

Abstract
Immunotherapy has been a remarkable clinical advance in the treatment of cancer. T cells are pivotal to the efficacy of current cancer immunotherapies, including immune-checkpoint inhibitors and adoptive cell therapies. However, cancer is associated with T cell exhaustion, a hypofunctional state characterized by progressive loss of T cell effector functions and self-renewal capacity. ‘Un-exhausting’ T cells in the tumour microenvironment is commonly regarded as a key mechanism of action for immune-checkpoint inhibitors, and T cell exhaustion is considered a pathway of resistance for cellular immunotherapies. Several elegant studies have provided important insights into the transcriptional and epigenetic programmes that govern T cell exhaustion. In this Review, we highlight recent discoveries related to the immunobiology of T cell exhaustion that offer a more nuanced perspective beyond this hypofunctional state being entirely undesirable. We review evidence that T cell exhaustion might be as much a reflection, as it is the cause, of poor tumour control. Furthermore, we hypothesize that, in certain contexts of chronic antigen stimulation, interruption of the exhaustion programme might impair T cell persistence. Therefore, prioritization of interventions that mitigate the development of T cell exhaustion, including orthogonal cytoreduction therapies and novel cellular engineering strategies, might ultimately confer superior clinical outcomes and the greatest advances in cancer immunotherapy.
ToC blurb
T cells are key effectors of immunotherapies that have revolutionized the treatment of cancer; however, chronic exposure to tumour-associated antigens can result in progressive loss of T cell effector functions and self-renewal capacity, a hypofunctional state termed ‘T cell exhaustion’ that is believed to limit the efficacy of immunotherapy. This Review synthesizes the new immunobiological insights that present a more nuanced view beyond T cell exhaustion being entirely undesirable and indicate that this hypofunctional state might be a reflection — rather than the cause — of poor tumour control. Hence, the authors describe how, in certain contexts, interruption of this programme could impair T cell persistence and discuss interventions to mitigate the development of T cell exhaustion that might ultimately improve clinical outcomes.
Introduction
Immunotherapies, particularly immune-checkpoint inhibitors (ICIs) and adoptive cell transfer, have revolutionized the treatment of cancer1,2. These advances have led to substantial research and clinical interest in T cell exhaustion, which is a cellular differentiation pathway associated with progressive declines in T cell effector functions in the setting of chronic antigen exposure (BOX 1). In this context, persistent stimulation of the T cell receptor (TCR) and downstream Ca2+ signalling activates the transcription factor NFAT, which in turn drives expression of transcription factors from the TOX3,4 and NR4A5 families. Thus, NFAT signalling in T cells leads to transcriptional upregulation of multiple inhibitory immune-checkpoint proteins, including PD-1, CTLA4, CD39 and LAG-36. The process of T cell exhaustion spans a spectrum from highly proliferative T cells with stem-like properties (termed ‘precursor exhausted T cells’) to T cells with complete loss of effector function and replicative capacity (frequently termed ‘terminally exhausted T cells’). While the precise definition of an exhausted T cell remains a subject of healthy debate7, we herein define T cell exhaustion as a state characterized by reduced effector function at the time of tumour cell encounter that is associated with expression of co-inhibitory markers and reduced production of inflammatory cytokines (such as IFNγ and TNF). Although parallels can be drawn with other hypofunctional T cell states, such as ignorance, anergy and tolerance (BOX 1)6, T cell exhaustion typically relates to T cells that have already been activated (primed) by antigen-presenting cells in secondary lymphoid organs and are undergoing secondary antigen encounter in the tumour microenvironment (TME). T cell exhaustion, as assessed by PD-1 expression on intratumoural lymphocytes, is frequently associated with poor patient survival across a number of different cancer types8–15; therefore, a broader understanding of factors underlying this T cell state could enable further improvement of cancer immunotherapies.
Box 1 |. Hypofunctional states of T cells.
Multiple mechanisms can result in T cell hypofunction, including ignorance, anergy, tolerance and exhaustion, defined as follows.
Ignorance: A lack of T cell response owing to a low level of antigen expression, usually early during tumorigenesis.
Tolerance: Central or peripheral inactivation of self-reactive T cells.
Anergy: A hypofunctional state of T cells following antigen-induced T cell receptor (TCR) and downstream NFAT signalling in the absence of co-stimulatory signals from professional antigen-presenting cells (APCs) and/or CD4+ T cell help. Anergic T cells can express the inhibitory immune-checkpoint receptors PD-1 and CTLA4, and thus generally are not distinguishable from exhausted T cells.
Exhaustion: Progressively declining T cell function owing to chronic TCR stimulation in the setting of persistent antigen exposure. What distinguishes this process from anergy is that these T cells have undergone productive initial activation (for example, with co-stimulation by professional APCs in secondary lymphoid organs). Exhausted T cells are characterized by reduced cytokine production and the expression of inhibitory receptors, such as PD-1, CTLA4 and LAG-3, as well as the immunosuppressive enzyme CD39. These receptors are also expressed by functional T cells following physiological, APC-mediated priming; however, when expressed intratumourally (particularly in patients with cancer in whom tumours typically form over the course of months to years), these markers denote exhausted T cells. A spectrum of T cell exhaustion states exists, ranging from precursor exhausted to terminally exhausted, with concordant reductions in effector function and proliferative capacity.
Notably, these mechanisms were first defined under specific experimental conditions. In a clinical context, no definitive phenotype has been identified that can distinguish between these T cell states. Moreover, rather than manifesting a specific T cell phenotype, each of these states can result in an absence of the hypofunctional T cell clones intratumorally, further complicating identification of the relevant mechanism. T cells found in the tumour might be hypofunctional owing to anergy or exhaustion, or both. Hence, the composite term ‘dysfunctional’ has been favoured by some groups6,7. Other hypofunctional states of T cells have been described that overlap with the four typical categories described above, including terminal differentiation234, activation-induced cell death235, senescence236, burned-out237 and impaired metabolism238. Indeed, the degree of overlap among these hypofunctional T cell states is an area of ongoing research, and further clarification of the transcriptional, epigenetic and metabolic pathways that underlie each of these cell programmes will lead to a better understanding of their relative contributions to inadequate tumour control.
The initial report of T cell exhaustion in the presence of high levels of antigen exposure dates back to the 1960s, prior to the development of immunological techniques enabling longitudinal analyses of T cell specificities (reviewed in16). In 1998, the groups of Rafi Ahmed and Rolf Zinkernagel used fluorochrome-labelled peptide–MHC tetramers to track lymphocytic choriomeningitis virus (LCMV)-specific CD8+ T cells following infection of mice with this virus17,18. This innovation facilitated the first description of a hypofunctional CD8+ T cell state associated with an inability to clear a viral pathogen17,18. In 2006, Ahmed’s group and their collaborators further demonstrated that even the markedly impaired responses of ‘helpless’ CD8+ T cell against LCMV (in mice depleted of CD4+ T helper cells) could be rescued through antibody-mediated blockade of the PD-1/PD-L1 immune checkpoint19. Concurrent with these discoveries of hypofunctional T cell states in the setting of chronic viral infections, the groups of James Allison and Jeffrey Bluestone discovered in the mid-1990s that CTLA4 acts as an co-inhibitory receptor in T cells20,21. Allison and colleagues also demonstrate that antibody-mediated blockade of CTLA4 enhances antitumour immunity in mice20. Work by the groups of Tasuku Honjo, Arlene Sharpe and Gordon Freeman subsequently identified the PD-1–PD-L1 axis as another immune checkpoint limiting T cell function22,23. Studies from the laboratories of Honjo and Lieping Chen demonstrated that abrogation of PD-L1 enhanced antitumour responses in mice24,25. In 2011, a gene-expression profiling study by the Speiser group showed that human melanoma-reactive tumour-infiltrating lymphocytes (TILs) have an ‘exhaustion profile’ similar to that previously reported for chronic viral infection, including expression of CTLA4 and PD-126. Pivotal preclinical and translational studies such as these paved the way to the first approved immunotherapies targeting CTLA4 and PD-1, initially for metastatic melanoma27–29 followed rapidly by an expanding list of approvals across a range of other cancer types2. Seminal work by multiple groups has demonstrated that T cell exhaustion is epigenetically encoded, for example, through reduced chromatin accessibility of genes involved in effector functions and increased accessibility of the genes encoding TOX transcription factors and co-inhibitory receptors, and that PD-1 inhibition can only partially reverse the transcriptional phenotype associated with exhaustion30–32. Taking these remarkable discoveries together, a straightforward interpretation emerges in which removal of the molecular ‘brakes’ from T cells using ICIs can reinvigorate clonally expanded CD8+ T cells and thereby result in clinical benefit, albeit only certain T cell subsets can be reinvigorated. Notwithstanding, logical hypotheses that extend from this conceptual understanding of immunological exhaustion include: (1) abrogation of additional immune checkpoints (such as LAG-3, TIM-3 and/or TIGIT) at the tumour–T cell interface might improve on the success of CTLA4 and PD-(L)1 inhibitors; and (2) interruption of the molecular exhaustion programme can yield therapeutic benefit in the setting of T cell-mediated immunotherapy.
Parallel to the rise of ICI therapy for solid tumours, adoptive transfer of genetically engineered chimeric antigen receptor (CAR) T cells has revolutionized therapy for patients with treatment-refractory lymphoid malignancies33. CAR T cells are autologous (or potentially allogeneic) T cell products with redirected antigen specificity, achieved through expression of a exogenous CAR construct consisting of an extracellular tumour-targeting motif (typically an antibody-derived single-chain variable fragment) coupled to intracellular signalling domains derived from the TCR CD3ζ chain and co-stimulatory receptors33,34. Initial clinical response rates are typically high with CAR T cells targeting CD19-expressing and BCMA-expressing B cell malignancies35–38,39,40, and these therapies have proven effective and even curative for a subset of patients41,42.
In spite of these important advances, most patients receiving the currently approved CAR T cell therapies will ultimately have disease relapse. As representative examples, the ZUMA-1 study of axicabtagene ciloleucel in adults with multiply-relapsed large B cell lymphoma (LBCL) showed a 42% continuing response rate at a median follow-up duration of 15 months35, and the ELIANA study of tisagenlecleucel in paediatric and young-adult patients with B cell acute lymphoblastic leukaemia (B-ALL) revealed relapse-free survival of <50% even among responders and despite a high rate of consolidation with allogeneic haematopoietic stem cell transplantation43. Disease relapse eventually occurs in almost all patients receiving BCMA-directed CAR T cell products for multiple myeloma39, and durable response rates with most investigational CAR T cell products for solid tumour types are low44. These data emphasize the need for a better understanding of disease relapse following CAR T cell therapy, including the potential contribution of T cell exhaustion, with the ultimate aim of enhancing the existing therapies and expanding this treatment modality to new diseases, including solid malignancies.
Herein, we review the important immunological insights into T cell exhaustion gleaned over the past decade. In light of these findings, we propose that T cell exhaustion is a reflection that the totality of antineoplastic efforts is insufficient to successfully reduce tumour antigen burden, rather than simply a cause of inadequate tumour control. Moreover, we discuss how abrogation of the exhaustion programme could, in certain contexts, be counterproductive for T cell-mediated antitumour immunity. We do not intend to imply that T cell exhaustion is not clinically actionable; instead, we propose that strategies focused on mitigating (or avoiding), as opposed to interrupting, T cell exhaustion might yield greater clinical efficacy. This shift in thinking will be informative for the design of next-generation immunotherapies that build on the clinical successes of ICIs and CAR T cell therapies. While CD4+ T cells also undergo T cell exhaustion in cancer45–47, CD8+ T cell exhaustion is better characterized, particularly in the setting of ICI treatment. Thus, the early sections of this Review are focused on CD8+ T cell exhaustion specifically, whereas the later sections on adoptive T cell therapies are subset-agnostic given that infusion of both CD4+ T cells and CD8+ T cells is crucial for the efficacy of such therapies48.
A balanced view of T cell exhaustion
Exhausted CD8+ T cells are enriched for tumour-reactive clones.
In 2014, Gros et al.49 reported that in patients with melanoma, CD8+ TILs expressing PD-1, LAG-3 and/or TIM-3 comprise an oligoclonal T cell population enriched for clonotypes capable of recognizing autologous tumour cells. Subsequent work by the same group demonstrated that circulating PD-1+ lymphocytes in patients with melanoma and gastrointestinal cancers are also enriched for tumour-reactive and neoantigen-specific T cells50,51. The preferential tumour-antigen reactivity of intratumoural PD-1highCD8+ T cells was also demonstrated in patients with non-small-cell lung cancer (NSCLC)52. Moreover, tumour antigen-linked MHC tetramers were reported to preferentially bind to CD39-expressing CD8+ TILs in lung and colorectal cancers (as opposed to bystander, mostly virus-specific CD8+ T cells lacking CD39 expression)53. Concordantly, CD39+CD103+CD8+ TILs from patients with melanoma or head and neck cancer demonstrated selective reactivity to autologous tumour cells and, for the one patient analysed, enhanced cytotoxicity compared with CD103− and/or CD39− CD8+ TILs from the same patient54. Of note, the CD39+CD103+CD8+ TILs also expressed high levels of PD-1, CTLA4 and TIM-354.
A subsequent single-cell RNA sequencing study of human melanomas revealed that greater than one-third of CD8+ TILs has a T cell dysfunction-related gene signature, characterized by high expression of PDCD1 (encoding PD-1), ENTPD1 (encoding CD39), LAG3, HAVCR2 (encoding TIM-3), ITGAE (encoding CD103), CXCL13, TNFRSF9 (encoding 4-1BB) and a number of cellular proliferation-related genes, which surprisingly suggested enhanced rather than reduced replicative capacity55. Moreover, CD8+ TILs obtained from patient biospecimens with enrichment for this transcriptional signature of T cell dysfunction preferentially demonstrated reactivity against autologous tumour digests55. In a Review published in 2020, the authors summarized these findings and identified PD-1, CD39, TIM-3, LAG-3, CXCL13, 4-1BB, GITR, proliferation markers and an oligoclonal TCR repertoire as hallmarks of tumour-reactive CD8+ T cells56. In several more-recent studies across multiple solid tumours, CD8+ TILs with empirically validated tumour reactivity were all marked by expression of PDCD1, ENTPD1 and CXCL1357–60. Notably, many of these markers of tumour reactivity are also generally consider to be markers of CD8+ T cell exhaustion. By contrast, bystander T cells, including those with specificity for viral pathogens, typically lack expression of these markers61, and thus have low expression of tumour-reactivity signatures. Also notable is that a high pre-treatment or early on-treatment abundance of CD8+ TILs expressing exhaustion markers such as PD-1, CD39 and/or CTLA4 is predictive of a clinical benefit from ICIs across tumour types52,62–68. Thus, exhausted CD8+ T cells are enriched for tumour reactivity and their presence intratumourally prior to or early after ICI treatment is associated with improved outcomes.
T cell exhaustion is inevitable in the setting of persistent tumour.
One interpretation of the aforementioned findings is that exhaustion of CD8+ TILs is inevitable in the context of chronic antigen exposure. Consistent with the ‘decreasing potential’ model posited two decades ago69, the picture that emerges is a linear diagram of progressive T cell exhaustion owing to successive stimulation with a persistent tumour antigen (FIG. 1a)70,71. Importantly, this model does not preclude a role for other factors, such as resistance of target tumour cells to cytotoxicity72, hypoxia73–75, immune contexture76 or TCR signal quality and/or quantity, in modifying the tempo of exhaustion.
Fig. 1 |. Tumour-reactive CD8+ T cells undergo progressive exhaustion in the TME.
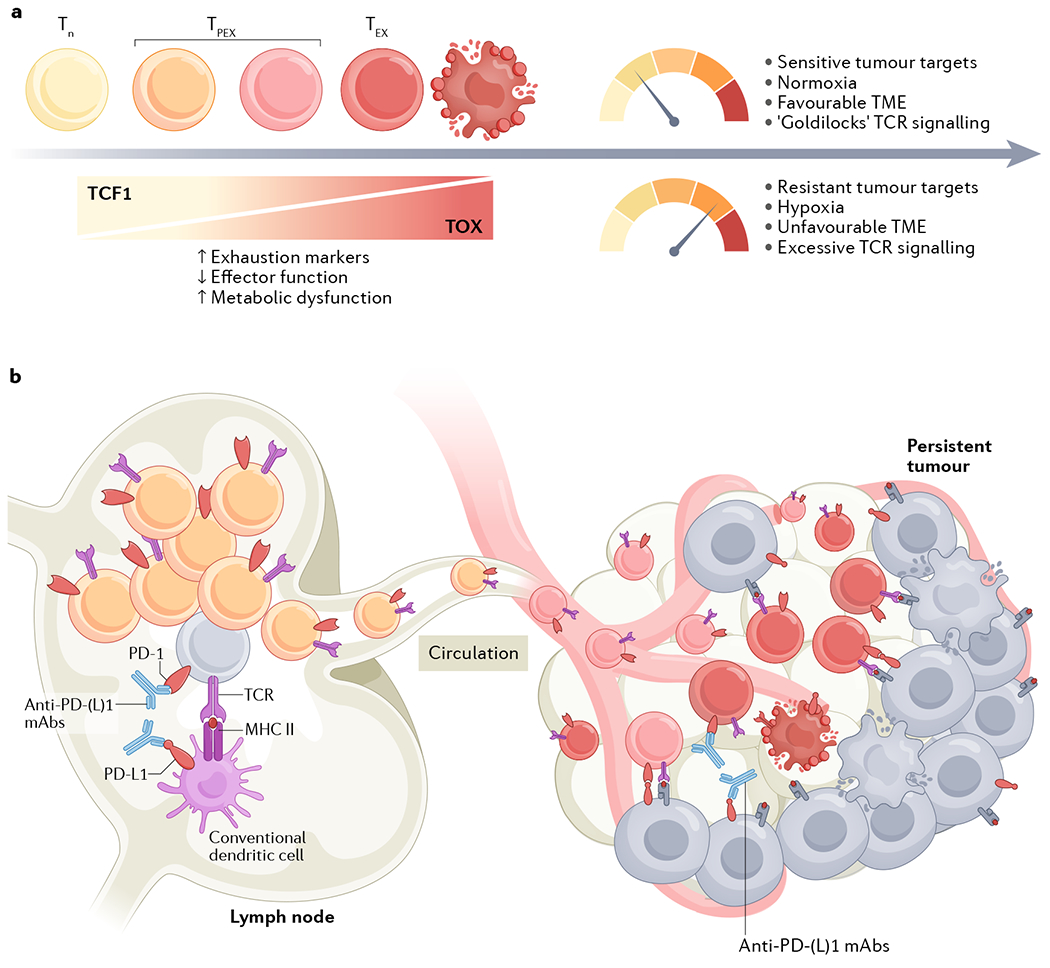
a | Naive CD8+ T cells (Tn) can differentiate along of spectrum of exhaustion states associated with progressively declining functional and proliferative capacity, spanning from stem-like precursor exhausted T cells (TPEX) to terminally exhausted T cells (TEX) that can ultimately undergo overstimulation-induced cell death. In the setting of persistent exposure to tumour antigen, expression of the transcription factor TCF1 in the T cell decreases while expression of TOX family transcription factors increases. As a result, expression of exhaustion markers (such PD-1, TIM-3, LAG-3 and CD39) will increase, effector function will decrease and the T cell will incur metabolic dysfunction. However, the tempo at which an individual T cell becomes exhausted is context-dependent (as indicated in the top right of the figure); the various T cell-intrinsic and T cell-extrinsic factors that modulate this process include the antigenic load, tissue oxygen concentration, immune contexture and target cell sensitivity to T cell-mediated cytotoxicity. b |Although the key mechanism of anti-PD-(L)1 antibody therapy was initially believed to be functional reinvigoration of exhausted T cells in the tumour microenvironment (TME), more recent data indicates that abrogation of the priming interaction between dendritic cells and precursor exhausted CD8+ T cells in tumour-draining lymph nodes is the crucial mechanistic target of such immune-checkpoint inhibitors.
A corollary of T cell exhaustion being inevitable in the setting of chronic antigen exposure is that the time of assessment in preclinical experiments and in clinical trials is crucial to the interpretation of exhaustion. This factor might partially explain the seeming paradox that evidence of CD8+ T cell exhaustion in pre-treatment and early on-treatment tumour samples predicts clinical benefit from ICIs52,62–68 (implying that it is indicative of a pre-existing antitumour T cell response that can be enhanced by therapeutic intervention), whereas the same signal observed in late on-treatment and post-treatment readouts portends unfavourable outcomes57,77 (given that exhaustion as an indicator of persistent cancer is still present despite therapeutic intervention).
One therapeutic implication emerging from this interpretation is that concomitant use of ICIs with orthogonal (that is, non-ICI) interventions that are cytotoxic to tumour cells might confer additive benefits. For example, ICIs combined with chemotherapy now constitute the standard-of-care front-line treatment for patients with advanced-stage lung cancers78–82. When such combinations were initially being tested in clinical trials, the pervasive belief was that chemotherapy could be detrimental to the benefit of ICIs owing to the elimination of effector T cells, either owing to direct cytotoxicity or potentially indirectly through the immunosuppressive influence of corticosteroid pre-treatment to mitigate the adverse effects of chemotherapy. Although the benefits of chemotherapy and immunotherapy might not be synergistic83, it is mechanistically intuitive that ICIs will have better activity in a setting in which chemotherapy has achieved some degree of tumour cytoreduction. Indeed, a high baseline tumour burden is associated with unfavourable response rates and overall survival in patients with metastatic melanoma or head and neck cancer receiving ICIs84–86. Using PD-1 or CD39 as markers of exhausted intratumoural CD8+ T cells, various cytotoxic chemotherapy agents have been shown to reduce T cell exhaustion in preclinical tumour models87,88. Similarly, patients receiving neoadjuvant chemotherapy have reductions in the abundance of exhausted intratumoural T cells89,90. Hence, orthogonal oncological approaches to reduce tumour burden (such as chemotherapy, receptor tyrosine kinase-targeted therapies, radiotherapy and/or surgical resection) might improve the activity of ICI and CAR T cells by reducing the antigenic load, which drives T cell exhaustion. This effect might partially explain why ICIs have shown substantial antitumour activity when added to neoadjuvant chemotherapy in patients with early stage NSCLC, potentially secondary to the cytoreductive effect of chemotherapy91,92. The common narrative is that T cell exhaustion is an important cause of poor tumour control, but perhaps it is equally true that T cell exhaustion is simultaneously a consequence of poor tumour control.
New insights on the mechanisms of ICIs
PD-L1+ cDCs in tumour-draining lymph nodes are a key target of ICIs.
If reversal of the exhaustion programme in tumour-reactive effector CD8+ T cells in the TME is the predominant therapeutic mechanism of ICIs, one would expect efficacy to be preserved in preclinical experiments in which tumour infiltration of new T cells is blocked. In multiple mouse models, however, blockade of T cell egress from secondary lymphoid organs using the sphingosine 1-phoshate receptor agonist FTY720 after tumour challenge but before treatment with anti-PD-(L)1 antibodies abrogates the therapeutic benefit93–98. Given that blockade of lymphoid egress effectively eliminates recruitment of new CD8+ T cells into the tumour, these findings suggest that reinvigoration of T cells in the TME alone is insufficient to mediate the preclinical efficacy of anti-PD-(L)1 antibodies. This hypothesis raises the question of which site outside the TME is the key target for blockade of the PD-1 axis.
Profiling of immune cells in the tumour-draining lymph nodes (TDLNs) of mice bearing peritoneal tumours revealed both enrichment for tumour-reactive PD-1+ T cells and high levels of PD-L1 expression on myeloid cell populations98. Notably, intrapleural infusion of anti-PD-L1 antibodies at a dose titrated to block PD-L1 on myeloid cells only in the mediastinal TDLN was sufficient to confer therapeutic benefit in this model98. Moreover, the quantity of interactions between PD-1 and PD-L1 in TDLNs, as opposed to in the primary tumour, was found to predict benefit from ICIs in patients with metastatic melanoma98. These findings were recapitulated in a separate study using mouse models of melanoma and breast cancer99. In these models, intradermal injection of anti-PD-L1 antibodies resulted in preferential targeting of TDLNs and superior antitumour activity, as compared with typical intraperitoneal (systemic) administration99. These results suggest that interactions between T cells and antigen-presenting cell in TDLNs, as opposed to solely in the primary tumour, are crucial for the efficacy of anti-PD-(L)1 antibodies, which implies that lymphoid tissues must harbour a population of PD-L1+ cells that can be perturbed by ICIs.
In the aforementioned study in mice bearing peritoneal tumours98, PD-1+ T cells in TDLNs were found in close proximity to PD-L1+ conventional dendritic cells (cDCs), with evidence suggesting that blockade of PD-L1 on these cDCs results in the induction of precursor-exhausted T cells that migrate to the tumour. Moreover, genetic deletion of PD-L1 in Clec9a-expressing cDCs, but not LysM (lysozyme M)-expressing macrophages, has been shown to abrogate the efficacy of anti-PD-L1 antibodies in a PD-L1-deficient MC38 colon adenocarcinoma model100. Similarly, genetic ablation of PD-L1 in cells expressing CD11c (predominantly found on DCs) impairs the growth of PD-L1-deficient MC38 tumours and abolishes the enhanced antitumour immunity conferred by PD-L1 inhibitors101. Furthermore, loss of PD-L1-inhibitor efficacy in Batf3-deficient mice, which lack cross-presenting cDC1, indicates that ICIs abrogate the co-inhibitory interactions between PD-L1+ cDCs and PD-1+CD8+ T cells, thereby enhancing T cell activation101. Together, these results indicate that cDCs in the TDLNs are a crucial target for blockade of the PD-1 axis.
ICI efficacy is dependent on TCF1+ precursor exhausted CD8+ T cells.
If the efficacy of anti-PD-(L)1 antibodies is dependent on cellular interactions in the TDLN, as opposed to the TME, another implication is that a lymphoid-resident tumour-reactive PD-1+CD8+ T cell population must be the key target of invigoration. Work from multiple groups has collectively demonstrated that a CXCR5+TCF1+PD-1+CD8+ T cell population residing in lymphoid tissues serves as the source of precursors for terminally exhausted CD8+ T cells in the setting of chronic viral infections102–105. The self-renewal of these precursor exhausted CD8+ T cells was found to be dependent on expression of TCF7, which encodes the transcription factor TCF1102–105. In both mouse and human tumours, infiltrating T cells undergo progressive exhaustion with concomitant loss of TCF1 expression owing to epigenetic silencing of the Tcf7/TCF7 locus (FIG. 1a)30. CXCR5+TCF1+PD-1+CD8+ T cells have variously been called ‘progenitor exhausted’, ‘memory-like’, ‘stem-like’ and ‘follicular cytotoxic’, but ‘precursor exhausted’ is likely the most accurate term (reviewed in106). Indeed, single-cell profiling of tumour-infiltrating CD8+ T cell from patients with melanoma or NSCLC revealed the presence of a partially exhausted population identified by expression of either TCF177 or CXCR5107. Subsequent data from mouse models definitively demonstrated that TCF1+PD-1+CD8+ T cells (that also express SLAMF6, but not TIM-3) served as precursors for exhausted CD8+ T cells in tumours, and importantly, it was this precursor population that underwent a proliferative burst after blockade of the PD-1 axis108–110. The crucial requirement of these precursor exhausted CD8+ T cells for the efficacy of ICIs has been demonstrated elegantly in multiple model systems, including through adoptive transfer experiments, depletion of the TCF1+ population using an inducible diphtheria toxin receptor-based ablation system (Tcf7DTR-GFP), or deletion of Tcf7 under the control of the E8i CD8-Cre promoter (E8i-Cre+ Tcf7fl/fl)108–110. For example, following adoptive transfer of LCMV gp33-specific Tcf7DTR-GFP CD8+ T cells (P14 T cells) into mice, selective depletion of Tcf7-expressing cells via diphtheria toxin administration reduced the total number of P14 T cells (including the abundance of the Tcf1− subset) in gp33-expressing melanomas and impaired tumour control108. Similarly, OT-1 CD8+ T cells (specific for an ovalbumin antigen) were found at a lower frequency in ovalbumin-expressing MC38 tumours from E8i-Cre+ Tcf7fl/fl mice (which lack TCF1 expression in CD8+ T cells) compared with their wild-type counterparts, and this deficiency was associated with impaired antitumour immunity109. Further heterogeneity within this TCF1+ precursor exhausted population is likely, with a CD62L+KIT− subset emerging as the subpopulation with the greatest progenitor capacity and anti-PD-1 responsiveness in a mouse chronic viral infection model111. Subsequent studies will further dissect the TCF1+ precursor exhausted population with the greatest progenitor capacity in the human tumour context.
The emergence of paired single-cell RNA and TCR sequencing data has confirmed that clonally-related CD8+ T cells can exist in the precursor exhausted and terminally differentiated states in human tumors112. Importantly, in a cohort of 14 patients with metastatic melanoma who derived clinical benefit from nivolumab and ipilimumab, a high proportion of TCF1+ cells among PD-1+CD8+ T cells (above the median of 14.9%) in pre-treatment biopsies was associated with substantially longer progression-free survival (median 681 days versus 392 days)110. Similarly, another analysis of pre-treatment tumour samples from 24 patients with metastatic melanoma demonstrated a higher ratio of TCF1+ to TCF1− CD8+ T cells in those who had a radiographical response following ICIs therapy77. Hence, TCF1+PD-1+CD8+ precursor exhausted T cells are a crucial target of ICIs.
If blockade of the PD-1 axis acts primarily in secondary lymphoid organs, one would expect to find evidence of a proliferative burst after anti-PD-(L)1 antibody treatment with subsequent trafficking of the expanded precursor exhausted cells through the circulation to tumour sites. Indeed, an increase in the abundance of Ki67+PD-1+CD8+ T cells in the peripheral blood of patients with melanoma occurs 3 weeks (and as early as 1 week) after the initiation of anti-PD-1 therapy; at least a subset of these PD-1+CD8+ T cells were CXCR5+ and TCF1+84,113. Notably, patients with metastatic melanoma with a high circulating Ki67+CD8+:tumour burden ratio (≥1.94) after one cycle of anti-PD-1 therapy had superior clinical outcomes84. Similarly, patients with NSCLC who have an increase in the abundance of peripheral blood Ki67+CD8+ T cells within 4 weeks of initiating anti-PD-1 therapy are more likely to have a clinical response114. These observations dovetail with reports of extensive clonal replacement of intratumoural CD8+ T cell populations after ICI therapy112,115,116, presumably from proliferating precursor exhausted T cells that originated in the TDLNs (FIG. 1b).
Implications of the new mechanistic insights on ICI efficacy.
Together, these findings challenge the commonly held view that the key target of PD-(L)1 inhibitors is contained exclusively within the TME. Of note, however, that these data are not wholly inconsistent with a direct effect of anti-PD-(L)1 antibodies on PD-1+CD8+ T cells in the TME. Indeed, CD8+ TILs isolated from patients with ovarian cancer retain ex vivo capacity for activation and cytotoxicity after blockade of the PD-1 axis117. Moreover, extensive single-cell RNA and TCR sequencing of longitudinal tumour samples from patients with NSCLC receiving ICIs in combination with chemotherapy revealed that, in addition to robust infiltration of new clonotypes, marked expansion of pre-existing CD8+ TIL clonotypes also occurs118. This dual source of antitumour T cells (that is, expansion of pre-existing TILs and clonal replacement by newly infiltrating T cells) was termed ‘clonal revival’, and it was theorized that different cancers have differential contributions from these two sources of tumour-reactive T cells118. We hypothesize that additional temporal and spatial heterogeneity in the contributions of these two T cell sources is likely, even within the same patient. For example, elegant studies using ex vivo tumour-fragment cultures have shown anti-PD-1 treatment can unleash a productive response by pre-existing CD8+ TILs that includes the release of cytotoxic markers (granzyme B, perforin, and granulysin), cytokines (IL-2, IFNγ, and TNF) and chemokine ligands (CXCL9, CXCL10, CCL5 and CXCL13)119, which might be crucial for recruitment of the circulating T cells that result in clonal replacement. Of note, this ex vivo response was preferentially observed in pre-treatment tissues obtained from patients who subsequently had a clinical response to ICIs. Consequently, in certain contexts, blockade of the PD-1 axis might modulate the activity of T cells directly in the TME.
Notwithstanding these results, the totality of the contemporary data suggests that the mechanistic ‘heavy lifting’ — at least in the context of ICI therapy — is accomplished by newly infiltrating CD8+ T cells. Thus, the picture that emerges is one in which the crucial target of anti-PD-(L)1 antibodies is the interaction between precursor exhausted PD-1+CD8+ T cells and PD-L1+ cDCs in TDLNs, which results in a proliferative burst of the precursor exhausted T cells that can be observed in the circulation and go on to differentiate towards terminally exhausted T cells in the TME (FIG. 1b). This paradigm broadens our understanding of why intratumoural expression of PD-L1 is an imperfect predictor of response to ICIs, and also explains the seeming paradox of why mice94,120–122 and patients2 with PD-L1-deficient tumours can still derive clinical benefit from such agents. Despite the methodological challenges of reliably measuring PD-L1 across pathologists, assays and disease types, NSCLC is one setting in which tumoural PD-L1 expression has consistently been associated with greater benefit from ICIs123. This correlation might reflect a scenario in which PD-L1 expression is induced by IFNγ derived from infiltrating tumour-reactive CD8+ T cells. In this scenario, tumoural PD-L1 might be predictive not because it is the crucial mechanistic target of PD-1–PD-L1 blockade, but rather because PD-L1 expression is a ‘canary in the coal mine’ associated with the presence of tumour-reactive CD8+ T cells in the periphery that can be mobilized with ICIs2. The pathways regulating PD-L1 expression are highly complex and can be influenced by genomic aberrations, transcriptional control, mRNA stability, oncogenic signalling and protein stability124. Hence, contexts exist in which tumoural PD-L1 expression is uncoupled from an antitumour CD8+ T cell response; therefore, some tumours with high levels of PD-L1 expression might not respond to inhibition of the PD-1 axis.
At least on the surface, the crucial importance of a peripherally-derived CD8+ T cell population might not mechanistically align with the findings presented above, for example, that the abundance of intratumoural exhausted T cells is predictive of ICI response. To elaborate, why would intratumoural expression of PD-1 and CD39 on infiltrating T cells predict a response if the therapeutic target of PD-(L)1 inhibition is remote from the tumour? One potential explanation is that the abundance of intratumoural tumour-reactive PD-1+CD39+CD8+ T cells is proportional to the number of tumour-reactive T cell clones in the periphery that can be invigorated upon blockade of the PD-1 axis. Indeed, tumour-reactive PD-1+ T cell clonotypes detected in peripheral blood are often also present within TILs51. Moreover, a study in 30 patients with triple-negative breast cancer revealed a correlation between tissue and blood levels of PD-1+CD8+ T cells and CD39+CD8+ T cells125. Conversely, the presence of few PD-1+ and CD39+ T cells in the tumour might indicate an overall paucity of CD8+ T cells in the patient that are capable of recognizing tumour antigen, and thus fewer tumour-reactive clones in the periphery that could be activated by ICIs.
Broadly, the new mechanistic insights discussed above have two implications for ICI therapy. First, caution should be exercised when the rationale for utilization of ICIs is upregulation of PD-L1 in the TME: certain cancer therapies might result in upregulation of PD-L1 on tumour cells, but this does not necessarily indicate that combination of these therapies with ICIs will yield greater clinical benefit. As stated above, this rationale might only be valid when PD-L1 expression is indicative of a pre-existing antitumour CD8+ T cell response, and not in contexts in which PD-L1 is upregulated by other tumour-intrinsic factors. Second, combinatorial ICI-based approaches should focus on candidate partner therapies that will amplify the total numbers of tumour-reactive T cells, rather than ‘un-exhausting’ terminally differentiated cells in the TME (which might provide limited therapeutic benefit given that these cells are in a fixed epigenetic state)30–32. By extension, further attention should be focused on therapies that lead to increases in the clone sizes of existing tumour-reactive T cell clonotypes and/or recruitment of CD8+ T cells with additional tumour-reactive TCR specificities. Approaches that might achieve these goals include vaccination with tumour antigens126, in situ vaccination via DC activation127, agonism of co-stimulatory receptors128,129, in vivo cytokine supplementation130,131 and adoptive T cell therapies.
Evidence of exhaustion in CAR T cells
As described previously, disease relapse after CAR T cell therapy remains a major challenge, motivating a desire to understand CAR T cell exhaustion as a potential mechanism of tumour escape39,41,132. In contrast to endogenous tumour-reactive T cells, CAR T cells are marked by their genetic modification, facilitating tracking and assessment of these tumour-targeted cells. Moreover, next-generation CAR T cell therapies can be further gene-edited to mitigate or interrupt exhaustion. These factors make CAR T cell therapy both a clinically relevant context and an important model in which to study T cell exhaustion.
Preclinical in vivo models have been crucial to understanding the CAR T cell exhaustion programme4,133,134. In a mouse model of melanoma, tumour-infiltrating CAR T cells have a PD-1+TIM-3high phenotype associated with increased expression of the canonical transcription factors of exhaustion, Tox and Tox24, and Nr4a1, Nr4a2 and Nr4a3133. Similarly, both chronic antigen-induced signalling135 and constitutive, low-level, basal (or ‘tonic’) signalling134 have been shown to drive CAR T cell exhaustion. These preclinical findings demonstrate that CAR T cells can have phenotypic, transcriptional and epigenetic profiles characteristic of exhaustion136,137.
Clinically, analyses of on-treatment or post-relapse biopsy samples would be informative to determine the extent to which CAR T cell exhaustion contributes to loss of disease control in patients. In patients with B-ALL, CD19-targeted 4-1BB-based CAR T cells acquire exhaustion-associated DNA-methylation signatures, including methylation of effector genes and demethylation of TOX, by 4 weeks after infusion138. Another study demonstrated persistent TIM-3+PD-1+ CAR T cells in the blood of patients with CLL several months after infusion, and these cells were enriched in the patients with inferior clinical outcomes139. Notwithstanding the comprehensive profiling of circulating exhausted CAR T cells in these two studies, there is a relative dearth of data on CAR T cell exhaustion in the TME, which is typically where exhaustion is most prominent. At least one group could not detect CAR T cells in post-treatment lymphoma biopsy samples by immunohistochemistry or flow cytometry as soon as 10 days after infusion140. A lack of persistence of the infused CAR T cells might limit the assessment of their exhaustion and explain the paucity of documented evidence of CAR T cell exhaustion intratumourally in patients.
Although the number of clinical reports characterizing post-infusion CAR T cells is low, the literature on in vitro CAR T cell phenotypes after manufacture and prior to infusion is comparatively robust. Products with higher proportions of less differentiated cells141–144, such as CD45RA+CCR7+ CAR T cells145, are associated with improved outcomes in a variety of haematological malignancies. In addition, CAR T cell products with higher levels of inhibitory receptors such as PD-1 and LAG-3 expand less following infusion and are associated with lower response rates in patients with chronic lymphocytic leukaemia (CLL)141, paediatric and young-adult CD19+ leukaemias146, or LBCLs147. Although T cells can show epigenetic hallmarks of exhaustion as early as 5–8 days post-stimulation148 (and therefore during CAR T cell manufacturing), co-inhibitory receptors are also expressed on highly functional, activated T cells. These pre-infusion data thus emphasize two important points: (1) other cell states beside exhaustion, including effector and memory differentiation, can explain T cell function and persistence, and (2) T cell exhaustion can be difficult to evaluate based on co-inhibitory receptor expression alone.
Interrupting CAR T cell exhaustion
Pre-infusion and post-infusion CAR T cell phenotypes taken together with data from preclinical in vivo modelling suggest, but do not conclusively demonstrate, a role for CAR T cell exhaustion in limiting clinical responses. If CAR T cells are indeed impaired by exhaustion, one would expect that interrupting this hypofunctional cell state would improve therapeutic efficacy. Interruption of CAR T cell exhaustion programme could potentially be achieved through abrogation of the canonical TOX and NR4A transcription factors or antibody-mediated blockade of co-inhibitory receptors, such as PD-1 (FIG. 2).
Fig. 2 |. Overview of strategies to mitigate or interrupt CAR T cell exhaustion.
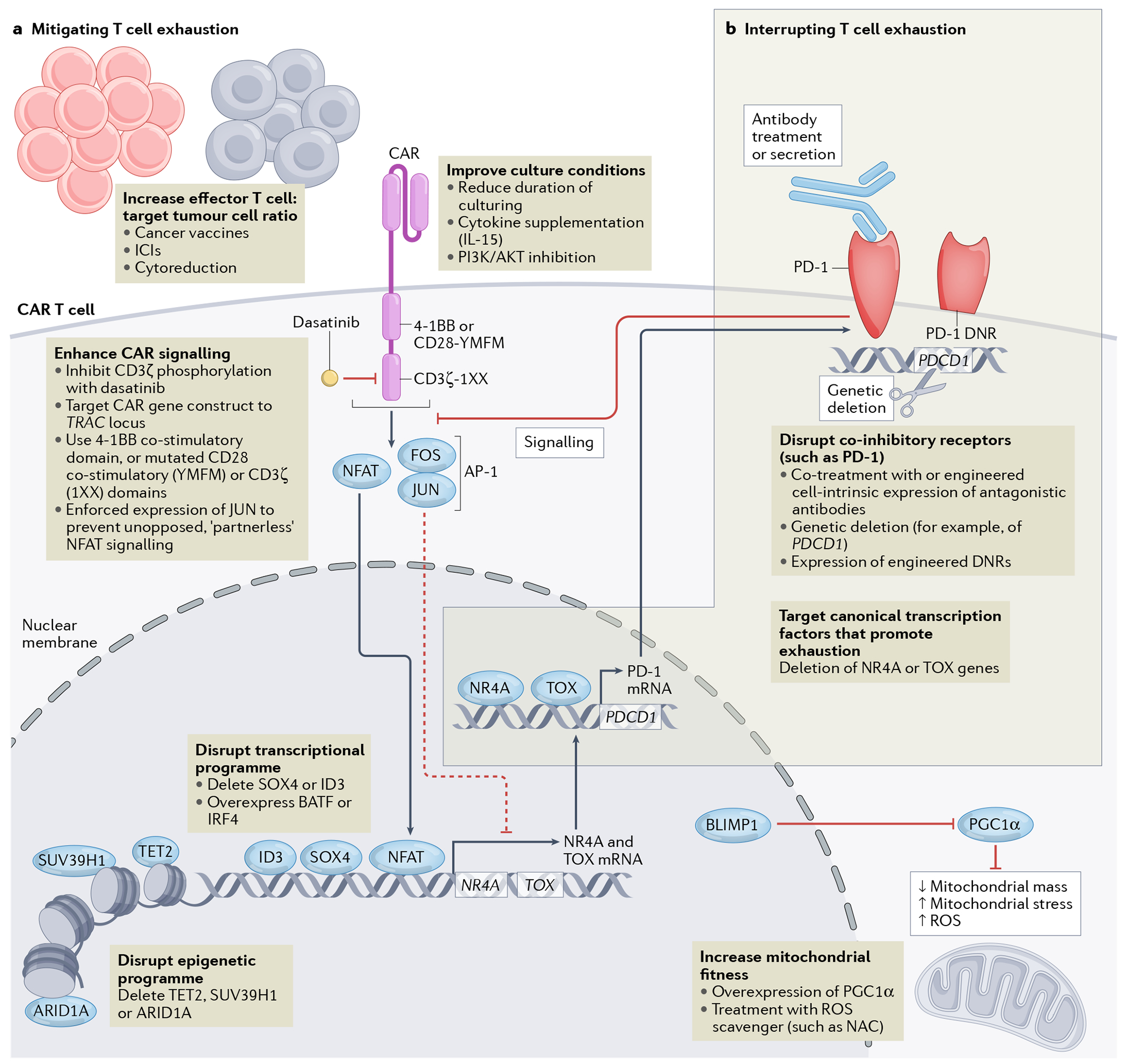
Downstream signalling induced by the chimeric antigen receptor (CAR) activates the transcription factor NFAT. Chronic NFAT signalling leads to expression of NR4A and TOX family transcription factors, which are crucial mediators of the epigenetic and transcriptional programme of T cell exhaustion. Cooperation between NR4A and TOX transcription factors results in the expression of genes encoding co-inhibitory receptors, such as PD-1, and also results in reduced cytokine production and effector function (not shown). Potential interventions to improve the function and thus therapeutic efficacy of CAR T cells are indicated in text boxes and are categorized as strategies that either mitigate T cell exhaustion (a) or more directly interrupt the exhaustion cascade downstream of NFAT signalling (b). a | Interventions to optimize effector-to-target ratios in the patient undergoing CAR T cell therapy, culture conditions during CAR T cell manufacturing, CAR signalling, transcriptional and/or epigenetic programmes and mitochondrial fitness might mitigate T cell exhaustion. b |More downstream interventions targeting the canonical transcription factors associated with T cell exhaustion (that is, NR4A and TOX) and co-inhibitory receptors are strategies to interrupt T cell exhaustion. DNR, dominant-negative receptor; ICIs, immune-checkpoint inhibitors; NAC, N-acetylcysteine; ROS, reactive oxygen species.
Numerous preclinical studies and at least 20 active clinical trials149 have been focused on blocking or eliminating signalling through PD-1 to enhance CAR T cell function. Functional abrogation of PD-1 in CAR T cells can be achieved in a variety of ways, with the most straightforward method being co-treatment with anti-PD-(L)1 antibodies. In patients with disease relapsed after CD19-directed CAR T cell therapy, treatment with such antibodies leads to re-expansion of CAR T cells in the peripheral blood150,151; however, clinical responses are infrequent, with one complete response and three partial responses in a cohort of 12 patients152. A larger, single-arm study in 28 patients anti-CD19 CAR T cells (axicabtagene ciloleucel) in combination with early PD-L1 blockade (atezolizumab initiated 1–21 days after cell infusion) for diffuse large B-cell lymphoma resulted in durable response rates similar to those of historical cohorts treated with CAR T cells alone, suggesting no added benefit from concomitant ICI therapy153. Data from a phase I trial testing the combination of a mesothelin-targeting CAR T cell product and the anti-PD-1 antibody pembrolizumab in 18 patients with malignant pleural mesothelioma has shown promising efficacy (median and 1-year overall survival of 23.9 months and 83%, respectively)154, although a subset of patients with mesothelioma will respond to single-agent anti-PD-1 therapy.
One aforementioned caveat of this approach is that blockade of the PD-1 axis is insufficient to epigenetically rewire the T cell exhaustion programme and, therefore, can only partially reverse the transcriptional phenotype of exhaustion30,31,155,156. Hence, whether co-inhibitory receptor blockade following CAR T cell infusion would be most effective early (when it could block inhibitory signalling in less-differentiated effector cells) or late (after the onset of exhaustion) is not clear. Whether CAR T cells develop a precursor exhausted state — which is most responsive to ICIs in other contexts — also remains unclear.
Among the several other methods to suppress signalling by PD-1 or other co-inhibitory receptors (beyond co-treatment with ICIs), one approach is to engineer CAR T cells themselves to secrete antagonistic antibodies (FIG. 2)157,158. Alternatively, overexpression of PD-1 dominant-negative receptors (DNRs)159 or gene-editing of the PDCD1 locus in CAR T cells can eliminate PD-1 signalling intrinsically160–163. Each approach has potential advantages and disadvantages. For example, in situ secretion of anti-PD-1 antibodies by CAR T cells might limit systemic antibody exposure and, therefore, toxicities. T cell-intrinsic gene-editing approaches can efficiently abrogate signalling in the edited cells, but do not potentiate endogenous T cell responses. Overexpression of a PD-1 DNR that lacks intracellular signalling domains can mitigate inhibitory signalling in the modified T cells by competing with endogenous PD-1 for binding to microenvironmental PD-L1. However, this effect of PD-1 DNR can theoretically be overcome by an excess of PD-L1 in the TME, whereas gene-edited T cells with a complete absence of PD-1 are insensitive to any level of PD-L1 expression. Each approach has been demonstrated to enhance the preclinical antitumour efficacy of CAR T cells across a variety of haematological and solid cancers.
More definitive conclusions will come from the multitude of ongoing clinical trials that are incorporating PD-1 abrogation in CAR T cell therapy. However, one early study in three patients who received CRISPR-Cas9-engineered T cells has yielded provocative results164. In these patients, CRISPR–Cas9-engineered cells expressing an NY-ESO-1-specific TCR and harbouring deletions of PDCD1 as well as TRAC and TRBC (encoding the TCR α-chain and β-chain constant regions, respectively) were tracked longitudinally; T cells harbouring a PD-1 deletion were found to be less persistent than PD-1-non-deleted T cells164. This finding might have alternate explanations unrelated to PD-1 biology, such as genotoxic stress from multiplex editing; however, it mirrors observations in mice in which Pdcd1-defiency and loss of inhibitory PD-1 signalling results in overstimulation-associated cell death of antigen-specific CD8+ T cells responding to a chronic viral infection165,166. Therefore, expression of co-inhibitory receptors during the effector phase might serve to restrain T cell overstimulation, and further studies will be needed to clarify the optimal method and timing of PD-1 abrogation that preserves T cell persistence and function.
Mitigating CAR T cell exhaustion
Consistent with the aforementioned findings indicating that terminal exhaustion of T cells is epigenetically fixed30–32 and the general agreement that these latter stages of exhaustion are therefore less amenable to therapeutic intervention7, interventions targeting earlier stages of the T cell differentiation pathway might be most effective in improving antitumour immunity. Rather than interrupting the T cell exhaustion programme, strategies to improve cell culture conditions and optimize CAR design and downstream signalling offer the potential to mitigate induction of this programme and thus enhance CAR T cell function (FIG. 2). Most of the strategies discussed below were developed for CAR T cell therapy, but many are also relevant for mitigating T cell exhaustion of TIL-based and TCR-engineered cell therapies (reviewed in167).
Increasing CAR T cell to tumour cell ratios.
Exhaustion is not purely a T cell-intrinsic process, and can depend on tumour and patient characteristics. T cell exhaustion can be modulated by metabolic73 and stromal168 factors in the TME, which might differ between cancer histologies. Moreover, features of the malignant cells can dictate the efficacy of CAR T cell therapies. For example, impaired death receptor signalling in leukaemia cells can induce CAR T cell exhaustion72, and the efficacy of CAR T cells against solid tumours, in contrast with haematological malignancies, has been found to be dependent on IFNγ receptor signalling169.
Beyond qualitative characteristics of the cancer cells, quantitative features might also determine the efficacy of CAR T cell therapy. Clinical benefit from CAR T cell therapy is inversely correlated with tumour burden across a variety of diseases, including ALL37 and LBCL170. One potential explanation for this finding is that higher antigen loads lead to sustained CAR stimulation and thus CAR T cell exhaustion, which further suggests the possibility of cytoreduction prior to CAR T cell treatment as a means of limiting exhaustion. This strategy can come in the form of surgery, radiotherapy, targeted therapies, immune checkpoint inhibitors, or chemotherapy. Notably, bridging chemotherapy might not always be effective171, given that many of the relapsed and/or refractory cancers historically treated with CAR T cells are chemo-refractory, but might be more effective as CAR T cells are used in earlier lines of therapy or novel agents are utilized for cytoreduction.
On the other side of the equation, one can consider increasing the numbers of CAR T cells infused. A retrospective study in 185 paediatric patients infused with CAR T cells found that event-free survival, relapse-free survival and overall survival were all increased with higher doses of tisagenlecleucel172. However, considering that prior treatment history and disease status can be confounders for CAR T cell yield, the question of whether higher CAR T cell doses would be beneficial needs to be addressed in a prospective clinical trial. Other approaches being studied that can potentially increase CAR T cell numbers in vivo include vaccination using the CAR-targeted antigen173.
Optimizing T cell culture conditions.
Manufacturing of clinical-grade CAR T cell products requires a collection of optimized and standardized operating procedures174. Substantial preclinical research has been focused on modifying this process to reduce exhaustion of CAR T cells. In a mouse xenograft model of B-ALL, CAR T cells that had been cultured for only 3 days as opposed to 9 days (7–14 days is standard) showed greater in vivo expansion and persistence as well as more robust tumour control, despite being infused at a six-fold lower dose175. Data indicated that the more-advanced differentiation of the 9-day CAR T cells limited their proliferative capacity and effector function175. The authors theorized that the 9-day CAR T cells were more exhausted175, suggesting that shorter culture durations might mitigate T cell exhaustion. Changing the cytokine composition of the culture might also limit T cell exhaustion; CAR T cells cultured in the presence of IL-15, as opposed to standard IL-2 supplementation, have reduced expression of exhaustion markers, enhanced antiapoptotic properties and increased proliferative capacity176. In addition, inhibition of the PI3K–AKT signalling pathway during CAR T cell manufacturing is associated with greater expansion, persistence and antitumour activity in mouse models177–179. Mechanistically, dual PI3Kδ/γ inhibition using duvelisib results in CAR T cells with dose-dependent decreases in levels of the exhaustion marker TIM-3, greater expression of TCF1 and increased mitochondrial fitness177. Hence, modifications in culture duration, cytokine supplementation and PI3K/AKT inhibition during CAR T cell manufacturing are promising avenues to test clinically with the aim of mitigating T cell exhaustion and thereby improving the efficacy of CAR T cell therapies.
CAR engineering to optimize signalling.
In endogenous T cells, exhaustion is driven by signals delivered upon repeated stimulation of the TCR180. In an elegant preclinical study by Shakiba et al.181, three different levels of TCR signal strength were achieved in tumour-specific CD8+ T cells through amino acid modifications of the target tumour antigen. Notably, high signal strength was associated with T cell exhaustion, but neither low-signal-strength or high-signal-strength TCRs led to optimal tumour control in mouse models181. Instead, an intermediate-signal-strength TCR resulted in the ‘Goldilocks’182 level of signalling for optimal in vivo antitumour activity (FIG. 1a)181. T cell persistence has also been correlated with TCR affinity, and thus signal strength, in humans. For example, the persistence of four different clonotypes of adoptively transferred T cells that recognized oncogenic KRASG12D in a patient with metastatic colorectal cancer was inversely correlated with the 3D binding affinity between their TCRs and cognate neoantigen–HLA-C complexes183,184. By analogy, the design and selection of CAR components can affect the signals that drive CAR T cell function, persistence and exhaustion.
The influence of CAR design on T cell exhaustion was first demonstrated for co-stimulatory domains. CARs designed with 4-1BB-based, compared to CD28-based, co-stimulation induce less T cell exhaustion185, with a corresponding metabolic signature of increased respiratory capacity, fatty acid oxidation and mitochondrial biogenesis (as opposed to enhanced glycolysis in T cells with CD28-based CARs)186. A phosphoproteomic analysis has demonstrated that both co-stimulatory domains induce similar signalling intermediates, although CD28-based CAR constructs activate faster and with greater potency187. Further studies identified a CD28 signalling motif (YMNM) responsible for greater VAV1 and PLCγ1 activation and calcium influx, which drives NFAT signalling and thus expression of the canonical transcription factors of exhaustion188. Modification of the asparagine in this motif to phenylalanine (YMFM) led to decreased exhaustion and enhanced persistence of the CAR T cells (FIG. 2)188. In line with these findings, 4-1BB-based CAR T cells demonstrate greater persistence than their CD28-based counterparts in some189 but not all190 models. However, this enhanced persistence might come at a cost, given that CD19-targeted CAR T cells with a CD28-based domain are more sensitive to low levels of antigen191,192. Reduction in antigen density has emerged as an important mechanism of immune escape from multiple engineered tumour-targeted agents191,193,194. Nonetheless, studies in mouse tumour models have demonstrated similar overall efficacy of CD28-based and 4-1BB-based CAR T cell products195. Furthermore, although direct comparisons are lacking, both co-stimulatory domains are in clinical use with similar efficacy196,197, despite the longer detectable persistence of 4-1BB-based CAR T cells198.
The strength of CAR signalling can also be modulated directly through editing of the TCR-derived CD3ζ signalling domain. In comparison with bead-based activation of the endogenous TCR, CAR activation induces more robust and rapid CD3ζ phosphorylation, and thus stronger signalling199. Modulation of CD3ζ signalling through elimination of two of three immunoreceptor tyrosine-based activation motifs (ITAMs) from this domain in a CD28-based CAR construct (called 1XX) leads to reduced T cell exhaustion and better persistence and tumour control in mouse models (FIG. 2)200.
Regulation of CAR expression can further control and optimize signalling. CRISPR-Cas9 editing of TRAC can enable homology-directed site-specific insertion of the CAR gene construct into this locus, placing CAR expression under the control of the endogenous TRAC promoter and its associated regulatory elements201. By contrast, standard retrovirally-transduced CARs integrate at semi-random sites within the genome, resulting in a broad range of expression levels (that is, variegation)201. CAR expression from the TRAC is uniform and recapitulates a TCR-like behaviour, such as transient suppression of signalling after stimulation owing to internalization, degradation and re-expression of the CAR201. TRAC-driven CAR expression engenders T cells with enhanced in vivo function and a less exhausted phenotype, resulting in greater efficacy compared with conventional CAR T cells in mouse models of B-ALL. Regulated, site-specific insertion of an engineered NY-ESO-1-specific TCR into T cells similarly leads to better tumour control, in association with lower expression of exhaustion markers202.
Tonic, antigen-independent signalling is an alternative source of chronic stimulation that can lead to excessive T cell exhaustion, with certain CAR constructs having an intrinsic propensity to drive constitutive, low-level downstream signalling185,203. This propensity for tonic signalling is determined by the CAR single-chain variable fragment, hinge and/or transmembrane domains and is thought to be mediated by CAR clustering on the cell membrane185,204. Tonic signalling manifests as CD3ζ phosphorylation and overexpression of transcription factors implicated in T cell exhaustion even in the absence of target antigen134. Notably, CAR constructs can be screened in vitro and in vivo for tonic signalling behaviour, enabling selection of constructs without this undesired behaviour205–207. The highly effective CD19-directed CAR constructs currently used in clinic have a low propensity for tonic signalling185, although some degree of tonic signalling might enhance responses in certain contexts208. Thus, a multitude of strategies can be used to engineer the CAR molecule and/or its expression to mitigate excessive signalling, reduce exhaustion and thereby improve the efficacy of CAR T cells.
Given that excessive signalling can result in CAR T cell exhaustion, multiple strategies to pharmacologically ‘turn off’ the signals have also been proposed. For example, the tyrosine-kinase inhibitor dasatinib has been identified as a pharmacological ‘on/off’ switch for CAR T cells owing to its ability to suppress CD3ζ phosphorylation209,210. In various mouse xenograft models, transient inhibition of CAR signalling with dasatinib was able to restore antitumour functionality in exhausted CAR T cells211. The investigators concluded that dasatinib treatment enhances CAR T cell efficacy by either mitigating or reversing exhaustion211. Given that the epigenetic readouts of exhaustion were performed on bulk CAR T cell populations evaluated at different times after dasatinib treatment, whether prevention or bona fide reversal of epigenetic features of exhaustion is the dominant mechanism of transient signalling cessation remains unknown. Nonetheless, this study presents a potential pharmacological approach to mitigate CAR T cell exhaustion and improve the efficacy of a broad range of existing CAR T cell therapies.
Engineering transcriptional programmes.
The balance between T cell function and exhaustion occurs downstream of TCR signalling based on an equilibrium between the transcription factor AP-1 and its binding partner NFAT212. Upstream TCR213 or CAR signalling187, including tonic CAR signalling, drives calcium-induced calcineurin activation, which subsequently causes NFAT dephosphorylation and nuclear translocation. Canonical AP-1 is heterodimeric complex consisting of FOS and JUN and is induced both by primary TCR signalling via MAPK and co-stimulatory signalling via CD28214. Cooperation between AP-1 with NFAT in T cells leads to robust IL-2 expression and effector function; however, expression of FOS and JUN is lost during sustained T cell signalling, leading to unopposed (or ‘partnerless’) NFAT signalling136. Accordingly, T cell exhaustion caused by tonically signalling GD2-specific CARs can be overcome by forced expression of JUN, leading to enhanced CAR T cell efficacy in mouse models134. Overexpression of the transcription factor BATF similarly counters NFAT-induced exhaustion, partnering with IRF4 to counter the transcriptional exhaustion programme and enhance CAR T cell function (FIG. 2)135.
Unopposed NFAT signalling leads to expression of the exhaustion-associated T cell transcription factors TOX and NR4A, thereby enforcing the epigenetic and transcriptional programme associated with exhaustion3,4,215. Indeed, murine CAR T cells with short-hairpin RNA (shRNA)-mediated knockdown of Tox are more persistent and have greater therapeutic activity than Tox-wild-type cells, particularly when also genetically deficient in Tox24. Similarly, triple deletion of Nr4a1, Nr4a2 and Nr4a3 restored a functional and epigenetic programme that enhanced CAR T cell efficacy in mice bearing solid tumours133. Intriguingly, other studies indicate that deletion of Tox results in increased apoptosis and impaired persistence of antigen-specific murine T cells in mice bearing tumours or with chronic viral infection3,215–217. This discrepancy might be explained by differential effects of complete genetic deletion of Tox, as opposed to partial reduction in expression conferred by shRNA-mediated knockdown. [Au: In figure 2, this engineering strategy is shown as a way to interupt, rather than mitigate exhaustion, unlike the previously approach of disrupting ID3 and SOX4. Therefore, the inclusion of TOX/NR4A in part b of figure 2 seems a little inconsistent. Please see my margin comment in response to your previous one.] The transcription factors ID3 and SOX4 have also been shown to be essential for CAR T cell exhaustion, with CRISPR-mediated genetic deletion of either ID3 or SOX4 resulting in improved in vitro cytotoxicity against tumour cells after chronic tumour antigen exposure218 (FIG. 2).
As described earlier, exhaustion is enforced in part by transcription factors via a canonical epigenetic programme. Indeed, the first clinical evidence that the disruption of this transcriptional programme could lead to dramatic changes in the functional behaviour of CAR T cells came from a case report of a CAR T cell clone with genetic loss of the epigenetic regulator TET2219. TET2 catalyzes the oxidation of 5-methylcytosine in DNA (thereby promoting demethylation), and TET2-disruption occurring through random integration of the CAR vector in a patient with a pre-existing hypomorphic TET2 allele led to a CAR T cell clone that accounted for 94% of circulating T cells at its peak. This preferential expansion of the TET2-deficient CAR+ T cell clone coincided with development of a delayed cytokine-release syndrome and tumour regression many months after initial cell infusion219. The TET2-disrupted CAR T cells were noted to have an epigenetic profile of reduced exhaustion219, suggesting that abrogation of an epigenetic regulator could mitigate T cell exhaustion (FIG. 2). Interestingly, further preclinical work has demonstrated that the effect of TET2 disruption is dependent on CAR signalling properties (for example, occurring with 4–1BB-based but not CD28-based CARs), which has been attributed to inherent variability in the level of effector differentiation induced by each construct220. Moreover, TET2 disruption eventually uncoupled proliferative capacity and effector function, leading to a hyperproliferative but poorly functional CAR T cells, again demonstrating the complexity of interrupting the broad exhaustion programme220.
In 2022, an elegant genome-wide CRISPR screen revealed that the INO80 and BAF chromatin remodelling complexes are essential for T cell exhaustion221. Importantly, functional inactivation of Arid1a, a component of the BAF complex, in adoptively transferred T cells resulted in epigenetic and transcriptional changes that impaired their exhaustion and improved their persistence and antitumour activity in mouse models221. Preliminary data from a second study highlighted a similar role for the histone methyl transferase SUV39H1, which has previously been implicated in control of T cell stemness222, with deletion of SUV39H1 resulting in enhanced anti-CD19 CAR T cell persistence and tumour control in a mouse model of B-ALL223. This result was associated with epigenetic changes that curtailed the expression of co-inhibitory receptors.
Mitigating metabolic exhaustion.
Persistent T cell stimulation is associated with substantial mitochondrial dysfunction73,224–226. Moreover, metabolic and tumour environment conditions such as hypoxia promote T cell exhaustion73 and trigger a dysfunctional metabolic programme driven the transcriptional repressor BLIMP1 (also known as PRDM1)227, leading to silencing of the transcriptional coactivator PGC1α. Indeed, overexpression of PGC1α in tumour-specific T cells results in enhanced mitochondrial fitness and tumour control in mouse models (FIG. 2)228.
Notably, impaired mitochondrial biogenesis has been associated with the reduced in vivo persistence of T cells transduced with CARs harbouring CD28, as opposed to 4–1BB, co-stimulatory domains186. This mitochondrial dysfunction leads to increased generation of reactive oxygen species (ROS), which limits survival and self-renewal of the adoptively transferred CAR T cells224–226,229; importantly, scavenging of ROS using N-acetylcysteine or nicotinamide could restore antitumour T cell responses (FIG. 2). Intriguingly, pre-infusion mitochondrial mass was found be higher in patients who subsequently had a complete response to autologous CD19-targeted CAR T cell therapy compared with those who did not230. Therefore, the exhaustion programme might have evolved as a mechanism to limit TCR overstimulation and the associated metabolic dysfunction, which if left unmitigated can result in T cell death and thus loss of persistence.
CAR T cell exhaustion in summary
Thus, a diversity of modifications to CAR T cells, from proximal to distal aspects of the signalling, transcriptional, epigenetic and metabolic programmes, have now been shown to potentially mitigate (and in some cases prevent) T cell exhaustion. We have attempted to provide a framework for understanding each of the approaches, and their respective strengths and weaknesses. Modern screening-based approaches will facilitate the identification of other novel candidates to engineer in order to mitigate CAR T cell exhaustion231–233, and perhaps even enable comparisons between different approaches. More importantly, many promising engineering strategies are being translated into the clinical setting and the upcoming clinical studies will provide information on how to best enhance the therapeutic efficacy of CAR T cells.
Conclusions
The advances discussed in this Review highlight nuances in the concept of T cell exhaustion. Non-exhausted tumour-reactive T cells are undoubtedly preferable to exhausted tumour-reactive T cells for the treatment of patients with cancer; however, this is perhaps a false choice, given the evidence we have highlighted that T cell exhaustion is inevitable in the setting of persistent tumour antigen. Adding further complexity, interruption of the exhaustion programme can impair T cell persistence in certain contexts. Therefore, we advocate that the greatest hope to improve the efficacy of immunotherapy is to focus on strategies that mitigate induction of T cell exhaustion in the first place.
Key points.
T cell exhaustion is a hypofunctional T cell state that is associated with decreased efficacy of immune-checkpoint inhibitors (ICIs) and adoptive T cell therapies.
Molecular features of T cell exhaustion are associated with tumour-reactive T cell receptor (TCR) clonotypes and can predict clinical benefit from ICIs.
The effectiveness of ICI therapy is dependent on peripheral expansion and subsequent infiltration of precursor-exhausted CD8+ T cells.
The T cell exhaustion programme protects CD8+ T cells from overstimulation-associated cell death; therefore, interruption of this programme might impair the persistence of tumour-reactive T cells in patients with cancer.
Engineering optimized receptors and downstream signalling, fine-tuning transcriptional and epigenetic states, and overcoming metabolic dysfunction can mitigate T cell exhaustion (rather than interrupting this programme per se), thereby enhancing the efficacy of chimeric antigen receptor (CAR) and TCR-engineered cell therapies.
Acknowledgements
We are grateful to Dr Andrea Schietinger of the Memorial Sloan Kettering Cancer Center (MSKCC; New York, NY, USA) for providing valuable feedback on the manuscript. We are also grateful for manuscript editing performed by Dr Clare Wilhelm of the MSKCC. The work of A.C. is supported by a Clinical Investigator Award from US NIH National Cancer Institute (NCI; K08 CA-248723), the Stony-Wold Herbert Fund, the International Association of Lung Cancer Research (IASLC)/International Lung Cancer Foundation (ILCF), and a Society of MSKCC Grant. The work of C.A.K. is supported by the NCI (grant R37 CA259177), Cancer Research Institute (grant CRI3176), the Damon Runyon Cancer Research Foundation (grant CI-96-18), and the Parker Institute for Cancer Immunotherapy. The work of J.D.W. is supported by the NCI (grant R01 CA056821), the Ludwig Collaborative and Swim Across America Laboratory, the Emerald Foundation, the Parker Institute for Cancer Immunotherapy, and a Stand Up To Cancer (SU2C)–American Cancer Society Lung Cancer Translational Research Dream Team grant (SU2C-AACR-DT17-15). The work of all authors is supported in part by a US NIH/NCI Cancer Center Support Grant to the MSKCC (P30 CA008748).
Competing interests
C.A.K. has received research grant support from Kite/Gilead and Intima Bioscience; is on the scientific and/or clinical advisory boards of Achilles Therapeutics, Aleta BioTherapeutics, Bellicum Pharmaceuticals, Catamaran Bio, Obsidian Therapeutics and T-knife; and has provided consulting services for Bristol Myers Squibb, PACT Pharma and Roche/Genentech. C.A.K. is also a co-inventor on patent-licensed applications related to T cell receptors targeting public neoantigens, unrelated to the current work. J.D.W. is a consultant for Adaptive Biotech, Amgen, Apricity, Ascentage Pharma, Arsenal IO, Astellas, AstraZeneca, Bayer, Beigene, Boehringer Ingelheim, Bristol Myers Squibb, Celgene, Chugai, Daiichi Sankyo, Dragonfly, Eli Lilly, Elucida, F Star, Georgiamune, Idera, Imvaq, Kyowa Hakko Kirin, Linneaus, Maverick Therapeutics, Merck, Neon Therapeutics, Polynoma, Psioxus, Recepta, Sellas, Serametrix, Surface Oncology, Syndax, Syntalogic, Takara Bio, Trieza, Truvax, Trishula, and Werewolf Therapeutics; has received grant/research support from Bristol Myers Squibb and Sephora; and has equity in Adaptive Biotechnologies, Apricity, Arsenal IO, Beigene, Imvaq, Linneaus, Georgiamune, and Tizona Pharmaceuticals. J.D.W. is also a co-inventor on patent applications related to heteroclitic cancer vaccines, recombinant poxviruses for cancer immunotherapy, and CD40 and in situ vaccination. A.C. and K.P. declare no competing interests.
References
- 1.Khalil DN, Smith EL, Brentjens RJ & Wolchok JD The future of cancer treatment: immunomodulation, CARs and combination immunotherapy. Nat Rev Clin Oncol 13, 394 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ribas A & Wolchok JD Cancer immunotherapy using checkpoint blockade. Science (80-. ). 359, 1350–1355 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scott AC et al. TOX is a critical regulator of tumour-specific T cell differentiation. Nature 571, 270–274 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Seo H et al. TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8+ T cell exhaustion. Proc. Natl. Acad. Sci. U. S. A 116, 12410–12415 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu X et al. Genome-wide analysis identifies NR4A1 as a key mediator of T cell dysfunction. Nature 567, 525–529 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Philip M & Schietinger A CD8+ T cell differentiation and dysfunction in cancer. Nat. Rev. Immunol 2021 224 22, 209–223 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blank CU et al. Defining ‘T cell exhaustion’. Nat. Rev. Immunol (2019) doi: 10.1038/s41577-019-0221-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kansy BA et al. PD-1 status in CD8+ T cells associates with survival and anti-PD-1 therapeutic outcomes in head and neck cancer. Cancer Res. 77, 6353 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mansfield AS et al. B7-H1 Expression in Malignant Pleural Mesothelioma is Associated with Sarcomatoid Histology and Poor Prognosis. J. Thorac. Oncol 9, 1036–1040 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thompson RH et al. PD-1 Is Expressed by Tumor-Infiltrating Immune Cells and Is Associated with Poor Outcome for Patients with Renal Cell Carcinoma. Clin. Cancer Res 13, 1757–1761 (2007). [DOI] [PubMed] [Google Scholar]
- 11.Sun S et al. PD-1+ immune cell infiltration inversely correlates with survival of operable breast cancer patients. Cancer Immunol. Immunother 63, 395–406 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kang MJ et al. Tumor-infiltrating PD1-Positive Lymphocytes and FoxP3-Positive Regulatory T Cells Predict Distant Metastatic Relapse and Survival of Clear Cell Renal Cell Carcinoma. Transl. Oncol 6, 282 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boorjian SA et al. T-Cell Coregulatory Molecule Expression in Urothelial Cell Carcinoma: Clinicopathologic Correlations and Association with Survival. Clin. Cancer Res 14, 4800–4808 (2008). [DOI] [PubMed] [Google Scholar]
- 14.Muenst S et al. The presence of programmed death 1 (PD-1)-positive tumor-infiltrating lymphocytes is associated with poor prognosis in human breast cancer. Breast Cancer Res. Treat 139, 667–676 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mao Y et al. B7-H1 and B7-H3 are independent predictors of poor prognosis in patients with non-small cell lung cancer. Oncotarget 6, 3452 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McLane LM, Abdel-Hakeem MS & Wherry EJ CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annual Review of Immunology (2019) doi: 10.1146/annurev-immunol-041015-055318. [DOI] [PubMed] [Google Scholar]
- 17.Zajac AJ et al. Viral immune evasion due to persistence of activated T cells without effector function. J. Exp. Med (1998) doi: 10.1084/jem.188.12.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gallimore BA et al. Induction and Exhaustion of Lymphocytic Choriomeningitis Virus – specific Cytotoxic T Lymphocytes Visualized Using Class I – Peptide Complexes. J. Exp. Med (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barber DL et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature (2006) doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- 20.Leach DR, Krummel MF & Allison JP Enhancement of antitumor immunity by CTLA-4 blockade. Science (80-. ). (1996) doi: 10.1126/science.271.5256.1734. [DOI] [PubMed] [Google Scholar]
- 21.Walunas TL et al. CTLA-4 can function as a negative regulator of T cell activation. Immunity (1994) doi: 10.1016/1074-7613(94)90071-X. [DOI] [PubMed] [Google Scholar]
- 22.Ishida Y, Agata Y, Shibahara K & Honjo T Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. (1992) doi: 10.1002/j.1460-2075.1992.tb05481.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Keir ME et al. Tissue expression of PD-L1 mediates peripheral T cell tolerance. J. Exp. Med (2006) doi: 10.1084/jem.20051776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dong H et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat. Med (2002) doi: 10.1038/nm730. [DOI] [PubMed] [Google Scholar]
- 25.Iwai Y et al. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc. Natl. Acad. Sci. U. S. A (2002) doi: 10.1073/pnas.192461099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baitsch L et al. Exhaustion of tumor-specific CD8+ T cells in metastases from melanoma patients. J. Clin. Invest 121, 2350–2360 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hodi FS et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med (2010) doi: 10.1056/nejmoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wolchok JD et al. Nivolumab plus Ipilimumab in Advanced Melanoma. N. Engl. J. Med (2013) doi: 10.1056/nejmoa1302369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Postow MA et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med 372, 2006–2017 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Philip M et al. Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature (2017) doi: 10.1038/nature22367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ghoneim HE et al. De Novo Epigenetic Programs Inhibit PD-1 Blockade-Mediated T Cell Rejuvenation. Cell (2017) doi: 10.1016/j.cell.2017.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mognol GP et al. Exhaustion-associated regulatory regions in CD8+ tumor-infiltrating T cells. Proc. Natl. Acad. Sci. U. S. A 114, E2776–E2785 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sadelain M, Rivière I & Riddell S Therapeutic T cell engineering. Nature (2017) doi: 10.1038/nature22395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.June CH & Sadelain M Chimeric Antigen Receptor Therapy. N. Engl. J. Med 379, 64–73 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Neelapu SS et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med NEJMoa1707447 (2017) doi: 10.1056/NEJMoa1707447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maude SL et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med 378, 439–448 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Park JH et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N. Engl. J. Med (2018) doi: 10.1056/nejmoa1709919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schuster SJ et al. Chimeric Antigen Receptor T Cells in Refractory B-Cell Lymphomas. N. Engl. J. Med NEJMoa1708566 (2017) doi: 10.1056/NEJMoa1708566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Raje N et al. Anti-BCMA CAR T-Cell Therapy bb2121 in Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med 380, 1726–1737 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Berdeja JG et al. Ciltacabtagene autoleucel, a B-cell maturation antigen-directed chimeric antigen receptor T-cell therapy in patients with relapsed or refractory multiple myeloma (CARTITUDE-1): a phase 1b/2 open-label study. Lancet 0, (2021). [DOI] [PubMed] [Google Scholar]
- 41.Locke FL et al. Axicabtagene Ciloleucel as Second-Line Therapy for Large B-Cell Lymphoma. N. Engl. J. Med 1–15 (2021) doi: 10.1056/NEJMoa2116133. [DOI] [PubMed] [Google Scholar]
- 42.Kamdar M et al. Lisocabtagene maraleucel versus standard of care with salvage chemotherapy followed by autologous stem cell transplantation as second-line treatment in patients with relapsed or refractory large B-cell lymphoma (TRANSFORM): results from an interim analysi. Lancet 399, 2294–2308 (2022). [DOI] [PubMed] [Google Scholar]
- 43.Rives S et al. Tisagenlecleucel in Pediatric and Young Adult Patients With Relapsed/Refractory (R/R) B-Cell Acute Lymphoblastic Leukemia (B-All): Final Analyses From the Eliana Study. HemaSphere 6, 13–14 (2022). [Google Scholar]
- 44.Hou AJ, Chen LC & Chen YY Navigating CAR-T cells through the solid-tumour microenvironment. Nat. Rev. Drug Discov 20, 531–550 (2021). [DOI] [PubMed] [Google Scholar]
- 45.Malandro N et al. Clonal Abundance of Tumor-Specific CD4+ T Cells Potentiates Efficacy and Alters Susceptibility to Exhaustion. Immunity 44, 179–193 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fu J et al. CD4+ T cell exhaustion leads to adoptive transfer therapy failure which can be prevented by immune checkpoint blockade. Am. J. Cancer Res 10, 4234 (2020). [PMC free article] [PubMed] [Google Scholar]
- 47.Balança CC et al. PD-1 blockade restores helper activity of tumor-infiltrating, exhausted PD-1hiCD39+ CD4 T cells. JCI Insight 6, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sommermeyer D et al. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia 30, 492–500 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gros A et al. PD-1 identifies the patient-specific CD8(+) tumor-reactive repertoire infiltrating human tumors. J Clin Invest 124, 2246–2259 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gros A et al. Selection of circulating PD-1+ lymphocytes from cancer patients enriches for tumor-reactive and mutation-specific lymphocytes. J. Immunother. Cancer (2015) doi: 10.1186/2051-1426-3-S2-O2. [DOI] [Google Scholar]
- 51.Gros A et al. Recognition of human gastrointestinal cancer neoantigens by circulating PD-1+ lymphocytes. J. Clin. Invest 129, 4992–5004 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thommen DS et al. A transcriptionally and functionally distinct PD-1(+) CD8(+) T cell pool with predictive potential in non-small-cell lung cancer treated with PD-1 blockade. Nat Med 24, 994–1004 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Simoni Y et al. Bystander CD8(+) T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature 557, 575–579 (2018). [DOI] [PubMed] [Google Scholar]
- 54.Duhen T et al. Co-expression of CD39 and CD103 identifies tumor-reactive CD8 T cells in human solid tumors. Nat Commun 9, 2724 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li H et al. Dysfunctional CD8 T Cells Form a Proliferative, Dynamically Regulated Compartment within Human Melanoma. Cell (2018) doi: 10.1016/j.cell.2018.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.van der Leun AM, Thommen DS & Schumacher TN CD8(+) T cell states in human cancer: insights from single-cell analysis. Nat Rev Cancer 20, 218–232 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Caushi JX et al. Transcriptional programs of neoantigen-specific TIL in anti-PD-1-treated lung cancers. Nat. 2021 5967870 596, 126–132 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Oliveira G et al. Phenotype, specificity and avidity of antitumour CD8+ T cells in melanoma. Nat. 2021 5967870 596, 119–125 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lowery FJ et al. Molecular signatures of antitumor neoantigen-reactive T cells from metastatic human cancers. Science eabl5447 (2022) doi: 10.1126/SCIENCE.ABL5447/SUPPL_FILE/SCIENCE.ABL5447_MDAR_REPRODUCIBILITY_CHECKLIST.PDF. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hanada K-I et al. A phenotypic signature that identifies neoantigen-reactive T cells in fresh human lung cancers. Cancer Cell 40, 479–493.e6 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Meier SL, Satpathy AT & Wells DK Bystander T cells in cancer immunology and therapy. Nat. cancer 3, 143–155 (2022). [DOI] [PubMed] [Google Scholar]
- 62.Kumagai S et al. The PD-1 expression balance between effector and regulatory T cells predicts the clinical efficacy of PD-1 blockade therapies. Nat Immunol (2020) doi: 10.1038/s41590-020-0769-3. [DOI] [PubMed] [Google Scholar]
- 63.Daud AI et al. Tumor immune profiling predicts response to anti-PD-1 therapy in human melanoma. J Clin Invest 126, 3447–3452 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Terranova-Barberio M et al. Exhausted T cell signature predicts immunotherapy response in ER-positive breast cancer. Nat Commun 11, 3584 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chalabi M et al. Neoadjuvant immunotherapy leads to pathological responses in MMR-proficient and MMR-deficient early-stage colon cancers. Nat Med 26, 566–576 (2020). [DOI] [PubMed] [Google Scholar]
- 66.Yeong J et al. Intratumoral CD39+CD8+ T Cells Predict Response to Programmed Cell Death Protein-1 or Programmed Death Ligand-1 Blockade in Patients With NSCLC. J. Thorac. Oncol 16, 1349–1358 (2021). [DOI] [PubMed] [Google Scholar]
- 67.Attrill GH et al. Higher proportions of CD39+ tumor-resident cytotoxic T cells predict recurrence-free survival in patients with stage III melanoma treated with adjuvant immunotherapy. J. Immunother. Cancer 10, e004771 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chow A et al. CD39 Identifies Tumor-Reactive CD8 T cells in Patients With Lung Cancer. bioRxiv 13, 2022.01.24.477554 (2022). [Google Scholar]
- 69.Kaech SM, Wherry EJ & Ahmed R Effector and memory T-cell differentiation: implications for vaccine development. Nat. Rev. Immunol 2002 24 2, 251–262 (2002). [DOI] [PubMed] [Google Scholar]
- 70.Beltra JC et al. Developmental Relationships of Four Exhausted CD8+ T Cell Subsets Reveals Underlying Transcriptional and Epigenetic Landscape Control Mechanisms. Immunity (2020) doi: 10.1016/j.immuni.2020.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hudson WH et al. Proliferating Transitory T Cells with an Effector-like Transcriptional Signature Emerge from PD-1+ Stem-like CD8+ T Cells during Chronic Infection. Immunity (2019) doi: 10.1016/j.immuni.2019.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Singh N et al. Impaired death receptor signaling in leukemia causes antigen-independent resistance by inducing CAR T-cell dysfunction. Cancer Discov. (2020) doi: 10.1158/2159-8290.CD-19-0813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Scharping NE et al. Mitochondrial stress induced by continuous stimulation under hypoxia rapidly drives T cell exhaustion. Nat. Immunol (2021) doi: 10.1038/s41590-020-00834-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Liu YN et al. Hypoxia Induces Mitochondrial Defect That Promotes T Cell Exhaustion in Tumor Microenvironment Through MYC-Regulated Pathways. Front. Immunol 11, 1906 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bannoud N et al. Hypoxia Supports Differentiation of Terminally Exhausted CD8 T Cells. Front. Immunol 12, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Thommen DS & Schumacher TN T Cell Dysfunction in Cancer. Cancer Cell 33, 547–562 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sade-Feldman M et al. Defining T Cell States Associated with Response to Checkpoint Immunotherapy in Melanoma. Cell 175, 998–1013 e20 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gandhi L et al. Pembrolizumab plus Chemotherapy in Metastatic Non-Small-Cell Lung Cancer. N Engl J Med 378, 2078–2092 (2018). [DOI] [PubMed] [Google Scholar]
- 79.Paz-Ares L et al. Durvalumab plus platinum-etoposide versus platinum-etoposide in first-line treatment of extensive-stage small-cell lung cancer (CASPIAN): a randomised, controlled, open-label, phase 3 trial. Lancet 394, 1929–1939 (2019). [DOI] [PubMed] [Google Scholar]
- 80.Paz-Ares L et al. Pembrolizumab plus Chemotherapy for Squamous Non–Small-Cell Lung Cancer. N. Engl. J. Med (2018) doi: 10.1056/nejmoa1810865. [DOI] [PubMed] [Google Scholar]
- 81.Socinski MA et al. Atezolizumab for First-Line Treatment of Metastatic Nonsquamous NSCLC. N Engl J Med 378, 2288–2301 (2018). [DOI] [PubMed] [Google Scholar]
- 82.Horn L et al. First-Line Atezolizumab plus Chemotherapy in Extensive-Stage Small-Cell Lung Cancer. N Engl J Med 379, 2220–2229 (2018). [DOI] [PubMed] [Google Scholar]
- 83.Palmer AC, Izar B, Hwangbo H & Sorger PK Predictable Clinical Benefits without Evidence of Synergy in Trials of Combination Therapies with Immune-Checkpoint Inhibitors. Clin. Cancer Res 28, 368–377 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Huang AC et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature 545, 60–65 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Joseph RW et al. Baseline Tumor Size Is an Independent Prognostic Factor for Overall Survival in Patients with Melanoma Treated with Pembrolizumab. Clin Cancer Res 24, 4960–4967 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Matoba T et al. Impact of tumor burden on survival in patients with recurrent or metastatic head and neck cancer treated with immune checkpoint inhibitors. (123AD) doi: 10.1038/s41598-022-18611-z. [DOI] [PMC free article] [PubMed]
- 87.Falvo P et al. Cyclophosphamide and Vinorelbine Activate Stem-Like CD8 þ T Cells and Improve Anti-PD-1 Efficacy in Triple-Negative Breast Cancer A C. doi: 10.1158/0008-5472.CAN-20-1818. [DOI] [PubMed] [Google Scholar]
- 88.Hanoteau A et al. Cyclophosphamide treatment regulates the balance of functional/exhausted tumor-specific CD8+ T cells. Oncoimmunology 6, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Liang H et al. Quantitative multiplex immunofluorescence analysis identifies infiltrating PD1+CD8+ and CD8+ T cells as predictive of response to neoadjuvant chemotherapy in breast cancer. Thorac. Cancer 11, 2941–2954 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chow A et al. Leveraging CD39 To Identify Tumor-Reactive CD8 T cells In Human Lung Cancer. bioRxiv 2022.01.24.477554 (2022) doi: 10.1101/2022.01.24.477554. [DOI] [Google Scholar]
- 91.Chaft JE et al. Evolution of systemic therapy for stages I–III non-metastatic non-small-cell lung cancer. Nat. Rev. Clin. Oncol 2021 189 18, 547–557 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Forde PM et al. Neoadjuvant Nivolumab plus Chemotherapy in Resectable Lung Cancer. N. Engl. J. Med 386, 1973–1985 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Spitzer MH et al. Systemic Immunity Is Required for Effective Cancer Immunotherapy. Cell (2017) doi: 10.1016/j.cell.2016.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tang H et al. PD-L1 on host cells is essential for PD-L1 blockade–mediated tumor regression. J. Clin. Invest (2018) doi: 10.1172/JCI96061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Fransen MF et al. Tumor-draining lymph nodes are pivotal in PD-1/PD-L1 checkpoint therapy. JCI insight (2018) doi: 10.1172/jci.insight.124507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chow MT et al. Intratumoral Activity of the CXCR3 Chemokine System Is Required for the Efficacy of Anti-PD-1 Therapy. Immunity (2019) doi: 10.1016/j.immuni.2019.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chamoto K et al. Mitochondrial activation chemicals synergize with surface receptor PD-1 blockade for T cell-dependent antitumor activity. Proc. Natl. Acad. Sci. U. S. A (2017) doi: 10.1073/pnas.1620433114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Dammeijer F et al. The PD-1/PD-L1-Checkpoint Restrains T cell Immunity in Tumor-Draining Lymph Nodes. Cancer Cell (2020) doi: 10.1016/j.ccell.2020.09.001. [DOI] [PubMed] [Google Scholar]
- 99.Francis DM et al. Blockade of immune checkpoints in lymph nodes through locoregional delivery augments cancer immunotherapy. Sci. Transl. Med (2020) doi: 10.1126/scitranslmed.aay3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Oh SA et al. PD-L1 expression by dendritic cells is a key regulator of T-cell immunity in cancer. Nat. Cancer (2020) doi: 10.1038/s43018-020-0075-x. [DOI] [PubMed] [Google Scholar]
- 101.Peng Q et al. PD-L1 on dendritic cells attenuates T cell activation and regulates response to immune checkpoint blockade. Nat. Commun (2020) doi: 10.1038/s41467-020-18570-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.He R et al. Follicular CXCR5-expressing CD8+ T cells curtail chronic viral infection. Nature (2016) doi: 10.1038/nature19317. [DOI] [PubMed] [Google Scholar]
- 103.Leong YA et al. CXCR5+ follicular cytotoxic T cells control viral infection in B cell follicles. Nat. Immunol (2016) doi: 10.1038/ni.3543. [DOI] [PubMed] [Google Scholar]
- 104.Utzschneider DT et al. T Cell Factor 1-Expressing Memory-like CD8+ T Cells Sustain the Immune Response to Chronic Viral Infections. Immunity (2016) doi: 10.1016/j.immuni.2016.07.021. [DOI] [PubMed] [Google Scholar]
- 105.Im SJ et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature (2016) doi: 10.1038/nature19330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kallies A, Zehn D & Utzschneider DT Precursor exhausted T cells: key to successful immunotherapy? Nature Reviews Immunology (2020) doi: 10.1038/s41577-019-0223-7. [DOI] [PubMed] [Google Scholar]
- 107.Brummelman J et al. High-dimensional single cell analysis identifies stemlike cytotoxic CD8+T cells infiltrating human tumors. J. Exp. Med (2018) doi: 10.1084/JEM.20180684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Siddiqui I et al. Intratumoral Tcf1 + PD-1 + CD8 + T Cells with Stem-like Properties Promote Tumor Control in Response to Vaccination and Checkpoint Blockade Immunotherapy. Immunity (2019) doi: 10.1016/j.immuni.2018.12.021. [DOI] [PubMed] [Google Scholar]
- 109.Kurtulus S et al. Checkpoint Blockade Immunotherapy Induces Dynamic Changes in PD-1 − CD8 + Tumor-Infiltrating T Cells. Immunity (2019) doi: 10.1016/j.immuni.2018.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Miller BC et al. Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol (2019) doi: 10.1038/s41590-019-0312-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Tsui C et al. MYB orchestrates T cell exhaustion and response to checkpoint inhibition. Nat. 2022 1–7 (2022) doi: 10.1038/s41586-022-05105-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Yost KE et al. Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat Med 25, 1251–1259 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Huang AC et al. A single dose of neoadjuvant PD-1 blockade predicts clinical outcomes in resectable melanoma. Nat. Med (2019) doi: 10.1038/s41591-019-0357-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kamphorst AO et al. Proliferation of PD-1+ CD8 T cells in peripheral blood after PD-1-targeted therapy in lung cancer patients. Proc Natl Acad Sci U S A 114, 4993–4998 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Li Z et al. In vivo labeling reveals continuous trafficking of TCF-1+ T cells between tumor and lymphoid tissue. J. Exp. Med 219, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wu TD et al. Peripheral T cell expansion predicts tumour infiltration and clinical response. Nature (2020) doi: 10.1038/s41586-020-2056-8. [DOI] [PubMed] [Google Scholar]
- 117.Duraiswamy J et al. Myeloid antigen-presenting cell niches sustain antitumor T cells and license PD-1 blockade via CD28 costimulation. Cancer Cell 39, 1623–1642.e20 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Liu B et al. Temporal single-cell tracing reveals clonal revival and expansion of precursor exhausted T cells during anti-PD-1 therapy in lung cancer. Nat. Cancer 2021 31 3, 108–121 (2021). [DOI] [PubMed] [Google Scholar]
- 119.Voabil P et al. An ex vivo tumor fragment platform to dissect response to PD-1 blockade in cancer. Nat. Med 2021 277 27, 1250–1261 (2021). [DOI] [PubMed] [Google Scholar]
- 120.Kleinovink JW et al. PD-L1 expression on malignant cells is no prerequisite for checkpoint therapy. Oncoimmunology (2017) doi: 10.1080/2162402X.2017.1294299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Lau J et al. Tumour and host cell PD-L1 is required to mediate suppression of anti-tumour immunity in mice. Nat. Commun (2017) doi: 10.1038/ncomms14572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Lin H et al. Host expression of PD-L1 determines efficacy of PD-L1 pathway blockade–mediated tumor regression. J. Clin. Invest (2018) doi: 10.1172/JCI96113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Doroshow DB et al. PD-L1 as a biomarker of response to immune-checkpoint inhibitors. Nat. Rev. Clin. Oncol 2021 186 18, 345–362 (2021). [DOI] [PubMed] [Google Scholar]
- 124.Sun C, Mezzadra R & Schumacher TN Regulation and Function of the PD-L1 Checkpoint. Immunity (2018) doi: 10.1016/j.immuni.2018.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Zahran AM et al. Overexpression of PD-1 and CD39 in tumor-infiltrating lymphocytes compared with peripheral blood lymphocytes in triple-negative breast cancer. PLoS One 17, e0262650 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Curran MA & Glisson BS New hope for therapeutic cancer vaccines in the era of immune checkpoint modulation. Annual Review of Medicine (2019) doi: 10.1146/annurev-med-050217-121900. [DOI] [PubMed] [Google Scholar]
- 127.Hammerich L et al. Systemic clinical tumor regressions and potentiation of PD1 blockade with in situ vaccination. Nat. Med (2019) doi: 10.1038/s41591-019-0410-x. [DOI] [PubMed] [Google Scholar]
- 128.Williams JB et al. The EGR2 targets LAG-3 and 4-1BB describe and regulate dysfunctional antigen-specific CD8+ T cells in the tumor microenvironment. J. Exp. Med 214, 381–400 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kraehenbuehl L, Weng CH, Eghbali S, Wolchok JD & Merghoub T Enhancing immunotherapy in cancer by targeting emerging immunomodulatory pathways. Nat. Rev. Clin. Oncol 2021 191 19, 37–50 (2021). [DOI] [PubMed] [Google Scholar]
- 130.West EE et al. PD-L1 blockade synergizes with IL-2 therapy in reinvigorating exhausted T cells. J. Clin. Invest 123, 2604–2615 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Pilipow K et al. IL-15 and T cell stemness in T cell-based cancer immunotherapy. Cancer Res 75, 5187 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Dimopoulos MA et al. Phase 3 Trial of Ibrutinib plus Rituximab in Waldenström’s Macroglobulinemia. N. Engl. J. Med NEJMoa1802917 (2018) doi: 10.1056/NEJMoa1802917. [DOI] [PubMed] [Google Scholar]
- 133.Chen J et al. NR4A transcription factors limit CAR T cell function in solid tumours. Nature 1 (2019) doi: 10.1038/s41586-019-0985-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Lynn RC et al. c-Jun overexpression in CAR T cells induces exhaustion resistance. Nature 576, 293–300 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Seo H et al. BATF and IRF4 cooperate to counter exhaustion in tumor-infiltrating CAR T cells. Nat. Immunol 983–995 (2021) doi: 10.1038/s41590-021-00964-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Martinez GJ et al. The Transcription Factor NFAT Promotes Exhaustion of Activated CD8+ T Cells. Immunity 42, 265–278 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Man K et al. Transcription Factor IRF4 Promotes CD8+ T Cell Exhaustion and Limits the Development of Memory-like T Cells during Chronic Infection. Immunity 47, 1129–1141.e5 (2017). [DOI] [PubMed] [Google Scholar]
- 138.Zebley CC et al. CD19-CAR T cells undergo exhaustion DNA methylation programming in patients with acute lymphoblastic leukemia. Cell Rep. 37, 110079 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kong W et al. BET bromodomain protein inhibition reverses chimeric antigen receptor extinction and reinvigorates exhausted T cells in chronic lymphocytic leukemia BET bromodomain protein inhibition reverses chimeric antigen receptor extinction and reinvigorates exhaust. 131, 1–16 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Chen PH et al. Activation of CAR and non-CAR T cells within the tumor microenvironment following CAR T cell therapy. JCI Insight 5, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Fraietta JA et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat. Med (2018) doi: 10.1038/s41591-018-0010-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Singh N, Perazzelli J, Grupp SA & Barrett DM Early memory phenotypes drive T cell proliferation in patients with pediatric malignancies. Sci. Transl. Med 8, 1–10 (2016). [DOI] [PubMed] [Google Scholar]
- 143.Bai Z et al. Single-cell antigen-specific landscape of CAR T infusion product identifies determinants of CD19-positive relapse in patients with ALL. Sci. Adv 8, eabj2820 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Monfrini C et al. Phenotypic Composition of Commercial Anti-CD19 CAR T Cells Affects In Vivo Expansion and Disease Response in Patients with Large B-cell Lymphoma . Clin. Cancer Res OF1–OF9 (2022) doi: 10.1158/1078-0432.ccr-22-0164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Locke FL et al. Tumor burden, inflammation, and product attributes determine outcomes of axicabtagene ciloleucel in large B-cell lymphoma. Blood Adv. 4, 4898–4911 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Gardner R et al. Starting T Cell and Cell Product Phenotype Are Associated with Durable Remission of Leukemia Following CD19 CAR-T Cell Immunotherapy. Blood 132, 4022–4022 (2018). [Google Scholar]
- 147.Deng Q et al. Characteristics of anti-CD19 CAR T cell infusion products associated with efficacy and toxicity in patients with large B cell lymphomas. Nat. Med (2020) doi: 10.1038/s41591-020-1061-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Pritykin Y et al. A unified atlas of CD8 T cell dysfunctional states in cancer and infection. Mol. Cell 81, 2477–2493.e10 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.McGowan E et al. PD-1 disrupted CAR-T cells in the treatment of solid tumors: Promises and challenges. Biomedicine and Pharmacotherapy (2020) doi: 10.1016/j.biopha.2019.109625. [DOI] [PubMed] [Google Scholar]
- 150.Chong EA et al. PD-1 blockade modulates chimeric antigen receptor (CAR)-modified T cells: refueling the CAR. Blood 129, 1039–1041 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Maude SL et al. The effect of pembrolizumab in combination with CD19-targeted chimeric antigen receptor (CAR) T cells in relapsed acute lymphoblastic leukemia (ALL). J. Clin. Oncol 35, 103 (2017). [Google Scholar]
- 152.Chong EA et al. Pembrolizumab for B-cell lymphomas relapsing after or refractory to CD19-directed CAR T-cell therapy. Blood 139, 1026–1038 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Jacobson CA et al. Abstract CT055: Phase 1/2 primary analysis of ZUMA-6: Axicabtagene ciloleucel (Axi-Cel) in combination With atezolizumab (Atezo) for the treatment of patients (Pts) with refractory diffuse large B cell lymphoma (DLBCL). Cancer Research vol. 80 CT055–CT055 (2020). [Google Scholar]
- 154.Adusumilli PS et al. A phase i trial of regional mesothelin-targeted car t-cell therapy in patients with malignant pleural disease, in combination with the anti–pd-1 agent pembrolizumab. Cancer Discov. 11, 2748–2763 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Sen DR et al. The epigenetic landscape of T cell exhaustion. Science (80-. ). (2016) doi: 10.1126/science.aae0491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Pauken KE et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science (80-. ). (2016) doi: 10.1126/science.aaf2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Rafiq S et al. Targeted delivery of a PD-1-blocking scFV by CAR-T cells enhances anti-tumor efficacy in vivo. Nat. Biotechnol 36, 847–858 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Nakajima M, Sakoda Y, Adachi K, Nagano H & Tamada K Improved survival of chimeric antigen receptor-engineered T (CAR -T) and tumor-specific T cells caused by anti-programmed cell death protein 1 single-chain variable fragment-producing CAR -T cells. Cancer Sci. 110, 3079–3088 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Cherkassky L et al. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J. Clin. Invest 126, 3130–3144 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Ren J et al. Multiplex Genome Editing to Generate Universal CAR T Cells Resistant to PD1 Inhibition. Clin. Cancer Res 23, 2255–2266 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Su S et al. CRISPR-Cas9 mediated efficient PD-1 disruption on human primary T cells from cancer patients. Sci. Rep 6, 1–14 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Rupp LJ et al. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci. Rep 7, 737 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Zhao Z et al. CRISPR knock out of programmed cell death protein 1 enhances anti-tumor activity of cytotoxic T lymphocytes. Oncotarget 9, 5208–5215 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Stadtmauer EA et al. CRISPR-engineered T cells in patients with refractory cancer. Science (80-. ). (2020) doi: 10.1126/science.aba7365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Odorizzi PM, Pauken KE, Paley MA, Sharpe A & John Wherry E Genetic absence of PD-1 promotes accumulation of terminally differentiated exhausted CD8+ T cells. J. Exp. Med 212, 1125–1137 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Pauken KE et al. The PD-1 Pathway Regulates Development and Function of Memory CD8+ T Cells following Respiratory Viral Infection. Cell Rep. (2020) doi: 10.1016/j.celrep.2020.107827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Chandran SS & Klebanoff CA T cell receptor-based cancer immunotherapy: Emerging efficacy and pathways of resistance. Immunol. Rev 290, 127–147 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Speiser DE, Ho PC & Verdeil G Regulatory circuits of T cell function in cancer. Nat. Rev. Immunol 16, 599–611 (2016). [DOI] [PubMed] [Google Scholar]
- 169.Larson RC et al. CAR T cell killing requires the IFNγR pathway in solid but not liquid tumours. Nat. 2022 6047906 604, 563–570 (2022). [DOI] [PubMed] [Google Scholar]
- 170.Vercellino L et al. Predictive factors of early progression after CAR T-cell therapy in relapsed/refractory diffuse large B-cell lymphoma. Blood Adv. 4, 5607–5615 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Perica K et al. Impact of bridging chemotherapy on clinical outcome of CD19 CAR T therapy in adult ALL. Leukemia Published, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Stefanski H et al. Higher doses of tisagenlecleucel associate with improved outcomes: a report from the pediatric real-world CAR consortium. Blood Adv. (2022) doi: 10.1182/BLOODADVANCES.2022007246/486120/HIGHER-DOSES-OF-TISAGENLECLEUCEL-ASSOCIATE-WITH. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Reinhard K et al. An RNA vaccine drives expansion and efficacy of claudin-CAR-T cells against solid tumors. Science (80-. ). 367, 446–453 (2020). [DOI] [PubMed] [Google Scholar]
- 174.Wang X & Rivière I Clinical manufacturing of CAR T cells: foundation of a promising therapy. Mol. Ther. — Oncolytics 3, 1–7 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Ghassemi S et al. Reducing ex vivo culture improves the antileukemic activity of chimeric antigen receptor (CAR) T cells. Cancer Immunol. Res 6, 1100–1109 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Alizadeh D et al. IL15 enhances CAR-T cell antitumor activity by reducing mTORC1 activity and preserving their stem cell memory phenotype. Cancer Immunol. Res 7, 759–772 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Funk CR et al. PI3Kδ/γ inhibition promotes human CART cell epigenetic and metabolic reprogramming to enhance antitumor cytotoxicity. Blood 139, 523–537 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Klebanoff CA et al. Inhibition of AKT signaling uncouples T cell differentiation from expansion for receptor-engineered adoptive immunotherapy. JCI Insight 2, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Petersen CT et al. Improving T-cell expansion and function for adoptive T-cell therapy using ex vivo treatment with PI3Kδ inhibitors and VIP antagonists. Blood Adv. 2, 210–223 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Bucks CM, Norton JA, Boesteanu AC, Mueller YM & Katsikis PD Chronic Antigen Stimulation Alone Is Sufficient to Drive CD8 + T Cell Exhaustion . J. Immunol 182, 6697–6708 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Shakiba M et al. TCR signal strength defines distinct mechanisms of T cell dysfunction and cancer evasion. J. Exp. Med 219, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Dada H & Dustin ML Goldilocks and the three TILs. J. Exp. Med 219, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Sim MJW et al. High-affinity oligoclonal TCRs define effective adoptive T cell therapy targeting mutant KRAS-G12D. Proc. Natl. Acad. Sci. U. S. A 117, 12826–12835 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Tran E et al. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N. Engl. J. Med 375, 2255–2262 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Long AH et al. 4–1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat. Med 21, 581–590 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Kawalekar OUU et al. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity 44, 380–390 (2016). [DOI] [PubMed] [Google Scholar]
- 187.Salter AI et al. Phosphoproteomic analysis of chimeric antigen receptor signaling reveals kinetic and quantitative differences that affect cell function. 6753, 1–18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Guedan S et al. Single residue in CD28-costimulated CAR T cells limits long-term persistence and antitumor durability. J. Clin. Invest (2020) doi: 10.1172/JCI133215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Carpenito C et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc. Natl. Acad. Sci 106, 3360–3365 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Zhong XS, Matsushita M, Plotkin J, Riviere I & Sadelain M Chimeric antigen receptors combining 4-1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and CD8+ T cell-mediated tumor eradication. Mol Ther 18, 413–420 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Hamieh M et al. CAR T cell trogocytosis and cooperative killing regulate tumour antigen escape. Nature vol. 568 112–116 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Majzner RG et al. Tuning the Antigen Density Requirement for CAR T-cell Activity. (2020) doi: 10.1158/2159-8290.CD-19-0945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Fry TJ et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat. Med 24, 20–28 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Mansilla-Soto J et al. HLA-independent T cell receptors for targeting tumors with low antigen density. Nat. Med 28, 345–352 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Zhao Z et al. Structural Design of Engineered Costimulation Determines Tumor Rejection Kinetics and Persistence of CAR T Cells. Cancer Cell 28, 415–428 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Posey AD et al. Distinct Signaling By Chimeric Antigen Receptors (CARs) Containing CD28 Signaling Domain Versus 4-1BB In Primary Human T Cells. Blood 122, (2013). [Google Scholar]
- 197.Song DG et al. In vivo persistence, tumor localization, and antitumor activity of CAR-engineered T cells is enhanced by costimulatory signaling through CD137 (4-1BB). Cancer Res. 71, 4617–4627 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Schuster SJ et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. NEJM 1–12 (2018) doi: 10.1056/NEJMoa1804980. [DOI] [PubMed] [Google Scholar]
- 199.Salter AI et al. Comparative analysis of TCR and CAR signaling informs CAR designs with superior antigen sensitivity and in vivo function. 2606, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Feucht J et al. Calibration of CAR activation potential directs alternative T cell fates and therapeutic potency. Nat. Med 25, 82–88 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Eyquem J et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 543, 113–117 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Roth theodore L. et al. Reprogramming human T cell function and specificity with non-viral genome targeting. Nature 13, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Frigault MJ et al. Identification of chimeric antigen receptors that mediate constitutive or inducible proliferation of T cells. Cancer Immunol. Res 3, 356–367 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Watanabe N et al. Fine-tuning the CAR spacer improves T-cell potency. Oncoimmunology 5, e1253656 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Smith EL et al. Development and Evaluation of an Optimal Human Single-Chain Variable Fragment-Derived BCMA-Targeted CAR T Cell Vector. Mol. Ther 26, 1447–1456 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Calderon H, Mamonkin M & Guedan S Analysis of CAR-Mediated Tonic Signaling. in Methods in Molecular Biology vol. 2086 223–236 (Humana Press Inc., 2020). [DOI] [PubMed] [Google Scholar]
- 207.Smith EL et al. GPRC5D is a target for the immunotherapy of multiple myeloma with rationally designed CAR T cells. Sci. Transl. Med 11, 7746 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Singh N et al. Antigen-independent activation enhances the efficacy of 4-1BB-costimulated CD22 CAR T cells. Nat. Med 27, 842–850 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Mestermann K et al. The tyrosine kinase inhibitor dasatinib acts as a pharmacologic on/off switch for CAR T cells. Sci. Transl. Med (2019) doi: 10.1126/scitranslmed.aau5907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Weber EW et al. Pharmacologic control of CAR-T cell function using dasatinib. Blood Adv 3, 711–717 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Weber EW et al. Transient rest restores functionality in exhausted CAR-T cells through epigenetic remodeling. Science (80-. ). 372, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Müller MR & Rao A NFAT, immunity and cancer: A transcription factor comes of age. Nat. Rev. Immunol 10, 645–656 (2010). [DOI] [PubMed] [Google Scholar]
- 213.Hogan PG Calcium–NFAT transcriptional signalling in T cell activation and T cell exhaustion. Cell Calcium 63, 66–69 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Smith-Garvin JE, Koretzky GA & Jordan MS T cell activation. Annu. Rev. Immunol 27, 591–619 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Khan O et al. TOX transcriptionally and epigenetically programs CD8+ T cell exhaustion. Nature (2019) doi: 10.1038/s41586-019-1325-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Alfei F et al. TOX reinforces the phenotype and longevity of exhausted T cells in chronic viral infection. Nature (2019) doi: 10.1038/s41586-019-1326-9. [DOI] [PubMed] [Google Scholar]
- 217.Yao C et al. Single-cell RNA-seq reveals TOX as a key regulator of CD8+ T cell persistence in chronic infection. Nat. Immunol 20, 890–901 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Good CR et al. An NK-like CAR T cell transition in CAR T cell dysfunction. Cell 184, 6081–6100.e26 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Fraietta JA et al. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature 1 (2018) doi: 10.1038/s41586-018-0178-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Jain N et al. Abstract LB153: Emergence of a hyper-proliferative phenotype in TET2 edited human CAR T cells . Cancer Res 81, LB153–LB153 (2021). [Google Scholar]
- 221.Belk JA et al. Genome-wide CRISPR screens of T cell exhaustion identify chromatin remodeling factors that limit T cell persistence. Cancer Cell (2022) doi: 10.1016/J.CCELL.2022.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Pace L et al. The epigenetic control of stemness in CD8+ T cell fate commitment. Science (80-. ). 359, 177–186 (2018). [DOI] [PubMed] [Google Scholar]
- 223.Jain N et al. Disruption of H3K9me3-mediated gene silencing augments CAR T cell functional persistence. Mol. Ther 30, (2022). [Google Scholar]
- 224.Yu YR et al. Disturbed mitochondrial dynamics in CD8+ TILs reinforce T cell exhaustion. Nat. Immunol (2020) doi: 10.1038/s41590-020-0793-3. [DOI] [PubMed] [Google Scholar]
- 225.Vardhana SA et al. Impaired mitochondrial oxidative phosphorylation limits the self-renewal of T cells exposed to persistent antigen. Nat. Immunol (2020) doi: 10.1038/s41590-020-0725-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Sukumar M et al. Mitochondrial Membrane Potential Identifies Cells with Enhanced Stemness for Cellular Therapy. Cell Metab. 23, 63–76 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Shin H et al. A Role for the Transcriptional Repressor Blimp-1 in CD8+ T Cell Exhaustion during Chronic Viral Infection. Immunity 31, 309–320 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Dumauthioz N et al. Enforced PGC-1α expression promotes CD8 T cell fitness, memory formation and antitumor immunity. Cell. Mol. Immunol 18, 1761–1771 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Pilipow K et al. Antioxidant metabolism regulates CD8+ T memory stem cell formation and antitumor immunity. JCI insight 3, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Van Bruggen JAC et al. Chronic Lymphocytic Leukemia Cells Impair Mitochondrial Fitness in CD8+ T Cells and Impede CAR T Cell Efficacy. Blood (2018) doi: 10.1182/blood-2018-99-115911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Legut M et al. A genome-scale screen for synthetic drivers of T cell proliferation. Nat. 2022 6037902 603, 728–735 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Schmidt R et al. CRISPR activation and interference screens decode stimulation responses in primary human T cells. Science (80-. ). 375, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Ye L et al. A genome-scale gain-of-function CRISPR screen in CD8 T cells identifies proline metabolism as a means to enhance CAR-T therapy. Cell Metab. 34, 595–614.e14 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Joshi NS & Kaech SM Effector CD8 T Cell Development: A Balancing Act between Memory Cell Potential and Terminal Differentiation. J. Immunol 180, 1309–1315 (2008). [DOI] [PubMed] [Google Scholar]
- 235.Tschumi BO et al. CART cells are prone to Fas- and DR5-mediated cell death. J. Immunother. Cancer 6, 71 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Kasakovski D, Xu L & Li Y T cell senescence and CAR-T cell exhaustion in hematological malignancies. J. Hematol. Oncol 11, 1–9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Sanmamed MF et al. A burned-out cd8+ t-cell subset expands in the tumor microenvironment and curbs cancer immunotherapy. Cancer Discov. 11, 1700–1715 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Franco F, Jaccard A, Romero P, Yu YR & Ho PC Metabolic and epigenetic regulation of T-cell exhaustion. Nat. Metab 2020 210 2, 1001–1012 (2020). [DOI] [PubMed] [Google Scholar]