

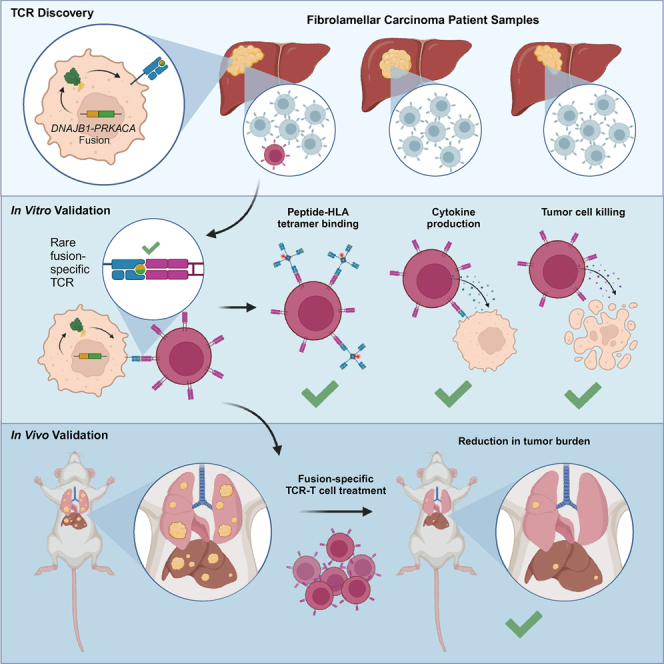
Summary
Fibrolamellar carcinoma (FLC) is a liver tumor with a high mortality burden and few treatment options. A promising therapeutic vulnerability in FLC is its driver mutation, a conserved DNAJB1-PRKACA gene fusion that could be an ideal target neoantigen for immunotherapy. In this study, we aim to define endogenous CD8 T cell responses to this fusion in FLC patients and evaluate fusion-specific T cell receptors (TCRs) for use in cellular immunotherapies. We observe that fusion-specific CD8 T cells are rare and that FLC patient TCR repertoires lack large clusters of related TCR sequences characteristic of potent antigen-specific responses, potentially explaining why endogenous immune responses are insufficient to clear FLC tumors. Nevertheless, we define two functional fusion-specific TCRs, one of which has strong anti-tumor activity in vivo. Together, our results provide insights into the fragmented nature of neoantigen-specific repertoires in humans and indicate routes for clinical development of successful immunotherapies for FLC.
Keywords: fibrolamellar carcinoma, DNAJB1-PRKACA, CD8 T cell, gene fusion, neoantigen, immunotherapy, cell therapy, T cell receptor, TCR repertoire
Graphical abstract
Highlights
-
•
Endogenous CD8 T cell responses against the FLC driver fusion are rare
-
•
FLC TCR repertoires lack characteristics of effective antiviral T cell responses
-
•
Rare fusion-specific TCRs are specific and functional and kill tumor cells in vitro
-
•
Fusion-specific TCR-T cell therapy can have potent anti-tumor activity in vivo
Kirk et al. identify rare endogenous CD8 T cell responses against conserved driver fusion neoantigens in fibrolamellar carcinoma, highlighting the potential for developing cell therapies for FLC. The rarity of these endogenous responses and fragmented nature of FLC TCR repertoires also provide insights into how TCR repertoires recognize cancer neoantigens.
Introduction
Fibrolamellar carcinoma (FLC) is a liver tumor with no effective systemic treatments.1,2,3 It occurs at an annual rate of 0.02 per 100,000 in the US4,5 and predominantly affects adolescents and young adults without underlying liver pathology.6,7,8 Surgical resection is typically the first line of treatment for FLC and can be curative for early-stage disease.9,10 However, patients with unresectable tumors or whose tumors recur after resection have a high risk of fatal outcomes.2 No standard-of-care systemic therapy is currently available, and FLC is often refractory to treatments used for other liver cancers.2,3 Consequently, an estimated 60%–80% of FLC patients will ultimately succumb to the disease.5,8,11
Among the most promising potential therapeutic vulnerabilities in FLC is its conserved driver mutation. More than 90% of FLC cases share a gene fusion arising from a 400-kb deletion on chromosome 19 that fuses the first exon of DNAJB1, which encodes heat shock protein 40, and the last 9 exons of PRKACA, which encodes a catalytic subunit of protein kinase A (PKA).12 This fusion protein is necessary for FLC tumorigenesis and survival13,14,15 and retains PKA activity,16 which is believed to underlie its oncogenic potential.17,18,19 However, attempts to directly inhibit this PKA activity with targeted therapy20 have led to unacceptable on-target toxicities in preclinical models. Targeting downstream signaling pathways has also been largely unsuccessful; recent clinical trials of the aurora kinase inhibitor ENMD-2076 (ClinicalTrials.gov: NCT02234986); combination everolimus, letrozole, and leuprolide therapy (ClinicalTrials.gov: NCT01642186); and combination neratinib, everolimus, and anti-PD-1 therapy (ClinicalTrials.gov: NCT01953926) in FLC reported response rates of 3% (1 of 35),21 0% (0 of 26),22 and 6% (1 of 15),23 respectively.
Although targeting the FLC fusion with small molecules has not yielded clinical benefits, it could also be targeted using immunotherapy. Importantly, the DNAJB1-PRKACA fusion breakpoint occurs in introns,12 resulting in a conserved amino acid sequence across patients and, therefore, an ideal shared neoantigen. Neoantigens are novel peptide antigens derived from genetic mutations, including gene fusions,24,25 which have been hypothesized to be a superior source of neoantigens.26 T cell recognition of neoantigens is well established to underlie the efficacy of several types of immunotherapy.27,28,29
Anecdotally, use of immune checkpoint blockade (ICB) to treat FLC has provided evidence that some FLC patients benefit from immunotherapy. A recent multicenter retrospective study considered 19 FLC patients treated with ICB and reported partial responses in 3 of 19 (15.8%) total patients and 2 of 15 (13.3%) patients who received ICB alone.30 Notably, two case reports have also described complete31 or near-complete32 responses to combination ipilimumab and nivolumab therapy. Like many other pediatric cancers, FLC is characterized by a very low tumor mutational burden (TMB) with a median of 1.85 mutations per megabase, suggesting a limited number of putative neoantigens aside from those derived from the fusion.30 We hypothesized that responses to ICB in FLC might be driven by endogenous T cell responses to the DNAJB1-PRKACA fusion. However, these endogenous responses have never been directly characterized.
Studies of endogenous fusion-specific T cell responses in FLC are critical to inform rational design of immunotherapy for this disease. The prevalence and extent of these endogenous responses dictate what proportion of the FLC patient population may respond to which types of immunotherapy. Furthermore, identification of even relatively few responses could benefit patients by supporting development of cellular therapies using T cell receptor-transgenic (TCR-T) cells.33,34,35,36 TCR-T therapy can both boost the magnitude of ineffective endogenous responses as well as offer an opportunity to equip engineered T cells to better survive the hostile tumor microenvironment.37,38,39,40 Finally, investigating the fusion-specific TCR repertoire may also illuminate general principles of neoantigen recognition that extend to cancers beyond FLC.
In this study, we aimed to characterize endogenous CD8 T cell responses to DNAJB1-PRKACA fusion neoantigens in FLC patients. We first explored the fusion neoantigen landscape in FLC to investigate how well DNAJB1-PRKACA neoantigens can be presented to CD8 T cells. We then measured endogenous T cell responses to the fusion in patients naive to immunotherapy. Interestingly, our TCR repertoire analyses highlighted potential differences in how T cells respond to FLC neoantigens compared with viral antigens, which may partially explain why endogenous T cell responses are not sufficient to clear tumors. Last, we evaluated fusion-specific TCRs defined in this study for potential use in TCR-T therapy for FLC and identified one TCR with superior anti-tumor activity in vivo. The results of these studies provide foundational data to inform rational development of neoantigen-targeted immunotherapies.
Results
T cell infiltration and sustained HLA expression in FLC tumors
We first investigated the immune microenvironment and likelihood of fusion neoantigen presentation in FLC patients. Others have previously studied immune cell infiltration in FLC tumor tissue via immunohistochemistry (IHC)41 and observed both CD8 T cells and myeloid cells in patient tumors. Consistent with these observations, we detected both CD3+CD8+ T cells (21% of CD3+ cells) and CD3+CD8− T cells (likely CD4+ T cells, 79% of CD3+ cells) in patient tumor tissue via immunofluorescence (Figure 1A). These T cells were present in small numbers overall, suggesting that ex vivo expansion could be necessary for in-depth characterization of tumor-infiltrating T cells in FLC. Also consistent with previous reports,42 we observed CD68+ cells, potentially tumor-infiltrating macrophages (Figure 1A).
Figure 1.

T cell infiltration and sustained HLA expression in FLC tumors
(A) Immunofluorescence of FLC-SJ5 tumor tissue. Blue, DAPI; red, CD3; green, CD8; white, CD68. Shown is a bar plot quantification of CD3+CD8+ T cells vs. CD3+CD8− T cells.
(B) HLA expression levels from 24 FLC patients, classified by sample type. NML, tumor-adjacent non-malignant liver; LIV, primary or recurrent liver tumor; MET, metastatic tumor. Samples from patients with matched samples across types are connected by colored lines, coded by patient ID. A dotted red line shows median HLA expression for healthy liver tissue reported by the GTEx Portal. Wilcoxon rank-sum test; ns, padj > 0.05.
(C) HLA expression levels from the patient cohort, plotted by patient and color-coded by sample location.
(D) Correlation between HLA expression and the number of predicted fusion neoepitopes for the patient cohort. R indicates the Pearson correlation coefficient.
Having confirmed that T cells are present in FLC tumor tissue, albeit in small numbers, we investigated potential neoantigen presentation via human leukocyte antigen (HLA) class I using publicly available RNA sequencing (RNA-seq) data from 24 FLC patients18,43 (Table S1). Tumor samples were taken from primary, recurrent (in liver), or metastatic FLC lesions. Four patients (FLC06, FLC09, FLC27, and FLC34) had matched tumor-adjacent non-malignant liver (NML) samples, and three patients (FLC06, FLC18, and FLC27) had matched samples from different tumor sites. We compared expression of HLA-A, -B, and -C across all samples and observed no significant differences in HLA expression between any tumor site in comparison with another tumor site or with NML (Figure 1B). To rule out any effect of proximity to tumor tissue in the NML data, we compared the FLC data with the median HLA-A, -B, and -C expression reported for healthy liver tissue in the GTEx Portal (www.gtexportal.org) (red dashed line in Figure 1B) and found that the FLC values were comparable with or even higher than those reported for healthy liver. There was greater variability in HLA expression in metastatic samples; however, we observed no trend in expression related to source tissue of the metastatic sample. Rather, variance in HLA expression appeared to arise from patient-to-patient variability (Figure 1C). Together, these results indicate that HLA class I expression in FLC tumors is not downregulated in comparison with normal liver tissue.
We then determined the HLA types of the 24 patients and predicted fusion neoantigen binding to their respective class I HLA alleles. While we might expect greater fusion neoantigen presentation to select for reduced HLA expression, we found no such correlation between the number of predicted fusion neoepitopes and HLA expression (Figure 1D). This result is in concordance with our comparison of expression across tumor and normal liver samples and, together with our immunofluorescence imaging, suggests that, while neoantigen-specific CD8 T cells may be present in FLC tumors, they do not instigate a sufficient endogenous response to clear tumors or exert strong selection on tumor clones.
Fusion-derived neoepitopes can be presented by diverse HLAs
We next explored potential immunogenicity of DNAJB1-PRKACA fusion peptides in a broader context. We first predicted binding of 84 peptides (8- to 15-mers, tiling across the fusion breakpoint) to 2,924 class I HLA alleles. 41 (48.8%) of these peptides were predicted to bind at least one allele at an affinity ≤500 nM (Figure S1A), while 1,837 alleles (62.8%) were predicted to bind at least one fusion peptide at ≤500 nM (Figure S1B). Notably, some class I HLA allotypes, including alleles of A24, A68, and B40, were predicted to bind 200 or more neopeptides. We also predicted binding of 75 fusion peptides (13- to 22-mers, tiling across the fusion breakpoint) to 54 class II HLA alleles. 35 (46.7%) of these peptides were predicted to bind at least one class II allele at ≤1,000 nM (Figure S1C), and 15 (27.8%) alleles were predicted to bind at least one fusion peptide at ≤1,000 nM (Figure S1D).
Due to the greater frequency of putative class I-restricted epitopes and higher likelihood of tumor cell neoantigen presentation via HLA class I, we then focused on two relevant sets of class I HLAs: alleles for which peptide-HLA multimer reagents are commercially available and alleles expressed by the above cohort of 24 FLC patients (Figure 2A). 16 commercially available alleles were predicted to bind at least one fusion peptide (Figure S1C; Table S2). We consulted the AlleleFrequencies.net database44 to determine the reported frequency of these alleles among US populations (Figure 2B) and biochemically verified 38 fusion neoepitopes that could bind and stabilize these HLA allotypes (Figures 2C and 2D). Based on reported HLA allelic frequencies, we estimate that these fusion neoepitopes could cover over 90% of the US population (sum of mean frequencies of two most frequent alleles for each class I locus = 99.5%), depending upon ancestry.
Figure 2.

Fusion neoantigens bind to diverse HLAs and can be detected among eluted HLA-bound peptides
(A) Frequency of class I HLAs expressed by 24 FLC patients.
(B) Frequency of 16 class I HLA alleles predicted to bind fusion neoepitopes, as reported by AlleleFrequencies.net for US populations, separated by six major racial/ethnic groups.
(C) Representative data from the biochemical HLA binding assay for fusion neoantigens predicted for HLA-A∗68:02.
(D) Summary of biochemical binding assay results across all 16 HLAs. See also Figure S1C.
(E) Number of fusion neoepitopes predicted for each patient in the cohort, color coded by results of the biochemical binding assay.
(F) Fragmentation spectrum of the EIFDRYGEEV peptide eluted from the A∗68:02 K562 cell line. See also Figure S2.
Next, we returned to our FLC patient cohort and classified each patient’s predicted fusion neoepitopes using the results of our peptide-HLA binding assays (Figure 2E). 22 of the 24 patients (91.7%) were predicted to present at least one fusion peptide, with some patients predicted to present 10 or more. Predictions for 20 of the 24 patients (83.3%) included at least one fusion peptide verified to bind HLA in our biochemical assays, including predictions of verified strong binders in 13 patients (54.2%). Together, these data indicate that a majority of FLC patients express class I HLA alleles that can bind to DNAJB1-PRKACA fusion-derived peptides and that this fusion could therefore be immunogenic in many patients.
To directly test whether predicted fusion neoantigens are presented by class I HLA allotypes, we chose three HLA alleles that are highly represented in US populations (A∗11:01, A∗24:02, and C∗07:02) and two alleles with high numbers of predicted fusion neoepitopes (A∗68:02 and B∗40:01) for validation via immunopeptidomics. A total of 19,095 peptides were identified among HLA class I eluates across monoallelic K562 cell lines stably expressing each of these HLAs and the DNAJB1-PRKACA fusion (Figure S2A). The identified peptides followed a canonical class I length distribution of 8–12 amino acids (Figure S2B) and agreed with reported consensus binding motifs for each HLA allotype (Figures S2C–S2G).45
Peptide elution data were also interrogated for the presentation of peptides from the DNAJB1-PRKACA fusion. Notably, a 10-mer peptide (EIFDRYGEEV) spanning the fusion junction was identified exclusively in the cell line expressing HLA-A∗68:02 (Figure 2F). This neopeptide matched the binding motif of A∗68:02 (Figure S2E; Asn at P1 and Val at P9). No additional bound fusion peptides were detected on the remaining four HLA alleles. Together, these data provide compelling evidence for presentation of A∗68:02-EIFDRYGEEV, in agreement with reports from Bauer et al.,46 who also detected this peptide in similar experiments.
Endogenous anti-fusion T cell responses are a source of rare fusion-specific TCRs
We then asked whether FLC patients could mount functional T cell responses to predicted neoantigens derived from the DNAJB1-PRKACA fusion. Having defined a bona fide fusion neoepitope presented on HLA-A∗68:02, we began our studies using samples from A∗68:02+ patient FLC-SJ1 (Figure 3A; Table S1). We predicted seven fusion neopeptides for this patient’s class I HLA alleles. Stimulating expanded tumor-infiltrating lymphocytes (TILs) with EIFDRYGEEV peptide, the same as detected by immunopeptidomics, resulted in robust interferon γ (IFNγ) and tumor necrosis factor alpha (TNFα) production by CD8+ TILs (Figure 3B), while none of the other peptides, including 13- to 15-mer peptides that could be presented by HLA class II, elicited functional responses in either CD8+ or CD4+ TILs (Figure S3D). These results confirm that FLC patients can mount endogenous T cell responses against the DNAJB1-PRKACA fusion.
Figure 3.

Functional responses and TCRs directed against A∗68:02-EIFDRYGEEV among FLC patient TILs and PBMCs
(A) Schematic for the experiments. Created using BioRender.
(B) Intracellular cytokine staining for IFNγ and TNFα of FLC-SJ1 TILs stimulated with the indicated fusion peptides or control reagents.
(C) UMAP plots highlighting expression of IFNG and SJ1-4 TCR in SJ1 TILs after stimulation with EIFDRYGEEV.
(D) Differential expression of select genes between TILs expressing SJ1-4 TCR vs. all others in the dataset. See also Table S3.
(E) A∗68:02-EIFDRYGEEV tetramer staining of SJ1 PBMCs after expansion (STAR Methods).
(F) Frequency of TCR clonotypes among tetramer-positive cells from (E).
We next aimed to identify fusion-specific TCRs in patient SJ1. Stimulation of patient TILs with EIFDRYGEEV peptide followed by single-cell gene expression and paired TCR sequencing identified a small cluster of cells with high expression of IFNG that shared the same TCR clonotype, designated SJ1-4 (Figure 3C). Compared with all other T cells in the dataset, cells expressing SJ1-4 upregulated multiple genes associated with an activated immune response, including genes recently reported by Lowery et al.47 to be associated with tumor-specific CD8 T cells (e.g., GALNT2, NELL2, and MIR155HG; Figure 3D; Table S3). From these results, we hypothesized that SJ1-4 was specific for the fusion neoantigen EIFDRYGEEV.
We then sought to identify additional fusion-specific TCRs from patient SJ1 first by single-cell sequencing unexpanded CD8+ T cells sorted from peripheral blood, including a population labeled with the A∗68:02-EIFDRYGEEV tetramer. Although we could detect tetramer-positive cells among unexpanded peripheral blood mononuclear cells (PBMCs; Figures S3D and S3E), clonal expansions in the overall dataset were small (Figure S3F), and we detected no clonal expansions among the tetramer-positive cells (Figure S3G). Because this result potentially indicated non-specific tetramer binding, we next aimed to more confidently identify fusion-specific cells after expanding patient SJ1 PBMCs, as described by Cimen Bozkus et al.,48 using a pool of fusion peptides predicted to bind HLA-A∗68:02. After staining expanded PBMCs with A∗68:02 tetramers, we again observed a population of T cells positive for the A∗68:02-EIFDRYGEEV tetramer (Figure 3E). 93% of these cells shared the same TCR, designated SJ1-12 (Figure 3F), which, we hypothesized, could also be specific for fusion neoantigen EIFDRYGEEV.
We applied similar approaches to search for fusion-specific T cells in other FLC patients, designated FLC-SJ2, FLC-SJ3, and FLC-SJ4 (Table S1). TILs from patients SJ2 and SJ3 were expanded ex vivo, while tumor-infiltrating T cells from patient SJ4 were sequenced without any prior expansion. For each patient, we stimulated T cells with fusion peptide to induce changes in gene expression in fusion-specific TILs as we did for patient SJ1 and then conducted single-cell gene expression and paired TCR sequencing. Across all 4 patients, we obtained 10,577 unique paired TCRs (Figure 4A) and selected multiple candidate fusion-specific TCRs from each patient based on clonal expansion and expression of the activation markers IFNG and TNFRSF9 (4-1BB).
Figure 4.

Rare endogenous CD8 T cell responses pose challenges to fusion-specific TCR identification in multiple FLC patients
(A) Summary of experiments conducted using expanded TIL and tumor samples from four FLC patients (see also Table 1). Samples were stimulated by the indicated fusion peptide prior to single-cell gene expression and paired TCR sequencing. Pie charts represent TCR clonal expansions; expanded pie charts (where present) represent the top 20 most frequent clonotypes and indicate the overall frequency of the most frequent clone (dark blue). UMAP plots highlight expression of IFNG. FLC-SJ1, 490 cells; FLC-SJ2, 5,619 cells; FLC-SJ3, 3,212 cells; FLC-SJ4, 3,016 cells.
(B) Frequency of A∗68:02-EIFDRYGEEV tetramer-positive Jurkat cells expressing SJ1 candidate fusion-specific TCRs.
(C) Normalized frequency of CD69-positive Jurkat cells expressing candidate fusion-specific TCRs from all patients in (A) (see STAR Methods for calculation).
(D) TCRdist network of 8,270 unique, paired TCRs from SJ1. Each node represents a unique TCR clonotype, and two nodes are connected by an edge when their TCRdist < 100; node size corresponds to node degree (number of neighbors). The large central cluster represents mucosal-associated invariant T (MAIT) cells. SJ1-4 and SJ1-12 TCRs are highlighted.
See also Figures S4 and S5.
A total of 25 TCRs derived from CD8+ T cells across all four patients were selected for additional validation (12 from SJ1, 6 from SJ2, 4 from SJ3, and 3 from SJ4; Table S4). We reconstructed the full-length TCR sequences and expressed each in TCR-null 2D3 Jurkat cells for preliminary validation using tetramer staining with A∗68:02-EIFDRYGEEV (Figure 4B) or a functional assay (Figure 4C). Surprisingly, of all 25 candidates, only SJ1-4 and SJ1-12 proved to be fusion specific. No TCRs from the three additional patients passed validation, and even many ostensibly tetramer-positive TCRs from patient SJ1 failed to bind tetramer upon reconstruction. These results highlight the importance of such validation experiments and indicate that endogenous responses to the DNAJB1-PRKACA fusion may be rare. Notably, patients SJ2, SJ3, and SJ4 had fewer and poorer fusion neoepitope predictions than patient SJ1 (Tables S1 and S2), which could explain why we were unable to identify fusion-specific responses in these patients.
We then attempted to discover additional fusion-specific TCRs in patient SJ1 using a TCR repertoire-based approach. It is well established that TCRs that recognize the same peptide-HLA epitope frequently share amino acid sequence features.49 Our group50,51 and others52 have developed metrics for quantifying this similarity and identifying closely related TCR sequences from TCR repertoires. Across all experiments, we obtained 8,270 unique paired TCR sequences from patient SJ1, so we sought to determine whether any other TCRs within this repertoire shared sequence features with SJ1-4 or SJ1-12. To this end, we applied TCRdist50,51 to quantify pairwise sequence similarity across all TCRs sequenced from patient SJ1. We then constructed a network in which each node represents a unique TCR sequence and two nodes are connected by an edge when their TCRdist similarity score is below a threshold of TCRdist = 100 (Figure 4D). While we did observe some clusters of TCRs with sequence similarities, we found that both SJ1-4 and SJ1-12 were structurally distinct, as neither had any close neighbors in TCR sequence space. We did select and reconstruct 2 additional TCRs belonging to a cluster of related sequences (SJ1-298 and SJ1-1460; Figure S4A; Table S4), neither of which proved to be fusion specific (Figure 4C). Although we have not exhaustively profiled all TCRs sequenced from this patient, the absence of any TCR sequences clustered with either SJ1-4 or SJ1-12 provides additional evidence that, among immunotherapy-naive patients, endogenous responses to the DNAJB1-PRKACA fusion are rare and challenging to detect, particularly within peripheral blood.
Interestingly, we have previously observed that, in viral infections,50,53,54,55 effective T cell responses against immunodominant epitopes typically involve large clusters of TCRs with closely related sequences. Indeed, large clusters of structurally similar TCRs are present among hepatitis C virus (HCV) tetramer-positive T cells from an HCV-infected patient who spontaneously resolved their infection56 (Figure S5A). Tetramer-positive TCRs from patient SJ1, on the other hand, form no clusters at all. Clustering of previously published TCR sequence data from peripheral blood of a severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)-infected donor57 also resulted in large clusters of SARS-CoV-2-specific TCRs, particularly at the peak of infection (day 15; Figures S5B–S5E). Clustering of SARS-CoV-2-specific TCRs was even more pronounced when examining repertoires from a site of antigen presentation (i.e., lymph nodes of SARS-CoV-2-vaccinated donors)54 (Figures S5F and S5H). However, clustering of tumor-associated TCR sequences from all four FLC patients sequenced (Figures S4, S5F, and S5G) demonstrated that none of these patients mounted a T cell response involving large clusters of related sequences, even at the site of tumor antigen presentation. Together, these results suggest that, while rare fusion-specific TCRs can be identified in certain patients, the endogenous fusion-specific response in these four patients seemed to lack features of effective antiviral responses, which might have prevented effective tumor clearance in these patients, and highlights a potential challenge in defining fusion-specific TCRs in immunotherapy-naive patients.
SJ1-4 and SJ1-12 TCRs are fusion specific, functional, and cytotoxic in vitro
We next sought to confirm the specificity and function of the SJ1-4 and SJ1-12 TCRs in vitro. When expressed in Jurkat cells, both TCRs specifically bound their cognate A∗68:02 tetramer (Figure 5A), although the intensity of tetramer staining was lower for SJ1-12. Both TCRs also mediated cytokine responses to A∗68:02 artificial antigen-presenting cells (aAPCs) pulsed with their cognate fusion peptide (Figures 5B and S6D). Notably, both SJ1-4 and SJ1-12 TCRs exhibited some cross-reactivity to the corresponding wild-type (WT) peptide (EIFDRYGEEG), which diminished as WT peptide concentration decreased (Figures 5B and S6D). We hypothesized that this result could be explained by differential affinity for HLA between the WT and fusion peptides. Indeed, when we tested the ability of each peptide to stabilize an empty A∗68:02 monomer, we found that the fusion peptide bound similarly to a known strong-binding peptide, while the WT peptide bound poorly (Figure S6E). Furthermore, the WT peptide sequence was not detected among A∗68:02-bound peptides in our immunopeptidomics experiments. The canonical peptide binding motif for A∗68:02 (Figure S2E) shows that Val is the preferred C-terminal anchor residue for this HLA, so the mutation from Gly to Val likely accounts for the stronger fusion peptide binding. Accordingly, when we stimulated cells expressing either TCR with aAPCs transduced to express the DNAJB1-PRKACA fusion (which needed to endogenously process and present the relevant peptides), we still observed a strong fusion-specific response but minimal reactivity to targets expressing the WT proteins (Figures 5C and S6F). Together, these results suggest that the tumor specificity of both TCRs depends less upon differential recognition of peptide and more upon differential fusion and WT peptide presentation.
Figure 5.

SJ1-4 and SJ1-12 TCRs are fusion specific, functional, and able to kill fusion-positive target cells in vitro
(A) A∗68:02-EIFDRYGEEV tetramer staining of SJ1-4 and SJ1-12 TCRs reconstructed and expressed in TCR-null Jurkat cells.
(B) Normalized frequency of IFNγ+TNFα+ cells among SJ1-4- or SJ1-12-transduced primary human T cells (donor Aph34) after stimulation with increasing doses of fusion (EIFDRYGEEV) or WT peptide (EIFDRYGEEG) (see STAR Methods for calculation). Data for each TCR were collected in separate single experiments, each using 3 PBMC donors. See also Figure S6D.
(C) Frequency of IFNγ+TNFα+ cells among SJ1-4 or SJ1-12 primary human T cells after stimulation with aAPCs expressing DNAJB1-PRKACA fusion or WT transgenes. All data were collected in the same single experiment using 3 PBMC donors. See also Figure S6F.
(D) xCelligence assay measuring SJ1-4 or SJ1-12 primary human T cell killing of fusion- or WT-expressing target cells. Target cells adhered for 24 h before addition of T cells at effector:target ratios of 20:1, 5:1, and 1.25:1 (mean ± SD across technical triplicates). Cell index was normalized to 1 at the time of T cell addition. Killing is indicated by a decrease in cell index as target cells die and lift from the plate. SJ1-12 and mock-transduced control data were collected in the same plate; SJ1-4 data were collected in a separate plate with an additional set of mock-transduced controls (comparable with those shown). A t test on normalized cell index values was performed at the final time point. ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001; ns, p > 0.05. See also Figure S6G.
(E) Frequency of cleaved caspase-3+ wells in the Berkeley Lights Lightning assay co-culturing a single SJ1-4 or SJ1-12 primary human T cell with a single fusion- or WT-expressing aAPC for 24 h. Data for SJ1-12 and mock-transduced controls were collected in the same experiment; data for SJ1-4 were collected in a separate experiment with an additional set of mock-transduced controls (comparable with those shown). Fisher’s exact test was performed on the proportion of caspase+ cells. ∗∗p < 0.01; ns, p >0 .0.05. See also Figure S6H.
(F) Frequency of IFNγ, IL-2, and TNFα production during same Berkeley Lights Lightning assay. Fisher’s exact test was performed on proportions of cytokine+ cells (i.e., single, double, or triple cytokine+). ∗∗p < 0.01; ns, p >0 .0.05.
We then performed in vitro cytotoxicity assays against target cells expressing A∗68:02 and either the DNAJB1-PRKACA fusion protein or WT DNAJB1 or PRKACA. In a bulk killing assay using the xCelligence platform, we observed minimal killing by mock-transduced T cells, regardless of whether targets expressed the WT or fusion transgenes. In agreement with functional assays, both SJ1-4 and SJ1-12 T cells killed fusion-expressing target cells at a variety of effector-to-target (E:T) ratios while largely sparing WT-expressing targets (Figures 5D and S6G). Notably, SJ1-12 appeared to be less effective than SJ1-4, particularly at low E:T ratios.
We also used the Berkeley Lights Lightning platform to assay cytotoxicity on an individual-cell level. In agreement with bulk killing assays, we observed low killing by mock-transduced T cells and preferential killing of fusion-expressing targets by both SJ1-4 and SJ1-12 T cells (Figures 5E and S6H), with a slightly lower frequency of killing by the SJ1-12 TCR. We also assessed cytokine production by individual T cells during the Lightning assay and found that, while few mock-transduced T cells produced cytokines, both SJ1-4 and SJ1-12 T cells produced multiple cytokines specifically in response to fusion-expressing targets (Figure 5F). Interestingly, SJ1-4 T cells appeared to be more polyfunctional than SJ1-12 T cells; approximately half of SJ1-4 cytokine-producing cells simultaneously produced IFNγ, TNFα, and interleukin-2 (IL-2), while no SJ1-12 cells were triple producers. Altogether, the in vitro studies confirm the specificity of SJ1-4 and SJ1-12 for fusion over the WT and demonstrate that these TCRs are both functional and cytotoxic in vitro. Further, comparisons between the two TCRs suggest that SJ1-4 may exhibit greater affinity for its cognate neoantigen and greater functionality than SJ1-12.
SJ1-4 T cells eliminate fusion-positive tumor cells in vivo
Finally, we tested whether our fusion-specific TCRs could control growth of fusion-expressing tumor cells in vivo (Figure 6A). Because no FLC cell lines are currently commercially available, and because lung metastases are a significant cause of morbidity and mortality in FLC,2 we chose to use a systemic A549 lung carcinoma tumor model58 modified to express luciferase and GFP to permit tumor visualization in vivo and ex vivo. We further modified the cells to express both HLA-A∗68:02 and DNAJB1-PRKACA fused to tagBFP to facilitate identification of neoantigen-positive cells. Notably, neither of these modifications was required for the tumor cells to grow in an immunodeficient mouse.
Table 1.
SJFLC patient cohort characteristics
| Patient ID | Sample type | Sample location | Age | Gender | Class I HLA type | Number of predicted fusion neoepitopes |
|---|---|---|---|---|---|---|
| FLC-SJ1 | expanded TILs | primary tumor |
21 | F | A∗31:01, A∗68:02, B∗35:02, B∗38:01, C∗04:01, C∗12:03 |
10 |
| PBMC | peripheral blood | |||||
| FLC-SJ2 | expanded TILs | primary tumor |
19 | F | A∗02:19, A∗23:01, B∗15:01, B∗14:01, C∗03:02, C∗05:01 |
5 |
| FLC-SJ3 | expanded TILs | ascites | 19 | F | A∗03:01, A∗23:01, B∗44:03, B∗55:01, C∗03:03, C∗04:01 |
6 |
| FLC-SJ4 | tumor | metastatic tumor | 15 | M | A∗01:01, A∗02:01, B∗37:01, B∗44:02, C∗05:01, C∗06:02 |
2 |
| FLC-SJ5 | tumor | primary tumor | 16 | F | A∗01:01, A∗32:01, B∗07:02, B∗44:02, C∗05:01, C∗07:02 |
5 |
Figure 6.

SJ1-4 T cells control growth of fusion-expressing tumors in vivo and drive fusion-negative recurrences
(A) Schematic for in vivo experiments. Created using BioRender.
(B) IVIS images of tumor burden in mice treated with SJ1-4 T cells, mock-transduced T cells, or PBS.
(C) Tumor radiance measured by in vivo bioluminescence imaging for the first 21 days of study; n = 5 animals/group. A dashed line represents background bioluminescence (approximately 106 photons/second). A red arrow indicates the day of T cell administration. 2-way ANOVA on log-transformed radiance values; ∗∗∗ padj < 0.001, ∗∗∗∗ padj < 0.0001.
(D) Survival curves for the duration of the study, n = 5 animals/group. Log rank test; ∗p < 0.05, ∗∗p < 0.01.
(E) Representative immunofluorescence of tumors harvested at euthanasia from mice treated with mock-transduced or SJ1-4 T cells. Blue, DAPI; green, GFP (tumor cells); red, tagBFP (cells expressing DNAJB1-PRKACA fusion); yellow, mCherry (transduced T cells).
(F) Quantification of GFP median fluorescence intensity (MFI) (left), tagBFP MFI (center), and number of mCherry+ T cells per μm2 × 104 (right). Wilcoxon rank-sum test; ∗ padj < 0.05, ∗∗ padj < 0.01, ∗∗∗ padj < 0.001, ∗∗∗∗ padj < 0.0001.
(G) Frequency of mCherry+ CD8 T cells in kidneys, livers, lungs, and spleens harvested at euthanasia from mice treated with mock-transduced or SJ1-4 T cells. Wilcoxon rank-sum test; ns, p > 0.05. See also Figures S7B–S7I.
After intravenous administration of tumor cells, imaging indicated that nearly all mice had established lung tumors by day 7 (the lack of a tumor in one mouse in the PBS group on day 7 is likely an imaging artifact). Mice were treated on day 8 with either SJ1-4 T cells, mock-transduced T cells, or vehicle. On day 12, we observed a striking decrease in tumor radiance among SJ1-4-treated mice (Figures 6B and 6C), while tumor growth continued unrestrained in both control groups (Figure 6B). By day 21, however, tumors recurred in all SJ1-4-treated mice, although tumor radiance continued to be significantly lower for these mice than for mice in either control group (Figure 6C), and survival was significantly extended in SJ1-4-treated mice compared with both vehicle-treated and mock T cell-treated groups (Figure 6D).
We explored several possibilities that could explain tumor recurrence in SJ1-4-treated mice. We first considered that there could be differences in T cell phenotype across treatment groups but observed no significant differences in expression of the activation marker 4-1BB or in the surrogate exhaustion markers PD-1 and TIM-3 (Figures S7B–S7E), ruling out T cell exhaustion as a likely cause of recurrence. We did observe a significantly greater frequency of effector memory T (TEM) cells (CCR7−CD45RO+) and a concomitant reduction in other phenotypes in tissues from SJ1-4-treated mice compared with controls (Figures S7F–S7I). However, this change likely only reflects that, as expected, SJ1-4 T cells recognized their cognate neoantigen, while mock-transduced T cells did not.
We also considered that recurrent tumors could consist of clones that lacked expression of the target neoantigen. Immunofluorescence imaging of tumors harvested from mice treated with mock-transduced T cells exhibited high (though somewhat variable) expression of the DNAJB1-PRKACA fusion neoantigen. Tumors from mice treated with SJ1-4 T cells, however, were strikingly devoid of neoantigen expression (Figures 6E and 6F). Recurrent lesions from SJ1-4-treated mice were also devoid of transduced T cells, although transduced T cells did persist in these mice both in tumor-bearing tissues and spleens (Figure 6G) and even induced graft versus host disease (GVHD) in some animals. These results demonstrate that, although SJ1-4 T cells did not completely clear tumors, they did exert sufficient selection pressure on fusion-expressing tumors to drive recurrences that lacked expression of the target neoantigen.
We then repeated the in vivo study with an expanded cohort, including a group of mice treated with SJ1-12 T cells and additional control groups of mice bearing tumors that lacked expression of HLA-A∗68:02 (no-HLA groups; Figure S7). Based on our observations of neoantigen-negative tumor recurrence in the first study, we also sorted tumor cells on tagBFP (i.e., DNAJB1-PRKACA fusion) expression, allowing approximately 2 weeks of post-sorting expansion prior to injection into mice. Unlike SJ1-4 T cells, SJ1-12 did not appear to provide any benefit over control treatments (Figures S7J and S7K), consistent with lower SJ1-12 tetramer binding (Figure 5A) and lower efficacy in in vitro killing assays (Figures 5D–5F). As expected, neither TCR mediated killing of A∗68:02-negative tumor cells, confirming that the anti-tumor T cell activity was HLA restricted.
As in the first experiment, we observed a striking reduction in tumor radiance among mice treated with SJ1-4 T cells (Figures S7J and S7K). Further, this reduction in tumor radiance persisted approximately 13 days longer than in the initial experiment (Figures 6B, 6C, S7J, and S7K; first evidence of recurrence on days 12–16 in the first experiment vs. day 25 in the second), likely due to injection of a purer population of neoantigen-positive tumor cells after sorting. After day 25 post tumor injection, we began to observe non-specific anti-tumor activity in mock-treated mice and later observed GVHD among both TCR-treated and mock-treated mice. Both phenomena are documented in the literature59,60,61 and resulted in non-significant differences in survival across groups. Nevertheless, these results together demonstrate that SJ1-4 is capable of thoroughly eliminating fusion-positive tumor cells in vivo and thereby provide support for development of cellular therapies for FLC.
Discussion
In this study, we define endogenous CD8 T cell responses to the DNAJB1-PRKACA driver fusion in FLC. There has been some speculation about the prevalence of endogenous neoantigen-specific immune responses in low-TMB tumors,62,63 including FLC. However, we have shown that, in a cohort of pediatric B cell acute lymphoblastic leukemia (B-ALL), patients, all patients mounted neoantigen-specific T cell responses, including responses to the ETV6-RUNX1 fusion.64 Similarly, our results indicate that anti-fusion CD8 T cell responses are theoretically possible in a large proportion of FLC patients. We defined 38 fusion neoantigens verified to bind to at least one of 16 class I HLA alleles, which could cover more than 90% of the US FLC patient population, depending upon ancestry. This estimate is concordant with HLA typing and neoepitope prediction for a cohort of 24 FLC patients, over 90% of whom were predicted to present at least one fusion neoepitope. Analysis of class I HLA expression in tumor and tumor-adjacent NML samples from the same cohort also indicated that there is little difference in the degree of HLA expression between FLC tumors and NML. This result implies that little endogenous immune selection occurs in FLC (in agreement with our observation of low CD8 T cell infiltration in patient tumor tissue) but that FLC tumors can, in principle, still present neoantigens and therefore be vulnerable to T cell killing.
Previous studies have demonstrated a positive correlation between predicted driver mutation presentation by class I and response to ICB,65 while others have shown that recurrent cancer mutations tend to be poorly presented by class I in general and that HLA alleles that can present recurrent mutations are underrepresented among affected patients.66 Together with our results, these prior observations suggest that the endogenous immune response to the DNAJB1-PRKACA driver fusion may be small, potentially due to low fusion presentation or a poor priming environment. We did not identify any other validated T cell responses in four FLC patients, although 25 candidates were screened. However, an important limitation of our study is that our analyses were focused on samples from only four patients at a single time point. Additional studies of larger FLC cohorts will be necessary to confirm whether the low fusion-specific CD8 T cell response we observed holds true in general. Our sample size in this study was also insufficient to determine whether fusion-presenting class I alleles are underrepresented in FLC. A larger study of HLA types in FLC patients would provide insight into whether HLA allele frequency correlates with poor fusion presentation and therefore might limit endogenous immune responses against the DNAJB1-PRKACA fusion.
Notably, neither of the fusion-specific TCRs identified in this study belonged to a cluster of related TCR sequences, indicating that, while patient SJ1 did mount an anti-fusion T cell response, that response lacked characteristics common in immunodominant antiviral T cell responses.50,54,55 The other patients sequenced in this study also did not mount detectable clustered responses. If recruitment of multiple structurally similar TCRs is a hallmark of effective T cell responses, this lack of fusion-specific TCR clustering could partially explain why endogenous anti-fusion responses failed to control tumor growth in the patients studied. Further work will be necessary to determine whether this lack of neoantigen-specific TCR clustering occurs in the broader FLC patient population or in other cancers. Interestingly, Puig-Saus et al.67 recently reported that the total number of neoantigen-specific TCR clonotypes correlated positively with response to PD-1 checkpoint blockade in metastatic melanoma patients, suggesting that more diverse neoantigen-specific T cell responses are beneficial. It remains to be determined whether other neoantigen-specific TCR repertoire features, including TCR clustering, also correlate with response to ICB or other therapies. However, if they do, then measures of TCR repertoire clustering could serve as novel biomarkers for response to immunotherapy.
Several factors could play a role in restraining endogenous fusion-specific T cell responses in FLC, including neoantigen density, neoantigen-specific precursor frequency, TCR affinity for cognate neoantigens, and factors in the FLC tumor microenvironment. Although FLC is reported to be a low-TMB tumor,30 it is possible that other neoantigens are immunodominant over fusion neoantigens, resulting in poor response to the fusion. However, lack of any large clusters of related TCRs (apart from mucosal-associated invariant T [MAIT] cell clusters) suggests an overall low anti-tumor response rather than a response against a different neoantigen. Alternatively, the precursor frequency of fusion-specific cells may be low in many patients, or fusion-specific precursors may lack sufficient avidity to effectively attack tumor cells. The FLC tumor microenvironment could also suppress anti-tumor immune responses. Importantly, our study also focused exclusively on CD8 T cell responses; however, CD4 T cells also play an important role in anti-tumor immunity.68,69 Indeed, a recent study by Bauer et al.46 identified a putatively fusion-specific CD4 T cell response in a single FLC patient treated with a DNAJB1-PRKACA peptide vaccine in combination with poly(ADP-ribose) polymerase (PARP) inhibition. While fusion specificity of the TCRs was not directly validated, these results suggest that class II-restricted fusion neoantigens might also elicit T cell responses, at least in the context of peptide vaccination. Interestingly, we did not observe any strong CD4 T cell responses to fusion peptides in our study, even when using 13- to 15-mer (Figure S3D) or 24-mer (Figure 4A) peptides that could stimulate CD4 T cells. Neoantigen peptide vaccines have frequently been reported in the literature to preferentially stimulate CD4 T cell responses,70,71,72 which could explain why Bauer et al.46 identified a predominant CD4 response as opposed to the endogenous T cell responses that were the focus of our study.
The two fusion-specific TCRs identified in this study showed strong anti-fusion activity in vitro, although SJ1-12 appeared to have lower affinity for cognate neoepitope A∗68:02-EIFDRYGEEV than SJ1-4. Accordingly, SJ1-4 showed very promising activity in vivo, while the subtly poorer performance of SJ1-12 in vitro translated to profoundly poorer activity in vivo. Notably, the SJ1-4 TCR was found among tumor-infiltrating T cells, while the SJ1-12 TCR was induced from patient PBMCs, suggesting that tumor-infiltrating cells could represent a superior population of tumor-specific T cells compared with those found in the blood. Indeed, the tumor-infiltrating T cell population is believed to be enriched for tumor-specific T cells, which are found at much lower frequencies in peripheral blood.73,74,75 However, some studies have observed that peripherally expanded T cell clones can be tracked to tumor tissue and suggest that these T cells play an important role in responsiveness to ICB.76,77 While tumor-specific T cells may be more rare in the blood, employing antigen-specific expansion (as we did in this study) or analyzing T cell clonal dynamics across longitudinal samples from the same patient78 may allow reliable detection of tumor-specific TCRs in PBMC samples as well as tumors.
Together, our studies and others indicate that at least some FLC patients mount an anti-DNAJB1-PRKACA T cell response but that a boost in the magnitude and function of this response is likely necessary for anti-tumor efficacy. Cell therapy is one possible avenue for providing a boost in response, and our in vivo results support possible development of TCR-T therapies for FLC. Although tumors did recur in our model system, recurrent tumors strikingly lacked expression of the DNAJB1-PRKACA fusion, rendering them effectively invisible to fusion-specific T cells. Unlike our model tumor, which did not require fusion expression to grow, genuine FLC tumors are believed to be dependent on DNAJB1-PRKACA15 and are therefore unlikely to escape immune recognition by its loss.
A neoantigen vaccine combined with ICB (or potentially other interventions) offers an alternative means for boosting magnitude and function of endogenous anti-fusion T cell responses. As highlighted above, one such combination, a peptide vaccine targeting the fusion added to ongoing therapy with a PARP inhibitor, has already been used to induce a putative fusion-specific CD4 T cell response in one FLC patient.46 An ongoing clinical trial (ClinicalTrials.gov: NCT04248569) testing a fusion peptide vaccine in combination with nivolumab and ipilimumab will soon provide additional insight into how immunotherapy can boost T cell responses directed against the DNAJB1-PRKACA fusion. As these clinical studies progress, it is also worth noting that any fusion-specific TCRs, which likely will be identified more readily after immunotherapy, can also be fed forward to develop cellular therapies and that even a relatively small number of fusion-directed TCRs could potentially cover and improve outcomes for a large portion of the FLC patient population.
Limitations of the study
Our study has several important limitations. First, our analyses of class I HLA expression levels in FLC tumors and NML include a limited number of matched NML samples available and lack truly healthy (i.e., not tumor-adjacent) liver samples from matched patients. These limitations may bias our interpretation of NML HLA expression levels relative to FLC tumor tissue. Our use of a non-FLC tumor model for our in vivo experiments is another important limitation of our study and highlights the need to develop better preclinical models for this disease. Another limitation of our study is our focus on endogenous CD8 T cell responses over CD4 T cell responses. We cannot exclude the possibility that strong anti-fusion T cell responses occur endogenously in the CD4 compartment, and future studies should explore this possibility in more detail. Finally, our study is limited by the small size of our FLC patient cohort. Our findings may provide insight into anti-tumor immune responses in not only FLC but other tumor types as well; however, due to the limited sample size of our study, we approach drawing broad conclusions with caution. Further work, which could include both experimental studies with larger cohorts and meta-analyses of existing data, will be necessary to determine whether our observations about the overall frequency of fusion-specific CD8 T cell responses and the sparseness of neoantigen-specific TCR repertoires hold true outside of our small cohort.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rat anti-human/mouse/rat CD3 (clone CD3-12) | Abcam | ab255972 |
| Mouse anti-human CD8 (clone C8/144B) | Abcam | ab17147 (RRID:AB_443686) |
| Rabbit anti-human CD68 (clone EPR20545) | Abcam | ab213363 (RRID:AB_2801637) |
| Donkey anti-rat Alexa Fluor 647 (polyclonal) | ThermoFisher Scientific | A48272 (RRID:AB_2893138) |
| Donkey anti-mouse Alexa Fluor 488 (polyclonal) | ThermoFisher Scientific | A32766 (RRID:AB_2762823) |
| Donkey anti-rabbit Alexa Fluor 555 (polyclonal) | ThermoFisher Scientific | A32794 (RRID:AB_2762834) |
| Anti-human β2m PE (clone BBM.1) | Santa Cruz Biotech | sc-13565 (RRID:AB_626748) |
| Anti-human HLA-A,B,C PE (clone W6/32) | Biolegend | 311406 (RRID:AB_314875) |
| Anti-human HLA-A,B,C APC (clone W6/32) | Biolegend | 311410 (RRID:AB_314879) |
| Goat anti-mouse IgG PE (polyclonal) | Southern Biotech | 1030-09 (RRID:AB_2794297) |
| Anti-human Fc block (clone Fc1) | BD Biosciences | 564220 (RRID:AB_2728082) |
| Anti-human CD3 FITC (clone SK7) | Biolegend | 344804 (RRID:AB_2043993) |
| Anti-human CD8 Brilliant Violet 785 (clone SK1) | Biolegend | 344740 (RRID:AB_2566202) |
| Anti-human CD4 PerCP/Cy5.5 (clone SK3) | Biolegend | 344608 (RRID:AB_1953236) |
| Anti-human CD28 (clone CD28.2) | BD Biosciences | 555725 (RRID:AB_396068) |
| Anti-human CD49d (clone 9F10) | BD Biosciences | 555501 (RRID:AB_2130052) |
| Human TruStain FcX | Biolegend | 422302 (RRID:AB_2818986) |
| Anti-human CD8 FITC (clone SK1) | Biolegend | 344704 (RRID:AB_1877178) |
| Anti-human CD3 Brilliant Violet 421 (clone SK7) | Biolegend | 344834 (RRID:AB_2565675) |
| TotalSeq™-C0251 anti-human hashtag 1 (clone LNH-94, 2M2) | Biolegend | 394661 (RRID:AB_2801031) |
| TotalSeq™-C0252 anti-human hashtag 2 (clone LNH-94, 2M2) | Biolegend | 394663 (RRID:AB_2801032) |
| TotalSeq™-C0253 anti-human hashtag 3 (clone LNH-94, 2M2) | Biolegend | 394665 (RRID:AB_2801033) |
| TotalSeq™-C0254 anti-human hashtag 4 (clone LNH-94, 2M2) | Biolegend | 394667 (RRID:AB_2801034) |
| Anti-human CD11b Brilliant Violet 421 (clone ICRF44) | Biolegend | 301324 (RRID:AB_11219589) |
| Anti-human CD19 PE/Dazzle594 (clone HIB19) | Biolegend | 302252 (RRID:AB_2563560) |
| Anti-mouse TCRβ chain PE (clone H57-597) | Biolegend | 109208 (RRID:AB_31343) |
| Anti-mouse TCRβ chain APC/Fire750 (clone H57-597) | Biolegend | 109246 (RRID:AB_2629697) |
| Anti-human CD3 (clone OKT3) | Miltenyi | 130-093-387 (RRID:AB_1036144) |
| Anti-human CD28 (clone 15E8) | Miltenyi | 130-093-375 (RRID:AB_1036134) |
| Anti-human CD3 APC/Cy7 (clone SK7) | Biolegend | 344818 (RRID:AB_10645474) |
| Anti-human CD279 (PD-1) PE (clone EH12.2H7) | Biolegend | 329906 (RRID:AB_940483) |
| Anti-human CD366 (TIM-3) PE/Cy7 (clone F38-2E2) | Biolegend | 345014 (RRID:AB_2561720) |
| Anti-human CD197 (CCR7) FITC (clone G043H7) | Biolegend | 353216 (RRID:AB_10916386) |
| Anti-human CD45RA Brilliant Violet 421 (clone HI100) | Biolegend | 304130 (RRID:AB_10965547) |
| Anti-human CD45RO Brilliant Violet 650 (clone UCHL1) | Biolegend | 304232 (RRID:AB_2563462) |
| Anti-human CD3 Brilliant Violet 785 (clone SK7) | Biolegend | 344842 (RRID:AB_2616891) |
| Anti-human IFNγ Alexa Fluor 647 (clone 4S.B3) | Biolegend | 502516 (RRID:AB_493031) |
| Anti-human TNFα Brilliant Violet 605 (clone MAb11) | Biolegend | 502936 (RRID:AB_2563884) |
| Anti-human CD69 PerCP/efluor710 (clone FN50) | ThermoFisher Scientific | 46-0699-42 (RRID:AB_2573694) |
| Anti-human IL-2 PE (clone MQ1-17H12) | Biolegend | 500307 (RRID:AB_315094) |
| FastImmune™ CD28/CD49d (clone L293, L25) | BD Biosciences | 347690 (RRID:AB_647457) |
| LEGENDplex™ human Th panel detection antibodies | Biolegend | 741041 |
| Anti-human CD8a FITC (clone RPA-T8) | Biolegend | 301006 (RRID:AB_314124) |
| Anti-mouse TCRβ chain APC (clone H57-597) | Biolegend | 109212 (RRID:AB_313435) |
| Rabbit anti-GFP (polyclonal) | Rockland Immunochemicals | 600-401-215 (RRID:AB_828167) |
| Goat anti-mCherry (polyclonal) | Biorbyt | orb11618 (RRID:AB_2687829) |
| Camelid anti-tagBFP Alexa Fluor 647 (clone 1H7) | Antibodies Online | ABIN6953244 |
| Donkey anti-rabbit Alexa Fluor 488 (polyclonal) | ThermoFisher Scientific | A32790 (RRID:AB_2762833) |
| Donkey anti-goat Alexa Fluor 555 (polyclonal) | ThermoFisher Scientific | A32816 (RRID:AB_2762839) |
| Anti-human CD4 Alexa Fluor 700 (clone SK3) | Biolegend | 344622 (RRID:AB_2563150) |
| Anti-human CD137 (4-1BB) APC (clone 4B4-1) | Biolegend | 309810 (RRID:AB_830672) |
| Anti-human CD197 (CCR7) APC/Cy7 (clone G043H7) | Biolegend | 353212 (RRID:AB_10916390) |
| Biological samples | ||
| FLC patient TILs (FLC-SJ1) | University of Maryland (approval #HP-00080263, Pro00008980) | N/A |
| FLC patient PBMCs (FLC-SJ1) | University of Maryland (approval #HP-00080263, Pro00008980) | N/A |
| FLC patient ascites fluid (FLC-SJ3) | University of Tennessee Health Science Center (approval #17-05064-XP, Pro00003992) | N/A |
| FLC patient tumor (FLC-SJ2, FLC-SJ4, FLC-SJ5) | Molecular Analysis of Solid Tumors (MAST, NCT01050296, approval #Pro00001240) | N/A |
| Healthy donor PBMCs | St. Jude Blood Donor Center (Dept. of Pathology protocol # BDC035) | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| FBS | Gibco | 16140–071 |
| OCT embedding matrix | Sakura | 4583 |
| Vector® trueVIEW™ autofluorescence quenching reagent with DAPI | Vector | SP-8500-15 |
| HLA class I easYmers | Immudex | Table S2 |
| DNAJB1-PRKACA fusion peptides | Genscript | Table S2 |
| Streptavidin beads, 6–8 μm | Spherotech | SVP-60-5 |
| Lenti-X concentrator | Takara | 631232 |
| Puromycin | Sigma-Aldrich | P9620 |
| G418 | Gibco | 10131–035 |
| Protease inhibitor tablets | ThermoFisher Scientific | A32963 |
| CaptivA® protein A resin | Repligen | CA-HF-0100 |
| Human serum AB | Gemini BioProducts | 100–512 |
| Human IL-2 | Stemcell Technologies | 78036 |
| X-VIVO 15 serum-free hematopoietic cell medium | Lonza | 04-418Q |
| Human GM-CSF | Miltenyi Biotec | 130-095-372 |
| Human IL-4 | R&D Systems | 204-IL-010 |
| Human Flt3-L | R&D Systems | 308-FKN-025 |
| R848 | InvivoGen | tlrl-r848-5 |
| Salmonella Minnesota lipopolysaccharide (LPS) | InvivoGen | tlrl-smlps |
| Human IL-1β | R&D Systems | 201-LB-010 |
| Human IL-2 | R&D Systems | 202-IL-050 |
| Human IL-7 | R&D Systems | 207-IL-025 |
| Human IL-15 | PeproTech | 200–15 |
| Dasatinib | Sigma-Aldrich | CDS023389 |
| Ghost Violet 510 viability dye | Tonbo Biosciences | 13-080-T100 |
| Collagenase IV | Worthington Biochemical | LS004188 |
| DNase I | Worthington Biochemical | LS002145 |
| TotalSeq™-C0954 PE streptavidin | Biolegend | 405267 |
| Human IL-7 | PeproTech | 200–07 |
| Retronectin | Takara | T100A |
| PE streptavidin | Biolegend | 405203 |
| APC streptavidin | Biolegend | 405207 |
| Brilliant Violet 421 streptavidin | Biolegend | 405226 |
| PE/Cy7 streptavidin | Biolegend | 405206 |
| GolgiPlug (containing brefeldin A) | BD Biosciences | 555029 |
| GolgiStop (containing monensin) | BD Biosciences | 554724 |
| Cell stimulation cocktail | ThermoFisher Scientific | 00-4970-93 |
| LEGENDplex™ human IFNγ capture beads B3 | Biolegend | 740545 |
| LEGENDplex™ human TNFα capture beads B7 | Biolegend | 740711 |
| LEGENDplex™ human IL-2 capture beads A5 | Biolegend | 740934 |
| Annexin V Brilliant Violet 421 | Biolegend | 640924 (RRID:AB_2893503) |
| Annexin V binding buffer | Biolegend | 422201 |
| Human IL-7 | Biolegend | 715302 |
| Human IL-15 | Biolegend | 715902 |
| NucView 530 caspase-3 substrate | Biotium | 10408 |
| LEGENDplex™ streptavidin-PE | Biolegend | 740452 |
| D-luciferin | Perkin Elmer | 122799 |
| Vectashield Vibrance® mounting media with DAPI | Vector | H-1800 |
| Critical commercial assays | ||
| 10x Chromium Single Cell 5′ Library & Gel Bead Kit v1 | 10x Genomics | 1000006 |
| 10x Chromium Single Cell 5′ Library Construction Kit | 10x Genomics | 1000020 |
| 10x Chromium Single Cell V(D)J Enrichment Kit, Human T cell | 10x Genomics | 1000005 |
| 10x Chromium Single Cell Chip A Kit | 10x Genomics | 1000009 |
| 10x Chromium i7 Multiplex Kit | 10x Genomics | 120262 |
| 10x Chromium Single Cell 5′ Feature Barcode Library Kit | 10x Genomics | 1000080 |
| 10x Chromium i7 Multiplex Kit N Set A | 10x Genomics | 1000084 |
| 10x Chromium Next GEM Single Cell 5′ Library & Gel Bead Kit v1.1 | 10x Genomics | 1000165 |
| 10x Chromium Next GEM Chip G Single Cell Kit | 10x Genomics | 1000127 |
| 10x Chromium Single Index Kit T Set A | 10x Genomics | 1000213 |
| 10x Chromium Single Index Kit N Set A | 10x Genomics | 1000212 |
| SuperScriptVILO cDNA synthesis kit | ThermoFisher Scientific | 11754250 |
| In-Fusion® snap assembly cloning kit | Takara | 638947 |
| Cytofix/Cytoperm fixation kit | BD Biosciences | 554714 |
| Deposited data | ||
| Raw single-cell GEX and TCR data | This paper | SRA: PRJNA1070700 |
| Immunopeptidomics data | This paper | PRIDE: PXD042316 |
| FLC patient bulk RNAseq | Dinh et al.18 | EGA: EGAS00001004169 |
| FLC patient bulk RNAseq | Francisco et al.43 | GEO: GSE181922 |
| IPD-IMGT/HLA database | Barker et al.79 | https://www.ebi.ac.uk/ipd/imgt/hla/ |
| Uniprot annotated proteome UP000005640 | Uniprot | https://www.uniprot.org/proteomes/UP000005640 |
| Human HLA allele frequencies in US populations | Gonzalez-Galarza et al.44 | http://allelefrequencies.net/hla.asp |
| Human reference genome GRCh38 | Genome Reference Consortium | https://www.ncbi.nlm.nih.gov/assembly/GCF_000001405.26/ |
| Cell Ranger human reference, GRCh38 (Ensembl 84) | 10x Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/release-notes/build |
| Cell Ranger human reference, GRCh38 (Ensembl 93) | 10x Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/release-notes/build |
| Cell Ranger human V(D)J reference vdj_IMGT_human | 10x Genomics | N/A |
| Cell Ranger human V(D)J reference vdj_IMGT_human-None | 10x Genomics | N/A |
| Cell Ranger human V(D)J reference vdj_GRCh38_alts_ensembl-3.1.0-3.1.0 | 10x Genomics | N/A |
| Single-chain TCR repertoire data from HCV-infected patient SR5 | Mazouz et al.56 | immuneACCESS: Mazouz_Shoukry_2021 https://clients.adaptivebiotech.com/pub/mazouz-2021-ji (https://doi.org/10.21417/SM2021JI) |
| Single-chain TCR repertoire data from SARS-CoV-2-infected donor M | Minervina et al.57 | Zenodo: 4065547 https://zenodo.org/record/4065547 |
| Single-chain TCR repertoire data from SARS-CoV-2-vaccinated donors 01a, 04, 20, 22 | Mudd et al.54 | GEO: GSE183393 |
| IMGT/GENE-DB | Giudicelli et al.80 | https://www.imgt.org/genedb/ |
| Experimental models: Cell lines | ||
| HEK 293T | ATCC | Cat#: CRL-3216 (RRID:CVCL_0063) |
| K562 | ATCC | Cat#: CCL-243 (RRID:CVCL_0004) |
| 2D3 Jurkat | A kind gift from Fumihiro Fujiki (Osaka University Graduate School of Medicine, Suita, Japan) | N/A |
| A549.eGFP.ffLuc | A kind gift from Christopher DeRenzo (St. Jude Children’s Research Hospital, Memphis, TN) | N/A |
| W6/32 hybridoma | ATCC | HB-95 (RRID:CVCL_7872) |
| Experimental models: Organisms/strains | ||
| Mouse: NSG (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) | St. Jude NSG colony | N/A |
| Oligonucleotides | ||
| Human TRAV/TRBV external primers | Wang et al.81 | See Table S5 |
| Human TRAC/TRBC external reverse primers | Wang et al.81 | See Table S5 |
| Human TRAV/TRBV internal forward primers | Wang et al.81 | See Table S5 |
| Human TRAC/TRBC internal reverse index primers | Wang et al.81 | See Table S5 |
| Recombinant DNA | ||
| pLVX-EF1α-IRES-Puro | Takara | 631253 |
| pSPAX2 | A kind gift from Didier Trono | Addgene plasmid #12260 (RRID:Addgene_12260) |
| pMD2.G | A kind gift from Didier Trono | Addgene plasmid #12259 (RRID:Addgene_12259) |
| HLA class I sequences | Genscript | See “Generation of aAPCs” for alleles |
| TCRαβ sequences | Genscript | Table S4 |
| pSFG | A kind gift from Stephen Gottschalk (St. Jude Children’s Research Hospital, Memphis, TN) | N/A |
| pEq-Pam3(-E) | A kind gift from Stephen Gottschalk (St. Jude Children’s Research Hospital, Memphis, TN) | N/A |
| pRD114 | A kind gift from Stephen Gottschalk (St. Jude Children’s Research Hospital, Memphis, TN) | N/A |
| pLVX- EF1α-IRES-G418 | This paper | N/A |
| Software and algorithms | ||
| NIS Elements software, version 5.30.05 | Nikon Instruments | N/A |
| HLApers | Aguiar et al.82 | github.com/genevol-usp/HLApers |
| kallisto | Bray et al.83 | http://pachterlab.github.io/kallisto/ |
| OptiType | Szolek et al.84 | github.com/FRED-2/OptiType |
| NetMHCcons v1.1 | Karosiene et al.85 | https://services.healthtech.dtu.dk/services/NetMHCcons-1.1/ |
| NetMHCpan v4.1b | Reynisson et al.45 | https://services.healthtech.dtu.dk/services/NetMHCpan-4.1/ |
| NetMHCIIpan v4.1 | Reynisson et al.45 | https://services.healthtech.dtu.dk/services/NetMHCIIpan-4.1/ |
| FlowJo v10.7.2 | BD Biosciences | https://www.flowjo.com/ |
| PEAKs Xpro v10.6 | Bioinformatic Solutions | N/A |
| Cell Ranger v2.2, v3.1.0, v4.0.0 | 10x Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/release-notes |
| Seurat v4.2.0 | Hao et al.86 | https://satijalab.org/seurat/ |
| conga v0.1.1 TCRdist implementation (tcrdist_cpp) | Schattgen et al.87 | https://github.com/phbradley/conga |
| data.table R package v1.14.6 | N/A | https://rdatatable.gitlab.io/data.table/ |
| dplyr R package v1.1.0 | N/A | https://dplyr.tidyverse.org |
| stringdist R package v0.9.10 | N/A | https://github.com/markvanderloo/stringdist |
| igraph R package v1.3.5 | N/A | https://r.igraph.org/ |
| gephi v0.9.7 | N/A | https://gephi.org/ |
| stitchr Python package v1.0.2 | Heather et al.88 | https://github.com/JamieHeather/stitchr |
| RTCA Software Pro | Agilent | N/A |
| Prism v9 | GraphPad | N/A |
| Assay Analyzer | Berkeley Lights | N/A |
| Living Image | Caliper Life Sciences | N/A |
| ggplot2 package v3.3.6 | N/A | https://ggplot2.tidyverse.org/ |
| rstatix package v0.7.0 | N/A | https://github.com/kassambara/rstatix |
| Other | ||
| 0.45 μm SFCA syringe filter | ThermoFisher Scientific | 723–9945 |
| Amicon 5 kDa MWCO filter | Millipore-Sigma | UFC8010 |
| Omix C18 stage tips | Agilent | A5700310 |
| 96-well RTCA E-plate | Agilent | 300600910 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Paul G. Thomas (paul.thomas@stjude.org)
Materials availability
This study did not generate new unique reagents.
Data and code availability
-
•
De-identified patient single-cell sequencing data have been deposited to the Sequence Read Archive at SRA: PRJNA1070700.
-
•
Immunopeptidomics data have been deposited to the PRoteomics IDEntifications (PRIDE) database at PRIDE: PXD042316 (https://doi.org/10.6019/PXD042316).
-
•
Publicly available data analyzed in this study were obtained from the European Genome-Phenome Archive (EGA) at EGA: EGAS00001004169, Gene Expression Omnibus (GEO) at GEO: GSE181922 and GEO: GSE183393, Zenodo at accession number Zenodo: 4065547, and immuneACCESS at immuneACCESS: Mazouz_Shoukry_2021 (https://doi.org/10.21417/SM2021JI).
-
•
Other data generated in this study are available within the article and its supplemental data or will be made available upon request.
Experimental model and study participant details
Human subjects
Written informed consent was obtained from all human participants who provided samples used in this study. Excess de-identified tumor material was collected from FLC patients at St. Jude Children’s Research Hospital (SJCRH) in agreement with local institutional ethical regulations and Institutional Review Board (IRB) approval. Patient consent for tissue acquisition was obtained under the guidelines of the MAST protocol (NCT01050296; IRB protocol Pro00001240, SJCRH) and distributed via the Childhood Solid Tumor Network (CSTN).89 Additional excess de-identified tumor material was collected from FLC patients at the University of Maryland (UoM) and University of Tennessee Health Science Center (UTHSC) under IRB-approved protocols HP-0008263 (UoM), 17-05064-XP and 19-06875-NHSR (UTHSC). De-identified patient samples from UoM and UTHSC were transferred to St. Jude under IRB-approved protocols Pro00003992 and Pro00008980 (SJCRH). Bulk RNAseq data from FLC patients were generated previously18,43 from samples collected under IRB-approved protocols 1802007780, 1811008421 (Cornell University) and/or 33970/1 (Fibrolamellar Cancer Foundation). All data were de-identified prior to analysis. Study participant details (age, gender, etc.) for FLC patients are provided in Table S1. Healthy donor PBMCs were isolated from apheresis rings obtained from the St. Jude Blood Donor Center under Department of Pathology protocol BDC035. All apheresis rings were de-identified before release.
Cell lines
HEK 293T cells (CRL-3216) and K562 cells (CCL-243) were obtained from ATCC. HEK 293T cells were cultured in complete DMEM (DMEM [Gibco 11965-092], 10% FBS [Gibco 16140-071], 2mM L-glutamine [Gibco 25030-081], 100 U/mL penicillin/streptomycin [Gibco 15140-122]). K562 cells were cultured in complete IMDM (IMDM [Gibco 12440-053], 10% FBS, 2mM L-glutamine, 100 U/mL penicillin/streptomycin). 2D3 Jurkat J76.7 cells90 (TCR-null, CD8+, NFAT-eGFP reporter) were a kind gift from Fumihiro Fujiki (Department of Cancer Immunology, Osaka University Graduate School of Medicine, Suita, Japan) and were cultured in complete RPMI (RPMI 1640 [Gibco 22400-089], 10% FBS, 2mM L-glutamine, 100 U/mL penicillin/streptomycin). Generation of A549.eGFP.ffLuc cells was described previously58; these cells were cultured in complete DMEM. All cell lines were maintained in humidified incubators at 37°C, 5% CO2.
Xenograft mouse model
All animal experiments followed protocol 640 approved by the St. Jude Children’s Research Hospital Institutional Animal Care and Use Committee (IACUC). All experiments utilized 12- to 16-week-old male NSG (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) mice obtained from the St. Jude NSG colony. For the systemic lung cancer model, 2 x 106 A549.eGFP.ffLuc.tDNAJB1-PRKACA.A6802 or A549.eGFP.ffLuc.tDNAJB1-PRKACA (no-HLA controls) cells in PBS were injected intravenously (i.v.). Seven days later, mice received a single i.v. dose of 1 x 107 SJ1-4-, SJ1-12-, or mock-transduced T cells in PBS, or PBS alone. Tumor burden was monitored via weekly bioluminescence imaging (see bioluminescence imaging). Mice were euthanized when they reached a bioluminescent flux endpoint of 1 x 1010 photons/s, or when they met physical euthanasia criteria (significant weight loss, signs of distress), or when recommended by the St. Jude veterinary staff. At euthanasia, spleens and tumor-bearing tissues (including lungs, liver, and kidneys) were harvested for further analysis.
Method details
Immunofluorescence imaging of FLC tissue
FLC tissue was frozen in isopentane chilled by liquid nitrogen, embedded in OCT embedding matrix (Sakura 4583), and stored at −80°C. 10 μm sections were obtained following methanol fixation and permeabilization, and blocking with 10% donkey serum in PBST. Sections were incubated at 4°C overnight with the following primary antibodies: rat anti-human/mouse/rat CD3 (1:200, Abcam ab255972, clone CD3-12), mouse anti-human CD8 (1:200, Abcam ab17147, clone C8/144B), rabbit anti-human CD68 (1:200, Abcam ab213363, clone EPR20545). Sections were washed with PBS, then incubated with the following secondary antibodies for 1 h at RT: donkey anti-rat Alexa Fluor 647 (1:500, ThermoFisher Scientific A48272), donkey anti-mouse Alexa Fluor 488 (1:500, ThermoFisher Scientific A32766), donkey anti-rabbit Alexa Fluor 555 (1:500, ThermoFisher Scientific A32794). Slides were then mounted with Vector trueVIEW autofluorescence quenching reagent with DAPI (Vector SP-8500-15) and imaged via Nikon Eclipse Ni-E scope. Images were acquired and analyzed using NIS Elements software, version 5.30.05 (Nikon Instruments).
Bulk RNAseq and HLA expression analysis
Bulk RNAseq data for patients FLC01-FLC34 (Supplmentary Table S1) were generated and published previously.18,43 HLA expression levels from these bulk RNAseq data were estimated using the HLApers pipeline82 with kallisto pseudoalignment.83
HLA typing
Inference of HLA types from bulk RNAseq data and single-cell (sc)RNAseq data was accomplished using OptiType.84 For patients FLC01-FLC34, FLC-SJ2, FLC-SJ4, and FLC-SJ5, bulk RNAseq data were available and were used for HLA inference. For patients FLC-SJ1 and FLC-SJ3, only scRNAseq generated during 10x experiments in this study were available. For those patients, 300k unique reads aligned to the HLA region were randomly selected from the relevant scRNAseq data and were used to infer HLA types.
Neoepitope prediction
To identify putative class I-restricted DNAJB1-PRKACA fusion neoepitopes, custom python scripts were used to generate all possible peptides of lengths 8–15 amino acids spanning the fusion junction. Peptides were subsequently screened for putative HLA binding affinity using NetMHCcons1.185 against all 2,924 HLA alleles included in the pipeline reference. As in previous assessments,64 we considered predicted peptide-HLA pairs with inferred IC50 of 500 nM or lower to be putative neoepitopes. To identify putative class II-restricted fusion neoepitopes, we used the web interface for NetMHCIIpan4.145 using default settings, including binding affinity predictions in output, and tested 75 13-22mer fusion peptides spanning the fusion breakpoint for binding against the 54 class I HLA alleles for which high quality binding data is available (i.e., designated as “high quality” alleles). For class II predictions, we considered predicted peptide-HLA pairs with inferred IC50 of 1000 nM or lower to be putative neoepitopes. Neoepitope prediction for class I HLA alleles available as peptide-HLA multimers was carried out using the web interfaces for NetMHCcons1.1 and NetMHCpan4.1b45 using default settings, including binding affinity predictions in output, and allowing for 8-15mer peptide predictions. For these predictions, we considered peptide-HLA pairs with inferred IC50 of 1000 nM or lower on at least one prediction platform to be putative neoepitopes and candidates for validation in biochemical fold tests. Results of neoepitope predictions for class I HLA alleles commercially available (positive predictions only) or expressed by patients in this study (positive predictions or “none” where no peptides were predicted) are available in Table S2.
Easymer assembly and fold tests
To biochemically validate predicted fusion peptide binding, easYmers (Immudex) were obtained for each available HLA allele predicted to bind one or more DNAJB1-PRKACA fusion peptide. The corresponding peptides (Table S2) were commercially synthesized (Genscript) and diluted to 1 mM in the manufacturer-recommended solvent (ddH2O or DMSO, depending on solubility). Each peptide was then loaded onto the corresponding easYmer allele(s) according to the manufacturer’s instructions. Peptide binding was evaluated using the manufacturer-recommended flow cytometry-based fold test according to the manufacturer-supplied protocol. Briefly, peptides of interest were loaded as described above; for each HLA allele, a no peptide negative control and a positive control using a known binding peptide for the HLA allele (supplied by the manufacturer) were also prepared. After loading, monomers were diluted to 40 nM (assuming a starting concentration of approximately 500 nM) in dilution buffer (PBS with 5% glycerol [Sigma-Aldrich G5516]), then further diluted to achieve three serial 3-fold dilutions with final concentrations of 9 nM, 3 nM, and 1 nM. These dilutions were incubated with streptavidin beads (Spherotech SVP-60-5, 6–8 μm) at 37°C for 1 h to allow binding of stable complexes to beads. Samples were then washed three times with FACS buffer (PBS, 0.5% BSA [Sigma-Aldrich A7030], 2 mM EDTA [ThermoFisher Scientific 15575-038]) to remove unbound peptide-HLA complexes. Bound, stable complexes were then detected by staining with PE-labeled anti-human β2m antibody (Santa Cruz Biotech sc-13565, clone BBM.1) at 1:200 for 30 min at 4°C. Samples were washed 3 times with FACS buffer then resuspended for analysis on a custom-configured BD Fortessa using FACSDiva software (Becton Dickinson). Flow cytometry data were analyzed using FlowJo version 10.7.2 software (BD Biosciences). The fluorescence intensity of anti-β2m staining observed in this assay depends on the proportion of monomer complexes where a peptide has bound to stabilize the peptide-HLA-β2m trimer; therefore, observed median fluorescence intensity (MFI) is proportional to the strength of peptide binding to HLA. We defined “strong binder” peptides as those whose anti-β2m MFI was at least 5x greater than the no-peptide negative control at 9 nM and “weak binder” peptides as those whose anti-β2m MFI was at least 1.25x greater than the no-peptide control at 9 nM. Peptides that fell below either threshold were defined as “non-binders.” Fold test results for each peptide-HLA combination tested are summarized in Table S2.
Artificial antigen-presenting cells
Monoallelic class I artificial antigen-presenting cells (aAPCs) were generated for HLA alleles A∗11:01, A∗23:01, A∗24:02, A∗68:02, B∗40:01, B∗44:02, B∗44:03, C∗03:02, C∗04:01, and C∗07:02. Full length coding sequences for these alleles were obtained from the IPD-IMGT/HLA database,79 then synthesized and cloned via restriction sites (Genscript) into lentiviral vector pLVX-EF1α-IRES-Puro (Takara 631253). Lentivirus was packaged by transfecting HEK 293T cells with the pLVX lentiviral vector containing the HLA insert, psPAX2 packaging plasmid (a kind gift from Didier Trono via Addgene; Addgene plasmid #12260), and the pMD2.G envelope plasmid (a kind gift from Didier Trono via Addgene; Addgene plasmid #12259) at a ratio of 4:3:1. Viral supernatant was harvested and filtered through a 0.45 μm SFCA syringe filter (ThermoFisher Scientific 723–9945) 24 and 48 h post-transfection, then concentrated using Lenti-X Concentrator (Takara 631232). Single-use aliquots of concentrated lentivirus in PBS were frozen at −80°C until use. On day of transduction, 2.5 x 105 K562 cells per well were plated in a 12-well tissue-culture-treated plate in complete IMDM (IMDM, 10% FBS, 2mM L-glutamine, 100 U/mL penicillin/streptomycin). Concentrated lentivirus was thawed rapidly at 37°C and added dropwise to each well to transduce K562s 48–72 h post-transduction, puromycin (Sigma-Aldrich P9620) was added at 2 μg/mL to antibiotic-select for transduced cells. After one week of antibiotic selection, transduction and surface HLA expression was confirmed via flow cytometry using a pan-HLA class I antibody (PE-conjugated, Biolegend 311406, clone W6/32; or APC-conjugated, Biolegend 311410, clone W6/32). Flow cytometry data were collected on a custom-configured BD Fortessa using FACSDiva software (Becton Dickinson) and analyzed using FlowJo version 10.7.2 software (BD Biosciences).
Culture of aAPC lines for immunopeptidomics
aAPC lines expressing the DNAJB1-PRKACA fusion and one of five HLA class I alleles (HLA-A∗11:01, -A∗24:02, -A∗68:02, -B∗40:01, or -C∗07:02) were cultured in IMDM supplemented with 2mM MEM non-essential amino acid solution (Gibco 11140-050), 100mM HEPES (Gibco 15630-080), 2mM L-glutamine, 100 U/mL penicillin/streptomycin, 50 μM 2-mercaptoethanol (Sigma-Aldrich M3148) and 10% heat inactivated FBS. Puromycin (2 μg/mL; Sigma-Aldrich P9620) and G418 (1 mg/mL; Gibco 10131-035) were added to obtain cells expressing high levels of both HLA gene of interest and fusion protein, respectively. HLA expression on the cell lines was monitored by flow cytometry. Cells were stained with hybridoma supernatant W6/32 produced in-house (pan-HLA class I [HB-95; ATCC]91) as primary antibody and goat anti-mouse IgG PE (Southern Biotech 1030-09) as the secondary antibody. The level of fusion protein expression was confirmed by monitoring BFP expression. Each cell line was expanded to generate a pellet of 5 x 107 cells, snap frozen, and stored in −80°C until further use.
Purification of peptide-HLA complexes
Small scale immunoaffinity purifications were performed for the 5 transfected K562 cell lines as described previously.92,93 Cells were lysed with 300 μL of lysis buffer (0.5% IGEPAL [Sigma-Aldrich I8896], 50mM Tris (pH 8.0) [ThermoFisher Scientific AM9856], 150 mM NaCl [Fisher Scientific S271-3], and 1X protease inhibitor tablet [ThermoFisher Scientific A32963]), mixed gently and left rolling at 4°C for 1 h. Following lysis, samples were centrifuged at 3724 x g, 15 min, 4°C. Each lysate was first incubated with only the pre-column (only protein A resin [PAS, CaptivA, Repligen CA-HF-0100]) for 1 h at 4°C to remove non-specific binding material, followed by affinity capture of class I peptides using W6/32 antibody. The columns including the pre-column and W6/32 affinity columns were washed with 5 CV of 1X PBS to remove unbound antibody, detergent, and other salts from the sample. Bound HLA-peptide complexes were eluted using 300 μL of 10% acetic acid (Sigma-Aldrich 695092). The eluate was warmed to 70°C for 10 min to disassociate any remaining peptide-HLA complex and then left to cool at RT. The eluate was passed through a 5 kDa molecular weight cut off (MWCO) filter (Amicon, Millipore-Sigma UFC8010) and centrifuged at 16,060 x g, 30 min at RT. Samples were desalted using C18 stage tips were washed with 0.1% Trifluoracetic acid (TFA; ThermoFisher Scientific 85183) and peptides were eluted with 80% acetonitrile (ACN; ThermoFisher Scientific 036423.K2) in 0.1% TFA. The filtered sample was centrifugally evaporated and desalted by reverse phase C18 stage tips (Omix, Agilent A5700310) and reconstituted in 2% ACN in 0.1% formic acid (FA; ThermoFisher Scientific 28905). A mixture of 11 iRT peptides were spiked into each sample to aid retention time alignment.94 Samples were interrogated by high resolution liquid chromatography-tandem mass spectrometry (LC-MS/MS).
Data acquisition by LC-MS/MS
Samples were analyzed using a timsTOF Pro mass spectrometer, where reconstituted peptides were loaded onto an IonOpticks Aurora 25 cm C18 column using a nanoElute liquid chromatography system (Bruker Daltonics). The peptides were separated using a gradient of buffer B (0.1% FA in ACN) against buffer A (0.1% FA, 2% ACN in water) initially to 17% over 60 min then to 25% over 30 min then to 37% over 10 min with a flow rate of 300 nL/min. Data dependent acquisition (DDA) was performed with the following settings: mz range: 100-1700mz, capillary voltage: 1600V, target intensity of 30000, TIMS ramp of 0.60–1.60 Vs/cm2 for 166 ms.
Identification of MHC ligand sequences
LC-MS/MS data was searched with PEAKs Xpro v10.6 (Bioinformatic Solutions) against the Uniprot annotated proteome (202006 release for human with 20,150 proteins) appended with the fusion protein and predicted binders for 5 different HLA class I alleles. The search was carried out using the following settings: Enzyme: none, parent mass tolerance 15 ppm, fragment mass tolerance 0.02 Da, Variable modifications: Deamidation (NQ), Oxidation (M). For data analysis a 5% false discovery rate (FDR) cut off was applied. For data processing, firstly iRT peptides were removed along with any duplicate peptides. Secondly, only peptides with 8–12 amino acids were retained. Finally, peptides found in blank and K562 parental samples were subtracted from peptides identified in transfectant samples to obtain the final list of peptides specific to HLA of interest.
TIL expansion
TILs were expanded from a variety of patient specimens, including tumor core-needle biopsies (patient FLC-SJ1), larger resected tumor specimens (patient FLC-SJ2), or ascites fluid (patient FLC-SJ3), according to previously published methods.95 Briefly, for solid tumor fragments (patients SJ1 and SJ2), each fragment was placed in a well of 24-well culture dish in 2 mL/well complete RPMI (RPMI 1640, 5% human serum AB [Gemini Bio-Products 100–512], 2mM L-glutamine, 100 U/mL/100 μg/mL penicillin/streptomycin) supplemented with 6000 IU IL-2 (Stemcell Technologies 78036). Fragments were left undisturbed for the first 5 days of culture, then monitored for growth and supplemented with additional IL-2 (6000 IU/mL) and fresh complete media every 2–3 days. When cultures had sufficiently expanded, TILs were harvested for analysis. The same approach was followed for TILs expanded from ascites fluid, except that rather than plating solid tumor fragments, cells collected from ascites fluid were initially plated at 2.5 x 105 cells/well in a 48-well culture dish.
PBMC expansion and single-cell sorting
FLC patient PBMCs were expanded according to published methods.48 Briefly, cryopreserved patient SJ1 PBMCs were thawed and plated in a 96-well plate at 1 x 105 cells/well in 200 μL X-VIVO 15 serum-free hematopoietic cell medium (Lonza 04-418Q). On day 0 (same day as thaw), cells were supplemented with human GM-CSF (Miltenyi Biotec 130-095-372) at 1000 IU/mL, human IL-4 (R&D Systems 204-IL-010) at 500 IU/mL, and human Flt3-L (R&D Systems 308-FKN-025) at 50 ng/mL in X-VIVO 15. On day 1, cells were stimulated with a pool of fusion peptides predicted to bind HLA-A∗68:02 (EVKEFLAKA, EIFDRYGEEV, EIFDRYGEEVKEFL, EIFDRYGEEVKEFLA, EVEKEFLAKAKEDFL; Genscript) at 1 μM each, R848 (InvivoGen tlrl-r848-5) at 10 μM, LPS (lipopolysaccharide, Salmonella minnesota; InvivoGen tlrl-smlps) at 100 ng/mL, and human IL-1β (R&D Systems 201-LB-010) at 10 ng/mL in X-VIVO 15. On days 2, 4, 7, and 9, cells were supplemented with human IL-2 (R&D Systems 202-IL-050) at 10 IU/mL, human IL-7 (R&D Systems 207-IL-025) at 10 ng/mL, and human IL-15 (PeproTech 200-15) at 10 ng/mL in R10 medium (RPMI 1640 supplemented with 10% human serum AB [Gemini Bio-Products 100–512], 10 mM HEPES, 0.1 mg/mL Gentamicin [ThermoFisher Scientific 15750-060], and 1X GlutaMAX [Gibco 35050-061]). On day 10, cells from all wells were pooled and washed with PBS. Cells were then treated with 50 nM dasatinib (Sigma-Aldrich CDS023389) in PBS for 30 min at 37°C, 5% CO2 to inhibit TCR internalization and enhance tetramer staining.96 Cells were then stained with 10 μL per million cells of each tetramer corresponding to each stimulating peptide in the pool (10 tetramers total, 2 corresponding to each stimulating peptide labeled with different fluorochromes [PE, APC, Brilliant Violet 421, or PE/Cy7] to permit combinatorial identification of cells positive for each tetramer) for 90 min at RT. After tetramer staining, cells were blocked using anti-human Fc block (BD Biosciences 564220) for 10 min at RT, then stained with 1 μL Ghost Violet 510 viability dye (Tonbo Biosciences 13-080-T100) and a cocktail of surface antibodies for 30 min at 4°C. Surface antibodies included 2 μL each of: anti-human CD3 (FITC-conjugated, Biolegend 344804, clone SK7), anti-human CD8 (Brilliant Violet 785-conjugated, Biolegend 344740, clone SK1), anti-human CD4 (PerCP/Cy5.5-conjugated, Biolegend 344608, clone SK3). After staining, cells were washed with MACS buffer (PBS, 0.5% BSA, 2 mM EDTA) before proceeding to single-cell sorting on a Sony SY3200 cell sorter. Individual live, CD3+, CD8+, tetramer+ cells (positive for either PE or APC, which were included in all five possible fluorochrome) combinations were sorted into individual wells of a 384-well plate loaded with Superscript VILO master mix (ThermoFisher Scientific 11754250). After sorting, plates were centrifuged at 500 x g and stored at −80°C until processing.
Paired TCRαβ amplification and sequencing
Single-cell paired TCR library preparation and sequencing was performed as previously described.81 Briefly, after reverse transcription, cDNA underwent two rounds of nested multiplex PCR amplification using a forward primer mix specific for human V-segments and reverse primers specific for human TRAC and TRBC segments (see Table S5 for primer sequences). The resulting amplicons were sequenced on Illumina MiSeq at 2x150bp read length.
Patient tumor dissociation
Solid tumor samples obtained from patients were first mechanically dissociated into small fragments, then further dissociated by incubation of tumor fragments in tumor digestion media (HBSS, 0.1% collagenase IV [Worthington Biochemical LS004188], 0.01% DNase I [Worthington Biochemical LS002145]) at 37°C for 30–60 min. After digestion, the resulting single cell suspension was filtered through a 100 μM nylon mesh, using a 1 mL plunger to break up any remaining solid tissue fragments, and washed with PBS. Samples with observable red blood cells were subjected to red blood cell lysis prior to a final wash in PBS and cryopreservation of single cell suspensions.
Preparation of TILs for 10x
Tumor-infiltrating T cells from FLC patients were first stimulated with fusion peptides to induce changes in gene expression in fusion-responsive cells. TILs from patient FLC-SJ1 were stimulated with 1 μM EIFDRYGEEV minimal peptide (Genscript), while TILs from patients FLC-SJ2, FLC-SJ3, and FLC-SJ4 were stimulated with 1 μM 24mer fusion peptide (RKREIFDRYGEE-VKEFLAKAKEDF; Genscript). For patients SJ1 and SJ4, all available TILs were stimulated, while for patients SJ2 and SJ3, half of cells were stimulated and half were used as unstimulated controls. All stimulations were carried out for 4–12 h in complete RPMI (RPMI 1640, 10% FBS, 2mM L-glutamine, 100 U/mL/100 μg/mL penicillin/streptomycin) with 1 μg/mL each of anti-human CD28 (BD Biosciences 555725) and CD49d (BD Biosciences 555501); unstimulated control media omitted peptide. After stimulation, TILs from patients SJ1 and SJ4 were washed with PBS, then stained with 1 μL Ghost Violet 510 viability dye (Tonbo Biosciences 13-080-T100) for 15 min at RT. Cells were washed again with FACS buffer (PBS, 2% FBS, 2 mM EDTA), blocked using anti-human Fc block (Biolegend 422302) for 10 min at 4°C, then stained with a cocktail of surface antibodies for 30 min at 4°C. Surface antibodies for patient SJ1 included 1 μL of anti-human CD8 (FITC-conjugated, Biolegend 344704, clone SK1). Surface antibodies for patient SJ4 included 1 μL each of: anti-human CD3 (Brilliant Violet 421-conjugated, Biolegend 344834, clone SK7), anti-human CD8 (Brilliant Violet 785-conjugated, Biolegend 344740, clone SK1), anti-human CD4 (PerCP/Cy5.5-conjugated, Biolegend 344608, clone SK3). TILs were then washed, and those from patient SJ1 were then sorted on viable, CD8+ cells using a Sony SY3200 cell sorter; TILs from patient SJ4 were sorted on viable cells only using a Sony SY3200 cell sorter. TILs from patients SJ2 and SJ3 were of sufficiently high viability to not require fluorescence-activated cell sorting; however, TILs from these patients were stained with human hashtag oligo antibodies to distinguish between separate TIL wells. After stimulation, cells were washed with MACS buffer (PBS, 0.5% BSA, 2 mM EDTA), blocked using anti-human Fc block (Biolegend 422302) for 10 min at 4°C, and separate populations each stained with 2 μL TotalSeq-C anti-human hashtag antibodies 1–4 (Biolegend 394661, 394663, 394665, 394667) for 30 min at 4°C. TILs were then washed and hashed populations combined in equal proportions. After sorting or combining, TILs were immediately loaded into 10x reactions as follows: Live, CD8+ TILs from patient SJ1 were loaded into a single 10x Chromium Single Cell 5′ V(D)J v1 reaction (10x Genomics PN: 1000006, 1000020, 1000005, 1000009, 120262). Live TILs from patient SJ4 were loaded into a single 10x Chromium Single Cell 5′ V(D)J v1 reaction (10x Genomics PN: 1000006, 1000020, 1000005, 1000009, 120262). TILs from patient SJ3 were loaded into two 10x Chromium Single Cell 5′ V(D)J v1 with Feature Barcoding reactions (10x Genomics PN: 1000006, 1000020, 1000080, 1000005, 1000009, 120262, 1000084), one for combined unstimulated populations and one for combined stimulated populations. TILs from patient SJ2 were loaded into two 10x Chromium NextGEM Single Cell V(D)J v1.1 with Feature Barcoding reactions (10x Genomics PN: 1000165, 1000020, 1000080, 1000005, 1000127, 1000213, 1000212), one for combined unstimulated populations and one for combined stimulated populations. For all samples, GEX, TCR, and surface feature (cell hashing for patients SJ2 and SJ3 only) libraries were prepared according to the manufacturer’s protocol. Libraries were sequenced on Illumina NovaSeq at read length 2x100bp for GEX libraries and 2x100-150bp for TCR and surface feature libraries.
Preparation of PBMCs for 10x
Cryopreserved patient SJ1 PBMCs were thawed and allocated to either peptide stimulation or barcoded tetramer staining. Cells allocated for peptide stimulation were split between stimulated and unstimulated conditions. Stimulation was carried out for 4 h using 1 μM EIFDRYGEEV peptide (Genscript) and 1 μg/mL each of anti-human CD28 (BD Biosciences 555725) and CD49d (BD Biosciences 555501) in complete RPMI (RPMI 1640, 10% FBS, 2mM L-glutamine, 100 U/mL/100 μg/mL penicillin/streptomycin); unstimulated control media omitted peptide. Cells allocated for tetramer staining were rested in complete RPMI in an incubator at 37°C, 5% CO2 during stimulation, then washed in MACS buffer (PBS, 0.5% BSA, 2 mM EDTA) and stained with an A∗68:02-EIFDRYGEEV tetramer labeled with TotalSeq-C0954 PE Streptavidin (Biolegend 405267) for 1 h at 4°C. After tetramer staining and stimulation, all cells were washed with PBS and stained with 1 μL Ghost Violet 510 viability dye (Tonbo Biosciences 13-080-T100) for 15 min at RT. Cells were washed again with MACS buffer, blocked using anti-human Fc block (Biolegend 422302) for 10 min at 4°C, then stained with a cocktail of surface antibodies for 30 min at 4°C. Surface antibodies included 1 μL each of: anti-human CD11b (Brilliant Violet 421-conjugated, Biolegend 301324, clone ICRF44), anti-human CD19 (PE/Dazzle594-conjugated, Biolegend 302252, clone HIB19), anti-human CD8 (Brilliant Violet 785-conjugated, Biolegend 344740, clone SK1), anti-human CD4 (PerCP/Cy5.5-conjugated, Biolegend 344608, clone SK3). During the surface staining step, cells from the peptide stimulation were also stained with 2 μL TotalSeq-C anti-human hashtag antibodies 1 or 2 (Biolegend 394661, 394663) to distinguish between stimulated and unstimulated cells. After surface staining, all cells were washed in MACS buffer and sorted on viable, CD11b-, CD19−, CD8+ cells using a Sony SY3200 cell sorter. Peptide stimulated and unstimulated CD8+ cells were combined in equal proportions, and all cells were immediately loaded into 10x reactions as follows: combined peptide-stimulated/unstimulated cells were loaded into a single 10x Chromium Single Cell 5′ V(D)J v1 with Feature Barcoding reaction (10x Genomics PN: 1000006, 1000020, 1000080, 1000005, 1000009, 120262, 1000084), and tetramer-labeled CD8+ cells were loaded into a single 10x Chromium Single Cell 5′ V(D)J v1 with Feature Barcoding reaction (10x Genomics PN: 1000006, 1000020, 1000080, 1000005, 1000009, 120262, 1000084) or a single 10x Chromium NextGEM Single Cell V(D)J v1.1 with Feature Barcoding reaction (10x Genomics PN: 1000165, 1000020, 1000080, 1000005, 1000127, 1000213, 1000212). For all samples, GEX, TCR, and surface feature (cell hashing or tetramer barcode) libraries were prepared according to the manufacturer’s protocol. Libraries were sequenced on Illumina NovaSeq at read length 2x100bp for GEX libraries and 2x100-150bp for TCR and surface feature libraries.
10x data analysis
Raw data from patient SJ1 and patient SJ3 TILs were processed using Cell Ranger version 2.2 and aligned to GRCh38 and vdj_IMGT_human Cell Ranger references. Raw data from patient SJ4 TILs were processed using Cell Ranger version 3.1.0 and aligned to GRCh38–3.0.0 and vdj_IMGT_human-None Cell Ranger references. Raw data from patient SJ2 TILs and patient SJ1 PBMCs were processed using Cell Ranger version 4.0.0 and aligned to GRCh38–3.0.0 and vdj_GRCh38_alts_ensembl_3.1.0–3.1.0 Cell Ranger references. The resulting GEX matrices were analyzed using the Seurat R package version 4.2.0,86 and associated TCR clonotypes were added to each Seurat object via the AddMetaData function. Following standard quality control filtering, we discarded low quality cells (nFeatures <200 or >3000, MT% >5%) and normalized the data via the NormalizeData function using the default parameters. We then found 2000 variable features via the FindVariableFeatures function and scaled the data via the ScaleData function. Cells were clustered by uniform manifold approximation and projection (UMAP) with the resolution parameter set to 0.5. Differentially expressed genes between SJ1-4 TCR-expressing cells and cells expressing other TCR clonotypes were identified using the FindMarkers function; the resulting differentially expressed genes can be found in Table S3.
Public HCV and SARS-CoV-2 TCR repertoire datasets
Bulk TCRα and TCRβ repertoire data from PBMCs of an HCV-infected donor (designated SR5) and a SARS-CoV-2-infected donor (designated Donor M) were reported previously.56,57 Bulk TCRα and TCRβ repertoire data from lymph node T follicular helper cells of four SARS-CoV-2-vaccinated donors (designated 01a, 04, 20, and 22) were reported previously.54 TCR cluster specificities were annotated based on SARS-CoV-2-specific clusters reported in the source publications54,57 and elsewhere.55
TCR repertoire analysis
TCR similarity networks were constructed as described previously.54,55 Briefly, to measure the distance between TCR α/β clonotypes, we used the TCRdist algorithm implementation from the conga python package.87 Further analysis was performed using the R language for statistical computing, with merging and subsetting of data performed using the data.table and dplyr packages. stringdist and igraph R packages were used to build TCR similarity networks, and gephi software was used for TCR similarity network layout and visualization.
TCR reconstruction and cloning
Full-length TCRαβ sequences (Table S4) were reconstructed from V/J gene usage and CDR3 sequences either manually from IMGT GENE-DB80 or using Stitchr.88 TCRα and β chain sequences were modified to use murine constant regions and joined by a 2A element from thosea asigna virus (T2A). A sequence encoding GFP or mCherry was additionally appended by a 2A element from porcine teschovirus (P2A) as a fluorescent marker of transduction. The full-length gene fragment encoding TCRα-T2A-TCRβ-P2A-mCherry/GFP was synthesized and cloned via restriction sites (Genscript) into lentiviral vector pLVX-EF1α-IRES-Puro (Takara 631253).
TCR expression in Jurkat cells
Lentivirus was packaged by transfecting HEK 293T cells with the pLVX lentiviral vector containing the TCRαβ-mCherry/GFP insert, psPAX2 packaging plasmid (a kind gift from Didier Trono via Addgene; Addgene plasmid #12260), and the pMD2.G envelope plasmid (a kind gift from Didier Trono via Addgene; Addgene plasmid #12259) at a ratio of 4:3:1. Viral supernatant was harvested and filtered through a 0.45 μm SFCA syringe filter (ThermoFisher Scientific 723–9945) 24 and 48 h post-transfection, then concentrated using Lenti-X Concentrator (Takara 631232). Single-use aliquots of concentrated lentivirus in PBS were frozen at −80°C until use. On day of transduction, 1 x 106 2D3 Jurkat J76.7 cells (TCR-null, CD8+, NFAT-eGFP reporter) per well were plated in a 12-well tissue-culture-treated plate in complete RPMI (RPMI 1640, 10% FBS, 2mM L-glutamine, 100 U/mL/100 μg/mL penicillin/streptomycin). Concentrated lentivirus was thawed rapidly at 37°C and added dropwise to each well to transduce Jurkats. 48–72 h post-transduction, puromycin was added at 1 μg/mL to antibiotic-select for transduced cells. After one week of antibiotic selection, transduction was confirmed by expression of mCherry/GFP, and surface TCR expression was confirmed via flow cytometry using antibodies against mouse TCRβ constant region (PE-conjugated, Biolegend 109208, clone H57-597; or APC/Fire750-conjugated, Biolegend 109246, clone H57-597) and human CD3 (Brilliant Violet 421-conjugated, Biolegend 344834, clone SK7). Flow cytometry data were collected on a custom-configured BD Fortessa using FACSDiva software (Becton Dickinson) and analyzed using FlowJo version 10.7.2 software (BD Biosciences).
TCR expression in primary human T cells
Gene fragments encoding full-length TCRαβ sequences modified to use murine constant regions (in the format TCRα-T2A-TCRβ-P2A-mCherry) were subcloned into pSFG retroviral vector (a kind gift from Stephen Gottschalk, Department of Bone Marrow Transplantation and Cellular Therapy, St. Jude Children’s Research Hospital, Memphis, TN) via In-Fusion cloning (Takara 638947). A control pSFG retroviral vector encoding only mCherry was also cloned. Retrovirus was packaged by transfecting HEK 293T cells with the pSFG lentiviral vector containing the TCRαβ-mCherry insert (or mock mCherry-only construct), pEq-Pam3(-E) packaging plasmid (a kind gift from Stephen Gottschalk), and the pRD114 envelope plasmid (a kind gift from Stephen Gottschalk) at a ratio of 3:3:2. Viral supernatant was harvested and filtered through a 0.45 μm SFCA syringe filter (ThermoFisher Scientific 723–9945) 48 and 72 h post-transfection. Single-use aliquots of retrovirus were snap frozen and stored at −80°C until use. Three days before transduction, 24-well non-tissue-culture-treated plates were coated with 0.25 μg each anti-human CD3 (Miltenyi 130-093-387) and anti-human CD28 (Miltenyi 130-093-375) per well in sterile ddH2O and incubated at 4°C overnight. Two days before transduction, human PBMCs collected from healthy donors (see human subjects) were plated in the anti-CD3/CD28 plate at 1 x 106 cells per well in 2 mL/well complete RPMI (RPMI 1640, 10% FBS, 2mM L-glutamine, 100 U/mL/100 μg/mL penicillin/streptomycin). One day before transduction, PBMCs were supplemented with human IL-7 (PeproTech 200-07) at 10 ng/mL and human IL-15 (PeproTech 200-15) at 5 ng/mL 24-well non-tissue-culture-treated plates were also coated with 10 μg Retronectin (Takara T100A) per well in PBS and incubated overnight at 4°C. On the day of transduction, retrovirus was rapidly thawed at 37°C and 0.5 mL/well was plated in retronectin-coated plates. Plates were sealed and centrifuged at 2000 x g for 90 min. During centrifugation, activated PBMCs were harvested and prepared at 1.25 x 105 cells/mL in complete RPMI supplemented with human IL-7 at 10 ng/mL and human IL-15 at 5 ng/mL. After centrifugation, supernatant was removed and PBMCs were plated at 2.5 x 105 cells/well. 48–72 h post-transduction, transduction was confirmed by expression of mCherry, and surface TCR expression was confirmed via flow cytometry using antibodies against mouse TCRβ constant region (PE-conjugated, Biolegend 109208, clone H57-597; or APC/Fire750-conjugated, Biolegend 109246, clone H57-597) and human CD3 (Brilliant Violet 421-conjugated, Biolegend 344834, clone SK7). Flow cytometry data were collected on a custom-configured BD Fortessa using FACSDiva software (Becton Dickinson) and analyzed using FlowJo version 10.7.2 software (BD Biosciences). Cells were fed with human IL-7 at 10 ng/mL and human IL-15 at 5 ng/mL every 2–3 days, and were used in experiments or cryopreserved within 7–14 days of transduction.
Tetramer assembly and staining
Peptide-HLA tetramers were assembled from easYmer monomers with confirmed peptide binding according to the manufacturer’s instructions. Briefly, fluorochrome-labeled streptavidin (PE, Biolegend 405203, 0.2 mg/mL; APC, Biolegend 405207, 0.2 mg/mL; Brilliant Violet 421, Biolegend 405226, 0.1 mg/mL; PE/Cy7, Biolegend 405206, 0.2 mg/mL) was added to loaded monomers at 8 ng per 1 μL peptide-HLA complex (assuming a concentration of approximately 500 nM peptide-HLA complexes); e.g., 0.48 μg fluorochrome-streptavidin was added to 60 μL peptide-HLA monomer. Samples were mixed well, then incubated for at least 1 h (typically overnight) at 4°C in the dark before use. Unless otherwise indicated, tetramer staining was performed by adding 5 μL tetramer/sample and incubating for 30 min at RT in MACS buffer (PBS, 0.5% BSA, 2 mM EDTA). After tetramer staining, cells were stained with 1 μL Ghost Violet 510 viability dye (Tonbo Biosciences 13-080-T100) for 15 min at RT. Cells were then washed and tetramer staining evaluated by flow cytometry on a custom-configured BD Fortessa using FACSDiva software (Becton Dickinson). Flow cytometry data were analyzed using FlowJo version 10.7.2 software (BD Biosciences). Unexpanded and expanded PBMCs from patient SJ1 were stained with additional surface antibodies as described in “Preparation of PBMCs for 10x” and “Antigen-specific PBMC expansion and single-cell sorting.”
Intracellular cytokine staining
For TILs expanded from primary patient tumor samples, 1 x 105 TILs per well were plated for each peptide or peptide pool being tested. For TCR-transduced Jurkat functional assays, 2.5 x 105 TCR Jurkats were co-cultured with 2.5 x 105 K562 aAPCs for each peptide being tested. For TCR-transduced primary T cell functional assays, 2 x 105 TCR-T cells were co-cultured with 1 x 105 K562 aAPCs for each peptide being tested. Each well was pulsed with a stimulation cocktail containing 1 μM fusion peptide of interest (Genscript; peptide pools contained 1 μM of each peptide), 1 μg/mL each of anti-human CD28 (BD Biosciences 555725) and CD49d (BD Biosciences 555501), brefeldin A (GolgiPlug, 1 μL/mL; BD Biosciences 555029), and monensin (GolgiStop, 0.67 μL/mL; BD Biosciences 554724), made up in complete RPMI (RPMI 1640, 10% FBS, 2mM L-glutamine, 100 U/mL/100 μg/mL penicillin/streptomycin). In peptide titration experiments, wells were pulsed with the same peptide stimulation cocktail, except that peptides were added in 10-fold serial dilutions to test concentrations ranging from 1 nM to 1 μM. In experiments testing processing and presentation of fusion neoantigens, 2 x 105 TCR-T cells were co-cultured with 1 x 105 K562 aAPCs expression fusion or control genes, and peptide was omitted from the stimulation cocktail. An unstimulated negative control (complete RPMI, CD28, CD49d, brefeldin A, monensin) and a PMA-ionomycin positive control (complete RPMI, 1X Cell Stimulation Cocktail [eBioscience 00-4970-93], CD28, CD49d, brefeldin A, monensin) were included in each peptide stimulation assay. Cells were incubated for 6 h (37°C, 5% CO2), washed with FACS buffer (PBS, 2% FBS, 2 mM EDTA), and blocked using anti-human Fc block (BD Biosciences 564220) for 10 min at RT. Cells were then stained with 1 μL Ghost Violet 510 viability dye (Tonbo Biosciences 13-080-T100) and a cocktail of surface antibodies for 20 min at RT. For TILs, the surface cocktail included 1 μL each of: anti-human CD3 (APC/Cy7-conjugated, Biolegend 344818, clone SK7), anti-human CD8 (Brilliant Violet 785-conjugated, Biolegend 344740, clone SK1), anti-human PD-1 (PE-conjugated, Biolegend 329906, clone EH12.2H7), anti-human TIM-3 (PE/Cy7-conjugated, Biolegend 345014, clone F38-2E2), anti-human CCR7 (FITC-conjugated, Biolegend 353216, clone G043H7), anti-human CD45RA (Brilliant Violet 421-conjugated, Biolegend 304130, clone HI100), and anti-human CD45RO (Brilliant Violet 650-conjugated, Biolegend 304232, clone UCHL1). For TCR-transduced Jurkats and primary T cells, the surface cocktail included 1 μL each of: anti-human CD3 (Brilliant Violet 785-conjugated, Biolegend 344842, clone SK7; or Brilliant Violet 421-conjugated, Biolegend 344834, clone SK7), anti-human CD8 (FITC-conjugated, Biolegend 344704, clone SK1; or Brilliant Violet 785-conjugated, Biolegend 344740, clone SK1), and anti-mouse TCRβ chain (APC/Fire750-conjugated, Biolegend 109246, clone H57-597). Cells from all samples were surface stained for 20 min at RT, then washed with FACS buffer. Then, cells were fixed and permeabilized using a Cytofix/Cytoperm Fixation kit (BD Biosciences 554714) according to the manufacturer’s instructions. After fixation and permeabilization, cells were washed with 1X Perm/Wash buffer and stained with a cocktail of intracellular antibodies. For TILs, this intracellular cocktail included 1.25 μL anti-human IFNγ (Alexa Fluor 647-conjugated, Biolegend 502516, clone 4S.B3) and 1.25 μL anti-human TNFα (Brilliant Violet 605-conjugated, Biolegend 502936, clone MAb11). For TCR-transduced Jurkats, this cocktail included 1.25 μL anti-human IFNγ (Alexa Fluor 647-conjugated, Biolegend 502516, clone 4S.B3), 1.25 μL anti-human TNFα (Brilliant Violet 605-conjugated, Biolegend 502936, clone MAb11), and 1 μL anti-human CD69 (PerCP/efluor710-conjugated, eBioscience 46-0699-42, clone FN50). For TCR-transduced primary T cells, this cocktail included 1.25 μL anti-human IFNγ (Alexa Fluor 647-conjugated, Biolegend 502516, clone 4S.B3), 1.25 μL anti-human TNFα (Brilliant Violet 605-conjugated, Biolegend 502936, clone MAb11), 1.25 μL anti-human IL-2 (PE-conjugated, Biolegend 500307, clone MQ1-17H12), and 1 μL anti-human CD69 (PerCP/efluor710-conjugated, eBioscience 46-0699-42, clone FN50). Cells were intracellular stained for 30 min at 4°C. After staining, cells were washed with 1X Perm/Wash buffer and analyzed by flow cytometry on a custom-configured BD Fortessa using FACSDiva software (Becton Dickinson). Flow cytometry data were analyzed using FlowJo version 10.7.2 software (BD Biosciences). All data from patient TILs were collected in the same experiment. For TCR-transduced Jurkat experiments, data from TCRs identified in the same patient were collected in the same experiments, and the frequency of CD69+ cells was normalized to the unstimulated and positive controls for each sample according to the following equation: Normalized Frequency = (Sample Frequency - Unstimulated Frequency)/(Positive Control Frequency - Unstimulated Frequency). For TCR-transduced primary cell experiments, SJ1-4 and SJ1-12 peptide pulse data were collected in separate experiments, while transduced target cell data for both TCRs were collected in the same experiment. For both TCRs, three PBMC donors were tested in each experiment. Frequency data were normalized to unstimulated and positive controls according to the same equation as above.
Fusion- and WT-expressing target cell lines
Full-length coding sequences were obtained for DNAJB1 (NM_006145.3) and PRKACA (NM_002730.4) from NCBI. The canonical fusion junction sequence12 was assembled from these references, then the fusion sequence was truncated after amino acid Asn100 in PRKACA to generate a non-functional fusion that still encoded the full fusion breakpoint. tagBFP was fused to each of the three sequences (DNAJB1, PRKACA, and truncated DNAJB1-PRKACA) as a direct marker of transgene expression. Constructs encoding DNAJB1-tagBFP, PRKACA-tagBFP, and tDNAJB1-PRKACA-BFP were synthesized and cloned via restriction sites (Genscript) into lentiviral vector pLVX-EF1α-IRES-G418 (modified in-house from pLVX-EF1α-IRES-Puro [Takara 631253]). Lentivirus was packaged by transfecting HEK 293T cells with the pLVX lentiviral vector containing the fusion or WT insert, psPAX2 packaging plasmid (a kind gift from Didier Trono; Addgene plasmid #12260), and the pMD2.G envelope plasmid (a kind gift from Didier Trono; Addgene plasmid #12259) at a ratio of 4:3:1. Viral supernatant was harvested and filtered through a 0.45 μm SFCA syringe filter (ThermoFisher Scientific 723–9945) 24 and 48 h post-transfection, then concentrated using Lenti-X Concentrator (Takara 631232). Single-use aliquots of concentrated lentivirus in PBS were frozen at −80°C until use. To generate fusion- or WT-expressing aAPCs, 2.5 x 105 K562 HLA aAPCs per well were plated in a 12-well tissue-culture-treated plate in complete IMDM (IMDM, 10% FBS, 2mM L-glutamine, 100 U/mL/100 μg/mL penicillin/streptomycin). Concentrated lentivirus was thawed rapidly at 37°C and added dropwise to each well to transduce K562s 48–72 h post-transduction, G418 (Gibco 10131-035) was added at 1 mg/mL to antibiotic-select for transduced cells. After one week of antibiotic selection, fusion or WT transgene expression was confirmed by BFP expression. To generate fusion- or WT-expressing A549.eGFP.ffLuc cells, 2.5 x 106 cells per well were plated in a 6-well tissue-culture-treated plate in complete DMEM (DMEM, 10% FBS, 2mM L-glutamine, 100 U/mL/100 μg/mL penicillin/streptomycin). Concentrated lentivirus was thawed rapidly at 37°C and added dropwise to each well to transduce A549s 48–72 h post-transduction, G418 was added at 1.5 mg/mL to antibiotic-select for transduced cells. After one week of antibiotic selection, fusion or WT transgene expression was confirmed by BFP expression. A549.eGFP.ffLuc.tDNAJB1-PRKACA and A549.eGFP.ffLuc.DNAJB1 cells were then further transduced with A∗68:02-encoding lentivirus (described above for generation of aAPCs) by plating 2.5 x 106 cells per well in a 6-well tissue-culture-treated plate in complete DMEM, then adding concentrated lentivirus dropwise to each well. 48–72 h post-transduction, puromycin (Sigma-Aldrich P9620) was added at 1.5 μg/mL to antibiotic-select for transduced cells. A549.eGFP.ffLuc.tDNAJB1-PRKACA.A6802 and A549.eGFP.ffLuc.DNAJB1.A6802 cells were sorted on BFP expression using a CytoFLEX SRT cell sorter (Beckman Coulter).
xCelligence assay
An xCelligence real-time cell analyzer (RTCA) MP instrument (Agilent) was used to assay in vitro cytotoxicity. All conditions were plated in technical triplicates and all experiments were performed without the addition of exogenous cytokines. 30,000 A549.eGFP.ffLuc.DNAJB1-PRKACA.A6802 (Fusion) or A549.eGFP.ffLuc.DNAJB1.A6802 (WT) cells per well were plated in a 96-well RTCA E-Plate (Agilent 300600910) in complete RPMI (RPMI 1640, 10% FBS, 2mM L-glutamine, 100 U/mL/100 μg/mL penicillin/streptomycin). Cells were incubated in the E-plate for 24 h at 37°C, 5% CO2 until cell index (relative cell impedance) reached a plateau. Then SJ1-4-, SJ1-12-, or mock-transduced primary human T cells were added at the following effector:target (E:T) ratios: 20:1, 10:1, 5:1, 2.5:1, 1.25:1, 0.625:1, 0.313:1. Fusion or WT A549 cells alone served as negative controls. Experiments were designed so that data collected from each plate included both WT and Fusion target cell killing by SJ1-4 or SJ1-12, each alongside a set of mock-transduced controls. SJ1-4 and SJ1-12 T cell data were collected from separate plates, with comparable results for mock-transduced controls across both plates. After addition of T cells, cell index was monitored every 15 min for 72 h. Cell index data were normalized to the maximum cell index immediately before T cell plating (i.e., at 24 h) using RTCA Software Pro (Agilent) and plotted using Prism version 9 (GraphPad).
Penning process and conditions on Berkeley Lights Lightning
Experiments were conducted on Berkeley Lights, Inc. Lightning platform and using OptoSelect 1500 chips. After priming, chips were washed and flushed with culture media. Cytokine capture beads (Human IFNγ B3 [Biolegend 740545], Human TNFα B7 [Biolegend 740711], and Human IL-2 A5 [Biolegend 740934]), T cells, and aAPCs were consecutively imported onto the Lightning OptoSelect 1500 chip (Berkeley Lights) and individual particles were loaded into NanoPens using opto-electropositioning (OEP) so that each NanoPen received one of each type of capture bead, one T cell, and one aAPC following manufacturer’s instructions. Briefly, cytokine capture beads were enumerated using a Luna Fx7 cell counter and brought to a concentration of 4.5 x 106 beads/mL for B-type beads and 5 x 106 beads/mL for A-type beads in bead dilution buffer (PBS, 0.2% w/v BSA, 0.1% w/v Tween 20 [Sigma-Aldrich P1379], and 0.09% w/v Sodium Azide [Sigma-Aldrich S2002]) prior to importing onto the Lightning OptoSelect 1500 chip. Next, 4 x 105 SJ1-4-mCherry-, SJ1-12-mCherry-, or mock-mCherry-transduced T cells were stained with Annexin V (Brilliant Violet 421-conjugated, Biolegend 640924) for 15 min at RT, washed with 400 μL of Annexin Binding Buffer (Biolegend 422201) and then resuspended in 100 μL of Loading Media (complete RPMI with 10% Loading Reagent [Berkeley Lights]) with 2.5mM CaCl2 (Sigma-Aldrich C4901) prior to loading. T cells that were mCherry-positive (i.e., transduced with TCR or mock), Annexin V-negative were selectively loaded into individual pens (SJ1-4- or SJ1-12-Transduced T cells were loaded into pens in field of views (FOVs) 2–9 and mock-transduced T cells were loaded into pens in FOVs 0–1 and 10–11). Last, fusion-tagBFP- or WT protein-tagBFP-transduced HLA-A∗68:02 aAPCs with a viability >90% were brought to a concentration of 4 x 105 cells/mL in 100 μL Loading Media with 2.5 mM CaCl2. After importing on to the chip, tagBFP-positive aAPC cells were selectively loaded into individual pens (fusion aAPCs were loaded into pens in FOVs 4–11, DNAJB1 WT aAPCs were loaded into pens in FOVs 0 and 2 and PRKACA WT aAPCs were loaded into pens in FOVs 1 and 3). Cytokine capture beads (BioLegend) were imported using the following parameters-nominal voltage: 7.0 V; cage shape: HomePlate; cage speed: 13 μm/s; cage line width: 23 μm. T cells and HLA-A∗68:02 aAPCs were imported using the following parameters-nominal voltage: 4.7 V; cage shape: HomePlate; cage speed: 8 μm/s; cage line width: 23 μm. Loading temperature was set to 37°C. Brightfield images of each chip were acquired automatically at the end of each step in the loading process and count algorithms were used to detect and count the number of beads (CnnBeadSegmentation algorithm) or cells (CnnBSegmentation algorithm for T cells and CnnJurkatSegmentation algorithm for aAPCs) in each pen.
Lightning culture conditions and staining
After T cell import, 1 μg of FastImmune CD28/CD49d (BD Biosciences 347690) was perfused through the chip followed by aAPCs import. SJ1-4-mCherry- or SJ1-12-mCherry-transduced T cells were co-cultured with fusion-tagBFP or WT (DNAJB1 or PRKACA)-tagBFP-transduced HLA-A∗68:02 aAPCs at a 1:1 ratio for 24 h in the presence of IFN-γ B3, TNF-α B7, and IL-2 A5 LEGENDplex cytokine capture beads (BioLegend 740545, 740711, 740934) and then imaged for phenotypic and functional analysis on the Lightning optofluidic system (Berkeley Lights). Mock-mCherry-transduced T cells from the same donor were used as negative controls. Cells were co-cultured on chip at 37°C and 5% CO2 in complete RPMI media (RPMI 1640, 10% FBS, 2mM L-glutamine, 100 U/mL/100 μg/mL penicillin/streptomycin) with recombinant human IL-7 and IL-15 cytokines (Biolegend 715302 and 715902) added to a final concentration of 10 ng/mL each and NucView 530 Caspase-3 substrate (Biotium 10408) added to a final concentration of 5 μM. Culture media was perfused through the chip at a flow rate of 0.01 μL/s. On-chip images were captured by the Lightning every 15 min during the co-culture in brightfield and fluorescence channels. Immediately following the 24-h co-culture, cytokine capture beads and cells were stained on-chip via perfusion of a detection cocktail containing LEGENDplex Human Th Panel Detection (BioLegend 741041), anti-human CD8a (FITC-conjugated, Biolegend 301006, clone RPA-T8), and anti-mouse TCR β chain (APC-conjugated, Biolegend 109212, clone H57-597) antibodies, followed by perfusion of LEGENDplex streptavidin-PE (BioLegend 740452) in PBS. After the final perfusions, on-chip images were taken again in brightfield and fluorescence channels. Data were analyzed using Assay Analyzer software (Berkeley Lights).
Bioluminescence imaging
Mice were injected intraperitoneally with 150 mg/kg of D-luciferin (PerkinElmer 122799) 5–10 min before imaging, anesthetized with isoflurane and imaged with a Xenogen In Vivo Imaging System (IVIS)-200 (PerkinElmer). The photons emitted from the luciferase-expressing tumor cells were quantified using Living Image software (Caliper Life Sciences). Total emitted photon flux (photons per second (p/s)) was used to determine tumor burden.
Immunofluorescence imaging of murine tumors
Tissues were fixed in 4% paraformaldehyde for 24 h prior to cryoprotection in 30% sucrose in PBS (w/v) and embedding in tissue freezing medium. 10 μm thick cryosections were blocked in PBS containing 2% bovine serum albumin, 5% normal donkey serum and 0.05% Tween 20 followed by incubation with the following primary antibodies, all at 1 μg/mL, overnight at 4°C: rabbit anti-GFP (Rockland Immunochemicals 600-401-215), goat anti-mCherry (Biorbyt orb11618), and camelid anti-tagBFP Alexa Fluor 647 (Antibodies Online ABIN6953244). Sections were washed in PBS followed by incubation with the following secondary antibodies for 1 h at RT: donkey anti-rabbit Alexa Fluor 488 (ThermoFisher Scientific A32790) and donkey anti-goat Alexa Fluor 555 (ThermoFisher Scientific A32816). Sections were mounted with Vectashield Vibrance mounting media with DAPI (Vector Laboratories H-1800) and imaged using an inverted Ti2 eclipse microscope (Nikon Instruments) equipped with a 20X 0.75 NA Plan Apo objective, SOLA light engine LED light source (Lumencorp) and Orca Fusion digital CMOS camera (Hamamatsu). Images were acquired and analyzed using NIS Elements software, version 5.30.05 (Nikon Instruments).
Murine tissue dissociation and flow cytometry
Tumor-bearing murine lungs, livers, and kidneys were first mechanically dissociated into small fragments, then further dissociated by incubation of tumor fragments in tumor digestion media (HBSS, 0.1% collagenase IV [Worthington Biochemical LS004188], 0.01% DNase I [Worthington Biochemical LS002145]) at 37°C for 30–60 min. After digestion, the resulting single cell suspensions were filtered through a 100 μM nylon mesh, using a 1 mL syringe plunger to break up any remaining solid tissue fragments, and washed with PBS. Murine spleens were mechanically dissociated using a 1 mL plunger to break up solid tissue fragments and filtered through a 100 μM nylon mesh, then washed with PBS. All tissue samples with observable red blood cells were subjected to red blood cell lysis prior to a final wash in PBS and cryopreservation of single cell suspensions. Single cell suspensions were later analyzed for expression of human T cell markers by flow cytometry. On the day of flow cytometry analysis, samples were thawed, washed with MACS buffer (PBS, 0.5% BSA, 2 mM EDTA), and blocked using anti-human Fc block (BD Biosciences 564220) for 10 min at RT. Cells were then stained with 1 μL Ghost Violet 510 viability dye (Tonbo Biosciences 13-080-T100) and a cocktail of surface antibodies for 20 min at RT, including 1 μL each of: anti-human CD3 (Brilliant Violet 421-conjugated, Biolegend 344834, clone SK7), anti-human CD8 (Brilliant Violet 785-conjugated, Biolegend 344740, clone SK1), anti-human CD4 (Alexa Fluor 700-conjugated, Biolegend 344622, clone SK3), anti-human CD137 (4-1BB) (APC-conjugated, Biolegend 309810, clone 4B4-1), anti-human CD279 (PD-1) (PE-conjugated, Biolegend 329906, clone EH12.2.H7), anti-human CD366 (TIM-3) (PE/Cy7-conjugated, Biolegend 345014, clone F38-2E2), anti-human CD197 (CCR7) (APC/Cy7-conjugated, Biolegend 353212, clone G043H7), anti-human CD45RO (Brilliant Violet 650-conjugated, Biolegend 304232, clone UCHL1). After staining, cells were washed with MACS buffer and analyzed by flow cytometry on a custom-configured BD Fortessa using FACSDiva software (Becton Dickinson). Flow cytometry data were analyzed using FlowJo version 10.7.2 software (BD Biosciences).
Statistics
Descriptive and comparative statistics were employed in the manuscript as described in the figure legends with the number of replicates indicated. Unless otherwise indicated, plots were generated using the ggplot2 R package and statistics were computed using the rstatix package. Within figure legends, ns indicates a not significant difference between comparator groups, and padj is the adjusted p-value.
Acknowledgments
We thank all study participants for their invaluable contributions to this work. We thank Greig Lennon (St. Jude Immunology Flow Core) for cell sorting, the St. Jude Hartwell Center for sequencing, and the St. Jude Animal Resource Center and Center for In Vivo Imaging and Therapeutics for assistance with in vivo studies and excellent animal care. We thank the Cell Therapy Lab Shared Resource (Medical College of Wisconsin Cancer Center) for Lightning runs. We thank the Childhood Solid Tumor Network, St. Jude Biorepository, and UTHSC Cancer Biorepository for providing primary human samples and associated data. Illustrations were created using BioRender.com. A.M.K. was supported by NCI Ruth L. Kirschstein Predoctoral Individual National Research Service Award F31CA254423 and the St. Jude Graduate School of Biomedical Sciences. A.W.P. was supported by NHMRC Investigator Fellowship 2016596. This work was funded by American Lebanese Syrian Associated Charities (ALSAC) at St. Jude, the Fibrolamellar Cancer Foundation (to P.S. and P.G.T.), NCI grant R01CA265009 (to P.G.T. and M.Y.), NIAID grant R01AI136514 (to P.G.T.), and the Assisi Foundation of Memphis (to C.D.).
Author contributions
Conceptualization, A.M.K., J.C.C., M.Y., A.E.Z., C.D., S.E.S., and P.G.T.; methodology, A.M.K., J.C.C., C.-H.C., C.G., K.P., P.N., M.B., R.C.M., E.K.A., M.V.P., H.K., A.A.M., E.A.F., N.P.C., A.W.P., A.E.Z., C.D., S.E.S., and P.G.T.; formal analysis, A.M.K., J.C.C., C.G., T.K., K.P., P.N., M.V.P., and H.K.; investigation, A.M.K., C.-H.C., C.G., K.P., T.K., R.K.S., S.C., P.N., X.Z., J.W., M.B., E.K.A., and W.A.; resources, E.K.A., R.B., T.W., B.G., M.M., E.S.G., A.J.M., Y.J., E.A.F., M.Y., P.S., S.M.F., E.S., S.G., A.E.Z., C.D., and S.E.S.; data curation, A.M.K., J.C.C., A.A.M., T.W., D.L., M.M., E.S., and P.S.; writing – original draft, A.M.K.; writing – review and editing, A.M.K., J.C.C., R.C.M., A.A.M., M.Y., P.S., A.W.P., S.G., A.E.Z., S.E.S., and P.G.T.; visualization, A.M.K., J.C.C., C.G., K.P., T.K., S.C., P.N., R.C.M., E.K.A., and S.G.; supervision, J.C.C., E.S.G., Y.J., E.A.F., P.S., N.P.C., A.W.P., E.S., S.G., A.E.Z., C.D., S.E.S., and P.G.T.; funding acquisition, A.M.K., M.Y., P.S., A.W.P., C.D., and P.G.T. All co-authors reviewed and approved the final manuscript.
Declaration of interests
J.C.C., A.M.K., A.E.Z., S.E.S., and P.G.T. have a patent application for TCRs for treating FLC. J.C.C. has additional patent applications in the field of immunotherapy. A.W.P. is a member of the scientific advisory board (SAB) of Bioinformatics Solutions Inc. (Canada), shareholder and SAB member of Evaxion Biotech (Denmark), consultant for Grey Wolf Therapeutics (UK), and cofounder of Resseptor Therapeutics (Melbourne). S.G. is a co-inventor on patent applications in the fields of cell/gene therapy for cancer, consultant of TESSA Therapeutics, member of the Data and Safety Monitoring Board of Immatics, and SAB member of Be Biopharma and has received honoraria from Tidal, Catamaran Bio, and Sanofi within the last 2 years. S.E.S. is a cofounder, stockholder, and paid consultant for Gliknik Inc., serves on the SAB and holds stock options for Virion Inc., and receives royalties from the Mayo Clinic for licensed IP surrounding manipulation of the PD-1:PD-L1 pathway for cancer treatment. P.G.T. is on the SAB of Immunoscape and Shennon Bio, received personal fees and research support from Elevate Bio, and consulted for 10×, Illumina, Pfizer, Cytoagents, and JNJ.
Published: March 19, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2024.101469.
Contributor Information
Scott E. Strome, Email: sstrome3@gmail.com.
Paul G. Thomas, Email: paul.thomas@stjude.org.
Supplemental information
References
- 1.Simon S.M. Fighting rare cancers: lessons from fibrolamellar hepatocellular carcinoma. Nat. Rev. Cancer. 2023;23:335–346. doi: 10.1038/s41568-023-00554-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.O’Neill A.F., Church A.J., Perez-Atayde A.R., Shaikh R., Marcus K.J., Vakili K. Fibrolamellar carcinoma: An entity all its own. Curr. Probl. Cancer. 2021;45 doi: 10.1016/j.currproblcancer.2021.100770. [DOI] [PubMed] [Google Scholar]
- 3.Dinh T.A., Utria A.F., Barry K.C., Ma R., Abou-Alfa G.K., Gordan J.D., Jaffee E.M., Scott J.D., Zucman-Rossi J., O’Neill A.F., et al. A framework for fibrolamellar carcinoma research and clinical trials. Nat. Rev. Gastroenterol. Hepatol. 2022;19:328–342. doi: 10.1038/s41575-022-00580-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.El-Serag H.B., Davila J.A. Is fibrolamellar carcinoma different from hepatocellular carcinoma? A US population-based study. Hepatol. Baltim. Md. 2004;39:798–803. doi: 10.1002/hep.20096. [DOI] [PubMed] [Google Scholar]
- 5.Eggert T., McGlynn K.A., Duffy A., Manns M.P., Greten T.F., Altekruse S.F. Fibrolamellar hepatocellular carcinoma in the USA, 2000-2010: A detailed report on frequency, treatment and outcome based on the Surveillance, Epidemiology, and End Results database. United Eur. Gastroenterol. J. 2013;1:351–357. doi: 10.1177/2050640613501507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Edmondson H.A. Differential diagnosis of tumors and tumor-like lesions of liver in infancy and childhood. AMA. J. Dis. Child. 1956;91:168–186. doi: 10.1001/archpedi.1956.02060020170015. [DOI] [PubMed] [Google Scholar]
- 7.Mavros M.N., Mayo S.C., Hyder O., Pawlik T.M. A systematic review: treatment and prognosis of patients with fibrolamellar hepatocellular carcinoma. J. Am. Coll. Surg. 2012;215:820–830. doi: 10.1016/j.jamcollsurg.2012.08.001. [DOI] [PubMed] [Google Scholar]
- 8.Stipa F., Yoon S.S., Liau K.H., Fong Y., Jarnagin W.R., D’Angelica M., Abou-Alfa G., Blumgart L.H., DeMatteo R.P. Outcome of patients with fibrolamellar hepatocellular carcinoma. Cancer. 2006;106:1331–1338. doi: 10.1002/cncr.21703. [DOI] [PubMed] [Google Scholar]
- 9.Kaseb A.O., Shama M., Sahin I.H., Nooka A., Hassabo H.M., Vauthey J.-N., Aloia T., Abbruzzese J.L., Subbiah I.M., Janku F., et al. Prognostic indicators and treatment outcome in 94 cases of fibrolamellar hepatocellular carcinoma. Oncology. 2013;85:197–203. doi: 10.1159/000354698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weeda V.B., Murawski M., McCabe A.J., Maibach R., Brugières L., Roebuck D., Fabre M., Zimmermann A., Otte J.B., Sullivan M., et al. Fibrolamellar variant of hepatocellular carcinoma does not have a better survival than conventional hepatocellular carcinoma--results and treatment recommendations from the Childhood Liver Tumour Strategy Group (SIOPEL) experience. Eur. J. Cancer. 2013;49:2698–2704. doi: 10.1016/j.ejca.2013.04.012. [DOI] [PubMed] [Google Scholar]
- 11.Berkovitz A., Migler R.D., Qureshi A., Rosemore C., Torbenson M.S., Vaughan R., Marcotte E., Simon S.M. Clinical and demographic predictors of survival for fibrolamellar carcinoma patients—A patient community, registry-based study. Hepatol. Commun. 2022;6:3539–3549. doi: 10.1002/hep4.2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Honeyman J.N., Simon E.P., Robine N., Chiaroni-Clarke R., Darcy D.G., Lim I.I.P., Gleason C.E., Murphy J.M., Rosenberg B.R., Teegan L., et al. Detection of a Recurrent DNAJB1-PRKACA Chimeric Transcript in Fibrolamellar Hepatocellular Carcinoma. Science. 2014;343:1010–1014. doi: 10.1126/science.1249484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kastenhuber E.R., Lalazar G., Houlihan S.L., Tschaharganeh D.F., Baslan T., Chen C.-C., Requena D., Tian S., Bosbach B., Wilkinson J.E., et al. DNAJB1-PRKACA fusion kinase interacts with β-catenin and the liver regenerative response to drive fibrolamellar hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA. 2017;114:13076–13084. doi: 10.1073/pnas.1716483114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Engelholm L.H., Riaz A., Serra D., Dagnæs-Hansen F., Johansen J.V., Santoni-Rugiu E., Hansen S.H., Niola F., Frödin M. CRISPR/Cas9 Engineering of Adult Mouse Liver Demonstrates That the Dnajb1–Prkaca Gene Fusion Is Sufficient to Induce Tumors Resembling Fibrolamellar Hepatocellular Carcinoma. Gastroenterology. 2017;153:1662–1673.e10. doi: 10.1053/j.gastro.2017.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Neumayer C., Ng D., Jiang C.S., Qureshi A., Lalazar G., Vaughan R., Simon S.M. Oncogenic Addiction of Fibrolamellar Hepatocellular Carcinoma to the Fusion Kinase DNAJB1-PRKACA. Clin. Cancer Res. 2023;29:271–278. doi: 10.1158/1078-0432.CCR-22-1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Riggle K.M., Turnham R., Scott J.D., Yeung R.S., Riehle K.J. Fibrolamellar Hepatocellular Carcinoma: Mechanistic Distinction From Adult Hepatocellular Carcinoma. Pediatr. Blood Cancer. 2016;63:1163–1167. doi: 10.1002/pbc.25970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Turnham R.E., Smith F.D., Kenerson H.L., Omar M.H., Golkowski M., Garcia I., Bauer R., Lau H.-T., Sullivan K.M., Langeberg L.K., et al. An acquired scaffolding function of the DNAJ-PKAc fusion contributes to oncogenic signaling in fibrolamellar carcinoma. Elife. 2019;8 doi: 10.7554/eLife.44187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dinh T.A., Sritharan R., Smith F.D., Francisco A.B., Ma R.K., Bunaciu R.P., Kanke M., Danko C.G., Massa A.P., Scott J.D., Sethupathy P. Hotspots of Aberrant Enhancer Activity in Fibrolamellar Carcinoma Reveal Candidate Oncogenic Pathways and Therapeutic Vulnerabilities. Cell Rep. 2020;31 doi: 10.1016/j.celrep.2020.03.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chan G.K.L., Maisel S., Hwang Y.C., Pascual B.C., Wolber R.R.B., Vu P., Patra K.C., Bouhaddou M., Kenerson H.L., Lim H.C., et al. Oncogenic PKA signaling increases c-MYC protein expression through multiple targetable mechanisms. Elife. 2023;12 doi: 10.7554/eLife.69521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blueprint Medicines Announces New Drug Discovery Program Targeting PRKACA Kinase Fusions for the Treatment of Fibrolamellar Carcinoma at 10th International Liver Cancer Association (ILCA) Annual Conference | Blueprint Medicines Corp. https://ir.blueprintmedicines.com/news-releases/news-release-details/blueprint-medicines-announces-new-drug-discovery-program.
- 21.Abou-Alfa G.K., Mayer R., Venook A.P., O’Neill A.F., Beg M.S., LaQuaglia M., Kingham P.T., Kobos R., Basturk O., Brennan C., et al. Phase II Multicenter, Open-Label Study of Oral ENMD-2076 for the Treatment of Patients with Advanced Fibrolamellar Carcinoma. Oncol. 2020;25:e1837–e1845. doi: 10.1634/theoncologist.2020-0093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.El Dika I., Mayer R.J., Venook A.P., Capanu M., LaQuaglia M.P., Kobos R., O’Neill A.F., Chou J.F., Ly M., Ang C., et al. A Multicenter Randomized Three-Arm Phase II Study of (1) Everolimus, (2) Estrogen Deprivation Therapy (EDT) with Leuprolide + Letrozole, and (3) Everolimus + EDT in Patients with Unresectable Fibrolamellar Carcinoma. Oncol. 2020;25:925–e1603. doi: 10.1634/theoncologist.2020-0367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abou-Alfa G.K., Meyer T., Zhang J., Sherrin S., Yaqubie A., Clemens O’Neill A., Xu F., Eli L.D., Harding J.J., O’Reilly E.M., et al. Evaluation of neratinib (N), pembrolizumab (P), everolimus (E), and nivolumab (V) in patients (pts) with fibrolamellar carcinoma (FLC) J. Clin. Oncol. 2021;39:310. doi: 10.1200/JCO.2021.39.3_suppl.310. [DOI] [Google Scholar]
- 24.Schumacher T.N., Scheper W., Kvistborg P. Cancer Neoantigens. Annu. Rev. Immunol. 2019;37:173–200. doi: 10.1146/annurev-immunol-042617-053402. [DOI] [PubMed] [Google Scholar]
- 25.Chang T.-C., Carter R.A., Li Y., Li Y., Wang H., Edmonson M.N., Chen X., Arnold P., Geiger T.L., Wu G., et al. The neoepitope landscape in pediatric cancers. Genome Med. 2017;9:78. doi: 10.1186/s13073-017-0468-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang W., Lee K.-W., Srivastava R.M., Kuo F., Krishna C., Chowell D., Makarov V., Hoen D., Dalin M.G., Wexler L., et al. Immunogenic neoantigens derived from gene fusions stimulate T cell responses. Nat. Med. 2019;25:767–775. doi: 10.1038/s41591-019-0434-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yarchoan M., Johnson B.A., Lutz E.R., Laheru D.A., Jaffee E.M. Targeting neoantigens to augment antitumour immunity. Nat. Rev. Cancer. 2017;17:209–222. doi: 10.1038/nrc.2016.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gubin M.M., Zhang X., Schuster H., Caron E., Ward J.P., Noguchi T., Ivanova Y., Hundal J., Arthur C.D., Krebber W.-J., et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature. 2014;515:577–581. doi: 10.1038/nature13988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ott P.A., Hu Z., Keskin D.B., Shukla S.A., Sun J., Bozym D.J., Zhang W., Luoma A., Giobbie-Hurder A., Peter L., et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. 2017;547:217–221. doi: 10.1038/nature22991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen K.Y., Popovic A., Hsiehchen D., Baretti M., Griffith P., Bista R., Baghdadi A., Kamel I.R., Simon S.M., Migler R.D., Yarchoan M. Clinical Outcomes in Fibrolamellar Hepatocellular Carcinoma Treated with Immune Checkpoint Inhibitors. Cancers. 2022;14:5347. doi: 10.3390/cancers14215347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Berger R., Dinstag G., Tirosh O., Schiff E., Kleiner D., Aldape K.D., Ruppin E., Beker T., Kurzrock R. Fibrolamellar carcinoma transcriptomic-based treatment prediction: complete response after nivolumab and ipilimumab. J. Immunother. Cancer. 2022;10 doi: 10.1136/jitc-2022-005620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.De Toni E.N., Roessler D. Using dual checkpoint blockade to treat fibrolamellar hepatocellular carcinoma. Gut. 2020;69:2056–2058. doi: 10.1136/gutjnl-2020-320604. [DOI] [PubMed] [Google Scholar]
- 33.Foy S.P., Jacoby K., Bota D.A., Hunter T., Pan Z., Stawiski E., Ma Y., Lu W., Peng S., Wang C.L., et al. Non-viral precision T cell receptor replacement for personalized cell therapy. Nature. 2023;615:687–696. doi: 10.1038/s41586-022-05531-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim S.P., Vale N.R., Zacharakis N., Krishna S., Yu Z., Gasmi B., Gartner J.J., Sindiri S., Malekzadeh P., Deniger D.C., et al. Adoptive Cellular Therapy with Autologous Tumor-Infiltrating Lymphocytes and T-cell Receptor–Engineered T Cells Targeting Common p53 Neoantigens in Human Solid Tumors. Cancer Immunol. Res. 2022;10:932–946. doi: 10.1158/2326-6066.CIR-22-0040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leidner R., Sanjuan Silva N., Huang H., Sprott D., Zheng C., Shih Y.-P., Leung A., Payne R., Sutcliffe K., Cramer J., et al. Neoantigen T-Cell Receptor Gene Therapy in Pancreatic Cancer. N. Engl. J. Med. 2022;386:2112–2119. doi: 10.1056/NEJMoa2119662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yamamoto T.N., Kishton R.J., Restifo N.P. Developing neoantigen-targeted T cell–based treatments for solid tumors. Nat. Med. 2019;25:1488–1499. doi: 10.1038/s41591-019-0596-y. [DOI] [PubMed] [Google Scholar]
- 37.DeRenzo C., Gottschalk S. Genetic Modification Strategies to Enhance CAR T Cell Persistence for Patients With Solid Tumors. Front. Immunol. 2019;10 doi: 10.3389/fimmu.2019.00218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bell M., Gottschalk S. Engineered Cytokine Signaling to Improve CAR T Cell Effector Function. Front. Immunol. 2021;12 doi: 10.3389/fimmu.2021.684642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Prinzing B., Zebley C.C., Petersen C.T., Fan Y., Anido A.A., Yi Z., Nguyen P., Houke H., Bell M., Haydar D., et al. Deleting DNMT3A in CAR T cells prevents exhaustion and enhances antitumor activity. Sci. Transl. Med. 2021;13 doi: 10.1126/scitranslmed.abh0272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lange S., Sand L.G.L., Bell M., Patil S.L., Langfitt D., Gottschalk S. A Chimeric GM-CSF/IL18 Receptor to Sustain CAR T-cell Function. Cancer Discov. 2021;11:1661–1671. doi: 10.1158/2159-8290.CD-20-0896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim A.K., Gani F., Layman A.J., Besharati S., Zhu Q., Succaria F., Engle E.L., Bhaijee F., Goggins M.B., Llosa N.J., et al. Multiple Immune-Suppressive Mechanisms in Fibrolamellar Carcinoma. Cancer Immunol. Res. 2019;7:805–812. doi: 10.1158/2326-6066.CIR-18-0499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ross H.M., Daniel H.D.J., Vivekanandan P., Kannangai R., Yeh M.M., Wu T.-T., Makhlouf H.R., Torbenson M. Fibrolamellar carcinomas are positive for CD68. Mod. Pathol. 2011;24:390–395. doi: 10.1038/modpathol.2010.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Francisco A.B., Kanke M., Massa A.P., Dinh T.A., Sritharan R., Vakili K., Bardeesy N., Sethupathy P. Multiomic analysis of microRNA-mediated regulation reveals a proliferative axis involving miR-10b in fibrolamellar carcinoma. JCI Insight. 2022;7 doi: 10.1172/jci.insight.154743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gonzalez-Galarza F.F., McCabe A., Santos E.J.M.D., Jones J., Takeshita L., Ortega-Rivera N.D., Cid-Pavon G.M.D., Ramsbottom K., Ghattaoraya G., Alfirevic A., et al. Allele frequency net database (AFND) 2020 update: gold-standard data classification, open access genotype data and new query tools. Nucleic Acids Res. 2020;48:D783–D788. doi: 10.1093/nar/gkz1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Reynisson B., Alvarez B., Paul S., Peters B., Nielsen M. NetMHCpan-4.1 and NetMHCIIpan-4.0: improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res. 2020;48:W449–W454. doi: 10.1093/nar/gkaa379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bauer J., Köhler N., Maringer Y., Bucher P., Bilich T., Zwick M., Dicks S., Nelde A., Dubbelaar M., Scheid J., et al. The oncogenic fusion protein DNAJB1-PRKACA can be specifically targeted by peptide-based immunotherapy in fibrolamellar hepatocellular carcinoma. Nat. Commun. 2022;13:6401. doi: 10.1038/s41467-022-33746-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lowery F.J., Krishna S., Yossef R., Parikh N.B., Chatani P.D., Zacharakis N., Parkhurst M.R., Levin N., Sindiri S., Sachs A., et al. Molecular signatures of antitumor neoantigen-reactive T cells from metastatic human cancers. Science. 2022;375:877–884. doi: 10.1126/science.abl5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cimen Bozkus C., Blazquez A.B., Enokida T., Bhardwaj N. A T-cell-based immunogenicity protocol for evaluating human antigen-specific responses. STAR Protoc. 2021;2 doi: 10.1016/j.xpro.2021.100758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bradley P., Thomas P.G. Using T Cell Receptor Repertoires to Understand the Principles of Adaptive Immune Recognition. Annu. Rev. Immunol. 2019;37:547–570. doi: 10.1146/annurev-immunol-042718-041757. [DOI] [PubMed] [Google Scholar]
- 50.Dash P., Fiore-Gartland A.J., Hertz T., Wang G.C., Sharma S., Souquette A., Crawford J.C., Clemens E.B., Nguyen T.H.O., Kedzierska K., et al. Quantifiable predictive features define epitope-specific T cell receptor repertoires. Nature. 2017;547:89–93. doi: 10.1038/nature22383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mayer-Blackwell K., Schattgen S., Cohen-Lavi L., Crawford J.C., Souquette A., Gaevert J.A., Hertz T., Thomas P.G., Bradley P., Fiore-Gartland A. TCR meta-clonotypes for biomarker discovery with tcrdist3 enabled identification of public, HLA-restricted clusters of SARS-CoV-2 TCRs. Elife. 2021;10 doi: 10.7554/eLife.68605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Glanville J., Huang H., Nau A., Hatton O., Wagar L.E., Rubelt F., Ji X., Han A., Krams S.M., Pettus C., et al. Identifying specificity groups in the T cell receptor repertoire. Nature. 2017;547:94–98. doi: 10.1038/nature22976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pogorelyy M.V., Rosati E., Minervina A.A., Mettelman R.C., Scheffold A., Franke A., Bacher P., Thomas P.G. Resolving SARS-CoV-2 CD4+ T cell specificity via reverse epitope discovery. Cell Rep. Med. 2022;3 doi: 10.1016/j.xcrm.2022.100697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mudd P.A., Minervina A.A., Pogorelyy M.V., Turner J.S., Kim W., Kalaidina E., Petersen J., Schmitz A.J., Lei T., Haile A., et al. SARS-CoV-2 mRNA vaccination elicits a robust and persistent T follicular helper cell response in humans. Cell. 2022;185:603–613.e15. doi: 10.1016/j.cell.2021.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Minervina A.A., Pogorelyy M.V., Kirk A.M., Crawford J.C., Allen E.K., Chou C.-H., Mettelman R.C., Allison K.J., Lin C.-Y., Brice D.C., et al. SARS-CoV-2 antigen exposure history shapes phenotypes and specificity of memory CD8+ T cells. Nat. Immunol. 2022;23:781–790. doi: 10.1038/s41590-022-01184-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mazouz S., Boisvert M., Abdel-Hakeem M.S., Khedr O., Bruneau J., Shoukry N.H. Expansion of Unique Hepatitis C Virus-Specific Public CD8+ T Cell Clonotypes during Acute Infection and Reinfection. J. Immunol. 2021;207:1180–1193. doi: 10.4049/jimmunol.2001386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Minervina A.A., Komech E.A., Titov A., Bensouda Koraichi M., Rosati E., Mamedov I.Z., Franke A., Efimov G.A., Chudakov D.M., Mora T., et al. Longitudinal high-throughput TCR repertoire profiling reveals the dynamics of T-cell memory formation after mild COVID-19 infection. Elife. 2021;10 doi: 10.7554/eLife.63502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kakarla S., Chow K.K.H., Mata M., Shaffer D.R., Song X.-T., Wu M.-F., Liu H., Wang L.L., Rowley D.R., Pfizenmaier K., Gottschalk S. Antitumor Effects of Chimeric Receptor Engineered Human T Cells Directed to Tumor Stroma. Mol. Ther. 2013;21:1611–1620. doi: 10.1038/mt.2013.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bendle G.M., Linnemann C., Hooijkaas A.I., Bies L., de Witte M.A., Jorritsma A., Kaiser A.D.M., Pouw N., Debets R., Kieback E., et al. Lethal graft-versus-host disease in mouse models of T cell receptor gene therapy. Nat. Med. 2010;16:565–570. doi: 10.1038/nm.2128. [DOI] [PubMed] [Google Scholar]
- 60.Mhaidly R., Verhoeyen E. Humanized Mice Are Precious Tools for Preclinical Evaluation of CAR T and CAR NK Cell Therapies. Cancers. 2020;12:1915. doi: 10.3390/cancers12071915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Duncan B.B., Dunbar C.E., Ishii K. Applying a clinical lens to animal models of CAR-T cell therapies. Mol. Ther. Methods Clin. Dev. 2022;27:17–31. doi: 10.1016/j.omtm.2022.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yarchoan M., Hopkins A., Jaffee E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017;377:2500–2501. doi: 10.1056/NEJMc1713444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.McGrail D.J., Pilié P.G., Rashid N.U., Voorwerk L., Slagter M., Kok M., Jonasch E., Khasraw M., Heimberger A.B., Lim B., et al. High tumor mutation burden fails to predict immune checkpoint blockade response across all cancer types. Ann. Oncol. 2021;32:661–672. doi: 10.1016/j.annonc.2021.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zamora A.E., Crawford J.C., Allen E.K., Guo X.-Z.J., Bakke J., Carter R.A., Abdelsamed H.A., Moustaki A., Li Y., Chang T.-C., et al. Pediatric patients with acute lymphoblastic leukemia generate abundant and functional neoantigen-specific CD8+ T cell responses. Sci. Transl. Med. 2019;11 doi: 10.1126/scitranslmed.aat8549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Goodman A.M., Castro A., Pyke R.M., Okamura R., Kato S., Riviere P., Frampton G., Sokol E., Zhang X., Ball E.D., et al. MHC-I genotype and tumor mutational burden predict response to immunotherapy. Genome Med. 2020;12:45. doi: 10.1186/s13073-020-00743-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Marty R., Kaabinejadian S., Rossell D., Slifker M.J., van de Haar J., Engin H.B., de Prisco N., Ideker T., Hildebrand W.H., Font-Burgada J., Carter H. MHC-I Genotype Restricts the Oncogenic Mutational Landscape. Cell. 2017;171:1272–1283.e15. doi: 10.1016/j.cell.2017.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Puig-Saus C., Sennino B., Peng S., Wang C.L., Pan Z., Yuen B., Purandare B., An D., Quach B.B., Nguyen D., et al. Neoantigen-targeted CD8+ T cell responses with PD-1 blockade therapy. Nature. 2023;615:697–704. doi: 10.1038/s41586-023-05787-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Oh D.Y., Fong L. Cytotoxic CD4+ T cells in cancer: Expanding the immune effector toolbox. Immunity. 2021;54:2701–2711. doi: 10.1016/j.immuni.2021.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kravtsov D.S., Erbe A.K., Sondel P.M., Rakhmilevich A.L. Roles of CD4+ T cells as mediators of antitumor immunity. Front. Immunol. 2022;13 doi: 10.3389/fimmu.2022.972021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kreiter S., Vormehr M., van de Roemer N., Diken M., Löwer M., Diekmann J., Boegel S., Schrörs B., Vascotto F., Castle J.C., et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature. 2015;520:692–696. doi: 10.1038/nature14426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sahin U., Derhovanessian E., Miller M., Kloke B.-P., Simon P., Löwer M., Bukur V., Tadmor A.D., Luxemburger U., Schrörs B., et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature. 2017;547:222–226. doi: 10.1038/nature23003. [DOI] [PubMed] [Google Scholar]
- 72.Keskin D.B., Anandappa A.J., Sun J., Tirosh I., Mathewson N.D., Li S., Oliveira G., Giobbie-Hurder A., Felt K., Gjini E., et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature. 2019;565:234–239. doi: 10.1038/s41586-018-0792-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Oliveira G., Stromhaug K., Klaeger S., Kula T., Frederick D.T., Le P.M., Forman J., Huang T., Li S., Zhang W., et al. Phenotype, specificity and avidity of antitumour CD8+ T cells in melanoma. Nature. 2021;596:119–125. doi: 10.1038/s41586-021-03704-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zheng L., Qin S., Si W., Wang A., Xing B., Gao R., Ren X., Wang L., Wu X., Zhang J., et al. Pan-cancer single-cell landscape of tumor-infiltrating T cells. Science. 2021;374:abe6474. doi: 10.1126/science.abe6474. [DOI] [PubMed] [Google Scholar]
- 75.Zheng C., Zheng L., Yoo J.-K., Guo H., Zhang Y., Guo X., Kang B., Hu R., Huang J.Y., Zhang Q., et al. Landscape of Infiltrating T Cells in Liver Cancer Revealed by Single-Cell Sequencing. Cell. 2017;169:1342–1356.e16. doi: 10.1016/j.cell.2017.05.035. [DOI] [PubMed] [Google Scholar]
- 76.Wu T.D., Madireddi S., de Almeida P.E., Banchereau R., Chen Y.-J.J., Chitre A.S., Chiang E.Y., Iftikhar H., O’Gorman W.E., Au-Yeung A., et al. Peripheral T cell expansion predicts tumour infiltration and clinical response. Nature. 2020;579:274–278. doi: 10.1038/s41586-020-2056-8. [DOI] [PubMed] [Google Scholar]
- 77.Nutsch K., Banta K.L., Wu T.D., Tran C.W., Mittman S., Duong E., Nabet B.Y., Qu Y., Patil N.S., Chiang E.Y., et al. Regular Abstracts – Part 2. BMJ Publishing Group Ltd; 2023. 579-B TIGIT and PD-L1 co-blockade promotes clonal expansion of non-exhausted anti-tumour CD8 + T cells by facilitating costimulation; p. A1758. [DOI] [Google Scholar]
- 78.Pogorelyy M.V., Minervina A.A., Shugay M., Chudakov D.M., Lebedev Y.B., Mora T., Walczak A.M. Detecting T cell receptors involved in immune responses from single repertoire snapshots. PLoS Biol. 2019;17 doi: 10.1371/journal.pbio.3000314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Barker D.J., Maccari G., Georgiou X., Cooper M.A., Flicek P., Robinson J., Marsh S.G.E. The IPD-IMGT/HLA Database. Nucleic Acids Res. 2023;51:D1053–D1060. doi: 10.1093/nar/gkac1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Giudicelli V., Chaume D., Lefranc M.-P. IMGT/GENE-DB: a comprehensive database for human and mouse immunoglobulin and T cell receptor genes. Nucleic Acids Res. 2005;33:D256–D261. doi: 10.1093/nar/gki010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang G.C., Dash P., McCullers J.A., Doherty P.C., Thomas P.G. T cell receptor αβ diversity inversely correlates with pathogen-specific antibody levels in human cytomegalovirus infection. Sci. Transl. Med. 2012;4:128ra42. doi: 10.1126/scitranslmed.3003647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Aguiar V.R.C., César J., Delaneau O., Dermitzakis E.T., Meyer D. Expression estimation and eQTL mapping for HLA genes with a personalized pipeline. PLoS Genet. 2019;15 doi: 10.1371/journal.pgen.1008091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bray N.L., Pimentel H., Melsted P., Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016;34:525–527. doi: 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- 84.Szolek A., Schubert B., Mohr C., Sturm M., Feldhahn M., Kohlbacher O. OptiType: precision HLA typing from next-generation sequencing data. Bioinforma. Oxf. Engl. 2014;30:3310–3316. doi: 10.1093/bioinformatics/btu548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Karosiene E., Lundegaard C., Lund O., Nielsen M. NetMHCcons: a consensus method for the major histocompatibility complex class I predictions. Immunogenetics. 2012;64:177–186. doi: 10.1007/s00251-011-0579-8. [DOI] [PubMed] [Google Scholar]
- 86.Hao Y., Hao S., Andersen-Nissen E., Mauck W.M., Zheng S., Butler A., Lee M.J., Wilk A.J., Darby C., Zager M., et al. Integrated analysis of multimodal single-cell data. Cell. 2021;184:3573–3587.e29. doi: 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Schattgen S.A., Guion K., Crawford J.C., Souquette A., Barrio A.M., Stubbington M.J.T., Thomas P.G., Bradley P. Integrating T cell receptor sequences and transcriptional profiles by clonotype neighbor graph analysis (CoNGA) Nat. Biotechnol. 2022;40:54–63. doi: 10.1038/s41587-021-00989-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Heather J.M., Spindler M.J., Alonso M.H., Shui Y.I., Millar D.G., Johnson D.S., Cobbold M., Hata A.N. Stitchr: stitching coding TCR nucleotide sequences from V/J/CDR3 information. Nucleic Acids Res. 2022;50:e68. doi: 10.1093/nar/gkac190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Stewart E., Federico S.M., Chen X., Shelat A.A., Bradley C., Gordon B., Karlstrom A., Twarog N.R., Clay M.R., Bahrami A., et al. Orthotopic patient-derived xenografts of paediatric solid tumours. Nature. 2017;549:96–100. doi: 10.1038/nature23647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Morimoto S., Fujiki F., Kondo K., Nakajima H., Kobayashi Y., Inatome M., Aoyama N., Nishida Y., Tsuboi A., Oka Y., et al. Establishment of a novel platform cell line for efficient and precise evaluation of T cell receptor functional avidity. Oncotarget. 2018;9:34132–34141. doi: 10.18632/oncotarget.26139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Brodsky F.M., Parham P. Monomorphic anti-HLA-A,B,C monoclonal antibodies detecting molecular subunits and combinatorial determinants. J. Immunol. 1982;128:129–135. [PubMed] [Google Scholar]
- 92.Purcell A.W., Ramarathinam S.H., Ternette N. Mass spectrometry-based identification of MHC-bound peptides for immunopeptidomics. Nat. Protoc. 2019;14:1687–1707. doi: 10.1038/s41596-019-0133-y. [DOI] [PubMed] [Google Scholar]
- 93.Pandey K., Ramarathinam S.H., Purcell A.W. Isolation of HLA Bound Peptides by Immunoaffinity Capture and Identification by Mass Spectrometry. Curr. Protoc. 2021;1:e92. doi: 10.1002/cpz1.92. [DOI] [PubMed] [Google Scholar]
- 94.Escher C., Reiter L., MacLean B., Ossola R., Herzog F., Chilton J., MacCoss M.J., Rinner O. Using iRT, a normalized retention time for more targeted measurement of peptides. Proteomics. 2012;12:1111–1121. doi: 10.1002/pmic.201100463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dudley M.E., Wunderlich J.R., Shelton T.E., Even J., Rosenberg S.A. Generation of tumor-infiltrating lymphocyte cultures for use in adoptive transfer therapy for melanoma patients. J. Immunother. 2003;26:332–342. doi: 10.1097/00002371-200307000-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Dolton G., Tungatt K., Lloyd A., Bianchi V., Theaker S.M., Trimby A., Holland C.J., Donia M., Godkin A.J., Cole D.K., et al. More tricks with tetramers: a practical guide to staining T cells with peptide–MHC multimers. Immunology. 2015;146:11–22. doi: 10.1111/imm.12499. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
De-identified patient single-cell sequencing data have been deposited to the Sequence Read Archive at SRA: PRJNA1070700.
-
•
Immunopeptidomics data have been deposited to the PRoteomics IDEntifications (PRIDE) database at PRIDE: PXD042316 (https://doi.org/10.6019/PXD042316).
-
•
Publicly available data analyzed in this study were obtained from the European Genome-Phenome Archive (EGA) at EGA: EGAS00001004169, Gene Expression Omnibus (GEO) at GEO: GSE181922 and GEO: GSE183393, Zenodo at accession number Zenodo: 4065547, and immuneACCESS at immuneACCESS: Mazouz_Shoukry_2021 (https://doi.org/10.21417/SM2021JI).
-
•
Other data generated in this study are available within the article and its supplemental data or will be made available upon request.