

Abstract
African American/Black adults are twice as likely to have Alzheimer’s disease (AD) compared to non-Hispanic White adults. Genetics partially contributes to this disparity in AD risk, among other factors, as there are several genetic variants associated with AD that are more prevalent in individuals of African or European ancestry. The phospholipid-transporting ATPase ABCA7 (ABCA7) gene has stronger associations with AD risk in individuals with African ancestry than in individuals with European ancestry. In fact, ABCA7 has been shown to have a stronger effect size than the apolipoprotein E (APOE) ε4 allele in African American/Black adults. ABCA7 is a transmembrane protein involved in lipid homeostasis and phagocytosis. ABCA7 dysfunction is associated with increased amyloid-beta production, reduced amyloid-beta clearance, impaired microglial response to inflammation, and endoplasmic reticulum stress. This review explores the impact of ABCA7 mutations that increase AD risk in African American/Black adults on ABCA7 structure and function and their contributions to AD pathogenesis. The combination of biochemical/biophysical and ‘omics-based studies of these variants needed to elucidate their downstream impact and molecular contributions to AD pathogenesis is highlighted.
Keywords: African Americans, Alzheimer’s disease, Blacks, health status disparities, human ABCA7 protein, proteomics, single nucleotide polymorphism, subfamily A of ATP binding cassette transporter
INTRODUCTION
Alzheimer’s disease (AD) is a neurodegenerative disease that leads to memory loss, cognitive decline, and activated immune response due to the increased presence of amyloid- (A) plaques and tau tangles in the brain [1]. In the United States, 6.2 million people are diagnosed with AD [2]; however, AD affects different racial/ethnic minorities disproportionately. AD occurs twice as frequently in African American/Black and one and a half times as frequently in Hispanic than non-Hispanic White adults [2–4], while it occurs less frequently in Asian American adults [5, 6]. Increased prevalence of AD in African American/Black adults could be related to differences in socioeconomics, comorbidities, and molecular factors between African American/Black and non-Hispanic White adults. For example, socioeconomic factors including education level and quality of education, local area/neighborhood, socioeconomic status, healthcare access, environmental factors, and willingness to seek care and treatment are not equitable between racial/ethnic subgroups [4, 7–10]. Comorbidities that increase AD risk such as hypertension and type 2 diabetes mellitus are also more prevalent in African American/Black adults than non-Hispanic White adults [5, 11–13].
Frequency and conferred risk of AD susceptibility genetic variants are also known to differ across racial/ethnic groups. It is important to note that existing genetic studies of AD have used both self-reported race/ethnicity and ancestry to distinguish racial/ethnic groups, which classify individuals based on distinct qualities. Racial groups are based on physical characteristics and skin color, which in turn influence many aspects of an individual’s life, while ethnicity groups individuals by a shared culture or language, though these terms are often used together or interchangeably [14–16]. Ancestry, on the other hand, is the genetic origin of a population, which does not reflect the socioeconomic impact encompassed under self-reported race/ethnicity [15, 17] and does not always correlate well with self-reported race/ethnicity [18]. In this review, we will use self-reported racial/ethnic groups, though previous studies of ancestry will be referenced to comprehensively review relevant literature. In non-Hispanic White adults, the genetic factor that causes the greatest risk for AD is the apolipoprotein E (APOE) ε4 allele. APOE ε4 causes 20–50% higher AD risk in non-Hispanic White adults [19]. This risk does not replicate in African American/Black adults [20, 21]. On the other hand, mutations in the phospholipid-transporting ATPase ABCA7 (ABCA7) gene have stronger associations with AD risk in African American/Black adults than non-Hispanic White adults [22–24]. The effect size of ABCA7 single nucleotide polymorphism (SNP) rs115550680 is comparable to that of APOE [19, 23]. Furthermore, mutations specific to or more frequent in African American/Black adults in ABCA7 have been discovered [3, 25].
ABCA7 is a transmembrane protein responsible for moving lipids across the cell membrane using energy from adenosine triphosphate (ATP) and it is involved in three major cellular processes: cholesterol metabolism, phospholipid regulation, and phagocytosis [26–29]. ABCA7 has implications in AD pathogenesis and is involved in Aβ clearance and amyloid-β protein precursor (AβPP) transport [23, 25, 30]. Mutations in the ABCA7 gene may disrupt one or more of the protein’s functions and therefore contribute to the development of AD neuropathology. However, not all mutations are equal and the specific changes to ABCA7 function due to these AD risk mutations remain unknown. These changes are particularly important to understand due to the strong effects of ABCA7-conferred AD risk for African American/Black adults. Several recent reviews have discussed ABCA7’s involvement in AD [25, 29, 31, 32], though only one [33] has previously examined this relationship in the context of African American/Black adults, which focused primarily on cognitive impacts of ABCA7 variants. This review, on the other hand, will specifically explore ABCA7 genetic variants which increase AD risk in African American/Black adults and their contributions to AD pathogenesis via impacts on ABCA7 structure and function that influence downstream cell processes. Though we focus on ABCA7 variants found in African American/Black adults throughout this review, we note here that much of the research presented about mechanistic roles of ABCA7 in AD is in other racial/ethnic groups, as such investigations in African American/Black adults are severely lacking.
ABCA7 AD GENETIC RISK VARIANTS IN AFRICAN AMERICANS
Mutations in ABCA7 have been associated with increased AD risk in various racial/ethnic groups, including European [34–36], Latin American [37], non-Hispanic White [23, 38], and African American/Black groups [19, 23, 25, 39, 40] (Table 1). Some of these mutations increase AD risk in multiple racial/ethnic groups, such as rs3764650, while others are specific to a given group, such as rs3764647 and rs115550680. Additionally, several of the risk mutations identified across groups have different effect sizes and/or frequencies. As a whole, ABCA7 risk variants have a greater impact on odds of AD diagnosis in African American/Black adults compared to other racial/ethnic groups [41]. ABCA7 variants have been found to increase AD risk 1.8-fold in individuals with African ancestry and 1.1–1.2-fold in individuals with European ancestry [23]. Interestingly, one ABCA7 variant (rs72973581) was predicted to be a potential protective factor against developing AD in a cohort of elderly adults from British and North American ancestry [42]. Further studies are needed to elucidate how this variant can impact ABCA7 function.
Table 1.
ABCA7 SNPs associated with AD in various racial/ethnic groupsa
| SNP | Mutation | Populations Associated with AD | Frequencyb | Effect Size | Sources |
|---|---|---|---|---|---|
|
| |||||
| rs3752232 | Thr319Ala | African American/Black | 27.2% AD | N’Songo et al. 2017 [40] | |
| 23.2% CN | 1.24 | Logue et al. 2018 [43] | |||
| rs3752239 | Asn718Thr | African American/Black | 1.8% AD 0.4% CN |
4.06 | N’Songo etal. 2017 [40] |
| rs3752246 | Gly1527Ala | Multiple racial groups | ND | 1.35 | Feheretal. 2019 [113]; Hollingworth et al. 2011 [44]; |
| 1.15 | Naj etal. 2011 [45] | ||||
| rs3764647 | His395Arg | African American/Black | 26.2–29.8% AD | 1.32 | Logue et al. 2011 [39]; |
| 21.6–23.1% CN | 1.29 | Logue et al. 2018 [43] | |||
| 1.47 | N’Songo etal. 2017 [40] | ||||
| rs3764650 | Intron variant | African American/Black | ND | Hohman et al. 2016 [24]; | |
| 1.27 | Logue et al. 2011 [39] | ||||
| Asian | 8.32 | Li etal. 2017 [114]; | |||
| 1.09 | Zhou etal. 2017 [115] | ||||
| Colombian | 1.7 | Moreno et al. 2017 [37] | |||
| Non-Hispanic White | 1.25 | Almeida et al. 2018 [38]; | |||
| 1.25 | Zhou etal. 2017 [115] | ||||
| Multiple racial groups | 1.23 | Hollingworth et al. 2011 [44] | |||
| rs4147929 | Danish | ND | 1.07 | Kjeldsen et al. 2017 [35] | |
| Non-Hispanic White | 1.66 | Monsell etal. 2017 [116] | |||
| Multiple racial groups | 1.15 | Lambert et al. 2013 [46] | |||
| rs59851484 | African American/Black | 14.8% AD 10.5% CN |
1.49 | Logue et al. 2018 [43] | |
| rs78117248 | Intron variant | Belgian | 3.8% AD | 2.07 | Cuyvers et al. 2015 [36] |
| 1.8% CN | |||||
| Non-Hispanic White | ND | 1.56 | Kunkle etal. 2017 [117] | ||
| rs115550680 | African American/Black | ND | 1.79 | Reitz et al. 2013 [23] | |
| rs142076058 | Arg578Alafs | African American/Black | 9.2–15.2% AD | 2.13 | Cukier et al. 2016 [3] |
| 7.4–9.7% CN | 1.27 | Logue et al. 2018 [43] | |||
| rs200538373 | Splice donor variant | Icelandic | ND | 1.91 | Steinberg et al. 2015 [34] |
| Non-Hispanic White | 2.12 | Kunkle etal. 2017 [117] | |||
| rs567222111 | Leu396fs | African American/Black | 1.1% AD 0.3% CN |
2.42 | Logue et al. 2018 [43] |
Updated from [118].
ND indicates that this information was not available for the denoted SNP. SNP, single nucleotide polymorphism; AD, Alzheimer’s disease; CN, cognitively normal.
ABCA7 mutations affecting African American/Black adults can be classified into common mutations with smaller effect sizes and rarer mutations with larger effect sizes. The more common mutations occur in ~10–25% of individuals with normal cognition and ~15–30% of individuals with AD and increase AD risk by up to 50% (Table 1). Most are missense mutations resulting in a single amino acid substitution in the ABCA7 protein. This includes rs3764650, located in intron 13 of the ABCA7 gene, which increases AD risk by 10–20% in African American/Black adults [39], though it has a larger effect size in non-Hispanic White [23, 38] and Colombian adults [37]. This variant only showed a trend of association with AD in a study of >500 African American/Black adults with normal cognition or who had AD, though it was in linkage disequilibrium with rs3764647, which did have a significant association with AD [40]. Higher percentages of African ancestry at this locus were associated with AD [24]. Rs3764647 increases AD risk by 30–50% (odds ratio (OR) = 1.32–1.47) [25, 39, 40], while rs3752232, which is in linkage disequilibrium with rs3764647 [40], increases AD risk by 25% in African American/Black adults (OR = 1.24) [43]. It is possible that the difference in conferred risk between these two SNPs, despite being in linkage disequilibrium, is due to the relatively small sample sizes of the supporting studies (538 [40] to 1,929 [43]). Another missense mutation, rs3752246, which is in linkage disequilibrium with rs3764650 [44] and rs4147929 [35], was associated with AD (OR = 1.15) in a meta-analysis of four large genome-wide association study (GWAS) datasets performed by the Alzheimer Disease Genetics Consortium that included data from >20,000 individuals of multiple racial backgrounds [45]; however, rs3752246 was not associated with AD in two studies of >500 African American/Black adults with normal cognition or who had AD [40]. Lastly, rs59851484 was recently associated with a 50% increase in AD risk in African American/Black adults (OR = 1.49) [43].
On the other hand, the rarer mutations tend to occur in < 1% of individuals with normal cognition and < 2% of individuals with AD but increase AD risk by > 70% (Table 1). This group includes rs3752239, which increases AD risk 4x (OR = 4.06) [40]. Interestingly, the C allele of this SNP that is associated with AD risk in African American/Black adults has been reported to have a protective effect against AD in non-Hispanic White adults [46]. Rs115550680 has also been associated with AD in African American/Black adults, with an effect size similar to that of the APOE ε4 allele (OR = 1.79) leading to a 70–80% increase in AD risk [19, 23]. This group also includes the frameshift mutations rs142076058 and rs567222111, both of which more than double AD risk in African American/Black adults (OR = 2.13 and 2.42, respectively) [25, 43]. Rs142076058 occurs more frequently in African American/Black adults than in non-Hispanic White adults [25].
CONTRIBUTIONS OF ABCA7 VARIANTS TO AD PATHOGENESIS
The potential contributions of these ABCA7 variants to AD pathogenesis are discussed in this section and are summarized in Fig. 1.
Fig. 1.
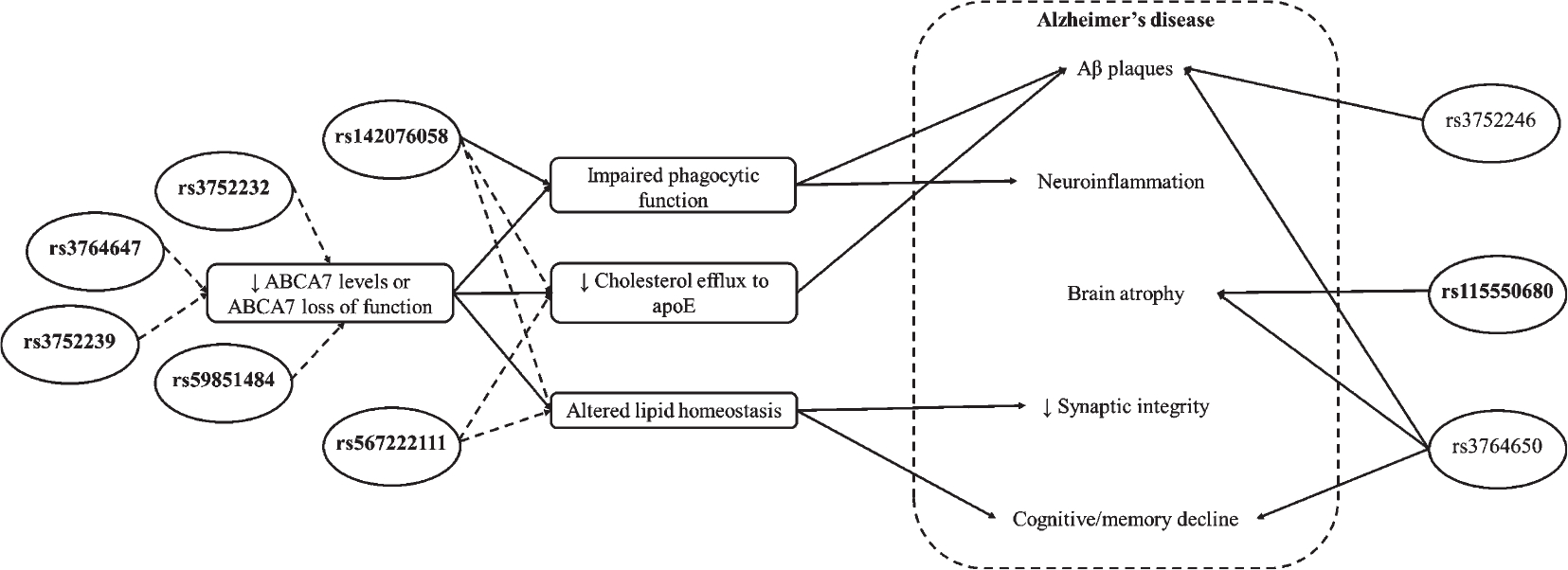
Summary of ABCA7 variant contributions to AD pathogenesis. ABCA7 SNPs that increase AD risk in African American/Black adults are shown in ovals, with bold text indicating risk SNPs specific to African American/Black adults. Solid arrows indicate proven effects of SNPs; dashed arrows indicate predicted or hypothesized effects of SNPs. ABCA7, phospholipid-transporting ATPase ABCA7; apoE, apolipoprotein E; A, amyloid-beta.
Impacts on ABCA7 structure
ATP-binding cassette (ABC) transporters are membrane-associated machines that transport a wide variety of substrates across extra- and intracellular membranes, including metabolic products, lipids, and sterols, as well as drugs [27, 47]. In general, ABC transporters contain two highly conserved nucleotide-binding domains (NBDs) which are connected to two integral transmembrane domains (TMDs), each containing 6–10 transmembrane-spanning helices (Fig. 2) [47]. High-resolution structures of ABC transporters have provided insight into how these proteins use ATP to transport molecules out of the cell [48–50]. ABC transporters utilize energy from intracellular ATP that induces conformational changes allowing various molecules, such as lipids or cholesterol, to be transported from the cell membrane to bind to extracellular proteins including apolipoprotein A-I (apoA-I) or apoE [51, 52].
Fig. 2.
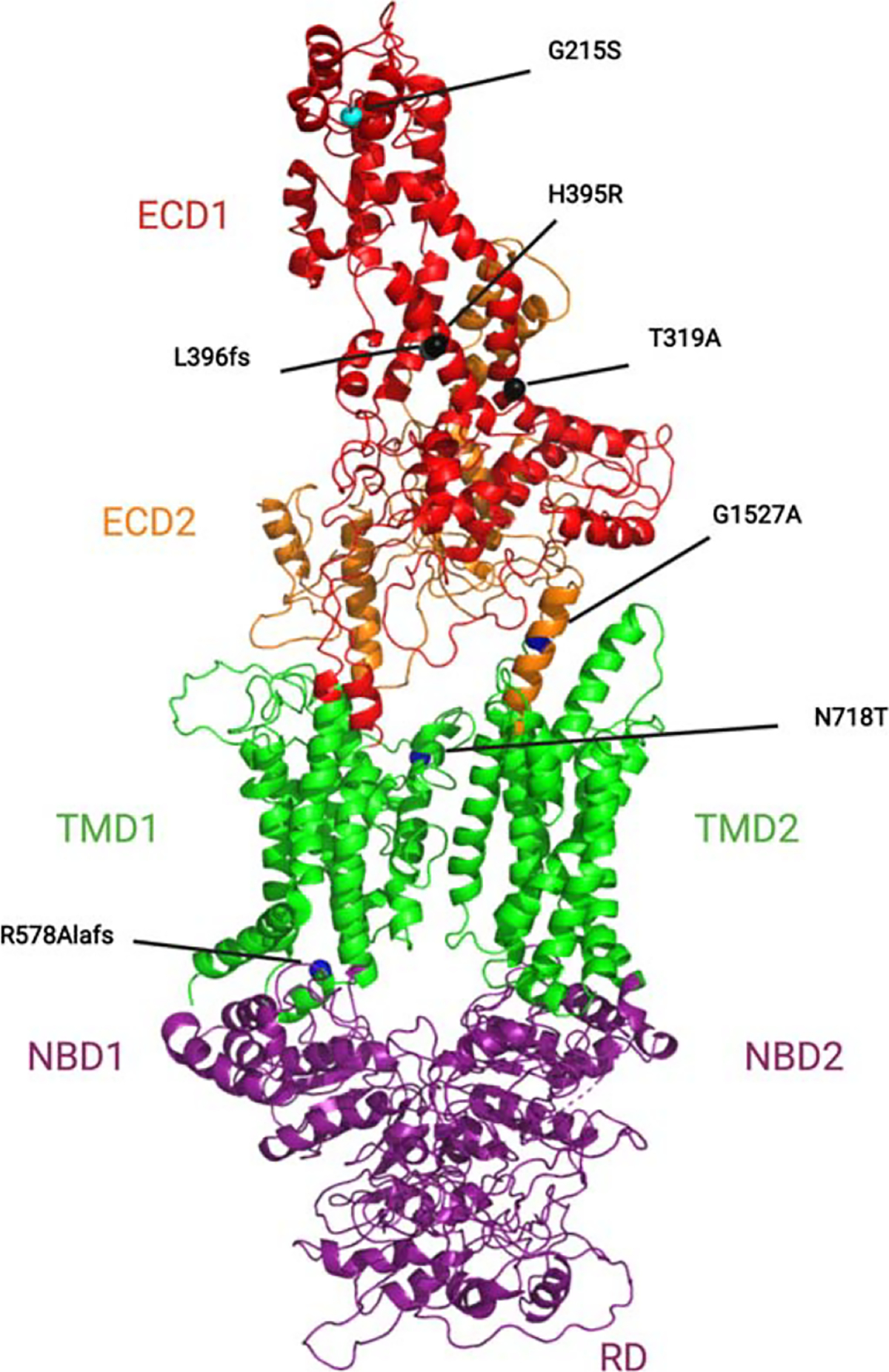
Cartoon homology model of full-length ABCA7. A homology model of full-length ABCA7 was generated using MODELLER [109] based on the alignment with the ABCA1 structure (PDB: 5xjy) and validated using the Structural Analysis and Verification Server (SAVES) server [110, 111]. AD-associated variants (Table 1) are noted and highlighted here as spheres. ECD, extracellular domain; TMD, transmembrane domain; NBD, nucleotide binding domain; RD, regulatory domain.
ABC transporter subfamily A (ABCA) transporters are distinct from other ABC subfamilies because they possess two large extracellular domains (ECDs) that are often glycosylated [53]. Current information about ABCA7 comes from our understanding of its closest homologue, ABCA1, which has been studied extensively and serves as the prototype for other ABCA transporters [54, 55]. The presence of ECD1 and ECD2 in ABCA1 and ABCA7 is an essential factor in their function due to their interactions with apolipoproteins (Fig. 2). Sequence alignment of the ECD1 domains between ABCA1 and ABCA7 shows 39.97% homology (Fig. 3). Structural/functional studies of ABCA1 show that these domains contain two intramolecular disulfide bonds (Cys75 and Cys309) needed for apoA-I-dependent cholesterol efflux [56]. ApoA-I is predicted to directly bind to ABCA1, which triggers signal transduction pathways to mediate lipid transport [57–59]. Nagao et al. (2012) demonstrated that the intracellular hydrolysis of ATP in both of the NBDs of ABCA1 induces conformational changes that signal the ECDs to bind apoA-I [60]. This conformational change couples ATP hydrolysis with apoA-I binding and cholesterol efflux. Despite the extensive information on ABCA1, the precise molecular details of its interaction with apoA-I are unknown. However, this interaction is necessary for apoA-I’s downstream signaling to activate cellular cyclic adenosine monophosphate (cAMP) signaling and mediate cellular lipid efflux [61].
Fig. 3.
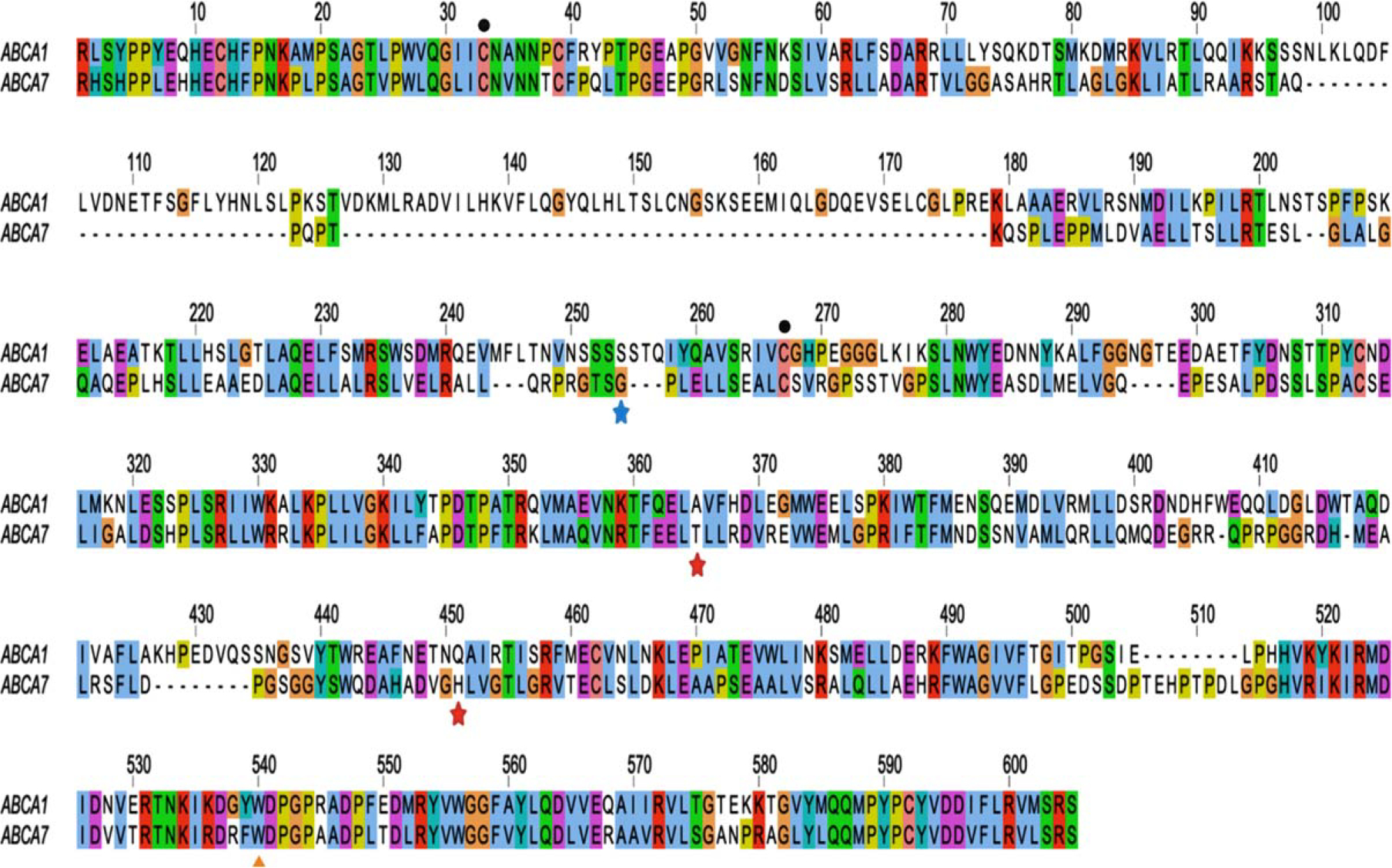
Sequence alignment of ECD1 domains of ABCA1 and ABCA7. Amino acid sequences for the corresponding ECD1s were obtained from UniProt.org (ABCA1 ID: O95477, residues 43–639; ABCA7 ID: Q8IZY2, residues 43–549). The numbering of the alignment starts with the beginning of the ECD1. The location of the missense variants that confer AD risk in African American/Black adults (T319A, H395R) and the missense variant predicted to have a protective effect (G215S) are indicated by stars. Intramolecular disulfide bonds implicated in apoA-I-dependent cholesterol efflux are denoted by circles. The Trp residue at position 590 linked to Tangier disease in ABCA1 is conserved in ABCA7 and highlighted by a triangle. Sequences were aligned using the Tcoffee multiple sequence alignment in Jalview [112]. The alignment was color coded using the ClustalX scheme. Colors represent categories of amino acids; white indicates gap/unconserved residues.
Mutations in ABCA1, including R587W, W590S, Q597R, C1477R, and S1506L, disrupt its interaction with apoA-I and have been linked to a rare lipoprotein metabolism disorder called Tangier disease [62–64]. Several of these mutations (R587W, W590S, Q597R) occur within the ECD1 of ABCA1, which spans residues 43–639. The W590S mutation is located at the end of ECD1, in close proximity to the cellular membrane. All three of these mutations reduce apoA-I-mediated lipid efflux and corresponding high-density lipoprotein (HDL) generation in human embryonic kidney (HEK) 293 cells [62]. Interestingly, the R587W and Q597R mutants were retained in the endoplasmic reticulum (ER), while the W590S mutant still localized to the plasma membrane similar to wild-type ABCA1, suggesting that the impact of the W590S mutation was functionally distinct from that of the R587W and Q597R mutations [62].
The ECDs of membrane proteins serve a distinct purpose to allow cell-cell communication. It is unclear why two missense mutations (rs3752232 and rs3764647 [40]) in the ECD1 of ABCA7 are strongly associated with AD in African American/Black adults, or why the rs72973581 variant, also located within ECD1, seems to have a protective effect [42]. Interestingly, the mutations identified in ABCA1 that disrupt binding of apoA-I are conserved in ABCA7. These mutations seem to cluster toward the end of ECD1. The missense ABCA7 mutations, however, are situated away from this small cluster of ABCA1 mutations, which appears to be a binding pocket and may alter ABCA7’s ability to bind lipid substrates instead. Rs3752232 causes a threonine to alanine substitution at position 319, and rs3764647 leads to a histidine to arginine substitution at position 395. Neither of these mutations is predicted to have harmful functional effects, such that they may contribute to AD risk via more subtle effects on ABCA7 protein’s structure or function [40]. Our recent study of rs3752232 supports this hypothesis, as we found that this mutation had subtle structural effects on ABCA7 protein’s ability to bind various lipids such as phosphatidylinositol 4,5-bisphosphate (PIP2) [65]. Another missense mutation located in ECD2, rs3752246, results in a glycine to alanine substitution at position 1527 and is also not predicted to affect ABCA7 protein’s function [66]. In the first intracellular domain of the protein, rs3752239 leads to a missense mutation changing an asparagine to a threonine at position 718 [40]. This mutation could lead to potentially damaging effects on the protein, though this remains to be investigated. Additionally, a low-frequency missense variant, rs72973581 (minor allele frequency = 4.3%), has been reported to confer modest but statistically significant protection against AD (p =0.024, OR = 0.57, 95% confidence interval = 0.41–0.80) [42]. This variant results in a glycine to serine substitution at position 215 (G215S), also located within ECD1. Taken together, mutations within the ECDs likely affect which lipid substrates get transported out of the cell to apolipoproteins. The mechanistic details of this function are critical to understand since lipoproteins such as apoA-I and apoE are cholesterol/lipid delivery carriers and alterations in their cargo due to ABCA7 variants can have significant downstream effects that promote AD.
On the other hand, ABCA7 frameshift mutations rs142076058 and rs567222111 are also associated with AD and cause significant structural changes to the ABCA7 protein. Rs142076058 is a 44 bp deletion that causes a frameshift (Arg578Alafs) and premature termination codon, producing a truncated protein missing many TMDs and both NBDs, likely with significant functional impact [3]. Rs567222111 leads to an 11 bp deletion and frameshift (Leu396fs) and is also hypothesized to lead to ABCA7 protein loss-of-function [43]. It is also possible that the shortened transcripts resulting from both of these mutations could undergo nonsense-mediated decay, which may be phenotypically comparable to a knockout, or lead to significantly decreased ABCA7 protein levels as have previously been associated with AD [25, 31, 67].
Impact on ABCA7 functions
Lipid metabolism and transport
ABCA7 is predicted to play a large role in regulating lipid metabolism and transporting newly synthesized lipids. ABCA7 transports primarily phospholipids such as phosphatidylcholines and phosphatidylserines across the cell membranes to apolipoproteins apoA-I and apoE, though it has a lesser ability to transport cholesterol [29, 68]. While the full repertoire of ABCA7 lipid substrates is not known, the preference of ABCA7 lipid export differs from that of ABCA1. ABCA7 preferred to export phosphatidylcholine (PC) ≥ lysophosphatidylcholine (lysoPC) > sphingomyelin (SM) = phosphatidylethanolamine (PE), whereas ABCA1 preferred to export PC >> SM > PE = lysoPC [69]. The authors of this study suggested that lysoPC export may be critical for ABCA7 function in the brain. However, ABCA7 expression is inversely correlated with cholesterol levels via the sterol regulatory element-binding protein 2 (SREBP2) pathway and reverse cholesterol transport [31]. The endogenous expression of ABCA7 stimulates cholesterol efflux to apoE, in turn suppressing Aβ production [30]. When ABCA7 function is lost (i.e., ABCA7 knockout), disruption in the microglial Aβ clearance pathway leads to a cholesterol deficiency, triggering accelerated Aβ production both in vitro and in vivo [25, 31]. This accelerated Aβ production causes Aβ plaques to form [70]. Therefore, it is well established that loss of ABCA7 function significantly increases the progression of AD [71].
Furthermore, decreased levels of ABCA7 can also lead to altered lipid homeostasis, which is linked to ER stress. In mice, ABCA7 knockout alters the brain phospholipid profile [71], decreases serum HDL and cholesterol levels [72], and disrupts lipid rafts on the plasma membrane [73]. ABCA7 knockdown induced ER stress in cultures of primary mouse neurons, as observed by increased protein kinase R-like ER kinase (PERK) levels which then activates eukaryotic initiation factor 2α (eIF2α) [71]. EIF2α phosphorylation increases β-secretase 1 (BACE1) expression, thus increasing Aβ generation [74]. Amyloid precursor protein/presenilin-1 double mutant (APP/PS1); ABCA7−/− mice also had increased levels of phosphorylated extracellular regulated kinase (ERK) in the brain, specifically in neurons, compared to APP/PS1 mice [71]. These findings suggest that ABCA7 deficiency, resulting from ABCA7 knockout, may be associated with abnormal activation of the ERK pathway, which leads to declines in cognition and synaptic integrity [75].
Mutations in the structurally similar ABCA1 protein disrupt efflux of phospholipids and cholesterol across cell membranes to extracellular apoA-I and apoE, and consequently HDL is unable to transport cholesterol and phospholipids from peripheral tissues, such as the heart, to the liver [29, 68]. Although ABCA7 knockout mice had altered lipid levels as described above, apoA-I-mediated phospholipid and cholesterol efflux were not affected [72]. Our study of rs3752232 in HEK 293 cells yielded similar results, wherein subtle structural changes in the mutant ABCA7 protein affected PIP2 binding compared to wild-type ABCA7, yet the impact on the cellular proteome was minimal without evidence for effects on lipid transport proteins [65]. Taken together, these findings suggest that ABCA7 risk mutations likely cause more significant functional changes outside of lipid metabolism and transport pathways that contribute to AD pathogenesis.
While rs3752246 is not predicted to adversely affect ABCA7 function, an in vitro study of Chinese hamster ovary (CHO) cells transfected with the human APP expressing the Swedish mutation (APPSwe) found that this SNP resulted in a modified form of ABCA7 with altered function, due to the lack of a myristoylation site [76]. This led to increased Aβ production via increased BACE1 activity, likely due to protein-protein interactions favoring BACE1 activity resulting from the lack of the myristate (14-carbon saturated fatty acid) posttranslational modification at position 1527. Rs3752246 has also been associated with increased amyloid deposition in individuals with normal cognition or mild cognitive impairment (MCI) in two different cohorts [77, 78], further supporting that this mutation contributes to AD via amyloid pathology, particularly early in disease progression. Contrary to the ABCA7 missense mutations, the frameshift mutations (rs142076058 and rs567222111) cause more significant protein structure changes that likely impair its lipid transport functions [3], thus leading to increased Aβ pathology and ER stress.
Immune functions and phagocytosis
ABCA7 plays an important role in immune response and synaptic function (specifically regarding phagocytosis), which are both affected by AD. The normal function of ABCA7 helps create the lipid rafts in the plasma membranes of antigen-presenting cells and thymocytes and promote phagocytosis of apoptotic debris [29]. In ABCA7 knockout mice the disruption of lipid rafts in thymocytes and antigen-presenting cells occurs [31], causing the plasma membrane of these cells to be malformed and ineffective. ABCA7 enhances phagocytosis [31, 79] and has high homology and similarity to the ABC transporter ced-7 (ced-7) gene in C. elegans involved in apoptotic cell engulfment [29]. In mouse macrophages, ABCA7 but not ABCA1 knockout affected phagocytosis both in vitro and in vivo [31, 80]. ABCA7 knockout creates AD-related phenotypes by decreasing phagocytic activity in macrophages, which results in larger A aggregates [25].
ABCA7 knockout and mutations have only shown modest effects on lipid homeostasis, as discussed in the previous section, suggesting that ABCA7 likely affects AD risk via its phagocytic functions [81]. ABCA7 is involved in the phagocytosis of apoptotic cells through the complement component 1q pathway in macrophages. Increased ABCA7 expression increases microglial phagocytosis as well as A uptake [82]. On the other hand, ABCA7 knockout mice showed reduced oligomeric uptake of Aβ proteins in macrophages and microglia, suggesting that suppression of ABCA7 negatively affects the phagocytosis of Aβ aggregates [31, 83]. ABCA7 knockdown in macrophages also results in incomplete phagocytosis of apoptotic debris [84]. Lack of clearance of such debris could potentially cause neuroinflammation and thus contribute to AD pathogenesis [29]. Additionally, ABCA7 haplodeficient mice had reduced proinflammatory responses to acute inflammation in the brain due to impaired monocyte differentiation antigen CD14 (CD14) expression [67]. CD14 deficit also suppresses microglial uptake of Aβ [85–87]. When ABCA7 haplodeficient mice were crossed with APPNL–G–F mice, these mice had increased Aβ accumulation in the brain and enlarged endosomes in microglia [67]. The observed disruption of cell membrane organization likely alters microglial responses to acute and chronic brain inflammation. Though one study in APP/PS1 mice found that ABCA7 knockout had no effect on microglial activation [71], the majority of this research supports ABCA7 microglial expression in AD [29, 82, 88] and provides further evidence that ABCA7 deficiency (from either ABCA7 knockout or haplodeficiency) contributes to AD pathogenesis.
The impact of ABCA7 risk mutations on its immune and phagocytic functions has been understudied to date. Cukier et al. recently studied rs142076058 in induced pluripotent stem cells (iPSCs) differentiated into cortical neurons and microglia [89]. In this study, the iPSCs were derived from African American/Black individuals with AD with this deletion and matched cognitively normal individuals. The deletion led to reduced Aβ clearance in microglia, along with increased Aβ production in cortical neurons. Additionally, some SNPs (rs3752246, rs3764650) have been associated with increased brain amyloid deposition [77, 78, 90], which could be due in part to disruption of ABCA7’s phagocytic functions. Mechanistic studies are necessary to elucidate the specific functional consequences of these mutations that lead to the observed increase in amyloid deposition.
Contributions to cognitive decline
The role of ABCA7 in cognitive decline has been studied in both mice and humans. ABCA7 knockout mice had impaired spatial memory [25, 71] and were unable to develop short-term novel object recognition [25]. On the other hand, in the J20 mouse model of AD, ABCA7 knockout did not cause any further cognitive deficits, despite doubling insoluble Aβ and plaques in the brain and reducing macrophage Aβ uptake by half [83]. These findings suggest that, in mice, ABCA7 knockout exerts specific (i.e., related to spatial memory) though minimal effects on cognition.
The effects of certain AD-associated ABCA7 SNPs on cognition and brain morphology changes associated with cognitive decline have been studied in humans, with rs3764650 being the most studied. In a study of the University of California, Los Angeles’ Imaging and Genetic Biomarkers for AD (ImaGene) cohort, rs3764650 was associated with increased atrophy in both hippocampus and cortex of individuals with normal cognition or MCI [91]. This mutation has also been associated with more postmortem amyloid plaques in the brain in a cohort of individuals with normal cognition, MCI, or AD [90] and faster rates of decline in memory in individuals with final diagnoses of MCI or AD [92]. Rs3764650 was also associated with lower baseline episodic memory scores in individuals with normal cognition over the age of 60 from the Personality and Total Health cohort in Australia [93], but not with decline in any cognitive domains by others [94]. It is important to note that these studies either were exclusively Caucasian or individuals of European ancestry [92, 93] or did not disclose racial/ethnic group information for their cohorts [90, 91].
Some cognitive effects of rs3764650 have only been identified in specific subpopulations. For example, this SNP resulted in a faster rate of cognitive decline only in individuals who were cognitively normal at baseline and became cognitively impaired longitudinally [95]. Additionally, rs3764650 was associated with cognitive decline in males [96]. Another study identified a significant APOE-ABCA7 interaction associated with memory scores and default mode network activity, and showed that, of individuals with ABCA7 rs3764650, APOE ε4 carriers had worse memory scores than APOE ε4 non-carriers [97]. However, both rs3764650 and rs3752246 have also been shown to be protective against memory decline in APOE ε4-positive individuals [98]. ABCA7 interactions with other genes such as clusterin (CLU) also impact cognition [99]. Specifically, interactions between ABCA7 rs3764650 and CLU rs9331888 resulted in decreased functional connectivity in different brain regions in healthy, middle-aged adults [99]. Interestingly, rs3764650 may prevent older African American/Black adults with normal cognition from receiving the benefits of aerobic exercise on brain health and cognition [33, 100, 101]. Specifically, rs3764650 moderated the association between aerobic fitness and hippocampus-related generalization, measured by a concurrent discrimination and generalization task, in older African American/Black adults [100]. Overall, rs3764650 has generally minimal effects on cognition, though these effects may vary in subgroups based on genetics (APOE ε4 positive versus negative), sex, and cognitive status [31].
The impact of other ABCA7 SNPs on cognitive decline has been much less studied. The ABCA7 SNP rs3752246 was associated with decreased gray matter density in dementia patients in a study of ~1500 individuals from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) cohort [102]. The ABCA7 SNP rs3752232 was associated with increased decline in visuospatial function, including working and visual memory, in a cohort of Korean AD patients [103], though this association has not been studied in African American/Black adults to date. This mutation did not correlate with other domains such as executive function, language, attention, and memory. Additionally, older African American/Black adults (ages 63–90 years) with rs115550680 had impaired functional connectivity of the medial temporal lobe with other brain regions and smaller anterolateral entorhinal cortices compared to matched African American/Black adults without the risk SNP [41]. The entorhinal cortex is among the first brain region to be affected by AD neuropathology and smaller volumes in this brain region have been associated with cognitive decline [104]. These differences may indicate an interaction between amyloid and tau pathology in the entorhinal cortex in individuals with this mutation, whereby increased amyloid leads to increased tau pathogenesis in this region [33, 105]. However, further studies of these ABCA7 SNPs are necessary to fully elucidate their contributions to cognitive decline, particularly in diverse racial/ethnic groups.
Summary
Overall, these findings implicate ABCA7 variants in multiple aspects of AD pathogenesis, including cognitive decline, brain morphology changes, amyloid pathology, neuroinflammation, and ER stress (Fig. 1). Many of these roles contribute to or are enhanced by amyloid pathology, either directly or indirectly, but interestingly, no relation of ABCA7’s functions to tau pathology have been noted. These variants result in various structural and functional impacts on the ABCA7 protein that likely contribute to AD pathogenesis by causing ABCA7 dysfunction [25] (particularly of its phagocytic functions [35]), reduced levels or expression, or loss of function [25, 31, 67]. A recent comprehensive review of ABCA7’s role in AD focusing on data from genomics, transcriptomics, and methylomics similarly concluded that ‘omics studies in humans point to ABCA7 loss or partial loss as the mechanism behind its contribution to AD pathogenesis [31]. However, further mechanistic studies of ABCA7 in AD are necessary to elucidate the specific roles of these variants.
Though significant research has been performed to identify and study the ABCA7 SNPs associated with AD at the genetic level, it is necessary to study the functional impacts of these genetic variants, i.e., at the protein level. In their review, De Roeck et al. stated that studying these SNPs at the gene and transcript level does not provide “sufficient insight” into how ABCA7 variants contribute to AD [31]. Other ‘omics, such as proteomics, lipidomics, and metabolomics, can be useful to interrogate the downstream effects of genetic variants in both model systems and human biospecimens. As ABCA7 has known lipid transport and metabolism functions ABCA7 variants have the potential to alter the profiles of these molecules, which ‘omics techniques are particularly poised to reveal. ‘Omics analyses can provide valuable insight into genetic variants both on their own and in combination with other methods. For example, we recently applied both proteomics and structural methods to study rs3752232. Combining these analyses allowed us to determine that slight structural differences caused by this mutation may impact ABCA7’s ability to bind lipids such as PIP2 and also that this mutation had subtle effects on the HEK 293 cell proteome [65]. Further ‘omics studies of ABCA7 AD variants are crucial to advance our understanding of the contributions of these variants to AD pathogenesis and AD risk in African American/Black adults.
Moreover, there is an overarching need for mechanistic studies of ABCA7 variants in AD in African American/Black adults. Development of preclinical models, such as the iPSC model described by Cukier et al. [89], that allow study of ABCA7 mutations in systems with African American/Black backgrounds will enhance understanding of the functional implications of these mutations and enable more informed studies in humans. Leveraging existing studies and biospecimens from African American/Black adults for ‘omics and other mechanistic studies of ABCA7 variants is also a key step to address this need. Recent studies have made significant efforts towards recruitment of individuals from underrepresented groups in research including African American/Black adults, such as the nationwide All of Us research program [106] and the African Ancestry Neuroscience Research Initiative [107], providing valuable resources to empower these necessary investigations. In addition to the need for sufficient numbers of biospecimens, it will also be important to expand the scope of data collected to characterize such existing and future samples to include more social, educational, cultural, and other factors that can impact health of African American/Black adults and thus need to be considered as confounding factors [108], towards which All of Us is also making efforts. Furthermore, generating additional functional data on ABCA7 variants in African American/Black adults from the described studies may lead to effective personalized therapeutics for individuals with specific variants. Beyond these necessary studies in African American/Black adults, investigations of ABCA7 variants and AD in non-American African populations could further expand understanding of ABCA7’s mechanisms in AD in the absence of Western cultural influences. These proposed studies are critical to address missing pieces in the puzzle of ABCA7 genetic risk of AD in African American/Black adults and advance fundamental molecular understanding of how ABCA7 variants contribute to AD pathogenesis.
Conclusions
Mutations within the ABCA7 gene account for a significant proportion of genetic risk for AD in African American/Black adults. The ABCA7 protein is involved in lipid metabolism and transport and phagocytosis processes, which may become dysfunctional in AD and lead to increased Aβ production and decreased Aβ clearance, impaired microglial response to inflammation, and ER stress. The impact of individual variants on ABCA7 function varies, as some AD-associated mutations are not predicted to affect protein function while others likely produce an aberrant, truncated protein. However, the implications of the majority of these ABCA7 mutations on protein function are predicted or hypothesized. Additional studies investigating the downstream effects of these mutations relative to ABCA7 protein function in both model systems and human samples are necessary to reveal the specific mechanisms by which these mutations are contributing to AD pathogenesis to understand their role in risk for AD among diverse racial/ethnic groups and the design of personalized therapeutics.
ACKNOWLEDGMENTS
The authors acknowledge funding from the Vanderbilt Interdisciplinary Training Program in Alzheimer’s Disease (T32-AG058524), RCMI Pilot funds from the National Institute on Minority Health and Health Disparities (U54MD007586, U54MD00 7586-34), Vanderbilt University Start-Up Funds, and the Vanderbilt University Chancellor’s Scholarship.
Footnotes
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/21-5306r1).
REFERENCES
- [1].Pan Y, Liu R, Terpstra E, Wang Y, Qiao F, Wang J, Tong Y, Pan B (2016) Dysregulation and diagnostic potential of microRNA in Alzheimer’s disease. J Alzheimers Dis 49, 1–12. [DOI] [PubMed] [Google Scholar]
- [2].Alzheimer’s Association (2021) 2021 Alzheimer’s disease facts and figures. Alzheimers Dement 17, 327–406. [DOI] [PubMed] [Google Scholar]
- [3].Cukier HN, Kunkle BW, Vardarajan BN, Rolati S, Hamilton-Nelson KL, Kohli MA, Whitehead PL, Dombroski BA, VanBooven D, Lang R, Dykxhoorn DM, Farrer LA, Cuccaro ML, Vance JM, Gilbert JR, Beecham GW, Martin ER, Carney RM, Mayeux R, Schellenberg GD, Byrd GS, Haines JL, Pericak-Vance MA, Alzheimer’s Disease Genetics Consortium (2016) ABCA7 frameshift deletion associated with Alzheimer disease in African Americans. Neurol Genet 2, e79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Chin AL, Negash S, Hamilton R (2011) Diversity and disparity in dementia: The impact of ethnoracial differences in Alzheimer disease. Alzheimer Dis Assoc Disord 25, 187–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Manly JJ, Mayeux R (2004) Ethnic differences in dementia and Alzheimer’s disease. In Critical Perspectives on Racial and Ethnic Differences in Health in Late Life, Anderson NB, Bulatao RA, Cohen B, eds. National Academies Press, Washington, DC. [PubMed] [Google Scholar]
- [6].Lines LM, Sherif NA, Wiener JM (2014) Racial and Ethnic Disparities Among Individuals with Alzheimer’s Disease in the United States: A Literature Review, RTI Press, Research Triangle Park, NC. [Google Scholar]
- [7].Mehta KM, Yeo GW (2017) Systematic review of dementia prevalence and incidence in United States race/ethnic populations. Alzheimers Dement 13, 72–83. [DOI] [PubMed] [Google Scholar]
- [8].Meeker KL, Wisch JK, Hudson D, Coble D, Xiong C, Babulal GM, Gordon BA, Schindler SE, Cruchaga C, Flores S, Dincer A, Benzinger TL, Morris JC, Ances BM (2021) Socioeconomic status mediates racial differences seen using the AT(N) framework. Ann Neurol 89, 254–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Walsemann KM, Gee GC, Ro A (2013) Educational attainment in the context of social inequality: New directions for research on education and health. Am Behav Sci 57, 1082–1104. [Google Scholar]
- [10].Weuve J, Barnes LL, Mendes de Leon CF, Rajan KB, Beck T, Aggarwal NT, Hebert LE, Bennett DA, Wilson RS, Evans DA (2018) Cognitive aging in Black and White Americans: Cognition, cognitive decline, and incidence of Alzheimer disease dementia. Epidemiology 29, 151–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Carnethon MR, Pu J, Howard G, Albert MA, Anderson CAM, Bertoni AG, Mujahid MS, Palaniappan L, Taylor HA Jr., Willis M, Yancy CW (2017) Cardiovascular health in African Americans: A scientific statement from the American Heart Association. Circulation 136, e393–e423. [DOI] [PubMed] [Google Scholar]
- [12].Burke SL, Cadet T, Maddux M (2018) Chronic health illnesses as predictors of mild cognitive impairment among African American older adults. J Natl Med Assoc 110, 314–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Gottesman RF, Fornage M, Knopman DS, Mosley TH (2015) Brain aging in African-Americans: The Atherosclerosis Risk in Communities (ARIC) experience. Curr Alzheimer Res 12, 607–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Wilkins CH, Schindler SE, Morris JC (2020) Addressing health disparities among minority populations: Why clinical trial recruitment is not enough. JAMA Neurol 77, 1063–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Borrell LN, Elhawary JR, Fuentes-Afflick E, Witonsky J, Bhakta N, Wu AHB, Bibbins-Domingo K, Rodríguez-Santana JR, Lenoir MA, Gavin JR, Kittles RA, Zaitlen NA, Wilkes DS, Powe NR, Ziv E, Burchard EG (2021) Race and genetic ancestry in medicine — a time for reckoning with racism. N Engl J Med 384, 474–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Kittles RA, Weiss KM (2003) Race, ancestry, and genes: Implications for defining disease risk. Annu Rev Genomics Hum Genet 4, 33–67. [DOI] [PubMed] [Google Scholar]
- [17].Fujimura JH, Rajagopalan R (2011) Different differences: The use of ‘genetic ancestry’ versus race in biomedical human genetic research. Soc Stud Sci 41, 5–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kittles RA, Santos ER, Oji-Njideka NS, Bonilla C (2007) Race, skin color, and genetic ancestry: Implications for biomedical research on health disparities. Calif J Health Promot 5, 9–23. [Google Scholar]
- [19].Reitz C, Mayeux R (2014) Genetics of Alzheimer’s disease in Caribbean Hispanic and African American populations. Biol Psychiatry 75, 534–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Tang M-X, Stern Y, Marder K, Bell K, Gurland B, Lantigua R, Andrews H, Feng L, Tycko B, Mayeux R (1998) The APOE-ε4 allele and the risk of Alzheimer disease among African Americans, Whites, and Hispanics. JAMA 279, 751–755. [DOI] [PubMed] [Google Scholar]
- [21].Mayeux R (2003) Apolipoprotein E, Alzheimer disease, and African Americans. Arch Neurol 60, 161–163. [DOI] [PubMed] [Google Scholar]
- [22].Barnes LL, Bennett DA (2014) Alzheimer’s disease in African Americans: Risk factors and challenges for the future. Health Aff 33, 580–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Reitz C, Jun G, Naj A, Rajbhandary R, Vardarajan BN, Wang LS, Valladares O, Lin CF, Larson EB, Graff-Radford NR, Evans D, De Jager PL, Crane PK, Buxbaum JD, Murrell JR, Raj T, Ertekin-Taner N, Logue M, Baldwin CT, Green RC, Barnes LL, Cantwell LB, Fallin MD, Go RC, Griffith P, Obisesan TO, Manly JJ, Lunetta KL, Kamboh MI, Lopez OL, Bennett DA, Hendrie H, Hall KS, Goate AM, Byrd GS, Kukull WA, Foroud TM, Haines JL, Farrer LA, Pericak-Vance MA, Schellenberg GD, Mayeux R (2013) Variants in the ATP-binding cassette transporter (ABCA7), apolipoprotein E e4,and the risk of late-onset Alzheimer disease in African Americans. JAMA 309s, 1483–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Hohman TJ, Cooke-Bailey JN, Reitz C, Jun G, Naj A, Beecham GW, Liu Z, Carney RM, Vance JM, Cuccaro ML, Rajbhandary R, Vardarajan BN, Wang L-S, Valladares O, Lin C-F, Larson EB, Graff-Radford NR, Evans D, De Jager PL, Crane PK, Buxbaum JD, Murrell JR, Raj T, Ertekin-Taner N, Logue MW, Baldwin CT, Green RC, Barnes LL, Cantwell LB, Fallin MD, Go RCP, Griffith P, Obisesan TO, Manly JJ, Lunetta KL, Kamboh MI, Lopez OL, Bennett DA, Hardy J, Hendrie HC, Hall KS, Goate AM, Lang R, Byrd GS, Kukull WA, Foroud TM, Farrer LA, Martin ER, Pericak-Vance MA, Schellenberg GD, Mayeux R, Haines JL, Thornton-Wells TA, Alzheimer Disease Genetics C (2016) Global and local ancestry in African-Americans: Implications for Alzheimer’s disease risk. Alzheimers Dement 12, 233–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Aikawa T, Holm ML, Kanekiyo T (2018) ABCA7 and pathogenic pathways of Alzheimer’s disease. Brain Sci 8, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Ikeda Y, Abe-Dohmae S, Munehira Y, Aoki R, Kawamoto S, Furuya A, Shitara K, Amachi T, Kioka N, Matsuo M, Yokoyama S, Ueda K (2003) Posttranscriptional regulation of human ABCA7 and its function for the apoA-I-dependent lipid release. Biochem Biophys Res Commun 311, 313–318. [DOI] [PubMed] [Google Scholar]
- [27].Takahashi K, Kimura Y, Nagata K, Yamamoto A, Matsuo M, Ueda K (2005) ABC proteins: Key molecules for lipid homeostasis. Med Mol Morphol 38, 2–12. [DOI] [PubMed] [Google Scholar]
- [28].Abe-Dohmae S, Ikeda Y, Matsuo M, Hayashi M, Okuhira K, Ueda K, Yokoyama S (2004) Human ABCA7 supports apolipoprotein-mediated release of cellular cholesterol and phospholipid to generate high density lipoprotein. J Biol Chem 279, 604–611. [DOI] [PubMed] [Google Scholar]
- [29].Zhao QF, Yu JT, Tan MS, Tan L (2015) ABCA7 in Alzheimer’s disease. Mol Neurobiol 51, 1008–1016. [DOI] [PubMed] [Google Scholar]
- [30].Chan SL, Kim WS, Kwok JB, Hill AF, Cappai R, Rye KA, Garner B (2008) ATP-binding cassette transporter A7 regulates processing of amyloid precursor protein in vitro. J Neurochem 106, 793–804. [DOI] [PubMed] [Google Scholar]
- [31].De Roeck A, Van Broeckhoven C, Sleegers K (2019) The role of ABCA7 in Alzheimer’s disease: Evidence from genomics, transcriptomics and methylomics. Acta Neuropathol 138, 201–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Dib S, Pahnke J, Gosselet F (2021) Role of ABCA7 in human health and in Alzheimer’s disease. Int J Mol Sci 22, 4603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Berg CN, Sinha N, Gluck MA (2019) The effects of APOE and ABCA7 on cognitive function and Alzheimer’s disease risk in African Americans: A focused mini review. Front Hum Neurosci 13, 387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Steinberg S, Stefansson H, Jonsson T, Johannsdottir H, Ingason A, Helgason H, Sulem P, Magnusson OT, Gudjonsson SA, Unnsteinsdottir U, Kong A, Helisalmi S, Soininen H, Lah JJ, Aarsland D, Fladby T, Ulstein ID, Djurovic S, Sando SB, White LR, Knudsen GP, Westlye LT, Selbaek G, Giegling I, Hampel H, Hiltunen M, Levey AI, Andreassen OA, Rujescu D, Jonsson PV, Bjornsson S, Snaedal J, Stefansson K (2015) Loss-of-function variants in ABCA7 confer risk of Alzheimer’s disease. Nat Genet 47, 445–447. [DOI] [PubMed] [Google Scholar]
- [35].Kjeldsen EW, Tybjaerg-Hansen A, Nordestgaard BG, Frikke-Schmidt R (2018) ABCA7 and risk of dementia and vascular disease in the Danish population. Ann Clin Transl Neurol 5, 41–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Cuyvers E, De Roeck A, Van den Bossche T, Van Cauwenberghe C, Bettens K, Vermeulen S, Mattheijssens M, Peeters K, Engelborghs S, Vandenbulcke M, Vandenberghe R, De Deyn PP, Van Broeckhoven C, Sleegers K (2015) Mutations in ABCA7 in a Belgian cohort of Alzheimer’s disease patients: A targeted resequencing study. Lancet Neurol 14, 814–822. [DOI] [PubMed] [Google Scholar]
- [37].Moreno DJ, Ruiz S, Rios A, Lopera F, Ostos H, Via M, Bedoya G (2017) Association of GWAS top genes with late-onset Alzheimer’s disease in Colombian population. Am J Alzheimers Dis Other Demen 32, 27–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Almeida JFF, Dos Santos LR, Trancozo M, de Paula F (2018) Updated meta-analysis of BIN1, CR1, MS4A6A, CLU, and ABCA7 variants in Alzheimer’s disease. J Mol Neurosci 64, 471–477. [DOI] [PubMed] [Google Scholar]
- [39].Logue MW, Schu M, Vardarajan BN, Buros J, Green RC, Go RC, Griffith P, Obisesan TO, Shatz R, Borenstein A, Cupples LA, Lunetta KL, Fallin MD, Baldwin CT, Farrer LA (2011) A comprehensive genetic association study of Alzheimer disease in African Americans. Arch Neurol 68, 1569–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].N’Songo A, Carrasquillo MM, Wang X, Burgess JD, Nguyen T, Asmann YW, Serie DJ, Younkin SG, Allen M, Pedraza O, Duara R, Greig Custo MT, Graff-Radford NR, Ertekin-Taner N (2017) African American exome sequencing identifies potential risk variants at Alzheimer disease loci. Neurol Genet 3, e141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Sinha N, Reagh ZM, Tustison NJ, Berg CN, Shaw A, Myers CE, Hill D, Yassa MA, Gluck MA (2019) ABCA7 risk variant in healthy older African Americans is associated with a functionally isolated entorhinal cortex mediating deficient generalization of prior discrimination training. Hippocampus 29, 527–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Sassi C, Nalls MA, Ridge PG, Gibbs JR, Ding J, Lupton MK, Troakes C, Lunnon K, Al-Sarraj S, Brown KS, Medway C, Clement N, Lord J, Turton J, Bras J, Almeida MR, Holstege H, Louwersheimer E, van der Flier WM, Scheltens P, Van Swieten JC, Santana I, Oliveira C, Morgan K, Powell JF, Kauwe JS, Cruchaga C, Goate AM, Singleton AB, Guerreiro R, Hardy J (2016) ABCA7 p.G215S as potential protective factor for Alzheimer’s disease. Neurobiol Aging 46, 235.e1–235.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Logue MW, Lancour D, Farrell J, Simkina I, Fallin MD, Lunetta KL, Farrer LA (2018) Targeted sequencing of Alzheimer disease genes in African Americans implicates novel risk variants. Front Neurosci 12, 592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Hollingworth P, Harold D, Sims R, Gerrish A, Lambert JC, Carrasquillo MM, Abraham R, Hamshere ML, Pahwa JS, Moskvina V, et al. (2011) Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33andCD2AP are associated with Alzheimer’s disease. Nat Genet 43, 429–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Naj AC, Jun G, Beecham GW, Wang LS, Vardarajan BN, Buros J, Gallins PJ, Buxbaum JD, Jarvik GP, Crane PK, et al. (2011) Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet 43, 436–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, Jun G, DeStefano AL, Bis JC, Beecham GW, et al. (2013) Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet 45, 1452–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Pereira CD, Martins F, Wiltfang J, da Cruz ESOAB, Rebelo S (2018) ABC transporters are key players in Alzheimer’s disease. J Alzheimers Dis 61, 463–485. [DOI] [PubMed] [Google Scholar]
- [48].Qian H, Zhao X, Cao P, Lei J, Yan N, Gong X (2017) Structure of the human lipid exporter ABCA1. Cell 169, 1228–1239. [DOI] [PubMed] [Google Scholar]
- [49].Shintre CA, Pike ACW, Li Q, Kim J-I, Barr AJ, Goubin S, Shrestha L, Yang J, Berridge G, Ross J, Stansfeld PJ, Sansom MSP, Edwards AM, Bountra C, Marsden BD, von Delft F, Bullock AN, Gileadi O, Burgess-Brown NA, Carpenter EP (2013) Structures of ABCB10, a human ATP-binding cassette transporter in apo- and nucleotide-bound states. Proc Natl Acad Sci U S A 110, 9710–9715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Jackson SM, Manolaridis I, Kowal J, Zechner M, Taylor NMI, Bause M, Bauer S, Bartholomaeus R, Bernhardt G, Koenig B, Buschauer A, Stahlberg H, Altmann K-H, Locher KP (2018) Structural basis of small-molecule inhibition of human multidrug transporter ABCG2. Nat Struct Mol Biol 25, 333–340. [DOI] [PubMed] [Google Scholar]
- [51].Tarling EJ, de Aguiar Vallim TQ, Edwards PA (2013) Role of ABC transporters in lipid transport and human disease. Trends Endocrinol Metab 24, 342–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Dean M, Hamon Y, Chimini G (2001) The human ATP-binding cassette (ABC) transporter superfamily. J Lipid Res 42, 1007–1017. [PubMed] [Google Scholar]
- [53].Bungert S, Molday LL, Molday RS (2001) Membrane topology of the ATP binding cassette transporter ABCR and its relationship to ABC1 and related ABCA transporters: Identification of N-linked glycosylation sites. J Biol Chem 276, 23539–23546. [DOI] [PubMed] [Google Scholar]
- [54].Zarubica A, Trompier D, Chimini G (2007) ABCA1, from pathology to membrane function. Pflügers Arch 453, 569–579. [DOI] [PubMed] [Google Scholar]
- [55].Broccardo C, Luciani M-F, Chimini G (1999) The ABCA subclass of mammalian transporters. Biochim Biophys Acta Biomembr 1461, 395–404. [DOI] [PubMed] [Google Scholar]
- [56].Hozoji M, Kimura Y, Kioka N, Ueda K (2009) Formation of two intramolecular disulfide bonds is necessary for ApoA-I-dependent cholesterol efflux mediated by ABCA1. J Biol Chem 284, 11293–11300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Ishigami M, Ogasawara F, Nagao K, Hashimoto H, Kimura Y, Kioka N, Ueda K (2018) Temporary sequestration of cholesterol and phosphatidylcholine within extracellular domains of ABCA1 during nascent HDL generation. Sci Rep 8, 6170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Vedhachalam C, Ghering AB, Davidson WS, Lund-Katz S, Rothblat GH, Phillips MC (2007) ABCA1-induced cell surface binding sites for apoA-I. Arterioscler Thromb Vasc Biol 27, 1603–1609. [DOI] [PubMed] [Google Scholar]
- [59].Kawanobe T, Shiranaga N, Kioka N, Kimura Y, Ueda K (2019) Apolipoprotein A-I directly interacts with extracellular domain 1 of human ABCA1. Biosci Biotechnol Biochem 83, 490–497. [DOI] [PubMed] [Google Scholar]
- [60].Nagao K, Takahashi K, Azuma Y, Takada M, Kimura Y, Matsuo M, Kioka N, Ueda K (2012) ATP hydrolysis-dependent conformational changes in the extracellular domain of ABCA1 are associated with apoA-I binding. J Lipid Res 53, 126–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Zhao G-J, Yin K, Fu Y-C, Tang C-K (2012) The interaction of ApoA-I and ABCA1 triggers signal transduction pathways to mediate efflux of cellular lipids. Mol Med 18, 149–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Tanaka AR, Abe-Dohmae S, Ohnishi T, Aoki R, Morinaga G, Okuhira K-i, Ikeda Y, Kano F, Matsuo M, Kioka N, Amachi T, Murata M, Yokoyama S, Ueda K (2003) Effects of mutations of ABCA1 in the first extracellular domain on subcellular trafficking and ATP binding/hydrolysis. J Biol Chem 278, 8815–8819. [DOI] [PubMed] [Google Scholar]
- [63].Haidar B, Denis M, Marcil M, Krimbou L, Genest J, Jr (2004) Apolipoprotein A-I activates cellular cAMP signaling through the ABCA1 transporter. J Biol Chem 279, 9963–9969. [DOI] [PubMed] [Google Scholar]
- [64].Singaraja RR, Visscher H, James ER, Chroni A, Coutinho JM, Brunham LR, Kang MH, Zannis VI, Chimini G, Hayden MR (2006) Specific mutations in ABCA1 have discrete effects on ABCA1 function and lipid phenotypes both in vivo and in vitro. Circ Res 99, 389–397. [DOI] [PubMed] [Google Scholar]
- [65].Stepler KE (2021) Proteomics to study racial disparities in Alzheimer’s disease. Vanderbilt University, Ph.D. dissertation. https://ir.vanderbilt.edu/handle/1803/16851. [Google Scholar]
- [66].Piehler AP, Ozcurumez M, Kaminski WE (2012) A-subclass ATP-binding cassette proteins in brain lipid homeostasis and neurodegeneration. Front Psychiatry 3, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Aikawa T, Ren Y, Yamazaki Y, Tachibana M, Johnson MR, Anderson CT, Martens YA, Holm M-L, Asmann YW, Saito T, Saido TC, Fitzgerald ML, Bu G, Kanekiyo T (2019) ABCA7 haplodeficiency disturbs microglial immune responses in the mouse brain. Proc Natl Acad Sci U S A 116, 23790–23796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Quazi F, Molday RS (2013) Differential phospholipid substrates and directional transport by ATP-binding cassette proteins ABCA1, ABCA7, and ABCA4 and disease-causing mutants. J Biol Chem 288, 34414–34426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Tomioka M, Toda Y, Manucat NB, Akatsu H, Fukumoto M, Kono N, Arai H, Kioka N, Ueda K (2017) Lysophos-phatidylcholine export by human ABCA7. Biochim Biophys Acta Mol Cell Biol Lipids 1862, 658–665. [DOI] [PubMed] [Google Scholar]
- [70].Rosenthal SL, Kamboh MI (2014) Late-onset Alzheimer’s disease genes and the potentially implicated pathways. Curr Genet Med Rep 2, 85–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Sakae N, Liu CC, Shinohara M, Frisch-Daiello J, Ma L, Yamazaki Y, Tachibana M, Younkin L, Kurti A, Carrasquillo MM, Zou F, Sevlever D, Bisceglio G, Gan M, Fol R, Knight P, Wang M, Han X, Fryer JD, Fitzgerald ML, Ohyagi Y, Younkin SG, Bu G, Kanekiyo T (2016) ABCA7 deficiency accelerates amyloid-beta generation and Alzheimer’s neuronal pathology. J Neurosci 36, 3848–3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Kim WS, Fitzgerald ML, Kang K, Okuhira K-i, Bell SA, Manning JJ, Koehn SL, Lu N, Moore KJ, Freeman MW (2005) Abca7 null mice retain normal macrophage phosphatidylcholine and cholesterol efflux activity despite alterations in adipose mass and serum cholesterol levels. J Biol Chem 280, 3989–3995. [DOI] [PubMed] [Google Scholar]
- [73].Nowyhed HN, Chandra S, Kiosses W, Marcovecchio P, Andary F, Zhao M, Fitzgerald ML, Kronenberg M, Hedrick CC (2017) ATP binding cassette transporter ABCA7 regulates NKT cell development and function by controlling CD1d expression and lipid raft content. Sci Rep 7, 40273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].O’Connor T, Sadleir KR, Maus E, Velliquette RA, Zhao J, Cole SL, Eimer WA, Hitt B, Bembinster LA, Lammich S, Lichtenthaler SF, Hébert SS, De Strooper B, Haass C, Bennett DA, Vassar R (2008) Phosphorylation of the translation initiation factor eIF2alpha increases BACE1 levels and promotes amyloidogenesis. Neuron 60, 988–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Sun J, Nan G (2017) The extracellular signal-regulated kinase 1/2 pathway in neurological diseases: A potential therapeutic target (Review). Int J Mol Med 39, 1338–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Bamji-Mirza M, Li Y, Najem D, Liu QY, Walker D, Lue LF, Stupak J, Chan K, Li J, Ghani M, Yang Z, Rogaeva E, Zhang W (2016) Genetic variations in ABCA7 can increase secreted levels of amyloid-beta40 and amyloid-beta42 peptides and ABCA7 transcription in cell culture models. J Alzheimers Dis 53, 875–892. [DOI] [PubMed] [Google Scholar]
- [77].Hughes TM, Lopez OL, Evans RW, Kamboh MI, Williamson JD, Klunk WE, Mathis CA, Price JC, Cohen AD, Snitz BE, Dekosky ST, Kuller LH (2014) Markers of cholesterol transport are associated with amyloid deposition in the brain. Neurobiol Aging 35, 802–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Apostolova LG, Risacher SL, Duran T, Stage EC, Goukasian N, West JD, Do TM, Grotts J, Wilhalme H, Nho K, Phillips M, Elashoff D, Saykin AJ (2018) Associations of the top 20 Alzheimer disease risk variants with brain amyloidosis. JAMA Neurol 75, 328–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Tanaka N, Abe-Dohmae S, Iwamoto N, Yokoyama S (2011) Roles of ATP-binding cassette transporter A7 in cholesterol homeostasis and host defense system. J Atheroscler Thromb 18, 274–281. [DOI] [PubMed] [Google Scholar]
- [80].Tanaka N, Abe-Dohmae S, Iwamoto N, Fitzgerald ML, Yokoyama S (2010) Helical apolipoproteins of high-density lipoprotein enhance phagocytosis by stabilizing ATP-binding cassette transporter A7. J Lipid Res 51, 2591–2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Vasquez JB, Fardo DW, Estus S (2013) ABCA7 expression is associated with Alzheimer’s disease polymorphism and disease status. Neurosci Lett 556, 58–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Villegas-Llerena C, Phillips A, Garcia-Reitboeck P, Hardy J, Pocock JM (2016) Microglial genes regulating neuroinflammation in the progression of Alzheimer’s disease. Curr Opin Neurobiol 36, 74–81. [DOI] [PubMed] [Google Scholar]
- [83].Kim WS, Li H, Ruberu K, Chan S, Elliott DA, Low JK, Cheng D, Karl T, Garner B (2013) Deletion of Abca7 increases cerebral amyloid-beta accumulation in the J20 mouse model of Alzheimer’s disease. J Neurosci 33, 4387–4394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Jehle AW, Gardai SJ, Li S, Linsel-Nitschke P, Morimoto K, Janssen WJ, Vandivier RW, Wang N, Greenberg S, Dale BM, Qin C, Henson PM, Tall AR (2006) ATP-binding cassette transporter A7 enhances phagocytosis of apoptotic cells and associated ERK signaling in macrophages. J Cell Biol 174, 547–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Wang J, Zhao D, Pan B, Fu Y, Shi F, Kouadir M, Yang L, Yin X, Zhou X (2015) Toll-like receptor 2 deficiency shifts PrP106–126-induced microglial activation from a neurotoxic to a neuroprotective phenotype. J Mol Neurosci 55, 880–890. [DOI] [PubMed] [Google Scholar]
- [86].Liu Y, Walter S, Stagi M, Cherny D, Letiembre M, Schulz-Schaeffer W, Heine H, Penke B, Neumann H, Fassbender K (2005) LPS receptor (CD14): A receptor for phagocytosis of Alzheimer’s amyloidpeptide. Brain 128, 1778–1789. [DOI] [PubMed] [Google Scholar]
- [87].Reed-Geaghan EG, Savage JC, Hise AG, Landreth GE (2009) CD14 and toll-like receptors 2 and 4 are required for fibrillar Aβ-stimulated microglial activation. J Neurosci 29, 11982–11992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Zhang M, Schmitt-Ulms G, Sato C, Xi Z, Zhang Y, Zhou Y, St George-Hyslop P, Rogaeva E (2016) Drug repositioning for Alzheimer’s disease based on systematic ‘omics’ data mining. PLoS One 11, e0168812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Cukier HN, Laverde-Paz J, Ramirez J, Adams LD, Starks TD, Vance JM, Cuccaro ML, Blurton-Jones M, Haines JL, Byrd GS, Pericak-Vance MA, Dykxhoorn DM (2020) iPSC-derived neurons and microglia with an African-specific ABCA7 frameshift deletion have impaired function. Alzheimers Dement 16, e046109. [Google Scholar]
- [90].Shulman JM, Chen K, Keenan BT, Chibnik LB, Fleisher A, Thiyyagura P, Roontiva A, McCabe C, Patsopoulos NA, Corneveaux JJ, Yu L, Huentelman MJ, Evans DA, Schneider JA, Reiman EM, De Jager PL, Bennett DA (2013) Genetic susceptibility for Alzheimer disease neuritic plaque pathology. JAMA Neurol 70, 1150–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Ramirez LM, Goukasian N, Porat S, Hwang KS, Eastman JA, Hurtz S, Wang B, Vang N, Sears R, Klein E, Coppola G, Apostolova LG (2016) Common variants in ABCA7 and MS4A6A are associated with cortical and hippocampal atrophy. Neurobiol Aging 39, 82–89. [DOI] [PubMed] [Google Scholar]
- [92].Carrasquillo MM, Crook JE, Pedraza O, Thomas CS, Pankratz VS, Allen M, Nguyen T, Malphrus KG, Ma L, Bisceglio GD, Roberts RO, Lucas JA, Smith GE, Ivnik RJ, Machulda MM, Graff-Radford NR, Petersen RC, Younkin SG, Ertekin-Taner N (2015) Late-onset Alzheimer’s risk variants in memory decline, incident mild cognitive impairment, and Alzheimer’s disease. Neurobiol Aging 36, 60–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Andrews SJ, Das D, Anstey KJ, Easteal S (2017) Late onset Alzheimer’s disease risk variants in cognitive decline: The PATH through life study. J Alzheimers Dis 57, 423–436. [DOI] [PubMed] [Google Scholar]
- [94].Vivot A, Glymour MM, Tzourio C, Amouyel P, Chene G, Dufouil C (2015) Association of Alzheimer’s related genotypes with cognitive decline in multiple domains: Results from the Three-City Dijon study. Mol Psychiatry 20, 1173–1178. [DOI] [PubMed] [Google Scholar]
- [95].Andrews SJ, Das D, Cherbuin N, Anstey KJ, Easteal S (2016) Association of genetic risk factors with cognitive decline: The PATH through life project. Neurobiol Aging 41, 150–158. [DOI] [PubMed] [Google Scholar]
- [96].Nettiksimmons J, Tranah G, Evans DS, Yokoyama JS, Yaffe K (2016) Gene-based aggregate SNP associations between candidate AD genes and cognitive decline. Age (Dordr) 38, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Chang Y-T, Hsu S-W, Huang S-H, Huang C-W, Chang W-N, Lien C-Y, Lee J-J, Lee C-C, Chang C-C (2019) ABCA7 polymorphisms correlate with memory impairment and default mode network in patients with APOEε4-associated Alzheimer’s disease. Alzheimers Res Ther 11, 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Engelman CD, Koscik RL, Jonaitis EM, Okonkwo OC, Hermann BP, La Rue A, Sager MA (2013) Interaction between two cholesterol metabolism genes influences memory: Findings from the Wisconsin Registry for Alzheimer’s Prevention. J Alzheimers Dis 36, 749–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Zhang XY, Wang YF, Zheng LJ, Zhang H, Lin L, Lu GM, Zhang LJ (2020) Impacts of AD-related ABCA7 and CLU variants on default mode network connectivity in healthy middle-age adults. Front Mol Neurosci 13, 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Berg CN, Sinha N, Gluck MA (2019) ABCA7 risk genotype diminishes the neuroprotective value of aerobic fitness in healthy older African Americans. Front Aging Neurosci 11, 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Sinha N, Berg CN, Shaw A, Gluck MA (2020) ABCA7 genotype moderates the effect of aerobic exercise intervention on generalization of prior learning in healthy older African Americans. J Alzheimers Dis 74, 309–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Stage E, Duran T, Risacher SL, Goukasian N, Do TM, West JD, Wilhalme H, Nho K, Phillips M, Elashoff D, Saykin AJ, Apostolova LG (2016) The effect of the top 20 Alzheimer disease risk genes on gray-matter density and FDG PET brain metabolism. Alzheimers Dement 5, 53–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Chung SJ, Kim MJ, Kim YJ, Kim J, You S, Jang EH, Kim SY, Lee JH (2014) CR1, ABCA7, and APOE genes affect the features of cognitive impairment in Alzheimer’s disease. J Neurol Sci 339, 91–96. [DOI] [PubMed] [Google Scholar]
- [104].Olsen RK, Yeung L-K, Noly-Gandon A, D’Angelo MC, Kacollja A, Smith VM, Ryan JD, Barense MD (2017) Human anterolateral entorhinal cortex volumes are associated with cognitive decline in aging prior to clinical diagnosis. Neurobiol Aging 57, 195–205. [DOI] [PubMed] [Google Scholar]
- [105].Pooler AM, Polydoro M, Maury EA, Nicholls SB, Reddy SM, Wegmann S, William C, Saqran L, Cagsal-Getkin O, Pitstick R, Beier DR, Carlson GA, Spires-Jones TL, Hyman BT (2015) Amyloid accelerates tau propagation and toxicity in a model of early Alzheimer’s disease. Acta Neuropathol Commun 3, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Denny JC, Rutter JL, Goldstein DB, Philippakis A, Smoller JW, Jenkins G, Dishman E (2019) The “All of Us” research program. N Engl J Med 381, 668–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Weinberger DR, Dzirasa K, Crumpton-Young LL (2020) Missing in action: African ancestry brain research. Neuron 107, 407–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Mapes BM, Foster CS, Kusnoor SV, Epelbaum MI, AuYoung M, Jenkins G, Lopez-Class M, Richardson-Heron D, Elmi A, Surkan K, Cronin RM, Wilkins CH, Pérez-Stable EJ, Dishman E, Denny JC, Rutter JL (2020) Diversity and inclusion for the All of Us research program: A scoping review. PLoS One 15, e0234962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Webb B, Sali A (2016) Comparative protein structure modeling using MODELLER. Curr Protoc Bioinformatics 54, 5.6.1–5.6.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Benkert P, Künzli M, Schwede T (2009) QMEAN server for protein model quality estimation. Nucleic Acids Res 37, W510–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Benkert P, Schwede T, Tosatto SCE (2009) QMEAN-clust: Estimation of protein model quality by combining a composite scoring function with structural density information. BMC Struct Biol 9, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Clamp M, Cuff J, Searle SM, Barton GJ (2004) The Jalview Java alignment editor. Bioinformatics 20, 426–427. [DOI] [PubMed] [Google Scholar]
- [113].Fehér Á, Juhász A, Pákáski M, Janka Z, Kálmán J (2019) Association study of the ABCA7 rs3752246 polymorphism in Alzheimer’s disease. Psychiatry Res 279, 376–377. [DOI] [PubMed] [Google Scholar]
- [114].Li H, Zhou J, Yue Z, Feng L, Luo Z, Chen S, Yang X, Xiao B (2017) A complex association between ABCA7 genotypes and blood lipid levels in Southern Chinese Han patients of sporadic Alzheimer’s disease. J Neurol Sci 382, 13–17. [DOI] [PubMed] [Google Scholar]
- [115].Zhou G, Mao X, Chu J, Chen G, Zhao Q, Wang L, Luo Y (2017) ATP binding cassette subfamily A member 7 rs3764650 polymorphism and the risk of Alzheimer’s disease. Pharmazie 72, 425–427. [DOI] [PubMed] [Google Scholar]
- [116].Monsell SE, Mock C, Fardo DW, Bertelsen S, Cairns NJ, Roe CM, Ellingson SR, Morris JC, Goate AM, Kukull WA (2017) Genetic comparison of symptomatic and asymptomatic persons with Alzheimer disease neuropathology. Alzheimer Dis Assoc Disord 31, 232–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Kunkle BW, Carney RM, Kohli MA, Naj AC, Hamilton-Nelson KL, Whitehead PL, Wang L, Lang R, Cuccaro ML, Vance JM, Byrd GS, Beecham GW, Gilbert JR, Martin ER, Haines JL, Pericak-Vance MA (2017) Targeted sequencing of ABCA7 identifies splicing, stop-gain and intronic risk variants for Alzheimer disease. Neurosci Lett 649, 124–129. [DOI] [PubMed] [Google Scholar]
- [118].Stepler KE, Robinson RAS (2019) The potential of ‘omics to link lipid metabolism and genetic and comorbidity risk factors of Alzheimer’s disease in African Americans. In Reviews on Biomarker Studies in Psychiatric and Neurodegenerative Disorders, Guest PC, ed. Springer Nature, Cham. [DOI] [PMC free article] [PubMed] [Google Scholar]