

Abstract
INTRODUCTION
Our population‐based study assessed whether clinically apparent Helicobacter pylori infection (CAHPI) is associated with the risk of Alzheimer's disease (AD).
METHODS
We assembled a population‐based cohort of all dementia‐free subjects in the United Kingdom's Clinical Practice Research Datalink (UK CPRD), aged ≥50 years (1988–2017). Using a nested case‐control approach, we matched each newly developed case of AD with 40 controls. Conditional logistic regression estimated odds ratios (ORs) with 95% confidence intervals (CIs) of AD associated with CAHPI compared with no CAHPI during ≥2 years before the index date. We also used salmonellosis as a negative control exposure.
RESULTS
Among 4,262,092 dementia‐free subjects, 40,455 developed AD after a mean 11 years of follow‐up. CAHPI was associated with an increased risk of AD (OR, 1.11; 95% CI, 1.01–1.21) compared with no CAHPI. Salmonellosis was not associated with the risk of AD (OR, 1.03; 95% CI, 0.82–1.29).
DISCUSSION
CAHPI was associated with a moderately increased risk of AD.
Highlights
CAHPI was associated with an 11% increased risk of AD in subjects aged ≥50 years.
The increase in the risk of AD reached a peak of 24% a decade after CAHPI onset.
There was no major effect modification by age or sex.
Sensitivity analyses addressing several potential biases led to consistent results.
Keywords: infectious disease, epidemiology, neurodegeneration, prevention
1. BACKGROUND
Alzheimer's disease (AD) is the most common form of dementia currently affecting roughly 40 million people globally. 1 In the past years, numerous pre‐clinical, serological, and post‐mortem studies suggested that a wide range of infectious pathogens such as Helicobacter pylori (HP) could play an important role in the development of AD. 2 , 3 , 4 , 5 , 6 , 7 , 8 This ‘infectious hypothesis’ was then tested in observational studies that showed an association between infectious disease burden and an increased risk of AD. 9 , 10
Recently, a population‐based study conducted by our group found an association between clinically apparent infections linked to pathogens implicated in AD pathophysiology and a small but statistically significant increase in the risk of this neurodegenerative disease (odds ratio [OR], 1.08; 95% confidence interval [CI], 1.07–1.10). 11 In exploratory analyses assessing the effects of specific infections, we observed a signal for clinically detected, unspecified gastritis (OR, 1.08; 95% CI, 1.03–1.13) but not for other clinical manifestations. 11 This signal is in line with prior observational studies showing an increased risk of AD associated with HP infection. 12 , 13 However, these studies had methodological limitations such as reverse causality and detection bias due to the lack of a lag period to account for the insidious nature of AD, and also important residual confounding. 12 , 13 Moreover, the findings were not consistent, with other studies failing to replicate them. 14
Given the limitations of available evidence, further, well‐designed population‐based studies are needed to address this important knowledge gap, that is whether HP infection is associated with an increased risk of AD. To this end, we conducted a large, population‐based study to assess the potential association between clinically apparent HP infection (CAHPI) and the risk of incident AD among individuals aged 50 years or older.
RESEARCH IN CONTEXT
Systematic review: The authors reviewed the literature using PubMed. They found that while observational studies have shown an increased risk of Alzheimer's disease (AD) associated with Helicobacter pylori (HP) infection, these studies had methodological limitations that render the interpretation of their findings difficult.
Interpretation: Our large population‐based study found an 11% increased risk of AD associated with exposure to clinically apparent HP infection (CAHPI) among subjects aged ≥50 years. The increase in the risk of AD reached a peak of 24% a decade after CAHPI onset. There was no major effect modification by demographic characteristics. Multiple sensitivity analyses corroborated the findings of the primary analysis.
Future directions: Our findings support the notion of HP infection as a potential modifiable risk factor of AD. They also pave the way for future randomized trials that would assess the impact and cost‐effectiveness of population‐based targeted interventions such as individualized HP eradication programs.
2. METHODS
2.1. Data source
We used the United Kingdom's (UK's) Clinical Practice Research Datalink (CPRD) GOLD, a large primary care database containing anonymized medical records from general practitioners (GPs). It covers >11 million patients across 674 UK general practices, and is broadly representative of the general UK population on demographics and ethnicity. 15 In CPRD GOLD, medical diagnoses and procedures are documented via Read codes. Medications prescribed by GPs are also documented. 15 In the UK, GPs serve as the first point of contact for non‐emergency health‐related issues; thus, information on routinely recorded symptoms, laboratory tests, diagnoses, treatments, health‐related behaviors, and referrals to secondary care are all recorded in the database. CPRD undergoes routine quality control assessments and recorded diagnoses are valid and of high quality. 15 , 16 We linked the CPRD to the Index of Multiple Deprivation (IMD), an area‐level measure of deprivation in the UK with seven domains: income, employment, education, health, crime, barriers to housing and services, and living environment. The study protocol was approved by the CPRD's Independent Scientific Advisory Committee (protocol 19_236R) and the Research Ethics Board of the Jewish General Hospital in Montreal, Canada.
2.2. Study population
We assembled a study cohort including all subjects ≥50 years of age enrolled in CPRD GOLD between January 1988, and December 2017, and followed until December 2019. Cohort entry was defined as the 50th birthday of the subject or 1‐year after their date of enrolment in the database, whichever occurred latest. We required all subjects to be dementia‐free at cohort entry. Thus, we excluded subjects with a prior diagnosis of dementia or mild cognitive impairment, early signs or symptoms suggestive of dementia (memory impairment, aphasia, apraxia, agnosia), or a prescription for AD medications (donepezil, rivastigmine, galantamine, memantine) at any time before cohort entry. A diagnosis of CAHPI before cohort entry was not an exclusion criterion. Subjects were followed from cohort entry until a diagnosis of AD (see below), end of registration with the CPRD's general practice, death from any cause, or end of the study period (December 2019), whichever occurred first.
2.3. Case definition
Within the study cohort, we identified all cases with a first‐time diagnosis of AD at any time after cohort entry. AD was defined based on a modified algorithm initially developed and validated by Imfeld et al. 17 and previously used by our group. 11 , 18 , 19 The algorithm required ≥1 of the following criteria to be met: (i) AD diagnosis and ≥1 prescription for AD medications in any sequence; (ii) unspecified dementia diagnosis followed by ≥2 prescriptions for AD medications; (iii) ≥2 records of an AD diagnosis; (iv) AD diagnosis following a dementia test (Mini‐Mental State Examination, clock drawing test, abbreviated mental test), referral to a specialist (neurologist, geriatrician, psychogeriatrician), or neuroimaging assessment (computed tomography, magnetic resonance imaging, single photon emission computed tomography); or (v) AD diagnosis and any dementia symptom (memory impairment, aphasia, apraxia, agnosia) in any sequence. The date of AD diagnosis (index date) corresponded to the date of the last event contributing to each criterion.
2.4. Control selection
Given the large size of the cohort, we implemented a nested case‐control approach within the cohort to facilitate the analyses. 20 Because cases and controls were selected from the same, well‐defined cohort, selection bias was minimized and the rare outcome assumption inherent in traditional case‐control studies was not required for the validity of the risk estimates. 20 We used risk‐set sampling to select appropriate controls. Each AD case was matched to up to 40 AD‐free controls randomly selected from the risk set defined by the case (those still being followed and event‐free at the date of the AD event). Given the use of risk‐set sampling, the ORs derived from our nested case‐control analysis calculated via conditional logistic regression could be interpreted as unbiased estimators of the hazard ratios derived from the underlying cohort analysis calculated via Cox regression with minimal loss in precision. 21 Matching criteria included sex, age (±1 year), cohort entry date (±90 days), and duration of follow‐up. The first three matching criteria minimized confounding due to demographics and calendar time. The latter matching criterion guaranteed that cases and controls have an equal time‐window to become ‘exposed’ to CAHPI, avoiding thus time‐window bias. 22 Controls were allowed to contribute to different risk sets and eventually become a case.
2.5. Exposure definition
Our exposure of interest was CAHPI detectable in the natural clinical setting. Given that the majority of HP infected individuals remain asymptomatic over time and may never develop any medical condition, 23 we focused on infected individuals presenting with symptoms or developing serious complications from the infection. Thus, for all cases and controls, we identified all CAHPI recorded after cohort entry. CAHPI was defined based on an algorithm using clinical guidelines and recommendations on the management of HP infection. 24 , 25 , 26 , 27
The algorithm required meeting ≥1 of the following criteria: (i) positive HP test (urea breath test, monoclonal stool antigen test) and eradication therapy (see below) within 30 days of the test; (ii) diagnosis of HP‐related disease or related complication (uninvestigated or functional dyspepsia, unspecified gastric or duodenal ulcer, unspecified gastritis, confirmed HP‐gastritis) and eradication therapy (see below) within 30 days of the diagnosis; or (iii) diagnosis of gastric mucosa‐associated lymphoid tissue (MALT) lymphoma or non‐cardia gastric cancer.
We defined eradication therapy as a prescription for triple or quadruple therapy including an acid‐suppressing drug (proton pump inhibitor or H2 receptor antagonist) and ≥2 of the antibiotics amoxicillin, clarithromycin, metronidazole, or tetracycline. CAHPI onset date was the last date contributing to this definition. To account for the non‐acute nature of AD and consider potential diagnostic delays, we imposed a 2‐year exposure lag period within the definition. 28 , 29 Thus, cases and controls with CAHPI occurring 2 years or more before the index date were considered as exposed, whereas cases and controls with no CAHPI within that time frame were considered as unexposed. Moreover, cases and controls with CAHPI that were detected within the 2‐year period before the index date were also considered as unexposed (Figure S1).
2.6. Primary analysis
Given the use of the lag period, only cases and matched controls with ≥2 years of follow‐up contributed to the analyses. We used conditional logistic regression to estimate OR and 95% CI of AD associated with exposure to CAHPI, as compared with no exposure to CAHPI. We adjusted for the following potential confounders (Figure S2), measured at cohort entry: body mass index (BMI) (<18.5 kg/m2, 18.5–24.9 kg/m2, 25–29 kg/m2, ≥30 kg/m2, unknown; last measurement before cohort entry), smoking (ever, never, unknown; last measurement before cohort entry), alcohol‐related disorders, hypertension, atrial fibrillation, heart failure, coronary artery disease, stroke or transient ischemic attack, peripheral vascular disease, cancer, chronic kidney disease, liver disease, hypothyroidism, dyslipidemia, diabetes mellitus, osteoporosis, depression, epilepsy, Parkinson's disease, and traumatic brain injury (all comorbidities measured ever before cohort entry). We also adjusted for use of angiotensin‐converting enzyme inhibitors, angiotensin II receptor blockers, thiazides, calcium channel blockers, beta‐blockers, lipid‐lowering drugs, oral anticoagulants, antiplatelet agents, non‐steroidal anti‐inflammatory drugs, opioids, proton pump inhibitors, antipsychotics, and antidepressants in the year before cohort entry.
FIGURE 1.
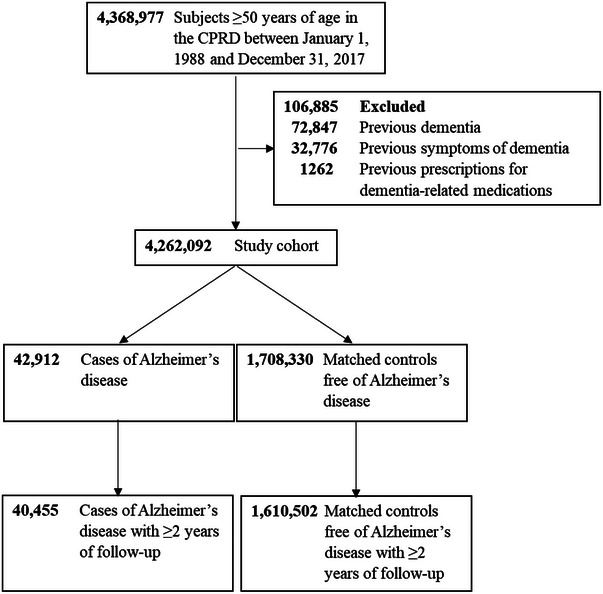
Flowchart illustrating the selection of cases of Alzheimer's disease and matched controls. CPRD, Clinical Practice Research Datalink.
2.7. Additional analyses
We conducted five secondary analyses. First, we assessed the association between time of CAHPI onset and diagnosis of AD (‘time‐response’ relation) by estimating ORs and 95% CIs for subgroups with different time intervals. The subgroups were based on the quartiles of the distribution of follow‐up durations among controls. To account for potential non‐linear associations, we also modelled time since CAHPI onset as a continuous variable using restricted cubic splines with five interior knots. 30 Second, we stratified by age (<65 vs. ≥65 years) and sex. Finally, we stratified by IMD quintiles to assess a potential effect measure modification by socioeconomic status.
We performed five pre‐specified sensitivity analyses. First, given the uncertainty regarding the delay between AD symptom onset and diagnosis, and considering the impact of the length of time between onset of CAHPI and AD, we repeated the primary analysis using alternate lag periods of 3, 5, and 10 years. Second, to restrict the cohort to subjects with AD exclusively, we censored follow‐up upon a diagnosis of non‐AD dementia (e.g., vascular, alcoholic, unspecified dementia). Finally, to assess the impact of residual confounding, we used salmonellosis, an infection with a common enterobacteria not previously associated with AD, 31 as negative control exposure. 32
We also conducted seven post‐hoc sensitivity analyses. First, we assessed the impact of exposure misclassification by applying alternate maximum time intervals between positive HP test or diagnosis of HP‐related disease or related complication and eradication therapy to define CAHPI (14, 60, 90 days). Second, we restricted our analyses to cases and controls without a CAHPI diagnosis before cohort entry. Third, we calculated the E‐value to assess the impact of residual confounding. 33 Fourth, we additionally adjusted for IMD quintiles. Finally, we used multiple imputation methods for missing values for smoking and BMI (Methods 1).
Prior studies assessing the role of infections in AD had also looked at dementia more broadly. Therefore, we conducted supplementary analyses including any diagnosis of dementia. Analyses were conducted using SAS version 9.4 (SAS Institute, Cary, NC).
3. RESULTS
Our study cohort included 4,262,092 dementia‐free subjects aged ≥50 years (Figure 1). Mean (standard deviation) age at cohort entry was 60.4 (11.5) years (52.1% female). Patient characteristics were overall similar between the 40,455 AD cases occurring during follow‐up and their 1,610,502 matched AD‐free controls (Table 1). As expected, depression and antidepressant use were more common among cases. The vast majority (>95%) of CAHPI in AD cases and their AD‐free matched controls was based on a diagnosis of HP‐related disease in combination with HP eradication therapy (Table S1). Moreover, >98% of AD cases and their AD‐free matched controls did not have a CAHPI diagnosis before cohort entry.
TABLE 1.
Characteristics of cases of Alzheimer's disease and matched controls.
| Characteristic | Cases n = 40,455 | Controls n = 1,610,502 |
|---|---|---|
| Age at cohort entry in years, mean (SD) | 69 (8.8) | 69 (8.8) |
| Age at index date in years, mean (SD) | 81 (7.7) | 81 (7.6) |
| Follow‐up in years, mean (SD) | 11 (5.5) | 11 (5.5) |
| Male sex, n (%) | 14,454 (35.7) | 573,799 (35.6) |
| Body mass index in kg/m2, n (%) a | ||
| <18.5 | 409 (1.0) | 13,552 (0.8) |
| 18.5–24.9 | 11,476 (28.4) | 402,659 (25.0) |
| 25–29.9 | 11,188 (27.7) | 442,651 (27.5) |
| ≥30 | 4737 (11.7) | 211,107 (13.1) |
| Unknown | 12,645 (31.3) | 540,533 (33.6) |
| Smoking, n (%) a | ||
| Never | 17,693 (43.7) | 689,978 (42.8) |
| Ever | 12,855 (31.8) | 490,181 (30.4) |
| Unknown | 9907 (24.5) | 430,343 (26.7) |
| Comorbidities, n (%) | ||
| Alcohol‐related disorders | 640 (1.6) | 23,027 (1.4) |
| Arterial hypertension | 11,814 (29.2) | 492,501 (30.6) |
| Atrial fibrillation | 964 (2.4) | 43,692 (2.7) |
| Congestive heart failure | 655 (1.6) | 29,028 (1.8) |
| Coronary artery disease | 4509 (11.1) | 173,760 (10.8) |
| Stroke or transient ischemic attack | 1242 (3.1) | 59,904 (3.7) |
| Peripheral vascular disease | 839 (2.1) | 33,160 (2.1) |
| Cancer | 2933 (7.3) | 114,238 (7.1) |
| Chronic kidney disease | 633 (1.6) | 24,959 (1.5) |
| Liver disease | 126 (0.3) | 5288 (0.3) |
| Hypothyroidism | 2405 (5.9) | 92,115 (5.7) |
| Dyslipidemia | 4134 (10.2) | 146,257 (9.1) |
| Diabetes mellitus | 2440 (6.0) | 93,420 (5.8) |
| Osteoporosis | 1356 (3.4) | 47,173 (2.9) |
| Traumatic brain injury | S | 32 (0.0) |
| Epilepsy | 555 (1.4) | 18,834 (1.2) |
| Parkinson's disease | 127 (0.3) | 4405 (0.3) |
| Depression | 5088 (12.6) | 173,333 (10.8) |
| Medications, n (%) b | ||
| Angiotensin‐converting enzyme inhibitors | 4196 (10.4) | 180,778 (11.2) |
| Angiotensin II receptor blockers | 1262 (3.1) | 53,633 (3.3) |
| Thiazide diuretics | 6000 (14.8) | 249,926 (15.5) |
| Calcium channel blockers | 5173 (12.8) | 208,028 (12.9) |
| Beta‐blockers | 6132 (15.2) | 251,650 (15.6) |
| Lipid‐lowering drugs | 5700 (14.1) | 210,066 (13.0) |
| Oral anticoagulants | 720 (1.8) | 34,485 (2.1) |
| Antiplatelet agents | 6767 (16.7) | 261,802 (16.3) |
| Non‐steroidal anti‐inflammatory drugs | 9051 (22.4) | 329,167 (20.4) |
| Opioids | 10,066 (24.9) | 359,806 (22.3) |
| Proton pump inhibitors | 4584 (11.3) | 173,994 (10.8) |
| Immunosuppressants or biologics | 2281 (5.6) | 88,841 (5.5) |
| Antidepressants | 4675 (11.6) | 142,454 (8.8) |
| Antipsychotic drugs | 1895 (4.7) | 65,655 (4.1) |
Note: S Numbers <5 were suppressed based on data protection regulations of the Clinical Practice Research Datalink.
Abbreviation: SD, standard deviation.
Last measurement before cohort entry.
Measured in the year before cohort entry.
Compared with no exposure to CAHPI, exposure to CAHPI was associated with a moderately increased risk of AD (OR, 1.11; 95% CI, 1.01–1.21) (Table 2). The increase in the risk peaked 7.3 to 10.8 years after CAHPI onset (OR, 1.24; 95% CI, 1.05–1.47); flexibly modelling time between CAHPI onset and AD diagnosis led to similar findings (Figure 2). There was no major effect modification by demographics or socioeconomic status (Table S2).
TABLE 2.
Crude and adjusted odds ratios of the association between CAHPI and the risk of incident Alzheimer's disease (primary analysis and time‐response analysis).
|
Cases n = 40,455 |
Controls n = 1,610,502 |
Crude OR (95% CI) | Adjusted a OR (95% CI) | |
|---|---|---|---|---|
| CAHPI, n (%) | ||||
| Exposed | 512 (1.3) | 17,522 (1.1) | 1.16 (1.06 to 1.27) | 1.11 (1.01 to 1.21) |
| Unexposed | 39,943 (98.7) | 1,592,980 (98.9) | Reference | Reference |
| Time since onset of CAHPI b in years, n (%) | ||||
| ≥10.9 | 126 (0.3) | 4381 (0.3) | 1.13 (0.95 to 1.36) | 1.09 (0.91 to 1.30) |
| 7.3–10.8 | 145 (0.4) | 4381 (0.3) | 1.31 (1.11 to 1.55) | 1.24 (1.05 to 1.47) |
| 4.5–7.2 | 114 (0.3) | 4383 (0.3) | 1.04 (0.86 to 1.25) | 0.99 (0.82 to 1.20) |
| 2.0–4.4 | 127 (0.3) | 4377 (0.3) | 1.16 (0.97 to 1.38) | 1.11 (0.93 to 1.32) |
| Unexposed | 110,170 (98.9) | 4,379,408 (99.1) | Reference | Reference |
Abbreviations: CAHPI, clinically apparent Helicobacter pylori infection; CI, confidence interval; OR, odds ratio.
Adjusted for body mass index, smoking, alcohol‐related disorders, arterial hypertension, atrial fibrillation, congestive heart failure, coronary artery disease, stroke or transient ischemic attack, peripheral vascular disease, cancer, chronic kidney disease, liver disease, hypothyroidism, dyslipidemia, diabetes mellitus, osteoporosis, depression, epilepsy, Parkinson's disease, traumatic brain injury, angiotensin‐converting enzyme inhibitors, angiotensin II receptor blockers, thiazide diuretics, calcium channel blockers, beta‐blockers, lipid‐lowering drugs, oral anticoagulants, antiplatelet agents, non‐steroidal anti‐inflammatory drugs, opioids, proton pump inhibitors, antipsychotics, and antidepressants.
The subgroups were based on the quartiles of the distribution of follow‐up durations among controls. The effect estimates for the different subgroups show the associations between CAHPI and dementia for those diagnosed with CAHPI 2.0–4.4, 4.5–7.2, 7.3–10.8, or ≥10.9 years prior to the diagnosis of dementia.
FIGURE 2.
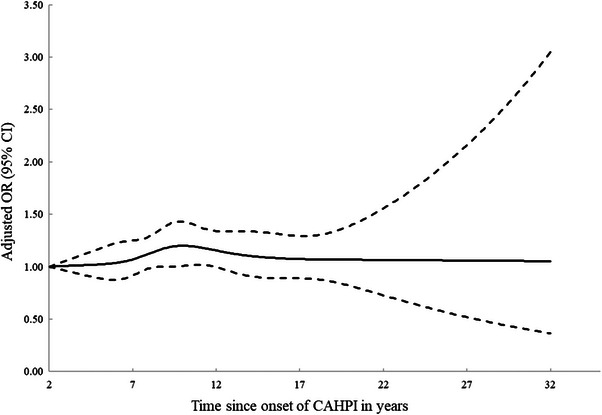
Time‐response analysis for the association between CAHPI and the risk of incident Alzheimer's disease. CAHPI, clinical apparent Helicobacter pylori infection; CI, confidence interval; OR, odds ratio.
Using 3‐ and 5‐year lag periods, censoring of follow‐up upon non‐AD dementia, using alternate CAHPI definitions, restricting to cases and controls without prior CAHPI, additionally adjusting for IMD, and using multiple imputation methods for missing values did not change the results (Table S3). The application of a 10‐year lag period led to a slight decrease in the OR and a lower number of exposed cases that resulted in loss in precision with wider CIs including the null (OR, 1.06; 95% CI, 0.90–1.25). Reassuringly, the analysis with salmonellosis as a negative control exposure showed no association with the risk of AD (OR, 1.03; 95% CI, 0.82–1.29) (Table S3). The E‐value was 1.46. Finally, the results remained highly consistent with any dementia as the outcome (case and control selection in Figure S3; characteristics of dementia cases and their matched dementia‐free controls in Table S4; analyses in Figure S4 and Tables S5–S7).
4. DISCUSSION
Our large population‐based nested case‐control study found an 11% increase in the risk of AD associated with exposure to CAHPI among subjects aged at least 50 years. The increase in the risk of AD reached a peak of 24% roughly a decade after the onset of CAHPI. There was no major effect modification by age or sex. Moreover, sensitivity analyses yielded findings that were overall consistent with those of the primary analysis.
A link between HP and the central nervous system has been proposed. HP can access the brain via the oral‐nasal‐olfactory axis or by infected circulating monocytes through a disrupted blood‐brain‐barrier, 34 potentially leading to neuroinflammation, neuronal damage, and neurodegeneration. 34 Another potential mechanism involves the gut‐brain axis; its disruption due to a HP infection could lead to the activation of multiple pathological pathways. 35 , 36 Alterations in gut microbiome can lead to amyloid and lipopolysaccharide oversecretion, production of pro‐inflammatory cytokines, and changes in the gut and the permeability of the blood‐brain‐barrier. 34 Finally, structural damage in gastric mucosa caused by chronic HP infection could affect the absorption of vitamin B12 and iron, a deficit of which is associated with dementia. 37 , 38 , 39 , 40
Observational studies have assessed the potential association between HP infection and AD risk. 12 , 13 Meta‐analyses of these studies showed numerically increased risks with a maximum pooled OR of 1.72. 12 , 13 However, most individual studies were based on small sample sizes and were at high risk of bias due to methodological limitations. For example, the ubiquitous absence of a lag period between HP infection and AD diagnosis possibly introduced reverse causality and detection bias. 12 , 13 Moreover, most studies either adjusted their models for few potential confounders or reported only crude results, probably introducing important residual confounding. 12 , 13
Our study found a moderate but statistically significant increase in the risk of AD associated with CAHPI. Capitalizing on the long follow‐up available, we were also able to characterize the time pattern of the association over many years. The increase in the risk of AD reached statistical significance roughly 8 years following CAHPI onset, peaked 2 years later, and decreased afterwards. The time interval from CAHPI onset to AD diagnosis could theoretically be divided into the induction period spanning from CAHPI onset to AD development, and the latent period spanning from AD development to its diagnosis. 29 Because the path from neuroinflammation to neurodegeneration may last several years, 41 and considering the known diagnostic delays associated with AD, 28 the observed time pattern could be compatible with a causal effect with a specific etiological time window. However, one could also envisage CAHPI “contributing” to AD development or the progression of underlying pathophysiological processes.
Our study has strengths. First, the large sample size and lengthy follow‐up allowed the calculation of precise effect estimates of the association between CAHPI and AD risk. Moreover, it allowed the assessment of potential effect modifiers. Second, the use of a lag period likely minimized reverse causality and early detection bias, which were important limitations of previous observational studies. However, given the unclear duration of the induction period in the association between CAHPI and AD, reverse causality cannot be excluded. Finally, the application of few exclusion criteria likely maximized the generalizability of our findings.
Our study also has limitations. First, given its observational nature, residual confounding is possible. To minimize confounding, we adjusted for many potential confounders. Moreover, we conducted a sensitivity analysis using salmonellosis as a negative control exposure. The absence of an increased risk of AD supports the validity of our primary analysis. That said, based on the calculated E‐value, an unmeasured confounder associated with both CAHPI and AD with an OR of 1.46 each, above and beyond the measured confounders, could also explain our findings; weaker confounding could not do so, though. Second, because our exposure definition was based on CAHPI recorded by GPs, exposure misclassification due to symptomatic patients not seeking primary care is possible. However, using alternate time intervals between HP‐related condition and prescription for eradication therapy to define CAHPI led to consistent findings. Third, outcome misclassification is possible. To alleviate this bias, we defined AD using a validated algorithm 17 that captures a wide range of criteria involved in AD diagnosis and treatment. Finally, we cannot rule out the possibility of an association between asymptomatic HP infection and AD risk. However, because asymptomatic infections were classified as “unexposed,” such association would lead to an underestimation of the true effect. In addition, our focus were infections easily detectable in the setting of routine clinical practice. Indeed, such conditions would be the most relevant target for prevention considering the current absence of population‐based screening programs for HP infection.
Overall, our large population‐based study showed a moderately increased risk of AD associated with CAHPI among individuals aged ≥50 years. These results support the notion of HP infection as a potential modifiable risk factor of AD. They also pave the way for future randomized controlled trials that would assess the impact and cost‐effectiveness of population‐based targeted interventions such as individualized HP eradication programs, 42 on the development of AD. Based on preliminary calculations for UK, HP eradication could lead to a 0.7% decrease in AD prevalence (Methods 2), which albeit small on the relative scale, would still translate to a reduction of roughly two hundred thousand cases globally. Such HP intervention and surveillance programs are already being tested for gastric cancer prevention in parts of the world, 43 , 44 , 45 therefore suggesting a public health benefit that has the potential to include gastric cancer, peptic ulcer, dyspepsia, and potentially AD prevention.
AUTHOR CONTRIBUTIONS
All authors contributed to the study concept and design, interpreted the data, and critically revised the manuscript. AD drafted the manuscript. ZA conducted the statistical analyses. PB acquired the data, obtained funding, and supervised the study. PB is the guarantor.
CONFLICT OF INTEREST STATEMENT
S.S. attended scientific advisory committee meetings or consulted for AstraZeneca, Atara, Bristol‐Myers‐Squibb, Merck, Novartis, Panalgo, Pfizer and Seqirus, and received speaking fees from Boehringer‐Ingelheim and Novartis. P.B. and CAF received consulting fees from Becton Dickinson and Pendopharm, respectively, on topics unrelated to this work. All other authors declare no conflict of interest. Author disclosures are available in the Supporting information.
Supporting information
Supplementary Information
Supplemental Information.
ACKNOWLEDGMENTS
AD is the recipient of Chercheur‐Boursier Junior 1 Award from the Fonds de Recherche du Québec – Santé (FRQS). The study was funded by a project grant from the Canadian Institutes of Health Research (CIHR #183856).
Douros A, Ante Z, Fallone CA, et al. Clinically apparent Helicobacter pylori infection and the risk of incident Alzheimer's disease: A population‐based nested case‐control study. Alzheimer's Dement. 2024;20:1716–1724. 10.1002/alz.13561
DATA AVAILABILITY STATEMENT
This study is based in part on data from the Clinical Practice Research Datalink obtained under license from the UK Medicines and Healthcare products Regulatory Agency. The data are provided by patients and collected by the UK National Health Service as part of their care and support. The interpretation and conclusions contained in this study are those of the authors alone. Because electronic health records are classified as “sensitive data” by the UK Data Protection Act, information governance restrictions (to protect patient confidentiality) prevent data sharing via public deposition. Data are available with approval through the individual constituent entities controlling access to the data. Specifically, the primary care data can be requested via application to the Clinical Practice Research Datalink (https://www.cprd.com).
REFERENCES
- 1. World Health Organization . Dementia. Accessed 05 May, 2020. https://www.who.int/news‐room/fact‐sheets/detail/dementia
- 2. Alonso R, Pisa D, Aguado B, Carrasco L. Identification of fungal species in brain tissue from Alzheimer's Disease by next‐generation sequencing. J Alzheimers Dis. 2017;58(1):55‐67. [DOI] [PubMed] [Google Scholar]
- 3. Balin BJ, Hammond CJ, Little CS, et al. Chlamydia pneumoniae: an etiologic agent for late‐onset dementia. Front Aging Neurosci. 2018;10:302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Beydoun MA, Beydoun HA, Elbejjani M, Dore GA, Zonderman AB. Helicobacter pylori seropositivity and its association with incident all‐cause and Alzheimer's disease dementia in large national surveys. Alzheimers Dement. 2018;14(9):1148‐1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lovheim H, Olsson J, Weidung B, et al. Interaction between cytomegalovirus and herpes simplex virus type 1 associated with the risk of Alzheimer's disease development. J Alzheimers Dis. 2018;61(3):939‐945. [DOI] [PubMed] [Google Scholar]
- 6. Itzhaki RF. Herpes simplex virus type 1 and Alzheimer's disease: possible mechanisms and signposts. FASEB J. 2017;31(8):3216‐3226. [DOI] [PubMed] [Google Scholar]
- 7. Singhrao SK, Harding A, Poole S, Kesavalu L, Crean S. Porphyromonas gingivalis periodontal infection and its putative links with Alzheimer's disease. Mediators Inflamm. 2015;2015:137357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhan X, Stamova B, Jin LW, DeCarli C, Phinney B, Sharp FR. Gram‐negative bacterial molecules associate with Alzheimer disease pathology. Neurology. 2016;87(22):2324‐2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mawanda F, Wallace RB, McCoy K, Abrams TE. Systemic and localized extra‐central nervous system bacterial infections and the risk of dementia among US veterans: a retrospective cohort study. Alzheimers Dement. 2016;4:109‐117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sipilä PN, Heikkilä N, Lindbohm JV, et al. Hospital‐treated infectious diseases and the risk of dementia: a large, multicohort, observational study with a replication cohort. Lancet Infect Dis. 2021;21(11):1557‐1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Douros A, Santella C, Dell'Aniello S, et al. Infectious disease burden and the risk of Alzheimer's disease: a population‐based study. J Alzheimers Dis. 2021;81(1):329‐338. [DOI] [PubMed] [Google Scholar]
- 12. Shindler‐Itskovitch T, Ravona‐Springer R, Leibovitz A, Muhsen K. A systematic review and meta‐analysis of the association between helicobacterpylori infection and dementia. J Alzheimers Dis. 2016;52(4):1431‐1442. [DOI] [PubMed] [Google Scholar]
- 13. Liu NY, Sun JH, Jiang XF, Li H. Helicobacter pylori infection and risk for developing dementia: an evidence‐based meta‐analysis of case‐control and cohort studies. Aging. 2021;13(18):22571‐22587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fani L, Wolters FJ, Ikram MK, et al. Helicobacter pylori and the risk of dementia: a population‐based study. Alzheimers Dement. 2018;14(10):1377‐1382. [DOI] [PubMed] [Google Scholar]
- 15. Herrett E, Gallagher AM, Bhaskaran K, et al. Data resource profile: clinical practice research datalink (CPRD). Int J Epidemiol. 2015;44(3):827‐836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Khan NF, Harrison SE, Rose PW. Validity of diagnostic coding within the General Practice Research Database: a systematic review. Br J Gen Pract. 2010;60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Imfeld P, Brauchli Pernus YB, Jick SS, Meier CR. Epidemiology, co‐morbidities, and medication use of patients with Alzheimer's disease or vascular dementia in the UK. J Alzheimers Dis. 2013;35(3):565‐573. [DOI] [PubMed] [Google Scholar]
- 18. Sinyavskaya L, Gauthier S, Renoux C, Dell'Aniello S, Suissa S, Brassard P. Comparative effect of statins on the risk of incident Alzheimer disease. Neurology. 2018;90(3):e179‐e187. [DOI] [PubMed] [Google Scholar]
- 19. Douros A, Ante Z, Suissa S, Brassard P. Common vaccines and the risk of incident dementia: a population‐based cohort study. J Infect Dis. 2023;227(11):1227‐1236. [DOI] [PubMed] [Google Scholar]
- 20. Schneeweiss S, Suissa S. Discussion of Schuemie et al: A plea to stop using the case‐control design in retrospective database studies. Stat Med. 2019;38(22):4209‐4212. [DOI] [PubMed] [Google Scholar]
- 21. Essebag V, Platt RW, Abrahamowicz M, Pilote L. Comparison of nested case‐control and survival analysis methodologies for analysis of time‐dependent exposure. BMC Med Res Methodol. 2005;5(1):5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Suissa S, Dell'aniello S, Vahey S, Renoux C. Time‐window bias in case‐control studies: statins and lung cancer. Epidemiology. 2011;22(2):228‐231. [DOI] [PubMed] [Google Scholar]
- 23. Abadi AT, Kusters JG. Management of Helicobacter pylori infections. BMC Gastroenterol. 2016;16(1):94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gastro‐oesophageal reflux disease and dyspepsia in adults: investigation and management. National Institute for Health and Care Excellence (NICE); Guidelines. London. October 2019. [PubMed]
- 25. Malfertheiner P, Megraud F, O'Morain CA, et al. Management of Helicobacter pylori infection‐the Maastricht V/Florence Consensus Report. Gut. 2017;66(1):6‐30. [DOI] [PubMed] [Google Scholar]
- 26. Fallone CA, Chiba N, van Zanten SV, et al. The Toronto consensus for the treatment of helicobacter pylori infection in adults. Gastroenterology. 2016;151(1):51‐69.e14. [DOI] [PubMed] [Google Scholar]
- 27. Chey WD, Leontiadis GI, Howden CW, Moss SF. ACG clinical guideline: treatment of Helicobacter pylori infection. Am J Gastroenterol. 2017;112(2):212‐239. [DOI] [PubMed] [Google Scholar]
- 28. Livingston G, Sommerlad A, Orgeta V, et al. Dementia prevention, intervention, and care. Lancet. 2017;390(10113):2673‐2734. [DOI] [PubMed] [Google Scholar]
- 29. Rothman KJ. Induction and latent periods. Am J Epidemiol. 1981;114(2):253‐259. [DOI] [PubMed] [Google Scholar]
- 30. Durrleman S, Simon R. Flexible regression models with cubic splines. Stat Med. 1989;8(5):551‐561. [DOI] [PubMed] [Google Scholar]
- 31. Kurtz JR, Goggins JA, McLachlan JB. Salmonella infection: Interplay between the bacteria and host immune system. Immunol Lett. 2017;190:42‐50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lipsitch M, Tchetgen Tchetgen E, Cohen T. Negative controls: a tool for detecting confounding and bias in observational studies. Epidemiology. 2010;21(3):383‐388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. VanderWeele TJ, Ding P. Sensitivity analysis in observational research: introducing the E‐value. Ann Intern Med. 2017;167(4):268‐274. doi: 10.7326/M16-2607 [DOI] [PubMed] [Google Scholar]
- 34. Doulberis M, Kotronis G, Thomann R, et al. Review: Impact of Helicobacter pylori on Alzheimer's disease: what do we know so far? Helicobacter. 2018;23(1) [DOI] [PubMed] [Google Scholar]
- 35. Zendehdel A, Roham M. Role of Helicobacter pylori infection in the manifestation of old age‐related diseases. MGG. 2020;8(4):e1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Budzyński J, Kłopocka M. Brain‐gut axis in the pathogenesis of Helicobacter pylori infection. World J Gastroenterol. 2014;20(18):5212‐5225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shindler‐Itskovitch T, Ravona‐Springer R, Leibovitz A, Muhsen K. A systematic review and meta‐analysis of the Association between Helicobacterpylori Infection and dementia. J Alzheimers Dis. 2016;52(4):1431‐1442. [DOI] [PubMed] [Google Scholar]
- 38. Jeong SM, Shin DW, Lee JE, Hyeon JH, Lee J, Kim S. Anemia is associated with incidence of dementia: a national health screening study in Korea involving 37,900 persons. Alzheimers Res Ther. 2017;9(1):94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Muhsen K, Cohen D. Helicobacter pylori infection and iron stores: a systematic review and meta‐analysis. Helicobacter. 2008;13(5):323‐340. [DOI] [PubMed] [Google Scholar]
- 40. Wang HX, Wahlin A, Basun H, Fastbom J, Winblad B, Fratiglioni L. Vitamin B(12) and folate in relation to the development of Alzheimer's disease. Neurology. 2001;56(9):1188‐1194. [DOI] [PubMed] [Google Scholar]
- 41. Fulop T, Witkowski JM, Bourgade K, et al. Can an infection hypothesis explain the beta amyloid hypothesis of Alzheimer's disease? Front Aging Neurosci. 2018;10:224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Reshetnyak VI, Burmistrov AI, Maev IV. Helicobacter pylori: commensal, symbiont or pathogen? World J Gastroenterol. 2021;27(7):545‐560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Liou J‐M, Malfertheiner P, Lee Y‐C, et al. Screening and eradication of Helicobacter pylori for gastric cancer prevention: the Taipei global consensus. Gut. 2020;69(12):2093. [DOI] [PubMed] [Google Scholar]
- 44. Ford AC, Yuan Y, Moayyedi P. Helicobacter pylori eradication therapy to prevent gastric cancer: systematic review and meta‐analysis. Gut. 2020;69(12):2113. [DOI] [PubMed] [Google Scholar]
- 45. Take S, Mizuno M, Ishiki K, et al. Risk of gastric cancer in the second decade of follow‐up after Helicobacter pylori eradication. J Gastroenterol. 2020;55(3):281‐288. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information
Supplemental Information.
Data Availability Statement
This study is based in part on data from the Clinical Practice Research Datalink obtained under license from the UK Medicines and Healthcare products Regulatory Agency. The data are provided by patients and collected by the UK National Health Service as part of their care and support. The interpretation and conclusions contained in this study are those of the authors alone. Because electronic health records are classified as “sensitive data” by the UK Data Protection Act, information governance restrictions (to protect patient confidentiality) prevent data sharing via public deposition. Data are available with approval through the individual constituent entities controlling access to the data. Specifically, the primary care data can be requested via application to the Clinical Practice Research Datalink (https://www.cprd.com).