

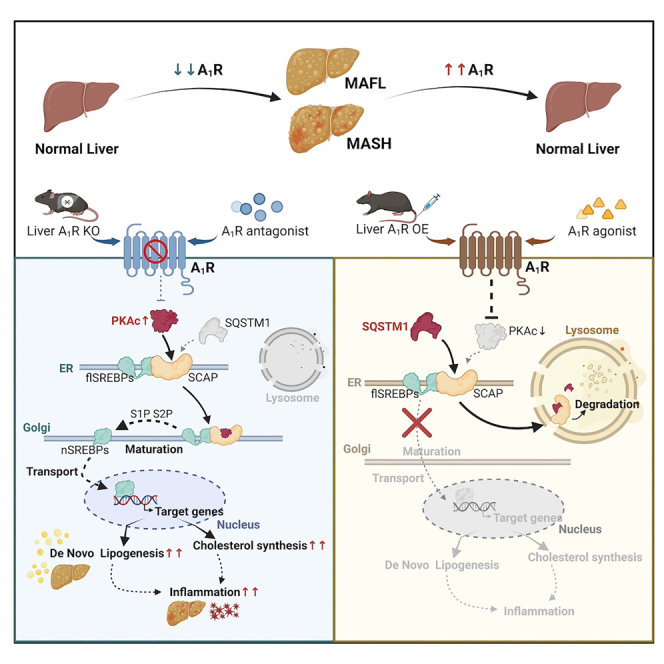
Summary
Metabolic (dysfunction)-associated steatohepatitis (MASH) is the advanced stage of metabolic (dysfunction)-associated fatty liver disease (MAFLD) lacking approved clinical drugs. Adenosine A1 receptor (A1R), belonging to the G-protein-coupled receptors (GPCRs) superfamily, is mainly distributed in the central nervous system and major peripheral organs with wide-ranging physiological functions; however, the exact role of hepatic A1R in MAFLD remains unclear. Here, we report that liver-specific depletion of A1R aggravates while overexpression attenuates diet-induced metabolic-associated fatty liver (MAFL)/MASH in mice. Mechanistically, activation of hepatic A1R promotes the competitive binding of sterol-regulatory element binding protein (SREBP) cleavage-activating protein (SCAP) to sequestosome 1 (SQSTM1), rather than protein kinase A (PKA) leading to SCAP degradation in lysosomes. Reduced SCAP hinders SREBP1c/2 maturation and thus suppresses de novo lipogenesis and inflammation. Higher hepatic A1R expression is observed in patients with MAFL/MASH and high-fat diet (HFD)-fed mice, which is supposed to be a physiologically adaptive response because A1R agonists attenuate MAFL/MASH in an A1R-dependent manner. These results highlight that hepatic A1R is a potential target for MAFL/MASH therapy.
Keywords: adenosine A1 receptor, MASH, SCAP-SREBPs pathway, de novo lipogenesis, inflammation
Graphical abstract
Highlights
-
•
Hepatic A1R activation ameliorates MAFL and MASH in mice
-
•
Activated hepatic A1R reduces de novo lipogenesis via inhibiting SREBP maturation
-
•
Activated hepatic A1R promotes the binding of SQSTM1 to SCAP, leading to SCAP degradation
-
•
Hepatic A1R is a potential therapeutic target for MASH
Zhu et al. show that hepatic A1R activation reduces the direct binding of SCAP with PKAc but increases the formation of an SCAP-SQSTM1 complex, facilitating SCAP degradation in the lysosome, and suppressing the maturation of SREBPs. Hepatic A1R is a promising target for MAFLD therapy.
Introduction
Metabolic (dysfunction)-associated fatty liver disease (MAFLD) was formerly known as nonalcoholic fatty liver disease (NAFLD)1,2,3,4 and is the predominant chronic liver disease with a broad spectrum consisting of steatosis (fatty liver [MAFL]), steatohepatitis (metabolic-associate steatohepatitis [MASH]), fibrosis, cirrhosis, and hepatocellular carcinoma (HCC).5,6 Although MAFL is the early and reversible stage of MAFLD, some MAFL patients will progress to MASH and even irreversible advanced stages.7,8 To date, there are no approved drugs for MASH therapy in clinic. Therefore, the identification of novel therapeutic targets of MASH is urgently needed.
Adenosine receptor (AR) belongs to the G-protein-coupled receptor (GPCR) superfamily, consisting of A1R, A2AR, A2BR, and A3R subtypes, which are widely distributed in central nervous system and major peripheral organs with different abundance.9 ARs are activated upon extracellular adenosine that derives from ATP/ADP hydrolysis catalyzed by CD39 and CD73. The CD39/CD73-adenosine-ARs axis is implicated in immune and inflammation-related diseases where the increased inflammation and cellular hypoxia stimulate the generation of adenosine, resulting in diversified outcomes depending on the activated subtype of ARs and cellular distribution.10,11 Recent study reveals that A2AR is a novel tumor suppressor of MASH-associated HCC12 and antagonism of A2AR enhances immunotherapy efficacy.13,14 A2BR has been implicated in regulatory T cell functions and is a contributing factor in adenosine-mediated protection against inflammatory disease and organ injury.15,16 A3R is involved in the inhibition of neutrophil degranulation in tissue injury,17 and activation of A3R reverses neuropathic pain via T cell-mediated production of interleukin (IL)-10.18 A1R is highly expressed in central nervous system, heart atria, kidney, pancreas, adipose tissue, etc.9,19 Therefore, A1R is a potential therapeutic target for diseases such as Parkinsonism, Alzheimer’s disease, diabetes, and obesity,20,21,22,23 and several specific agonists or antagonists of A1R are under phase II/III trials.24,25,26
In contrast to the well-established distribution and role of A1R in central nervous system and other peripheral organs,9 hepatic A1R expression is supposed to be weak in liver under physiological condition.27 Nevertheless, studies suggested that dysregulated hepatic A1R signaling was associated with some liver diseases, such as liver fibrosis, hepatic ischemia-reperfusion injury, and intrahepatic cholestasis.28,29,30 Notably, it was reported that global A1R knockout mice were resistant to alcohol-induced liver disease, and the underlying mechanism was supposed to be associated with the suppression of sterol-regulatory element binding protein 1c (SREBP1c)-mediated fatty acid de novo lipogenesis (DNL).31 However, this effect was not consistently observed in another study, which was supposed to be due to the low expression of A1R in hepatocytes.27 Paradoxically, the presence of the four subtypes of ARs in hepatocytes in either human beings or rodents has also been demonstrated, with the A1R sequence being the most conserved.9,32 Thus, the exact role of hepatic A1R in MAFLD development and progression remains uncertain.
In this study, we first investigated the role of hepatic A1R by generating liver-specific A1R knockout (A1RLiver−/−) and overexpressed (A1RLiver OE) mice in high-fat diet (HFD) and choline-deficient (CD)-HFD-induced MAFL and MASH models. Liver-specific deletion of A1R aggregated diet-induced MAFL or MASH, which was attenuated in A1RLiver OE mice. Mechanistically, our results revealed that activated A1R suppressed the maturation of SREBPs (SREBP1c/2), leading to the inhibition of DNL via protein kinase A catalytic subunit (PKAc)/SREBP cleavage-activating protein (SCAP) pathway. Activated A1R accelerated the degradation of SCAP protein in lysosome and reduced its anchoring at Golgi apparatus by forming SCAP-sequestosome 1 (SQSTM1) complex. Ultimately, we demonstrated that activation of hepatic A1R by pharmacological agonist 2-chloro-N6-cyclopentyladenosine (CCPA) or screened natural compound timosaponin AⅢ (TA3) with potent activating ability on A1R ameliorated diet-induced MAFL and MASH in mice in an A1R-dependent manner. Our current findings revealed a previously undetermined role of hepatic A1R as a promising therapeutic target for MAFL and MASH.
Results
Liver-specific A1R knockout exaggerates MAFL and MASH in mice
To investigate the exact role of hepatic A1R in MAFLD formation, we first generated liver-specific A1R knockout (LKO) mice with no impact on the expression of other AR subtypes (Figures S1A–S1D). In the context of the normal chow diet (NCD), liver-specific A1R deletion did not affect body weight gain, liver function, or histology (Figures S1E–S1H). Nevertheless, NCD-fed LKO mice showed increased hepatocyte triglyceride (TG), impaired glucose tolerance, and elevated protein expression of fatty acid synthase (FASN) and acetyl-coenzyme A (CoA) carboxylase (ACC) (Figures S1I–S1K), implying that liver-specific A1R ablation might activate hepatic DNL. Then, we observed that liver-specific ablation of A1R aggravated hepatic steatosis, increased liver index and serum alanine aminotransferase (ALT) level, induced dyslipidemia, and impaired glucose homeostasis in HFD-induced MAFL mice (Figures 1A–1E and S1L–S1S). Meanwhile, LKO mice showed increased liver pro-inflammatory cytokine (IL-β, tumor necrosis factor [TNF]-α) levels and enhanced protein expression of hepatic FASN and ACC but not CPT1α or CD36 (Figures S1T and 1F).
Figure 1.

Liver-specific A1R knockout exaggerates MAFL and MASH in mice
(A–F) Control mice (A1RFlox/Flox without Cre, Flox) and liver-specific A1R knockout mice (A1RLiver−/−, LKO) were fed an HFD (60 kcal%) for 16 weeks (n = 6). (A) Diagram of experimental design. (B) Representative image of liver and liver H&E staining. (C) Hepatic steatosis scores and liver index. (D) Quantification of hepatic triglycerides (TGs). (E) The level of serum alanine aminotransferase (ALT). (F) Relative protein expression of CPT1α, CD36, FASN, and ACC.
(G–K) Flox mice and LKO mice were fed a calorie-restricted HFD (CD-HFD) for 9 weeks (n = 5∼7). (G) Diagram of experimental design. (H) H&E, F4/80, and Sirius red staining of liver sections, with histological evaluation. (I) Liver injury indicators, including serum ALT and AST. (J) mRNA expression in liver. Results are normalized for 18S. (K) Quantification of hepatic triglycerides (TGs).
(L) Relative protein expression of α-SMA, pP65, P65, and IκB. GAPDH functioned as a reference protein. Results are representative of one biological replicate. Data are depicted as mean ± SEM. Student’s unpaired t test; ∗p < 0.05, ∗∗p < 0.01.
To test whether liver-specific ablation of A1R would aggravate MASH formation, we fed mice with CD-HFD, a commonly used diet for inducing a MASH model in relatively short time33 (Figure 1G). Histopathological examination revealed elevated hepatic steatosis, fibrosis, macrophage marker F4/80 and CD11b, and NAFLD activity score (NAS) in LKO mice (Figures 1H and S1U). These changes were accompanied with increased serum ALT and aspartate transaminase (AST) activities and serum inflammatory cytokines (IL-1β, IL-6, and TNF-α), as well as increased gene expression of fibrosis markers, chemotactic factors, and inflammatory cytokines, suggesting enhanced liver damage and inflammation (Figures 1I, 1J, and S1V). The liver TG of LKO mice on CD-HFD was higher than Flox mice (Figure 1K). In addition, elevated protein expression of myofibroblast markers alpha smooth muscle actin (α-SMA) and inflammation pathway phosphorylated P6534 but reduced IκB35 level were also observed in LKO mice (Figure 1L). We also measured the expression of TGF-β and phosphorylated Smad2 (p-Smad2)/Smad2 that are critically involved in fibrosis development.36 Our results showed that hepatic A1R knockout enhanced the expression of TGFβ and the phosphorylation of Smad2 (Figure S1W). Quantification of 13C-incorporated fatty acids was conducted with liver tissue of mice in the context of short-term amylin liver NASH (AMLN) diet feeding. The results showed DNL was obviously higher in LKO than Flox mice (Figure S1X), which consolidated the observation of increased DNL in hepatic A1R deletion mice. These results indicated that absence of hepatic A1R exaggerated the diet-induced MAFL and MASH in mice.
Liver-specific A1R overexpression protects mice from MAFL and MASH
The physiological role of hepatic A1R was further validated by adeno-associated virus (AAV)-mediated liver-specific A1R overexpression in mice (A1RLiver OE, Figure S2A). In contrast to the observations in LKO mice, A1RLiver OE mice showed dramatic improvements in HFD-induced metabolic disorders including reduced hepatic steatosis and dyslipidemia, improved liver function, and glucose homeostasis (Figures 2A–2E and S2A–S2H). In addition, A1RLiver OE mice also showed decreased hepatic inflammatory cytokines, including IL-1β, IL-6, and TNF-α (Figure S2I). Moreover, overexpression of hepatic A1R significantly suppressed CD36, FASN, and ACC protein expression (Figure 2F). In line with the protection on MAFL in wild-type (WT) C57BL/6J mice, the protective effect of hepatic A1R on MAFL was also validated in LKO mice, in which the hepatic A1R expression was rescued by AAVs-mediated A1R overexpression (LKORes, Figure S2J). Rescue of hepatic A1R protected mice from HFD-induced MAFL, including improved hepatic steatosis, and reduced hepatic TG accumulation and liver injury (Figures S2K–S2N). Beside the preventive effect, overexpression of hepatic A1R also exerted a therapeutic effect on the established MAFL in HFD-fed mice (Figures S2O–S2R). Collectively, these results indicated that hepatic A1R activation attenuated diet-induced MAFL in mice.
Figure 2.

Liver-specific A1R overexpression protects mice from MAFL and MASH in mice
(A–F) C57BL/6J mice were inoculated with vehicle-AAV (vehicle) or A1R-overexpression-AAV (A1RLiver OE) intravenously, and then were fed an HFD for 12 weeks (n = 5∼9). (A) Diagram of experimental design. (B) Representative image of liver and liver H&E staining. (C) Hepatic steatosis scores. (D) Quantification of hepatic TG. (E) Liver injury indicators, including serum ALT and AST. (F) Relative protein expression of CPT1α, CD36, FASN, and ACC.
(G–K) Vehicle mice and A1RLiver OE mice were fed a CD-HFD for 9 weeks (n = 10). (G) Diagram of experimental design. (H) H&E (n = 10), F4/80 (n = 6), and Sirius red staining (n = 10) of liver sections, with histological evaluation. (I) Liver injury indicators, including serum ALT and AST. (J) mRNA expression in liver. (K) Relative protein expression of α-SMA, pP65, P65, and IκB. Results are representative of one biological replicate. Data are depicted as mean ± SEM. Student’s unpaired t test; ∗p < 0.05, ∗∗p < 0.01.
We then tested whether overexpression of hepatic A1R could also ameliorate MASH formation by feeding CD-HFD in mice. Our results showed that A1RLiver OE mice had reduced lipid accumulation, fibrosis, and macrophage infiltration in liver, as well as reduced serum ALT and AST levels (Figures 2G–2I, S2S, and S2T). Furthermore, hepatic A1R activation suppressed the mRNA expression of fibrosis markers, chemotactic markers, and inflammatory cytokines, as well as the protein expression of α-SMA and pP65/P65, but it increased IκB expression (Figures 2J and 2K). In addition, the anti-MASH effect by activating hepatic A1R was further confirmed in a 28-week AMLN diet-induced MASH mouse model,37,38 as indicated by reduced hepatic steatosis, NAS, Sirius red staining, and improved liver function (Figures S2U–S2Z). Altogether, these results demonstrated activation of hepatic A1R protected mice from diet-induced MAFL and MASH.
A1R controls the maturation of SREBPs via PKAc
SREBPs (mainly SREBP1c and SREBP2) are critical nuclear transcriptional factors that regulating the DNL, cholesterol ,and inflammation signaling pathways in hepatocytes that are involved in MAFLD formation.39 To investigate whether the hepatic A1R regulates the MAFL and MASH development and progression through modulation on SREBPs, we measured the hepatic expression of precursor SREBPs (full-length SREBPs [flSREBPs]) in whole-cell and mature SREBPs (cleaved form of flSREBPs) in the nucleus (nSREBPs) in both LKO and A1RLiver OE mice under HFD or CD-HFD feeding. LKO mice showed reduced flSREBP1c and flSREBP2 but increased nSREBP1c and nSREBP2 in the nucleus compared to Flox mice, while A1RLiver OE mice showed the opposite trend in diet-induced MAFL or MASH mice (Figure 3A), implying A1R negatively controlled the maturation of SREBPs in hepatocytes.
Figure 3.

A1R controls the maturation of SREBPs via PKAc
(A) Relative hepatic protein expression of flSREBP1c and nSREBP1c from LKO mice and A1RLiver OE mice on HFD and CD-HFD.
(B) Hepatic PKAc protein expression in LKO and A1RLiver OE mice.
(C) PKAc protein expression in AML-12 cells treated with CPA (A1R activator, 1 μM) and DPCPX (A1R inhibitor, 1 μM) for 48 h. GAPDH or tubulin was used as total protein control, and lamin B1 was used as nuclear protein control.
(D) AML-12 cells were treated with CPA (1 μM, 48 h), DbcAMP (PKA activator, 200 μM, 12 h), or co-treated with CPA and DbcAMP, respectively. Fluorescent staining of SREBP1c or SREBP2 (red) in each group was performed. The nuclei were stained by Hoechst (blue).
(E) Overview of A1R intracellular signaling pathways. Activated A1R decreases adenylate cyclase (AC) activity to suppress the activity of cAMP/PKAc in cytoplasm, resulting in modulation of the intracellular signaling molecules. However, the exact modulation on SREBPs by PKAc remains unclear. Image created using BioRender. Results are representative of one biological replicate. Data are depicted as mean ± SEM. Student’s unpaired t test; ∗p < 0.05, ∗∗p < 0.01.
Activated A1R modulates the intracellular signaling molecules through decreasing adenylate cyclase (AC) activity in cytomembrane,9 resulting in suppression of activity of cAMP/PKAc in cytoplasm controlling the downstream targets, including SREBPs. The negative control of PKAc by hepatic A1R was confirmed in both LKO and A1RLiver OE mice as well as in AML-12 cells treated by 8-cyclopentyl-1,3-dipropylxanthine (DPCPX) and CPA, specific antagonist and agonist of A1R, respectively9 (Figures 3B and 3C). However, the exact modulation on SREBPs by PKAc remains unclear because both positive and negative regulation of SREBPs by cAMP/PKAc were previously reported.40,41 Thus, the nuclear translocation of mature SREBP1c and SREBP2 was measured in AML-12 cells under the treatment of either DPCPX or CPA alone, or in combination with H89, a selective and potent inhibitor of PKA,42 or dibutyryl-cAMP sodium salt (DbcAMP), a cell-permeable PKA activator by mimicking the action of endogenous cAMP.43 The results showed the translocation of SREBP1c and SREBP2 to nucleus was stimulated by DPCPX but suppressed by CPA, whereas the combined treatment of H89 or DbcAMP reversed the SREBP nuclear translocation under DPCPX or CPA, as shown by consistent observations with either western blot or immunofluorescence. These results suggested the controlling of SREBP endonuclear content by A1R was dependent on PKAc (Figures 3C–3E, S3A, and S3B).
To further evaluate the contribution of increased PKAc to MAFL formation in LKO mice, H89 was administered to either Flox or LKO mice during the 8 weeks of HFD feeding (Figure S3C). H89 treatment significantly improved hepatic steatosis and reduced ALT activity in both Flox and LKO mice (Figures S3D–S3I). In addition, the nuclear level of mature SREBP1c (nSREBP1c) was dramatically reduced by H89 in both Flox and LKO groups (Figure S3J). In summary, hepatic A1R inhibited the nuclear translocation of SREBPs by decreasing PKAc, resulting in the attenuation of MAFLD.
A1R reduces the cellular content of SCAP and its anchoring at Golgi membrane
The maturation of SREBPs is tightly controlled by SCAP and insulin-induced gene 1 protein (INSIG1) in endoplasmic reticulum (ER) escorting SREBPs from ER to Golgi apparatus where SREBPs is sequentially cleaved by two proteases: S1P and S2P44,45 (Figure 4A). To explore how A1R controlled the maturation of SREBPs, we tested the expression of these proteins in liver tissue of either LKO or A1RLiver OE mice. We found only SCAP and S2P protein were significantly increased in LKO mice and decreased in A1RLiver OE mice compared to their counterparts (Figures 4B; S4A).
Figure 4.

A1R reduces the cellular content of SCAP and its anchoring at Golgi membrane
(A) Maturation process of SREBPs. SREBPs are exported from the ER to the Golgi apparatus by SCAP, and then transcriptional activation domain of SREBPs is released from the membrane by S1P and S2P. The released domain migrates into the nucleus and activates their transcription. Image created using BioRender.
(B) Protein expression of SCAP in the liver of A1RLiver OE or LKO mice.
(C) SCAP colocalizes with the Golgi apparatus. AML-12 cells were treated with DPCPX (1 μM) or CPA (1 μM) for 48 h. Fluorescent staining of nuclei (Hoechst, blue), Golgi (green), and SCAP (red) in each group was performed.
(D) Protein expression of SCAP in the AML-12 cells treated or co-treated with CPA and DbcAMP (200 μM, 12 h), or DPCPX and H89 (20 μM, 4 h). Results are representative of one biological replicate. Cell experiments performed n = 3. Data are depicted as mean ± SEM. Student’s unpaired t test; ∗p < 0.05, ∗∗p < 0.01.
Because the transport of SCAP from ER to Golgi apparatus and anchoring is the critical step for facilitating the maturation of SREBPs,46 we then further explored the anchoring status of SCAP at Golgi apparatus by using 3D modeling of confocal image. As expected, inhibition of A1R by DPCPX not only increased the cellular content of SCAP protein but also increased its anchoring at Golgi apparatus (yellow color in merged image and co-localization in 3D image), while CPA showed the opposite effect (Figure 4C), implying the negative regulation of SCAP content and function by A1R. Given the established regulation of PKAc-SREBPs by the A1R and SCAP-SREBPs axis,46 we further asked whether the regulation of SCAP by A1R was dependent on PKAc. The observations in vitro showed that the effects of CPA and DPCPX on SCAP protein content were abolished by co-treatment with H89 or DbcAMP, respectively (Figure 4D), suggesting A1R might control the cellular content of SCAP protein through PKAc. Meanwhile, the binding between PKAc and SCAP was facilitated by DPCPX but suppressed by CPA (Figure S4B), implying A1R inhibited the direct binding between PKAc and SCAP. Altogether, these results indicated that A1R reduced intracellular SCAP content as well as its anchoring at Golgi membrane through PKAc.
A1R activation accelerates SCAP protein degradation through SQSTM1 in lysosome
The cellular protein content is determined by the balance between synthesis and degradation. To test whether A1R regulated the degradation of SCAP protein, AML-12 cells were treated with either CPA or DPCPX in the presence of cycloheximide (CHX), an inhibitor of protein biosynthesis. The results showed that CPA dramatically accelerated the degradation of SCAP, which was maintained stable by DPCPX in the presence of CHX treatment (Figure 5A), implying A1R may control the cellular content of SCAP protein by accelerating its degradation. In line with the impact of PKAc on SCAP content, we found that the degradation of SCAP protein was accelerated by H89 but maintained by DbcAMP (Figure S4C), suggesting PKAc also controlled the degradation of SCAP. There are two major pathways regulating protein catabolism: the ubiquitin-proteasome system and the autophagy-lysosomal system.47 To determine how A1R controlled the SCAP degradation, bafilomycin A1 (Baf.A1), blocking autophagosome-lysosome fusion and inhibiting protein degradation in lysosomes, or MG132, a potent proteasome and calpain inhibitor,48 was co-treated with CPA in AML-12 cells, respectively. Results showed that coculture of Baf.A1, but not MG132, with CPA prevented the accelerated SCAP degradation induced by CPA (Figure 5B), suggesting that CPA may mainly accelerate SCAP degradation in lysosome. Further evidence was consistently observed by immunofluorescence, in which CPA increased the co-localization of SCAP with lysosome (yellow color in merged image), but not in DPCPX-treated cells (Figure 5C). These results indicated activated A1R promoted the degradation of SCAP in lysosome.
Figure 5.

A1R activation accelerates SCAP protein degradation through SQSTM1 in lysosome
(A) Relative protein expression of SCAP in the AML-12 cells treated with CPA or DPCPX in the presence of cycloheximide (CHX, 50 μM) for 0, 2, 4, and 8 h.
(B) Relative protein expression of SCAP in the AML-12 cells treated or co-treated with CPA and bafilomycin A1 (Baf.A1, lysosomal inhibitor, 1 μM, 8 h) or MG132 (proteasome inhibitor, 10 μM, 8 h).
(C) SCAP colocalizes with the lysosome. AML-12 cells were treated with DPCPX or CPA for 48 h. Fluorescent staining of nuclei (Hoechst, blue), lysosome (red), and SCAP (green) was performed.
(D) Co-immunoprecipitation (coIP) of PKAc or SQSTM1 with SCAP in the AML-12 cells treated with DPCPX or CPA for 12 h.
(E and F) Representative results and quantification of the proximity ligation assay (PLA) analysis of (E) PKAc or (F) SQSTM1 with SCAP in the AML-12 cells treated with CPA or DPCPX. Results are representative of one biological replicate. Cell experiments performed n = 3. Data are depicted as mean ± SEM. Student’s unpaired t test; ∗p < 0.05, ∗∗p < 0.01.
We next investigated how A1R controlled SCAP degradation. Recently, Zheng et al. revealed that SCAP was captured by SQSTM1 after it exited from ER and underwent a novel SQSTM1-mediated autophagy-independent lysosomal degradation.48 Given the role of PKAc in maintaining cellular content of SCAP (Figures 4D and S4C) and the physical binding with SCAP directly (Figure S4B), we hypothesized that the increased degradation of SCAP by activated A1R was due to the competitive binding with SCAP between PKAc and SQSTM1. The co-immunoprecipitation (Co-IP) test showed that CPA stimulated the formation of SCAP-SQSTM1 and reduced SCAP-PKAc complex, whereas the opposite effect was observed in DPCPX-treated cells (Figure 5D). Moreover, in agreement with the coIP observation, proximity ligation assay (PLA) revealed that CPA reduced the direct interaction between PKAc and SCAP but increased SQSTM1 and SCAP interaction, which was oppositely regulated by DPCPX (Figures 5E and 5F). Therefore, the results indicated that activated A1R promoted the competitive binding of SCAP with SQSTM1 rather than PKAc, leading to SCAP degradation in lysosome.
Pharmacological hepatic A1R activation attenuates MAFL and MASH in mice
Since the exact status of hepatic A1R in MAFLD remains unclear, we examined the expression of A1R protein in liver specimens from clinical patients. A total of 30 liver samples from a group of patients who underwent hepatectomy were retrospectively studied with immunohistology, in which 22 of them were diagnosed with the presence of hepatic steatosis, and eight of them were used as control without hepatic steatosis. The patient information is described in the supplementary methods (Table S1). The results showed universal positive expression of A1R protein in hepatocytes in patients with hepatic steatosis but not in control patients (Figures 6A–6C). To further characterize the status of hepatic A1R in MASH patients, the expression of hepatic A1R was evaluated by using immunofluorescence in specimens from nine patients with MASH and four controls who underwent liver biopsy (Table S2). In agreement with the observation in patients with hepatic steatosis retrospectively, hepatic A1R expression was obviously triggered in patients diagnosed with MASH but not in controls (Figures 6D–6F). Consistently, public clinical data also revealed the increased mRNA expression of A1R in liver of patients with MAFLD49,50 (Figure S4D, GSE89632, GSE135251). In agreement with these clinical findings, elevated hepatic A1R protein expression was also present in HFD-fed mice time dependently or CD-HFD-fed mice compared to their chow diet controls (Figures S4E and S4F). As a result, we hypothesized that the elevated expression of hepatic A1R in MAFLD patients or diet-induced MAFLD mice might be an adaptively protective response against MAFLD, but it is not sufficient.
Figure 6.

Hepatic A1R negatively correlates with the advance of MAFLD
(A) Representative images showing H&E staining and A1R immunohistochemical (IHC) staining of liver tissue from individuals with or without hepatic steatosis (n = 8∼22).
(B) Hepatic steatosis score.
(C) A1R IHC-positive staining intensity.
(D) Representative images showing H&E staining and A1R immunofluorescence staining of livers from healthy control or MASH individuals (n = 9).
(E) NAFLD activity score (NAS).
(F) A1R fluorescence-positive staining intensity. Results are representative of one biological replicate. Data are depicted as mean ± SEM. Student’s unpaired t test; ∗p < 0.05, ∗∗p < 0.01.
To determine whether activation of A1R signaling could protect against MAFLD, specific A1R agonist CCPA (intraperitoneal [i.p.], 1 mg/kg once per day) was administered in both HFD-induced MAFL and CD-HFD-induced MASH mice (Figures S5A and S5L). The results showed CCPA treatment substantially improved the diet-induced MAFL and MASH in mice, including attenuation in hepatic steatosis and fibrosis in histology, and improvement in dyslipidemia, glucose intolerance (fasting blood glucose, insulin, and IGTT), liver function (serum ALT, AST, or alkaline phosphatase [ALP]), and inflammation (levels of serum IL-1β, IL-6 and TNF-α, and gene expression of chemokines), without affecting energy intake (Figures S5B–S5J and S5M–S5O). Moreover, CCPA treatment also suppressed the expression of FASN, ACC, PKAc, and SCAP protein; reduced maturation of SREBP1c/2 in MAFL mice; inhibited α-SMA; and enhanced IκB proteins in MASH mice (Figures S5K and S5Q). Collectively, these results demonstrated that pharmacological activation of hepatic A1R was effective for MAFL and MASH therapy. To further test whether adenosine administration could also attenuate MAFLD in mice, we treated HFD-induced MAFLD mice with either adenosine (10 mg/kg, i.p.) or CCPA (1 mg/kg, i.p.) in parallel. As shown in Figures S6A–S6F, we found that even high doses of adenosine did not attenuate the extent of MAFLD, while specific A1R agonist, CCPA, was effective in reducing lipid accumulation in hepatocytes and serum ALT/AST levels. These results suggested that the anti-MAFLD effect was due to specific activation of A1R but not in non-specific ligand of ARs.
Hepatic A1R is an applicable target for screening MAFLD therapy compounds
Based on our current observations, we further asked whether hepatic A1R was applicable for screening potential MAFLD therapy compounds. For this purpose, we then screened natural compounds with activating capacity on A1R among 3,600+ natural compounds library by using computer-aid molecular docking and followed by in vitro test on inhibition of cAMP production and TG accumulation. A total of 200 compounds stood out based on their binding free energy with A1R, then 12 of them were selected with stronger activating capacity on A1R than CCPA in A1R-overexpressed HEK293T cells by measuring production of cAMP. At last, a natural component, timosaponin AIII (TA3), was identified based on its potent activating A1R capacity, inhibition of intracellular TG production in primary hepatocytes from WT mice, and AML-12 and HepG2 cells dose dependently but not in primary hepatocytes from LKO mice (Figure 7A, S6G-J). TA3 also dose-dependently inhibited the expression of PKAc, FASN, ACC, and nuclear localization of nSREBP1c/2 in AML-12 cells, which were consistent with those of CCPA treatment or A1RLiver OE mice (Figures S6K, S5K, 3A, and 3B). Additionally, ligand activation assay indicated that TA3 was strongest in reducing intracellular cAMP content in AML-12, HEK293T or A1R-overexpressed HEK293T cells compared to CCPA or adenosine, endogenous ligand for ARs (Figure S6L). Moreover, we observed that adenosine had no effect on cAMP content, intracellular TG accumulation, or protein expression of ACC and FASN in AML-12 or HEK293T cells, while reduced cAMP content was only observed in A1R-overexpressed HEK293T cells (Figures S6L–S6N), implying selectivity on A1R was necessary for suppressing TG accumulation.
Figure 7.

Timosaponin AIII, an identified potent A1R activator, inhibits diet-induced MAFL and MASH in mice
(A) Schematic of the drug screen.
(B–G) C57BL/6J mice were fed with HFD for 16 weeks and injected intraperitoneally with 0.9% NaCl solution (MAFL) or 5 or 10 mg/kg timosaponin AIII (TA3) daily from ninth week (n = 7–∼10). (B) Diagram of experimental design. (C) Representative image of liver H&E staining. (D) Liver weight. (E) Hepatic steatosis scores. (F) Quantification of hepatic TG. (G) Relative protein expression of PKAc, SCAP, SREBPs (flSREBP1c, flSREBP2, nSREBP1c, nSREBP2), and SREBP1c regulated proteins (FASN, ACC) (n = 6).
(H–N) C57BL/6J mice were fed a normal chow diet (NCD, Con) or CD-HFD for 9 weeks; during the process, CD-HFD mice were divided into three subgroups and injected intraperitoneally with 0.9% NaCl solution (CD-HFD) or 5 or 10 mg/kg TA3 daily from fourth week. (H) Diagram of experimental design.
(I−N) C57BL/6J mice were fed with CD-HFD for 9 weeks and injected intraperitoneally with 0.9% NaCl solution (MASH) or 5 or 10 mg/kg timosaponin AIII (TA3) daily from fourth week (n = 8). (I) H&E and Sirius red staining of liver sections. (J and K) Histological evaluation. (L) Serum ALT, AST, and ALP. (M) Serum inflammatory cytokines. (N) Relative protein expression of α-SMA and IκB. GAPDH was used as total protein control; lamin B1 was used as nuclear protein control. Results are representative of one biological replicate. Data are depicted as mean ± SEM. One-way ANOVA analysis; ∗p < 0.05, ∗∗p < 0.01.
To evaluate the potential of TA3 as a novel MAFLD therapeutic candidate, the anti-MAFLD effect of TA3 was evaluated in HFD-induced MAFL and CD-HFD-induced MASH mice (5 or 10 mg/kg, i.p., once per day) (Figures 7B and 7H). As expected, TA3 treatment obviously attenuated HFD-induced MAFL, as shown by improved hepatic steatosis in histology, liver TG, and serum total cholesterol (TC), ALT, and AST (Figures 7C–7F and S7A–S7D). In line with the effects of CCPA (Figure S5K), TA3 suppressed the expression of PKAc, SCAP, ACC, and FASN as well as the maturation of SREBP1c/2, resulting in inhibition of DNL (Figure 7G). In parallel, TA3 treatment also effectively ameliorated CD-HFD-induced MASH, as shown by reduced hepatic steatosis, liver injury, and fibrosis based on histology; reduced serum inflammation cytokines; as well as related gene and protein expressions (Figures 7I–7N, S7E, and S7F). Taken together, these results indicated that the screened TA3 by targeting A1R was effective for MAFL and MASH therapy, highlighting the potential of hepatic A1R as MAFLD therapy target.
To further determine whether hepatic A1R was the key therapeutic target for MAFLD, the anti-MASH effect of either CCPA (1 mg/kg, i.p., once per day) or TA3 (10 mg/kg, i.p., once per day) was tested in LKO mice that were fed with CD-HFD and treated with an identical dosage to that in WT mice (Figure S7G). To our expectation, both CCPA and TA3 treatment did not improve the histological change or the scores of hepatic steatosis and NAS, and they had minor impacts on serum parameters in LKO mice (Figures S7G–S7J), in contrast to their significant effects in WT mice (Figures 7 and S5). Consistently, neither CCPA nor TA3 reversed the expression of serum inflammation cytokines or hepatic α-SMA and IκB in CD-HFD-fed LKO mice (Figures S7K and S7L).
Altogether, these results revealed a novel role of hepatic A1R as an anti-MAFLD therapeutic target, especially for MASH therapy.
Discussion
We report here that hepatic A1R activation protected mice from diet-induced MAFL and MASH. Activated hepatic A1R reduced the cellular content of SCAP protein and its anchoring at Golgi apparatus due to increased lysosomal degradation by forming SCAP-SQSTM1 complex rather than SCAP-PKAc. The reduced cellular SCAP resulted in less formation of mature SREBP1c/2 and nuclear SREBP1c/2 translocation, leading to reduced DNL and inflammation. The anti-MAFLD role of hepatic A1R was further confirmed by specific A1R agonist CCPA and screened natural compound TA3 with potent activating capacity on A1R activity, which implied that the observed increase of A1R expression in liver of MAFL/MASH patients or MAFLD mice might be an adaptive response against MAFLD development and progression.
The four subtypes of ARs are widely distributed with diversified functions and involved in various pathophysiological processes upon activation by extracellular adenosine.9 Extracellular adenosine can be released from intracellular stores, and is predominantly derived from the metabolism of precursor nucleotides ATP/ADP by CD39 and CD73 catalyzation, especially in pathological conditions such as inflammation and hypoxia.11,51 The adenosine signaling could be terminated by adenosine uptake from the extracellular compartment into cytoplasm through equilibrative nucleoside transporter 1 (ENT1) and equilibrative nucleoside transporter 2 (ENT2) where adenosine is converted to AMP through adenosine kinase or to inosine through the adenosine deaminase.52,53,54 A1R is mainly expressed in central nervous system and some peripheral organs where extracellular adenosine performs different functions in a cell-type-dependent way.9,55,56 Blocking A1R-mediated signaling could abolish heart-rate-slowing effects of intravascular adenosine. Since the four subtypes of ARs are present in hepatocytes, or partly expressed in stellate cells, Kupffer cells, and sinusoidal endothelial cells,9,57 previous study suggested that the number and affinity of A1R and A3R did not change in cirrhotic and fatty livers but the numbers of A2AR and A2BR increase in cirrhotic and fatty livers.31 In agreement with the observation that disrupted A2AR exacerbated MAFLD development in A2AR-deficient mice,58 our findings showed a higher ratio of hepatocytes with positive A1R protein expression in liver of patients with MAFL/MASH or MAFLD mice than their healthy controls. In addition, we observed the expression of these four ARs in mouse liver, in which A1R was the highest among them in both male and female (Figure S4G). We therefore questioned whether the increased adenosine and hepatic A1R signaling contributed to the development of MAFLD or was an adaptive response for combating MAFLD development instead. To answer this question, genetic deletion of hepatic A1R in LKO mice served as an ideal model, rather than global A1R knockout mice, which makes it hard to exclude the impacts derived from A1R deletion in other organs. To our surprise, LKO mice were more prone to diet-induced MAFL and MASH than Flox mice, which was also reversely validated in A1RLiver OE mice, and, moreover, rescue of hepatic A1R attenuated hepatic steatosis in LKO mice. In contrast to the report by Peng et al. that global knockout of either A1R or A2BR protected mice against ethanol-induced fatty liver disease,31 our results demonstrated that hepatic A1R activation would attenuate diet-induced MAFL/MASH based on liver-specific A1R knockout or overexpression mice. Meanwhile, the other three subtypes of ARs in liver of mice were not altered by the deletion or overexpression of hepatic A1R. Therefore, our current findings revealed a previously unreported function of hepatic A1R against MAFLD development, implying that increased expression of A1R in liver in MAFLD patients or mice might be an adaptive response instead of causative for MAFLD development.
Sterol-regulatory element binding proteins (mainly SREBP1c and SREBP2) are essential transcription factors activating the expression of genes in DNL and cholesterol synthesis59,60 and promoting inflammation, and they are crucial for MAFL and MASH formation.61 In agreement with the phenotypes in LKO or A1RLiver OE mice, the maturation and nuclear translocation process of SREBPs was stimulated by A1R deletion and inhibited by A1R overexpression, as well as consistent observation by A1R antagonist DPCPX or agonist CPA in vitro. The premature SREBPs are synthesized as ER membrane-bound proteins that are activated upon sterol through the escorting from ER to Golgi apparatus by SCAP.46,61,62 Consistent with the changes of nSREBPs upon A1R activation or inhibition either in vivo or in vitro, cellular content of SCAP protein was increased by A1R deletion and decreased by A1R activation, as well as its anchoring at Golgi apparatus. Previous studies showed liver-specific deletion of SCAP caused low SREBPs protein and mRNA expression, resulting in marked reduction of both the precursor and nSREBPs.63,64,65 In contrast, our results suggested that activated A1R blocked the maturation and translocation to nucleus of SREBPs by reducing cellular SCAP content, instead of the suppression of SREBP generation because of the observed accumulation of flSREBPs in cytoplasm. This discrepancy between ours and a previous report might be due to the altered cellular SCAP content controlled by A1R in our model, which was different from the direct impact on SREBPs by liver-specific Scap knockout.
There are several other signal pathways that are tightly controlled by A1R, including cAMP/PKA, PLCβ-IP3-PKC, and MAPK family such as ERK, p38, and JNK.66,67 Nevertheless, the role of cAMP/PKA in A1R-controlled TG accumulation was also validated by intervention of A1R agonist CCPA in the presence or absence of PKA agonist or inhibitors of PKC, ERK, p38, and JNK in vitro. These results showed that only PKA agonist abolished the inhibitory effect of cellular TG accumulation upon A1R activation, while inhibitors of PKC, ERK, p38, and JNK did not (data not shown). So, it was concluded that cAMP/PKA might mediate the subsequent intracellular signal upon A1R activation. cAMP/PKA is known to coordinate with the SCAP-SREBPs pathway through controlling SCAP phosphorylation.68 In our study, although we observed that specific PKA inhibitor, H89, attenuated hepatic steatosis and nSREBP1c content in liver of LKO mice, our phosphoproteomic analyses of PKA-dependent phosphorylated targets excluded the possibility of SCAP phosphorylation in response to A1R suppression (Figure S4H) but facilitated the co-localization of SCAP protein in lysosome instead. PKA is a heterotetrametric holoenzyme in which two catalytic subunits from two major families, Cα/β, combine with homodimers formed by any of four R-subunits (RIα, RIβ, RIIα, RIIβ) to form a number of R2:C2 holoenzymes.69,70,71,72 Canonically, PKA is activated when cAMP binds to the regulatory subunits, triggering release of the catalytic subunit PKAc.73 We observed that the catalytic subunit PKAc was reduced upon A1R activation or increased by A1R inhibition, which was consistently accompanied by SCAP-SREBP alteration. Since we did not observe the evidence of SCAP phosphorylation by PKA upon A1R activity inhibition, we then postulated that the SCAP content might be modulated by A1R through direct binding with PKAc, which was shown by the following coIP test.
The cellular content of protein is tightly controlled by the balance between biosynthesis and degradation. We demonstrated that activation of A1R by CPA increased the degradation of SCAP protein in the context of inhibiting protein biosynthesis with CHX. Moreover, CPA increased SCAP co-localization and degradation in lysosome, instead of the ubiquitin-proteasome system, which are the two major pathways regulating protein catabolism.47 In agreement with our current finding, Zheng et al. revealed an autophagy-independent SCAP degradation in lysosome through binding with SQSTM1 induced by the small molecule lycorine.48 It is unclear whether A1R controlled the lysosomal SCAP degradation via SQSTM1. We observed that A1R activation resulted in increased binding of SCAP with SQSTM1 or promoted the formation of SCAP-PKAc complex upon A1R inhibition, as shown by both co-IP and PLA tests. These results implicated an interestingly competitive binding of SCAP with SQSTM1 rather than PKAc upon A1R activation, facilitating SCAP degradation in lysosome and then reducing maturation of SREBPs. This is a new mechanism underlying the process of A1R controlling the PKAc-SCAP-SREBPs pathway.
Finally, in agreement with the protection of hepatic A1R against MAFLD development based on liver-specific knockout or overexpression of A1R mice, pharmacological activation of A1R by specific agonist CCPA, a lead compound for improving lung function, ischemia reperfusion, neuro-protection, and anticonvulsant activity,74,75,76,77 also effectively attenuated diet-induced MAFL and MASH in mice. Moreover, the potential of A1R as an anti-MAFLD target was validated by the screened natural compound TA3 based on its potent ability for A1R activation. TA3 is a well-established lead compound with multiple-pharmacological activities, including anti-cancer, anti-neuronal disorders, anti-inflammation, and anti-coagulant,78 while our results demonstrated its anti-MAFLD effect by activating A1R. Notably, both CCPA and TA3 were effective in treating diet-induced MAFL and MASH in WT mice, but not in LKO mice, implicating that hepatic A1R is the target of pharmacological agonists for MAFLD therapy instead of other tissues.
In summary, our current study reveals a previously undetermined role of hepatic A1R as anti-MAFLD drug target and elucidates the underlying mechanism by controlling the PKAc-SCAP-SREBPs pathway. The increased expression of hepatic A1R in MAFLD patients might be an adaptive response to combating MAFLD development, but it is not sufficient. Pharmacological activation of hepatic A1R is practical for MAFLD treatment, in particular MASH.
Limitations of the study
In the current study, we demonstrated that genetic or pharmacological activation of hepatic A1R inhibited MAFL/MASH development. The underlying mechanism was that activated A1R promoted the competitive binding of SCAP with SQSTM1 rather than PKAc, leading to SCAP degradation in lysosome, which reduced SREBP maturation and its regulated DNL and inflammation. It should be noted that the main limitations of the study were summarized follows.
First, although our results demonstrated the control by A1R on SCAP degradation through PKAc, the mechanistic insight into how A1R regulates the competitive binding between PKAc and SQSTM1 with SCAP remains unclear; for example, the key binding site of SCAP with PKAc or SQSTM1 needs to be fully characterized.
Second, A1R agonists of CCPA and TA3 were effective in treating MAFL/MASH; however, CCPA inhibited locomotor activity in mice but TA3 did not. Because both CCPA and TA3 could be detected in brain, we postulated that these two agonists might activate the A1R in different ways, such as different binding structural domains of A1R. Currently, the binding characteristics of these two agonists on A1R were under in-depth exploration in cooperation with experts in the structural biology group. We believe the elucidation of the binding characteristics by agonists of CCPA or TA3 would accelerate the research and development of anti-MASH drugs by targeting activation of A1R with more safety and fewer side effect in inhibition of central nervous system.
Third, although our results demonstrated that liver-specific deletion A1R exacerbated the severity of MAFL/MASH, which was relieved by its liver-specific overexpression in vivo and in vitro, we could not completely rule out whether the observed effects might be partly attributed to the endogenous adenosine action on other subtypes of ARs, which are also implicated in metabolic diseases such as MAFLD.58
Fourth, the underlying mechanism of the elevated expression of hepatic A1R both in MAFLD/MASH patients and mouse model was not addressed in our current report. Further investigation is warranted to uncover the exact mechanism that regulates the expression of hepatic A1R during the development of MAFLD/MASH.
Finally, Stagg et al. recently demonstrated that A2AR protected against MASH-HCC development,12 so it would be of interest to explore whether A1R is also involved in the development of MASH-HCC in the future. Since TA3 has been reported to have anti-tumor effects on various tumor cells, including HCC,79 we speculated that hepatic A1R may be involved in the development of MASH-HCC. It should be noted that the presence and correlation of hepatic A1R with pathological stages of MASH was only evaluated on nine liver specimens from MASH patients with liver biopsy in our current study. Further study is urgently needed by including a clinical cohort with large-scale MASH patients, which is of vital significance to translate current findings to the clinic.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| A1R antibody | Abcam, UK | ab151523 |
| A1R antibody | Abcam, UK | ab82477, RRID: AB_2049141 |
| SREBP1c antibody | Abcam, UK | ab28481, RRID: AB_778069 |
| SREBP2 antibody | Abclonal, China | A13049, RRID, AB_2759897 |
| FASN antibody | Cell signaling, USA | #3180, RRID: AB_2100796 |
| ACC antibody | Cell signaling, USA | #3676, RRID: AB_2219397 |
| CPT1 antibody | Proteintech, USA | ag7202 |
| CD36 antibody | Abcam, UK | ab133625, RRID, AB_2716564 |
| F4/80 antibody | Servicebio, China | GB113373, RRID, AB_2938980 |
| CD11B antibody | Boster, China | BM3925, RRID: AB_2832991 |
| TGF-β antibody | Abclonal, China | A2124, RRID: AB_2764143 |
| Smad2 antibody | Affinity, China | AF6449, RRID: AB_2835272 |
| pSmad2 antibody | Affinity, China | AF3449, RRID: AB_2834844 |
| PKAc antibody | Cell signaling, USA | #4782, RRID: AB_2170170 |
| SCAP antibody | Cell signaling, USA | #13102, RRID: AB_2798121 |
| SCAP antibody | Abmart, China | TD13713 |
| SCAP antibody | SantaCruz, USA | SC-13553, RRID: AB_628237 |
| S2P antibody | Abcam, UK | ab140594 |
| S2P antibody | Affinity, USA | DF13911 |
| S1P antibody | Abcam, UK | ab140592, RRID: AB_2811283 |
| Insig1 antibody | Abcam, UK | ab70784, RRID: AB_1269181 |
| SQSTM1 antibody | Abcam, UK | ab109012, RRID: AB_2810880 |
| NF-κB P65 antibody | Proteintech, USA | 66535–1, RRID: AB_2881898 |
| Phospho-NF-κB p65 antibody | Cell signaling, USA | #3033s, RRID: AB_331284 |
| α-SMA antibody | Cell signaling, USA | #19245, RRID: AB_2734835 |
| IκBα antibody | Cell signaling, USA | #4814s, RRID: AB_390781 |
| LAMIN B1 antibody | Cell signaling, USA | #13435, RRID: AB_2737428 |
| β-TUBULIN antibody | Cell signaling, USA | #2146, RRID: AB_2210545 |
| GAPDH antibody | Proteintech, USA | 60004-1, RRID: AB_2107436 |
| ATP1A1 antibody | PTM Bio, China | PTM-6271 |
| Bacterial and virus strains | ||
| AAV8-TBG-A1R | This paper | N/A |
| AAV8-TGB-GFP | This paper | N/A |
| Biological samples | ||
| Human liver paraffin blocks | This paper | N/A |
| Mouse liver paraffin blocks | This paper | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| CCPA | Aladdin, China | C170001 |
| DPCPX | Sigma, USA | C101 |
| CPA | Sigma, USA | C8301 |
| H89 | Selleck, China | S1582 |
| DbcAMP | Selleck, China | S7858 |
| Cycloheximide | Selleck, China | S7418 |
| MG132 | MCE, USA | HY-13259 |
| Bafilomycin A1 | MCE, USA | HY-100558 |
| Timosaponin AIII | MCE, USA | HY-N0810 |
| PMSF | Beyotime, China | ST506 |
| Critical commercial assays | ||
| IL-1β ELISA Kit | Mlbio, China | ml301814 |
| IL-6 ELISA Kit | Mlbio, China | ml063159 |
| TNFα ELISA Kit | Mlbio, China | ml002095 |
| Insulin ELISA Kit | Mlbio, China | ml001983V |
| PKA kinase activity kit | Enzo Life Sciences, USA | ADI-EKS-390A |
| cAMP Gi Kit | Cisbio, France | 62a.m.9PEB |
| Hoechst | Beyotime, China | P0133 |
| LysoTracker red | Beyotime, China | C1046 |
| Golgi Staining Kit | Abcam, UK | ab139483 |
| Mouse genotyping kit | Vazyme, China | PD101-01 |
| FastKing gDNA Dispelling RT SuperMix | TIANGEN, China | KR118 |
| qPCR SYBR Green Master Mix | Vazyme, China | Q131 |
| RIPA Lysis Buffer | Beyotime, China | P0013B |
| Nuclear and Cytoplasmic Protein Extraction Kit | Beyotime, China | P0028 |
| Triglycerides Assay Kit | Njjcbio, China | A110-1-1 |
| Total cholesterol assay kit | Njjcbio, China | A111-1-1 |
| Alanine aminotransferase Assay Kit | Njjcbio, China | C009-2-1 |
| Aspartate aminotransferase Assay Kit | Njjcbio, China | C010-2-1 |
| Alkaline phosphatase assay kit | Njjcbio, China | A059-2-2 |
| Low-density lipoprotein cholesterol assay kit | Njjcbio, China | A113-1-1 |
| High-density lipoprotein cholesterol assay kit | Njjcbio, China | A112-1-1 |
| Nonesterified Free fatty acids assay kit | Njjcbio, China | A042-2-1 |
| Oil Red O Stain Kit | Njjcbio, China | D027-1-2 |
| Duolink In Situ Wash Buffers, Fluorescence | Sigma, USA | DUO82049 |
| Duolink In Situ Detection Reagents Red | Sigma, USA | DUO92008 |
| Duolink In Situ PLA Probe Anti-Rabbit PLUS | Sigma, USA | DUO92002 |
| Duolink In Situ PLA Probe Anti-Mouse MINUS | Sigma, USA | DUO92004 |
| Immunoprecipitation Kit | Invitrogen, USA | 10007D |
| Membrane and Cytosol Protein Extraction Kit | Beyotime, China | P0033 |
| EZ-press RNA purification kit | EZBio, USA | B0004DP |
| DMEM | Meilunbio, China | MA0212 |
| DMEM/F-12 | Meilunbio, China | MA0214 |
| Deposited data | ||
| Human NAFLD expression profiling data | GEO portal | GSE89632 |
| Human NAFLD transcriptomic data | GEO portal | GSE135251 |
| Experimental models: Cell lines | ||
| AML12 cells | ATCC | CRL-2254 |
| HepG2 cells | ATCC | HB-8065 |
| Experimental models: Organisms/strains | ||
| C57BL/6 Adora1liver−/− mice | Shanghai Model Organisms Center Inc. | N/A |
| Hepatic ADORA1-OE C57BL/6 mice | This paper | N/A |
| C57BL/6 mice | Shanghai SLAC Laboratory Animal Co., Ltd | N/A |
| Oligonucleotides | ||
| Primes for mouse see Table S3 | This paper | N/A |
| Software and algorithms | ||
| Prism, version 10 | GraphPad | https://www.graphpad.com/ |
| LAS X, version 3.5.7 | Leica | https://www.leica.com/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Houkai Li (hk_li@shutcm.edu.cn).
Materials availability
This study did not generate new unique regents.
Data and code availability
-
•
This paper analyzes existing, publicly available data. These accession numbers for the datasets are listed in the key resources table. All data reported in this paper will be shared by the lead contact upon reasonable request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and study partipant details
Human samples and approval
The patients’ paraffin sections for A1R IHC staining were collected from Huzhou Central Hospital (Zhejiang, China). The study protocol was approved by Medical Ethics Committee of Huzhou Central Hospital (No.20190401-02). The human liver samples for A1R immunofluorescence that patients with biopsy-proven metabolic (dysfunction)-associate steatohepatitis (MASH) were recruited from Huzhou Central Hospital. This study procedures were approved by Medical Ethics Committee of Huzhou Central Hospital (No.20201201-02). All tissue samples were collected in compliance with informed consent policy. Clinical information is summarized in Tables S1 and S2. Each specimen was evaluated by experienced pathologists without knowledge of the clinical findings. The NAS consists of liver steatosis (scale of 0–3), lobular inflammation (scale of 0–3), hepatocellular ballooning (scale of 0–2).
Mice study and approval
Male C57BL/6J mice were housed under a 12:12-h light/dark cycle at controlled temperature conditions in specific-pathogen-free (SPF) condition. All animals were purchased from the Shanghai Model Organisms Center Inc, Shanghai SLAC Laboratory Animal Co., Ltd, and bred at the experimental animal center, Shanghai University of Traditional Chinese Medicine. All experimental procedures were approved by the Institutional Animal Care and Use Committee of Shanghai University of Traditional Chinese Medicine. All experiments were conducted in strict accordance with the Guidelines for the Investigation of Pain in Conscious Animal Experiments in order to minimize animal suffering and improve animal welfare (Ethical Approval Number: PZSHUTCM220221003, PZSHUTCM201030006, PZSHUTCM190426007, PZSHUTCM2304190012, PZSHUTCM2304190013).
HFD/CD-HFD diet feeding, AAV8 injection, and pharmacological treatment
Diet intervention started at 6-week-old. For diet induced MAFLD, mice were fed with high fat diet (HFD, 60% kcal from fat; D12492; Research Diets, New Brunswick, NJ, USA) for 12 or 16 weeks. To establish the NASH model, mice were fed with CD-HFD diet (A06071302, Research Diets, New Brunswick, NJ, USA) for 9 weeks or AMLN diet (Research Diet, Cat. D09100301, consisting of 40% fat, 20% fructose and 2% cholesterol) for 28 weeks.37
Hepatic A1R overexpression mice (A1R Liver OE) were conducted via AAV8 mediated A1R overexpression purchased from Shandong Vigene, vehicle-injected mice (pAV-TBG-P2A-GFP, Vehicle) were used as controls.
For PKA inhibition in vivo, PKA inhibitor H89 (1 mg/kg body weight, in 0.9% NaCl solution) was intraperitoneally injected into Flox and LKO mice daily with HFD feeding.41 For CCPA treatment in vivo, CCPA (1 mg/kg body weight, in 0.9% NaCl solution) was injected into mice daily from 9th week of the 16-week HFD feeding, or from 4th week of the 9-week CD-HFD feeding, or from 13th week of the 28-week AMLN diet feeding. For adenosine treatment in vivo, adenosine (10 mg/kg, ip) or CCPA (1 mg/kg, ip) were treated from 13th week to 18th. For timosaponin AIII (TA3) treatment in vivo, TA3 (5, 10 mg/kg body weight, in 0.9% NaCl solution) was injected into mice daily from 4th week of the 9-week CD-HFD feeding.
All mice were fasted 12 h before the collection of blood and tissues. Serum was distributed into several centrifuge tubes for further analysis after being centrifuged at 4000 rpm for 15 min at 4°C. Mice tissue samples were used instantly or snap-frozen in liquid nitrogen and stored at −80°C after collection and being weighted.
Method details
Generation of genetically modified mice
Hepatic A1R knockout mice (A1Rliver −/−, LKO) was generated using the CRISPR/Cas9 system and Cre-loxP-mediated recombination technology. First, two single guide RNAs (sgRNA1 and sgRNA2) were used to target a fragment of A1R exon3. Cas9 mRNA and gRNA were obtained by in vitro transcription. The donor vector was constructed by In-Fusion cloning, which contained a 3.0kb 5 'homology arm, a 1.0kb flox region and a 3.0kb 3' homology arm. Microinjection of Cas9 mRNA, gRNA, and donor vector into zygotes of C57BL/6J mice. We selected one founder mouse and crossed it with a C57BL/6J mouse to generate A1R-Flox mice. Finally, the A1R LKO mice were generated by crossing albumin-Cre recombinase (Alb-Cre) transgenic mice with A1Rflox/flox mice. Littermates Alb-Cre negative, A1Rflox/flox mice (Flox) were used as controls. Mice were housed in barrier facility with free access to drinking water and fed standard chow diet.
To generate hepatic A1R over expression mice (A1Rliver OE), AAV8-TBG-A1R adenoviruses were injected intravenously (mousetail) at 10ˆ12 vg per mice, single injection. The gene ID number is 11539, and transcript is NM_001008533.3 from Mus musculus. And control mice were injected with AAV8-TBG-GFP adenoviruses (Vehicle).
Cell lines
HepG2 cells were purchased from the Chinese Academy of Science (Shanghai, China), and were cultured in DMEM medium (Gibco, USA) supplemented with 10% FBS (BI, Israel), 100 U/ml penicillin and 100 mg/mL streptomycin (Meilunbio, China). AML-12 cells were cultured in DMEM medium (Gibco, USA) supplemented with 10% FBS (BI, Israel), 100 U/ml penicillin and 100 mg/mL streptomycin (Gibco, USA), 1% Insulin-Transferrin-Selenium-Sodium pyruvate (ITS-A) (Gibco, USA), 40 ng/mg dexamethasone (Sigma, USA).
For the stable A1R over expression cell lines, AAV8-A1R was purchased from Vigene Biosciences (Shandong). A1R was over expressed in AML-12 cell line using pAV-TBG-P2A-GFP, packaged in Adeno-associated virus. AAV infection was carried out in 6-well plates by mixing 10 μL virus supernatant plus 10 mL complete medium with a final concentration of 1011 vg/ml. Then, the selection via fluorescence-activated cell sorter was performed 48 h after infection, and then infected cells were further grown for about 2 weeks in the medium. Identification of over expression was carried out by RT-qPCR and western blotting.
Mice primary hepatocytes were isolated from 8 to 12-week-old Flox or LKO mice by using collagenase perfusion and gradient centrifugation, as previously described.80 Briefly, the mice liver was perfused with reperfusion buffer (135 mmol/L NaCl, 5.3 mmol/L KCl, 28.2 mmol/L NaHCO3, 0.12 mmol/L Na2HPO4, 0.56 mmol/L NaH2PO4, 0.5 mmol/L EGTA, 9.0 g/L glucose, and 0.06 g/L Phenol red, PH 7.4) and enzyme buffer (135 mmol/L NaCl, 5.3 mmol/L KCl, 28.2 mmol/L NaHCO3, 0.12 mmol/L Na2HPO4, 0.56 mmol/L NaH2PO4, 0.5 mmol/L EGTA, 10 mmol/L HEPES, and 0.06 g/L phenol red, 200 mg/L collagenase D and 3.8 mmol/L CaCl2, PH7.4). The isolated primary hepatocytes were cultured in DMEM medium (Gibco, USA) supplemented 10% FBS (BI, Israel). Cells were grown at 37°C in a 5% CO2 and 95% air atmosphere.
In vitro, cells were treated or co-treated with DPCPX (1 μM) for 48 h, CPA (1 μM) for 48 h, H89 (20 μM) for 4 h, DbcAMP (200 μM) for 12 h, and TA3 (0.5–10 μM) for 48 h, respectively. For mechanistic insight, cells were treated with cycloheximide (CHX, 50 μM) for 0, 2, 4, 8 h, bafilomycin A1 (Baf.A1, 1 μM) for 8 h, or MG132 (10 μM) for 8 h, in the presence of drugs.48
Intraperitoneal glucose tolerance test (ipGTT) and insulin tolerance test (ipITT)
For ipGTT, mice were fasted for 15 h overnight with free access to water, and then were injected intraperitoneally with glucose solution (1 g/kg body weight).81 Fasting glucose levels were first measured as time 0, then blood glucose was examined at 15th, 30th, 60th, 90th, and 120th min after glucose injection. ipITT was performed by intraperitoneal injection with human insulin (0.75 units/kg body weight, Novo Nordisk) without fasting.82
Serum insulin level test and HOMA-IR
Homeostatic model assessment of insulin resistance (HOMA-IR) was employed to measure the fasting insulin levels in serum samples using a mouse Insulin ELISA kit. The following equation was used to determine the homeostasis model assessment of insulin resistance (HOMA-IR): HOMA-IR=(fasting glucose × fasting insulin)/22.5.83
Serum biochemistry index test
Serum cholesterol (TC), high-density lipoprotein cholesterol (HDL-C), low-density lipoprotein cholesterol (LDL-C), triglyceride (TG), alanine aminotransferase (ALT), aspartate aminotransferase (AST), and alkaline phosphatase (ALP) were tested according to the instruction manual. List of reagents (including catalog numbers) can be found in “Critical Commercial Assays” list.
Inflammatory cytokines test
For liver samples, the tissue was rinsed with pre-cold PBS to remove residual blood. Twenty mg of liver tissue was added to 200 μL PBS containing protease inhibitor PMSF, and further ground by tissue grinding instrument at 4°C. The homogenate was centrifuged at 5000 g for 10 min and the supernatant was collected for assay. The concentrations of proinflammatory factors (TNFα, IL-6, and IL-1β) from serum or liver samples were measured using ELISA according to the manufacturer’s instructions.
Liver triglycerides examination
For the hepatic lipid extraction, each 12 mg of liver tissue was added to 500 μL extraction solvent mixed with chloroform: methanol (v: v = 2:1), homogenized by tissue grinding instrument, then fully shaken for 1 min, centrifuged at 4000 rpm at 4°C for 10 min, 10 μL supernatant were transferred into a clean centrifugal tube and dried at room temperature. Then, 10 μL pure water was added to the precipitates for redissolution. After adding with 200 μL working solution supplied by the TG test kit, the reaction mixture was incubated at 37°C for 10 min. After briefly vertexing, the absorbance at 510 nm was measured in 96-well plate. The concentration of TG was calculated according to the standard curve, and the results were presented as mmol/g liver weight.84
Tissues staining
Tissues were fixed with 10% formalin for over 24 h, embedded in paraffin, and then stained with hematoxylin-eosin staining (H&E) and Sirius Red using a standard protocol. Hematoxylin staining was used for nuclear counterstaining (blue), and eosin was used to stain the cytoplasm red. Sirius Red was used for collagen fibers staining (red). For Oil red O staining, fresh large lobes of the liver were embedded in OCT and flash-frozen in liquid nitrogen, then stored at −80°C. The embedded tissues were sectioned into 8 μm pieces after equilibrated at −20°C, and then stained with Oil red O to determine the extent of hepatic steatosis.
Histopathological evaluation
NAFLD activity score (NAS, 0 to 8) including separate scores for steatosis (0–3), lobular inflammation (0–3) and hepatocellular ballooning scale (0–2). The criteria for hepatic steatosis scoring include grade 0, steatosis involved <5%; grade 1, steatosis involved between 5 and 33%; grade 2, steatosis involved between 34 and 66%; and grade 3, steatosis involved >66%. Hepatic lobular inflammation was grade based on overall assessment of all inflammatory foci per 200 field, including grade 0, no foci; grade 1, <2 foci per 200 field; grade 2, 2–4 foci per 200 field; grade 3, >4 foci per 200 field. Ballooning degeneration was scored by the severity based on numbers of hepatocytes: 0 points assigned as none, 1 point as mild, 2 points as marked.85
Liver F4/80 and CD11b immunofluorescence staining
Tissue paraffin sections were permeabilized in PBS supplemented with 0.4% Triton X-100 (Sigma-Aldrich) and 3% BSA (Sigma-Aldrich) for 1 h and preincubated in blocking buffer (PBS supplemented with 3% BSA) for 1 h. Tissues were then labeled with anti-F4/80 or anti-CD11b primary antibody and the corresponding secondary antibody for 2 h at room temperature. Then, immunofluorescence imaging was performed.
Quantification of 13C incorporated fatty acids
Flox and LKO mice (n = 6) were fed an AMLN diet for 7 days, fasted from 9 a.m. to 7 p.m., refed for 2 h, and force-fed 13C-fructose. The mice were fed overnight and killed the next morning. LC-MS was performed to examine the amount of 13C label incorporation into hepatic fatty acids.
Oil red O staining for cells
Cells were gently washed 2 times with PBS. 10% formalin was added to each well and incubated for 30 min. Then formalin was removed, and cells were gently washed 2 times with ddH2O. Each well was added 60% isopropanol and incubated for 5 min. After removing isopropanol, Oil Red O working solution was added to cover the cells completely and evenly and incubated for 10–20 min, then cells were washed 2–5 times with ddH2O as needed. Hematoxylin was added to the cells and incubated for 1 min, and washed 2–5 times as needed. Cells was viewed under microscope. Lipid droplets appear red and nucleal appear blue.86
Immunohistochemical staining and quantification
After deparaffinization and dehydration, antigen retrieval was performed using a microwave for 5 min, before peroxidase quenching with 3% H2O2 in phosphate-buffered saline (PBS) was done for 15 min. Subsequently, the sections were blocked with 5% bovine serum albumin (BSA) for 30 min and then incubated with a primary antibody at a dilution ratio of 1:500 in PBS overnight at 4°C. The negative control was treated in the same way, as were the sample groups, except that primary antibody were omitted in the incubation steps. The percentage of the area of the section occupied by positive staining was determined using ImageJ 1.53 software.
Immunofluorescence
For multiple immunofluorescence, Firstly, after washing twice with PBS, the cells were fixed in 4% paraformaldehyde in PBS for 15 min and then washed three times in PBS with Tween 20 (PBST). After removing supernatant, the PBS solution containing 0.5% Triton X-100 was used for permeation for 15–20 min. The permeation solution was discarded, PBS solution containing 5% donkey serum was added to seal the cells at room temperature for 1h. After removing the blocking solution, a primary antibody diluted with 5% BSA and 0.1% Triton X-100 PBS solution (1:150 dilution) was added and incubated overnight at 4°C. The next day, cells were washed 3 times with PBST. Fluorescent secondary antibody was added (diluted with PBS solution containing 5%BSA, 1:200), and incubated for 1 h at room temperature in dark. After washing with PBST, the sealing solution containing Hoechst dye was added under dark conditions and stored at 4°C. Images was detected and captured by Fluorescent Confocal Microscopy (Leica, TCS SP-8, Germany). To generate three-dimensional images, images were recorded as vertical z stacks and processed using Leica LAS X 3.5.7 software.
Co-immunoprecipitation (Co-IP)
AML-12 cells were lysed with IP lysis buffer (P0013, Beyotime, China) containing an inhibitor cocktail. After centrifugation, the supernatant containing proteins was subjected to IP with protein G agarose beads (10007D, invitrogen, USA) and the indicated antibodies overnight at 4°C. The beads were washed with NaCl buffer and boiled with 2×SDS loading buffer prior to analysis by Western blotting.
Proximity ligation assay (PLA)
AML-12 cells were seeded on coverslips and allowed to attach overnight. After fixation in 4% paraformaldehyde, PLA was performed using Duolink In Situ Proximity Ligation Assay reagent (Sigma-Aldrich) according to the manufacturer’s instructions. Briefly, blocking was performed with blocking solution for 30 min at 37°C, and primary antibodies: anti-SCAP(1:100 dilution), anti-SQSTM1(1:100 dilution), and anti-PKAc(1:100 dilution) were incubated overnight at 4°C. The PLA Probe anti-mouse MINUS and PLA Probe anti-rabbit PLUS were incubated for 1 h at 37°C. Ligation and amplification were performed using Detection Reagents Red. Duolink Mounting Medium with DAPI was used for nuclear staining and mounting. The average PLA signal intensity of each cell was calculated by ImageJ software.87 List of reagents (including catalog numbers) can be found in “Critical Commercial Assays” list. List of antibodies can be found in “Antibodies” list.
Natural compound database screening
Natural compounds screening with activating capacity on A1R activity was performed based on a commercial Natural Product Library Plus (website: https://www.medchemexpress.cn/screening/natural-product-library-plus.html) by using computer-aid molecular docking. The Virtual Screening Workflow module was used to perform virtual screening by importing the prepared compounds and using the Glide module to perform molecular docking, i.e., docking between receptor and ligand molecules by geometric and energy matching. The 3600 prepared small molecules in the Natural Product Library Plus were first screened using the High Throughput Screening (HTVS) mode in the Glide module, and the top 20% of the scored small molecules were selected for a second round of screening using the Standard (SP) mode; then the top 20% of the scored compounds were selected for a third round of screening using the High Precision (XP) mode to obtain the ranking of the small molecules. The top 200 compounds screened from Natural Product Library Plus, was further analyzed using cAMP test in vitro.
Protein isolation and western blot analyses
Tissue/Cells were lysed in RIPA Lysis Buffer (Beyotime, China) supplemented with PMSF (Beyotime, China) and protease inhibitors and phosphatase inhibitors (Sigma, USA). Nuclear or membrane extracts were prepared with a nuclear and cytoplasmic protein extraction kit (Beyotime, China) according to the manufacturer’s instructions. Protein concentrations were measured with the BCA reagent (YEASEN, China) by using a Microplate Reader Spark System (TECAN, Switzerland). Equal amounts of protein were resolved by SDS–PAGE and immunoblotted with different antibodies as described previously. The immunoblots were detected using an Image 600 ECL System (GE, USA).
Extraction of total RNA and real-time quantitative PCR
Total RNA was isolated using an RNA purification kit (EZBioscience, USA), and then used to synthesize cDNA with the FastKing gDNA Dispelling RT SuperMix (TIANGEN, China). Real-time qPCR was performed using SYBR Green (Vazyme, China), 96-well plates and the CFX connect Real-Time System. Each well was loaded with a total of 20 μL containing 2 μL of cDNA, 0.5 μL of target primers, 7.5 μL of water and 10 μL of SYBR Select Master Mix. Real-time qPCR was performed for 40 cycles, with each cycle consisting of denaturation for 15 s at 94°C, annealing for 30 s at 60°C and elongation for 30 s at 72°C. Relative quantification was done using the 2- ΔΔCT method. Expression was normalized against 18s. Mean expression levels of chow-fed mice were set as 100%. The primers used are shown below.
Quantification and statistical analysis
Data representation and statistical analysis
Data are presented as mean ± SEM (Standard Error of Mean). p values were obtained using unpaired Student’s t test (between two groups) or One-way ANOVA analysis (during over two groups) for continuous variables via prism 9 (GraphPad Software Inc., La Jolla, CA). Significant differences among groups are indicated as ∗p < 0.05, ∗∗p < 0.01.
Acknowledgments
We sincerely acknowledge Prof. Jiayi Wang at Shanghai Chest Hospital for his generous contribution of the PLA probe kit (DUO92008), which helped us finish Figures 5E and 5F on time. We also acknowledge Prof. Gonghong Wei at Fudan University for his help in analyzing the GWAS summary of MAFLD/MASH. This work was supported by the National Natural Science Foundation of China (nos. 81873059, 82374129 to H.L.), the Joint Funds of National Natural Science Foundation of China (no. U21A20413 to H.L.), Shanghai Excellent Academic Leaders Program (no. 21XD1403500 to H.L.), and Youth Qi Huang Scholar by State Administration of TCM (to H.L.).
Author contributions
W.Z. and Y.H. performed all experiments, data analysis, and figure arrangement and took part in manuscript writing. Z.T., X.Z., X.W., and Z.T. were responsible for clinical sample and patient information collection. P.S. was responsible for pathological diagnosis of MAFLD patients. X.H., Y.L., H.W., X.G., W.H., Z.L., Y.B., J.M., and N.Z. were involved in most of the animal experiments and data analysis. C.X. guided the mechanistic study and helped in the manuscript writing. X.K. guided the PLA experiment and data analysis. W.Z. assisted in screening and identification of TA3 compound from dozens of candidates. W.J. helped in the study design. J.Z. coordinated the clinical study of the project, including sample and patient information collection and A1R analysis of clinical samples. L.S. took part in the study design, data analysis, and manuscript writing. M.L. participated in manuscript revision and data analysis. H.L. organized the studies, supervised the project, and revised the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: March 19, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2024.101477.
Contributor Information
Mingxiao Li, Email: mingxiaoli0928@126.com.
Jing Zhong, Email: Zhongjing1003@126.com.
Lili Sheng, Email: llsheng@shutcm.edu.cn.
Houkai Li, Email: hk_li@shutcm.edu.cn.
Supplemental information
References
- 1.Méndez-Sánchez N., Bugianesi E., Gish R.G., Lammert F., Tilg H., Nguyen M.H., Sarin S.K., Fabrellas N., Zelber-Sagi S., Fan J.G., et al. Global multi-stakeholder endorsement of the MAFLD definition. Lancet. Gastroenterol. Hepatol. 2022;7:388–390. doi: 10.1016/S2468-1253(22)00062-0. [DOI] [PubMed] [Google Scholar]
- 2.Eslam M., Newsome P.N., Sarin S.K., Anstee Q.M., Targher G., Romero-Gomez M., Zelber-Sagi S., Wai-Sun Wong V., Dufour J.F., Schattenberg J.M., et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020;73:202–209. doi: 10.1016/j.jhep.2020.03.039. [DOI] [PubMed] [Google Scholar]
- 3.Zhang X.L., Fan J.G., Wei L., Shi J.P., Zheng M.H., Chinese MAFLD Clinical Research Network Promoting the term MAFLD: China in action. Lancet. Gastroenterol. Hepatol. 2022;7:598. doi: 10.1016/S2468-1253(22)00127-3. [DOI] [PubMed] [Google Scholar]
- 4.Shiha G., Korenjak M., Eskridge W., Casanovas T., Velez-Moller P., Högström S., Richardson B., Munoz C., Sigurðardóttir S., Coulibaly A., et al. Redefining fatty liver disease: an international patient perspective. Lancet. Gastroenterol. Hepatol. 2021;6:73–79. doi: 10.1016/S2468-1253(20)30294-6. [DOI] [PubMed] [Google Scholar]
- 5.Powell E.E., Wong V.W.S., Rinella M. Non-alcoholic fatty liver disease. Lancet. 2021;397:2212–2224. doi: 10.1016/S0140-6736(20)32511-3. [DOI] [PubMed] [Google Scholar]
- 6.Eslam M., Sanyal A.J., George J., International Consensus Panel MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology. 2020;158:1999–2014.e1. doi: 10.1053/j.gastro.2019.11.312. [DOI] [PubMed] [Google Scholar]
- 7.James O.F., Day C.P. Non-alcoholic steatohepatitis (NASH): a disease of emerging identity and importance. J. Hepatol. 1998;29:495–501. doi: 10.1016/s0168-8278(98)80073-1. [DOI] [PubMed] [Google Scholar]
- 8.Sawada K., Chung H., Softic S., Moreno-Fernandez M.E., Divanovic S. The bidirectional immune crosstalk in metabolic dysfunction-associated steatotic liver disease. Cell Metabol. 2023;35:1852–1871. doi: 10.1016/j.cmet.2023.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Borea P.A., Gessi S., Merighi S., Vincenzi F., Varani K. Pharmacology of Adenosine Receptors: The State of the Art. Physiol. Rev. 2018;98:1591–1625. doi: 10.1152/physrev.00049.2017. [DOI] [PubMed] [Google Scholar]
- 10.Jain S., Jacobson K.A. Purinergic Signaling in Liver Pathophysiology. Front. Endocrinol. 2021;12 doi: 10.3389/fendo.2021.718429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Le T.T.T., Berg N.K., Harting M.T., Li X., Eltzschig H.K., Yuan X. Purinergic Signaling in Pulmonary Inflammation. Front. Immunol. 2019;10:1633. doi: 10.3389/fimmu.2019.01633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Allard B., Jacoberger-Foissac C., Cousineau I., Bareche Y., Buisseret L., Chrobak P., Allard D., Pommey S., Ah-Pine F., Duquenne S., et al. Adenosine A2A receptor is a tumor suppressor of NASH-associated hepatocellular carcinoma. Cell Rep. Med. 2023;4 doi: 10.1016/j.xcrm.2023.101188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Slaats J., Wagena E., Smits D., Berends A.A., Peters E., Bakker G.J., van Erp M., Weigelin B., Adema G.J., Friedl P. Adenosine A2a Receptor Antagonism Restores Additive Cytotoxicity by Cytotoxic T Cells in Metabolically Perturbed Tumors. Cancer Immunol. Res. 2022;10:1462–1474. doi: 10.1158/2326-6066.CIR-22-0113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Giuffrida L., Sek K., Henderson M.A., Lai J., Chen A.X.Y., Meyran D., Todd K.L., Petley E.V., Mardiana S., Mølck C., et al. CRISPR/Cas9 mediated deletion of the adenosine A2A receptor enhances CAR T cell efficacy. Nat. Commun. 2021;12:3236. doi: 10.1038/s41467-021-23331-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ehrentraut H., Westrich J.A., Eltzschig H.K., Clambey E.T. Adora2b adenosine receptor engagement enhances regulatory T cell abundance during endotoxin-induced pulmonary inflammation. PLoS One. 2012;7 doi: 10.1371/journal.pone.0032416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ehrentraut H., Clambey E.T., McNamee E.N., Brodsky K.S., Ehrentraut S.F., Poth J.M., Riegel A.K., Westrich J.A., Colgan S.P., Eltzschig H.K. CD73+ regulatory T cells contribute to adenosine-mediated resolution of acute lung injury. Faseb. J. 2013;27:2207–2219. doi: 10.1096/fj.12-225201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fishman P., Bar-Yehuda S. Pharmacology and therapeutic applications of A3 receptor subtype. Curr. Top. Med. Chem. 2003;3:463–469. doi: 10.2174/1568026033392147. [DOI] [PubMed] [Google Scholar]
- 18.Durante M., Squillace S., Lauro F., Giancotti L.A., Coppi E., Cherchi F., Di Cesare Mannelli L., Ghelardini C., Kolar G., Wahlman C., et al. Adenosine A3 agonists reverse neuropathic pain via T cell-mediated production of IL-10. J. Clin. Invest. 2021;131 doi: 10.1172/JCI139299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koeppen M., Eckle T., Eltzschig H.K. Selective deletion of the A1 adenosine receptor abolishes heart-rate slowing effects of intravascular adenosine in vivo. PLoS One. 2009;4 doi: 10.1371/journal.pone.0006784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jakova E., Moutaoufik M.T., Lee J.S., Babu M., Cayabyab F.S. Adenosine A1 receptor ligands bind to alpha-synuclein: implications for alpha-synuclein misfolding and alpha-synucleinopathy in Parkinson's disease. Transl. Neurodegener. 2022;11:9. doi: 10.1186/s40035-022-00284-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Badimon A., Strasburger H.J., Ayata P., Chen X., Nair A., Ikegami A., Hwang P., Chan A.T., Graves S.M., Uweru J.O., et al. Negative feedback control of neuronal activity by microglia. Nature. 2020;586:417–423. doi: 10.1038/s41586-020-2777-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peleli M., Carlstrom M. Adenosine signaling in diabetes mellitus and associated cardiovascular and renal complications. Mol. Aspect. Med. 2017;55:62–74. doi: 10.1016/j.mam.2016.12.001. [DOI] [PubMed] [Google Scholar]
- 23.Dhalla A.K., Chisholm J.W., Reaven G.M., Belardinelli L. A1 adenosine receptor: role in diabetes and obesity. Handb. Exp. Pharmacol. 2009:271–295. doi: 10.1007/978-3-540-89615-9_9. [DOI] [PubMed] [Google Scholar]
- 24.Laties A., Rich C.C., Stoltz R., Humbert V., Brickman C., McVicar W., Baumgartner R.A. A Randomized Phase 1 Dose Escalation Study to Evaluate Safety, Tolerability, and Pharmacokinetics of Trabodenoson in Healthy Adult Volunteers. J. Ocul. Pharmacol. Therapeut. 2016;32:548–554. doi: 10.1089/jop.2015.0147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lu L.J., Tsai J.C., Liu J. J. Novel Pharmacologic Candidates for Treatment of Primary Open-Angle Glaucoma. Yale J. Biol. Med. 2017;90:111–118. [PMC free article] [PubMed] [Google Scholar]
- 26.Thursz M.R., Richardson P., Allison M., Austin A., Bowers M., Day C.P., Downs N., Gleeson D., MacGilchrist A., Grant A., et al. Prednisolone or pentoxifylline for alcoholic hepatitis. N. Engl. J. Med. 2015;372:1619–1628. doi: 10.1056/NEJMoa1412278. [DOI] [PubMed] [Google Scholar]
- 27.Yang M., Chu R., Chisholm J.W., Doege H., Belardinelli L., Dhalla A.K. Adenosine A(1) receptors do not play a major role in the regulation of lipogenic gene expression in hepatocytes. Eur. J. Pharmacol. 2012;683:332–339. doi: 10.1016/j.ejphar.2012.03.012. [DOI] [PubMed] [Google Scholar]
- 28.Yang P., Han Z., Chen P., Zhu L., Wang S., Hua Z., Zhang J. A contradictory role of A1 adenosine receptor in carbon tetrachloride- and bile duct ligation-induced liver fibrosis in mice. J. Pharmacol. Exp. Therapeut. 2010;332:747–754. doi: 10.1124/jpet.109.162727. [DOI] [PubMed] [Google Scholar]
- 29.Kim J., Kim M., Song J.H., Lee H.T. Endogenous A1 adenosine receptors protect against hepatic ischemia reperfusion injury in mice. Liver Transplant. 2008;14:845–854. doi: 10.1002/lt.21432. [DOI] [PubMed] [Google Scholar]
- 30.Yang P., Chen P., Wang T., Zhan Y., Zhou M., Xia L., Cheng R., Guo Y., Zhu L., Zhang J. Loss of A1 Adenosine Receptor Attenuates Alpha-naphthylisothiocyanate-Induced Cholestatic Liver Injury in Mice. Toxicol. Sci. 2013;131:128–138. doi: 10.1093/toxsci/kfs263. [DOI] [PubMed] [Google Scholar]
- 31.Peng Z., Borea P.A., Varani K., Wilder T., Yee H., Chiriboga L., Blackburn M.R., Azzena G., Resta G., Cronstein B.N. Adenosine signaling contributes to ethanol-induced fatty liver in mice. J. Clin. Invest. 2009;119:582–594. doi: 10.1172/JCI37409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ramkumar V., Stiles G.L., Beaven M.A., Ali H. The A3 adenosine receptor is the unique adenosine receptor which facilitates release of allergic mediators in mast cells. J. Biol. Chem. 1993;268:16887–16890. [PubMed] [Google Scholar]
- 33.Matsumoto M., Hada N., Sakamaki Y., Uno A., Shiga T., Tanaka C., Ito T., Katsume A., Sudoh M. An improved mouse model that rapidly develops fibrosis in non-alcoholic steatohepatitis. Int. J. Exp. Pathol. 2013;94:93–103. doi: 10.1111/iep.12008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gao X.M., Dong W.H., Xia C.L., Ma Z.Y., Wang Y., Sarra S., Xu H.M., Qi W.Y. Centranthera grandiflore alleviates alcohol-induced oxidative stress and cell apoptosis. Chin. J. Nat. Med. 2022;20:572–579. doi: 10.1016/S1875-5364(22)60181-X. [DOI] [PubMed] [Google Scholar]
- 35.Zhou S., Fan K.K., Gu L.F., Yu B.Y., Chai C.Z. Anti-inflammatory effects of Abelmoschus manihot (L.) Medik. on LPS-induced cystitis in mice: potential candidate for cystitis treatment based on classic use. Chin. J. Nat. Med. 2022;20:321–331. doi: 10.1016/S1875-5364(22)60140-7. [DOI] [PubMed] [Google Scholar]
- 36.Zhang X., Sharma P., Maschmeyer P., Hu Y., Lou M., Kim J., Fujii H., Unutmaz D., Schwabe R.F., Winau F. GARP on hepatic stellate cells is essential for the development of liver fibrosis. J. Hepatol. 2023;79:1214–1225. doi: 10.1016/j.jhep.2023.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhao P., Sun X., Chaggan C., Liao Z., In Wong K., He F., Singh S., Loomba R., Karin M., Witztum J.L., Saltiel A.R. An AMPK-caspase-6 axis controls liver damage in nonalcoholic steatohepatitis. Science. 2020;367:652–660. doi: 10.1126/science.aay0542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Clapper J.R., Hendricks M.D., Gu G., Wittmer C., Dolman C.S., Herich J., Athanacio J., Villescaz C., Ghosh S.S., Heilig J.S., et al. Diet-induced mouse model of fatty liver disease and nonalcoholic steatohepatitis reflecting clinical disease progression and methods of assessment. Am. J. Physiol. Gastrointest. Liver Physiol. 2013;305:G483–G495. doi: 10.1152/ajpgi.00079.2013. [DOI] [PubMed] [Google Scholar]
- 39.Horton J.D., Goldstein J.L., Brown M.S. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Invest. 2002;109:1125–1131. doi: 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee J.H., Lee G.Y., Jang H., Choe S.S., Koo S.H., Kim J.B. Ring finger protein20 regulates hepatic lipid metabolism through protein kinase A-dependent sterol regulatory element binding protein1c degradation. Hepatology. 2014;60:844–857. doi: 10.1002/hep.27011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yan F., Wang Q., Lu M., Chen W., Song Y., Jing F., Guan Y., Wang L., Lin Y., Bo T., et al. Thyrotropin increases hepatic triglyceride content through upregulation of SREBP-1c activity. J. Hepatol. 2014;61:1358–1364. doi: 10.1016/j.jhep.2014.06.037. [DOI] [PubMed] [Google Scholar]
- 42.Chijiwa T., Mishima A., Hagiwara M., Sano M., Hayashi K., Inoue T., Naito K., Toshioka T., Hidaka H. Inhibition of forskolin-induced neurite outgrowth and protein phosphorylation by a newly synthesized selective inhibitor of cyclic AMP-dependent protein kinase, N-[2-(p-bromocinnamylamino)ethyl]-5-isoquinolinesulfonamide (H-89), of PC12D pheochromocytoma cells. J. Biol. Chem. 1990;265:5267–5272. [PubMed] [Google Scholar]
- 43.Coqueret O., Demarquay D., Lagente V. Role of cyclic AMP in the modulation of IgE production by the beta 2-adrenoceptor agonist, fenoterol. Eur. Respir. J. 1996;9:220–225. doi: 10.1183/09031936.96.09020220. [DOI] [PubMed] [Google Scholar]
- 44.Kim J.Y., Garcia-Carbonell R., Yamachika S., Zhao P., Dhar D., Loomba R., Kaufman R.J., Saltiel A.R., Karin M. ER Stress Drives Lipogenesis and Steatohepatitis via Caspase-2 Activation of S1P. Cell. 2018;175:133–145.e15. doi: 10.1016/j.cell.2018.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Osborne T.F., Espenshade P.J. Evolutionary conservation and adaptation in the mechanism that regulates SREBP action: what a long, strange tRIP it's been. Genes Dev. 2009;23:2578–2591. doi: 10.1101/gad.1854309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brown M.S., Radhakrishnan A., Goldstein J.L. Retrospective on Cholesterol Homeostasis: The Central Role of Scap. Annu. Rev. Biochem. 2018;87:783–807. doi: 10.1146/annurev-biochem-062917-011852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Goldberg A.L. In: Second Edition. Lennarz W.J., Daniel Lane M., editors. Academic Press; 2013. Encyclopedia of Biological Chemistry; pp. 617–624. [Google Scholar]
- 48.Zheng Z.G., Zhu S.T., Cheng H.M., Zhang X., Cheng G., Thu P.M., Wang S.P., Li H.J., Ding M., Qiang L., et al. Discovery of a potent SCAP degrader that ameliorates HFD-induced obesity, hyperlipidemia and insulin resistance via an autophagy-independent lysosomal pathway. Autophagy. 2021;17:1592–1613. doi: 10.1080/15548627.2020.1757955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Arendt B.M., Comelli E.M., Ma D.W.L., Lou W., Teterina A., Kim T., Fung S.K., Wong D.K.H., McGilvray I., Fischer S.E., Allard J.P. Altered Hepatic Gene Expression in Nonalcoholic Fatty Liver Disease Is Associated With Lower Hepatic n-3 and n-6 Polyunsaturated Fatty Acids. Hepatology. 2015;61:1565–1578. doi: 10.1002/hep.27695. [DOI] [PubMed] [Google Scholar]
- 50.Govaere O., Cockell S., Tiniakos D., Queen R., Younes R., Vacca M., Alexander L., Ravaioli F., Palmer J., Petta S., et al. Transcriptomic profiling across the nonalcoholic fatty liver disease spectrum reveals gene signatures for steatohepatitis and fibrosis. Sci. Transl. Med. 2020;12 doi: 10.1126/scitranslmed.aba4448. [DOI] [PubMed] [Google Scholar]
- 51.Bowser J.L., Lee J.W., Yuan X., Eltzschig H.K. The hypoxia-adenosine link during inflammation. J. Appl. Physiol. 1985;123:1303–1320. doi: 10.1152/japplphysiol.00101.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Aherne C.M., Collins C.B., Rapp C.R., Olli K.E., Perrenoud L., Jedlicka P., Bowser J.L., Mills T.W., Karmouty-Quintana H., Blackburn M.R., Eltzschig H.K. Coordination of ENT2-dependent adenosine transport and signaling dampens mucosal inflammation. JCI Insight. 2018;3 doi: 10.1172/jci.insight.121521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Eckle T., Hughes K., Ehrentraut H., Brodsky K.S., Rosenberger P., Choi D.S., Ravid K., Weng T., Xia Y., Blackburn M.R., Eltzschig H.K. Crosstalk between the equilibrative nucleoside transporter ENT2 and alveolar Adora2b adenosine receptors dampens acute lung injury. Faseb. J. 2013;27:3078–3089. doi: 10.1096/fj.13-228551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ruan W., Li J., Choi S., Ma X., Liang Y., Nair R., Yuan X., Mills T.W., Eltzschig H.K. Targeting myocardial equilibrative nucleoside transporter ENT1 provides cardioprotection by enhancing myeloid Adora2b signaling. JCI Insight. 2023;8 doi: 10.1172/jci.insight.166011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen J.F., Eltzschig H.K., Fredholm B.B. Adenosine receptors as drug targets--what are the challenges? Nat. Rev. Drug Discov. 2013;12:265–286. doi: 10.1038/nrd3955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dunwiddie T.V., Masino S.A. The role and regulation of adenosine in the central nervous system. Annu. Rev. Neurosci. 2001;24:31–55. doi: 10.1146/annurev.neuro.24.1.31. [DOI] [PubMed] [Google Scholar]
- 57.Jacobson K.A., Gao Z.G. Adenosine receptors as therapeutic targets. Nat. Rev. Drug Discov. 2006;5:247–264. doi: 10.1038/nrd1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cai Y., Li H., Liu M., Pei Y., Zheng J., Zhou J., Luo X., Huang W., Ma L., Yang Q., et al. Disruption of adenosine 2A receptor exacerbates NAFLD through increasing inflammatory responses and SREBP1c activity. Hepatology. 2018;68:48–61. doi: 10.1002/hep.29777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Buyl K., Vrints M., Fernando R., Desmae T., Van Eeckhoutte T., Jans M., Van Der Schueren J., Boeckmans J., Rodrigues R.M., De Boe V., et al. Human skin stem cell-derived hepatic cells as in vitro drug discovery model for insulin-driven de novo lipogenesis. Eur. J. Pharmacol. 2023;957 doi: 10.1016/j.ejphar.2023.175989. [DOI] [PubMed] [Google Scholar]
- 60.Xia Q., Lu F., Chen Y., Li J., Huang Z., Fang K., Hu M., Guo Y., Dong H., Xu L., Gong J. 6-Gingerol regulates triglyceride and cholesterol biosynthesis to improve hepatic steatosis in MAFLD by activating the AMPK-SREBPs signaling pathway. Biomed. Pharmacother. 2024;170 doi: 10.1016/j.biopha.2023.116060. [DOI] [PubMed] [Google Scholar]
- 61.Shimano H., Sato R. SREBP-regulated lipid metabolism: convergent physiology - divergent pathophysiology. Nat. Rev. Endocrinol. 2017;13:710–730. doi: 10.1038/nrendo.2017.91. [DOI] [PubMed] [Google Scholar]
- 62.Lee S.H., Lee J.H., Im S.S. The cellular function of SCAP in metabolic signaling. Exp. Mol. Med. 2020;52:724–729. doi: 10.1038/s12276-020-0430-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tlapak-Simmons V.L., Baggenstoss B.A., Clyne T., Weigel P.H. Purification and lipid dependence of the recombinant hyaluronan synthases from Streptococcus pyogenes and Streptococcus equisimilis. J. Biol. Chem. 1999;274:4239–4245. doi: 10.1074/jbc.274.7.4239. [DOI] [PubMed] [Google Scholar]
- 64.Moon Y.A., Liang G., Xie X., Frank-Kamenetsky M., Fitzgerald K., Koteliansky V., Brown M.S., Goldstein J.L., Horton J.D. The Scap/SREBP Pathway Is Essential for Developing Diabetic Fatty Liver and Carbohydrate-Induced Hypertriglyceridemia in Animals. Cell Metabol. 2012;15:240–246. doi: 10.1016/j.cmet.2011.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kober D.L., Xu S., Li S., Bajaj B., Liang G., Rosenbaum D.M., Radhakrishnan A. Identification of a degradation signal at the carboxy terminus of SREBP2: A new role for this domain in cholesterol homeostasis. Proc. Natl. Acad. Sci. USA. 2020;117:28080–28091. doi: 10.1073/pnas.2018578117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schulte G., Fredholm B.B. Human adenosine A(1), A(2A), A(2B), and A(3) receptors expressed in Chinese hamster ovary cells all mediate the phosphorylation of extracellular-regulated kinase 1/2. Mol. Pharmacol. 2000;58:477–482. [PubMed] [Google Scholar]
- 67.Schulte G., Fredholm B.B. Signalling from adenosine receptors to mitogen-activated protein kinases. Cell. Signal. 2003;15:813–827. doi: 10.1016/s0898-6568(03)00058-5. [DOI] [PubMed] [Google Scholar]
- 68.Shimizu-Albergine M., Van Yserloo B., Golkowski M.G., Ong S.E., Beavo J.A., Bornfeldt K.E. SCAP/SREBP pathway is required for the full steroidogenic response to cyclic AMP. Proc. Natl. Acad. Sci. USA. 2016;113:E5685–E5693. doi: 10.1073/pnas.1611424113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kornev A.P., Aoto P.C., Taylor S.S. Calculation of centralities in protein kinase A. Proc. Natl. Acad. Sci. USA. 2022;119 doi: 10.1073/pnas.2215420119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yang J., Ten Eyck L.F., Xuong N.H., Taylor S.S. Crystal structure of a cAMP-dependent protein kinase mutant at 1.26A: new insights into the catalytic mechanism. J. Mol. Biol. 2004;336:473–487. doi: 10.1016/j.jmb.2003.11.044. [DOI] [PubMed] [Google Scholar]
- 71.Sastri M., Barraclough D.M., Carmichael P.T., Taylor S.S. A-kinase-interacting protein localizes protein kinase A in the nucleus. Proc. Natl. Acad. Sci. USA. 2005;102:349–354. doi: 10.1073/pnas.0408608102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bastidas A.C., Deal M.S., Steichen J.M., Guo Y., Wu J., Taylor S.S. Phosphoryl Transfer by Protein Kinase A Is Captured in a Crystal Lattice. J. Am. Chem. Soc. 2013;135:4788–4798. doi: 10.1021/ja312237q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dell'Acqua M.L., Scott J.D. Protein kinase A anchoring. J. Biol. Chem. 1997;272:12881–12884. doi: 10.1074/jbc.272.20.12881. [DOI] [PubMed] [Google Scholar]
- 74.Cheng P., Zuo X., Ren Y., Bai S., Tang W., Chen X., Wang G., Wang H., Huang W., Xie P. Adenosine A1-Receptors Modulate mTOR Signaling to Regulate White Matter Inflammatory Lesions Induced by Chronic Cerebral Hypoperfusion. Neurochem. Res. 2016;41:3272–3277. doi: 10.1007/s11064-016-2056-0. [DOI] [PubMed] [Google Scholar]
- 75.Fernandez L.G., Sharma A.K., LaPar D.J., Kron I.L., Laubach V.E. Adenosine A1 receptor activation attenuates lung ischemia-reperfusion injury. J. Thorac. Cardiovasc. Surg. 2013;145:1654–1659. doi: 10.1016/j.jtcvs.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Meads K.L., Thomas T.P., Langston J.L., Myers T.M., Shih T.M. Evaluation of adenosine A1 receptor agonists as neuroprotective countermeasures against Soman intoxication in rats. Toxicol. Appl. Pharmacol. 2021;416 doi: 10.1016/j.taap.2021.115466. [DOI] [PubMed] [Google Scholar]
- 77.Fabera P., Parizkova M., Uttl L., Vondrakova K., Kubova H., Tsenov G., Mares P. Adenosine A1 Receptor Agonist 2-chloro-N6-cyclopentyladenosine and Hippocampal Excitability During Brain Development in Rats. Front. Pharmacol. 2019;10:656. doi: 10.3389/fphar.2019.00656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lin Y., Zhao W.R., Shi W.T., Zhang J., Zhang K.Y., Ding Q., Chen X.L., Tang J.Y., Zhou Z.Y. Pharmacological Activity, Pharmacokinetics, and Toxicity of Timosaponin AIII, a Natural Product Isolated From Anemarrhena asphodeloides Bunge: A Review. Front. Pharmacol. 2020;11:764. doi: 10.3389/fphar.2020.00764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhang L., Zhang S., Jiang M., Lu L., Ding Y., Ma N., Zhao Y., Xuchen S., Zhang N. Novel Timosaponin AIII-Based Multifunctional Liposomal Delivery System for Synergistic Therapy Against Hepatocellular Carcinoma Cancer. Int. J. Nanomed. 2021;16:5531–5550. doi: 10.2147/IJN.S313759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hu M., Zhang D., Xu H., Zhang Y., Shi H., Huang X., Wang X., Wu Y., Qi Z. Salidroside Activates the AMP-Activated Protein Kinase Pathway to Suppress Nonalcoholic Steatohepatitis in Mice. Hepatology. 2021;74:3056–3073. doi: 10.1002/hep.32066. [DOI] [PubMed] [Google Scholar]
- 81.Hong Y., Sheng L., Zhong J., Tao X., Zhu W., Ma J., Yan J., Zhao A., Zheng X., Wu G., et al. Desulfovibrio vulgaris, a potent acetic acid-producing bacterium, attenuates nonalcoholic fatty liver disease in mice. Gut Microb. 2021;13:1–20. doi: 10.1080/19490976.2021.1930874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mansuy-Aubert V., Zhou Q.L., Xie X., Gong Z., Huang J.Y., Khan A.R., Aubert G., Candelaria K., Thomas S., Shin D.J., et al. Imbalance between neutrophil elastase and its inhibitor alpha1-antitrypsin in obesity alters insulin sensitivity, inflammation, and energy expenditure. Cell Metabol. 2013;17:534–548. doi: 10.1016/j.cmet.2013.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Matthews D.R., Hosker J.P., Rudenski A.S., Naylor B.A., Treacher D.F., Turner R.C. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28:412–419. doi: 10.1007/BF00280883. [DOI] [PubMed] [Google Scholar]
- 84.Folch J., Lees M., Sloane Stanley G.H. A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 1957;226:497–509. [PubMed] [Google Scholar]
- 85.Kleiner D.E., Brunt E.M., Van Natta M., Behling C., Contos M.J., Cummings O.W., Ferrell L.D., Liu Y.C., Torbenson M.S., Unalp-Arida A., et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41:1313–1321. doi: 10.1002/hep.20701. [DOI] [PubMed] [Google Scholar]
- 86.Mehlem A., Hagberg C.E., Muhl L., Eriksson U., Falkevall A. Imaging of neutral lipids by oil red O for analyzing the metabolic status in health and disease. Nat. Protoc. 2013;8:1149–1154. doi: 10.1038/nprot.2013.055. [DOI] [PubMed] [Google Scholar]
- 87.Guo S., Chen Y., Yang Y., Zhang X., Ma L., Xue X., Qiao Y., Wang J. TRIB2 modulates proteasome function to reduce ubiquitin stability and protect liver cancer cells against oxidative stress. Cell Death Dis. 2021;12:42. doi: 10.1038/s41419-020-03299-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
This paper analyzes existing, publicly available data. These accession numbers for the datasets are listed in the key resources table. All data reported in this paper will be shared by the lead contact upon reasonable request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.