

Abstract
BACKGROUND
Models for forecasting individual clinical progression trajectories in early Alzheimer's disease (AD) are needed for optimizing clinical studies and patient monitoring.
METHODS
Prediction models were constructed using a clinical trial training cohort (TC; n = 934) via a gradient boosting algorithm and then evaluated in two validation cohorts (VC 1, n = 235; VC 2, n = 421). Model inputs included baseline clinical features (cognitive function assessments, APOE ε4 status, and demographics) and brain magnetic resonance imaging (MRI) measures.
RESULTS
The model using clinical features achieved R 2 of 0.21 and 0.31 for predicting 2‐year cognitive decline in VC 1 and VC 2, respectively. Adding MRI features improved the R 2 to 0.29 in VC 1, which employed the same preprocessing pipeline as the TC. Utilizing these model‐based predictions for clinical trial enrichment reduced the required sample size by 20% to 49%.
DISCUSSION
Our validated prediction models enable baseline prediction of clinical progression trajectories in early AD, benefiting clinical trial enrichment and various applications.
Keywords: clinical trial enrichment, disease progression, machine learning, mild cognitive impairment, prognosis
1. BACKGROUND
The heterogeneity of individual disease progression trajectories between patients poses a major challenge for designing efficient clinical trials to detect meaningful benefits of candidate treatments 1 , 2 , 3 , 4 and for physicians, caregivers, and patients to make appropriate decisions and plans on treatment and long‐term care. 5 , 6 , 7 , 8 This is especially true of Alzheimer's disease (AD) where patients initially diagnosed with mild cognitive impairment (MCI) and confirmed to be brain amyloid beta (Aβ) positive (Aβ+) experience different rates of clinical progression, depending on their baseline clinical and biological characteristics. 4 , 9 , 10 , 11 , 12 , 13 Several unique demographics, clinical, genetic, proteomic, imaging, and disease pathological patient characteristics may help explain some of this heterogeneity and more accurately determine their disease prognosis 14 , 15 , 16 , 17 .
Clinical trials on Aβ+ patients with early AD, that is, patients with either MCI due to AD or mild AD dementia, tend to employ cutoffs and selection criteria on cognitive function, apolipoprotein E (APOE) ε4 genetic status (defined in Table 1), and disease pathological factors such as the amyloid burden and tau assessed via positron emission tomography (PET) scan, cerebrospinal fluid (CSF), or blood. 18 , 19 , 20 , 21 , 22 , 23 , 24 , 25 Despite the consideration of such factors for screening patients in clinical trials, there is usually considerable variability in the rates of clinical progression of randomized patients. 26 Including patients with a low likelihood of progression or the failure to account for this heterogeneity may reduce the power to detect clinically meaningful treatment effects and thus require a larger sample size in clinical trials. 17 , 26 , 27
TABLE 1.
Demographic and clinical data summary of training and validation cohorts.
| Training cohort | Validation cohort 1 | Validation cohort 2 | ||
|---|---|---|---|---|
| Patient characteristics | N = 934 | N = 235 | N = 421 | |
| Diagnosis | MCI; N (%) | 761 (81%) | 153 (65%) | 286 (68%) |
| Mild AD; N (%) | 173 (19%) | 82 (35%) | 135 (32%) | |
| Gender | Female; N (%) | 431 (46%) | 136 (58%) | 189 (45%) |
| Male; N (%) | 503 (54%) | 99 (42%) | 232 (55%) | |
| Race | White; N (%) | 706 (76%) | 213 (91%) | 394 (94%) |
| Non‐White; N (%) | 228 (24%) | 22 (9%) | 27 (6%) | |
| ApoE4 status | E4 homozygous; N (%) | 140 (15%) | 39 (17%) | 83 (20%) |
| E4 heterozygous; N (%) | 458 (49%) | 127 (54%) | 210 (50%) | |
| Non‐E4; N (%) | 336 (36%) | 69 (29%) | 128 (30%) | |
| Education | Mean (SD) | 13.4 (3.6) | 14.0 (3.7) | 16.0 (2.7) |
| Age | Mean (SD) | 72.1 (7.3) | 71.2 (8.8) | 73.4 (7.0) |
| BMI | Mean (SD) | 25.9 (4.3) | 25.7 (3.8) | 26.6 (5.0) |
| MMSE | Mean (SD) | 25.8 (2.5) | 26.0 (2.3) | 26.1 (3.0) |
| CDR‐SB | Mean (SD) | 2.6 (1.2) | 2.9 (1.5) | 2.5 (1.8) |
Note: All the demographic and clinical characteristics are significantly different (p < .05) between the cohorts. The training cohort has a significantly greater proportion of MCI and ApoE4‐positive subjects. The first validation cohort (VC 1) has a greater proportion of males. Subjects in the second validation cohort (VC 2) are older and have higher body mass index (BMI). These differences among early AD patients across different clinical trials and observational cohorts help to provide a more generalizable assessment of the performance of the prediction models between the training and validation cohorts.
Abbreviations: BMI, body mass index; CDR‐SB, sum of boxes of clinical dementia rating scale; MCI, mild cognitive impairment; MMSE, Mini‐Mental State Examination; SD, standard deviation.
RESEARCH IN CONTEXT
Systematic review: Heterogeneity of clinical progression is a common phenomenon in early Alzheimer's disease (AD). Given recent advances in novel therapeutics and the growing AD population, models that can predict individual progression trajectories using common baseline clinical assessments are needed for drug development and real‐world applications.
Interpretation: Prediction models trained via machine learning in a clinical trial cohort using common baseline clinical assessments accurately predicted the 24‐month clinical progression trajectories in two validation cohorts. Key baseline predictors include cognitive status, particularly in memory, praxis, language, and the volume of medial temporal and associated cortical regions. Applying these model‐based predictions for prognostic clinical trial enrichment reduced the required sample size by 20% to 49%.
Future directions: The added value of various fluid‐based biomarkers to these prediction models should be explored. For real‐world applications, these models should be adapted to include assessments that are more feasible in clinical practice.
Some of the variability in the rates of clinical progression between patients may be explained by the differences in their baseline cognitive function in specific domains such as word recall, ideational praxis, orientation, and executive function, in addition to differences in disease pathological characteristics such as brain magnetic resonance imaging (MRI) measures or other biomarkers. 28 , 29 , 30 , 31 , 32 If such baseline patient characteristics can be used collectively to build prognostic prediction models to predict the future clinical decline of each patient, such predictions during initial patient screening can be used to design optimal clinical trials and as a guide for treatment, monitoring, and patient care decisions in clinical practice and real‐world situations. 17 , 27 , 33 Current AD clinical trials tend to be 18 to 24 months in duration. Prognostic prediction of clinical progression over such a short time frame in Aβ+ early AD patients tends to be less accurate than predicting progression over, say, 3 to 5 years (eg, tab. 2 in Franzmeier et al.). 17
Thus, in this report, we present the development of such prognostic prediction models via a machine learning algorithm for predicting the future longitudinal clinical decline of Aβ+ early AD patients over a typical 18‐ to 24‐month duration of a clinical trial using historical placebo subject data from two clinical trials. We then demonstrate and validate the prediction performance of these models in an independent cohort of placebo subjects from another clinical trial and in another cohort of subjects from the Alzheimer's Disease Neuroimaging Initiative (ADNI) database. Finally, we study the utility of using the prognostic predictions of clinical progression for increasing the efficiency of clinical trials via clinical trial simulations.
2. METHODS
2.1. Database
The training cohort (TC) for constructing the prediction models comprised 934 Aβ+ early AD subjects from the placebo arm of two identical clinical studies that were part of the same Elenbecestat phase‐3 program (A Placebo‐Controlled, Double‐Blind, Parallel‐Group, 24 Month Study with an Open‐Label Extension Phase to Evaluate the Efficacy and Safety of Elenbecestat [E2609]) in Subjects with Early Alzheimer's Disease; NCT02956486, MissionAD1 and NCT03036280, MissionAD2). Over 81% of the placebo subjects in TC had MCI due to AD, and the rest had mild AD. The trial was approved by the Institutional Review Board or independent ethics committee at each center, and all the participants provided written informed consent.
The first validation cohort (VC 1) for testing the performance of the prediction models included 235 Aβ+ early AD subjects from the placebo arm of an 18‐month clinical study of another program (A Study to Evaluate Safety, Tolerability, and Efficacy of Lecanemab in Subjects with Early Alzheimer's Disease; NCT01767311). The trial was approved by the Institutional Review Board or independent ethics committee at each center, and all the participants provided written informed consent.
The second validation cohort (VC 2) for further assessment of the prediction models included 421 Aβ+ subjects diagnosed as either MCI or AD with at least 1 year of clinical follow‐up and the relevant clinical and MRI assessments from the ADNI 2 and ADNI 3 phases of the ADNI database (adni.loni.usc.edu). The ADNI was launched in 2003 as a public–private partnership, led by Principal Investigator Michael W. Weiner, MD. The primary goal of ADNI has been to test whether serial MRI, PET, other biological markers, and clinical and neuropsychological assessment can be combined to measure the progression of MCI and AD. For up‐to‐date information, see www.adni‐info.org. The study was approved by the Institutional Review Boards of all of the participating institutions and informed written consent was obtained from all participants. Data used for the analyses presented here were accessed on December 15, 2021.
Data on the longitudinal clinical decline in the TC for constructing the prediction models were based on the change from baseline in the sum of boxes of the clinical dementia rating scale (CDR‐SB) at months 3, 6, 9, 12, 15, 18, 21, and 24. The clinical follow‐up times considered for evaluating the prediction models in VC 1 and VC 2 were months 3, 6, 9, 12, 15, and 18 and months 6, 12, and 24, respectively. A summary of some key demographics and clinical characteristics of the subjects in these three cohorts is included in Table 1, and a summary of the longitudinal change in CDR‐SB at each time point is provided in Table S1.
2.2. Cognitive function assessments
While the clinical decline was defined in terms of the change from baseline in CDR‐SB, other baseline cognitive function assessments at baseline were also considered as potential predictors for constructing the prediction models. These include the composite endpoints, Mini‐Mental State Examination (MMSE), Alzheimer's Disease Assessment Scale–Cognitive Subscale (ADAS‐Cog‐13), CDR‐SB, and all their subscores.
2.3. Imaging data
All subjects in TC and VC 1 received a 3.0 Tesla (T) structural MRI at baseline. Approximately 75% of subjects in VC 2 received 1.5T, and the rest received 3T MRI. Brain MRI data (volume, area, and cortical thickness) in all three cohorts were generated for various brain regions of interest using the Desikan‐Killiany atlas, 34 resulting in 207 regional measures. Image processing for TC and VC 1 was carried out by Clario using their proprietary imaging pipeline. For VC 2, this was done using FreeSurfer software at the University of California San Francisco. Further details can be found in the “UCSF FreeSurfer Methods” PDF document under “MR Image Analysis” in the ADNI section of https://ida.loni.usc.edu/ as well as in Dale et al. and Fischl et al. 35 , 36 , 37 Cortical thickness values are represented in millimeters (mm). The volume (cubic millimeters) and area (square millimeters) were normalized (divided) by the intracranial volume to reduce intersubject variability and account for variance due to head size within each cohort. While the performance of prediction models that include MRI features was assessed and reported for both VC 1 and VC 2, it is important to emphasize that the reliable ascertainment of the performance of these models with MRI features can only be achieved in VC 1 due to the inability to reprocess MRI images in VC 2 using the same preprocessing method used for TC and VC 1.
2.4. Data analysis
Structural brain network (SBN) modules and hubs were derived using 207 MRI regional measures (volume, area, and cortical thickness) in TC via an algorithm from genomics called “multiscale embedded gene co‐expression network analysis.” 38 This algorithm first entails the calculation of the correlation of MRI measures across all pairs of regions. Regions with significant correlations were embedded on a spherical surface and representative edges (regions that are correlated with multiple other regions) were extracted to create planar‐filtered networks. Finally, a hierarchy of network modules was constructed by recursively clustering the regional measures with coherent structures into network modules. This resulted in a total of 18 SBN modules (labeled SBN.1 to SBN.18) and 45 hub regional measures. Some regions may be present in more than one network module depending on the nature of correlations between the neighboring regions. The regional measures in each of the SBN modules were aggregated into a single composite eigenvalue for each subject by the MEGENA algorithm. Subsequent prediction modeling efforts using the MRI data focused only on these 18 SBN modules and 45 hub regional measures as this helped to reduce the redundancy and dimensionality across the 207 regional measures and were more likely to yield meaningful and interpretable insights on the key prognostic predictors of clinical progression. A complete listing of all the SBN modules and hub regional measures is provided in Tables S2 and S3, respectively.
A prognostic model for predicting longitudinal cognitive trajectory for each patient was first constructed from the training cohort using baseline cognitive function assessments, APOE ε4 status, demographics such as gender, age, and body mass index (BMI), and the measurement time of cognitive function as predictors and the change in CDR‐SB as the outcome of interest via the Stochastic Gradient Boosting Machine (SGBM) algorithm. 39 For simplicity, we do not account for the within‐subject correlation in the training process and only include visits where all predictors and CDR‐SB are measured for the patient in the TC. This is analogous to using the working independence correlation matrix in the generalized estimating equation (GEE) approach to analyzing longitudinal data. An expanded version of this model was then developed by adding the baseline SBNs to the baseline clinical features. A complete listing of all the features (predictors) considered in these two models is provided in Tables S4 and S5, respectively. SGBM is an ensemble tree‐based model that efficiently combines the predictions from multiple decision trees to generate the final predictions. It learns from the data to automatically model the inherent non‐linearity and interactions between predictors, without prior assumptions on the distribution or specific mathematical forms of the relationships between predictors and outcomes. Overfitting is avoided by internally tuning the model with hold‐out datasets and cross‐validation. In our implementation of the prediction model, we consider assembling up to 1000 decision trees with up to three‐way interactions among predictors. The ranking and relative influence of each predictor in the prognostic models were derived by assessing the reduction in the mean squared error (MSE) each time the predictor was used as a root node to split the decision trees in the SGBM algorithm, and these were then normalized to range from 0 to 100%. 40 Insights into the relationships between predictors and outcomes and the interaction between predictors were derived via individual conditional expectation (ICE) profiles and partial dependence plots of the prediction profiles. 41
The prediction performance of our SGBM models underwent rigorous evaluation. Initially, a comprehensive assessment was conducted through 10 iterations of 10‐fold cross‐validation within the TC. 42 Subsequently, the models were evaluated in two independent validation cohorts, VC 1 and VC 2. This evaluation involved measuring the coefficient of determination (R 2), MSE, and mean absolute error (MAE) for observed versus predicted cognitive decline (CDR‐SB change from baseline) at each time point.
One of the applications studied in this research is the use of prognostic predictions of individual cognitive trajectories from our proposed prediction models as an enrichment tool to screen and select patients for a clinical trial. To that end, 500 clinical trials were simulated via the bootstrap approach (sampling with replacement) based on the data from the placebo arm of the clinical trial used for VC 1, with a 1:1 random allocation of active treatment and placebo. The clinical trial duration was set at 18 months. The treatment effect, defined as the difference in the change from baseline in CDR‐SB between the treatment and placebo groups at month 18, was set at 30%. The impact of selecting only patients with predicted 18‐month CDR‐SB change of at least 0.5 and 1 (enrichment scenarios 1 and 2, respectively) was then evaluated for each simulated clinical trial by comparing the sample size requirement and power between the non‐enriched and enriched clinical trials for these different enrichment scenarios. The sample size evaluations were based on the two‐sample t test. The rationale for the thresholds used in the two enrichment scenarios is to exclude subjects that are progressing more slowly over the 18‐month period where treatment effects may be more difficult to detect. In addition, these thresholds are likely to represent a clinically meaningful progression from MCI to mild AD. 43
All analyses were performed using R version 4.2.2 (R Foundation for Statistical Computing), 44 along with the packages MEGENA, 45 gbm, 46 and pdp. 47
3. RESULTS
3.1. Demographics
Data in the training and two validation cohorts (TC, VC 1, and VC 2) included 934, 235, and 421 Aβ+ subjects, respectively. Most of these subjects had MCI due to AD and the rest had mild AD. A summary of some key demographic and clinical characteristics (gender, APOE ε4 status, age, BMI, diagnosis, MMSE, and CDR‐SB) is shown in Table 1. All demographic and clinical characteristics were significantly different (p < .05) between the three cohorts. The training cohort had a significantly greater proportion of MCI and ApoE4‐positive subjects. The first validation cohort (VC 1) had a greater proportion of males. Subjects in the second validation cohort were older and had a higher BMI. These differences among early AD patients across different clinical trials and observational cohorts help to provide a robust assessment of the performance of the prediction models between the training and validation cohorts. The Kruskal‐Wallis test was used for comparing age, education, BMI, and MMSE, and the chi‐squared test was used for comparing gender and ApoE4 status. A summary of CDR‐SB change from baseline at each time point in the three cohorts is shown in Table S1. The longitudinal trajectory of clinical progression (CDR‐SB change from baseline) for individual subjects in the three cohorts shown in Figure 1 reflects the heterogeneity in clinical progression over a typical 18‐ to 24‐month duration of AD clinical trials.
FIGURE 1.
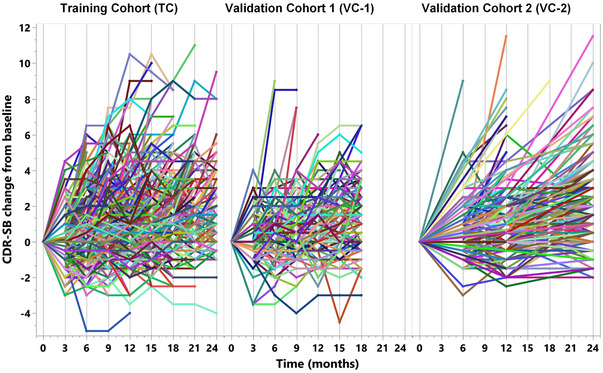
Longitudinal cognitive trajectory. The longitudinal profiles of the change from baseline of CDR‐SB of 934, 235, and 421 subjects in the training and the two validation cohorts, respectively, reflect the considerable heterogeneity in progression over a typical 18‐ to 24‐month duration of AD clinical trials.
3.2. Prediction models
The relative influence of the top 10 predictors in the prediction model constructed via the SGBM algorithm using only the baseline clinical features (cognitive function assessments and demographics) are shown in Figure 2A, and the top 10 predictors from the expanded model that includes the SBN modules and hub regional measures based on the MRI features are shown in Figure 2B. In addition to time, the key baseline clinical predictors in these models were ADAS‐Cog‐13, MMSE, word recall and recognition, ideational praxis, CDR‐SB, and word‐finding difficulty, along with BMI and age. Ideational praxis refers to the ability to perform multilevel tasks, for example, the sequence of steps needed for brushing teeth. Some of the key MRI‐based predictors include features from the SBN hub regions, middle temporal cortical area, and inferior parietal cortical volume, along with the network module (SBN.11) comprising the inferior parietal gyri, inferior temporal gyri, middle temporal gyri, and banks of the superior temporal sulci (Figure 2C), the network module (SBN.15) comprising the entorhinal cortices and temporal poles (Figure 2D), and the network module (SBN.3) comprising the superior parietal gyri, precunei, isthmus of the cingulate gyri, lateral occipital gyri, postcentral gyri, supramarginal gyri, superior temporal gyri, fusiform gyri, lingual gyri, and transverse temporal gyri, plus the regions in SBN.11 (Figure 2E). A complete listing of the ranking and relative influence of all the predictors used in these two prognostic models is provided in Tables S4 and S5, respectively.
FIGURE 2.
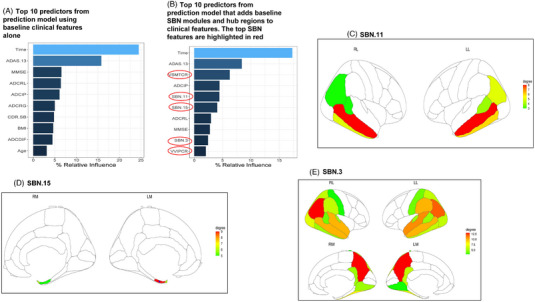
(A‐E) Top 10 predictors in prediction models and brain heatmaps of key SBN modules and hub regions. The relative influence of the top 10 predictors in the prediction model based on the baseline clinical features alone (A) and in the model that also includes MRI‐based SBN modules and hub regional measures (B) are shown here. The key baseline clinical predictors in these models include ADAS‐Cog‐13 (ADAS.13), Mini‐Mental State Examination (MMSE), word recall (ADCRL), ideational praxis (ADCIP), word recognition (ADCRG), CDR‐SB, and word‐finding difficulty (ADCDIF), along with demographic features, body mass index (BMI), and age. The key MRI‐based predictors include the SBN hub regional measures, middle temporal cortical area (VSMTCR), and inferior parietal cortical volume (VVIPCR), along with the SBN modules, SBN.11 (C) that includes the inferior parietal gyri, inferior temporal gyri, middle temporal gyri, and banks of the superior temporal sulci, SBN.15 (D) that includes the entorhinal cortices and temporal poles, and SBN.3 (E) that includes the superior parietal gyri, precunei, isthmus of the cingulate gyri, lateral occipital gyri, postcentral gyri, supramarginal gyri, superior temporal gyri, fusiform gyri, lingual gyri, transverse temporal gyri; the regions in SBN.11. RL, LL, RM, LM refer to right lateral, left lateral, right medial, and left medial, respectively. Degree refers to the number of spatially connected (correlated) neighboring regions.
The nature of the relationship between each of the key baseline predictors versus the predicted clinical progression from these two prediction models is examined at the individual subject level and for an average subject via ICE profiles, as shown in Figure 3. This is accomplished by plotting the individual and average predicted outcomes for different values of each predictor while holding the values of other predictors constant. These ICE prediction profiles reveal strong sigmoidal‐like non‐linear relationships between each baseline predictor and clinical decline with floor and ceiling effects and a region of linear impact in between. The flexibility of the SGBM algorithm allows for such relationships and inflection nodes of the predictors to be modeled without prior specifications or assumptions. The heterogeneity of profiles across subjects reflects the interaction between the predictors that are examined further in Figure 4.
FIGURE 3.
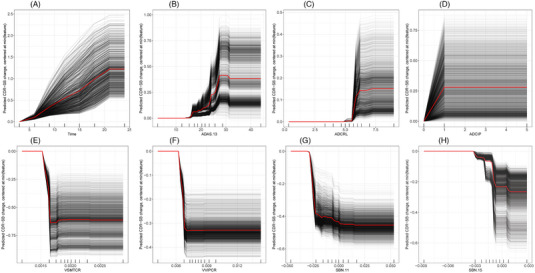
(A‐H) Individual conditional expectation profiles of some key predictors. The nature of the relationship between some of the key baseline clinical and MRI‐based SBN predictors versus the predicted clinical decline (CDR‐SB change from baseline) is shown here for each subject (in black) and the average subject (in red) via these individual conditional expectations (ICE) profiles. The prediction profile of each subject was centered by subtracting from the predicted CDR‐SB change corresponding to the lowest value of the predictor. The intersubject heterogeneity in these ICE profiles is mostly due to the strong interaction between the predictors, which is evident in Figure 4. The SGBM algorithm accounts for these non‐linear relationships and interactions without prior assumptions on the distribution or mathematical forms of the relationships.
FIGURE 4.
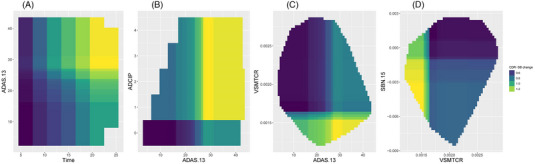
(A‐D) Interaction prediction profiles between some key predictors. These interaction prediction profiles reveal the strong dependence between some key predictors that were accounted for by the SGBM algorithm.
The interaction prediction profiles shown in Figure 4 reveal the strong dependence between some of the key predictors that have been accounted for by the SGBM algorithm. For example, greater cognitive decline (CDR‐SB change from baseline) over time is seen in subjects with higher baseline ADAS‐Cog‐13 (Figure 4A). Subjects with high ADAS‐Cog‐13 and worsening ideational praxis (ADCIP) at baseline are likely to have greater cognitive decline (Figure 4B). In addition, subjects with high ADAS‐cog‐13 and with lower middle temporal cortical area (VSMTCR) or lower area, volume, or thickness in the entorhinal cortex and temporal pole (SBN.15) at baseline are also expected to experience a greater cognitive decline (Figures 4C‐D).
3.3. Performance evaluation of prediction models
The prediction performance of the two prediction models, one based on the baseline clinical features alone and the other that adds the baseline MRI features in the form of SBNs, was evaluated in the two independent validation cohorts, VC1 and VC 2. As is evident from the overlapping 95% confidence intervals in Figures 5A and 5B, the average predicted clinical decline from these two models with respect to the change from baseline in CDR‐SB tracks well and does not significantly differ from the average observed decline in both validation cohorts. Predictions of the clinical decline of individual subjects from the two prediction models are significantly correlated (p < .001) with the observed clinical decline (Figures 5C and 5D). Table 2 presents a detailed breakdown of performance metrics, including the coefficient of determination (R 2), MSE, and MAE, for each time point. Furthermore, Table S6 provides Pearson correlation coefficients as an additional performance metric.
FIGURE 5.
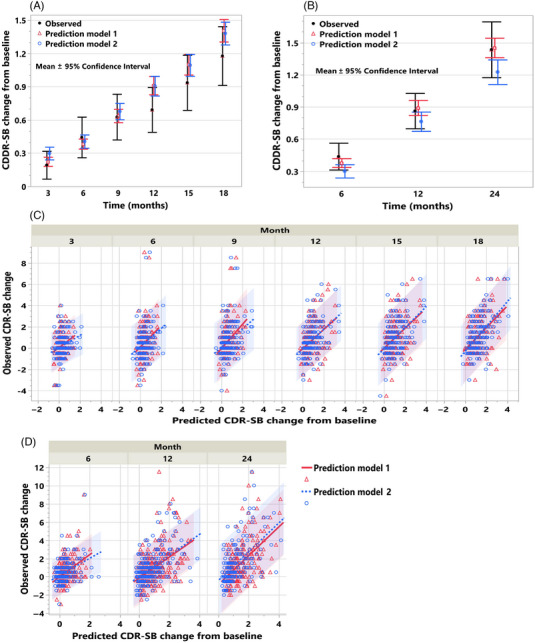
(A‐D) Prognostic prediction of average cognitive decline and cognitive decline of individual subjects in the two validation cohorts. Mean and 95% confidence interval of the observed and predicted CDR‐SB change from baseline using the models based on the baseline clinical features (model 1) alone and with the addition of MRI‐based SBN features (model 2) are shown in (A) for validation cohort (VC) 1 and in (B) for VC 2. The observed versus predicted CDR‐SB change from baseline for individual subjects at each time point from models 1 and 2 along with the 95% prediction intervals are shown for VC 1 in (C) and VC 2 in (D).
TABLE 2.
Prediction performance summary in the two validation cohorts.
| VC 1 | VC 2 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| R 2 | MSE | MAE | R 2 | MSE | MAE | |||||||
| Month | Model 1 | Model 2 | Model 1 | Model 2 | Model 1 | Model 2 | Model 1 | Model 2 | Model 1 | Model 2 | Model 1 | Model 2 |
| 3 | 0.07 | 0.07 | 0.88 | 0.91 | 0.68 | 0.69 | – | – | – | – | – | – |
| 6 | 0.06 | 0.08 | 1.76 | 1.74 | 0.88 | 0.87 | 0.16 | 0.14 | 1.09 | 1.16 | 0.72 | 0.73 |
| 9 | 0.13 | 0.11 | 1.95 | 1.99 | 0.94 | 0.97 | – | – | – | – | – | – |
| 12 | 0.17 | 0.2 | 1.64 | 1.59 | 0.96 | 0.94 | 0.25 | 0.28 | 2.15 | 2.03 | 1.01 | 0.99 |
| 15 | 0.17 | 0.22 | 2.26 | 2.14 | 1.16 | 1.14 | – | – | – | – | – | – |
| 18 | 0.21 | 0.29 | 2.28 | 2.08 | 1.16 | 1.15 | – | – | – | – | – | – |
| 24 | – | – | – | 0.31 | 0.29 | 3.34 | 3.88 | 1.35 | 1.38 | |||
Note: The coefficient of determination (R 2), mean squared error (MSE), and mean absolute error (MAE) of the predicted versus observed clinical decline (CDR‐SB change from baseline) from the prognostic clinical progression model based on baseline clinical features alone (model 1) and the model that adds baseline MRI‐based SBNs (model 2) is shown here for the two validation cohorts (VC 1 and VC 2). Model 1 predictions are mostly on par with those of model 2, except at later time points with respect to R 2 and MSE in VC 1, which employed the same image processing pipeline as the training cohort (TC).
The model utilizing baseline clinical features demonstrated a moderate performance, achieving an R 2 of 0.21 and 0.31, along with MSE values of 2.28 and 3.34 and MAE values of 1.16 and 1.35 for predicting cognitive decline at 18 and 24 months in VC 1 and VC 2, respectively. Adding MRI features to this model resulted in a notable improvement. In VC 1, which employed the same image processing pipeline as TC, the R 2 value increased to 0.29, and the MSE decreased to 2.08. However, this enhancement in model performance was not replicated in VC 2, where differences in the image preprocessing methods between TC and VC 2 likely had an impact. While the model relying solely on baseline clinical features performs reasonably well in predicting cognitive decline, the inclusion of baseline MRI features, processed using the same pipeline, is recommended when MRI assessments are available. This holds true particularly in the context of patient selection and clinical trial enrichment, as illustrated below.
3.4. Use of prognostic predictions for clinical trial enrichment
The impact on the sample size reduction and power increase is shown in Figures 6A and 6B when enriching the clinical trial for subjects predicted to experience at least mild to moderate levels of clinical decline, that is, subjects with predicted 18‐month CDR‐SB change of at least 0.5 and 1. Approximately 88% and 65% of the clinical trial subjects used in VC 1 met these criteria for the two enrichment scenarios. The results for these two enrichment scenarios are presented using the prediction model based on the baseline clinical features alone (cognitive function assessments and demographics; model 1) and the model with the addition of baseline MRI features in the form of SBNs (model 2).
FIGURE 6.
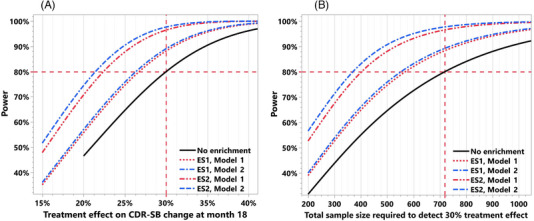
(A and B) Application of prognostic prediction of longitudinal cognitive decline for clinical trial enrichment. The impact on the sample size reduction and power increase is shown here when enriching the clinical trial for subjects with predicted 18‐month CDR‐SB change of at least 0.5 and 1. 500 clinical trial simulations based on the placebo arm data from a clinical trial (validation cohort 1) were used to study the impact of this enrichment. In panel (A), when using the predictions from the model based on baseline clinical features alone for enrichment (model 1), the power increases from 80% to 88.3% and 96.5%, respectively, for the two enrichment scenarios (ES1, ES2). Fixing the power at 80%, these enrichment scenarios with model 1 improve the ability to detect the treatment effect from 30% to 26.7% and 22.3%, respectively. In panel (B), the total sample size required to detect a 30% treatment effect reduces from 718 to 568 and 398 (20.9% and 44.6% reduction), respectively, for the two enrichment scenarios with model 1. Modest improvement in these numbers is seen when using the model based on both the baseline clinical features and the MRI‐based SBN features (model 2) for enrichment; for the two enrichment scenarios the power increases from 80% to 89.2% and 97.6%, respectively, and the minimum treatment effect that can be detected with 80% power improves from 30% to 26.3% and 21.3% (Figure 6A). The total sample size required to detect a 30% treatment effect reduces from 718 to 552 and 364 (23.2% and 49.4% reduction), respectively, for the two ESs using model 2 predictions.
A clinical trial without these enrichment scenarios would require a total sample size of 718 subjects (359 per group) to detect a 30% treatment effect with respect to the change from baseline in CDR‐SB at month 18 with 80% power. As evident in Figure 6A, when using the predictions from the model based on only the baseline clinical features for enrichment (model 1), the power increases to 88.3% and 96.5%, respectively, for the two enrichment scenarios. Fixing the power at 80%, these enrichment scenarios with model 1 improve the ability to detect the treatment effect from 30% to 26.7% and 22.3%, respectively. In Figure 6B, we see that the total sample size required to detect a 30% treatment effect reduces from 718 to 568 and 398 (20.9% and 44.6% reduction), respectively, for the two enrichment scenarios with model 1. Modest improvement in these numbers is seen when using the model based on both the baseline clinical features and the MRI‐based SBN features (model 2) for enrichment; for the two enrichment scenarios, the power increases to 89.2% and 97.6%, respectively, and the minimum treatment effect that can be detected with 80% power improves from 30% to 26.3% and 21.3% (Figure 6A). The total sample size required to detect a 30% treatment effect reduces from 718 to 552 and 364 (23.2% and 49.4% reduction), respectively, for the two enrichment scenarios using model 2 predictions (Figure 6B).
One of the questions that may arise with an enrichment strategy is whether it will require screening a significant number of additional patients to satisfy the enrichment criteria. For the VC 1 clinical trial population and using the prediction model 2, approximately 89% and 62% of patients met the enrichment criteria of the predicted 18‐month CDR‐SB change of at least 0.5 and 1, respectively. We also observed that the total sample size required to detect a 30% treatment effect reduced from 718 to 552 and 364 (23.2% and 49.4% reduction), respectively. Therefore, instead of screening 718 subjects, we will need to screen 620 subjects (552 divided by 0.89) and 587 subjects (364 divided by 0.62), respectively, to apply these enrichment strategies. That is, in addition to significant reductions in sample size requirements and an increase in power, these enrichment strategies will entail screening 13.6% and 18.2% fewer subjects compared to the no‐enrichment strategy. More importantly, there may be other practical benefits/needs for such enrichment strategies in clinical trials, for example, if the candidate treatment is expected to benefit only subjects that are likely to experience mild to moderate cognitive decline.
4. DISCUSSION
In this study, prediction models were developed to forecast the individual clinical progression trajectories of Aβ+ early AD patients over a typical 18‐month duration of a clinical trial using only their baseline clinical and structural imaging characteristics. A machine learning algorithm called stochastic gradient boosting machine was used to build these models in the training cohort of 934 placebo subjects pooled from two clinical trials. We found that the prediction profiles of the resulting models account for the inherent non‐linearity of the key predictors and their interactions (Figures 3 and 4), without any prior assumptions on the distribution or parametric specifications.
Some recent publications have proposed models for predicting individual progression trajectories. 17 , 48 Our prediction models are different in the following ways. Our models were (1) developed using clinical trial cohorts and validated in both clinical trial and observational research cohorts, (2) optimized for predicting near‐term clinical progression (2 years), which is more relevant for clinical trial and real‐world applications, and (3) based on commonly used baseline clinical assessments and optionally structural MRI.
The inclusion of individual subscores of CDR‐SB and ADAS‐Cog‐13 in our prediction models rather than just the composite scores turned out to be quite useful, as evident from the relative influence of the top predictors in the models and provided valuable insights (Figures 2A,B); baseline deficits in word recall, word recognition, and ideational praxis have a greater impact on clinical progression relative to the other subscores of these composite measures.
A comprehensive performance evaluation of our models in the validation cohorts demonstrates that the use of standard clinical assessments and demographics that are routinely assessed during the screening phase of AD clinical trials can be used to predict the longitudinal clinical progression trajectories of individual subjects. It also suggests that adding MRI‐based SBNs to the model could improve the performance of the predictions and therefore would be preferable if MRI data were available and preprocessed using the same method. A prediction model using the baseline MRI‐based SBN features alone was also developed. As the predictions from this model did not perform as well as the model based on baseline clinical features alone, and as the clinical assessments at baseline are more readily available than MRI, the model based on MRI features alone is not reported here in detail.
As these prognostic progression models provide a forecast of the individual clinical progression trajectories for Aβ+ early AD patients over a typical duration of AD clinical trials of 18 to 24 months, there are wide‐ranging applications for these models across clinical drug development and real‐world clinical practice. One such application that we studied in depth was related to clinical trial prognostic enrichment. 49 , 50 Clinical trial simulations using the placebo arm data of the clinical trial used in VC 1 were carried out to study the impact of different enrichment scenarios based on the predicted future clinical progression of subjects at baseline. These analyses revealed that enriching a clinical trial with subjects predicted to experience mild to moderate clinical decline (as defined by the CDR‐SB change from baseline) using our prediction models led to an approximately 20% to 49% reduction in the sample size requirement for the two enrichment scenarios based on predicted 18‐month CDR‐SB change of at least 0.5 and 1 (ES 1 and ES 2) and an increase in power from 80% to approximately 88% to 98% to detect a 30% treatment effect on the clinical decline at month 18. The rationale for the thresholds used in ES 1 and ES 2 was to exclude subjects who are progressing more slowly over the 18‐month period where treatment effects may be more difficult to detect.
Similar to Tam et al., 51 we compared non‐enriched and enriched populations in training and validation cohorts using the clinical assessment‐based prediction model (Table S7). While we observed differences in MCI and mild AD patient distribution and baseline MMSE and CDR‐SB for ES 2 (p < .05), demographic and APOE ε4 status distributions remained mostly unaffected. This trend held in the subgroup with complete CDR‐SB scores at month 18 (TC and VC 1) and month 24 (VC 2) as detailed in Table S8. In this subgroup, the prevalence of individuals meeting ES 1 and ES 2 enrichment criteria and the average CDR‐SB decline was higher (though not significant) in TC and VC 1 when enriched based on ES 1 and significantly higher for all three cohorts when enriched based on ES 2 (p < .05). This suggests the prediction model met the intended goal of enriching more decliners. However, given the propensity of these model‐based enrichment tools to favor mild AD over MCI, additional screening may be required when greater representation of MCI subjects is needed in clinical trials.
Predictions of CDR‐SB change may also be used as a prognostic covariate for increasing the precision and power of treatment effect evaluation 52 , 53 , 54 , 55 and can be considered either with or without enrichment. Enrichment is appropriate if the treatment is expected to be effective only for a subpopulation (eg, patients with mild to moderate cognitive decline), and the prognostic covariate approach is particularly useful if there is no justification to exclude a subgroup of patients via enrichment. Other potential applications in clinical trials include precision medicine investigations via patient stratification, subgroup identification of treatment responders and non‐responders, and targeted post hoc evaluations of treatment effects and adverse events. 56 , 57 In real‐world settings, these predictions could be used as a supplemental tool for patient diagnosis and monitoring and for decisions related to treatment and care 8 , 58 ; however, this would require clinical assessments that are more feasible in clinical practice, and the predictions may need to be more accurate.
It is important to understand whether the proposed progression models predict well for under‐represented populations such as non‐White and those with lower education levels. While the training cohort used for constructing our prediction models included 128 (13.7%) subjects with lower education level (<10 years of education) and 228 (24.4%) non‐White subjects, the prediction performance could not be reliably ascertained for these subpopulations in the two validation cohorts due to limited sample size (7.7% and 0.4% of the subjects had lower education level in VC 1 and VC 2, respectively, and 9.4% and 6.4% of the subjects were non‐Whites). Further evaluation in other cohorts is needed to verify the applicability of our progression models for these subpopulations. In addition, as none of the subjects in the training cohort (TC) had other dementias that combine with AD, our progression models may be suitable for people with AD pathology only.
Although our progression models utilize all the clinical and MRI‐based SBN features, these features are weighted proportional to their relative influence on the prediction of progression, and the features that had zero influence are practically not used by the models (Table S4). Thus, our algorithm allowed for the unbiased identification of predictive features without any preselection. Although more parsimonious models based on only a subset of the most important predictors can be developed via methods such as regularization, 59 , 60 this was not necessary because the predictors considered are part of routine screening assessments in AD clinical trials, and such parsimonious models did not perform better than our models.
While the application of the proposed prediction models in clinical trials or real‐world applications is limited by a specific set of baseline clinical and optionally MRI‐based features, these models are readily amenable to the inclusion of other types of baseline patient data. This may include other cognitive function assessments such as the functional activity questionnaire (FAQ), convenient real‐world digital instruments such as the CogState Brief Battery, 61 computer‐administered neuropsychological screens for MCI, 62 Cambridge neuropsychological test automated battery paired associates learning, 63 , 64 and various fluid biomarkers in the ATN research framework that may help improve the prediction of clinical progression. 24 , 32 , 65 , 66 , 67 , 68 , 69 , 70 , 71
Lastly, we did not consider the within‐subject correlation in training the prediction model, which may cause a loss in prediction accuracy. On the other hand, such a loss is usually limited, and it avoids the need to consider a parametric model or assumptions on the specific mathematical form of the relationship between predictors and outcomes (eg, linear, non‐linear) and about the interactions among predictors or explicit modeling of the correlation structure, which is often difficult. Thus, the advantages of our machine learning approach may outweigh the use of parametric models that consider the within‐subject correlation for developing the prediction models. In validating the model performance, we separately summarize the prediction performance at different visits to avoid this correlation issue.
In summary, we show that the individual cognitive progression trajectories of Aβ+ early AD patients over a typical 18‐month duration of AD clinical trials can be predicted using commonly measured clinical and demographic characteristics at baseline or initial screening. These models can readily accommodate other types of baseline assessments such as those from MRI, PET, and fluid biomarkers to improve prediction performance. These prognostic predictions have a wide‐ranging impact across clinical drug development, clinical practice, and real‐world applications. 33 , 57
CONFLICT OF INTEREST STATEMENT
V.D., Y.Y., A.C., E.A., P.S., L.Z., H.H., L.K., S.D., and MI are employees of Eisai Inc. HH is also the Senior Associate Editor for the journal Alzheimer's & Dementia. No competing disclosures to report for DAL and LT. Author disclosures are available in the supporting information.
CONSENT STATEMENT
All the participants in the studies used for the training and validation cohorts in this manuscript provided written informed consent.
CLINICALTRIALS.GOV IDENTIFIERS
NCT02956486 (MissionAD1) and NCT03036280 (MissionAD2): A Placebo‐Controlled, Double‐Blind, Parallel‐Group, 24 Month Study with an Open‐Label Extension Phase to Evaluate the Efficacy and Safety of Elenbecestat (E2609) in Subjects with Early Alzheimer's Disease. NCT01767311: A Study to Evaluate Safety, Tolerability, and Efficacy of Lecanemab in Subjects with Early Alzheimer's Disease
Supporting information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
ACKNOWLEDGMENTS
The authors thank the patients, their families, and the sites who participated in these studies. The authors also thank Leema Krishna Murali, Todd Nelson, Xin Qi, Kiran Putti, Saad Anbari, Huihong Chen, Malathi Reddy, and Michio Kanekiyo for their help with data curation and preparation. The authors are grateful to the anonymous reviewers for their constructive and helpful comments, which have improved the quality and clarity of the manuscript. Editorial support, funded by Eisai Inc., was provided by JD Cox, Ph.D., Mayville Medical Communications. Funding for the studies and analyses was provided by Eisai Inc. Collection and sharing of one of the datasets used in this project was funded by the Alzheimer's Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and Department of Defense (DOD) ADNI (DOD Award No. W81XWH‐12‐2‐0012). ADNI is funded by the National Institute on Aging and the National Institute of Biomedical Imaging and Bioengineering and through generous contributions from the following: AbbVie, Alzheimer's Association; Alzheimer's Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol‐Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann‐La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study was coordinated by the Alzheimer's Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California. Funding for the studies, analyses, and editorial support was provided by Eisai Inc.
Devanarayan V, Ye Y, Charil A, et al. Predicting clinical progression trajectories of early Alzheimer's disease patients. Alzheimer's Dement. 2024;20:1725–1738. 10.1002/alz.13565
One of the datasets used in the preparation of this article was obtained from the Alzheimer's Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in the analysis or writing of this report. A complete listing of ADNI investigators can be found at:http://adni.loni.usc.edu/wp‐content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf
REFERENCES
- 1. Mills EA, Begay JA, Fisher C, Mao‐Draayer Y. Impact of trial design and patient heterogeneity on the identification of clinically effective therapies for progressive MS. Mult Scler. 2018;24:1795‐1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. van Eijk RPA, Nikolakopoulos S, Roes KCB, et al. Innovating clinical trials for amyotrophic lateral sclerosis: challenging the established order. Neurology. 2021;97:528‐536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Catenacci DV. Next‐generation clinical trials: novel strategies to address the challenge of tumor molecular heterogeneity. Mol Oncol. 2015;9:967‐996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Duara R, Barker W. Heterogeneity in Alzheimer's disease diagnosis and progression rates: implications for therapeutic trials. Neurotherapeutics. 2022;19:8‐25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Manton KG, Vertrees JC, Woodbury MA. Functionally and medically defined subgroups of nursing home populations. Health Care Financ Rev. 1990;12:47‐62. [PMC free article] [PubMed] [Google Scholar]
- 6. Dammann M, Staudacher S, Simon M, Jeitziner MM. Insights into the challenges faced by chronically critically ill patients, their families and healthcare providers: an interpretive description. Intensive Crit Care Nurs. 2022;68:103135. [DOI] [PubMed] [Google Scholar]
- 7. Abdelnour C, Agosta F, Bozzali M, et al. Perspectives and challenges in patient stratification in Alzheimer's disease. Alzheimers Res Ther. 2022;14:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hampel H, Au R, Mattke S, et al. Designing the next‐generation clinical care pathway for Alzheimer's disease. Nature Aging. 2022;2:692‐703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mann UM, Mohr E, Gearing M, Chase TN. Heterogeneity in Alzheimer's disease: progression rate segregated by distinct neuropsychological and cerebral metabolic profiles. J Neurol Neurosurg Psychiatry. 1992;55:956‐959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hampel H, Lista S. Dementia: the rising global tide of cognitive impairment. Nat Rev Neurol. 2016;12:131‐132. [DOI] [PubMed] [Google Scholar]
- 11. Goyal D, Tjandra D, Migrino RQ, Giordani B, Syed Z, Wiens J. Characterizing heterogeneity in the progression of Alzheimer's disease using longitudinal clinical and neuroimaging biomarkers. Alzheimers Dement. 2018;10:629‐637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Devi G, Scheltens P. Heterogeneity of Alzheimer's disease: consequence for drug trials? Alzheimers Res Ther. 2018;10:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hampel H, Hardy J, Blennow K, et al. The Amyloid‐beta pathway in Alzheimer's disease. Mol Psychiatry. 2021;26:5481‐5503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hersi M, Irvine B, Gupta P, Gomes J, Birkett N, Krewski D. Risk factors associated with the onset and progression of Alzheimer's disease: a systematic review of the evidence. Neurotoxicology. 2017;61:143‐187. [DOI] [PubMed] [Google Scholar]
- 15. Fleisher AS, Sowell BB, Taylor C, et al. Clinical predictors of progression to Alzheimer disease in amnestic mild cognitive impairment. Neurology. 2007;68:1588‐1595. [DOI] [PubMed] [Google Scholar]
- 16. Iddi S, Li D, Aisen PS, et al. Predicting the course of Alzheimer's progression. Brain Inform. 2019;6:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Franzmeier N, Koutsouleris N, Benzinger T, et al. Predicting sporadic Alzheimer's disease progression via inherited Alzheimer's disease‐informed machine‐learning. Alzheimers Dement. 2020;16:501‐511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Doody RS, Raman R, Farlow M, et al. A phase 3 trial of semagacestat for treatment of Alzheimer's disease. N Engl J Med. 2013;369:341‐350. [DOI] [PubMed] [Google Scholar]
- 19. Siemers ER, Sundell KL, Carlson C, et al. Phase 3 solanezumab trials: secondary outcomes in mild Alzheimer's disease patients. Alzheimers Dement. 2016;12:110‐120. [DOI] [PubMed] [Google Scholar]
- 20. Vandenberghe R, Rinne JO, Boada M, et al. Bapineuzumab for mild to moderate Alzheimer's disease in two global, randomized, phase 3 trials. Alzheimers Res Ther. 2016;8:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Honig LS, Vellas B, Woodward M, et al. Trial of solanezumab for mild dementia due to Alzheimer's disease. N Engl J Med. 2018;378:321‐330. [DOI] [PubMed] [Google Scholar]
- 22. Wessels AM, Tariot PN, Zimmer JA, et al. Efficacy and Safety of Lanabecestat for Treatment of Early and Mild Alzheimer Disease: the AMARANTH and DAYBREAK‐ALZ Randomized Clinical Trials. JAMA Neurol. 2020;77:199‐209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Budd Haeberlein S, Aisen PS, Barkhof F, et al. Two randomized phase 3 studies of aducanumab in early Alzheimer's disease. J Prev Alzheimers Dis. 2022;9:197‐210. [DOI] [PubMed] [Google Scholar]
- 24. Hampel H, O'Bryant SE, Molinuevo JL, et al. Blood‐based biomarkers for Alzheimer disease: mapping the road to the clinic. Nat Rev Neurol. 2018;14:639‐652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hampel H, Shaw LM, Aisen P, et al. State‐of‐the‐art of lumbar puncture and its place in the journey of patients with Alzheimer's disease. Alzheimers Dement. 2022;18:159‐177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jutten RJ, Sikkes SAM, Van der Flier WM, et al. Finding treatment effects in alzheimer trials in the face of disease progression heterogeneity. Neurology. 2021;96:e2673‐e2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mattsson‐Carlgren N, Salvado G, Ashton NJ, et al. Prediction of longitudinal cognitive decline in preclinical Alzheimer disease using plasma biomarkers. JAMA Neurol. 2023;80(4):360‐369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hampel H, Frank R, Broich K, et al. Biomarkers for Alzheimer's disease: academic, industry and regulatory perspectives. Nat Rev Drug Discov. 2010;9:560‐574. [DOI] [PubMed] [Google Scholar]
- 29. Llano DA, Laforet G, Devanarayan V, Alzheimer's Disease Neuroimaging I . Derivation of a new ADAS‐cog composite using tree‐based multivariate analysis: prediction of conversion from mild cognitive impairment to Alzheimer disease. Alzheimer Dis Assoc Disord. 2011;25:73‐84. [DOI] [PubMed] [Google Scholar]
- 30. Desikan RS, Cabral HJ, Settecase F, et al. Automated MRI measures predict progression to Alzheimer's disease. Neurobiol Aging. 2010;31:1364‐1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Llano DA, Bundela S, Mudar RA, Devanarayan V, Alzheimer's Disease Neuroimaging I . A multivariate predictive modeling approach reveals a novel CSF peptide signature for both Alzheimer's Disease state classification and for predicting future disease progression. PLoS One. 2017;12:e0182098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Devanarayan P, Devanarayan V, Llano DA, Alzheimer's Disease Neuroimaging I . Identification of a simple and novel cut‐point based cerebrospinal fluid and MRI signature for predicting Alzheimer's disease progression that reinforces the 2018 NIA‐AA research framework. J Alzheimers Dis. 2019;68:537‐550. [DOI] [PubMed] [Google Scholar]
- 33. Wattmo C, Wallin AK, Minthon L. Progression of mild Alzheimer's disease: knowledge and prediction models required for future treatment strategies. Alzheimers Res Ther. 2013;5:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Desikan RS, Segonne F, Fischl B, et al. An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. Neuroimage. 2006;31:968‐980. [DOI] [PubMed] [Google Scholar]
- 35. Dale AM, Fischl B, Sereno MI. Cortical surface‐based analysis. I. Segmentation and surface reconstruction. Neuroimage. 1999;9:179‐194. [DOI] [PubMed] [Google Scholar]
- 36. Fischl B, Sereno MI, Dale AM. Cortical surface‐based analysis. II: inflation, flattening, and a surface‐based coordinate system. Neuroimage. 1999;9:195‐207. [DOI] [PubMed] [Google Scholar]
- 37. Fischl B, Sereno MI, Tootell RB, Dale AM. High‐resolution intersubject averaging and a coordinate system for the cortical surface. Hum Brain Mapp. 1999;8:272‐284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Song WM, Zhang B. Multiscale embedded gene co‐expression network analysis. PLoS Comput Biol. 2015;11:e1004574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Friedman JH. Greedy function approximation: a gradient boosting machine. Annals of Statistics. 2001;29:1189‐1232. [Google Scholar]
- 40. Natekin A, Knoll A. Gradient boosting machines, a tutorial. Front Neurorobot. 2013;7:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Goldstein A, Kapelner A, Bleich J, Pitkin E. Peeking inside the black box: visualizing statistical learning with plots of individual conditional expectation. J Comput Graph Statist. 2015;24:44‐65. [Google Scholar]
- 42. Shi L, Campbell G, Jones WD, et al. The MicroArray Quality Control (MAQC)‐II study of common practices for the development and validation of microarray‐based predictive models. Nat Biotechnol. 2010;28:827‐838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. O'Bryant SE, Waring SC, Cullum CM, et al. Staging dementia using clinical dementia rating scale sum of boxes scores: a Texas Alzheimer's research consortium study. Arch Neurol. 2008;65:1091‐1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. R Core Team . R: A language and environment for statistical computing. R Foundation for Statistical Computing; 2022. [Google Scholar]
- 45. Song W, Zhang B, MEGENA: Multiscale Clustering of Geometrical Network. 2018. R package, version 1.3.7, https://CRAN.R‐project.org/package=MEGENA
- 46. Greenwell B, Boehmke B, Cunningham J, Developers GBM, gbm: Generalized Boosted Regression Models. 2022. R package version 2.1.8.1, https://CRAN.R‐project.org/package=gbm
- 47. Greenwell BM. pdp: an R Package for constructing partial dependence plots. R Journal. 2017;9:421‐436. [Google Scholar]
- 48. Maheux E, Koval I, Ortholand J, et al. Forecasting individual progression trajectories in Alzheimer's disease. Nat Commun. 2023;14:761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kuhnel L, Bouteloup V, Lespinasse J, et al. Personalized prediction of progression in pre‐dementia patients based on individual biomarker profile: a development and validation study. Alzheimers Dement. 2021;17:1938‐1949. [DOI] [PubMed] [Google Scholar]
- 50. U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research, Center for Biologics Evaluation and Research . Guidance for Industry: Enrichment Strategies for Clinical Trials to Support Approval of Human Drugs and Biological Products. 2019. https://www.fda.gov/media/121320/download
- 51. Tam A, Laurent C, Gauthier S, Dansereau C. Prediction of cognitive decline for enrichment of Alzheimer's disease clinical trials. J Prev Alzheimers Dis. 2022;9:400‐409. [DOI] [PubMed] [Google Scholar]
- 52. Kahan BC, Jairath V, Dore CJ, Morris TP. The risks and rewards of covariate adjustment in randomized trials: an assessment of 12 outcomes from 8 studies. Trials. 2014;15:139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhang Z, Ma S. Machine learning methods for leveraging baseline covariate information to improve the efficiency of clinical trials. Stat Med. 2019;38:1703‐1714. [DOI] [PubMed] [Google Scholar]
- 54. Schuler A, Walsh D, Hall D, et al. Increasing the efficiency of randomized trial estimates via linear adjustment for a prognostic score. Int J Biostat. 2022;18:329‐356. [DOI] [PubMed] [Google Scholar]
- 55. U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research, Center for Biologics Evaluation and Research . Guidance for Industry: Adjusting for Covariates in Randomized Clinical Trials for Drugs and Biological Products. 2023. https://www.fda.gov/media/148910/download
- 56. Huang X, Sun Y, Trow P, et al. Patient subgroup identification for clinical drug development. Stat Med. 2017;36:1414‐1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hampel H, Gao P, Cummings J, et al. The foundation and architecture of precision medicine in neurology and psychiatry. Trends Neurosci. 2023;46:176‐198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Llano DA, Devanarayan P, Devanarayan V, Alzheimer's Disease Neuroimaging I . CSF peptides from VGF and other markers enhance prediction of MCI to AD progression using the ATN framework. Neurobiol Aging. 2023;121:15‐27. [DOI] [PubMed] [Google Scholar]
- 59. Friedman J, Hastie T, Tibshirani R. Regularization paths for generalized linear models via coordinate descent. J Stat Softw. 2010;33:1‐22. [PMC free article] [PubMed] [Google Scholar]
- 60. Hong F, Tian L, Devanarayan V. Improving the robustness of variable selection and predictive performance of regularized generalized linear models and cox proportional hazard models. mathematics. 2023;11:557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Maruff P, Lim YY, Darby D, et al. Clinical utility of the cogstate brief battery in identifying cognitive impairment in mild cognitive impairment and Alzheimer's disease. BMC Psychol. 2013;1:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Saxton J, Morrow L, Eschman A, Archer G, Luther J, Zuccolotto A. Computer assessment of mild cognitive impairment. Postgrad Med. 2009;121:177‐185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Barnett JH, Blackwell AD, Sahakian BJ, Robbins TW. The paired associates learning (PAL) test: 30 years of CANTAB translational neuroscience from laboratory to bedside in dementia research. Curr Top Behav Neurosci. 2016;28:449‐474. [DOI] [PubMed] [Google Scholar]
- 64. Staffaroni AM, Tsoy E, Taylor J, Boxer AL, Possin KL. Digital Cognitive Assessments for Dementia: digital assessments may enhance the efficiency of evaluations in neurology and other clinics. Pract Neurol (Fort Wash Pa). 2020;2020:24‐45. [PMC free article] [PubMed] [Google Scholar]
- 65. Jack CR Jr, Bennett DA, Blennow K, et al. NIA‐AA research framework: toward a biological definition of Alzheimer's disease. Alzheimers Dement. 2018;14:535‐562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Hampel H, Cummings J, Blennow K, Gao P, Jack CR Jr, Vergallo A. Developing the ATX(N) classification for use across the Alzheimer disease continuum. Nat Rev Neurol. 2021;17:580‐589. [DOI] [PubMed] [Google Scholar]
- 67. Leuzy A, Mattsson‐Carlgren N, Palmqvist S, Janelidze S, Dage JL, Hansson O. Blood‐based biomarkers for Alzheimer's disease. EMBO Mol Med. 2022;14:e14408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Varesi A, Carrara A, Pires VG, et al. Blood‐Based biomarkers for Alzheimer's disease diagnosis and progression: an overview. Cells. 2022;11:1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Stevenson‐Hoare J, Heslegrave A, Leonenko G, et al. Plasma biomarkers and genetics in the diagnosis and prediction of Alzheimer's disease. Brain. 2023;146:690‐699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Shi L, Buckley NJ, Bos I, et al. Plasma proteomic biomarkers relating to Alzheimer's disease: a Meta‐analysis based on our own studies. Front Aging Neurosci. 2021;13:712545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Jiang Y, Zhou X, Ip FC, et al. Large‐scale plasma proteomic profiling identifies a high‐performance biomarker panel for Alzheimer's disease screening and staging. Alzheimers Dement. 2022;18:88‐102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information