

Abstract
INTRODUCTION
Recent data suggest that distinct prion‐like amyloid beta and tau strains are associated with rapidly progressive Alzheimer's disease (rpAD). The role of genetic factors in rpAD is largely unknown.
METHODS
Previously known AD risk loci were examined in rpAD cases. Genome‐wide association studies (GWAS) were performed to identify variants that influence rpAD.
RESULTS
We identified 115 pathology‐confirmed rpAD cases and 193 clinical rpAD cases, 80% and 69% were of non‐Hispanic European ancestry. Compared to the clinical cohort, pathology‐confirmed rpAD had higher frequencies of apolipoprotein E (APOE) ε4 and rare missense variants in AD risk genes. A novel genome‐wide significant locus (P < 5×10−8) was observed for clinical rpAD on chromosome 21 (rs2832546); 102 loci showed suggestive associations with pathology‐confirmed rpAD (P < 1×10−5).
DISCUSSION
rpAD constitutes an extreme subtype of AD with distinct features. GWAS found previously known and novel loci associated with rpAD.
Highlights
Rapidly progressive Alzheimer's disease (rpAD) was defined with different criteria.
Whole genome sequencing identified rare missense variants in rpAD.
Novel variants were identified for clinical rpAD on chromosome 21.
Keywords: Alzheimer's disease, genetic risk, rapid progression
1. BACKGROUND
Alzheimer's disease (AD) is the most common cause of dementia. Typically, AD is characterized by slowly progressive deficits in behavioral and cognitive functions; however, there can be considerable variation in clinical phenotype and disease progression across the AD population. 1 , 2 One extreme subtype of AD is rapidly progressive AD (rpAD), which demonstrates a very rapid progression of dementia and/or a shortened lifespan. 3 , 4 , 5 , 6 Currently, there is no consensus on the definition of rpAD. In previous studies, the rapid progression was either described by the rate of cognitive decline or by the length of survival. 7 , 8 , 9 Depending on which definition is used, ≈ 10% to 30% of AD cases are classified as rpAD. 5
rpAD cases can often be identified through post mortem examinations conducted at prion disease surveillance centers. Prion diseases are caused by the accumulation of misfolded prion protein (PrPSc) in the brain. 10 The most common form of human prion disease is sporadic Creutzfeldt–Jakob disease (sCJD). Typical clinical symptoms of sCJD include rapidly progressive dementia, cerebellar ataxia, myoclonus, and a short disease duration of 5 to 6 months. 11 , 12 Given the partial overlap in clinical presentation, the early distinction between rpAD and prion disease can be challenging. Moreover, similarly elevated cerebrospinal fluid (CSF) biomarkers of total tau and 14‐3‐3 proteins occur in both disorders. 13 , 14 Due to the inconsistent definition of rpAD and difficulties distinguishing it from other types of rapidly progressive dementia, 6 , 15 , 16 individuals with rpAD can be hard to identify for clinical management and research studies.
Factors responsible for the heterogeneity in progression rate of AD are poorly understood, and it is possible that different pathophysiology, genetic background, medical and psychiatric comorbidity, or other factors are involved. Recent data comparing the neuropathology of typical AD (tAD; having a relatively slow progression) to rpAD found no differentiating patterns in the morphology of neurofibrillary tangles and amyloid plaques, nor their distribution in different anatomic areas. 4 Emerging findings indicate that a unique molecular signature involving the conformational characteristics and size distribution of amyloid beta (Aβ), aggregates of misfolded 4R‐Tau protein, 4 , 17 , 18 , 19 , 20 , 21 , 22 and their interactomes 23 , 24 might differentiate rpAD from tAD and suggest prion‐like propagation mechanism.
Although genetic variation has a significant impact on the risk of developing AD, its role in rpAD has not been extensively examined. Genome‐wide association studies (GWAS) of late‐onset AD have revealed at least 70 loci, among which apolipoprotein E (APOE) ε4 is the dominating major risk factor. 25 , 26 , 27 Conversely, previous studies for rpAD did not find convincing evidence of an effect of the APOE ε4 allele on rapid disease progression. 3 , 4 , 5 , 28 , 29 Sherva et al. 30 conducted a GWAS on the rate of cognitive decline in AD patients and found several suggestive genes in neural development, apoptosis, memory, and inflammatory pathways. However, this study was not designed to compare rpAD to tAD. There is a lack of knowledge on genetic factors contributing specifically to rapid disease progression or different formations of clinicopathological phenotypes in AD. Moreover, previous studies only relied on cognitive function or survival to define rpAD; little is known about rpAD cases identified through pathological evidence and whether they differ from clinically defined rpAD. By analyzing data from the National Prion Disease Pathology Surveillance Center (NPDPSC) and the National Alzheimer's Coordinating Center (NACC), we constructed two data sets of rpAD cases based on either pathological findings or cognitive measurements. This gives us a unique opportunity to gain a more comprehensive understanding of rpAD. The primary goal of this study was to identify genetic variations that contribute to the risk for rpAD. By comparing rpAD cases to individuals with tAD and to individuals with normal cognition, our findings demonstrate genetic variants and genes associated with different AD phenotypes.
RESEARCH IN CONTEXT
Systematic review: The authors conducted a literature review using PubMed. Although genetics play an important role in Alzheimer's disease (AD), the genetic factors that affect AD progression have not been extensively studied. Relevant publications are cited properly.
Interpretation: The authors found no strong effect of apolipoprotein E (APOE) ε4 on rapidly progressive AD (rpAD). Individuals with pathology‐confirmed rpAD had a higher frequency of rare missense variants in known AD risk genes than the general population. Most importantly, this study revealed novel genetic variants associated with clinical rpAD on chromosome 21. These findings may inform the design of future studies that seek to identify genetic factors and treatments for rpAD.
Future directions: A larger sample size of rpAD is needed to validate and expand the findings in this study. Future research is necessary to determine the regulatory functions of the identified loci on chromosome 21.
2. METHODS
2.1. Study population
2.1.1. NPDPSC pathology‐confirmed rpAD (NPDPSC rpAD)
The pathologically confirmed rpAD data set was identified from individuals examined at NPDPSC at Case Western Reserve University from 2009 to 2017. The inclusion and exclusion criteria for NPDPSC rpAD have been previously described. 4 Briefly, all cases were referred with rapidly progressive dementia resembling prion disease; neuropathology examinations, immunohistochemistry, molecular typing of the PrPSc protein, sequencing of the prion protein gene (PRNP), and medical records review to rule out sporadic and familial prion diseases, other dementias, and neurologic comorbidities were performed. After carefully assessing the decline in cognitive testing and duration, individuals with neuropathology and immunohistochemistry evidence of unequivocal classification as sporadic AD using the National Institute on Aging–Alzheimer's Association (NIA‐AA) criteria 31 were included in the analysis.
2.1.2. NACC clinical rpAD (NACC rpAD)
The data from participants enrolled in Alzheimer's Disease Research Centers is deposited into the NACC database. Data from 2005 to 2021 were screened for clinically defined rpAD. We analyzed the longitudinal Uniform Data Set (UDS) data 32 , 33 to identify eligible NACC rpAD cases by diagnosis and cognitive measurements at each visit. The criteria for NACC rpAD include: (1) carrying a diagnosis of AD at baseline or incident AD at a follow‐up visit according to the NIA‐AA criteria; 31 (2) rapid decline in cognitive function, defined by a decrease in Mini–Mental State Examination (MMSE) ≥ 6 points per year 5 , 34 and/or an increase in Clinical Dementia Rating (CDR) global score from ≤0.5–2 or 3 within 3 years; 35 (3) for deceased individuals, survival since the onset of symptoms ≤ 3 years. 5
2.1.3. NACC tAD and normal controls
We formed two groups of individuals as controls from those who contributed more than five visits to NACC. Individuals were classified as having tAD if they presented with: (1) a diagnosis of AD at baseline or incident AD on follow‐up visits; (2) a slow decline in cognitive function, characterized by a drop of MMSE score < 6 points per year and/or CDR global score < 2; (3) more than 5 years of disease duration since the onset of symptoms for individuals who were deceased. The group of normal controls consisted of individuals who did not receive any dementia‐related diagnosis and had relatively intact cognitive function throughout NACC visits (MMSE ≥ 24 and/or CDR global rating ≤ 0.5).
If available in NACC, additional information from the Neuropathology Data Set was also reviewed. Among a small subset of NACC rpAD (n = 35) and NACC tAD (n = 142) individuals who went to the autopsy, 97.7% of them (34 NACC rpAD and 139 NACC tAD) had neuropathological changes of AD determined by the Aβ deposition, neurofibrillary tangles, and neuritic plaques. 36 The four individuals with inconsistent neuropathology were dropped from the analysis.
2.2. Genotyping, sequencing, and quality control
DNA was extracted from the frozen brain tissue of pathology‐confirmed AD cases from the NPDPSC. Whole genome sequencing (WGS) was performed on the Illumina platform, using the Isaac alignment 37 and Strelka variant caller for single nucleotide variants and small indels. 38 For NACC samples, DNA was extracted from blood or saliva for WGS. Sequencing data were obtained from the Alzheimer's Disease Sequencing Project (ADSP) and followed the ADSP workflow. 39 In brief, WGS data were processed using the single nucleotide polymorphism (SNP)/indel variant calling pipeline and data management tool (VCPA), 40 then followed ADSP quality control (QC) to generate project‐level VCF files. 41
Before merging, samples and sequencing data from NPDPSC and NACC underwent independent downstream QC, and a second round of QC was performed on the combined data. Using PLINK 1.9, only biallelic variants were retained for QC, including steps to filter samples and SNPs based on sex discrepancy, genotyping rate, heterozygosity rate, sample relatedness, minor allele frequency (MAF), and Hardy–Weinberg equilibrium (Figure S1A in supporting information). In the NPDPSC rpAD data set, 114 samples and 6,298,451 variants passed initial QC, while in the NACC cohort, 2901 samples and 4,807,186 variants passed QC. After the second round of QC on the merged data, a total of 4,466,093 variants and 2964 samples were kept for GWAS analyses, including 114 NPDPSC rpAD cases, 2162 NACC normal controls, 507 NACC tAD cases, and 181 NACC rpAD cases (Figure S1B). The samples that passed QC were from individuals with a diverse race/ethnicity background, including those reported to have a non‐Hispanic European (n = 2149), African American (n = 788), Asian (n = 4), American Indian/Alaska Native (n = 3), and other (n = 3) ancestry. When the frequency of genetic variants differs among subpopulations, combining individuals with different ancestries may lead to spurious associations. However, our sample size was too small for ancestry‐specific GWAS, so principal component analysis (PCA) was used to account for population stratification. We performed linkage disequilibrium (LD) pruning to generate a set of independent genetic variants with an LD threshold of r 2 < 0.2 for PCA. Pruned SNPs were used to create principal components (PCs). We generated the scree plot and examined the elbow point of the curve, where the slope begins to level off, to determine the optimum number of PCs for GWAS analyses. We calculated the cumulative sum of the variance to check if the PCs retained explained the majority of the variation.
2.3. Statistical analysis
In the comparison of demographic and clinical features between different groups, P values were calculated by the Kruskal–Wallis or Fisher exact test. Spaghetti plots were used to visualize the individual trajectory of cognitive function measured by MMSE and CDR Sum of Boxes scores. The non‐genetic analyses were conducted in R (version 4.1.2).
2.3.1. Examination of known AD risk genes
We performed a thorough examination of rare variants (MAF < 0.01) within the region of known early‐onset AD risk genes, including Presenilin 1 (PSEN1), Presenilin 2 (PSEN2), and amyloid precursor protein (APP). The frequency of rare variants in our samples was compared to the population frequency published in the Genome Aggregation Database (gnomAD), 42 1000 Genomes Project, 43 and Trans‐Omics for Precision Medicine (TOPMed) 44 database. Additionally, we determined the APOE ε2/ε3/ε4 polymorphisms by two SNPs at rs429358 and rs7412, yielding six genotypes (ε2/ε2, ε2/ε3, ε2/ε4, ε3/ε3, ε3/ε4, and ε4/ε4). 45 The APOE genotypes from WGS data were cross‐checked with genotypes reported from the source data.
2.3.2. Association tests
Individuals included in the GWAS were those aged 60 to 90 years old, based on age at visit for NACC normal controls and age of onset for individuals with an AD diagnosis in NPDPSC and NACC. Figure 1 shows the workflow to ascertain different AD subtypes and individuals in each association test. Analyses 1 through 3 compared pathology‐confirmed rpAD in NPDPSC to normal controls, typical AD, or clinical rpAD in NACC. Analyses 4 through 6 were pairwise comparisons between normal controls, typical AD, and clinical rpAD in the NACC database. The goal of the GWAS was to identify genetic variants that influence pathology‐confirmed or clinical rpAD compared to controls. For each case in the association test, we randomly selected three age‐ and sex‐matched controls. Given that the NACC normal and tAD groups had enough individuals for matching, we used different random seeds in R to generate three sets of normal and tAD individuals as controls to ensure the consistency of GWAS results. For Analyses 1 and 2 and 4 through 6, we conducted GWAS using logistic regression models, adjusting for the first three PCs. In Analysis 3, due to insufficient clinical rpAD cases in NACC, we compared the NPDPSC rpAD to NACC rpAD in a logistic regression model adjusting for age, sex, and the first three PCs. Genetic variants were considered genome‐wide suggestive with a P value ≤ 1 × 10−5 and genome‐wide significant with a P value ≤ 5 × 10−8. Quantile–quantile (Q‐Q) plots were used to compare the genome‐wide distribution of the test statistic to the expected null distribution. Lambda (λ), defined as the median of the resulting chi‐squared test statistics divided by the expected median of the chi‐squared distribution, was calculated to estimate inflation in each GWAS. Association tests were performed in PLINK 1.9.
FIGURE 1.
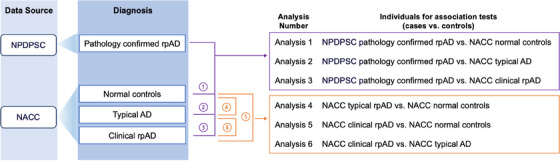
Ascertainment of AD subtypes and analysis workflow for genetic association tests. AD, Alzheimer's disease; NACC, National Alzheimer's Coordinating Center; NPDPSC, National Prion Disease Pathology Surveillance Center; rpAD, rapidly progressive Alzheimer's disease
2.3.3. Post‐GWAS analysis
By searching the online Genotype–Tissue Expression (GTEx) project 46 database, we investigated the regulatory potential for the most significant SNPs from the association tests to see whether they represent expression/splicing quantitative trait loci (eQTLs/sQTLs). While GTEx provides data on gene expression in 54 tissues, we emphasized genes expressed in the brain.
To summarize, for SNP associations at the gene level, we conducted gene‐based analyses using the Multimarker Analysis of GenoMic Annotation (MAGMA, v.1.08) tool in FUMA, accounting for the number of variants and LD between them. 47 , 48 The LD between SNPs was estimated with the European reference panel in 1000 Genomes phase 3. Positional mapping was used to map SNPs to genes based on their physical location, with 10 kb windows as the default. MAGMA runs an SNP‐wise model to obtain gene‐based P values. The significance level was corrected by the Bonferroni method.
3. RESULTS
3.1. Demographics and clinical features
We identified 115 pathology‐confirmed cases of rpAD in NPDPSC. Of the 2931 subjects in NACC with WGS data available, 2213 individuals had normal cognition, 525 had tAD, and 193 had clinical rpAD (Table 1). In the NPDPSC data set, most subjects were female with reported non‐Hispanic European (NHE) ancestry. There was more diversity in race/ethnicity among NACC individuals; ≈ 70.5% had an NHE ancestry, and 25.6% reported African American ancestry. Only 1.3% of all individuals reported being Hispanic. NPDPSC rpAD cases had a considerably younger age of onset (71.0 ± 10.8, P < 0.001) and age at death (72.4 ± 10.1, P < 0.001), as well as the shortest median survival of 0.6 years (interquartile range: 0.17–1.4, P < 0.001) than the deceased individuals with tAD or clinical rpAD in NACC. Compared to NACC normal controls, NACC rpAD cases had significantly lower education (14.5 ± 3.4, P < 0.001). The average age of onset was 69.3 among non‐deceased NACC rpAD cases and 80.7 among deceased NACC rpAD cases. Among the deceased individuals in NACC, clinical rpAD had a significantly younger age at death (83.2 ± 11.4) compared to tAD (87.5 ± 8.2, P = 0.004) and a shorter median survival of 3 years than tAD (P < 0.05). While examining the progression of cognitive measures in NACC, the MMSE score among clinical rpAD cases decreased by 3.37 ± 2.71 points/year, faster than the annual rate of decline in tAD (0.77 ± 0.94) and normal controls (0.03 ± 0.57). Meanwhile, clinical rpAD cases had an increase of 2.44 ± 1.51 points/year in the CDR Sum of Boxes scores, higher than the annual rate of increase in tAD (1.02 ± 0.70) and normal controls (0.00 ± 0.08). The pattern of changes in cognitive functions between NACC groups is shown in Figure 2.
TABLE 1.
Demographic and clinical characteristics.
| NPDPSC | NACC | ||||
|---|---|---|---|---|---|
| Pathology‐confirmed rpAD | Normal controls | tAD | Clinical rpAD | ||
| n = 115 | n = 2213 | n = 525 | n = 193 | P | |
| Male (%) | 57 (49.6) | 700 (31.6) | 234 (44.6) | 79 (40.9) | <0.001 |
| Race (%) | <0.001 * | ||||
| Non‐Hispanic European | 92 (80.0) | 1522 (68.8) | 412 (78.5) | 133 (69.3) | |
| African American | 2 (1.7) | 686 (31.0) | 112 (21.3) | 52 (27.1) | |
| Asian | 0 (0.0) | 2 (0.1) | 1 (0.2) | 6 (3.1) | |
| American Indian/Alaska Native | 0 (0.0) | 3 (0.1) | 0 (0.0) | 0 (0.0) | |
| Other | 2 (1.7) | 0 (0.0) | 0 (0.0) | 1 (0.5) | |
| Unknown | 17 (14.8) | 0 (0.0) | 0 (0.0) | 0 (0.0) | |
| Hispanic (%) | 2 (1.7) | 24 (1.1) | 9 (1.7) | 5 (2.6) | 0.159 * |
| Years of education, mean (SD) | – | 15.8 (3.0) | 15.4 (3.0) | 14.5 (3.4) | <0.001 |
| Age of onset, mean (SD) | |||||
| Not deceased | – | – | 73.7 (11.1) | 69.3 (11.6) | <0.001 |
| Deceased | 71.0 (10.8) | – | 76.9 (8.8) | 80.7 (11.6) | <0.001 |
| Age at death, mean (SD) | 72.4 (10.1) | 84.3 (7.4) | 87.5 (8.3) | 83.2 (11.4) | <0.001 |
| Survival in years, median [IQR] ** | 0.6 [0.17–1.4] | – | 10.0 [8.0–13.0] | 3.0 [2.0–3.0] | <0.001 |
Abbreviations: IQR, interquartile range; NACC, National Alzheimer's Coordinating Center; NPDPSC, National Prion Disease Pathology Surveillance Center; rpAD, rapidly progressive Alzheimer's disease; SD, standard deviation; tAD, typical Alzheimer's disease.
P value was obtained from the Fishes exact test.
In NACC, survival was defined as the disease duration from the onset of symptoms until death.
FIGURE 2.
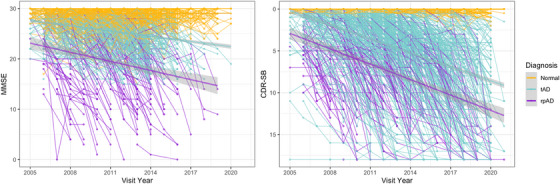
The progression of cognitive function for NACC individuals. Longitudinal changes of MMSE and CDR Sum of Boxes scores are colored in yellow for individuals with normal cognition, blue for tAD, and purple for rpAD cases. CDR‐SB, Clinical Dementia Rating Sum of Boxes scores; MMSE, Mini–Mental State Examination; NACC, National Alzheimer's Coordinating Center; rpAD, rapidly progressive Alzheimer's disease; tAD, typical Alzheimer's disease
3.2. APOE status and rare variants
In the NPDPSC rpAD data set, the allele frequency of APOE ε2 was 4.8%, ε3 was 63.0%, and ε4 was 32.2%. Among clinical rpAD cases in NACC, the frequency of ε2, ε3, and ε4 was 2.1%, 73.0%, and 24.9%, respectively. Table S1 in supporting information displays a detailed breakdown of APOE allele frequencies and genotypes by diagnostic and ancestry group.
From the NPDPSC and NACC rpAD WGS data, we identified two rare missense variants (rs63750082 and rs17125721) in the PSEN1 gene, one rare missense variant (rs140501902) in the PSEN2 gene, and two rare missense variants (rs202074408 and rs202198008) in the APP gene (Table 2). Within the NPDPSC rpAD cohort, 11 people carried a rare missense variant in PSEN1 (n = 9) or APP (n = 2). Across all three NACC diagnostic groups, a total of 117 individuals carried a rare missense variant, mostly in the PSEN1 gene (n = 88), followed by PSEN2 (n = 16), then APP (n = 13). Compared to the publicly available MAF, the MAF of these rare variants was often increased in the NPDPSC rpAD. Carriers with NPDPSC rpAD generally had an earlier age of onset and age at death compared to NACC rpAD (Table S2 in supporting information).
TABLE 2.
Rare missense variants in PSEN1, PSEN2, and APP gene.
| PSEN1 | PSEN2 | APP | |||
|---|---|---|---|---|---|
| Variant | rs63750082 | rs17125721 | rs140501902 | rs202074408 | rs202198008 |
| chr14: 73192712:G > C | chr14: 73206470:A > G | chr1: 226883774:C > T | chr21: 26000152:G > A | chr21: 26021879: T > A | |
| p.G206A | p.E318G | p.R71W | – | – | |
| Number of carriers, n (MAF) | |||||
| NPDPSC pathology confirmed rpAD | 2 (0.01304) | 7 (0.03043) | 0 (0) | 1 (0.00434) | 1 (0.00434) |
| Total in NACC | 3 (0.00051) | 85 (0.01450) | 16 (0.00273) | 0 (0) | 13 (0.00221) |
| NACC clinical rpAD | 2 (0.00045) | 6 (0.00136) | 2 (0.00045) | 0 (0) | 0 (0) |
| NACC tAD | 1 (0.00259) | 14 (0.03627) | 2 (0.00518) | 0 (0) | 2 (0.00518) |
| NACC normal controls | 0 (0) | 65 (0.06190) | 12 (0.01143) | 0 (0) | 11 (0.01143) |
| Population MAF * | |||||
| 0.00012 | 0.01497 | 0.00349 | 0.00001 | 0.00176 | |
Abbreviations: APP, amyloid precursor protein; MAF, minor allele frequency; NACC, National Alzheimer's Coordinating Center; NPDPSC, National Prion Disease Pathology Surveillance Center; PSEN1, presenilin 1; PSEN2, presenilin 2; rpAD, rapidly progressive Alzheimer's disease; tAD, typical Alzheimer's disease.
Population MAF was obtained from the GnomAD database, 1000 Genomes Project, or TOPMed database.
3.3. Association tests
Of the 2964 samples that passed two rounds of QC, we selected those from subjects aged 60 to 90 for association testing, which included 96 NPDPSC pathology‐confirmed rpAD cases, 2122 individuals with normal cognition, 452 tAD individuals, and 142 clinical rpAD cases in NACC. Using a 1:3 case–control ratio for matching, we selected three sets of controls to compare to NPDPSC rpAD or NACC rpAD. The exact number of individuals in each comparison group is shown in Table S3 in supporting information.
No SNPs demonstrated genome‐wide significant associations with NPDPSC pathology‐confirmed rpAD compared to individuals with normal cognition in NACC (Analysis 1, Figure 3A). From regression models using three different sets of normal controls, we found 51 genome‐wide suggestive loci with P < 1 × 10−5 (Table S4.1 in supporting information). Among these, five SNPs were repeatedly observed in two comparison groups, three on chromosome 19 overlapping with the APOE (rs429358) and AC011481.3 (rs483082 and rs438811) genes, and two on chromosome 1 close to the High Mobility Group Box 1 Pseudogene 26 (HMGB1P26) and Long Intergenic Non‐Protein Coding RNA 1682 (LINC01682) genes (rs36017930 and rs67678913). In the comparisons between NPDPSC rpAD and NACC tAD (Analysis 2), 47 SNPs showed genome‐wide suggestive associations (Figure 3B). Four of these SNPs (rs7116599, rs7129262, rs4753706, and rs10831453) were identified using two different sets of tAD individuals as controls and are located near the Myotubularin Related Protein 2 (MTMR2) and AP000870.1 genes on chromosome 11 (Table S4.2). Because there were only 142 individuals with clinical rpAD in NACC, we applied a logistic regression model adjusting for age, sex, and the first three PCs (Analysis 3). Five SNPs showed genome‐wide suggestive association with NPDPSC rpAD (Figure 3C), including rs6506688 overlapping with the RAB31 gene on chromosome 18, and four SNPs (rs874828, rs13056658, rs13054402, and rs13056139) on chromosome 22 overlapping with the Cadherin EGF LAG Seven‐Pass G‐Type Receptor 1 (CELSR1) gene (Table S4.3). Although P values for these loci in Analyses 1 through 3 are only suggestive across all three sets of controls, the effect sizes trended in the same direction. In these seven association tests comparing NPDPSC rpAD to NACC diagnostic groups, the genomic inflation factor λ ranged between 0.99 and 1.01 (Figure S2). We conducted sensitivity analyses with both six PCs and eight PCs to assess the robustness of the results. The main findings from Analyses 1 through 3 remained unchanged.
FIGURE 3.
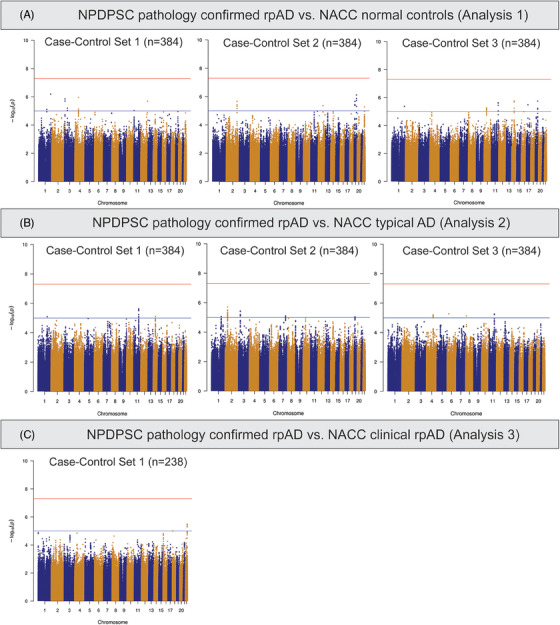
Manhattan plots of genome‐wide association tests comparing NPDPSC pathology‐confirmed rpAD cases to NACC diagnostic groups. NACC, National Alzheimer's Coordinating Center; NPDPSC, National Prion Disease Pathology Surveillance Center; rpAD, rapidly progressive Alzheimer's disease
We also conducted GWAS analyses using just the NACC subjects. Comparing individuals with tAD to those with normal cognition (Analysis 4), rs429358 in the APOE gene showed a genome‐wide significant association with NACC tAD across all three sets of normal controls (Figure 4A). Meanwhile, three SNPs (rs7670598, rs13161859, and rs7338612 on chromosomes 4, 5, and 13, respectively) consistently showed suggestive associations with NACC tAD, and 37 more suggestive SNPs were observed in at least one GWAS model (Table S5.1 in supporting information). In the comparisons between clinical rpAD and normal controls in NACC (Analysis 5), rs2832546 on chromosome 21 had a genome‐wide significant association with NACC clinical rpAD (Figure 4B), along with a set of suggestive loci (n = 81) on the same chromosome that associated with the increased risk for NACC clinical rpAD (Table S5.2). This SNP remained significant while comparing NACC rpAD to NACC tAD (Analysis 6, Figure 4C). Additionally, on chromosome 21, we observed 6 genome‐wide significant loci and 94 suggestive loci for NACC rpAD across all GWAS analyses using different sets of tAD individuals as controls (Table S5.3). In the association tests comparing the NACC diagnostic groups, λ ranged from 0.99 to 1.01 (Figure S3 in supporting information). Sensitivity analyses with six PCs in Analyses 4 through 6 found consistent results. The most significant SNP on chromosome 21, rs2832546, is 52 kbp downstream of the long intergenic non‐coding RNA gene AF096876.1 and ≈ 97.7 kbp upstream of the Glutamate Ionotropic Receptor Kainate Type Subunit 1 (GRIK1) gene (Table S5.2‐S5.3). From a further examination of the region containing rs2832546, variants in the region are in high LD (r 2 ≥ 0.8) with each other and can be mapped to the AF165147.1 gene (Figure 5). No significant eQTLs or sQTLs were found in the GTEx database for rs2832546.
FIGURE 4.
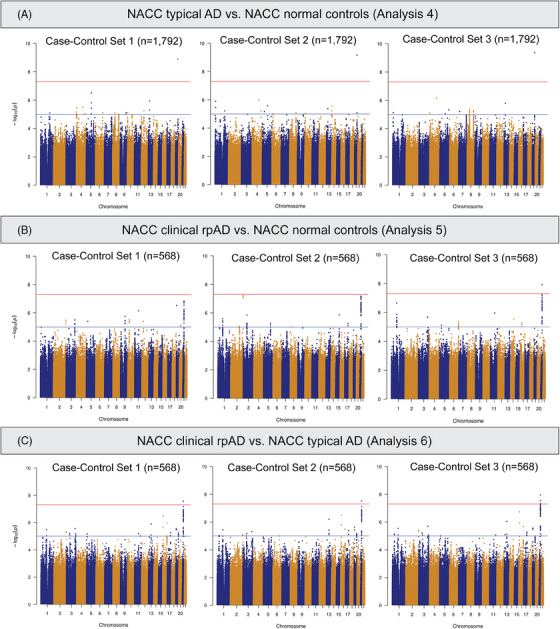
Manhattan plots of genome‐wide association tests comparing NACC diagnostic groups. rpAD, rapidly progressive Alzheimer disease; NACC, National Alzheimer's Coordinating Center.
FIGURE 5.
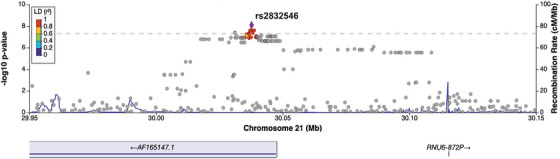
Regional plot for genome‐wide significant loci associated with NACC clinical rpAD. The lead genetic variant (rs2832546) was identified in the association tests comparing NACC clinical rpAD to normal or typical AD individuals. SNPs are color coded according to their LD with the lead SNP in the region. AD, Alzheimer's disease; LD, linkage disequilibrium; NACC, National Alzheimer's Coordinating Center; NPDPSC, National Prion Disease Pathology Surveillance Center; rpAD, rapidly progressive Alzheimer's disease; SNP, single nucleotide polymorphism
3.4. Gene‐based analysis
We performed a gene‐based analysis using the GWAS statistics derived from comparing clinical rpAD to tAD in NACC, as the lead SNP demonstrated the most significant association with NACC clinical rpAD in this comparison group. Because MAGMA can adjust for LD between SNPs, all variants included in the GWAS were used in the gene‐based analysis; 4,466,093 post‐QC variants were mapped to 8919 protein‐coding genes. The Eyes Shut Homolog (EYS), Zinc Finger Matrin–Type 4 (ZMAT4), and Tenascin N (TNN) genes were significantly associated with NACC clinical rpAD with Bonferroni‐adjusted P < 5.61E‐6 (Figure 6).
FIGURE 6.
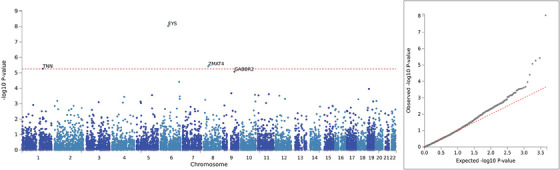
Gene‐based analysis comparing clinical rpAD and typical AD in NACC individuals. AD, Alzheimer's disease; EYS, The Eyes Shut Homolog; GABBR2, Gamma–Aminobutyric Acid Type B Receptor Subunit 2; NACC, National Alzheimer's Coordinating Center; rpAD, rapidly progressive Alzheimer's disease; TNN, Tenascin N; ZMAT4, Zinc Finger Matrin–Type 4
4. DISCUSSION
There has been an enormous interest in understanding the heterogenous clinical phenotypes and disease progression in AD. Emerging evidence suggests that different populations of amyloid and tau deposits may explain some of the variability in disease state. 49 , 50 We focused on an extreme subgroup of AD that experienced a rapid disease progression and/or short survival. The overarching goal was to decipher the genetic architecture of rpAD and identify genetic variation that might contribute to its pathophysiology.
Clinically, rpAD exhibits distinct features compared to tAD. In most cases of late onset AD, progression is gradual and slow. Individuals > age 65 have an average survival of 4 to 8 years after diagnosis, while some live up to 20 years. 51 We defined pathology‐confirmed rpAD in NPDPSC and clinical rpAD in NACC as having significantly shorter median survival, with 0.6 years in NPDPSC rpAD and 3 years in the NACC rpAD. This definition aligns with the median survival of 10 months observed in 96 rpAD cases collected independently at prion centers in Spain, France, Germany, and Japan. 5 , 29 , 52 Meanwhile, both NPDPSC and NACC rpAD cases present with noticeably younger age of onset and earlier age at death than individuals with tAD (Table 1). These findings support the argument that rpAD is a distinct subtype of AD. Finally, the different conformers (strains) of Aβ and misfolded tau protein interacting with distinct sets of proteins in rpAD suggest involvement of variable genetic factors. 4 , 19 , 20 , 21 , 23 , 24
To understand the effect of genetic factors on rpAD, we first examined the APOE status in our study. In an AD population with NHE ancestry, the allele frequency of APOE ε4 was ≈ 40%. 53 We observed lower APOE ε4 NHE allele frequencies (33.2% in NPDPSC rpAD and 19.5% in NACC rpAD). Previous studies of pathology‐confirmed rpAD cases reported NHE APOE ε4 allele frequencies ranging from 22.2% to 38%. 3 , 29 , 54 The role of APOE in predicting the rate of disease progression is an ongoing debate as Cosentino et al. 55 found individuals with mild AD declined rapidly if APOE ε4 was present, whereas others observed a weak to no effect of APOE ε4 on rpAD. 28 , 29 These conflicting results suggest there might be other genetic factors influencing rapid disease progression in AD.
Overall, we found five rare missense coding variants in known AD risk genes with a higher frequency in NPDPSC rpAD cases than observed in the general population. The G206A mutation (rs63750082) in PSEN1 was found in two NPDPSC rpAD cases, two NACC rpAD cases, and one NACC tAD. The G206A mutation is a pathogenic variant 56 and associated with variable age at onset. 57 One of the NPDPSC rpAD cases was a homozygous carrier of G206A with a ‐reported Hispanic ethnicity. This person had an early onset at 42 and a duration of < 1 year. The other rare coding variant in PSEN1, E318G (rs17125721), is associated with high levels of total tau, phosphorylated tau, and a faster cognitive decline under the influence of APOE ε4. 58 , 59 There is not yet strong evidence for the pathogenicity of the rare variants in PSEN2 and APP.
Because rpAD was ascertained using different criteria in the NPDPSC and NACC cohorts, we performed independent GWAS analysis of these rpAD cases and found several genes potentially influencing pathology‐confirmed or clinical rpAD. This study highlights genome‐wide significant GWAS findings on chromosome 21 comparing clinical rpAD to tAD or normal controls in NACC (Analyses 5 and 6, Figure 4B, C). The most significant SNP, rs2832546, is an intergenic variant close to the protein‐coding gene GRIK1. GRIK1 encodes the kainate family of glutamate receptors that function as a ligand‐gated ion channel. The downregulation of GRIK1 is consistent with the hypothesis that excess Aβ may eventually suppress long‐term potentiation in AD. 60 rs2832546 can be mapped to the lncRNA gene AF165147.1 on the regional plot (Figure 5). Because lncRNAs can exhibit a wide range of regulatory functions, SNPs within lncRNA‐containing loci might interfere with their biological function and contribute to AD pathology. 61 Unfortunately, no significant eQTLs or sQTLs were found for this SNP in the GTEx database, and there is a lack of information in public databases about the corresponding lncRNA and its expression pattern.
Due to discordant patterns of LD, GWAS at the SNP level may produce less consistent results in diverse populations. 62 In our GWAS analysis, PCs adjusted for population stratification; however, there could be a residual impact of different MAFs and LD patterns across populations. An alternative would be to perform ancestry‐specific analyses; however, our limited sample sizes make that untenable. Compared to single SNP association tests, gene‐based analysis is advantageous because genes are more robust across populations, and it provides greater power by evaluating the aggregated effect of multiple SNPs than that of individual SNPs. Gene‐based analyses identified three genes that were significantly associated with NACC rpAD. No previous studies have linked EYS or TNN to rpAD. The product of EYS is expressed in the photoreceptor layer of the retina, and mutation in EYS is a major cause of recessive retinitis pigmentosa. 63 Meanwhile, TNN encodes the gene that plays a major role in developing skeletal and cardiac muscles. ZMAT4 is involved in myopia development. Steffens et al. 64 reported a synonymous SNP (rs17851751) in ZMAT4 associated with cognitive decline in late‐life depression. Although the P value in the gene‐based analysis for the GABBR2 gene is slightly below the statistical significance threshold, GABBR2 encodes the primary inhibitory neurotransmitter in the human brain. Rare variants in GABBR2 might affect synaptic functioning, 65 and the downregulation of GABA receptors is associated with AD by potentially disrupting the excitation/inhibition balance. 66
A few limitations should be noted in this work. The relevant clinical data in NPDPSC were very limited so we could not determine whether environmental or social conditions might influence the differences in clinical characteristics (Table 1). Although NACC is designed to collect longitudinal clinical measurements, the quality of some variables, such as age of onset, may suffer from recall bias and inaccurate self‐report. Bias may also exist in NACC data due to the loss of follow‐up for unclear reasons.
We used different criteria to define rpAD in two national cohorts, which allowed a larger sample size and a unique opportunity to assess rpAD defined pathologically versus clinically. However, NPDPSC primarily enrolls decreased individuals with a rapid progression of dementia and NACC encourages collecting longitudinal data, but neither cohort represents the complete rpAD population. rpAD in these two cohorts may represent biologically different AD mechanisms. Using conformation‐sensitive immunoassays and fluorescent ligands, our earlier findings indicate a major conformational diversity of Aβ42 accumulating in the neocortex, and at least three distinctly misfolded 4R Tau conformers associated with pathology‐confirmed rpAD in NPDPSC cohorts. 4 , 17 , 19 , 20 , 21 Additionally, we observed a higher APOE ε4 allele frequency in NPDPSC pathology‐confirmed rpAD cases, which might contribute to different biological mechanisms.
Last, compared to genetic studies of typical AD, the sample size of rpAD is small for GWAS and thus will have limited power. For example, while comparing NPDPSC rpAD to NACC normal controls (Analysis 1), rs429358 in APOE was suggestively significant (highest odds ratio [OR] = 2.68, P = 4.9 × 10−6). In a meta‐analysis, the OR for AD in individuals carrying one APOE ε4 allele is 3.68 (95% confidence interval: 3.30–4.11). 67 With the weaker effect size of 2.68 that we found, a minimum of 250 NPDPSC rpAD cases would be needed for this variant to be genome‐wide significant. If limited to our current sample size, rs429358 would need at least an OR of 5 to reach genome‐wide significance. We tried to minimize this concern by using a case–control ratio of 1:3 for each association test, but a larger sample size is needed to validate our findings.
Despite these issues, our findings are interesting for several reasons. First, the WGS data empowered the identification of rare missense variants that were not detected by the genotyping array. The presence of rare missense variants may play a role in the variability of the age of onset and survival in rpAD. The significance of the top SNPs identified in each GWAS was commensurate with a consistent trend of effect sizes and directions across different case–control sets. The genome‐wide significant SNPs on chromosome 21 were novel variants associated with clinical rpAD in NACC. Future investigations are needed to understand the regulatory functions of these SNPs.
In conclusion, rpAD cases from two different cohorts were identified to understand an enduring puzzle of heterogenous disease progression in AD. Our analyses confirmed that rpAD is a distinct subtype of AD phenotypically and has started to shed light on possible genetic risks associated with variable rates of progression in AD.
CONFLICT OF INTEREST STATEMENT
Ping Wang, Audrey Lynn, Kristy Miskimen, Yeunjoo E. Song, Mark Cohen, Jiri G. Safar, Jonathan L. Haines: none. Thomas Wisniewski: Dr. Wisniewski is a scientific adviser to Alzamend Neuro Inc. and Amylonix, and serves on the Data Safety Monitoring Board (DSMB) for LIFE‐DSR Bio. Brian S. Appleby: Dr. Appleby has received research funding from CDC, NIH, Ionis, Alector, and the CJD Foundation. He has provided consultation to Ionis, Acadia, Sangamo, and Merck. He receives royalties from Wolters Kluwer. Author disclosures are available in the supporting information.
DATA AND CODE AVAILABILITY
Clinical data are available upon request through the National Alzheimer's Coordinating Center (https://naccdata.org/). Genetic data are available through the National Institute on Aging Genetics of Alzheimer's Disease Data Storage Site (NIAGADS; https://www.niagads.org/). The code used in the genetic analysis is available on GitHub at https://github.com/PingWang‐github/Genetic_Architecture_rpAD
CONSENT STATEMENT
All study procedures were approved by the institutional review boards at Case Western Reserve University and University Hospitals Cleveland Medical Centre in Cleveland, OH. Written informed consent was obtained from the participant or legal guardian. For the sake of confidentiality, clinical and genetic data were de‐identified according to the study protocol.
Supporting information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
ACKNOWLEDGMENTS
This work was supported by the Centers for Disease Control and Prevention (grant number UR8/CCU515004) and National Institutes of Health (grant numbers AG058267, AG032984, AG058654, AG060882, AG066512, and AG061797). The authors thank Drs. Will Bush and Nicholas Wheeler for their help in preparing the whole genome sequencing data for our analysis. This work used the High Performance Computing Resource in the Core Facility for Advanced Research Computing at Case Western Reserve University. The whole genome sequencing data was supported by the Alzheimer's Disease Sequencing Project (ADSP). The full acknowledgment statement for the Alzheimer's Disease Sequencing Project data can be found here: https://adsp.niagads.org/acknowledgment/. The NACC database is funded by NIA/NIH Grant U24 AG072122. NACC data are contributed by the NIA‐funded ADRCs: P30 AG062429 (PI James Brewer, MD, PhD), P30 AG066468 (PI Oscar Lopez, MD), P30 AG062421 (PI Bradley Hyman, MD, PhD), P30 AG066509 (PI Thomas Grabowski, MD), P30 AG066514 (PI Mary Sano, PhD), P30 AG066530 (PI Helena Chui, MD), P30 AG066507 (PI Marilyn Albert, PhD), P30 AG066444 (PI John Morris, MD), P30 AG066518 (PI Jeffrey Kaye, MD), P30 AG066512 (PI Thomas Wisniewski, MD), P30 AG066462 (PI Scott Small, MD), P30 AG072979 (PI David Wolk, MD), P30 AG072972 (PI Charles DeCarli, MD), P30 AG072976 (PI Andrew Saykin, PsyD), P30 AG072975 (PI David Bennett, MD), P30 AG072978 (PI Neil Kowall, MD), P30 AG072977 (PI Robert Vassar, PhD), P30 AG066519 (PI Frank LaFerla, PhD), P30 AG062677 (PI Ronald Petersen, MD, PhD), P30 AG079280 (PI Eric Reiman, MD), P30 AG062422 (PI Gil Rabinovici, MD), P30 AG066511 (PI Allan Levey, MD, PhD), P30 AG072946 (PI Linda Van Eldik, PhD), P30 AG062715 (PI Sanjay Asthana, MD, FRCP), P30 AG072973 (PI Russell Swerdlow, MD), P30 AG066506 (PI Todd Golde, MD, PhD), P30 AG066508 (PI Stephen Strittmatter, MD, PhD), P30 AG066515 (PI Victor Henderson, MD, MS), P30 AG072947 (PI Suzanne Craft, PhD), P30 AG072931 (PI Henry Paulson, MD, PhD), P30 AG066546 (PI Sudha Seshadri, MD), P20 AG068024 (PI Erik Roberson, MD, PhD), P20 AG068053 (PI Justin Miller, PhD), P20 AG068077 (PI Gary Rosenberg, MD), P20 AG068082 (PI Angela Jefferson, PhD), P30 AG072958 (PI Heather Whitson, MD), P30 AG072959 (PI James Leverenz, MD).
Wang P, Lynn A, Miskimen K, et al. Genome‐wide association studies identify novel loci in rapidly progressive Alzheimer's disease. Alzheimer's Dement. 2024;20:2034–2046. 10.1002/alz.13655
Ping Wang will handle correspondence at all stages of refereeing and publication, also post‐publication.
Contributor Information
Ping Wang, Email: pxw287@case.edu.
Jonathan L. Haines, Email: jlh213@case.edu.
REFERENCES
- 1. Cummings JL. Cognitive and behavioral heterogeneity in Alzheimer's disease: seeking the neurobiological basis. Neurobiol Aging. 2000;21(6):845‐861. doi: 10.1016/s0197-4580(00)00183-4 [DOI] [PubMed] [Google Scholar]
- 2. Komarova NL, Thalhauser CJ. High degree of heterogeneity in Alzheimer's disease progression patterns. PLoS Comput Biol. 2011;7(11):e1002251. doi: 10.1371/journal.pcbi.1002251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Abu‐Rumeileh S, Capellari S, Parchi P. Rapidly progressive Alzheimer's disease: contributions to clinical‐pathological definition and diagnosis. J Alzheimers Dis. 2018;63(3):887‐897. doi: 10.3233/jad-171181 [DOI] [PubMed] [Google Scholar]
- 4. Cohen ML, Kim C, Haldiman T, et al. Rapidly progressive Alzheimer's disease features distinct structures of amyloid‐β. Brain. 2015;138(4):1009‐1022. doi: 10.1093/brain/awv006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schmidt C, Wolff M, Weitz M, Bartlau T, Korth C, Zerr I. Rapidly progressive Alzheimer disease. Arch Neurol. 2011;68(9):1124‐1130. doi: 10.1001/archneurol.2011.189 [DOI] [PubMed] [Google Scholar]
- 6. Chitravas N, Jung RS, Kofskey DM, et al. Treatable neurological disorders misdiagnosed as Creutzfeldt‐Jakob disease. Ann Neurol. 2011;70(3):437‐444. doi: 10.1002/ana.22454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Soto ME, Andrieu S, Arbus C, et al. Rapid cognitive decline in Alzheimer's disease. Consensus paper. J Nutr Health Aging. 2008;12(10):703‐713. doi: 10.1007/BF03028618 [DOI] [PubMed] [Google Scholar]
- 8. Ito K, Ahadieh S, Corrigan B, French J, Fullerton T, Tensfeldt T. Disease progression meta‐analysis model in Alzheimer's disease. Alzheimers Dement. 2010;6(1):39‐53. doi: 10.1016/j.jalz.2009.05.665 [DOI] [PubMed] [Google Scholar]
- 9. Josephs KA, Ahlskog JE, Parisi JE, et al. Rapidly progressive neurodegenerative dementias. Arch Neurol. 2009;66(2):201‐207. doi: 10.1001/archneurol.2008.534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Puoti G, Bizzi A, Forloni G, Safar JG, Tagliavini F, Gambetti P. Sporadic human prion diseases: molecular insights and diagnosis. Lancet Neurol. 2012;11(7):618‐628. doi: 10.1016/s1474-4422(12)70063-7 [DOI] [PubMed] [Google Scholar]
- 11. Ladogana A, Puopolo M, Croes EA, et al. Mortality from Creutzfeldt‐Jakob disease and related disorders in Europe, Australia, and Canada. Neurology. 2005;64(9):1586‐1591. doi: 10.1212/01.Wnl.0000160117.56690.B2 [DOI] [PubMed] [Google Scholar]
- 12. Hermann P, Appleby B, Brandel J‐P, et al. Biomarkers and diagnostic guidelines for sporadic Creutzfeldt‐Jakob disease. Lancet Neurol. 2021;20(3):235‐246. doi: 10.1016/S1474-4422(20)30477-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jayaratnam S, Khoo AKL, Basic D. Rapidly progressive Alzheimer's disease and elevated 14‐3‐3 proteins in cerebrospinal fluid. Age Ageing. 2008;37(4):467‐469. doi: 10.1093/ageing/afn094 [DOI] [PubMed] [Google Scholar]
- 14. Dorey A, Tholance Y, Vighetto A, et al. Association of cerebrospinal fluid prion protein levels and the distinction between Alzheimer disease and Creutzfeldt‐Jakob disease. JAMA Neurol. 2015;72(3):267‐275. doi: 10.1001/jamaneurol.2014.4068 [DOI] [PubMed] [Google Scholar]
- 15. Hermann P, Zerr I. Rapidly progressive dementias — aetiologies, diagnosis and management. Nat Rev Neurol. 2022;18(6):363‐376. doi: 10.1038/s41582-022-00659-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Reinwald S, Westner IM, Niedermaier N. Rapidly progressive Alzheimer's disease mimicking Creutzfeldt‐Jakob disease. J Neurol. 2004;251(8):1020‐1022. doi: 10.1007/s00415-004-0480-6 [DOI] [PubMed] [Google Scholar]
- 17. Cohen M, Appleby B, Safar JG. Distinct prion‐like strains of amyloid beta implicated in phenotypic diversity of Alzheimer's disease. Prion. 2016;10(1):9‐17. doi: 10.1080/19336896.2015.1123371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Qiang W, Yau WM, Lu JX, Collinge J, Tycko R. Structural variation in amyloid‐β fibrils from Alzheimer's disease clinical subtypes. Nature. 2017;541(7636):217‐221. doi: 10.1038/nature20814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu H, Kim C, Haldiman T, et al. Distinct conformers of amyloid beta accumulate in the neocortex of patients with rapidly progressive Alzheimer's disease. J Biol Chem. 2021;297(5):101267. doi: 10.1016/j.jbc.2021.101267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hromadkova L, Siddiqi MK, Liu H, Safar JG. Populations of tau conformers drive prion‐like strain effects in Alzheimer's disease and related dementias. Cells. 2022;11(19):2997. doi: 10.3390/cells11192997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kim C, Haldiman T, Kang SG, et al. Distinct populations of highly potent TAU seed conformers in rapidly progressing Alzheimer's disease. Sci Transl Med. 2022;14(626):eabg0253. doi: 10.1126/scitranslmed.abg0253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kelley S, Perez‐Urrutia N, Morales R. Misfolded amyloid‐β strains and their potential roles in the clinical and pathological variability of Alzheimer's disease. Neural Regen Res. 2023;18(1):119‐120. doi: 10.4103/1673-5374.340403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Drummond E, Nayak S, Faustin A, et al. Proteomic differences in amyloid plaques in rapidly progressive and sporadic Alzheimer's disease. Acta Neuropathol. 2017;133(6):933‐954. doi: 10.1007/s00401-017-1691-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Drummond E, Pires G, MacMurray C, et al. Phosphorylated tau interactome in the human Alzheimer's disease brain. Brain. 2020;143(9):2803‐2817. doi: 10.1093/brain/awaa223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Corder EH, Saunders AM, Strittmatter WJ, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science. 1993;261(5123):921‐923. doi: 10.1126/science.8346443 [DOI] [PubMed] [Google Scholar]
- 26. Kunkle BW, Grenier‐Boley B, Sims R, et al. Genetic meta‐analysis of diagnosed Alzheimer's disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat Genet. 2019;51(3):414‐430. doi: 10.1038/s41588-019-0358-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bellenguez C, Küçükali F, Jansen IE, et al. New insights into the genetic etiology of Alzheimer's disease and related dementias. Nat Genet. 2022;54(4):412‐436. doi: 10.1038/s41588-022-01024-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. van der Vlies AE, Koedam EL, Pijnenburg YA, Twisk JW, Scheltens P, van der Flier WM. Most rapid cognitive decline in APOE epsilon4 negative Alzheimer's disease with early onset. Psychol Med. 2009;39(11):1907‐1911. doi: 10.1017/s0033291709005492 [DOI] [PubMed] [Google Scholar]
- 29. Schmidt C, Redyk K, Meissner B, et al. Clinical features of rapidly progressive Alzheimer's disease. Dement Geriatr Cogn Disord. 2010;29(4):371‐378. doi: 10.1159/000278692 [DOI] [PubMed] [Google Scholar]
- 30. Sherva R, Gross A, Mukherjee S, et al. Genome‐wide association study of rate of cognitive decline in Alzheimer's disease patients identifies novel genes and pathways. Alzheimers Dement. 2020;16(8):1134‐1145. doi: 10.1002/alz.12106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7(3):263‐269. doi: 10.1016/j.jalz.2011.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Besser L, Kukull W, Knopman DS, et al. Version 3 of the National Alzheimer's coordinating center's uniform data set. Alzheimer Dis Assoc Disord. 2018;32(4):351‐358. doi: 10.1097/wad.0000000000000279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Beekly DL, Ramos EM, Lee WW, et al. The National Alzheimer's Coordinating Center (NACC) database: the uniform data set. Alzheimer Dis Assoc Disord. 2007;21(3):249‐258. doi: 10.1097/WAD.0b013e318142774e [DOI] [PubMed] [Google Scholar]
- 34. Tosto G, Zimmerman ME, Carmichael OT, Brickman AM. Predicting aggressive decline in mild cognitive impairment: the importance of white matter hyperintensities. JAMA Neurol. 2014;71(7):872‐877. doi: 10.1001/jamaneurol.2014.667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ito K, Ahadieh S, Corrigan B, et al. Disease progression meta‐analysis model in Alzheimer's disease. Alzheimers Dement. 2010;6(1):39‐53. [DOI] [PubMed] [Google Scholar]
- 36. Hyman BT, Phelps CH, Beach TG, et al. National Institute on Aging‐Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease. Alzheimers Dement. 2012;8(1):1‐13. doi: 10.1016/j.jalz.2011.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Raczy C, Petrovski R, Saunders CT, et al. Isaac: ultra‐fast whole‐genome secondary analysis on Illumina sequencing platforms. Bioinformatics. 2013;29(16):2041‐2043. doi: 10.1093/bioinformatics/btt314 [DOI] [PubMed] [Google Scholar]
- 38. Kim S, Scheffler K, Halpern AL, et al. Strelka2: fast and accurate variant calling for clinical sequencing applications. Biorxiv. 2017:192872. doi: 10.1101/192872 [DOI] [Google Scholar]
- 39. Beecham GW, Bis J, Martin E, et al. The Alzheimer's disease sequencing project: study design and sample selection. Neurol Genet. 2017;3(5):e194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Leung YY, Valladares O, Chou Y‐F, et al. VCPA: genomic variant calling pipeline and data management tool for Alzheimer's Disease Sequencing Project. Bioinformatics. 2018;35(10):1768‐1770. doi: 10.1093/bioinformatics/bty894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Naj AC, Lin H, Vardarajan BN, et al. Quality control and integration of genotypes from two calling pipelines for whole genome sequence data in the Alzheimer's disease sequencing project. Genomics. 2019;111(4):808‐818. doi: 10.1016/j.ygeno.2018.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Genome Aggregation Database. https://gnomad.broadinstitute.org
- 43. Abecasis GR, Altshuler D, Auton A, et al. A map of human genome variation from population‐scale sequencing. Nature. 2010;467(7319):1061‐1073. doi: 10.1038/nature09534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Taliun D, Harris DN, Kessler MD, et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed program. Nature. 2021;590(7845):290‐299. doi: 10.1038/s41586-021-03205-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mahley R, Nathan B, Pitas R. Apolipoprotein E. Structure, function, and possible roles in Alzheimer's disease. Ann NY Acad Sci. 1996;777(1):139‐145. [DOI] [PubMed] [Google Scholar]
- 46. Lonsdale J, Thomas J, Salvatore M, et al. The genotype‐tissue expression (GTEx) project. Nat Genet. 2013;45(6):580‐585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. de Leeuw CA, Mooij JM, Heskes T, Posthuma D. MAGMA: generalized gene‐set analysis of GWAS data. PLoS Comput Biol. 2015;11(4):e1004219. doi: 10.1371/journal.pcbi.1004219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Watanabe K, Taskesen E, van Bochoven A, Posthuma D. Functional mapping and annotation of genetic associations with FUMA. Nat Commun. 2017;8(1):1826. doi: 10.1038/s41467-017-01261-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Murphy MP, LeVine H. Alzheimer's disease and the amyloid‐beta peptide. J Alzheimers Dis. 2010;19(1):311‐323. doi: 10.3233/jad-2010-1221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kang S‐G, Eskandari‐Sedighi G, Hromadkova L, Safar JG, Westaway D. Cellular biology of tau diversity and pathogenic conformers. Review. Front Neurol . 2020;11:590199. doi: 10.3389/fneur.2020.590199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.2022 Alzheimer's disease facts and figures. Alzheimers Dement. 2022;18(4):700‐789. doi: 10.1002/alz.12638 [DOI] [PubMed] [Google Scholar]
- 52. Schmidt C, Haïk S, Satoh K, et al. Rapidly progressive Alzheimer's disease: a multicenter update. J Alzheimers Dis. 2012;30:751‐756. doi: 10.3233/JAD-2012-120007 [DOI] [PubMed] [Google Scholar]
- 53. Farrer LA, Cupples LA, Haines JL, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease: a meta‐analysis. JAMA. 1997;278(16):1349‐1356. [PubMed] [Google Scholar]
- 54. Pillai JA, Appleby BS, Safar J, Leverenz JB. Rapidly progressive Alzheimer's disease in two distinct autopsy cohorts. J Alzheimers Dis. 2018;64:973‐980. doi: 10.3233/JAD-180155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Cosentino S, Scarmeas N, Helzner E, et al. APOE ε4 allele predicts faster cognitive decline in mild Alzheimer disease. Neurology. 2008;70(19):1842‐1849. Part 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lee JH, Kahn A, Cheng R, et al. Disease‐related mutations among Caribbean Hispanics with familial dementia. Mol Genet Genomic Med. 2014;2(5):430‐437. doi: 10.1002/mgg3.85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Arnold SE, Vega IE, Karlawish JH, et al. Frequency and clinicopathological characteristics of presenilin 1 Gly206Ala mutation in Puerto Rican Hispanics with dementia. J Alzheimers Dis. 2013;33(4):1089‐1095. doi: 10.3233/jad-2012-121570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Benitez BA, Karch CM, Cai Y, et al. The PSEN1, p.E318G variant increases the risk of Alzheimer's disease in APOE‐ε4 carriers. PLoS Genet. 2013;9(8):e1003685. doi: 10.1371/journal.pgen.1003685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Nho K, Horgusluoglu E, Kim S, et al. Integration of bioinformatics and imaging informatics for identifying rare PSEN1 variants in Alzheimer's disease. BMC Med Genom. 2016;9(1):30. doi: 10.1186/s12920-016-0190-9. Suppl. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sheng M, Sabatini BL, Südhof TC. Synapses and Alzheimer's disease. Cold Spring Harb Perspect Biol. 2012;4(5):a005777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Policarpo R, d'Ydewalle C. Missing lnc(RNAs) in Alzheimer's Disease? Genes (Basel). 2021;13(1):39. doi: 10.3390/genes13010039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Charles BA, Shriner D, Rotimi CN. Accounting for linkage disequilibrium in association analysis of diverse populations. Genet Epidemiol. 2014;38(3):265‐273. doi: 10.1002/gepi.21788 [DOI] [PubMed] [Google Scholar]
- 63. Numa S, Oishi A, Higasa K, et al. EYS is a major gene involved in retinitis pigmentosa in Japan: genetic landscapes revealed by stepwise genetic screening. Sci Rep. 2020;10(1):20770. doi: 10.1038/s41598-020-77558-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Steffens DC, Garrett ME, Soldano KL, McQuoid DR, Ashley‐Koch AE, Potter GG. Genome‐wide screen to identify genetic loci associated with cognitive decline in late‐life depression. Int Psychogeriatr. 2020;27(4):1990‐1999. doi: 10.1017/s1041610220001143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Neumann A, Küçükali F, Bos I, et al. Rare variants in IFFO1, DTNB, NLRC3 and SLC22A10 associate with Alzheimer's disease CSF profile of neuronal injury and inflammation. Mol Psychiatry. 2022;27(4):1990‐1999. doi: 10.1038/s41380-022-01437-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Govindpani K, Turner C, Waldvogel HJ, Faull RLM, Kwakowsky A. Impaired expression of GABA signaling components in the Alzheimer's disease middle temporal gyrus. Int J Mol Sci. 2020;21(22):8704. doi: 10.3390/ijms21228704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Yamazaki Y, Zhao N, Caulfield TR, Liu CC, Bu G. Apolipoprotein E and Alzheimer disease: pathobiology and targeting strategies. Nat Rev Neurol. 2019;15(9):501‐518. doi: 10.1038/s41582-019-0228-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Data Availability Statement
Clinical data are available upon request through the National Alzheimer's Coordinating Center (https://naccdata.org/). Genetic data are available through the National Institute on Aging Genetics of Alzheimer's Disease Data Storage Site (NIAGADS; https://www.niagads.org/). The code used in the genetic analysis is available on GitHub at https://github.com/PingWang‐github/Genetic_Architecture_rpAD