is in line with the lower physiological concentrations usually found for this metabolite compared to others. Cysteine is highly sensitive to oxidation (manifested as white flake precipitation), then a high-concentrated stock solution [2500mM] (Sigma, Cat. C7477) dissolved in ddH2O with pH slightly acid (∼6) was prepared to prevent this issue. We corroborated a pH always close to 7.4 in the cells maintained in vitro (i.e., after adding the 1C-MIM as a cocktail) before and after treating them with cocktails containing the diluted solution of cysteine. Because the growth factor bFGF was added to improve survival (only for astrocytes), we also perform a curve of different concentrations of bFGF; of note, the selected concentration of 20 ng/mL did not inhibit Gfap, and lower concentrations even potentiate the expression of Gfap (this effect is different from the overall effect of the 1C-MIM cocktail repressing Gfap-expression). We tested the mixture of all elements at 1mM, 2.5mM, and 5mM, except for SAM, which was provided at 0.1mM, 0.25mM, and 0.5mM, respectively (i.e., in a ten-times less concentrated compared with the other metabolites). This cocktail represents the 1C-MIM6 with 6 metabolites. Despite that the range of 1mM with separate metabolites like putrescine or threonine (Sigma, Cat. P5780 and T8441), repressed Gfap, when those were added in combination, the synergic result did not inhibit Gfap. From these readouts, we selected the concentration of 5mM for all metabolites, except for SAM, which was added at 0.5mM, because of the toxicity explained above. We evaluated the 1C-MIM of 6 metabolites versus the elimination of SAM (component lethal at high concentrations) and cysteine (component with higher susceptibility to oxidation, but is the only one that potentiated the Gfap expression in a dose-fashion). This cocktail represents the 1C-MIM4 with 4 metabolites. For astrocytes, the combination without SAM or cysteine achieved more inhibition of the Gfap marker. However, considering that the metabolites were a priori selected because they are critical components of the one-carbon network, we tested both combinations in the experiments with other cells. Finally, we evaluated the effect of the addition of a scramble condition of metabolites not related to 1C-metabolism, including arginine, creatine, fructose, histidine, leucine, and valine, as a scramble cocktail of 6 metabolites, added at 5mM (i.e., with similar concentrations than 1C-MIM).
For myoblasts, differentiation was done by reducing the percentage of serum, from 20% to 2%. Myoblasts well supported the addition of a cocktail with six metabolites (1C-MIM6). The individual metabolites were tested in dose-response by Cell Titer Glo as described below. For RNA sequencing C2C12 myoblasts were cultured under growth medium conditions until they reached 90–100% confluence. Then, the medium was changed to a differentiation medium (DMEM supplemented with 2% horse serum) for four days and returned to medium 10% FBS for another four days, for a total of 8 days in culture to reach a differentiated phenotype. On day 8, cells were treated for 72h with or without 1C-MIM cocktail. For the in vitro 1CMIM concentration: methionine, threonine, putrescine, glycine, and cysteine were used at 5mM and SAM at 0.5mM. For the scramble combination was prepared with bicine, histidine, glucose, sucrose, raffinose, and sodium bicarbonate at equimolar concentrations as the 1CMIM cocktail described before. After the 72h treatment, the cells were collected in TRIzol reagent for RNA extraction.
CellTiter-Glo Luminescent Cell assay
Cells were seeded on adherent conditions and used when reached a confluence of 80%. As a control, a killing solution with 1% puromycin was added in each assay together with the range of each metabolite tested. Assays were performed in 96-well plates, where each concentration was run in four wells per assay. Then, each assay was run at least 3 independent times. Cell titer Glo solution was prepared according to the manufacturer’s instructions, then incubated over the cells for 15 min (cells previously treated with metabolites for 48h), and luminescence-Glo was quantified on a ClarioStar Plate reader.
Proteasome 20S activity
The assay ab112154 was used to determine by fluorometric methods the activity of the proteasome. Briefly, the LLVY-R110 Substrate was prepared with assay buffer according to the manufacturer’s instructions. Myoblasts treated with metabolites were incubated with the reagent protected from light. Controls included the reagent without cells, and cells seeded without adding the reagent. Fluorescence intensity by top read was measured at Ex/Em = 490/525nm on a ClarioStar Plate reader.
Immunocytochemistry
Required cells were seeded on Poly-D-Lysine coverslips, washed with PBS, and fixed in 4% PFA (15min). Samples were permeabilized and blocked for 1h in 5% BSA +0.02% Triton X-100; afterward, the primary antibody solution was added in PBS, and samples were kept in a wet chamber overnight. The next day, samples were washed with PBS +0.2% Tween 20 and incubated with a secondary antibody solution in PBS for 1h. DAPI-Vectashield was used to mount the samples. For myoblasts, after fixation, cells were blocked with 5% goat serum, 2% BSA, 0.2% Triton X-100, and 0.1% NaN3 in PBS for at least 1h; then the samples were incubated with primary antibodies overnight. After washing with PBS, the samples were incubated with respective secondary antibodies and DAPI for 45 min at room temperature. Images were acquired using a Zeiss LSM 710 Laser Scanning Confocal Microscope (Zeiss). For quantification purposes, the percentage of cells positive to each marker was calculated regarding the total cell number identified by DAPI nuclei, from at least five pictures obtained from each sample. Images were processed with NIH ImageJ software (https://imagej.nih.gov/ij/index.html). Primary antibodies used include anti-Nestin from Millipore (MAB353), anti-Pax7 and anti-MF20 from DSHB (Cat. AB_528428 and AB_2147781), anti-MyoD from Santa Cruz (Cat. sc-377460), anti-Ki67 from Cell Signaling (Cat. 12202), Alexa Fluor 568 and 488 (Cat. A10042, A21134, A11039, and A21206).
Relative measurement of methionine and S-adenosylmethionine
Cells treated under the respective conditions were collected (at least 250,000 cells per each technical replicate). Pellets were flash-frozen and stored in LN2-tank until processed. We lysed and processed the cell pellets according to kit’s manufacturer instructions either for methionine quantification (Methionine Assay Kit, fluorometric, Abcam, ab234041) or for SAM quantification (S-Adenosylmethionine ELISA Kit, Cell Biolabs, STA-672).
RNA isolation and gene expression analysis by RT-PCR
Total RNA was isolated at the needed time points, using the RNeasy Plus Mini kit QIAGEN (Cat. 74106), according to the manufacturer’s protocol, including a DNA-removal step with DNAseI. Amount and purity of RNA were assessed using a NanoDrop spectrophotometer (Nanodrop Technologies); at least 500ng of total RNA was used to synthesize cDNA by reverse transcription, using Maxima H Minus cDNA Synthesis Master Mix (Thermo Fisher Scientific, Cat. M1662). 2.5–10ng of cDNA was used in the following qPCR performed on a CFX384 thermal cycler (Bio-Rad) using the SsoAdvanced Universal SYBR Green Supermix (BIO-RAD, Cat. 1725274). Results were normalized to at least one reference genes (β-Actin, RPL38, GAPDH, Gus, CTCF, and Nat1, specified per figure), selected for their highest stability among a pool of common housekeeping genes. Primers sequences in Table S5. Statistical analysis of the results was performed using the 2ΔCt method. Results were expressed relative to the expression values of the experimental control.
Transdifferentiation experiments with 1C-MIM treatment
For transdifferentiate MSCs to myofibers, we constructed a MyoD overexpression AAV vector by inserting MyoD-2A-GFP cDNA into an AAV vector (AAV2 inverted terminal repeat vector) under the control of CAG promoter (Vector Biolabs Cat#230116). We initiated the transdifferentiation of MSCs to myocytes by adding 1x10ˆ9 GC AAV-MyoD-2A-GFP and metabolites to the differentiation medium (DMEM with 2% FBS). The control cells were only treated with 1x10ˆ9 GC AAV-MyoD-2A-GFP. At day 4, 6, and 8 post-differentiation, the cells were fixed with 4% PFA and processed for immunofluorescence. Myocytes were recognized by the myosin-heavy chain, which was labeled by MF20.
For transdifferentiate BJ-fibroblast into neurons, the first cell type was transduced with lentivirus containing doxycycline-inducible Neurogenin and rTA. The recombinant AAV vector was generated following the procedures of the Gene Transfer Targeting and Therapeutics Core at the Salk Institute for Biological Studies. Two days post-transduction cells were plated in desired density and treated with metabolites cocktail (MIM4 or MIM6) for 5-day followed by the addition of doxycycline at a concentration of 0.5 μg/mL for induction of Neurogenins for 3-day, then collecting the cells for RNA analysis.
In vivo intervention
C57BL or ICR mice were allocated into groups according to their age, gender, and strain, where possible siblings were split into control and treatment to reduce differences. All mice used in the experiment were wild-type, with no evident health problems. We used mice between 3-4-months-old for experiments for the young phenotype, and mice between 17 and 20-months-old for experiments for the old phenotype. All animals were weighed at the beginning and end of the intervention. The drinking solution was prepared from the stock of water provided by the animal facility, for both controls and supplemented animals. A volume of 150mL of water was provided every other day for each cage containing 5 mice maximum. The remaining volumes of control and supplemented water were measured every time before discarding the remaining volumes and the volume consumed was correlated to the number of mice present in each cage, to determine the drinking volume. The treated water supplementation was delivered in sterile red bottles for a period of one to three months. After these interventions, mice were habituated to walking around an open field test every other day for a week before the muscle injury.
Muscle injury by cardiotoxin
A solution of cardiotoxin (Latoxan S.A.S. Cat. L8102) was prepared at a concentration of 10μM. For old-phenotype, a volume of 50μL was injected into both back legs in the tibialis anterior (TA) and in gastrocnemius (GAS) muscles, using a 29-gauge insulin syringe. For the young phenotype the injection was in TA, GAS, and quadriceps. To provide the injection mice were anesthetized using an isoflurane chamber, afterward, legs were shaved to facilitate the visualization of muscle at the injection time. Mice were allowed to recover over a warm bed set at 38°C for 30 min and then returned to their usual housing when they were awake.
Open field tests
For pre-conditioning and for evaluation after muscle injury, animals were set in the behavioral room for 1 h in advance to habituate them. Next, each animal was placed in its respective chamber to monitor its physical activity. The chamber for recording was connected to the Activity Monitor Software to quantify distance and movements including distance, velocity, resting time, and jumps. Representative snapshots in real-time were taken for a visual representation of the distance traveled, and the raw data was further analyzed using the Activity Monitor and Excel Software.
Immunostaining and HE-staining
After euthanasia muscles from the injury site tibialis anterior (TA) and gastrocnemius (GAS) were collected and frozen in OCT for further sectioning. Transversal sections from both control and treated were obtained using a Leica cryostat set at 10μm thickness. We arranged three slices of muscle per glass slide for downstream experiments. Briefly, muscle slides were fixed with 4% PFA solution, washed with PBS, and cleared with incubation of 100mM Glycine (Sigma, Cat. G5417) for 15 min. Then, samples were blocked and permeabilized (5% goat serum +0.02%Triton X-100 + 0.01% NaN3 + 2% BSA, in PBS) for 1h at room temperature followed by an overnight incubation with the needed primary antibody at 4°C. The next day, samples were washed with PBS+0.02%tween 20 at least three times, afterward, were incubated with the respective secondary antibodies and DAPI for 1h at room temperature. All secondary antibodies were diluted at a ratio of 1:500. Fluorescent images were captured using a confocal scanning microscope. Quantification using the ImageJ software. For H&E staining we incubated slides into hematoxylin for 5-min and eosin for 3-min, with clearance with ammonium hydroxide solution (1:500), followed by standard washes. Images were captured using bright field microscopy.
Myofibers area measurement
Muscles processed by cryostat as described above were utilized to capture images in Axioscan. For analysis of area of individual myofibers the software ImageJ was used and area of at least 50 center-nucleated myofibers per slice were measured. Then an automatic subdivision per ranges was performed by the Prisma software and using those ranges the averages were estimated per condition.
Single myofiber isolation
EDL muscles were extracted and carefully digested with 2 mg/mL collagenase type I in DMEM (Worthington Biochem) for 45 min at 37°C. Digestion was stopped with a medium containing horse serum. Myofibers were released by gently flushing with a large bore glass pipette. Released single myofibers were treated with 1C-MIM or control medium and 3 days later fixed with 4%PFA for subsequent immunostaining.
DNA-methylation clock from quadriceps
After euthanasia muscles quadriceps (i.e., from non-injury-site) were extracted and flash-frozen and minced in liquid nitrogen, then samples from 25 to 50 mg of ground frozen tissue were separated for genomic DNA extraction. DNA was processed by the Clock Foundation (https://clockfoundation.org/data-tools/),43 which retrieved the averaged results of three-biological replicates per condition.
RNA-sequencing from quadriceps
After euthanasia muscles quadriceps (i.e., from non-injury-site) were extracted and flash-frozen, and minced in liquid nitrogen, then samples from 50 to 100 mg of ground frozen tissue were separated for RNA extraction. The processing from total-RNA extraction, library prep, sequencing, and analysis was performed at the company Active Motif (https://www.activemotif.com) according to their standard procedures.
RNA-sequencing from in vitro cells
Cells were collected at required times and treatments. Cell pellets were lysed with 1mL of Trizol and stored at −80°C for subsequent RNA extraction. Bulk-RNA analysis of astrocytes was developed at Salk Institute; analysis of myoblast at Altos Labs. Analyses of data were performed in BasePair software following the pipelines ‘DESeq2, compare 2 groups’ or ‘DESeq2, 3+groups comparisons’. Further analysis like differential mRNA genes, we tested whether each had enriched GO terms in biological processes and molecular functions using the ToppGene Suite and Gene Set Enrichment Analysis (GSEA) software. For ToppGene, only those functional annotation terms associated with the various sets of differentially expressed genes were clustered that were significantly enriched (Bonferroni correction, p < 0.01) compared with the function annotation terms associated with the total population of genes. Biological interpretations and visualizations of differentially expressed genes by using EnrichR and gProfiler.
Re-analysis of in vitro canonical partial reprogrammed astrocytes
We re-analyzed RNA-seq data from a previous study tracking the induction of pluripotency in astrocytes through a neural stem cell-like state NCBI Database GEO: GSE69237. We acquired the raw sequencing data and performed differential expression analysis to compare to our 1C-MIM treatment in astrocytes.
Quantification of histone modifications
Bulk histones were acid-extracted from cell pellets, propionylated, and subjected to trypsin digestion. Histone peptides were resuspended in 0.1% TFA in H2O for mass spectrometry analysis. Samples were analyzed on a triple quadrupole (QqQ) mass spectrometer (Thermo Fisher Scientific TSQ Quantiva) directly coupled with an UltiMate 3000 Dionex nano-liquid chromatography system. Targeted analysis of unmodified and various modified histone peptides was performed. This entire process was repeated three separate times for each sample. Data were imported and analyzed in Skyline with Savitzky-Golay smoothing. Each modification was represented as a percentage of the total pool of modifications. The process from the histone extraction to the analysis was carried out by Active Motif, Inc.
Quantification and statistical analysis
Statistical analysis applied, exact n values, and precision measures are indicated in each figure legend. All readouts derived from metabolomics are derived from n = 5 biological replicates. For other experiments, refer to each figure legend. No statistical methods were used to predetermine the sample size. Randomization or stratification was not applied. In general, data is shown as averages ±S.D. or S.E.M, as indicated in each figure legend. Statistics were performed using GraphPad Prism Software. Comparisons between two groups were analyzed using t-Test Two-tailed, or one-way ANOVA followed by either Bonferroni or Tukey’s post hoc test, as appropriate. Frequency distribution plots were obtained by analysis tool in Graph Pad Prism (version 9). The statistics from metabolome analyses were obtained under the Metabolon Portal Software as provided for the company (www.portal.metabolon.com/en). For transcriptomic analyses, R Software (R version 3.5.1) was used for statistics, other details about these analyses are in their respective methods' section. For all experiments, values of p ≤ 0.05 were considered statistically significant.
Acknowledgments
We thank Ruben Rabadan-Ros, Javier Prieto, Paloma Martinez-Redondo, and Nicola Allen for helpful discussions; David O’Keefe and Mallory Shields for manuscript editing; Saskia Linder, Amy Nemeth, Jingkai Wang, Paola Moreno, Mariana Morales, and Audrey Kristin for technical assistance; May Schwarz and Peter Schwarz for administrative assistance; Dr. Zhiqiu Man for providing human astrocyte cultures; and Dr. Hitoshi Niwa for providing the ZHBTcH4 ESCs. Special thanks to Greg Palczewski for his valuable help in metabolomics. Thanks to Nasun Han and Rachel Feiring for supporting genomics and cell culture work, respectively, at Altos Labs. Work in the laboratories of J.Q. and G.-H.L. was supported by the National Natural Science Foundation of China (82125011 and 81921006), the National Key Research and Development Program of China (2020YFA0804000 and 2022YFA1103700), and the CAS Project for Young Scientists in Basic Research (YSBR-076). This work was partially supported by King Abdullah University of Science and Technology (OSR-2015-CRG4-2631), Universidad Católica San Antonio de Murcia, Inventium, and Fundación Dr. Pedro Guillen.
Author contributions
Conceptualization, R.H.-B., P.M., and J.C.I.B.; methodology, R.H.-B., E.A.M., C.Z., R.D.S., C.W., V.L., S.M., E.N.D., C.R.E., T.H., H.-K.L., and I.G. (for general research and development), S.S. and C.W. (for transdifferentiation experiments), Y.S. (on human fibroblast cultures), V.L. and A.A.-V. (histochemical evaluations), and F.H., M.Y., I.G., C.W., and R.H.-B. (in vivo experiments); formal analysis, L.S. and Y.O. (transcriptomic analyses), R.H.-B. and Y.O. (metabolome analysis), R.H.-B., C.F., S.M., A.A.-V., and H.-K.L. (gene expression analysis), and S.H. (DNA methylation for Clock-mDNAge analyses); data interpretation, A.A.-V., T.H., M.Y., H.-K.L., E.N.D., P.G., C.R.E., P.R., J.Q., C.Z., and G.-H.L.; visualization and writing, R.H.-B., J.C.I.B., and P.M. (with the input of all other authors).
Declaration of interests
Patent applications have been filed related to the subject matter of this publication. R.H.-B., C.W., C.Z., S.M., F.H., V.L., I.G., S.S., M.Y., Y.S., A.A.-V., C.R.E., P.R., S.H., and J.C.I.B. are employees of Altos Labs.
Published: March 19, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2024.101449.
Contributor Information
Pierre Magistretti, Email: pierre.magistretti@kaust.edu.sa.
Juan Carlos Izpisua Belmonte, Email: jcbelmonte@altoslabs.com.
Supplemental information
References
- 1.Moran J.L., Li Y., Hill A.A., Mounts W.M., Miller C.P. Gene expression changes during mouse skeletal myoblast differentiation revealed by transcriptional profiling. Physiol. Genomics. 2002;10:103–111. doi: 10.1152/physiolgenomics.00011.2002. [DOI] [PubMed] [Google Scholar]
- 2.Riuzzi F., Sorci G., Sagheddu R., Donato R. HMGB1-RAGE regulates muscle satellite cell homeostasis through p38-MAPK- and myogenin-dependent repression of Pax7 transcription. J. Cell Sci. 2012;125:1440–1454. doi: 10.1242/jcs.092163. [DOI] [PubMed] [Google Scholar]
- 3.Furusawa C., Kaneko K. A Dynamical-Systems View of Stem Cell Biology. Science. 2012;338:215–217. doi: 10.1126/science.1224311. [DOI] [PubMed] [Google Scholar]
- 4.Bracha A.L., Ramanathan A., Huang S., Ingber D.E., Schreiber S.L. Carbon metabolism-mediated myogenic differentiation. Nat. Chem. Biol. 2010;6:202–204. doi: 10.1038/nchembio.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Magistretti P.J., Allaman I. Lactate in the brain: from metabolic end-product to signalling molecule. Nat. Rev. Neurosci. 2018;19:235–249. doi: 10.1038/nrn.2018.19. [DOI] [PubMed] [Google Scholar]
- 6.Niwa H., Miyazaki J., Smith A.G. Quantitative expression of Oct-3/4 defines differentiation, dedifferentiation or self-renewal of ES cells. Nat. Genet. 2000;24:372–376. doi: 10.1038/74199. [DOI] [PubMed] [Google Scholar]
- 7.Chen J., Liu J., Chen Y., Yang J., Chen J., Liu H., Zhao X., Mo K., Song H., Guo L., et al. Rational optimization of reprogramming culture conditions for the generation of induced pluripotent stem cells with ultra-high efficiency and fast kinetics. Cell Res. 2011;21:884–894. doi: 10.1038/cr.2011.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li X., Xu J., Deng H. Small molecule-induced cellular fate reprogramming: promising road leading to Rome. Curr. Opin. Genet. Dev. 2018;52:29–35. doi: 10.1016/j.gde.2018.05.004. [DOI] [PubMed] [Google Scholar]
- 9.Lei H., Leong D., Smith L.R., Barton E.R. Matrix metalloproteinase 13 is a new contributor to skeletal muscle regeneration and critical for myoblast migration. Am. J. Physiol. Cell Physiol. 2013;305:C529–C538. doi: 10.1152/ajpcell.00051.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Filvaroff E.H., Guillet S., Zlot C., Bao M., Ingle G., Steinmetz H., Hoeffel J., Bunting S., Ross J., Carano R.A.D., et al. Stanniocalcin 1 Alters Muscle and Bone Structure and Function in Transgenic Mice. Endocrinology. 2002;143:3681–3690. doi: 10.1210/en.2001-211424. [DOI] [PubMed] [Google Scholar]
- 11.Nakajima-Koyama M., Lee J., Ohta S., Yamamoto T., Nishida E. Induction of Pluripotency in Astrocytes through a Neural Stem Cell-like State. J. Biol. Chem. 2015;290:31173–31188. doi: 10.1074/jbc.M115.683466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu L., Michowski W., Kolodziejczyk A., Sicinski P. The cell cycle in stem cell proliferation, pluripotency and differentiation. Nat. Cell Biol. 2019;21:1060–1067. doi: 10.1038/s41556-019-0384-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu J., Ocampo A., Belmonte J.C.I. Cellular Metabolism and Induced Pluripotency. Cell. 2016;166:1371–1385. doi: 10.1016/j.cell.2016.08.008. [DOI] [PubMed] [Google Scholar]
- 14.Ramadasan-Nair R., Gayathri N., Mishra S., Sunitha B., Mythri R.B., Nalini A., Subbannayya Y., Harsha H.C., Kolthur-Seetharam U., Srinivas Bharath M.M. Mitochondrial alterations and oxidative stress in an acute transient mouse model of muscle degeneration: implications for muscular dystrophy and related muscle pathologies. J. Biol. Chem. 2014;289:485–509. doi: 10.1074/jbc.M113.493270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lin C., Han G., Ning H., Song J., Ran N., Yi X., Seow Y., Yin H. Glycine Enhances Satellite Cell Proliferation, Cell Transplantation, and Oligonucleotide Efficacy in Dystrophic Muscle. Mol. Ther. 2020;28:1339–1358. doi: 10.1016/j.ymthe.2020.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Summermatter S., Bouzan A., Pierrel E., Melly S., Stauffer D., Gutzwiller S., Nolin E., Dornelas C., Fryer C., Leighton-Davies J., et al. Blockade of Metallothioneins 1 and 2 Increases Skeletal Muscle Mass and Strength. Mol. Cell Biol. 2017;37:e00305-16. doi: 10.1128/MCB.00305-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Peng M., Yin N., Chhangawala S., Xu K., Leslie C.S., Li M.O. Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science. 2016;354:481–484. doi: 10.1126/science.aaf6284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carey B.W., Finley L.W.S., Cross J.R., Allis C.D., Thompson C.B. Intracellular α-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature. 2015;518:413–416. doi: 10.1038/nature13981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ryall J.G., Cliff T., Dalton S., Sartorelli V. Metabolic Reprogramming of Stem Cell Epigenetics. Cell Stem Cell. 2015;17:651–662. doi: 10.1016/j.stem.2015.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wellen K.E., Hatzivassiliou G., Sachdeva U.M., Bui T.V., Cross J.R., Thompson C.B. ATP-citrate lyase links cellular metabolism to histone acetylation. Science. 2009;324:1076–1080. doi: 10.1126/science.1164097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Browder K.C., Reddy P., Yamamoto M., Haghani A., Guillen I.G., Sahu S., Wang C., Luque Y., Prieto J., Shi L., et al. In vivo partial reprogramming alters age-associated molecular changes during physiological aging in mice. Nat. Aging. 2022;2:243–253. doi: 10.1038/s43587-022-00183-2. [DOI] [PubMed] [Google Scholar]
- 22.Hishida T., Yamamoto M., Hishida-Nozaki Y., Shao C., Huang L., Wang C., Shojima K., Xue Y., Hang Y., Shokhirev M., et al. In vivo partial cellular reprogramming enhances liver plasticity and regeneration. Cell Rep. 2022;39 doi: 10.1016/j.celrep.2022.110730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gill D., Parry A., Santos F., Okkenhaug H., Todd C.D., Hernando-Herraez I., Stubbs T.M., Milagre I., Reik W. Multi-omic rejuvenation of human cells by maturation phase transient reprogramming. Elife. 2022;11 doi: 10.7554/eLife.71624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ocampo A., Reddy P., Martinez-Redondo P., Platero-Luengo A., Hatanaka F., Hishida T., Li M., Lam D., Kurita M., Beyret E., et al. In Vivo Amelioration of Age-Associated Hallmarks by Partial Reprogramming. Cell. 2016;167:1719–1733.e12. doi: 10.1016/j.cell.2016.11.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang C., Rabadan Ros R., Martinez-Redondo P., Ma Z., Shi L., Xue Y., Guillen-Guillen I., Huang L., Hishida T., Liao H.-K., et al. In vivo partial reprogramming of myofibers promotes muscle regeneration by remodeling the stem cell niche. Nat. Commun. 2021;12:3094. doi: 10.1038/s41467-021-23353-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lehmann M., Canatelli-Mallat M., Chiavellini P., Cónsole G.M., Gallardo M.D., Goya R.G. Partial Reprogramming As An Emerging Strategy for Safe Induced Cell Generation and Rejuvenation. Curr. Gene Ther. 2019;19:248–254. doi: 10.2174/1566523219666190902154511. [DOI] [PubMed] [Google Scholar]
- 27.Xu B., Siehr A., Shen W. Functional skeletal muscle constructs from transdifferentiated human fibroblasts. Sci. Rep. 2020;10 doi: 10.1038/s41598-020-78987-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yagi M., Ji F., Charlton J., Cristea S., Messemer K., Horwitz N., Di Stefano B., Tsopoulidis N., Hoetker M.S., Huebner A.J., et al. Dissecting dual roles of MyoD during lineage conversion to mature myocytes and myogenic stem cells. Genes Dev. 2021;35:1209–1228. doi: 10.1101/gad.348678.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sarkar T.J., Quarta M., Mukherjee S., Colville A., Paine P., Doan L., Tran C.M., Chu C.R., Horvath S., Qi L.S., et al. Transient non-integrative expression of nuclear reprogramming factors promotes multifaceted amelioration of aging in human cells. Nat. Commun. 2020;11 doi: 10.1038/s41467-020-15174-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Love A., Cotter M.A., Cameron N.E. Nerve function and regeneration in diabetic and galactosaemic rats: antioxidant and metal chelator effects. Eur. J. Pharmacol. 1996;314:33–39. doi: 10.1016/S0014-2999(96)00528-6. [DOI] [PubMed] [Google Scholar]
- 31.Wu C.-T., Liao J.-M., Ko J.-L., Lee Y.-L., Chang H.-Y., Wu C.-H., Ou C.-C. D-Methionine Ameliorates Cisplatin-Induced Muscle Atrophy via Inhibition of Muscle Degradation Pathway. Integr. Cancer Ther. 2019;18 doi: 10.1177/1534735419828832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fu Y., Yang M., Yu H., Wang Y., Wu X., Yong J., Mao Y., Cui Y., Fan X., Wen L., et al. Heterogeneity of glial progenitor cells during the neurogenesis-to-gliogenesis switch in the developing human cerebral cortex. Cell Rep. 2021;34 doi: 10.1016/j.celrep.2021.108788. [DOI] [PubMed] [Google Scholar]
- 33.Beyret E., Martinez Redondo P., Platero Luengo A., Izpisua Belmonte J.C. Elixir of Life: Thwarting Aging With Regenerative Reprogramming. Circ. Res. 2018;122:128–141. doi: 10.1161/CIRCRESAHA.117.311866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim Y., Jeong J., Choi D. Small-molecule-mediated reprogramming: a silver lining for regenerative medicine. Exp. Mol. Med. 2020;52:213–226. doi: 10.1038/s12276-020-0383-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu Y., Song Z., Zhao Y., Qin H., Cai J., Zhang H., Yu T., Jiang S., Wang G., Ding M., Deng H. A novel chemical-defined medium with bFGF and N2B27 supplements supports undifferentiated growth in human embryonic stem cells. Biochem. Biophys. Res. Commun. 2006;346:131–139. doi: 10.1016/j.bbrc.2006.05.086. [DOI] [PubMed] [Google Scholar]
- 36.Shi Y., Hou L., Tang F., Jiang W., Wang P., Ding M., Deng H. Inducing Embryonic Stem Cells to Differentiate into Pancreatic β Cells by a Novel Three-Step Approach with Activin A and All- Trans Retinoic Acid. STEM CELLS. 2005;23:656–662. doi: 10.1634/stemcells.2004-0241. [DOI] [PubMed] [Google Scholar]
- 37.Xia J., Psychogios N., Young N., Wishart D.S. MetaboAnalyst: a web server for metabolomic data analysis and interpretation. Nucleic Acids Research. 2009;37:W652–W660. doi: 10.1093/nar/gkp356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Heberle H., Meirelles G.V., Da Silva F.R., Telles G.P., Minghim R. InteractiVenn: a web-based tool for the analysis of sets through Venn diagrams. BMC Bioinformatics. 2015;16:169. doi: 10.1186/s12859-015-0611-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen E.Y., Tan C.M., Kou Y., Duan Q., Wang Z., Meirelles G.V., Clark N.R., Ma’ayan A. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics. 2013;14:128. doi: 10.1186/1471-2105-14-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Raudvere U., Kolberg L., Kuzmin I., Arak T., Adler P., Peterson H., Vilo J. g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update) Nucleic Acids Research. 2019;47:W191–W198. doi: 10.1093/nar/gkz369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mauri, M., Elli, T., Caviglia, G., Uboldi, G., and Azzi, M. (2017). RAWGraphs: A Visualisation Platform to Create Open Outputs. In Proceedings of the 12th Biannual Conference on Italian SIGCHI Chapter (ACM), pp. 1–5. 10.1145/3125571.3125585. [DOI]
- 42.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mozhui K., Lu A.T., Li C.Z., Haghani A., Sandoval-Sierra J.V., Wu Y., Williams R.W., Horvath S. Genetic loci and metabolic states associated with murine epigenetic aging. Elife. 2022;11 doi: 10.7554/eLife.75244. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Metabolomics.
-
•
Data depository, Mendeley Data : https://doi.org/10.17632/ghjgddwfzz.1.
-
•
RPC strategy for metabolomics, https://doi.org/10.17632/zgftdrtrxx.1.
Transcriptomics.
-
•
Data depositories: assigned GEO accession numbers as appended below: ∗ Database GEO: GSE155193 for myoblast differentiation. ∗ Database GEO: GSE145897 for neural stem cell transitions
-
•
Database GEO: GSE229533 Effect of metabolite intervention in mice muscle gene expression
-
•
Database GEO: GSE229534 Effect of metabolites supplementation on C2C12 gene expression
No original code was generated in this study.
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.