

Abstract
Myoclonus classically presents as a brief (10–50 ms duration), non‐rhythmic jerk movement. The etiology could vary considerably ranging from self‐limited to chronic or even progressive disorders, the latter falling into encephalopathic pictures that need a prompt diagnosis. Beyond the etiological classification, others evaluate myoclonus' body distribution (i.e., clinical classification) or the location of the generator (i.e., neurophysiological classification); particularly, knowing the anatomical source of myoclonus gives inputs on the observable clinical patterns, such as EMG bursts duration or EEG correlate, and guides the therapeutic choices. Among all the chronic disorders, myoclonus often presents itself as a manifestation of epilepsy. In this context, myoclonus has many facets. Myoclonus occurs as one, or the only, seizure manifestation while it can also present as a peculiar type of movement disorder; moreover, its electroclinical features within specific genetically determined epileptic syndromes have seldom been investigated. In this review, following a meeting of recognized experts, we provide an up‐to‐date overview of the neurophysiology and nosology surrounding myoclonus. Through the dedicated exploration of epileptic syndromes, coupled with pragmatic guidance, we aim to furnish clinicians and researchers alike with practical advice for heightened diagnostic management and refined treatment strategies.
Plain Language Summary
In this work, we described myoclonus, a movement characterized by brief, shock‐like jerks. Myoclonus could be present in different diseases and its correct diagnosis helps treatment.
Keywords: electroclinical features, epilepsy, myoclonus, neurophysiology, nosology
Key Points.
Myoclonus represents a clinical manifestation of heterogeneous neurological disorders.
Correct classification of myoclonus is mandatory to guide treatment.
Disease‐modifying drugs will gain increasing importance in the treatment of specific underlying genetic disorders.
1. INTRODUCTION
Myoclonus is defined as an involuntary, brief (shock‐like) movement caused by muscular contraction (positive myoclonus) or inhibition (negative myoclonus or asterixis). 1 , 2 Myoclonus could be either physiological (i.e., hypnic jerks, hiccough, myoclonus induced by anxiety or exercise, and benign infantile myoclonus with feeding), essential (i.e., nonprogressive, both familiar or sporadic) or pathological, the latter showing an estimated annual incidence of about 1.3 cases per 100 000 persons/year. 1
Given the wide range of underlying etiologies, different groups could be identified. The symptomatic myoclonus group, which includes conditions in which encephalopathy dominates, is the most represented, right followed by the epileptic myoclonus group. 2 Myoclonus may be a component of a seizure, it could be the solely seizure type or one of the seizure types within an epileptic syndrome. Furthermore, myoclonus could also occur separately from seizures, as in the case of action‐ and stimulus‐induced myoclonus in progressive myoclonus epilepsies (PMEs). 3
Therefore, ranging from benign to potentially life‐threatening causes, newly‐onset myoclonus poses a challenge to clinicians and a stepwise approach becomes essential. The initial assessment includes a thorough medical history, encompassing medications taken, toxin exposure, recent infections, and familiar predisposition, and a careful physical examination, to clinically characterize myoclonus (i.e., distribution, temporal and activation profile). Then basic, easily accessible, exams are carried out including blood, urine, and antibody testing, as well as brain magnetic resonance imaging (MRI) to tighten the list of possible causes. 2 , 4 Then neurophysiologic testing (e.g., surface EMG or EEG–EMG polygraphy) and, eventually, advanced and emerging testing could be performed based on the clinical scenario. 4
In this Review, which follows a meeting of recognized experts in the field, we seek to delve into the neurophysiology and electroclinical features of myoclonus, giving a State of the Art with suggestions and hints for clinical practice.
2. NEUROPHYSIOLOGY OF MYOCLONUS
Myoclonus can originate at different levels in the central nervous system (CNS), ranging from the neocortex to the spinal cord, and it has been historically classified considering the location of its generators and etiology. 5 According to the location of jerks’ generators, the associated pathological conditions and the electromyographic features greatly vary, identifying completely different pictures illustrated in Table 1.
TABLE 1.
Schematic representation of myoclonic‐presenting disorders and neurophysiological features based on origin. Obviously, other brain areas/structures (e.g., cerebellum, thalamus) are also involved, but the myoclonus characteristics are mainly due to its main generator.
| Main structure | Most common pathological condition | Definition | Presentation | EMG burst duration; EEG correlates; Needed tests |
|---|---|---|---|---|
| Neocortex & thalamus | “Idiopathic” generalized epilepsies | “Epileptic myoclonus” | Spontaneous myoclonic seizures, PPR |
|
| Neocortex & thalamus | “Symptomatic” epileptic syndromes; Progressive myoclonus epilepsies | “Epileptic myoclonus” | Spontaneous myoclonic seizures, PPR |
|
| Neocortex | Progressive myoclonus epilepsies | “Reflex” myoclonus | Movements activated, sensory stimuli, posture |
|
| Neocortex | Focal (rarely multifocal) symptomatic seizures, EPC | Focal motor seizures | Spontaneous seizures, rarely also reflex seizures |
|
| Brainstem & Neocortex | Post‐hypoxic myoclonus | Reticular & cortical “reflex” myoclonus | Mostly reflex, local, and bilateral |
|
| Basal ganglia (Neocortex) |
Cortico‐basal degeneration Myoclonic dystonia |
Multifocal myoclonus, often asymmetric & movement‐induced | Spontaneous multifocal, can be reflex |
|
| Guillain‐Mollaret triangle | Palatal myoclonus | Rhythmic jerks of the palatal (roof of the mouth) muscles | Spontaneous |
|
| Spinal cord | Propriospinal myoclonus | Axial flexion involving trunk and hip muscles, spreading caudally and rostrally | Spontaneous & reflex |
|
| Spinal cord | Spinal myoclonus | Local rhythmic jerks with variable frequency | Spontaneous |
|
Abbreviations: BAEP, brainstem auditory‐evoked potentials; JLBA, jerk‐locked back‐averaging; PPR, photoparoxysmal response; PSW, polyspikes‐and‐waves; SSEP, somatosensory evoked potentials; SW, spike‐and‐waves.
The early clinical presentation of myoclonus is certainly revealing since acute or subacute onset should activate targeted evaluations of infectious, inflammatory/immune, paraneoplastic, and toxic‐metabolic etiologies, whereas a “progressive” onset of diffuse or multifocal myoclonus generally indicates neurodegenerative and genetic etiologies. As well, the physiopathological classification is critical to avoid confusion between different conditions, in terms of both causes and prognosis. Typical disorders needing an early and solving evaluation reside in the differentiation of “epileptic myoclonus”, characterizing many benign epilepsies or primary nonprogressive symptomatic syndromes, from progressive/genetic disorders, namely progressive myoclonus epilepsies (PME). 6
The EMG characteristic of the myoclonic bursts, their EEG (or MEG) correlates, and the results of neurophysiological tests also allow differentiation of myoclonus associated with cortical dysfunction from myoclonus generated by progressive (e.g., cerebellar or basal ganglia diseases) or nonprogressive subcortical dysfunctions.
The neurophysiological tests could include (1) a polygraphic (EEG–EMG) assessment of the characteristics and location of the jerks on activated antagonist muscles couples; (2) a study of the waveform components of somatosensory‐evoked potentials (SSEPs), suitable to assess sensory cortex excitability; (3) an evaluation of C‐reflex, to reveal cortical reflex myoclonus. Moreover, the averaging of multiple epochs preceding the jerks, a rather simple procedure called “jerk‐locked back‐averaging” (JLBA), or moderately more complex analytical methods (i.e., corticomuscular coherence), may help to discern the cortical versus noncortical involvement. Yet, the SSEP characteristics, JLBA, and C‐reflex are diagnostic for cortical myoclonus and psychogenic jerks. 7
Conversely, in myoclonus of supposed subcortical origin (such as jerks generated by brainstem or spinal cord damage), neurophysiological assessments could help avoid errors and monitor the disease course, but are not diagnostic. 8 Indeed, the definition of propriospinal myoclonus may be challenging requiring extensive EMG analysis and evaluation of premotor potential (“bereitschafts‐potential” or “readiness‐potential”) to differentiate this phenomenon from psychogenic jerks. 8
Sometimes, myoclonus could appear transiently or permanently in pathologies in which it is not “typical” (e.g., degenerative diseases such as some dementias, nondegenerative pathological conditions such as metabolic diseases, or in the presence of the toxic effect of drugs). Also in these cases, a precise electrophysiological characterization is appropriate to classify the phenomenon and evaluate an appropriate treatment.
Multiple tests are often needed, to explore the generators but also to avoid incorrect information. Furthermore, it is necessary to have the appropriate caution and precision in carrying out the various tests. Polygraphy is often poorly applied or not applied; however, it is essential to identify myoclonus, the EMG burst duration, and the synchronous occurrence of antagonist muscles couple. Polygraphy is playable in each laboratory and needs to be considered mandatory, with a “personalized” application in each patient in terms of muscle choice and number. When jerks are not associated with obvious transient or epileptic discharge, the application of JLBA is a simple task even if it needs a sufficient number of jerks and the exclusion of EEG artifacts. 9 SSEPs need to be evoked with low‐frequency stimuli (e.g., 1 Hz) to avoid the extinction of the enlarged middle and late components, due to high‐frequency stimulation. Evaluation of C‐reflex is a rather elementary procedure but needs to be applied both at rest and during motor activation. These tests can in any case be applied in every hospital or laboratory and nominally on all electrophysiological instruments. Other methods, such as the assessment of cortico‐muscular coherence need some signal post‐processing.
Drug treatments may reduce cortical hyperexcitability reducing the amplitude of the SSEPs components or C‐reflex, as well as “enlarged” (even no giant) SSEPs may occur in some patients with “benign” epilepsies; therefore, the application of multiple electrophysiological tests avoids errors in the specific patient condition. It appears that neurophysiological features and treatment of the myoclonus are better identified in some conditions, while “loosely defined” in others. Namely, subcortical pathways involving the thalamus, cerebellum, and brainstem need further clarification.
3. CLASSIFICATION OF MYOCLONUS
Myoclonus may be classified according to clinical signs, etiology, and neurophysiological characteristics. 5 , 10 , 11 , 12 , 13 These classification systems are strictly interrelated: clinical features are important to provide insights into etiology and anatomic origin, and neurophysiological features and anatomic origin are essential for an aetiological diagnosis. Understanding physiological categories and etiology guides the treatment approach. 10 , 11 , 12 , 13
3.1. Clinical classification
According to body distribution, myoclonus may have a focal, multifocal, segmental, or generalized distribution. Myoclonus usually shows irregular (arrhythmic) temporal patterns but can at times be rhythmic; moreover, it can occur at rest, during an action, or while maintaining a posture and it may be provoked by tactile, acoustic, or visual stimuli (reflex or stimulus‐sensitive myoclonus). 10 , 12
3.2. Etiological classification
Myoclonus recognizes a wide and heterogeneous spectrum of etiologies. The most accepted scheme of classification 5 distinguishes five categories:
physiologic, i.e., myoclonic jerks that occur as normal phenomena (sleep jerks, startle response, hiccups);
essential, characterized by nonprogressive, isolated, minimally disabling myoclonus. Essential myoclonus is idiopathic, sporadic, or hereditary (mutation in the epsilon‐sarcoglycan gene in the myoclonus‐dystonia syndrome);
epileptic myoclonus, characterized by the presence of an EEG correlate (sometimes detected only by jerk‐locked back‐averaging);
symptomatic or secondary, due to an underlying neurodegenerative, toxic, metabolic, infectious, inflammatory, degenerative, or structural disorder; and
psychogenic myoclonus, which occurs in the setting of functional disorders. 5 , 11 , 13
3.3. Neurophysiological classification
The neurophysiological classification of myoclonus relies on the identification of pathophysiologic generators and mechanisms of propagation 12 and is mainly based on the results of neurophysiologic tests. 14 , 15 Five categories of myoclonus may be recognized: cortical, cortical–subcortical, subcortical‐nonsegmental, segmental, and peripheral.
Cortical myoclonus is due to abnormal neuronal discharges in the sensorimotor cortex. Motoneurons may be hyperexcitable themselves or may be activated by other hyperexcitable brain regions (e.g., parietal or occipital areas). 12 , 16 , 17 The abnormal firing may remain localized (determining focal myoclonus) or spread to contiguous motoneurons (multifocal myoclonus) and through cortico‐cortical and transcallosal pathways (activating bilateral muscles synchronously). 3 , 12 Clinically, cortical myoclonus is typically arrhythmic, frequently triggered by action, and usually involves the face or distal extremities. Polygraphy shows brief (usually <50 ms) EMG myoclonic jerks preceded by time‐locked cortical transient within a very short interval, ranging from 10–20 (arm muscles) to 30 ms (leg muscles). Enlarged cortical SSEPs, enhanced C‐reflex, and increased cortico‐muscular coherence are supportive findings. 12 , 15 , 18 Cortical myoclonus may be observed in progressive myoclonic epilepsies (PMEs), familial adult myoclonic epilepsy, or epilepsia partialis continua. 10 , 12 , 19 , 20 , 21 , 22 It may also occur in other diseases such as Rett syndrome or neurodegenerative disease (e.g., Alzheimer's disease, corticobasal syndrome, dementia with Lewy bodies, Creutzfeldt–Jakob disease). 10 , 12
Cortical–subcortical myoclonus is characteristic of primary generalized seizures, such as juvenile myoclonic epilepsy and different generalized epilepsy syndromes. 13 Myoclonus arises from paroxysmal abnormal excessive oscillation in bidirectional connections between cortical and subcortical areas (particularly the thalamus), resulting in simultaneously widespread excitation over the sensorimotor cortex. 12 , 13 Surface EMG bursts are brief (typically 25–100 ms) and time‐locked to spike/polyspike and wave discharges at EEG. 12 , 13 , 14 , 15
Subcortical–nonsegmental myoclonus represents a heterogeneous entity. Abnormal activity may originate from multiple sites and circuits (ranging from the basal ganglia to the spinal cord) and then transmitted to ascending and descending motor pathways. Typically, EMG discharges are long (up to 300 ms) in duration and additional neurophysiologic tests (EEG, polygraphy with inclusion of rostral and caudal muscles, SSEPs) exclude the presence of a cortical generator. Two major subtypes can be recognized based on clinical and EMG discharge recruitment patterns. In the first, EMG demonstrates the simultaneous rostral and caudal distribution of muscle recruitment spreading from a localized source. An example is reticular reflex myoclonus, in which early activation of muscles (sternocleidomastoid and trapezius) innervated by the XI cranial nerve is followed by rostrocaudal diffusion with involvement of muscles supplied by VII (facial) and V (masseter) cranial nerves and, at the same time, bilateral limbs and trunks muscles. Jerks are present at rest, usually heightened by voluntary movements and triggered by multisensory stimuli. It has been mainly reported in the setting of hypoxic encephalopathy. 23 Another example of subcortical–nonsegmental myoclonus is propriospinal myoclonus, which recognizes its primary source located in the cervical or thoracic spinal cord. 24 , 25 In a second subtype of subcortical–nonsegmental myoclonus, a multifocal recruitment pattern is observed. Jerks mostly occur during muscle activation, while stimuli sensitivity is uncommon. Myoclonus–dystonia syndrome (formerly named “essential myoclonus”) 26 and opsoclonus‐myoclonus syndrome 27 fit in this category.
Segmental myoclonus is characterized by jerks limited to the muscles corresponding to one or just contiguous spinal or brainstem segments but may sometimes occur bilaterally. Jerks are usually unaffected by motor activity, sensory stimuli, or state of consciousness (persisting in sleep). Surface EMG characteristically demonstrates rhythmic (typically with a 1–3 Hz frequency, but a broad frequency range of 0.2–8 Hz has been reported), widely variable in duration (lasting from 50 up to 500 ms) myoclonus discharges. 12 Palatal myoclonus (also termed palatal tremor) and spinal segmental myoclonus are the most common type. 13 A pathological lesion either in the brainstem or spinal cord is observed. 28
Peripheral myoclonus refers to intermittent, semi‐rhythmic or rhythmic focal jerks arising from a peripheral nervous system generator (a specific root, plexus, or peripheral nerve). Ectopic excitation, ephaptic transmission and central relay reorganization mechanisms contribute to its development. 29 EMG shows nearly synchronous myoclonic discharges of muscles sharing the same peripheral nerve distribution; a marked variability of muscular bursts duration (ranging from 50 to 200 ms) may be observed. 12 , 15 Hemifacial spasm is the best‐documented example of peripheral myoclonus. 13
4. SYMPTOMATIC MYOCLONUS
Symptomatic myoclonus occurs in many different acute and chronic conditions, notably in metabolic disorders, infectious diseases, post‐hypoxic cerebral damage, reactions to drugs or toxic substances, and neurodegenerative disorders. 1 Despite different etiologies, the clinical presentation might be similar, hence a careful anamnestic and semiology approach is strongly recommended for etiological classification. Hematological, urinary, and cerebrospinal fluid examinations often provide prompt and useful information to rule out infectious, toxic, and metabolic disorders.
EEG with polygraphic (EMG) recording remains the mainstay in the definition of the origin of the myoclonus. Some EEG patterns may be highly suggestive of specific disorders (i.e., triphasic waves, or lateralized or generalized periodic discharges); however, similar features can be also present in different etiological conditions, suggesting shared physiopathological mechanisms, see Table 2.
TABLE 2.
Main features of symptomatic myoclonus associated with the underlying etiology. Different etiologies may share a common myoclonus type, as well as different types of myoclonus may be present in the same condition.
| Symptomatic myoclonus | EEG pattern | Myoclonus features |
|---|---|---|
| ||
| Post‐hypoxic cerebral damage | ||
| Acute: MSE | Burst suppression with PSW (4 stages) |
|
| Chronic: LAS | Normal/nonspecific /PSW or focal spikes |
|
| Infectious diseases | ||
| SSPE | Periodic—pseudo periodic complexes |
|
| HSV Encephalitis | Lateralized periodic discharges |
|
| Metabolic disorders | ||
| Acute and chronic kidney disease | Nonspecific |
|
| Hepatic encephalopathy | Diffuse triphasic waves |
|
| Reactions to drugs or toxic substances | Nonspecific |
|
| Progressive myoclonus epilepsies (PMEs) | ||
|
||
| Dementia/neurodegenerative | ||
| CJD | Diffuse periodic triphasic waves |
|
| AD | SW/ (central focal transient) |
|
| Huntington disease | SW/ (central focal transient) |
|
| Corticobasal degeneration | Not specific |
|
Abbreviations: AD, Alzheimer's disease; CJD, Creutzfeldt–Jakob disease; HSV, Herpes simplex encephalitis; LAS, Lance–Adams syndrome; MSE, myoclonic status epilepticus; SSPE, subacute sclerosing panencephalitis; SW, sharp and wave complexes.
Severe kidney disease often shows reticular, stimulus‐sensitive myoclonus because of the toxic effect exerted by uremia on the medulla oblongata. 30 Hepatic encephalopathy shows a typical EEG pattern with diffuse triphasic waves, often associated with negative myoclonus. Among infectious diseases, subacute sclerosing panencephalitis displays a typical EEG pattern with symmetrical periodic delta wave complexes at long intervals, associated with generalized jerks. 31 In Herpes simplex virus encephalitis, lateralized periodic discharges can be related to myoclonus.
Post‐hypoxic myoclonus (PHM) deserves special mention, due to its importance in prognostic evaluation. PHM might present in the acute phase (up to 72 h after cardiac arrest) as a myoclonic status epilepticus, or as a chronic condition (from days to years after cardiac arrest), constituting the so‐called “Lance–Adams Syndrome” (LAS). The early appearance of the myoclonus is associated with a high mortality rate or a worse neurological outcome. Myoclonus can affect the face, limbs, or trunk with a generalized or multifocal distribution. 32 The LAS may include both myoclonus of subcortical (reticular reflex, namely in the acute phase) and cortical origin (action or stimulus‐induced myoclonus).
Many drugs, including antiseizure medications (ASMs), especially at high doses, can induce myoclonus. Drugs or toxic substances may induce different myoclonus types, with probably distinct neuro‐anatomical generators. It is always important to consider drugs as a cause of myoclonus regardless of its features. 33
In the setting of chronic diseases featuring myoclonus we can distinguish neurodegenerative, genetically determined, syndromes that can be included in the progressive myoclonus epilepsy phenotypes and that typically show worsening myoclonus and associated epileptic seizures starting from infancy to adulthood. In this context, it may be useful to consider the main associated neurological deficits, including dementia. Dementias with myoclonus should be distinguished into two main groups: (1) Creutzfeldt–Jakob disease (JCD); and (2) Alzheimer's disease (AD). JCD often has a distinctive EEG pattern characterized by diffuse period activity, which is probably generated by a cortical–subcortical loop. Periodic activity is commonly linked to positive or negative myoclonus. 34 The fast‐progressive AD form, which is typically due to genetic mutations of the presenilin‐1, is most frequently associated with myoclonus. In these cases, EEG can be nonspecific, and the cortical correlate of the myoclonus should be investigated by JLBA.
Juvenile Huntington's disease combines extrapyramidal symptoms with behavioral issues, epilepsy, and myoclonus. Early onset of the disorder correlates with very long CAG repeats within the HTT gene. Myoclonus may be either generalized or associated with spike and wave complexes or multifocal with features compatible with cortical reflex myoclonus. In a few cases, a picture reminding progressive myoclonus epilepsy may also occur, and rigidity may be replaced by hypotonia. 35 Corticobasal degeneration has a peculiar presentation due to hyperexcitability of the motor cortex often prevalent on one side, and degeneration of the ipsilateral parietal cortex. Myoclonus can sometimes be lateralized, and stimulus sensitive. Although the final effector producing the myoclonus is the motor cortex, the pathway does not fully represent that of cortical reflex myoclonus because giant evoked potentials are lacking, and the C‐reflex has short latencies, which prevents a transcortical reflex circuit.
4.1. Myoclonus in PMEs
The syndromic category of progressive myoclonus epilepsies (PMEs) comprises a group of heterogeneous and rare disorders typically presenting with a variable combination of action myoclonus, epileptic seizures, and progressive neurologic deterioration, which can include cognitive decline, ataxia, and sometimes neuropathy, and myopathy.
The first PMEs were identified by Lafora (1911), based on the pathological inclusion found in the brain and other tissues, and by Unverricht (1891), and Lundborg (1903), whose cases were initially called Baltic myoclonus, based on the clinical phenotype and the geographic area. Moreover, Hunt (1921) reported a similar phenotype associated with signs of Friedreich's ataxia. 36
The more recent definition of PMEs starts with a “consensus” expressed at the Marseille PME workshop in 1989. 37 This consensus helped to define the various types of PMEs known at that time and to start a new era of genetic research, which soon led to the discovery of many PME genes. Over the years, many PMEs have been reported, bearing in mind that the forms now defined as PMEs have highly variable phenotypes, Table 3. 38
TABLE 3.
Simplified sum‐up of the syndromic conditions defined as PMEs. 38
| Name | Gene/inheritance | Seizures | Commonly associated signs | Onset age | Progression |
|---|---|---|---|---|---|
| Lafora; EPM2 (EPM2A& NHLRC1) |
6p22.3 6p24.3 AR |
Generalized tonic–clonic, myoclonic, and visual (frequent) | Frequently severe dementia | Late childhood, adolescence | Fast (less severe in some cases) |
| Unverricht‐ Lundborg: EPM1 (CSTB) |
21q22.3 AR |
Myoclonic, tonic–clonic (rare, often responsive to treatment) | Ataxia (variable but not severe cognitive decay) | Late childhood, adolescence | Slow, often stabilization |
| Sialidoses, I (NEU1) |
6p21.33 AR |
Myoclonic, tonic–clonic (rare) | Cherry red spots, visual loss (can be lacking), urinary sialyloligosaccharides | Second to third decades of life | Slow |
| Sialidoses, II (NEU1) |
6p21.33 AR |
Myoclonic, tonic–clonic | Dysmorphisms, hepatosplenomegaly, early cognitive impairment, ataxia | Early childhood (rarely later) | Variable, often fast |
|
NCL AR (KCTD7) |
7q11.21 AR |
Myoclonic, tonic–clonic | Mental retardation, dysarthria, ataxia | Early childhood | Variable |
|
NCL AR (CLN6) |
15q23 AR |
Myoclonic, tonic–clonic | Cognitive deterioration, extrapyramidal, ataxia | Adults | Intermediate |
|
EPM1B PRICKLE1 |
12q12 AR |
Myoclonic, tonic–clonic | Ataxia, mild mental retardation | Late childhood–adolescence | Slow |
|
EPM4 SCARB2 |
4q21.1 AR |
Rare (or absent) tonic–clonic | Ataxia; with or without renal failure | Adolescence‐juvenile | Fast (death at around 30 years) |
|
EPM6 GOSR2 |
17q21.32 AR |
Absence, or tonic–clonic (not predominant) | Ataxia, preceding myoclonus, neuropathy dementia | Early childhood | Variable |
|
EPM7 MEAK KCNC1 |
11p15.1 AD |
Tonic–clonic Severe myoclonus |
Ataxia, mild cognitive impairment | Childhood to adolescence | Fast (wheel‐chair bound since late teen‐age) |
|
EPM8 CERS1 |
19p13.11 AR |
Tonic–clonic | Ataxia, other movement disorders, dementia | Early childhood | Variable (few affected) |
|
EPM9 LMNB2 |
19p13.3 AR |
Tonic–clonic | Cognitive delay, scoliosis, muscle atrophy | Childhood | Variable (few affected) |
|
EPM10 PRDM8 |
4q21.21 AR |
Undefined | Ataxia, spasticity, and cognitive decay | Childhood | Variable (few affected) |
|
EPM11 SEMA6B |
19p13.3 AR |
Various types | Developmental regression | Childhood | Variable (few affected) |
|
EPM12 SLC7A6OS |
16q22.1 AR |
Tonic–clonic | Ataxia, mild cognitive impairment | Juvenile | Slow (few affected) |
|
MERRF MERRF/MELAS overlap |
Various mitochondrial genes Mutations at nucleotide 8344 in most Maternal |
Mostly Tonic–clonic | Ataxia, migraine, hearing loss, (short stature, pes cavus, ophthalmoparesis, myopathy) | Adolescent‐adult | Variable |
|
FAME BAFME |
Intronic TTTCA and TTTTA repeat in different loci AD |
Rare (or absent) tonic–clonic | Late mild cognitive deficits or psychiatric symptoms | Adult (juvenile) | Very slow |
Abbreviations: AD, autosomal dominant; AR, autosomal recessive; BAFME, Benign adult familial myoclonic epilepsy; FAME, adult familial myoclonic epilepsy; MEAK, Myoclonic epilepsy and ataxia due to KCNC1 mutation; NCL, neuronal Ceroid Lipofuscinoses.
In most conditions, the EEG demonstrates abnormal EEG background activity and the presence of epileptic activity, mostly generalized, occurring as spike and wave with variable frequency, or polyspike and wave, rarely focal (i.e., in Lafora disease, possibly showing occipital spikes). Often marked photosensitivity is present, classically this activity weakens with the disease progression and the use of ASMs. Neuroradiological “Imaging” may reveal atrophic changes or features associated with the causative genetic factors, but there are nonspecific.
The “myoclonus” observed in the PME phenotypes can have various features and uneven generators and includes “epileptic” myoclonus, associated with EEG paroxysms, and multifocal jerks at rest, with or without obvious EEG correlate. “Reflex” myoclonus in response to somatic afferent mechanisms, namely the intention to move or active movement, is the fundamental symptom in all PMEs, 39 indicating a specific hyperexcitability of the sensorimotor cortex. On the other hand, the severity of seizures is highly variable, as well as the severity of cognitive impairment, onset age, and severity of the progression. Other neurological symptoms can coexist depending on the specific disorder, preceding or following the early seizure presentation.
The clear demonstration of the “reflex” origin of the myoclonus needs to be supported by the demonstration of cortical fast spikes on sensory‐motor EEG derivations, preceding myoclonic jerks without obvious paroxysmal correlate (by applying JLBA or other post‐analyses), while enlarged middle components of somatosensory evoked potentials can demonstrate the hyperexcitability of the sensory cortex. 6 However, it is necessary to take into account limitations possibly influencing the neurophysiological findings in patients with onset in early childhood and the possible influence of ASM.
The increased opportunity to identify a gene mutation in PMEs with previously unrecognized genetic determinants (with AR, AD, maternal transmission, or “de novo”) considerably increased the variability of the “genetic” origin. This is reflected in increased variability of associated clinical signs and age of onset, but above all, in the possibility of a relatively delayed presentation of myoclonus for the earliest appearance of other neurological symptoms, including seizures. This implies that the phenotypic definition of a PME must be kept sufficiently precise, in order not to mix different syndromic pictures, and to offer a guide for the diagnosis and treatment. 40
Recent papers, 41 , 42 reporting the genetic factors of various “new” PMEs, proposed, to face this problem, to classify different phenotypes: (1) “Unverricht‐Lundborg‐like”, with the occurrence and presentation similar to that of the more “common” (even rare) EPM1, with adolescent‐onset, prominent movement‐activated myoclonus, and no or minimal dementia, (2) PME plus dementia, (3) PME plus developmental delay, when myoclonus manifests after the occurrence of early signs of developmental encephalopathy, and (4) Late‐onset PMEs, starting in adulthood.
Indeed, a subclassification of the PME phenotypes can be further refined but it is needed not only to maintain “ordered” syndromic differentiation, but also to be of support in the identification of disease mechanisms resulting from the gene mutation, possibly shared by several forms, and to explore new treatments.
The most “common” PMEs are Unverricht–Lundborg (EPM1) and Lafora (EPM2) diseases, discernible thanks to genetic investigations but also their different clinical‐electroencephalographic picture. Patients with EPM1 often have generalized seizures and massive myoclonus at the onset, responding to “adequate” ASMs. Thereafter, they maintain myoclonus during voluntary movements (clinically presenting as a prominent movement disorder). EPM1A patients classically have preservation of cognitive skills. The differential diagnosis between EPM1 and EPM2 is easy as EPM2 patients present frequent generalized seizures that are difficult to control with ASMs; moreover, “visual” seizures are often part of the clinical picture. Epileptic myoclonus is common and associated with slow spike‐and‐waves EEG discharges. Patients show early and progressive cognitive deterioration.
Special care must be paid in the differential diagnosis when EPM1 is due to a compound heterozygous mutation coupling the classic CSTB gene promoter expansion with a different mutation in the same gene. In this case, the severity of the phenotype relies on the effect of the coupled mutation; although, most of the cases present with difficult‐to‐control epilepsy, giving rise to severe “epileptic” myoclonus that requires to be differentiated from that due to EPM2 mutations. 43
5. MYOCLONUS IN GENETICALLY DETERMINED EPILEPTIC SYNDROMES
The occurrence of myoclonic seizures, as well as cortical myoclonus has been described in several conditions in which epilepsy can be genetically determined. In these clinical situations, myoclonic seizures may present as one of the different seizure types experienced by the patient. 3 Moreover, myoclonic seizures can occur at a typical age for the specific condition and may disappear during the evolution or appear later on.
Polygraphic‐video‐EEG recordings are extremely important in these individuals to obtain ictal recording of the events. Indeed, the ictal registration of the episodes may differentiate epileptic versus nonepileptic events, may allow the recognition of the specific seizure type and consequently pave the way to a proper treatment.
Myoclonic seizures can be the hallmark of early infantile developmental and epileptic encephalopathy (EIDEE), characterized by early onset seizures (within the first 3 months of life or even earlier), usually very frequent and drug‐resistant. Abnormal neurological signs and developmental impairment are usually evident even before seizure onset. Infants with EIDEE may experience different seizure types: focal/generalized tonic, myoclonic, focal clonic, epileptic spasms, and sequential seizures. Interictal EEG shows abnormal background activity with burst‐suppression, multifocal spikes/spike waves/sharp waves with or without slowing, discontinuity and/or diffuse slowing. The ictal EEG pattern depends on seizure type.
Etiologically, causative pathogenic gene variants can be identified in more than half of the patients with EIDEE. Even metabolic studies should be carried out, particularly if a clear structural abnormality is not found on the imaging. 44
Among DEEs occurring in infancy, Dravet Syndrome is the condition in which myoclonic seizures, although not the first seizure type presenting at onset, are very typical, specially between the 1st and 4th year of life. Seizures are usually intractable and, from the second year of life, children demonstrate cognitive and behavioral impairments; subsequrently, gait abnormalities do also appear, such as a characteristic “crouch gait”. The clinical diagnosis is supported by the identification of pathogenic variants in the sodium channel gene SCN1A (found in over 80% of cases). Infants may present myoclonic seizures along with other seizure types and developmental impairment in the context of etiology‐specific syndrome, involving different genes, e.g. KCNQ2, CDKL5, NEXMIF, SLC2A1. 44
Patients with classic Rett Syndrome may show a distinctive pattern of cortical reflex myoclonus, clinically characterized by multifocal, arrhythmic, and asynchronous jerks mainly involving distal limbs. Burst‐locked EEG averaging generated a contralateral centroparietal premyoclonus transient preceding the burst. 45 Motor‐evoked potentials showed normal latencies, while cortical hyperexcitability was confirmed by enlarged somatosensory evoked potentials and prolonged C‐reflex.
In girls with Rett syndrome, myoclonic status is difficult to distinguish from movement disorders, such as hand stereotypies, tremors, and dystonia. The importance of identifying this epileptic condition and treating it with antimyoclonic medications has been recently highlighted. 46
Myoclonic seizures are also common in Angelman Syndrome, typically occurring in young children and associated with spike‐and‐wave discharges on the EEG. Myoclonic status is often associated with developmental regression. In contrast, nonepileptic myoclonus typically develops in adolescence or early adulthood and has no EEG correlation, alteration in consciousness, or regression, but can significantly impact the overall quality of life. 47
Even children with Pallister–Killian syndrome (PKS) may show myoclonic seizures by 18 months of age: these seizures may occur spontaneously but can be also triggered by low frequency (1–6 Hz) intermittent photic stimulation. In addition, children with PKS may present epileptic spasms and focal seizures in their evolution, so the identification of the ictal pattern may be fundamental for treatment choices. 48
Frequently observable in the clinical practice, individuals with Down syndrome over the age of 40 years may develop a neurologic condition called Late Onset Myoclonic Epilepsy in Down's syndrome (LOMEDS), with a distinct pattern of evolution. Initially, patients present with spatial–temporal and language deficits, diffuse EEG abnormalities during sleep, and cerebral atrophy at neuroimaging. After a few months, myoclonic seizures involving the upper limbs at awakening develop. The myoclonic jerks are time‐locked to diffuse poly‐spikes on EEG, and seizures are controlled by ASMs. Afterward, photosensitivity develops, epileptic and nonepileptic myoclonus become persistent, cerebellar signs, severe dementia, and global decline become evident. 49
5.1. Familial adult myoclonic epilepsy (FAME)
FAME is an autosomal dominant condition characterized by cortical tremor and myoclonus usually manifesting within the second decade of life, and infrequent seizures developing in the third or fourth decade. Cortical tremor is the core feature of FAME and is considered part of a spectrum of cortical myoclonus. Neurophysiological investigations such as JLBA and cortico‐muscular coherence analysis, giant SSEPs, and the presence of C‐reflex at rest support cortical tremor as the result of sensorimotor cortex hyperexcitability. 50
FAME has been described worldwide as geographic aggregates, especially in Japan and Europe and initially linked to four main loci. The specific genetic defect underlying FAME consists of the expansion of similar noncoding pentanucleotide repeats, TTTCA and TTTTA, in different genes (SAMD12 for FAME1, STARD7 for FAME 2, MARCH6 for FAME3, YEATS2 for FAME4, TNRC6a for FAME 5 and RAPGEF2 for FAME6) rather than the altered gene function. 51
The clinical management is essentially symptomatic and based on ASMs that have both an antiseizure and an antimyoclonic effect, such as valproate, levetiracetam, benzodiazepines, and perampanel. 52 The clinical course is nonprogressive or slowly progressive: epilepsy is commonly controlled with medications and individuals have a normal life expectancy. However, the myoclonus severity increases with age and leads to some degree of disability in the elderly. 50
6. DIFFERENTIAL DIAGNOSIS
Non epileptic manifestations are more frequent than epileptic, especially during infancy and childhood. Myoclonic manifestations (true or supposed) are often referred by parents and differential diagnosis can be challenging. Many factors can interfere with diagnosis (e.g., mental retardation, EEG abnormalities). We can find nonepileptic myoclonic manifestations since the neonatal period (neonatal benign hypnic myoclonus, myoclonus/jitteriness), but in infancy occurs the most known non epileptic myoclonic phenomenon (Fejerman myoclonus). Subsequently, it was clarified that some of these conditions are quite different from myoclonus and described as separate entities (shuddering, head atonic attacks, shaking attacks). Other conditions occurring in infancy and childhood that can be reported as myoclonic are Marcus‐Gunn phenomenon and febrile myoclonus. Many of these conditions have more than one feature in common: occurrence in the first months or years of life, normal psychomotor development, normal neurological examination, and spontaneous regression generally after some months without sequelae. The burden of misdiagnosis can be severe for children and their parents. Epileptologists often overdiagnose and over‐treat thus its early recognition would avoid unnecessary therapies. Often Fejerman myoclonus or head atonic attacks are misdiagnosed as epileptic fits. 53 , 54 , 55
Nonepileptic manifestations do not respond to ASMs, and some children are incorrectly considered not only epileptic but also drug‐resistant. Hitherto, the burden of therapy can be increased. When the introduction of an ASM coincides with spontaneous regression of the paroxysms this effect can be incorrectly related to ASM and a nonnecessary therapy can be continued for many years. Differential diagnosis versus epilepsy and per se can be achieved with a deeper anamnesis, and with video captured by parents or video‐polygraphy recordings.
Despite allowing level identification, neurophysiology is not specific thus it cannot provide clues on the etiology underlying the myoclonus. 7 , 56
Neuroimaging has always been neglected as a diagnostic tool investigating such movement disorders, indeed, being transient and often stimulus‐dependent, it results particularly difficult to tackle with available MRI techniques. However, conventional MRI may be used in conjunction with phenomenology and neurophysiology to disentangle the etiology of myoclonus. On the other hand, advanced MRI techniques may shed light on the pathophysiology of such movement disorders. If applied to patients showing isolated myoclonus, or with myoclonus‐dystonia, MRI usually does not hold real diagnostic value. Conversely, when myoclonus is combined with other movement disorders and/or neurological symptoms, MRI may show signs suggestive of specific etiologies. When presenting with dementia and additional neurologic symptoms, myoclonus may be suggestive of atypical parkinsonism or severe/advanced forms of Parkinson's and Alzheimer's disease. If dementia is rapidly progressive, depending on the age onset, one may suspect the presence of a prion disease or a progressive myoclonic epilepsy. Myoclonus presenting with ataxia and other neurological symptoms may suggest the presence of ataxia telangiectasia or other spinocerebellar ataxias. The presence of myoclonus, opsoclonus, and ataxia with an unraveling MRI, hints at an autoimmune/paraneoplastic etiology. MRI may provide insights into myoclonus pathophysiology through advanced neuroimaging methods such as structural and functional connectivity. Converging evidence from studies conducted on patients with myoclonus‐dystonia and familial cortical tremor with epilepsy suggest a cerebellar involvement in both cortical and subcortical myoclonus. 56 However, studies using such techniques on larger patient cohorts are needed to corroborate such hypotheses.
7. CURRENT TREATMENTS AND FUTURE PERSPECTIVES
Treatment of the underlying disorder is the best therapeutic strategy. Some toxic‐metabolic states or surgically resectable lesions can be reversible; similarly, psychogenic jerks can be solved by psychotherapy and pharmacological psychiatric treatment at the first attempt. 1 , 2 However, in most cases, multiple drug trials and co‐treatment are needed. 2
One useful path to guide treatment is firstly to establish the origin of myoclonus. A gamma‐aminobutyric acid (GABA) enhancing or glutamate‐lowering approach has historically been of choice given the recognized abnormal excitatory state of the corticospinal output. Although, levetiracetam (LEV) and piracetam, pyrrolidone derivatives, can be effective in this owing to a different mechanism to modulate cortical hyperexcitability; that is the affinity for synaptic vesicle protein 2A (SV2A) and ions current (i.e., calcium and potassium currents). 57 Also, brivaracetam which has a 10‐ to 30‐fold times higher affinity for the SV2A protein than LEV, showed consistent anti‐myoclonic activity in animal models; although, in‐human studies have shown discrepant results 58 with some cases of acquired myoclonus (i.e., post‐anoxic), 59 , 60 difficulties in untying the bold with LEV, given in the majority of cases BRV was added to a therapeutic scheme already including LEV. Hence, the first‐line choice remains LEV at a dose between 1000 and 3000 mg/day. Valproate (VPA) strengthens inhibitory neurotransmission in the cortex and remains another drug of choice for treating cortical myoclonus. Daily dosage hovers around 1200–2000 mg/day. Other medications such as clonazepam, zonisamide, and primidone could be added‐on to a treatment including LEV or VPA. 4 Of note sodium channel blockers, such as phenytoin and carbamazepine, have the potential to exacerbate seizures in some patients and, therefore, found no place in the current therapeutic algorithm of myoclonus. 4
For cortico‐subcortical myoclonus, VPA remains superior to LEV and particular effectiveness is seen in juvenile myoclonic epilepsy (JME). 61 , 62 BRV has also shown high response rates (up to 75%) as adjuncts in the treatment of JME. 63 Phenytoin and carbamazepine should be considered carefully also in this case.
Subcortical myoclonus (i.e., nonsegmental or segmental) treatment should be diagnosis driven. Although, some general hints could be made; standard ASMs, such as VPA and LEV are not consistently useful, conversely clonazepam, a facilitator of GABAergic transmission, and carbamazepine may be useful at different levels. Also, perampanel (a blocker of the AMPA receptors) has recently proven effective on myoclonus in myoclonus‐dystonia syndrome. 64 The usefulness of botulinum toxin injection should also be considered in both segmental and peripheral myoclonus (see Figure 1).
FIGURE 1.
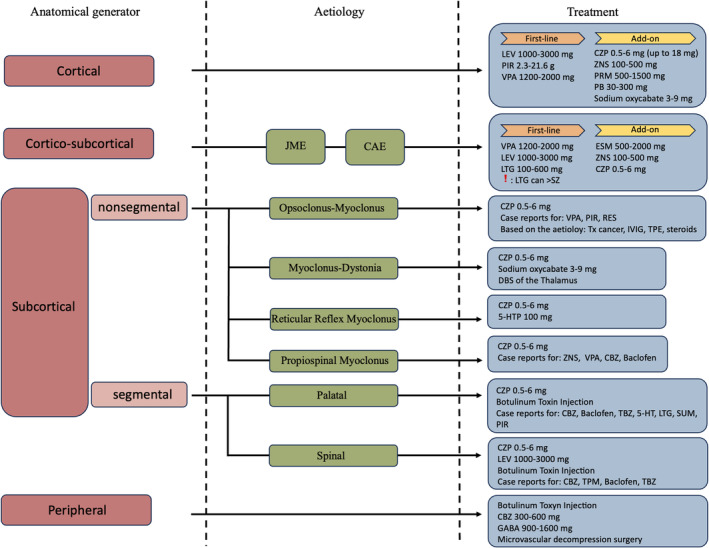
Schematic representation of the neurophysiological classification‐driven treatment of myoclonus. CZP, clonazepam; DBS, deep brain stimulation; ESM, ethosuximide; GABA, gabapentin; 5‐HTP, 5‐hydroxytryptophan; IVIG, intravenous immunoglobuline; LEV, levetiracetam; LTG, lamotrigine; PB, phenobarbital; PIR, piracetam; PRM, primidone; RES, reserpine; SUM, sumatriptan; SZ, seizures; TBZ, tetrabenazine; TPE, therapeutic plasma exchange; Tx, treatment; VPA, valproic acid; ZNS, zonisamide. Note, that doses are reported *daily.
New therapeutic agents are currently being studied. To cite: Pro‐Drug T2000 (1,3‐dimethoxymethyl‐5,5‐diphenyl‐barbituric acid) or zonisamide in myoclonus‐dystonia syndrome (NCT00506012; NCT01806805); or VAL‐1221, a fusion Fab‐rhGAA recombinant protein, for Lafora disease (NCT05930223). 65 , 66 Finally, given the advent and progress in the use CRISP/Cas9 and antisense oligonucleotide technologies to correct genetically determined disorders, their role as disease‐modifying drugs will gain increasing importance in the treatment of specific genetic disorders. 67 , 68
8. CONCLUSIONS
Myoclonus represents a complex, polyfaceted, phenomenon with broad etiologies and clinical correlates. Quick discard of some unlikely diagnoses may help in fastening the management and therapeutic decisions. Although, when routine exams do not bring results or the clinical picture remains poorly discernible some more “specific” assessments should be undertaken with the involvement of specialists in the field. Treatment is mainly based on prospective and retrospective studies, with little evidence from randomized clinical trials. Valproate is commonly the first choice alone or in combination with some benzodiazepines or levetiracetam. There is, however, sufficient evidence for the use of newer drugs in monotherapy or add‐on. Nevertheless, it remains pivotal to avoid medication that may aggravate the disorder. A better understanding of the pathophysiologic mechanisms of myoclonus could yield great improvement in the treatment and quality of life of patients.
AUTHOR CONTRIBUTIONS
A.R, collection of data, drafting; G. D'O, E. F, A. P. E. P, S. F, F. P, L. C, A. V, A. C, V. B, F. B, A. L, A. G, A. R, G. C, and R. M drafting; P. S and V. B conception and design of the study, revision and final approval of the manuscript. All authors agree to be accountable for all aspects of the work.
ACKNOWLEDGEMENTS
This study was supported by the PNRR‐MUR‐M4C2 PE0000006 Research Program “MNESYS”—A multiscale integrated approach to the study of the nervous system in health and disease. IRCCS ‘G. Gaslini’ is a member of ERN‐Epicare.
CONFLICT OF INTEREST STATEMENT
Neither of the authors has any conflict of interest to disclose. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
ETHICS STATEMENT
All methods were performed in accordance with the ethical standards as laid down in the Declaration of Helsinki and its later amendments or comparable ethical standards.
PATIENT CONSENT STATEMENT
Not applicable as no human subjects were involved in the study.
Riva A, D’Onofrio G, Ferlazzo E, Pascarella A, Pasini E, Franceschetti S, et al. Myoclonus: Differential diagnosis and current management. Epilepsia Open. 2024;9:486–500. 10.1002/epi4.12917
Contributor Information
Pasquale Striano, Email: pasquale.striano@gaslini.org.
Vincenzo Belcastro, Email: vincenzo.belcastro@asst-lodi.it.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available in the article. If additional data were required, they might be requested from the corresponding author.
REFERENCES
- 1. Caviness JN, Brown P. Myoclonus: current concepts and recent advances. Lancet Neurol. 2004;3(10):598–607. 10.1016/S1474-4422(04)00880-4 [DOI] [PubMed] [Google Scholar]
- 2. Kojovic M, Cordivari C, Bhatia K. Myoclonic disorders: a practical approach for diagnosis and treatment. Ther Adv Neurol Disord. 2011;4(1):47–62. 10.1177/1756285610395653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Guerrini R, Takahashi T. Myoclonus and epilepsy. Handb Clin Neurol. 2013;111:667–679. 10.1016/B978-0-444-52891-9.00069-5 [DOI] [PubMed] [Google Scholar]
- 4. Caviness JN. Treatment of myoclonus. Neurotherapeutics. 2014;11(1):188–200. 10.1007/s13311-013-0216-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Marsden CD, Hallet M, Fahn S. The nosology and pathophysiology of myoclonus. In: Marsden CD, Fahn S, editors. Movement disorders. London: Butterworth Scientific; 1981. p. 196–248. [Google Scholar]
- 6. Avanzini G, Shibasaki H, Rubboli G, Canafoglia L, Panzica F, Franceschetti S, et al. Neurophysiology of myoclonus and progressive myoclonus epilepsies. Epileptic Disord. 2016;18(S2):11–27. 10.1684/epd.2016.0835 [DOI] [PubMed] [Google Scholar]
- 7. Shibasaki H. Electrophysiologic studies of myoclonus. Muscle Nerve. 2000;23(3):321–335. [DOI] [PubMed] [Google Scholar]
- 8. Erro R, Bhatia KP, Edwards MJ, Farmer SF, Cordivari C. Clinical diagnosis of propriospinal myoclonus is unreliable: an electrophysiologic study. Mov Disord. 2013;28(13):1868–1873. 10.1002/mds.25627 [DOI] [PubMed] [Google Scholar]
- 9. Hill AT, Briggs BA, Seneviratne U. Simultaneous recording of EEG and electromyographic polygraphy increases the diagnostic yield of video‐EEG monitoring. J Clin Neurophysiol. 2014;31(3):203–207. 10.1097/WNP.0000000000000059 [DOI] [PubMed] [Google Scholar]
- 10. Dijk JM, Tijssen MA. Management of patients with myoclonus: available therapies and the need for an evidence‐based approach. Lancet Neurol. 2010;9:1028–1036. 10.1016/S1474-4422(10)70193-9 [DOI] [PubMed] [Google Scholar]
- 11. Zutt R, van Egmond ME, Elting JW, van Laar PJ, Brouwer OF, Sival DA, et al. A novel diagnostic approach to patients with myoclonus. Nat Rev Neurol. 2015;11:687–697. [DOI] [PubMed] [Google Scholar]
- 12. Caviness JN. Myoclonus. Continuum (MinneapMinn). 2019;25:1055–1080. 10.1212/CON.0000000000000750 [DOI] [PubMed] [Google Scholar]
- 13. Pena AB, Caviness JN. Physiology‐based treatment of myoclonus. Neurotherapeutics. 2020;17:1665–1680. 10.1007/s13311-020-00922-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Merchant SHI, Vial‐Undurraga F, Leodori G, van Gerpen JA, Hallett M. Myoclonus: an electrophysiological diagnosis. Mov Disord Clin Pract. 2020;7:489–499. 10.1002/mdc3.12986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shibasaki H, Hallett M. Electrophysiological studies of myoclonus. Muscle Nerve. 2005;31:157–174. 10.1002/mus.20234 [DOI] [PubMed] [Google Scholar]
- 16. Deuschl G, Ebner A, Hammers R, Lücking CH. Differences of cortical activation in spontaneous and reflex myoclonias. Electroencephalogr Clin Neurophysiol. 1991;80:326–328. 10.1016/0168-5597(91)90117-g [DOI] [PubMed] [Google Scholar]
- 17. Kanouchi T, Yokota T, Kamata T, Ishii K, Senda M. Central pathway of photic reflex myoclonus. J NeurolNeurosurg Psychiatry. 1997;62:414–417. 10.1136/jnnp.62.4.414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cassim F, Houdayer E. Neurophysiology of myoclonus. Neurophysiol Clin. 2006;36:281–291. 10.1016/j.neucli.2006.10.001 [DOI] [PubMed] [Google Scholar]
- 19. Hallett M. Physiology of human posthypoxic myoclonus. Mov Disord. 2000;15(suppl 1):8–13. [DOI] [PubMed] [Google Scholar]
- 20. Michelucci R, Pasini E, Riguzzi P, Andermann E, Kälviäinen R, Genton P. Myoclonus and seizures in progressive myoclonus epilepsies: pharmacology and therapeutic trials. Epileptic Disord. 2016;18(S2):145–153. [DOI] [PubMed] [Google Scholar]
- 21. Ferlazzo E, Trenite DK, Haan GJ, Felix Nitschke F, Ahonen S, Gasparini S, et al. Update on pharmacological treatment of progressive myoclonus epilepsies. Curr Pharm des. 2017;23:5662–5666. 10.2174/1381612823666170809114654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lagorio I, Zara F, Striano S, Striano P. Familial adult myoclonic epilepsy: a new expansion repeats disorder. Seizure. 2019;67:73–77. 10.1016/j.seizure.2019.03.009 [DOI] [PubMed] [Google Scholar]
- 23. Hallett M, Chadwick D, Adam J, Marsden CD. Reticular reflex myoclonus: a physiological type of human post‐hypoxic myoclonus. J Neurol Neurosurg Psychiatry. 1977;40(3):253–264. 10.1136/jnnp.40.3.253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Brown P, Thompson PD, Rothwell JC, Day BL, Marsden CD. Axial myoclonus of propriospinal origin. Brain. 1991;114(pt 1A):197–214. 10.1093/oxfordjournals.brain.a101857 [DOI] [PubMed] [Google Scholar]
- 25. Antelmi E, Provini F. Propriospinal myoclonus: the spectrum of clinical and neurophysiological phenotypes. Sleep Med Rev. 2015;22:54–63. 10.1016/j.smrv.2014.10.007 [DOI] [PubMed] [Google Scholar]
- 26. Roze E, Apartis E, Clot F, Dorison N, Thobois S, Guyant‐Marechal L, et al. Myoclonus‐dystonia: clinical and electrophysiologic pattern related to SGCE mutations. Neurology. 2008;70(13):1010–1016. 10.1212/01.wnl.0000297516.98574.c0 [DOI] [PubMed] [Google Scholar]
- 27. Oh SY, Kim JS, Dieterich M. Update on opsoclonus‐myoclonus syndrome in adults. J Neurol. 2019;266:1541–1548. 10.1007/s00415-018-9138-7 [DOI] [PubMed] [Google Scholar]
- 28. Deuschl G, Mischke G, Schenck E, Schulte‐Monting J, Lucking CH. Symptomatic and essential rhythmic palatal myoclonus. Brain. 1990;113:1645–1672. [DOI] [PubMed] [Google Scholar]
- 29. Shin H‐W, Ye BS, Kim J, Kim SM, Sohn YH. The contribution of a spinal mechanism in developing peripheral myoclonus: a case report. Mov Disord. 2007;22:1350–1352. [DOI] [PubMed] [Google Scholar]
- 30. Safarpour Y, Vaziri ND, Jabbari B. Movement disroders in chronic kidney disease – a descriptive review. J Stroke Cerebrovasc Dis. 2021;30(9):105408. 10.1016/j.jstrokecerebrovasdis.2020.105408 [DOI] [PubMed] [Google Scholar]
- 31. Ser MH, Gündüz A, Demirbilek V, Yalçınkaya C, Nalbantoğlu M, Coşkun T, et al. Progression of myoclonus subtypes in subacute sclerosing panencephalitis. Neurophysiol Clin. 2021;51(6):533–540. 10.1016/j.neucli.2021.07.001 [DOI] [PubMed] [Google Scholar]
- 32. Freund B, Kaplan PW. Post‐hypoxic myoclonus: differentiating benign and malignant etiologies in diagnosis and prognosis. Clin Neurophysiol Pract. 2017;2:98–102. 10.1016/j.cnp.2017.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Janssen S, Bloem BR, van de Warrenburg BP. The clinical heterogeneity of drug‐induced myoclonus: an illustrated review. J Neurol. 2017;264(8):1559–1566. 10.1007/s00415-016-8357-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Binelli S, Agazzi P, Canafoglia L, Scaioli V, Panzica F, Visani E, et al. Myoclonus in Creutzfeldt‐Jakob disease: polygraphic and video‐electroencephalography assessment of 109 patients. Mov Disord. 2010;25(16):2818–2827. 10.1002/mds.23397 [DOI] [PubMed] [Google Scholar]
- 35. Rossi Sebastiano D, Soliveri P, Panzica F, Moroni I, Gellera C, Gilioli I, et al. Cortical myoclonus in childhood and juvenile‐onset Huntington's disease. Parkinsonism Relat Disord. 2012;18(6):794–797. 10.1016/j.parkreldis.2012.03.011 [DOI] [PubMed] [Google Scholar]
- 36. Genton P, Striano P, Minassian BA. The history of progressive myoclonus epilepsies. Epileptic Disord. 2016;18(S2):3–10. 10.1684/epd.2016.0834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Marseille Consensus Group . Classification of progressive myoclonus epilepsies and related diseases. Ann Neurol. 1990;28:113–116. [DOI] [PubMed] [Google Scholar]
- 38. Kälviäinen R. Progressive myoclonus epilepsies. Semin Neurol. 2015;35(3):293–299. 10.1055/s-0035-1552620 [DOI] [PubMed] [Google Scholar]
- 39. Koskiniemi M, Donner M, Majuri H, Haltia M, Norio R. Progressive myoclonus epilepsy. A clinical and histopathological study. Acta Neurol Scand. 1974;50(3):307–332. [PubMed] [Google Scholar]
- 40. Orsini A, Valetto A, Bertini V, Esposito M, Carli N, Minassian BA, et al. The best evidence for progressive myoclonic epilepsy: a pathway to precision therapy. Seizure. 2019;71:247–257. 10.1016/j.seizure.2019.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Courage C, Oliver KL, Park EJ, Cameron JM, Grabińska KA, Muona M, et al. Progressive myoclonus epilepsies‐residual unsolved cases have marked genetic heterogeneity including dolichol‐dependent protein glycosylation pathway genes. Am J Hum Genet. 2021;108(4):722–738. 10.1016/j.ajhg.2021.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Canafoglia L, Franceschetti S, Gambardella A, Striano P, Giallonardo AT, Tinuper P, et al. Progressive myoclonus epilepsies: diagnostic yield with next‐generation sequencing in previously unsolved cases. Neurol Genet. 2021;7(6):e641. 10.1212/NXG.0000000000000641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Franceschetti S, Michelucci R, Canafoglia L, Striano P, Gambardella A, Magaudda A, et al. Progressive myoclonic epilepsies: definitive and still undetermined causes. Neurology. 2014;82(5):405–411. 10.1212/WNL.0000000000000077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zuberi SM, Wirrell E, Yozawitz E, Wilmshurst JM, Specchio N, Riney K, et al. ILAE classification and definition of epilepsy syndromes with onset in neonates and infants: position statement by the ILAE task force on nosology and definitions. Epilepsia. 2022;63(6):1349–1397. 10.1111/epi.17239 [DOI] [PubMed] [Google Scholar]
- 45. Guerrini R, Bonanni P, Parmeggiani L, Santucci M, Parmeggiani A, Sartucci F. Cortical reflex myoclonus in Rett syndrome. Ann Neurol. 1998;43:472–479. [DOI] [PubMed] [Google Scholar]
- 46. d'Orsi G, Demaio V, Minervini MG. Myoclonic status misdiagnosed as movement disorders in Rett syndrome: a video‐polygraphic study. Epilepsy Behav. 2009;15(2):260–262. 10.1016/j.yebeh.2009.03.033 [DOI] [PubMed] [Google Scholar]
- 47. Pollack SF, Grocott OR, Parkin KA, Larson AM, Thibert RL. Myoclonus Angelman syndrome. Epilepsy Behav. 2018;82:170–174. 10.1016/j.yebeh.2018.02.006 [DOI] [PubMed] [Google Scholar]
- 48. Ricci E, Bonfatti R, Rocca A, Sperti G, Cagnazzo V, Vignoli A, et al. Myoclonic epilepsy with photosensitivity in infants with Pallister‐Killian syndrome. Eur J Paediatr Neurol. 2019;23(4):653–656. 10.1016/j.ejpn.2019.05.012 [DOI] [PubMed] [Google Scholar]
- 49. d'Orsi G, Specchio LM, Apulian Study Group on Senile Myoclonic Epilepsy . Progressive myoclonus epilepsy in down syndrome patients with dementia. J Neurol. 2014;261(8):1584–1597. 10.1007/s00415-014-7376-x [DOI] [PubMed] [Google Scholar]
- 50. Giraldez BG, Serratosa JM, Striano S, Ikeda A, Striano P, Coppola A. Familial adult myoclonus epilepsy: clinical findings, disease course, and comorbidities. Epilepsia. 2023;64(Suppl 1):S9–S13. [DOI] [PubMed] [Google Scholar]
- 51. Corbett MA, Depienne C, Veneziano L, Klein KM, Brancati F, Guerrini R, et al. Genetics of familial adult myoclonus epilepsy: from linkage studies to noncoding repeat expansions. Epilepsia. 2023;64(Suppl 1):S14–S21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Coppola A, Dubbioso R, Cuccurullo C, Licchetta L, Carreno M, Hirsch E, et al. Current treatment options for familial adult myoclonus epilepsy. Epilepsia. 2023;64(Suppl 1):S58–S63. [DOI] [PubMed] [Google Scholar]
- 53. Caraballo RH, Capovilla G, Vigevano F, Beccaria F, Specchio N, Fejerman N. The spectrum of benign myoclonus of early infancy: clinical and neurophysiologic features in 102 patients. Epilepsia. 2009;50(5):1176–1183. [DOI] [PubMed] [Google Scholar]
- 54. Capovilla G. Shaking body attacks: a new type of benign non‐epileptic attack in infancy. Epileptic Disord. 2011;13(2):140–144. [DOI] [PubMed] [Google Scholar]
- 55. Capovilla G, Montagnini A, Peruzzi C, Beccaria F. Head atonic attacks: a new type of benign non‐epileptic attack in infancy strongly mimicking epilepsy. Epileptic Disord. 2013;15(1):44–48. [DOI] [PubMed] [Google Scholar]
- 56. Menozzi E, Balint B, Latorre A, Valente EM, Rothwell JC, Bhatia KP. Twenty years on: myoclonus‐dystonia and ε‐sarcoglycan — neurodevelopment, channel, and signaling dysfunction. Mov Disord. 2019;34:1588–1601. [DOI] [PubMed] [Google Scholar]
- 57. Genton P, Van Vleymen B. Piracetam and levetiracetam: close structural similarities but different pharmacological and clinical profiles. Epileptic Disord. 2000;2(2):99–105. [PubMed] [Google Scholar]
- 58. Ferlazzo E, Russo E, Mumoli L, Sueri C, Gasparini S, Palleria C, et al. Profile of brivaracetam and its potential in the treatment of epilepsy. Neuropsychiatric Dis Treat. 2015;11:2967–2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Houston DM, Chen E, Schusse CM. Use of brivaracetam in cases of acquired pathologic myoclonus. Abstract 2406. Aesnet.org
- 60. Chandak R, Gudlavalleti A, Medin K. Refractory myoclonus in lance Adams syndrome responding to Brivaracetam (P6.046). Neurology. 2018;90(15 Supplement). [Google Scholar]
- 61. Christe W, Jacob R, Janz D. Juvenile myoclonic epilepsy: response to valproate monotherapy in 27 previously untreated patients. Epilepsia. 1995;36(Suppl. 3):S65. [Google Scholar]
- 62. Calleja S, Sala‐Puig J, Ribacoba R, Lahoz CH. Evolution of juvenile myoclonic epilepsy treated form the outset with sodium valproate. Seizure. 2001;10(6):424–427. 10.1053/seiz.2000.0530 [DOI] [PubMed] [Google Scholar]
- 63. Fonseca E, Guzmán L, Quintana M, Abraira L, Santamarina E, Salas‐Puig X, et al. Efficacy, retention, and safety of brivaracetam in adult patients with genetic generalized epilepsy. Epilepsy Behav. 2020;102:106657. 10.1016/j.yebeh.2019.106657 [DOI] [PubMed] [Google Scholar]
- 64. Belli E, Del Prete E, Mazzucchi S, Frosini D, Siciliano G, Ceravolo R. Perampanel as a novel treatment for myoclonus in myoclonus‐dystonia syndrome. P147. www.parkinsonlimpedismov.it [DOI] [PubMed]
- 65. Clinicaltrials.gov
- 66. Kishnani P, Lachmann R, Mozaffar T, Walters C, Case L, Appleby M, et al. Safety and efficacy of VAL‐1221, a novel fusion protein targeting cytoplasmic glucogen, in patients with late‐onset Pompe disease. Mol Genet Metabol. 2019;126:S85–S86. [Google Scholar]
- 67. Minassian BA. Post‐modern therapeutic approaches for progressive myoclonus epilepsy. Epileptic Disord. 2016;18(Suppl 2):154–158. 10.1684/epd.2016.0862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Riva A, Guglielmo A, Balagura G, Marchese F, Amadori E, Iacomino M, et al. Emerging treatments for progressive myoclonus epilepsies. Expert Rev Neurother. 2020;20(4):341–350. 10.1080/14737175.2020.1741350 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available in the article. If additional data were required, they might be requested from the corresponding author.