

Abstract
Genetic variants in relevant genes coexisting with MRI lesions in children with drug‐resistant epilepsy (DRE) can negatively influence epilepsy surgery outcomes. Still, presurgical evaluation does not include genetic diagnostics routinely. Here, we report our presurgical evaluation algorithm that includes routine genetic testing. We analyzed retrospectively the data of 68 children with DRE operated at a mean age of 7.8 years (IQR: 8.1 years) at our center. In 49 children, genetic test results were available. We identified 21 gene variants (ACMG III: n = 7, ACMG IV: n = 2, ACMG V: n = 12) in 19 patients (45.2%) in the genes TSC1, TSC2, MECP2, DEPDC5, HUWE1, GRIN1, ASH1I, TRIO, KIF5C, CDON, ANKD11, TGFBR2, ATN1, COL4A1, JAK2, KCNQ2, ATP1A2, and GLI3 by whole‐exome sequencing as well as deletions and duplications by array CGH in six patients. While the results did not change the surgery indication, they supported counseling with respect to postoperative chance of seizure freedom and weaning of antiseizure medication (ASM). The presence of genetic findings leads to the postoperative retention of at least one ASM. In our cohort, the International League against Epilepsy (ILAE) seizure outcome did not differ between patients with and without abnormal genetic findings. However, in the 7/68 patients with an unsatisfactory ILAE seizure outcome IV or V 12 months postsurgery, 2 had an abnormal or suspicious genetic finding as a putative explanation for persisting seizures postsurgery, and 3 had received palliative surgery including one TSC patient. This study highlights the importance of genetic testing in children with DRE to address putative underlying germline variants as genetic epilepsy causes or predisposing factors that guide patient and/or parent counseling on a case‐by‐case with respect to their individual chance of postoperative seizure freedom and ASM weaning.
Plain Language Summary
Genetic variants in children with drug‐resistant epilepsy (DRE) can negatively influence epilepsy surgery outcomes. However, presurgical evaluation does not include genetic diagnostics routinely. This retrospective study analyzed the genetic testing results of the 68 pediatric patients who received epilepsy surgery in our center. We identified 21 gene variants by whole‐exome sequencing as well as deletions and duplications by array CGH in 6 patients. These results highlight the importance of genetic testing in children with DRE to guide patient and/or parent counseling on a case‐by‐case with respect to their individual chance of postoperative seizure freedom and ASM weaning.
Keywords: epilepsy, epilepsy surgery, outcome, pediatrics, variant
1. INTRODUCTION
Epilepsy surgery is currently the only curative treatment option in children with structural drug‐resistant epilepsy (DRE). 1 , 2 Depending on the etiology and surgery type, seizure freedom (ILAE class I outcome) is achieved in 60%–90% of patients. 3 Surprisingly, the number of patients with an Engel class I outcome has not increased over time between 2008 and 2015 despite advances in the presurgical workup. 3
Coexisting genetic causes are beginning to emerge as a possible reason for the lack of seizure freedom in some patients. Such genetic causes or predisposing factors include germline variants and/or somatic variants identified mainly through analysis of blood samples and/or brain tissue, respectively. In a recent study, channelopathies or mTORopathies were reported as factors influencing negatively surgical outcome. 4 , 5 The authors further highlighted that in one individual with a poor outcome (Engel class IV) who underwent epilepsy surgery, only postoperatively genetic testing revealed a pathogenic variant in the sodium voltage‐gated channel alpha subunit 1 (SCN1A), confirming the diagnosis of Dravet syndrome. 4 Other germline variants in candidate genes revealed in epilepsy surgery patients were the DEP domain containing 5, GATOR1 subcomplex subunit (DEPDC5) associated with familial focal epilepsy with variable foci, collagen type IV alpha 1 chain (COL4A1) associated with focal epilepsy and intracerebral hemorrhage and NPR3 like, GATOR1 complex subunit (NPRL3) associated with autosomal dominant focal epilepsy. 6 , 7
In addition, somatic variants of various genes in brain tissues are being increasingly identified in developmental lesions such as malformations of cortical development (MCD), focal cortical dysplasia (FCD), hemimegalencephaly (HME), and low‐grade developmental tumors like gangliogliomas (GG). However, potential genetic factors such as germline variants and brain‐restricted somatic variants are currently not systematically analyzed within the presurgical valuation. 4 Here, we retrospectively analyzed the genetic findings of the patients undergoing epilepsy surgery in our center and introduced our current algorithm of genetic testing for germline variants for epilepsy surgery counseling with respect to postsurgery chances of seizure freedom and reduction of antiseizure medication (ASM).
2. METHODS
We performed a retrospective study of all children <18 years who underwent epilepsy surgery at Charité – Universitätsmedizin Berlin between 10/2017 and 12/2022 with a minimum of 6 months follow‐up. Data were compiled from medical records. Seizure outcome was classified using the ILAE outcome classification 6 and 12 months after surgery. 8 The ASM reduction (ASM diff.) was calculated as the difference between the number of ASM before surgery and 12 months after surgery. Satisfactory seizure outcome was defined as an outcome class between class I to class III, whereas class IV and V were defined as unsatisfactory. Genetic testing (including chromosomal analysis, array CGH, and whole‐exome sequencing) was performed either by our Institute of Human Genetics or in other commercial labs. Variants were categorized using the ACMG guidelines. Patients were included in case they carry a likely pathogenic (class IV) or a pathogenic variant (class V). As a separate category, we also incorporate patients with variants of unknown significance (class III).
Statistical analysis was performed with R version 4.2.1 using the packages psych, crosstable, dplyr, resphape2, and ggpubr. The retrospective study was approved by the local ethics committee (approval no. EA2/084/18).
We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
3. RESULTS
The study comprised 68 children (male:female = 38:30) with a median follow‐up of 26 months (range 6–69 months, IQR: 27 months). Median age at seizure onset was 1 year (range 0 months to 15 years, IQR: 3.8), and the epilepsy surgery was performed at a median age of 7.3 years (0.2 year to 18.8 years, IQR: 8.1 years), resulting in a median seizure duration of 3.8 years (range: 0.16 to 18.8 years, IQR: 5.3 years). The underlining structural etiology was stroke in 21 patients (n = 8 media stroke), focal cortical dysplasia (FCD) in 13 patients, in 2 patients multiple FCD types (ILAE FCD types IIa: n = 3, IIb: n = 6, IIIa: n = 1, IIIb: n = 4, IIIc: n = 1). Some patients had hemispheric lesions including malformation of cortical development (n = 6), hemimegalencephaly (n = 3), schizenzephaly (n = 1), angiomatosis due to sturge–weber syndrome (n = 2), defects after hypoglycemia (n = 2), or HSV‐encephalitis (n = 1), and various tumors (dysembryoplastric neuroepithelial tumor: n = 4, ganglioglioma: n = 3, infantile glioma: n = 1, low‐grade glioma: n = 3). Five patients had Rasmussen encephalitis, and two patients had tuberous sclerosis complex (TSC) with one tuber being resected. Epilepsy surgery procedures applied were hemispherotomies (n = 35), temporal lobe resections with (amygdala)hippocampectomy (n = 8) and without (n = 1), lesionectomies (n = 20). Five patients received surgeries with palliative indication, that is, with the goal to reduce the seizure load rather than aiming for seizure freedom (lesionectomy in TSC, n = 1, anterior disconnection and callosotomy: n = 1, callosotomy: n = 1, parieto‐occipital disconnection: n = 1, temporo‐parieto‐occipital disconnection: n = 1).
Preoperatively, patients had been treated with a median lifetime number of 4 ASM (range 1–9 ASM), and just before surgery with a median of 3 ASM (range 1–5 ASM). Six months after surgery, 56 patients (82%) were seizure free and 12 patients (18%) were not. Data regarding seizure freedom were available 12 months after surgery for 59 of the 68 patients. In these, 48 patients were seizure free (81%, ILAE class 1), and 4 patients had up to three seizure days a year (ILAE class 3), resulting in a satisfactory seizure outcome in 52 patients (88%). Genetic testing was performed in 49 patients, in 19 patients genetic testing could not be performed due to lack of parental consent. In our cohort, we identified 6 patients with a pathological array CGH result and 17 patients with an abnormal WES result (Table 1). In total, 21 patients of 49 who received genetic testing had an abnormal genetic finding: 7/21 patients had an ACMG class III variant, 2/21 class IV variants, and 12/21 class V variants (Table 1; Figure 1A). The identified genes included Ankyrin repeat domain‐containing protein 11 (ANKRD11, ACMG V, MIM_611192), transforming growth factor‐beta receptor type II (TGFBR2, ACMG V, MIM_190182), collagen type IV alpha 1 (COL4A1, ACMG V, MIM_120130), tuberous sclerosis complex subunit 1 (TSC1, ACMG V, MIM_605284), tuberous sclerosis complex subunit 2 (TSC2, ACMG V, MIM_191092), Gli‐Kruppel Familiy member 3 (Gli3, ACMG V, MIM_165240), DEP domain‐containing protein 5 (DEPDC5, ACMG V, MIM_614191), methyl‐CpG‐binding protein 2 (MECP2, ACMG III, and V, MIM_300005), Na+/K+ transporting alpha‐2 polypeptide (ATP1A2, ACMG IV, MIM_182340), Atrophin 1 (ATN1, ACMG III, MIM_607462), HECT, UBA, and WWE domains‐containing protein 1 (HUWE1, ACMG III, MIM_300697), glutamate receptor, ionotropic, N‐Methyl‐D‐Aspartate, subunit 1 (GRIN1, ACMG III, MIM_138249), cell adhesion molecule‐related/downregulated by oncogenes (CDON, ACMG III, MIM_608707), Janus kinase 2 (JAK2, ACMG III, MIM_147796), potassium channel KQT‐like subfamiliy, member 2 (KCNQ2, ACMG III, MIM_602235), ASH1‐like histone lysine methyltransferase (ASH1L, ACMG III, MIM_607999), trio rho guanine nucleotide exchange factor (TRIO, ACMG III, MIM_609761), and kinesin family member 5C (KIF5C, ACMG III, MIM_604593) (Table 1)
TABLE 1.
Genetic findings in our cohort.
| Pat. | Etiology | Surgery | ILAE class | No. ASM | Chrom. analyses | Array CGH | Whole‐exome sequencing | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene name | NM | DNA change | AA change | ACMG | Linked | |||||||
| 12/6* mo. | 12/6* mo. | Phenotype | ||||||||||
| 15 | Perinatal media stroke | Hemispherotomy | I | 3 | Normal | Normal | ANKRD11 | 013275 | c.5375_5376delCT | p.Ser1792CysFS*4 | V | KGB |
| 16 | Perinatal media stroke | Hemispherotomy | I | 0 | Normal | Normal | TGFBR2 | 003242 | c.1587C > T | p.Leu529* | V | LDS2 |
| 25 | Intrauterine bleeding | Hemispherotomy | I | 1 | Normal | Normal | COL4A1 | 001845 | c.2317G > A | p.Gly773Arg | V | BSVD1 |
| 37 | FCD IIa, IIIb | Temporal lobe + amgydala‐hippectocampy | I | 1 | Normal | Dupl.20p12.1p11.21 | MECP2 | 00111079 | c.1208delC | p.Pro403Lfs*8 | V | RTT |
| 38 | MCD, polymicrogyria | Hemispherotomy | I | 1 | Normal | Microdel. 20p13 | NP | ‐ | ‐ | V | 20p13 microdel.‐syndrome | |
| 51 | Ganglioglioma | Lesionectomy | I | 1 | Normal | Del.15q11.2. 429 kb | Normal | ‐ | ‐ | ‐ | V | Chrom. 15q11.2 del.‐syndrome |
| 52 | Tubera | Lesionectomy | V | 5 | Normal | Normal | TSC2 | 000548 | c.4606C > T | p.Gln1536* | V | TSC |
| 53 | FCD IIa | Lesionectomy | IV | 2 | Normal | Normal | ATP1A2 | 000702 | c.2827C > T | p.Gln943* | IV | DEE98 |
| 54 | MCD, polymicrogyria | Hemispherotomy | I | 1 | Normal |
Del.2p15 280 kb |
NP | ‐ | ‐ | ‐ | IV | Chrom. 2p15 del.‐syndrome |
| 59 | Hemimegaencephaly | Hemispherotomy | I | 2 | Normal | Normal | GLI3 | 000168 | c.4430_4439delCTGAGTTACT | p.Ser1477Phefs*8 | V | GCPS |
| 61 | Stroke | Anterior disconnection +callosotomy | I | 2 | Normal |
Del.7p21.3 5.91 Mb |
DEPDC5 | 001242896 | Deletion exons 7–9 | ‐ | V | FFEVF1 |
| 63 | Tubera | Lesionectomy | I* | 4* | Normal | Normal | TSC1 | 000368 | c.2257dup | p.Ser753Lysfs*8 | V | TSC |
| 65 | Perinatal media stroke | Hemispherotomy | I* | 2* | Normal | Normal | COL4A1 | 001845 | c.3307G > A | p.Gly1103Arg | V | BSVD1 |
| 66 | MCD | Hemispherotomy | I* | 2* | Normal | Del.10q24.32, 3.2 Mb | NP | ‐ | ‐ | ‐ | V | Contiguous gene syndrome |
| 1 | Rasmussen | Temporal lobe + amgydala‐hippectocampy | I | 4 | Normal | Normal | HUWE1 | 031407 | c.5542A > G | p. Ser1848Gly | III | MRXST |
| 4 | MCD | Temporal lobe + amgydala‐hippectocampy | I | 1 | Normal | Normal | GRIN1 | 007327 | c.2122A > C | p.Lys708Gln | III | NDHMSD |
| 5 | Perinatal media stroke | Hemispherotomy | V | 2 | Normal | Normal | ASH1L | 018489 | c.4264C > T | p.Pro1422Ser | III | MRD52 |
| TRIO | 007118 | c.3288C > T | p.Pro1096Ser | MRD44 | ||||||||
| KIF5C | 004522 | c.2731A > G | p. Lys911Glu | CDCBM2 | ||||||||
| 6 |
Schizencephaly, FCD IIb |
Lesionectomy | I | 2 | Normal | Normal | CDON | 001378964 | c.2083A > G | p.Lys695Glu | III | HPE11 |
| 23 | Perinatal stroke | Hemispherotomy | I | 1 | Normal | Normal | ATN1 | 001940 | c.165A > G | p.Lys55Arg | III | CHEDDA |
| MECP2 | 00111079 | c.1219G > A | p.Asp407Asn | RTT | ||||||||
| 30 | Defects after HSV‐encephalitis | Temporal lobe + amgydala‐hippectocampy | V | 3 | Normal | Normal | JAK2 | 004972 | c.2171 T > C | p.Ile724Thr | III | Immune disorders |
| 33 | Perinatal stroke | Hemispherotomy | I | 3 | Normal | Normal | KCNQ2 | 172 107 | c.2329C > T | p.pro777Ser | III | DEE7 |
Abbreviations: BSVD1, brain small vessel disease 1; CDCBM2, cortical dysplasia, complex, with other brain malformations 2; CHEDDA, congenital hypotonia, epilepsy developmental delay and digital anomalies; DEE, developmental epileptic encephalopathy; FFEVF1, Epilepsy, familial focal, with variable foci 1; GCPS, Greig cephalopolysyndactyly syndrome; HPE; Holoprosencephaly; LDS2, Loeys‐Dietz Syndrome 2; MRD, intellectual developmental disorder; MRXST, intellectual developmental disorder, X‐linked, syndromic, Turner Type; NDHMSD, neurodevelopmental disorder with or without hyperkinetic movements and seizures, autosomal dominant; NP, not performed; RTT, Rett syndrome; TSC, tuberous sclerosis complex.
FIGURE 1.
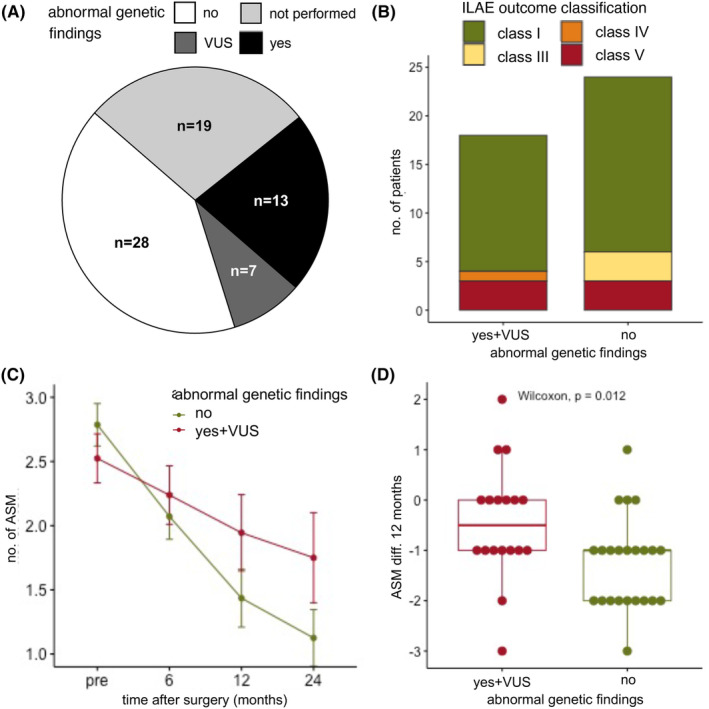
Seizure outcome and reduction of antiseizure medication (ASM) based on genetic findings. (A) In our cohort, a total of 49 patients had genetic testing, of these, 28 patients (57%) had no abnormal genetic finding, 7 patients (12%) had a variant of unknown significance (VUS), and 14 patients (31%) had a pathological genetic finding. Two patients an ACMG classification of IV (likely pathogenic) and 12 patients of V (pathogenic). (B) There was no statistical difference between the ILAE outcome classes after 12 months between patients with a normal (total = 24, ILAE class I: n = 18 (75%), class III: n = 3 (12.5%), class V: n = 3 (12.5%)) and abnormal genetic testing (incl. VUS, total = 18, ILAE class I: n = 14 (77.8%), class IV: n = 1 (5.6%), class V: n = 3 (16.7%) chi‐square test, p = 0.31)). (C) The number of ASM could be significantly reduced when comparing presurgery (n = 49, mean ± SD: 2.67 ± 0.88 ASM, range 1–4 ASM), 6 months after surgery (n = 49, mean ± SD: 2.14 ± 0.98 ASM, range 0–4), 12 months after surgery (n = 41, mean ± SD: 1.66 ± 1.17 ASM, range 0–5 ASM), and 24 months after surgery (n = 28, mean ± SD: 1.39 ± 1.07 ASM, range 0–5 ASM, Kruskal–Wallis test, p < 0.0001). (D) The absolute reduction of ASM (ASM diff.) between presurgery and after 12 months was significantly higher in patients with abnormal genetic testing (n = 41, Wilcoxon test, p = 0.012, normal genetic testing, n = 23: mean ± SD: −1.26 ± 0.92 ASM, range −3 to 1 ASM, abnormal genetic testing incl. VUS, n = 18: mean ± SD: −0.44 ± 1.15 ASM, range −3 to 2 ASM).
The median seizure duration was the same in patients with (median 3.6 years, IQR 5 years) and without an abnormal genetic finding (median 4 years, IQR 5.5 years). There was no statistically significant difference between the ILAE outcome classes after 12 months between the patients with a normal and abnormal genetic testing (chi‐square test, p = 0.31, Figure 1B) or when comparing satisfactory seizure (n = 35) outcomes against unsatisfactory seizure outcomes (n = 7) (Chi‐square test, p = 0.44). The number of ASM was significantly reduced over the 24‐month follow‐up period (Kruskal–Wallis test, p < 0.0001, Figure 1C). However, when comparing the absolute reduction of ASM (ASM diff.) after 12 months in patients with an abnormal genetic finding, ASM were significantly less reduced (Wilcoxon test, p = 0.019, Figure 1D). A total of 7 patients had an ILAE outcome class of IV and V (Figure S1). Of these, 3 patients had a palliative surgery indication: (i) patient 52 (P52) had ongoing seizures after a palliative lesionectomy in TSC, (ii) patient 44 (P44) had ongoing seizures after a palliative callosotomy after bilateral defects after hypoglycemia, (iii) and patient 30 (P30) had a palliative temporal lobe resection with amygdalahippocampectomy after herpes‐simplex encephalitis with bilateral lesions. In two patients with ongoing seizures postoperatively, no genetic finding was identified: (i) patient 12 (P12) after hemispherotomy due to a unilateral stroke and (ii) patient 47 (P47) after temporal lobe resection of a hippocampus malformation and cortical gliosis. In two patients with ongoing seizures despite complete resection or deafferentation of the structural epilepsy cause, a likely additional genetic finding was identified: (i) patient 5 (P5) with a perinatal unilateral middle cerebral artery stroke was not seizure free 12 months after hemispherotomy. Genetic testing revealed three variants of unknown significance in the ASH1L gene (NM_018489, c.3288C > T, p.Pro1422Ser9) associated with intellectual developmental disorder 52 (MRD52, MIM_617796), the TRIO gene (NM_007118, c.3288C > T, p.Pro1096Ser) associated with intellectual developmental disorder 44 and 63 (MRD44, MIM_617061 and MRD63, MIM_618825), and the KIF5C gene (NM_004522, c.2731A > G, p.Lys911Glu) associated with cortical dysplasia, complex, with other brain malformations 2 (CDCBM2, MIM_615282) likely unrelated to the structural epilepsy etiology. Some MRD52 and MRD44 patients have seizures, this could be a possible explanation for the ongoing seizures after surgery in this patient. (ii) Patient 53 had ongoing seizures after a lesionectomy of an FCD IIA. Genetic testing revealed a likely pathogenic variant in the ATP1A2 gene (NM_000702, c.2827C > T, p.Gln943*) likely unrelated to the structural epilepsy etiology associated with developmental and epileptic encephalopathy 98 (DEE98), thus confirming an underlying genetic cause in this patient.
4. DISCUSSION
Germline variants are currently not systematically analyzed within the presurgical evaluation. 4 In our cohort, we identified abnormal variants in the genes HUWE1, CDON, ANKRD11, DEPDC5, TGFBR2, ATN1, MECP2, COL4A1, KCNQ2, TSC1/2, and GLI3 as well as multiple copy number variants as additional genetic causes for their epilepsy. Additionally, we identified three patients with an unexpected bad outcome (ILAE outcome IV, V) despite clear structural lesions that were completely resected or differentiated (Figure S1). Two of these patients carried rare variants in the following genes (i) P5: ASH1L (MRD52), TRIO (MRD44), KIF5C (CDCBM2), and (ii) ATP1A2 (DEE98). We show that in our cohort, ASM were reduced to a lesser degree in patients with an abnormal genetic finding and that the ILAE outcome is the same in patients with and without an abnormal genetic finding. Limitations of this study are the small patient cohort with a low statistic power, the retrospective character of our study, and the missing genetic testing in 19/61 patients.
This and recent studies support the importance of genetic testing also in seemingly isolated structural epilepsies. In these patients, genetic findings could contribute to the overall phenotype. 9 As a result, we adapted our preoperative algorithm including genetic testing also in seemingly solely structural epilepsies. In line with the current ILAE guideline, we propose to first perform in all patients with DRE and parental consent as part of the presurgical workup a next‐generation sequencing (NGS) testing, preferably WES/Trio‐WES or alternatively an epilepsy panel if WES is not available. 10 , 11 Given that genetic testing is not always covered by insurance in some countries and is still expensive, we suggest to perform NGS, especially in those individuals with a positive family history of epilepsy, intellectual disability, autism spectrum disorder, or dysmorphic features. Chromosomal analysis or array CGH has a lower diagnostic yield and cost efficacy and are not recommended to be performed as a first genetic test (Figure 2). 10 , 11 , 12
FIGURE 2.
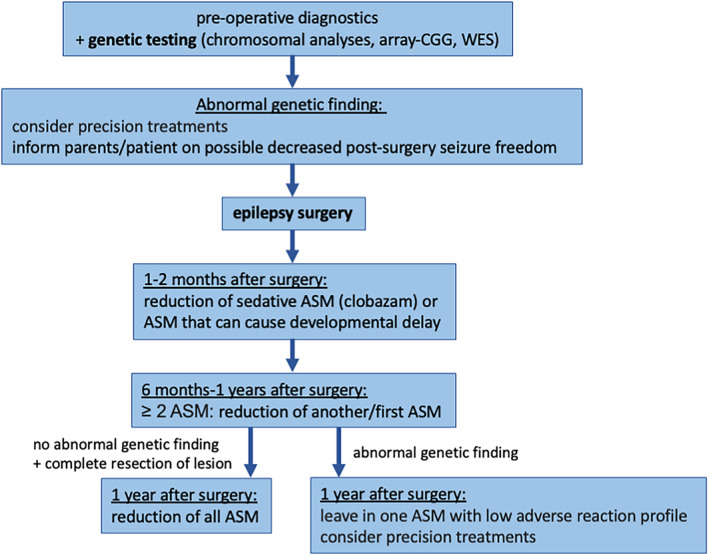
Genetic counseling algorithm pre‐ and postepilepsy surgery.
In line with recommendations by others, a pathological result does not exclude patients with a structural lesion from evaluation of an epilepsy surgery or surgery itself but is rather used for the preoperative counseling with respect to chances of postoperative seizure freedom and reduction of ASM. The genetic results offer better consultation for parents in terms of outcome and reduction of ASM postoperatively. Children with positive genetic results and their families receive an individualized treatment plan with a slower and possibly not complete discontinuation of ASM.
In addition to germline variants, recently, multiple studies on resected brain tissue during epilepsy surgery have identified an increasing number of brain‐restricted variants. 6 , 7 Brain‐restricted variants include variants in the solute carrier family 35 member A2 (SCL35A2) and genes of the phosphatidylinositol‐3‐kinase (PI3K)–protein kinase B (AKT)–mechanistric target of rapamycin kinase (MTOR) pathway (including protein tyrosine phosphatase nonreceptor type 11, PTPN11) and gains of chromosome 1q in focal cortical dysplasia. 6 , 7 These genetic findings can help to delineate the underlining mechanism of structural abnormalities with similar pathways, linking histological findings previously regarded as separate entities, such as variants in the mTOR pathway causing both hemimegalencephaly and FCDs, especially FCD IIb. Complementary, precise neuropathological classifications 13 , 14 and methods in vitro and in resected brain tissue can deepen our understanding of the mechanism of structural and genetic epilepsies, for example, developmental dendritopathy in KCNC1‐associated progressive myoclonus epilepsy 15 and dysbalanced synaptic ratio of excitatory/inhibitory in malformations. 16 These novel findings highlight the need for germline and somatic genetic analyses to offer patients and their families a better consultation and possibly improve surgery outcomes through the identification of a pathogenic variant as an additional cause for seizures and thereby the possibility of offering targeted treatments for these patients. 3 Further multicenter studies with larger cohorts are needed to evaluate the outcome of patients with an underlining genetic etiology and to determine if adapting the treatment options regarding surgery or reduction of ASM postsurgery patients have a better outcome regarding life quality and seizure freedom.
AUTHOR CONTRIBUTIONS
AMK was involved in conceptualization and supervision. LLB, KM, and AAP were involved in investigation, collection of patient data, and validation. LLB was involved in writing the original draft. All authors were involved in the review and editing process.
FUNDING INFORMATION
Our research is supported by the German Research Foundation (DFG; SFB1315, FOR3004) and the Einstein Stiftung Fellowship through the Günter Endres Fund.
CONFLICT OF INTEREST STATEMENT
None of the authors has any conflict of interest to disclose.
Supporting information
Figure S1
ACKNOWLEDGMENTS
We thank the patients and their families. Open Access funding enabled and organized by Projekt DEAL.
Becker L‐L, Makridis KL, Abad‐Perez AT, Thomale U‐W, Tietze A, Elger CE, et al. The importance of routine genetic testing in pediatric epilepsy surgery. Epilepsia Open. 2024;9:800–807. 10.1002/epi4.12916
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Makridis KL, Atalay DA, Thomale UW, Tietze A, Elger CE, Kaindl AM. Epilepsy surgery in the first six months of life: a systematic review and meta‐analysis. Seizure. 2022;96:109–117. 10.1016/j.seizure.2022.02.009 [DOI] [PubMed] [Google Scholar]
- 2. Makridis KL, Prager C, Atalay DA, Triller S, Rosenstock T, Thomale UW, et al. Ictal EEG recording is not mandatory in all candidates for paediatric epilepsy surgery with clear MRI lesions and corresponding seizure semiology. Epileptic Disord. 2022;24(4):657–666. 10.1684/epd.2022.1436 [DOI] [PubMed] [Google Scholar]
- 3. Barba C, Cross JH, Braun K, Cossu M, Klotz KA, de Masi S, et al. Trends in pediatric epilepsy surgery in Europe between 2008 and 2015: country‐, center‐, and age‐specific variation. Epilepsia. 2020;61(2):216–227. 10.1111/epi.16414 [DOI] [PubMed] [Google Scholar]
- 4. Bosselmann CM, San Antonio‐Arce V, Schulze‐Bonhage A, et al. Genetic testing before epilepsy surgery – an exploratory survey and case collection from German epilepsy centers. Seizure. 2022;95:4–10. 10.1016/j.seizure.2021.12.004 [DOI] [PubMed] [Google Scholar]
- 5. Stevelink R, Sanders MW, Tuinman MP, et al. Epilepsy surgery for patients with genetic refractory epilepsy: a systematic review. Epileptic Disord. 2018;20(2):99–115. 10.1684/epd.2018.0959 [DOI] [PubMed] [Google Scholar]
- 6. Bedrosian TA, Miller KE, Grischow OE, Schieffer KM, LaHaye S, Yoon H, et al. Detection of brain somatic variation in epilepsy‐associated developmental lesions. Epilepsia. 2022;63(8):1981–1997. 10.1111/epi.17323 [DOI] [PubMed] [Google Scholar]
- 7. Lopez‐Rivera JA, Leu C, Macnee M, et al. The genomic landscape across 474 surgically accessible epileptogenic human brain lesions. Brain. 2022;146:1342–1356. 10.1093/brain/awac376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wieser HG, Blume WT, Fish D, Goldensohn E, Hufnagel A, King D, et al. ILAE commission report. Proposal for a new classification of outcome with respect to epileptic seizures following epilepsy surgery. Epilepsia. 2001;42(2):282–286. [PubMed] [Google Scholar]
- 9. Helbig I, Swinkels ME, Aten E, et al. Structural genomic variation in childhood epilepsies with complex phenotypes. Eur J Hum Genet. 2014;22(7):896–901. 10.1038/ejhg.2013.262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Krey I, Platzer K, Esterhuizen A, Berkovic SF, Helbig I, Hildebrand MS, et al. Current practice in diagnostic genetic testing of the epilepsies. Epileptic Disord. 2022;24:765–786. 10.1684/epd.2022.1448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sanchez Fernandez I, Loddenkemper T, Gainza‐Lein M, Sheidley BR, Poduri A. Diagnostic yield of genetic tests in epilepsy: a meta‐analysis and cost‐effectiveness study. Neurology. 2019;92(5):e418–e428. 10.1212/WNL.0000000000006850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schwarze K, Buchanan J, Taylor JC, Wordsworth S. Are whole‐exome and whole‐genome sequencing approaches cost‐effective? A systematic review of the literature. Genet Med. 2018;20(10):1122–1130. 10.1038/gim.2017.247 [DOI] [PubMed] [Google Scholar]
- 13. Blumcke I, Coras R, Busch RM, et al. Toward a better definition of focal cortical dysplasia: an iterative histopathological and genetic agreement trial. Epilepsia. 2021;62(6):1416–1428. 10.1111/epi.16899 [DOI] [PubMed] [Google Scholar]
- 14. Najm I, Lal D, Alonso Vanegas M, Cendes F, Lopes‐Cendes I, Palmini A, et al. The ILAE consensus classification of focal cortical dysplasia: an update proposed by an ad hoc task force of the ILAE diagnostic methods commission. Epilepsia. 2022;63(8):1899–1919. 10.1111/epi.17301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Carpenter JC, Mannikko R, Heffner C, et al. Progressive myoclonus epilepsy KCNC1 variant causes a developmental dendritopathy. Epilepsia. 2021;62(5):1256–1267. 10.1111/epi.16867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sarnat HB, Flores‐Sarnat L. Excitatory/inhibitory synaptic ratios in polymicrogyria and down syndrome help explain epileptogenesis in malformations. Pediatr Neurol. 2021;116:41–54. 10.1016/j.pediatrneurol.2020.11.001 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.