

Abstract

Isoaspartic acid (isoAsp) is a common protein modification that spontaneously arises from asparagine or aspartic acid and has been linked to various diseases and health conditions. However, current methods for identifying isoAsp sites in proteins often suffer from ambiguity and have not gained widespread adoption. We developed a novel method that exclusively labels isoAsp with deuterium. This method capitalizes on the unique structural characteristics of isoAsp residues, which possess a free α-carboxyl group and can form an oxazolone ring. Once the oxazolone ring forms, it facilitates racemization at the Cα-position, incorporating a deuteron from a D2O solvent. The sites of deuterium-incorporated isoAsp in proteins can be unequivocally determined by comparing the precursor and product ion masses of the peptides from proteins reacted in H2O and D2O. The effectiveness of this method has been demonstrated through its application to model proteins lysozyme and rituximab. Furthermore, we have confirmed that the isoAsp deuterium-labeling reaction efficiently labels both l- and d-isoAsp without distinction, as well as isoglutamic acid (isoGlu), for which no effective detection methods currently exist.
Isoaspartic acid (isoAsp) formation from Asn or Asp is a common protein modification, occurring spontaneously (nonenzymatically) through a succinimide intermediate (Scheme 1).1 The rate of isoAsp formation varies significantly depending on the adjacent amino acid on the C-terminal side of Asp or Asn. For instance, the half-life (t1/2) of the isomerization reaction for the -Asn-Gly- sequence is 1.4 days at pH 7.4 and 37 °C, whereas it exceeds 100 days for the -Asn-Phe-sequence.2 The formation of isoAsp introduces a methylene group into the protein backbone, potentially altering the three-dimensional structure crucial for the physicochemical and functional properties of proteins. This alteration can have significant biological consequences. For example, research has demonstrated that the formation of isoAsp in proteins associated with Alzheimer’s disease, such as amyloid-β and tau, increases their aggregation propensity.3,4 Furthermore, this modification is one of the most frequent modifications in therapeutic proteins during production and storage,5,6 potentially impairing their efficacy. Consequently, considerable interest exists in identifying isoAsp sites in proteins.
Scheme 1. Pathways for Spontaneous Deamidation, Isomerization, and Racemization of l-Asn and l-Asp Residues in Proteins.

Deamidation of l-Asn begins with the backbone nitrogen of the C-terminal neighboring residue attacking the side-chain carbonyl carbon of Asn, forming succinimide intermediate I. Hydrolysis of intermediate I leads to l-Asp or l-isoAsp. The intermediate I can also undergo racemization via its enol form II, which produces the D configuration of succinimide intermediate III. Hydrolysis of intermediate III yields d-Asp or d-isoAsp. Isomerization and racemization of l-Asp begin similarly with the backbone nitrogen of the C-terminal neighboring residue attacking the side-chain carbonyl carbon of Asp, forming succinimide intermediate I. Hydrolysis of intermediate I leads to either the original l-Asp or l-isoAsp. l-Asp can also be converted to d-isoAsp or d-Asp via intermediates II and III as l-Asn.
Various strategies have been developed to identify the sites of isoAsp residues within proteins. These strategies are based on (1) liquid chromatography (LC) or ion mobility mass spectrometry (IM-MS) separation, (2) diagnostic fragment ion analysis, or (3) protein l-isoaspartyl methyltransferase (PIMT)-mediated labeling. The first strategy involves the separation of isoAsp-containing peptides from their unmodified counterparts, either through LC or IM-MS techniques.7−11 The second strategy monitors the diagnostic fragment ions unique to isoAsp produced by electron-capture/-transfer dissociation,12−14 photodissociation,15 or matrix-assisted laser desorption/ionization time-of-flight/time-of-flight with collision-induced dissociation.16,17 The third strategy involves using PIMT to label isoAsp residues. PIMT methylates the β-carboxyl group of isoAsp and facilitates the formation of succinimide, which can react with a nucleophile, such as H218O, to generate labeled isoAsp residues.18−23 The first approach falls short in achieving the required chemical specificity to differentiate between those of isoAsp and Asp. Consequently, the preparation of peptide isomers is imperative for thorough verification. The second approach has the capability to directly pinpoint isoAsp sites without the need for labeling of isoAsp residues. However, it heavily depends on high-quality tandem mass spectrometry data, introducing the possibility of ambiguity, especially in situations in which isoAsp formation is minimal. As a result, this approach may also necessitate the synthesis of peptide isomers to ensure validity of the results. The third approach produces two labeled products, labeled isoAsp and Asp, depending on which carbonyl carbon in the succinimide is attacked by the nucleophile used (see Scheme 1). This complexity hinders the accurate quantitative assessment of the isoAsp formation. Furthermore, these PIMT-mediated labeling approaches cannot be applied to label the d-enantiomer of isoAsp (d-isoAsp) due to a lack of d-isoAsp recognition by PIMT.24,25 Thus, overcoming these limitations is essential to enhancing our capacity to characterize isoAsp residues within proteins.
In the field of peptide synthesis, researchers have long been aware of the racemization of C-terminal amino acids. This process is well-understood and involves the formation of an oxazolone intermediate,26 which is accelerated when the free α-carboxy group of the C-terminal amino acid is activated using acylation agents.27,28 Leveraging this unique feature, methods have been developed to label the C-terminal amino acids with tritium at the Cα-position29,30 or with nucleophiles at the C-terminal carboxyl group.31,32 We realized that isoAsp residues, unlike other amino acids in proteins (excluding C-terminal amino acids), also have a free α-carboxyl group, as shown in Scheme 1. This distinctive characteristic suggests that isoAsp residues might undergo oxazolone ring formation. If confirmed, we could develop a method to label isoAsp residues with deuterium selectively.
Based on oxazolone chemistry, we used model peptides to optimize selective deuterium-labeling conditions for isoAsp, in which we ensured exclusive deuterium labeling of isoAsp, without any isoAsp formation from Asn or Asp. We confirmed that the deuterium labeling reaction can label both l- and d-isoAsp. We subsequently developed a protocol to detect isoAsp sites in proteins using lysozyme, and this protocol was validated using rituximab. Our investigations demonstrated the reliability of our approach in selectively labeling and identifying isoAsp residues, even in cases with low isoAsp formation. Furthermore, we also confirmed that the same isoAsp labeling reaction can efficiently deuterate isoglutamic acid (isoGlu).
Experimental Section
Materials
The following peptides were used in the study: VYPNGA-NH2, VYPDGA-NH2, VYPL-isoDGA-NH2 (L-isoD is l-isoaspartic acid), PGPQGFQ-NH2, PGPEGFQ-NH2, and PGPL-isoEGFQ-NH2 (L-isoE is l-isoglutamic acid). These peptides were custom-synthesized by Biomatik Corp. (Kitchener, ON, Canada). Additionally, delta-sleep-inducing peptide (DSIP, WAGGDASGE), l-isoAsp5-delta-sleep-inducing peptide (L-isoD-DSIP, WAGGL-isoDASGE), and d-isoAsp5-delta-sleep-inducing peptide (D-isoD-DSIP, WAGGD-isoDASGE) were obtained from Bachem America (Torrance, CA). Chicken egg lysozyme, rituximab, pyridine, acetic anhydride, deuterium oxide (99.9 atom % D), and hydrazine (35 wt % in H2O) were purchased from Sigma-Aldrich (St. Louis, MO). All other chemicals were either of reagent grade or the highest commercially available quality.
Aging of Lysozyme and Rituximab
Chicken egg lysozyme (12 nmol) was dissolved in 50 μL of 100 mM ammonium bicarbonate containing 8 M urea. Next, 5 μL of 100 mM DTT (dithiothreitol) was added to reduce the disulfide bonds, and the mixture was incubated at room temperature for 15 min. Then, 5 μL of 250 mM iodoacetamide was added to alkylate the thiol group of the cysteine residues, and the solution was incubated further at room temperature for 15 min. The resulting protein solution was diluted four times by adding 100 mM ammonium bicarbonate and then divided into two parts. One part was allowed to age at room temperature for 16 h, and the other part was stored at −20 °C to serve as an unaged protein control. The aged and unaged protein solutions were stored at −20 °C until analyzed.
The preparation of aged rituximab was conducted as follows. First, 3.45 nmol rituximab was dissolved in 500 μL of 100 mM N-ethylmorpholine-acetate buffer (pH 8.0). This solution was then divided into two parts. One part was allowed to age at 50 °C for 5 days, while the other was stored at −20 °C for the unaged protein control. The aged and unaged rituximab was stored at −20 °C until analyzed.
Deuterium Labeling of Peptides
The following peptides were subjected to the deuterium-labeling reaction: VYPNGA-NH2, VYPDGA-NH2, VYPL-isoDGA-NH2, DSIP, L-isoD-DSIP, D-isoD-DSIP, PGPQGFQ-NH2, PGPEGFQ-NH2, and PGPL-isoEGFQ-NH2. To each peptide (1 nmol), 10 μL of D2O and 30 μL of pyridine were added, and the solution was vortexed to ensure the peptide’s complete solubilization. Then, 30 μL of acetic anhydride (Ac2O) was added to initiate the reaction, and the mixture was incubated at room temperature for for 30 min to deuterate the isoAsp or isoGlu residue. As a control, the peptide was also reacted in an H2O solvent, where the D2O portion in the reaction mixture was replaced with H2O. After the incubation, the reaction mixture was dried using a Speed vac concentrator at 25 °C. Then, the dried peptide was further incubated in 50 μL of a hydrazine solution (0.35 wt % in H2O) at room temperature for 30 min to ensure the complete removal of the acetyl moiety that could have formed on Ser/Thr/Tyr hydroxyl groups. Subsequently, the reaction mixture was dried in a Speed vac concentrator at 40 °C to remove hydrazine. The peptides, treated in both D2O and H2O, were then solubilized in 50 μL of 0.1% formic acid and subjected to liquid-chromatography–mass spectrometry (LC–MS) analysis as described below.
Deuterium Labeling of Proteins
Essentially, the same chemical reaction used for the peptides was employed to label the isoAsp residues in proteins, except that the volume of the reaction was 3 times larger than that for the peptides, but the ratio of D2O/pyridine/Ac2O was maintained to be 1:3:3.
Labeling of rituximab was carried out as follows. Aged and unaged rituximab (1.7 nmol of each) was dissolved in 50 μL of 100 mM ammonium bicarbonate containing 8 M urea. Next, 5 μL of 100 mM DTT (dithiothreitol) was added to reduce disulfide bonds, and the mixture was incubated at room temperature for 15 min. Then, 5 μL of 250 mM iodoacetamide was added, and the solution was incubated at room temperature for 15 min. The protein was then desalted using a reverse-phase C4 spin column (BioPureSPN Mini, PROTO 300 C4 cartridge, Nest Group) following the manufacturer’s instructions. The collected protein fraction was divided into two microcentrifuge tubes and dried in a Speed vac concentrator at 25 °C. As described below, one tube was subjected to the isoAsp labeling reaction in D2O and the other in H2O. To the dried protein, 30 μL of D2O and 90 μL of pyridine were added, and the solutions were vortexed to solubilize the protein. Then, 90 μL of acetic anhydride (Ac2O) was added, and the mixture was incubated at room temperature for 30 min to deuterate the protein’s isoAsp residues. As a control, the protein was also made to react in a H2O solvent. After the incubation, the reaction mixture was dried using a Speed vac concentrator at 25 °C. The dried protein was further incubated in 50 μL of a hydrazine solution (0.35 wt % in H2O) at room temperature for 30 min to remove any acetyl moieties that could have formed on Ser/Thr/Tyr hydroxyl groups. Subsequently, hydrazine was removed by drying the protein solution in a Speed vac concentrator at 40 °C. The dried deuterium-labeled protein was initially solubilized in 20 μL of 100 mM ammonium bicarbonate containing 8 M urea and then diluted five times with 100 mM ammonium bicarbonate. Then, 8 μg of immobilized chymotrypsin (ProteoChem, Hurricane, UT) was added to the solution and incubated at room temperature for 30 min. The protein digest was then desalted using a reverse-phase C18 spin column (BioPureSPN Mini, FastEq, TARGA C18 cartridge, Nest Group) following the manufacturer’s instructions. The eluted peptide fraction was dried in a Speed vac concentrator at 25 °C, dissolved in 50 μL of 0.1% formic acid, and analyzed by liquid-chromatography–tandem mass spectrometry (LC–MS/MS) as described below.
Labeling of aged and unaged lysozyme was carried out with the same procedures described above to label rituximab, except that the reduction and S-alkylation steps were skipped because they were already reduced and S-alkylated in their aging process.
Mass Spectrometry Analysis
LC–MS analysis of peptide samples was conducted using an Agilent 1290 Infinity HPLC system (Agilent, Santa Clara, CA) coupled to an Agilent 6460 Triple-Quadrupole mass spectrometer equipped with an electrospray ion source. After the sample preparation described above, 5 μL of the sample was injected into a Kinetex 2.6 μm Polar C18 100 Å (100 × 2.1 mm) column (Phenomenex, Torrance, CA). The chromatography used a linear acetonitrile gradient from 0 to 30% over 30 min in aqueous 0.1% formic acid with a 200 μL/min flow rate. The ion source parameters were set as follows: capillary voltage (2000 V), nozzle voltage (2000 V), nebulizer gas pressure (45 psi), sheath gas flow (11 L/min), and sheath gas temperature (300 °C). Nitrogen was used as both the source and the collision gas. The mass spectrometer monitored the ions in the m/z 300 to 1800 range. The acquired LC–MS data were analyzed using Agilent MassHunter Qualitative Analysis software.
LC–MS/MS analyses of peptides and protein digests were performed using the Fusion LumosTM Orbitrap Mass Spectrometer (Thermo Fisher Scientific, Waltham, MA), coupled with a Dionex Ultimate 3000 RSLCnano LC system (Thermo Fisher Scientific). LC was carried out using a Dionex 25 cm × 75 μm id Acclaim Pepmap C18, 2 μm, 100 Å reversed-phase capillary chromatography column (Thermo Fisher Scientific). Peptides eluted from the column in an acetonitrile/0.1% formic acid gradient (flow rate = 0.3 μL/min) were introduced into the microelectrospray ion source of the mass spectrometer and analyzed using a data-dependent method with CID fragmentation.
Peptide Identification
Peptides were identified by comparing experimental peptide MS/MS spectra with the amino acid sequences of chicken egg lysozyme or rituximab using the MassMatrix search engine (Mass Matrix, Columbus, OH). Carbamidomethylation of cysteine (+57 Da) was considered a fixed modification, while variable modifications included oxidation of methionine to methionine sulfoxide (+16 Da), deamidation of Asn to Asp (+1 Da), conversion of Asp to isoAsp, followed by the incorporation of a deuteron (+1 Da), and conversion of Asn to isoAsp, followed by the incorporation of a deuteron (+2 Da). Chymotrypsin was allowed to perform cleavages at any amino acids.
Results and Discussion
Optimization of IsoAsp-Labeling Conditions with Deuterium
To investigate the deuterium incorporation conditions in isoAsp residues, we prepared three hexapeptides: isoDG (VYPL-isoDGA-NH2), DG (VYPDGA-NH2), and NG (VYPNGA-NH2). isoDG contained an isoAsp residue, DG replaced it with Asp, and NG replaced it with Asn. DG was a negative control to ensure that no deuterium incorporation into its Asp occurred. NG was included to verify that no Asn to isoAsp conversion occurred. Our primary goal was to identify conditions for deuterium labeling in the isoDG’s isoAsp residue, keeping DG’s Asp unaffected and preventing Asn to isoAsp conversion in NG.
We initially followed Matsuo and Narita’s method for tritium labeling of the protein C-terminal amino acids.33 We found that pyridine was most satisfactory as a base in comparison with the other bases such as triethylamine because it led to the highest deuteration of isoAsp in the range of temperature from 0 to 37 °C. The pH of the H2O/pyridine (1:3) solution used for dissolving the peptides was initially 7.3 and then rapidly dropped to 3.8 within 1 min after the addition of Ac2O, followed by a gradual increase to 4.3 in 30 min at room temperature. This pH shift could suggest that the initial pH drop is attributed to the rapid consumption of pyridine by the reaction with Ac2O, resulting in the formation of acetic acid and acetylpyridinium, which could react gradually with the α-carboxy group of isoAsp, forming a mixed anhydride IV and regenerating pyridine, causing the gradual pH rise.34 The possible reaction scheme for the deuterium labeling of isoAsp is shown in Scheme 2.
Scheme 2. Deuterium Labeling of isoAsp and isoGlu.

In the presence of pyridine and acetic anhydride, acetylpyridinium is formed, which then reacts with the α-carboxyl group of isoX (X = Asp when n = 1; X = Glu when n = 2) to form mixed anhydride IV. The α-hydrogen (α-H) of VI in the keto form is abstracted by pyridine in step a1 to form an enolate anion represented by two resonance hybrids VII and VIII. The subsequent reaction of this enolate with deuterium (red) in D2O in step a2 gives a racemic oxazolone VIV incorporating a deuteron at the α-position. The yield of deuterium labeling is significantly enhanced by the repetitive process of generating an oxazolone VI from IsoX through step b, as long as a large excess of acetic anhydride remains in the reaction mixture. The labile amide, hydroxy, and carboxy protons (blue) being exchanged with deuterons in D2O are exchanged back to protons in H2O, but the deuterons incorporated into the α-position of isoX remain unexchanged.
Figure 1a shows the representative LC–MS profiles of isoDG, DG, and NG underwent a reaction in D2O/pyridine/Ac2O (1:3:3, v/v/v) or H2O/pyridine/Ac2O (1:3:3, v/v/v) at room temperature for 30 min, followed by treatment with 0.35% hydrazine at the same temperature for another 30 min. Note that the isoAsp labeling reaction acetylates the amino groups of Lys and N-termini of proteins as well as the hydroxyl groups of Ser/Thr/Tyr. Following hydrazine treatment, the acetyl groups are removed from the hydroxyl groups of Ser/Thr/Tyr while remaining unaffected on the Lys and proteins’ N-termini. Scheme S1 provides a summary of amino acid residues susceptible to acetylation during the isoAsp labeling reaction and outlines their subsequent outcomes following hydrazine treatment. The reaction product of isoDG, i.e., N-acetyl-isoDG, yielded two chromatographic peaks with identical mass, presumably representing N-acetyl-l- and d-isoAsp resulting from racemization of isoAsp. Its mass reacted in D2O was 662.4 (observed ion: m/z 663.4, z = 1), which was 1 Da larger than that reacted in H2O (661.4 Da, observed ion: m/z 662.4, z = 1), indicating the incorporation of one deuterium atom. Additionally, the MS/MS spectra of isoDG reacted in D2O clearly showed that all of the isoAsp-containing fragment ions were 1 Da heavier than those originating from the same peptide reacted in H2O, indicating that the isoAsp residue was the one labeled with a deuterium atom (Figure 1b).
Figure 1.

(a) LC–MS analysis of isoDG, DG, and NG subjected to the deuterium-labeling reaction. Each peptide was dissolved in 40 μL of D2O/pyridine (1:3, v/v) or H2O/pyridine (1:3, v/v) and reacted with 30 μL of acetic anhydride (Ac2O) at room temperature for 30 min. After the reaction, the peptide was dried and further incubated in 50 μL of a hydrazine solution (0.35 wt % in H2O) at room temperature for 30 min. The resulting peptide was then analyzed by LC–MS. The total ion chromatograms (m/z 300–1800) and mass spectra of the three peptides reacted in D2O (red) or H2O (blue) are shown. (b) MS/MS spectra of isoDG reacted in D2O (upper MS/MS spectrum, precursor ions: m/z 663.3, z = 1) or H2O (lower MS/MS spectrum, precursor ions: m/z 662.3, z = 1). The fragment ions that contain a deuterated isoAsp residue are indicated with an asterisk.
The corresponding experiment on DG showed no incorporation of deuterium as its mass incubated in D2O (661.4 Da, observed ion: m/z 662.4, z = 1) remained the same as the mass incubated in H2O (Figure 1a), demonstrating the selectivity of this labeling reaction to isoAsp. Similarly, the corresponding experiment on NG showed no incorporation of deuterium as its mass incubated in D2O (660.4 Da, observed ion: m/z 661.4, z = 1) remained the same as the masses incubated in H2O (Figure 1a). In addition, no evidence of the conversion of Asn to isoAsp or Asp was observed. Thus, these results with the model peptides demonstrated that isoAsp-specific deuterium labeling can be achieved with the conditions thus established, and the condition does not induce the deamidation of Asn.
Verification of Deuterium Labeling in Both l- and d-isoAsp
According to the oxazolone chemistry concerning the racemization of the isoAsp residue, both the l- and d-enantiomers of isoAsp could be deuterated without differentiation. To validate this prediction, we conducted experiments using L-isoD-DSIP (WAGGL-isoDASGE), D-isoD-DSIP (WAGGD-isoDASGE), and a negative control peptide DSIP (WAGGDASGE). These peptides were subjected to the same deuterium-labeling reaction described above, and the reaction products were analyzed by LC–MS/MS.
The observed masses for the L-isoD-DSIP and D-isoD-DSIP reacted in D2O were 2 Da higher (892.4 Da, observed ion: m/z 893.4, z = 1) compared to those reacted in H2O (890.4 Da, observed ion: m/z 891.4, z = 1) (Figure S1a,b, respectively), indicating the incorporation of two deuterium atoms likely to the isoAsp and C-terminal Glu residues. The MS/MS spectra of those peptides reacted in D2O and H2O confirmed the deuteration in these specific sites. In contrast, the mass for the negative control peptide DSIP reacted in D2O was only 1 Da higher (891.4 Da, observed ion: m/z 890.4, z = 1) than the peptide reacted in H2O (890.4 Da, observed ion: m/z 891.4, z = 1) (Figure S1c), indicating that only the C-terminal amino acid incorporated a deuterium atom. The MS/MS spectra of the peptides further supported this finding. These results demonstrated that the oxazolone-based isoAsp-labeling method effectively labels both l- and d-isoAsp residues without distinction. This method has an advantage over PIMT-mediated labeling, which targets the l-enantiomers of isoAsp residues exclusively.
Strategy to Identify isoAsp Residues in Proteins
The oxazolone-based chemistry was then tested with aged and unaged lysozyme prepared as described in the experimental section. We chose lysozyme because the protein contains an Asn103-Gly104 sequence, the most deamidation-prone amino acid sequence.1 The aged and unaged lysozymes were desalted by a reverse-phase C4 spin column and subjected to the isoAsp-labeling reaction in D2O or H2O. After the labeling, the protein was treated with hydrazine to remove the acetyl groups on the hydroxy groups of Ser/Thr/Tyr residues. Then, the protein was digested by immobilized chymotrypsin and analyzed by LC–MS/MS. The entire sample-processing workflow is shown in Figure S2.
The unaged lysozyme yielded only the peptide with the original sequence (CAKKIVSDGNGMNAW, where Asn103 is underlined; Figure 2a, peak 1). In contrast, the aged lysozyme produced three peptide species: one with the original sequence, another with Asn103 converted to Asp, and a third with Asn103 converted to isoAsp (Figure 2a, peaks 1–3, respectively). The masses of peak 1 (1749.8 Da, observed ion: m/z 875.9, z = 2) and peak 2 (1750.8 Da, observed ion: m/z 876.4, z = 2) remained the same regardless of the labeling solvent, whether D2O or H2O (Figure 2b, peaks 1 and 2), indicating that no deuterium was incorporated into these peptides. However, the mass of peak 3 reacted in D2O (1751.8 Da, observed ion: m/z 876.9, z = 2) was 1 Da heavier than that reacted in H2O (1750.8 Da, observed ion: m/z 876.4, z = 2), indicating deuterium incorporation during the labeling reaction in D2O and therefore the presence of an isoAsp residue. The MS/MS spectra of peak 3 from the peptide reacted in D2O showed that all of the fragment ions containing residue 103 (isoAsp103) (y9, y10, y11, y12, b13, and b142+) were shifted by 1 Da compared to the corresponding fragment ions from the peptide reacted in H2O, unequivocally indicating that residue 103 was the site of deuterium incorporation and, thus, the location of isoAsp.
Figure 2.

LC–MS/MS analyses of aged and unaged lysozyme subjected to the predigestion isoAsp-labeling reaction. Aged and unaged lysozymes were dissolved in 120 μL of D2O/pyridine (1:3, v/v) or H2O/pyridine (1:3, v/v) and reacted with 90 μL of acetic anhydride (Ac2O) at room temperature for 30 min. After the reaction, the mixture was dried and further incubated in 50 μL of a hydrazine solution (0.35 wt % in H2O) at room temperature for 30 min. Subsequently, the protein was digested by chymotrypsin and analyzed by LC–MS/MS. (a) Extracted ion chromatograms (m/z 875.89–877.91) encompassing the doubly charged ions of the following three peptides: peak 1: CAKKIVSDGNGMNAW, peak 2: CAKKIVSDGDGMNAW, and peak 3: CAKKIVSDGisoDGMNAW. (b) Mass spectra of peaks 1–3 from the lysozyme reacted in D2O or H2O. (c) MS/MS spectra of peak 3 from the aged lysozyme reacted in D2O or H2O. The fragment ions incorporating a deuterium atom are indicated with an asterisk.
The proportions of Asn, Asp, and isoAsp variants at residue 103 were determined by analyzing the chromatographic peak areas of distinct peptide variants (Figure 2a, peak 1–3). In a mere 16 h of incubation at room temperature and pH 8, approximately 60% of Asn103 within lysozyme was converted to isoAsp (47.5%) or Asp (12.4%). This rapid conversion can be attributed to the fact that the lysozyme subjected to the aging process had been entirely denatured (reduced and S-alkylated), leading to full exposure of Asn103 to the surrounding solvent during the aging process. This observation implies that a significant Asn to isoAsp or Asp conversion could occur for Asn–Gly sequences during the commonly employed overnight protein digestion procedure in proteomics. To address this, we carefully fine-tuned the oxazolone-based deuterium-labeling procedure and subsequent protein digestion conditions under which the conversion of labile Asn to isoAsp does not occur. As verified by the results shown in Figure 2a, no chromatographic peaks corresponding to Asp or isoAsp species were observed in unaged-lysozyme samples subjected to the same isoAsp-labeling reaction and subsequent protein digestion protocols, demonstrating that the optimized isoAsp characterizing protocol shown in Figure 2 does not promote the formation of isoAsp from the most labile Asn–Gly sequence.
The strategy described above labeled isoAsp before digesting proteins. It is also possible to label isoAsp after digesting proteins. This postdigestion labeling approach allows the use of trypsin and Lys-C for protein digestion since the Nε-amino groups of Lys residues are not yet acetylated. However, it is important to note that in this postdigestion labeling approach, acetylation of N-terminus and incorporation of a deuteron occur for all the peptides subjected to the isoAsp deuterium-labeling reaction. In addition, the reaction leads to the racemization of isoAsp residues and the C-terminal amino acids, generating both l- and d-forms, and thus could complicate the LC–MS profiles and data interpretation. Given these complexities, we recommend a predigestion labeling strategy.
Identification of isoAsp Sites in an Antibody, Rituximab
Subsequently, our investigation focused on pinpointing isoAsp sites within the rituximab samples subjected to a 5 day aging process at 50 °C. The aged and unaged rituximab samples underwent the same isoAsp-labeling protocol for proteins, as illustrated in Figure 2.
Figure 3a shows the chromatographic profile of the peptides containing one of the isoAsp sites (Asn55 in the heavy chain). The unaged rituximab gave one dominant species with the original sequence (IGAIYPGNGDTSY, where Asn55 is underlined; Figure 3a, peak 1). On the other hand, the aged rituximab produced three peptide species: one with the original sequence, another with Asn55 converted to Asp, and a third with Asn55 converted to isoAsp (Figure 3a, peaks 1–3, respectively). The masses of peak 1 (1326.6 Da, observed ion: m/z 664.3, z = 2) and peak 2 (1327.6 Da, observed ion: m/z 664.8, z = 2) remained the same regardless of the labeling solvent (Figure 2b, peaks 1 and 2), indicating that no deuterium was incorporated into these peptides. The mass of peak 3 reacted in D2O (1328.6 Da, observed ion: m/z 665.3, z = 2), however, was 1 Da heavier than that reacted in H2O (1327.6 Da, observed ion: m/z 664.8, z = 2), indicating that the peptide incorporated a deuteron and, therefore, the presence of an isoAsp. The MS/MS spectra of peak 3 from the peptide reacted in D2O showed that all the fragment ions containing residue 55 (isoAsp103) (y8, y9, b8, b11, and b12) were shifted by 1 Da compared to the corresponding fragment ions from the peptide reacted in H2O, indicating that residue 55 was the site of deuterium incorporation and therefore the site of isoAsp.
Figure 3.
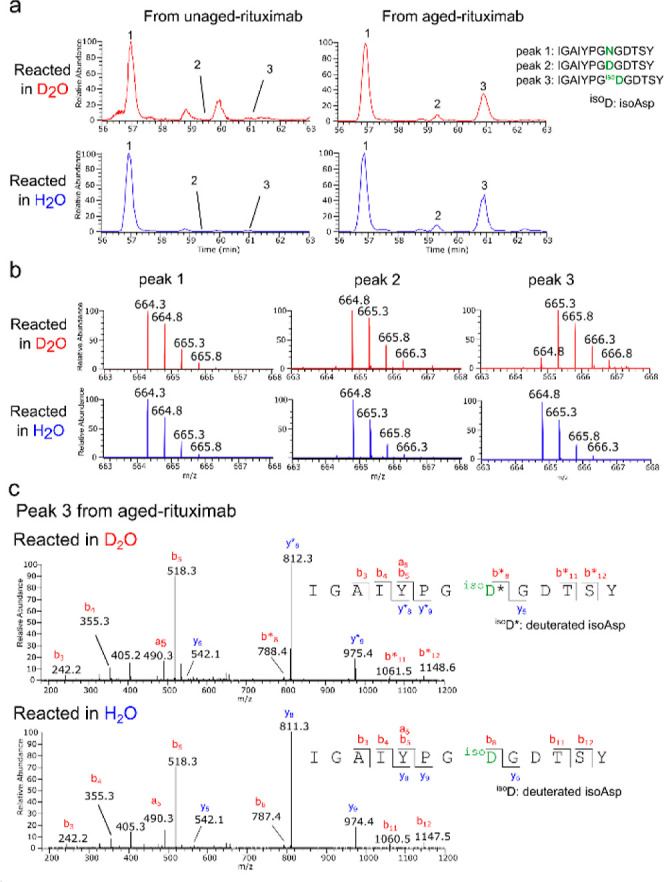
Representative LC–MS/MS data for one of the isoAsp sites identified in rituximab. Aged and unaged rituximab were dissolved in 120 μL of D2O/pyridine (1:3, v/v) or H2O/pyridine (1:3, v/v) and reacted with 90 μL of acetic anhydride (Ac2O) at room temperature for 30 min. After the reaction, the mixture was dried and further incubated in 50 μL of a hydrazine solution (0.35 wt % in H2O) at room temperature for 30 min. Subsequently, the protein was digested by chymotrypsin and analyzed by LC–MS/MS. (a) Extracted ion chromatograms (m/z 664.29–666.31) encompassing the doubly charged ions of the following three peptides containing amino acid residue 55 of the heavy chain of rituximab: peak 1: IGAIYPGNGDTSY, peak 2: IGAIYPGDGDTSY, and peak 3: IGAIYPGisoDGDTSY. Residue 55 is underlined. (b) Mass spectra of peaks 1–3 from rituximab reacted in D2O or H2O. (c) MS/MS spectra of peak 3 from aged rituximab reacted in D2O or H2O. The fragment ions incorporating a deuterium atom are indicated with an asterisk.
We identified four other isoAsp sites (Asn52 and 157 within the light chain and Asn163 and 319 within the heavy chain). Their chromatographic profiles and the MS and MS/MS spectra are shown in Figures S3–S6. The proportions of Asn, Asp, and isoAsp variants at individual sites within aged rituximab were determined by analyzing the chromatographic peak areas of distinct peptide variants (Figures 3a and S3a–6a). The analysis revealed varying levels of isoAsp conversion, ranging from 0.2 to 26%, at five isoAsp sites in rituximab (Table 1). This demonstrates the effectiveness of the oxazolone-based deuterium-labeling method in detecting and quantifying isoAsp, even when its conversion is less than 1%. No Asp to isoAsp conversion was observed in these experiments. This may be due to the fact that the conversion of Asp to isoAsp favors mildly acidic conditions when the β-carboxyl group of Asp is protonated.35,36 However, since we aged the protein at pH 8, which is alkaline, this conversion from Asp to isoAsp is known to proceed more slowly.37
Table 1. Estimated isoAsp Formation at Each isoAsp Site in Aged Rituximaba.
| fraction
of Asn/Asp/isoAsp residues (%) |
||||
|---|---|---|---|---|
| position | peptide sequence | Asn | conversion to Asp | conversion to isoASP |
| Rituximab_Light chain | ||||
| Asn52 | ATSNLASGVPVRF | 99.8 | n.o. | 0.2 |
| Asn157 | KVDNALQSGNSQESVTEQDSKDSTY | 94.4 | 1.2 | 4.4 |
| Rituximab_Heavy chain | ||||
| Asn55 | IGAIYPGNGDTSY | 69.3 | 4.4 | 26.3 |
| Asn163 | SWNSGALTSGVH | 69.7 | 4.0 | 26.3 |
| Asn319 | TVLHQDWLNGKEY | 97.2 | n.o. | 2.8 |
n.o.: Not observed. The amino acid residues converted to isoAsp are underlined.
Identification of C-Terminal Amino Acid Residues of Rituximab
In addition to its primary application of identifying isoAsp residues, this method demonstrated an added capability: the precise identification of C-terminal amino acids within proteins. Shown in Figure S7a are the MS spectra of the C-terminal peptide of rituximab’s light chain, namely, SSPVTKSFNRGEC (where K is Nε-Ac-Lys and C is carbamidemethyl-Cys), following a labeling reaction in D2O or H2O. The mass of the peptide originated from the D2O-labeled rituximab was 1510.8 Da (observed ion: m/z 756.4, z = 2), which was 1 Da higher than that originated from the H2O-labeled rituximab (1509.8 Da, observed ion: m/z 755.9, z = 2), indicating the incorporation of one deuterium atom into the peptide. The MSMS spectra (Figure S7b) clearly showed that the C-terminal amino acid, Cys, was the one that incorporated a deuteron since all the fragment ions from the peptide originated from the D2O-labeled rituximab were 1 Da heavier than those originated from the peptide originated from the H2O-labeled rituximab, unequivocally showing that the Cys is the C-terminal amino acid of the light chain.
Similarly, the C-terminal amino acid of the rituximab heavy chain was determined to be Gly (Figure S8). Interestingly, it was found that C-terminal Gly incorporated two deuterium atoms. This phenomenon can be attributed to Gly residue’s possession of two α-protons, both susceptible to deuterium exchange. Incorporating two deuterons into Gly highlights that the oxazolone-based deuterium-labeling reaction occurs iteratively during the labeling process. Consequently, this methodology effectively extends its utility to encompass the determination of proteins’ C-terminal amino acids.
Deuterium Labeling of isoGlu
We predicted that isoGlu residues also form an oxazolone ring and thus can be deuterated through the same mechanism shown in Scheme 2. This possibility was investigated by subjecting isoEG (PGPL-isoEGFQ-NH2), as well as EG (PGPEGFQ-NH2) and QG (PGPQGFQ-NH2) to the same labeling reaction as that used for labeling isoAsp. The reaction product of isoEG, N-acetyl-isoEG, gave 772.4 Da (m/z 773.4, z = 1), which was 1 Da larger than that reacted in H2O (771.4 Da, observed ion: m/z 772.4, z = 1), indicating the incorporation of one deuterium atom (Figure 4a). The MS/MS spectra of isoEG reacted in D2O showed that all of the isoGlu-containing fragment ions were 1 Da heavier than those originating from the same peptide reacted in H2O, confirming that the isoGlu residue was the one labeled with a deuterium atom (Figure 1b). No deuterium was incorporated into EG regardless of the reaction in D2O or H2O (Figure 4a), indicating that the labeling reaction did not label Glu residues. In addition, the results for QG demonstrated that the conversion of Gln to isoGlu did not occur during the reaction, as the observed peak corresponded to the (M + H)+ ion of the original QG peptide (m/z 771.4) (Figure 4a). Thus, the study with those peptides demonstrated that the same reaction for labeling isoAsp can also be used to label isoGlu.
Figure 4.

(a) LC–MS analysis of isoEG, EG, and QG peptides subjected to the deuterium-labeling reaction. Each peptide was dissolved in 40 μL of D2O/pyridine (1:3, v/v) or H2O/pyridine (1:3, v/v) and reacted with 30 μL of acetic anhydride (Ac2O) at room temperature for 30 min. After the reaction, the peptide was dried and further incubated in 50 μL of a hydrazine solution (0.35 wt % in H2O) at room temperature for 30 min. The resulting peptide was then analyzed by LC–MS. The total ion chromatograms (m/z 300–1800) and mass spectra of the three peptides reacted in D2O (red) or H2O (blue) are shown. (b) MS/MS spectra of isoEG reacted in D2O (upper MS/MS spectrum, precursor ions: m/z 773.3, z = 1) or H2O (lower MS/MS spectrum, precursor ions: m/z 772.3, z = 1). The m/z values of the fragment ions containing deuterated isoGlu residues in the upper spectrum are indicated with an asterisk.
The mechanism of Gln or Glu to isoGlu conversion occurs also through the succinimide intermediate as the formation of isoAsp does (Scheme 1) but at a significantly slower rate (t1/2 = 2–50 years depending on the amino acid residues following Gln) compared to that of isoAsp formation.38−40 Despite the considerably slower rate of isoGlu formation, it may still occur, especially in long-lived proteins. The biological significance of isoGlu formation is currently not well understood, primarily due to the lack of effective methods for identifying this modification. We postulate that this oxazolone-based deuterium-labeling approach could prove valuable for studying the consequences of isoGlu formation in various biological systems.
Conclusions
We have demonstrated that oxazolone-chemistry-based deuterium labeling can specifically target isoAsp residues. The presented protocol is simple and takes only half a day to execute, including the reduction and alkylation of Cys residues, deuterium labeling of isoAsp, hydrazine treatment, and protein digestion, as shown in Scheme S1. Importantly, our experiments did not observe any deuterium labeling occurring for the Asp residues. The remarkable selectivity of this labeling reaction for isoAsp suggests that the deuterium-labeling conditions employed favor the oxazolone formation of isoAsp while preventing the formation of the succinimide intermediate not only from isoAsp but also from Asp, as depicted in Figure S9.
The inherent strengths of this method are (1) capable of determining unambiguously the sites of isoAsp in proteins by comparing the differences in precursor and product ion masses of the peptides from proteins reacted in H2O and D2O, (2) unlike PIMT-mediated labeling approaches, labeled Asp is not produced from isoAsp during the labeling reaction and is capable of labeling both l- and d-isoAsp, making the quantitative estimation of isoAsp formation straightforward and accurate, and (3) in addition to isoAsp, isoGlu and the C-terminal amino acids of proteins can be determined.
This method would also be useful when combined with techniques based on LC separation, ion mobility separation, and diagnostic fragment ion analysis. The validation of results using these techniques often requires the synthesis of possible peptide isomers. The use of our method with these techniques would simplify the verification step by eliminating the preparation of possible peptide isomers, thereby further enhancing the utility of those techniques.
As we have demonstrated, this method can be applied to simple protein samples, such as isolated proteins or simple protein mixtures, without encountering difficulties in data interpretation. However, detecting a 1 Da mass shift solely through a database search poses a considerable challenge, particularly when dealing with peptides exceeding 2000 Da in size, as M + 1 peaks tend to be higher than their monoisotopic peaks. Applying this method to complex protein mixtures, especially when the protein components are not known, adds an additional layer of difficulty. Nevertheless, a notable advantage of this method lies in its versatility, allowing the reaction to take place in both H2O and D2O solvents. As demonstrated, the 1 Da mass shift can be readily identified by comparing the mass spectra from both samples. The comparison of MS/MS spectra is also feasible. Currently, a computational tool for comparing mass spectrometry data and detecting deuterium-incorporated peptides is lacking. The development of such a tool would significantly enhance the method’s applicability to complex protein samples.
Acknowledgments
High-resolution MS data were acquired by the Proteomics and Metabolomics Core at the Lerner Research Institute of the Cleveland Clinic Foundation. We thank Belinda Willard and Ling Li for acquiring MS data. The mass spectrometer was purchased via an NIH-shared instrument grant, S10 OD023436. The National Institutes of Health partly supported this study to M.M. (RF1 AG066578). Part of this study was also supported by the Ministry of Education, Culture, Sports, Science, and Technology of Japan to T.N. (17H05130, 19H04525, 22K00986, and 23H03921).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.3c05194.
Amino acid residues susceptible to acetylation; MS and MS/MS spectra of L-isoD-DSIP, D-isoD-DSIP, and DSIP reacted in D2O or H2O; workflow of deuterium labeling of isoAsp in proteins; further LC–MS/MS data for isoAsp sites in rituximab; LC–MS/MS data for the C-terminal peptides of rituximab’s light and heavy chain; and the mechanism of the selective isoAsp deuterium labeling (PDF)
Author Contributions
M.M. conceived and designed the experiments, performed the experiments, analyzed and interpreted the data, and wrote the first draft. E.K. performed the experiments, analyzed the data, and reviewed and edited the manuscript. K.N. performed the experiments, analyzed the data, and reviewed and edited the manuscript. T.N. conceived and designed the experiments and reviewed and edited the manuscript. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Geiger T.; Clarke S. Deamidation, isomerization, and racemization at asparaginyl and aspartyl residues in peptides. Succinimide-linked reactions that contribute to protein degradation. J. Biol. Chem. 1987, 262, 785–794. 10.1016/S0021-9258(19)75855-4. [DOI] [PubMed] [Google Scholar]
- Stephenson R. C.; Clarke S. Succinimide Formation from Aspartyl and Asparaginyl Peptides as a Model for the Spontaneous Degradation of Proteins. J. Biol. Chem. 1989, 264, 6164–6170. 10.1016/S0021-9258(18)83327-0. [DOI] [PubMed] [Google Scholar]
- Roher A. E.; Lowenson J. D.; Clarke S.; Wolkow C.; Wang R.; Cotter R. J.; Reardon I. M.; Zurcher-Neely H. A.; Heinrikson R. L.; Ball M. J.; Greenberg B. D. Structural alterations in the peptide backbone of beta-amyloid core protein may account for its deposition and stability in Alzheimer’s disease. J. Biol. Chem. 1993, 268, 3072–3083. 10.1016/s0021-9258(18)53661-9. [DOI] [PubMed] [Google Scholar]
- Watanabe A.; Takio K.; Ihara Y. Deamidation and Isoaspartate Formation in Smeared Tau in Paired Helical Filaments. J. Biol. Chem. 1999, 274, 7368–7378. 10.1074/jbc.274.11.7368. [DOI] [PubMed] [Google Scholar]
- Nowak C.; Cheung J. M.; M Dellatore S.; Katiyar A.; Bhat R.; Sun J.; Ponniah G.; Neill A.; Mason B.; Beck A.; Liu H. Forced degradation of recombinant monoclonal antibodies: A practical guide. mAbs 2017, 9, 1217–1230. 10.1080/19420862.2017.1368602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins N.; Murphy L.; Tyther R. Post-translational Modifications of Recombinant Proteins: Significance for Biopharmaceuticals. Mol. Biotechnol. 2008, 39, 113–118. 10.1007/s12033-008-9049-4. [DOI] [PubMed] [Google Scholar]
- Gibson K.; Cooper-Shepherd D. A.; Pallister E.; Inman S. E.; Jackson S. E.; Lindo V. Toward Rapid Aspartic Acid Isomer Localization in Therapeutic Peptides Using Cyclic Ion Mobility Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2022, 33, 1204–1212. 10.1021/jasms.2c00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler K. E.; Dodds J. N.; Flick T.; Campuzano I. D. G.; Baker E. S. High-Resolution Demultiplexing (HRdm) Ion Mobility Spectrometry–Mass Spectrometry for Aspartic and Isoaspartic Acid Determination and Screening. Anal. Chem. 2022, 94, 6191–6199. 10.1021/acs.analchem.1c05533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomczyk N.; Giles K.; Richardson K.; Ujma J.; Palmer M.; Nielsen P. K.; Haselmann K. F. Mapping Isomeric Peptides Derived from Biopharmaceuticals Using High-Resolution Ion Mobility Mass Spectrometry. Anal. Chem. 2021, 93, 16379–16384. 10.1021/acs.analchem.1c02834. [DOI] [PubMed] [Google Scholar]
- Wu H. T.; Julian R. R. Two-dimensional identification and localization of isomers in crystallin peptides using TWIM-MS. Analyst 2020, 145, 5232–5241. 10.1039/D0AN01036G. [DOI] [PubMed] [Google Scholar]
- Dick L. W.; Qiu D.; Cheng K. C. Identification and measurement of isoaspartic acid formation in the complementarity determining region of a fully human monoclonal antibody. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2009, 877, 3841–3849. 10.1016/j.jchromb.2009.09.031. [DOI] [PubMed] [Google Scholar]
- Ni W.; Dai S.; Karger B. L.; Zhou Z. S. Analysis of Isoaspartic Acid by Selective Proteolysis with Asp-N and Electron Transfer Dissociation Mass Spectrometry. Anal. Chem. 2010, 82, 7485–7491. 10.1021/ac101806e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeGraan-Weber N.; Zhang J.; Reilly J. P. Distinguishing Aspartic and Isoaspartic Acids in Peptides by Several Mass Spectrometric Fragmentation Methods. J. Am. Soc. Mass Spectrom. 2016, 27, 2041–2053. 10.1007/s13361-016-1487-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sargaeva N. P.; Lin C.; O’Connor P. B. Identification of Aspartic and Isoaspartic Acid Residues in Amyloid β Peptides, Including Aβ1–42, Using Electron–Ion Reactions. Anal. Chem. 2009, 81, 9778–9786. 10.1021/ac901677t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashyal A.; Hui J. O.; Flick T.; Dykstra A. B.; Zhang Q.; Campuzano I. D. G.; Brodbelt J. S. Differentiation of Aspartic and Isoaspartic Acid Using 193 nm Ultraviolet Photodissociation Mass Spectrometry. Anal. Chem. 2023, 95, 11510–11517. 10.1021/acs.analchem.3c02025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekov S. I.; Ivanov D. G.; Bugrova A. E.; Indeykina M. I.; Zakharova N. V.; Popov I. A.; Kononikhin A. S.; Kozin S. A.; Makarov A. A.; Nikolaev E. N. Evaluation of MALDI-TOF/TOF Mass Spectrometry Approach for Quantitative Determination of Aspartate Residue Isomerization in the Amyloid-β Peptide. J. Am. Soc. Mass Spectrom. 2019, 30, 1325–1329. 10.1007/s13361-019-02199-2. [DOI] [PubMed] [Google Scholar]
- Hui J. O.; Flick T.; Loo J. A.; Campuzano I. D. G. Unequivocal Identification of Aspartic Acid and isoAspartic Acid by MALDI-TOF/TOF: From Peptide Standards to a Therapeutic Antibody. J. Am. Soc. Mass Spectrom. 2021, 32, 1901–1909. 10.1021/jasms.0c00370. [DOI] [PubMed] [Google Scholar]
- Silzel J. W.; Lambeth T. R.; Julian R. R. PIMT-Mediated Labeling of l-Isoaspartic Acid with Tris Facilitates Identification of Isomerization Sites in Long-Lived Proteins. J. Am. Soc. Mass Spectrom. 2022, 33, 548–556. 10.1021/jasms.1c00355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M.; Cheetham J.; Cauchon N.; Ostovic J.; Ni W.; Ren D.; Zhou Z. S. Protein Isoaspartate Methyltransferase-Mediated 18O-Labeling of Isoaspartic Acid for Mass Spectrometry Analysis. Anal. Chem. 2012, 84, 1056–1062. 10.1021/ac202652z. [DOI] [PubMed] [Google Scholar]
- Wang S.; Kaltashov I. A. An 18O-Labeling Assisted LC/MS Method for Assignment of Aspartyl/Isoaspartyl Products from Asn Deamidation and Asp Isomerization in Proteins. Anal. Chem. 2013, 85 (13), 6446–6452. 10.1021/ac400984r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao G.; Bondarenko P. V.; Jacob J.; Chu G. C.; Chelius D. 18O Labeling Method for Identification and Quantification of Succinimide in Proteins. Anal. Chem. 2007, 79, 2714–2721. 10.1021/ac0617870. [DOI] [PubMed] [Google Scholar]
- Alfaro J. F.; Gillies L. A.; Sun H. G.; Dai S.; Zang T.; Klaene J. J.; Kim B. J.; Lowenson J. D.; Clarke S. G.; Karger B. L.; Zhou Z. S. Chemo-Enzymatic Detection of Protein Isoaspartate Using Protein Isoaspartate Methyltransferase and Hydrazine Trapping. Anal. Chem. 2008, 80, 3882–3889. 10.1021/ac800251q. [DOI] [PubMed] [Google Scholar]
- Klaene J. J.; Ni W.; Alfaro J. F.; Zhou Z. S. Detection and Quantitation of Succinimide in Intact Protein via Hydrazine Trapping and Chemical Derivatization. J. Pharm. Sci. 2014, 103, 3033–3042. 10.1002/jps.24074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray E. D.; Clarke S. Synthetic peptide substrates for the erythrocyte protein carboxyl methyltransferase. Detection of a new site of methylation at isomerized L-aspartyl residues. J. Biol. Chem. 1984, 259, 10722–10732. 10.1016/S0021-9258(18)90571-5. [DOI] [PubMed] [Google Scholar]
- Mcfadden P. N.; Clarke S. Methylation at D-aspartyl residues in erythrocytes: possible step in the repair of aged membrane proteins. Proc. Natl. Acad. Sci. U.S.A. 1982, 79, 2460–2464. 10.1073/pnas.79.8.2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman M.; McGahren W. J. Mechanistic studies of peptide oxazolone racemization. Tetrahedron 1967, 23, 2031–2050. 10.1016/0040-4020(67)80037-1. [DOI] [PubMed] [Google Scholar]
- Goodman M.; Levine L. Peptide Synthesis via Active Esters. IV. Racemization and Ring-Opening Reactions of Opitcally Active Oxazolones. J. Am. Chem. Soc. 1964, 86, 2918–2922. 10.1021/ja01068a030. [DOI] [Google Scholar]
- Neuberger A. Stereochemistry of Amino Acids. Adv. Protein Chem. 1948, 4, 297–383. 10.1016/S0065-3233(08)60009-1. [DOI] [PubMed] [Google Scholar]
- Matsuo H.; Fujimoto Y.; Tatsuno T. A novel method for the determination of C-terminal amino acid in polypeptides by selective tritium labelling. Biochem. Biophys. Res. Commun. 1966, 22, 69–74. 10.1016/0006-291X(66)90604-8. [DOI] [PubMed] [Google Scholar]
- Matsuo H.; Fujimoto Y.; Tatsuno T. A novel approach to the C-terminal determination of peptides: selective 2H- and 3H-labelling reaction of C-terminal amino acids through oxazolone. Tetrahedron Lett. 1965, 6, 3465–3457. 10.1016/S0040-4039(01)89329-1. [DOI] [PubMed] [Google Scholar]
- Nakazawa T.; Yamaguchi M.; Nishida K.; Kuyama H.; Obama T.; Ando E.; Okamura T. A.; Ueyama N.; Tanaka K.; Norioka S. Enhanced responses in matrix-assisted laser desorption/ionization mass spectrometry of peptides derivatized with arginine via a C-terminal oxazolone. Rapid Commun. Mass Spectrom. 2004, 18, 799–807. 10.1002/rcm.1409. [DOI] [PubMed] [Google Scholar]
- Nakajima C.; Kuyama H.; Nakazawa T.; Nishimura O. C-Terminal sequencing of protein by MALDI mass spectrometry through the specific derivatization of the α-carboxyl group with 3-aminopropyltris-(2,4,6-trimethoxyphenyl)phosphonium bromide. Anal. Bioanal. Chem. 2012, 404, 125–132. 10.1007/s00216-012-6093-5. [DOI] [PubMed] [Google Scholar]
- Matsuo H.; Narita K.. Protein Sequence Determination; Needleman S. B., Ed.; Springer-Verlag: Berlin—Heidelberg—New York, 1975; pp 104–109. [Google Scholar]
- Fersht A. R.; Jencks W. P. Acetylpyridinium ion intermediate in pyridine-catalyzed hydrolysis and acyl transfer reactions of acetic anhydride. Observation, kinetics, structure-reactivity correlations, and effects of concentrated salt solutions. J. Am. Chem. Soc. 1970, 92, 5432–5442. 10.1021/ja00721a023. [DOI] [Google Scholar]
- Rehder D. S.; Chelius D.; McAuley A.; Dillon T. M.; Xiao G.; Crouse-Zeineddini J.; Vardanyan L.; Perico N.; Mukku V.; Brems D. N.; Matsumura M.; Bondarenko P. V. Isomerization of a Single Aspartyl Residue of Anti-Epidermal Growth Factor Receptor Immunoglobulin γ2 Antibody Highlights the Role Avidity Plays in Antibody Activity. Biochemistry 2008, 47, 2518–2530. 10.1021/bi7018223. [DOI] [PubMed] [Google Scholar]
- Tomizawa H.; Yamada H.; Ueda T.; Imoto T. Isolation and Characterization of 101-Succinimide Lysozyme That Possesses the Cyclic Imide at Asp101-Gly102. Biochemistry 1994, 33, 8770–8774. 10.1021/bi00195a019. [DOI] [PubMed] [Google Scholar]
- Shalitin Y.; Bernhard S. A. Cooperative Effects of Functional Groups in Peptides. II. Elimination Reactions in Aspartyl-(O-acyl)-serine Derivatives1. J. Am. Chem. Soc. 1966, 88, 4711–4721. 10.1021/ja00972a035. [DOI] [PubMed] [Google Scholar]
- Riggs D. L.; Silzel J. W.; Lyon Y. A.; Kang A. S.; Julian R. R. Analysis of Glutamine Deamidation: Products, Pathways, and Kinetics. Anal. Chem. 2019, 91, 13032–13038. 10.1021/acs.analchem.9b03127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capasso S.; Mazzarella L.; Sica F.; Zagari A. First evidence of spontaneous deamidation of glutamine residue via cyclic imide to α- and γ-glutamic residue under physiological conditions. J. Chem. Soc. Chem. Commun. 1991, 1667–1668. 10.1039/C39910001667. [DOI] [Google Scholar]
- Robinson N. E.; Robinson Z. W.; Robinson B. R.; Robinson A. L.; Robinson J. A.; Robinson M. L.; Robinson A. B. Structure–dependent nonenzymatic deamidation of glutaminyl and asparaginyl pentapeptides. J. Pept. Res. 2004, 63, 426–436. 10.1111/j.1399-3011.2004.00151.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.